
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Trop. Dis., 11 March 2025
Sec. Neglected Tropical Diseases
Volume 6 - 2025 | https://doi.org/10.3389/fitd.2025.1450908
This article is part of the Research TopicFrontiers in Tropical Diseases Webinar SeriesView all 3 articles
 Mebratu Tamir1*
Mebratu Tamir1* Bisrat Birke Teketelew2
Bisrat Birke Teketelew2 Dereje Mengesha Berta2
Dereje Mengesha Berta2 Abiy Ayele Angelo3Amare Mekuanint Terekegne4
Abiy Ayele Angelo3Amare Mekuanint Terekegne4 Negesse Cherie5Gebeyaw Getnet Mekonnen1Aberham Abere1
Negesse Cherie5Gebeyaw Getnet Mekonnen1Aberham Abere1 Tegegne Eshetu1
Tegegne Eshetu1Leishmaniasis are a group of neglected tropical vector-borne diseases caused by an obligate intracellular protozoan parasite of the genus Leishmania. Currently, standard chemotherapy has challenges due to its cytotoxicity, cost, painful route of administration, long treatment duration, resultant partial efficacy, and high risk of resistance. To overcome this issue, new intervention approaches have been formulated to treat leishmaniasis. Host-directed immunotherapy is a novel approach that involves the adoptive transfer of host-derived biomolecules to enhance the natural power of protective cellular immunity. This restores the function of effector cells, enabling them to clear intracellular amastigotes and leads to the recovery of patients from infections. The advantages of this modality over routine treatment include less cytotoxicity, short hospitalization, affordability, and better efficacy for drug-resistant parasite strains. Several studies have reported better efficacy of this treatment model for drug-resistant Leishmania species. However, current knowledge and evidence are highly insufficient to implement this agent to treat any form of leishmaniasis. This review aims to show the efficacy of this immunotherapeutic agent against leishmaniasis. The discussion has focused on major pro-inflammatory cytokines (interferon-gamma, interleukin-12, and granulocyte-macrophage colony-stimulating factors), immune cells (dendritic and mesenchymal stem cells), and monoclonal-antibodies (anti-interleukin-10, anti-interleukin-4, and immune checkpoint inhibitory molecules). Our finding shows that this treatment approach has the potential to be a successful treatment and improve clinical outcomes by reducing the adverse effects of routine therapy. This suggests the future deployment of this treatment modality as an alternative strategy. However, it needs extensive pre-clinical trials using local animal models that reflect typical host immunological profiles against leishmaniasis in order to select the most protective candidate agents.
Leishmaniasis are a group of neglected tropical diseases caused by obligate intracellular protozoan parasites of the genus Leishmania (1). Over 20 different Leishmania species have been reported to be pathogenic for humans (2, 3). These are transmitted to the host through the bite of infected female sand-flies of genera Lutzomyia and Phelbotomus in both developing and developed countries (4, 5). Leishmaniasis remains a serious major public health problem, resulting in substantial morbidity and mortality (1, 3). Globally, it was estimated to affect over 12 million people with an annual incidence of 2 to 2.5 million new cases, resulting in 774,000 disability-adjusted life years (DALYs), 60,000 deaths, and 350 million people at risk (1, 6). Of this burden, 90% of cases disproportionally occur in the Indian subcontinent, Latin America, and Africa. Ethiopia is one of the African countries highly affected by leishmaniasis where L. donovani and L. aethiopica are etiological agents for all clinical forms of the disease (7, 8). Environmental and behavioral aspects, low socioeconomic conditions, population mobility, an increase in HIV co-infection, a lack of vaccines, and the expansion of resistance to current chemotherapy are some of the factors behind the persistent rise in leishmaniasis in endemic regions (9).
Depending on the pathogenic parasite species and the immune status of the infected host, the clinical manifestations of leishmaniasis range from self-healing localized cutaneous (LCL) and diffused cutaneous leishmaniasis (DCL) to severe deep visceral forms (10). Cutaneous leishmaniasis (CL) is caused by L. tropica complex species in Europe, Asia, and Africa, and L. mexicana complex in the Americas and the Caribbean. It is self-resolving with long-life immunity and features ulcerative skin lesions on the face, hand, and feet (3, 7). Nevertheless, among immunocompromised individuals, including those with human immune deficiency virus (HIV) and patients with defective cellular immune responses, CL is not self-healing (11). Since it can cause long-lasting chronic and diffuse forms. A mucocutaneous leishmaniasis (MCL) is a severe form of LCL infection that affects the lips and nostrils. This is usually provoked by L. brasilliensis, L. panamensis, or L. guyanensis agents (12). This spectrum is associated with high morbidity and is potentially life-threatening if untreated (13). Remarkably, current genomic studies findings have documented the emergence of L. donovani, a causal agent for CL in different regions including Ethiopia (14, 15).
Visceral leishmaniasis (VL) is the most dangerous and damaging form of infection that attacks the reticuloendothelial system and is caused by the L. donovani complex. Hepatosplenomegaly, weight loss, fever, and pancytopenia are the clinical features of VL, and it is 100% lethal if left untreated (16, 17). Furthermore, 10% to 20% of VL patients still die while receiving standard chemotherapy (5). It is clear that the outcome of leishmaniasis pathology is linked to an imbalance in the T helper-1/T helper 2 (Th1/Th2) immunological makeup (11). Thus, the presence of pro-inflammatory cytokines is protective, and the key to treatment and recovery. Leishmania involves heteroxenic life stages of the metacyclic promastigote stage in the sand fly’s midgut and ovoid spherical intracellular amastigote forms in the mammalian host (1).
In humans, a lifecycle begins when an infective promastigote is injected into the skin by female sand flies during a blood meal. Professional phagocytes including neutrophils, dendritic cells (DCs), and macrophages (MQs) ingest the promastigote and transform it into a round, non-flagellated amastigote. In these cells, amastigotes survive and multiply by binary fission, rupture of infected cells, and are released into the circulation to infect new phagocytic cells. The sand flies feed on the infected blood, taking amastigotes, and the lifecycle continues in a circular fashion (7). Moreover, this intracellular amastigote is a target for host immunity and different therapeutic models for disease management.
Once infective metacyclic promastigotes are inoculated into human skin, the parasite needs rapid host cell entry where they differentiate into amastigotes which survive and propagate in the harsh/acidic cell environment (18). The parasites are challenged by complement proteins prior to their cell entry into the infection site. These proteins are part of innate immunity that kills promastigotes by classical activation, and the amastigotes by alternative pathways (13). Resident/recruited neutrophils, macrophages, and dendritic cells are initial innate responses that identify pathogen components via pattern recognition receptors (PRR) (19). Yet, some studies state that neutrophils cannot kill promastigotes since the parasite inhibits apoptotic signals, delaying the lifespan of cells and thereby spreading to macrophages using the Trojan horse principle (20). In contrast, MQs and DCs are key for the rapid clearance of invading parasites and initiating adaptive immunity that produces a specific response. In this, toll-like receptors (TLRs) are vital cellular receptors used to identify pathogen-associated molecular patterns (PAMPs) for downstream signal transduction and activation in the innate response (13, 21). Particularly, TLR2 and TLR4 are surface receptors in MQs that detect specific ligands of parasites from outside, while TLR3 and TLR9 recognize them from vacuoles and become activated. Activated TLRs recruit adapter proteins such as myeloid differentiation 88 (MyD88) to stimulate Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription factors which migrate into the nucleus and initiate DNA transcription for the synthesis of pro-inflammatory cytokines/chemokines and toxic free radicals that kill amastigotes (22, 23).
Effective anti-leishmanial immunity requires the synergetic interaction of immune cells, cytokine microenvironment, and chemokines (24). Pro-inflammatory cytokines produced by DCs (IL-12) prime cluster of differentiation 4 (CD4+) naive T-cells to differentiate into the Th1 phenotype, the main source of interferon-gamma (IFN-γ) that is used for MQ stimulation/activation (5). The activation of MQs by endogenously produced IFN-γ upregulates the inducible nitric oxide synthetase and mediates nitric/oxygen radical-dependent killing of intracellular amastigotes (17, 20). Among immunocompetent hosts, natural killer cells, a Th1 cytokine (IFN-γ, IL-12, or TNF-α), DCs, and MQs are major sources of protective immunity against leishmaniasis (25). In contrast, disease progression is linked with a Th2 cytokine milieu that is primarily featured by the presence of interleukin-4 (IL-4), interleukin-10 (IL-10), IL-13, and TGF-β (5, 26). Understanding the complicated involvement of host immune molecules with critical activity for leishmaniasis prevention is an important method for establishing exogenous host-directed immunotherapy.
In resistant hosts, classically activated MQs are pioneer cells that kill intracellular parasites by secretion of toxic radicals and the synthesis of pro-inflammatory cytokines (5, 25). However, Leishmania evade and modulate the physiological functions of MQs by glycoprotein 63 and lipophosphoglycan effectors expressed on the parasite’s surface. These are released into the cytoplasm, altering the most desirable host cell signaling pathways (27). The Janus tyrosine kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway of MQs is modulated by Leishmania. This downregulates the synthesis of protective cytokines, chemokines, and toxic nitric (NOS-)/oxygen radicals. The production of Th2 (IL-10, IL-4, IL-5, and TGF-β) become highly upregulated during this time (5, 28).
Of the Th2 cytokines, IL-10 and IL-4 downregulate human protective immune arms (Th1) by blocking the activity of IFN-γ, deactivating MQs, inhibiting the proliferation of lymphocytes, reducing expression of major histocompatibility complex II (MHC II) molecules, and hindering the biogenesis and maturation of phagolysosome, and oxidative stress guarantees host immunosuppression, and parasite survival, proliferation, and disease progression (22, 29). Furthermore, immunosuppression is the main cause of poor treatment outcomes and frequent disease relapses among patients treated with conventional chemotherapy (5). Thus, immunomodulation therapy that restores the ordinary function of MQ cells and neutralizes immunosuppressive Th2 cytokines using monoclonal antibodies (mAbs) is now a growing therapeutic approach for Leishmania infection (4).
Pentavalent antimonial, miltefosine, deoxycholate amphotericin B, paromomycin, and liposomal amphotericin B (AmBisome) are the currently available first and second-line anti-leishmanial drugs. The route of administration for these drugs is painful systemic intravenous and intramuscular infusion except for miltefosine (30–32). The activation of impaired effector cells to produce parasiticidal toxic radicals and cytokines that enhance host defense is one of their mechanisms (11, 33). However, cytotoxicity, cost, painful route of administration with prolonged treatment course, and poor patient compliance are challenges in chemotherapy (5, 34). Additionally, their efficacy is affected by variations in parasite species, drug pharmacokinetic features, patient immune/nutritional status, and regional features (9, 35). The evolution of drug-resistant parasite strains against pentavalent antimony is one of the public health threats in endemic regions of developing countries. According to an Indian report, nearly 60% to 90% of antimony-treated patients were found to be unresponsive, which pushed them to decide on the utilization of second-line drugs (36–38). Chemotherapy cannot provide a complete cure in zoonotic cases, which might be the main obstacle in the control of leishmaniasis since this disease is spread by the sand-fly (5).
To circumvent all the challenges of conventional therapy, there was an extensive clinical trial to design vaccine therapy using mouse, dog, and primate animal models. However, there is a paucity of effective licensed vaccines for human leishmaniasis partly due to the complexity of the host-parasite interaction and/or human’s delicate immune modulation system (37). Therefore, a change in drug policy is required to escape the impact of drug and immune-resistant strain infections (20). At this point, the immunomodulatory treatment approach might offer vital solutions with the prospect of breaking parasite transmission and the risk of drug resistance (4, 39). Immunopathology treatment using immune factors has been suggested as an appropriate therapeutic system in chronic and acute infectious diseases (12, 34). Some studies have focused on applying major cytokines, monoclonal antibodies, immune cells, and inhibitory immune checkpoints for the targeted treatment of leishmaniasis to restore the functional activity of impaired MQs and other immune cells (4, 34). These immunomodulation treatment approaches are collectively called immunotherapy (40).
The term immunotherapy, often known as biotherapy, refers to the use of biological molecules that modulate immune responses to achieve preventative/therapeutic goals (41). This term can be used to describe pathogen-directed/vaccine immunotherapy that delivers parasite antigens to induce cell-mediated immunity, known as active immunotherapy. Additionally, the term can be used for other types of therapy that comprise passive or adoptive transfer of host-derived biomolecules such as cytokines, immune cells, and monoclonal antibodies to enhance the natural power of immunity in leishmaniasis, called host-directed immunotherapy (HDT) (2, 19). This therapeutic model is based on the basic concept that the immune system can protect against different infections but disease occurs when there is a failure of or suboptimal and excessive immune reactions. This might be remedied by using a suitable immunomodulation intervention that redirects the effector function of cells and/or reduces an inflated response using biological modifiers (20).
For two decades, immunotherapy with and without chemotherapy has been developed and applied as an additional approach for the treatment of leishmaniasis (4, 42). Different reviews of this therapy have shown its ability to accelerate the targeted specific effector cell response to the parasiticidal state and its efficacy in the clearance of parasites, including drug-resistant strains (42). However, most of the reviews of immunotherapy have focused on pathogen-directed (vaccine immunotherapy) with very limited information regarding HDT against leishmaniasis. Moreover, the available reviews of HDT are only a part of either cellular therapy/cytokine or monoclonal antibody therapies. Thus, review efforts that deliver comprehensive data on the therapeutic potential of HDT against leishmaniasis are required. This review aimed to provide comprehensive evidence of the therapeutic potential of major HDT candidate agents (cytokine, cellular, monoclonal antibody, and immune checkpoint inhibitor) therapy against leishmaniasis from different pre-clinical, experimental animal model and/or human-based studies.
Host-directed immunotherapy refers to a novel treatment modality that uses host-derived biomolecules for the treatment of leishmaniasis (43). It is a broad-spectrum treatment that could treat infectious diseases with high mortality rates (12). The historical background of HDT dates back to a large early clinical trial in influenza, hepatitis, HIV, systemic respiratory syndrome, and tuberculosis with promising efficacy (44, 45). HDT remains a superior treatment modality to treat several immunopathological conditions including allergy, autoimmune disease, cancer, hepatitis, and leishmaniasis (5).
HDT enhances the natural power of the innate and adaptive immune system, restoring the function of covert effector cells, and reducing the immunopathology, leading to host recovery from infection (5). It contains different multi-potent host-derived immunostimulatory or neutralizing agents, namely, cytokines, immune cells, mAbs, and immune checkpoint inhibitors. These host-derived products have ultimately been found to augment host cellular defense capacity and improve clinical outcomes in patients with leishmaniasis measured by reduced parasite load, morbidity, mortality rate, and recovery. This makes HDT the suggested practical alternative for intervention in leishmaniasis with and without routine chemotherapy (4, 43). The general mechanism of action of HDT against Leishmania parasites involves the re-direction of pro-active effector or memory responses, activation of autophagy/apoptosis, the induction of oxidative/nitrosative stress, and enhancement of antigen processing/presentation by antigen-presenting cells (APCs) which triggers an efficient clearance of intracellular amastigotes (43).
Additionally, HDT works by shifting non-protective Th2 immune arms into the protective Th1 phenotype, ameliorating the pathological pathway of leishmaniasis (12). Thus, major mAbs, immune cells, cytokines, and immune checkpoint inhibitors that trigger the restoration of diverted effector cells to the parasiticidal state seem promising candidates for the treatment of leishmaniasis (2). The advantages of HDT over conventional chemotherapy include less cytotoxicity, more therapeutic success, short hospitalization, affordability, lower teratogenicity risk, avoidance of drug-resistant strains, and species-dependent efficacy (12, 39). The next section will focus on potential HDT candidates and their treatment efficacy in different forms of leishmaniasis in animal and/or human models.
Following the identification of the normal function of Th1/Th2 cytokines, mAbs, and different immune cells in the natural course of leishmaniasis, several immunotherapeutic approaches have been implicated in the treatment of the infection (4). The enhancement of Th1 immune responses and neutralization of Th2 cytokines are the main mechanisms of action and were suggested as a promising alternative treatment for leishmaniasis (1, 34). This review addresses the therapeutic effect of selected Th1 cytokines, mAbs, innate immune cells, and immune checkpoint inhibitory molecules against leishmaniasis from animal models and human studies (43).
Cytokines are messenger molecules that mediate intercellular communication in the immune system and are produced by different cell types. They have pleiotropic and regulatory effects involving host defense processes (46). In leishmaniasis, cytokines are critical decision-makers that facilitate disease progression or host resistance and they are key targets for diagnosis and/or immunotherapy (4). The history of cytokine therapy dates back to an experiment by Murray et al. in which the effect of anti-IL10 mAbs for the treatment of leishmaniasis was explored (47). Th1 cytokines (IFN-γ and IL-12) are protective immune arms for leishmaniasis, while Th2/T-reg cytokines (IL-10, IL-4,IL-5, and IL-13) facilitate the parasite’s progression by downregulating the Th1 response (4). Thus, in the center of cytokine immunotherapy, IFN-γ and IL-12 have received great attention as targets for host-directed immunotherapy (1). Thus, the therapeutic potential of both pro-inflammatory cytokines is reviewed in the next section.
IFN-γ is a homodimer glycoprotein consisting of two subunits each approximately 21 to 24 kDa. Of the several anti-leishmanial cytokines, IFN-γ is the most potent cytokine in host protection and plays a key role in macrophage activation to a leishmanicidal state (48–50). It is a monocyte-activating factor that augments the release of oxygen radicals, secretion of pro-inflammatory cytokines, expression of major histocompatibility class II (MHC class-II), and antigen presentation (49, 51). Moreover, IFN-γ blocks the production of IL-10, which shifts the Th1 to the Th2 response (52). The main cellular sources of IFN-γ production are activated CD4+ T cells, CD8+ T-cells, and natural killer (NK) cells after IL-12 signaling (48). With this activation and blockage role against parasites, IFN-γ has been suggested to be a promising candidate for the treatment of leishmaniasis.
A study elsewhere used lymphokines (IFN-γ) collected from murine spleen cell culture supernatant encapsulated in liposomes for the treatment of VL. The treated mice had a reduction of parasite burden in their livers compared with the untreated control, indicating the protective effect of lymphokines against leishmaniasis. Another study treated VL infection using recombinant Th1 cytokines (r IFN-γ and rIL-2), after challenging mice with L. donovani. The therapeutic efficacy was observed with a substantial reduction of parasite burden among the treated mice (34). In another study on the treatment of VL in C57BL/6 and Balb/c mice, rIFN-γ and muramyl tripeptide (MTP-PE) were administered by several intravenous injections in varying doses, and the treated mice had a decreased parasite burden in the spleen (53). Furthermore, a combination therapy of recombinant human IFN-γ and pentavalent antimonials has been reported to provide better parasitological and clinical treatment compared to drug monotherapy in VL patients from Brazil, Kenya, and India. This indicates a short course of IFN-γ can have adequate synergetic effects to activate/stimulate macrophages, thereby accelerating the immune-induced efficacy of drugs (4).
Furthermore, 13 VL patients were enrolled in an open-label trial in an Indian hospital to examine the treatment efficacy of cytokines against L. donovani infection. These patients previously failed antimonial and pentamidine treatments and had repeatedly relapsed. For these patients, a combination of IFN -γ plus antimony was administrated, and 69% of them were cured of the VL infection (54). This suggests the beneficial effect of IFN-γ for patients with standard treatment and primarily reflects its capacity to activate monocytes and macrophages to kill intracellular L. donovani. IFN- γ therapy influences immunopathogenic mechanisms by promoting a Th1 response and by inhibiting a suppressive Th2-associated response (54).Similarly, IFN-γ was found to be effective for the treatment of cutaneous leishmaniasis in certain trials (12). Moreover, a study on L. donovani-infected mice continuously infused them with IFN- γ via intraperitoneal routes. The result showed that 50% of the treated mice had a reduction in liver parasite burden, indicating that the effect of IFN- γ on MQ activation exploits antimicrobial effects with or without conventional chemotherapy (55) (Table 1).
IL-12 is a pro-inflammatory heterodimer cytokine with two subunits (35 and 40 kDa) connected by a disulfide bond and it is primarily produced by activated macrophages and DCs. The production of IL-12 is often linked to protective immunity against leishmaniasis and it bridges innate and adaptive immune responses (56, 57). It promotes the Th1 response and synthesis of IFN- γ and other lymphokines from NK and T cells, and increases nitric oxide synthetase 2(NOS2) expression and NO generation. This mediates T-cell proliferation, upregulating the leshimanicidal activity of macrophages to eliminate amastigotes. Moreover, IL-12 limits the synthesis of IL-4 by CD4+ T cells, controlling the growth of Th2 arms and clearing leishmaniasis (58–60). As a result, it is one of the key target cytokines inhibited by Leishmania to escape immunity. Because of these hopeful in vivo protective roles of IL-12, it has been suggested as a promising candidate agent for host-direct immunotherapeutic approaches.
To prove its protective role, some experiments have removed IL-12 cytokines from L. major and L. donovani-infected hosts. The outcome was a rapid progression of disease. The addition of IL-12 to the lymphocyte culture of patients with VL restored IFN-γ synthesis and improved the cytotoxic activity of NK cells, resulting in recovery from infection (60–62). Furthermore, exogenous administration of the rIL-12 cytokine for the treatment of leishmaniasis had been found to give rise to resistance among treated susceptible mice (63), indicating its value in clinical outcomes. Similarly, treatment of L. major-infected BALB/c mice with IL-12 cytokines was reported to reduce parasite load and lesion size. This treatment is linked to the pronounced effect of IL-12 on T cell growth together with the IFN-stimulatory effects while suppressing IL-4 production. In another experiment, L. major-infected BALB/c mice were treated with mouse rIL-12 via the intraperitoneal route of injection at the first week of infection to determine its therapeutic role against leishmaniasis. The outcome revealed that 89% of BALB/c mice were cured, as measured by the reduction lesion size and there was a 1,000 to 10,000-fold decrease in parasite burden after the treatments. Furthermore, the mice were found to be resistant to subsequent infections after a 4-month follow-up (63) (Table 1).
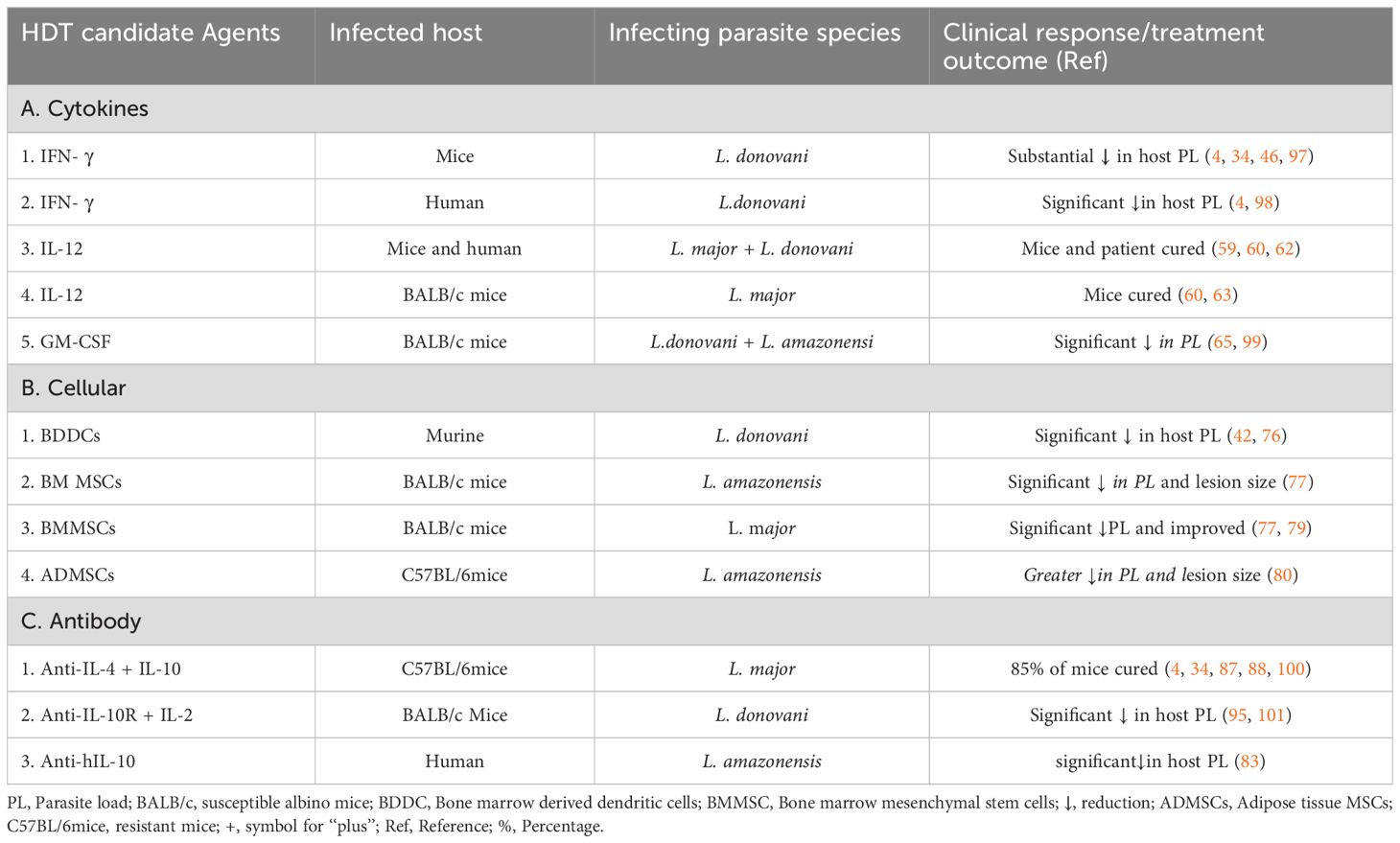
Table 1. Summary of the HDT candidate agents and post-treatment clinical response in animal models and human populations.
A granulocyte-macrophage colony-stimulating factor (GM-CSF) is a key humoral growth factor glycoprotein that stimulates the production of neutrophilic granulocytes and macrophages from bone marrow precursor cells. Additionally, it upregulates the proliferation and clonal expansion of blood monocytes and tissue macrophages (64). Interleukin 3 (IL-3) and CSF-1 are fundamental components of GM-CSF that stimulate the proliferation, activation, and chemotaxis of T lymphocytes, keratinocytes, endothelial, and professional phagocytic cells. This is a critical role in developing functional, robust innate immunity towards a range of intracellular pathogens including Leishmania, suggested GM-CSF is a candidate for immunomodulatory therapy (65, 66). A clinical trial study on GM-CSF treatment in AIDS, cancer, aplastic anemia, myelodysplastic syndromes, and patients with Leishmania was undertaken and the finding shows a significant increase in the number of myeloid cells and resultant recovery from disease after GM-CSF treatments (67–69).
Leishmania infection compromises the effector function of MQs by inhibiting cellular signaling pathways, transcription factors, and protease-dependent cleavages of host immune factors, thereby reducing the ability of MQs to secret pro-inflammatory cytokines and toxic anti-parasitic molecules and resulting in the commencement of infection (70). GM-CSF stimulates both Th1 and Th2 cell subsets and it activates macrophages to destroy Leishmania. In addition to parasiticidal activities, GM-CSF is known to promote fibrosis and tissue wound healing, which plays a key role in the recovery of lesions in patients with CL (71). Thus, administration of GM-CSF in patients with leishmaniasis appears a promising HDT agent since it enhances MQ phagocytic activity, secretion of anti-parasitic molecules, and destruction of the intracellular stage of parasites, improving patient outcomes while reducing the risk of drug resistance (40, 72). This review aims to summarize the HDT effect of GM-CSF against leishmaniasis.
In a randomized, double-blind study on patients with CL, one group received a combination of stibogluconate and GM-CSF whereas the other group received stibogluconate alone. The study demonstrated that the patients with CL who received the combination therapy had a higher proportion of recovery and quicker wound healing than those who received stibogluconate monotherapy. This suggests that GM-CSF may have a greater impact on effective antigen presentations, overall parasiticidal activity of MQ, rapid scar formation, and better immunological maturation than chemotherapy alone (72). Another experimental work was carried out on VL-infected BALB/c mice to evaluate the therapeutic role of GM-CSF against leishmaniasis. In this study, L. donovani-infected mice were treated with murine anti-GM-CSF antibodies, which may neutralize endogenous GM-CSF cytokines and change the maturation of mice immunity against infection. Thus, the study found severe disease progression and a threefold increase in liver parasite load following anti-GM-CSF medication. This validated the therapeutic role of GM-CSF against Leishmania, making it a promising HDT candidate (65) (Table 1).
Immune cell therapy is a novel modality to combat different infectious diseases and cancer (73). A dendritic cell is a network of diverse cell types that develop from hematopoietic stem cells in the bone marrow (74). It is a highly specialized APC of the immune system capable of priming naïve T cells and mounting a T cell response upon pathogen entry in the body. This is a significant cell bridging innate and adaptive immunity since it uptakes/processes antigens, matures and upregulates MHCII and co-stimulatory molecules, migrates to lymph nodes, and activates T cells to differentiate into effector Th1 cells through the synthesis of IL-12 (73, 75). For therapy, DCs act by boosting antigen-specific T-cell immunity, activating T cells, B cells, and NK cells. Furthermore, DCs inhibit the early secretion of immunosuppressive IL-10 cytokines, conversely stimulating the synthesis of Th1 cytokines. Thus, DCs are a desirable candidate for immunotherapy because of their role in the induction of memory T-cell differentiation and host protection (4, 74).
In experiments, DC-based immunotherapy combined with an antimony drug was very effective against murine VL (76). Bone marrow-derived DCs pulsed with soluble parasite antigens were given in combination with antimonial to mice infected with L. donovani, and the results showed a significant reduction in the parasite load in both liver and spleen organs (42, 76) (Table 1). Thus, the future of DC-based therapy appears promising as a prospective treatment against VL.
Mesenchymal stem cells (MSCs) are a subset of adult stem cells that can differentiate into a wide range of functional cells as well as proliferate and self-renewal. These are usually found in bone marrow, but they can also be found in the blood, dermis, cartilage, muscle, tendons, spleen, adipose tissue, thymus, tooth pulp, and embryonic tissues (77). Due to their capacity to move rapidly to the site of infections, and other characteristics of cell proliferation, multi-potent differentiation, and cytokine production/immune modulation, MSCs have been postulated to be used in cell therapy for a variety of infections including leishmaniasis (78).
In certain experiments, BALB/c mice were infected with L. major and treated with an intra-lesion injections of mesenchymal stem cells, glucantime, or glucantime + mesenchymal stem cells. The result showed that the mice that were treated with mesenchymal stem cells revealed significant regression and healing of the lesions. Furthermore, they found that a proliferation of splenocytes stimulated with soluble Leishmania antigen, the efficacy of phagocytosis in MQs of the mice treated with mesenchymal stem cells was considerably higher, and the clinical outcome of infection was improved (77).
In another experiment, bone marrow MSCs were injected intravenously and intra-lesionally into a BALB/c mice model that had been infected with L. amazonensis. The therapeutic outcomes indicated that there was no appreciable difference in lesion progression regardless of the MSC delivery method but the analysis of spleen cellular profiles revealed that only mice given intravenous MSCs had higher levels of IL-10-producing CD4+ and CD8+ T cells. This cell upregulates macrophage phagocytosis and splenocyte multiplication, inhibiting parasite duplication and lesions (77, 79).
Furthermore, adipose tissue-sourced mesenchymal stromal cells (AD-MSCs) were evaluated in vivo in C57BL/6 mice infected with L.amazonosis and two doses of AD-MSCs were injected into the jugular vein and the vein was clamped for a few seconds to avoid any loss of blood or injected cells. When compared to the untreated control, the AD-MSC treatment conferred partial protection against infection. Moreover, the same infected mice were treated with a combination of antimonial + MSCs and antimonials only. In comparison to the mice that only received antimonials, the study demonstrated that the combination therapy offered a greater reduction in lesion size and parasite burden, demonstrating the ability of AD-MSC therapy to activate mouse macrophages to produce parasiticidal effects (80). Once MSCs are injected into an infected host, they will migrate to the infection site using homing receptors, performing multiple immunological functions. Initially, MSC signals recruitment of blood monocyte to the site of infection, allowing cells to be differentiated to MQs. These phagocytic cells ingest the parasite and are activated to secret toxic oxygen and nitrogen radicals which enables destruction of intracellular pathogens and results in infection recovery. For the detailed mechanism, please refer to (81) (Table 1).
During the infection, the ability of IL-10 and IL-4 to suppress Th1 and upregulate Th2 immune responses has been established as the key component of parasite biology. The overproduction of IL-4 and IL-10 favors parasite persistence and disease progression (73). IL-10 can inhibit the function of DCs and renders MQs unresponsive for protective cell signaling. This meant that mice deficient in IL-10 cytokines were found to be highly resistant to L. donovani infection (82).Thus, a reduction in IL-10 and IL-4 cytokines by the adoptive transfer of neutralizing mAbs (anti-IL-10, anti-IL-10R, and anti-IL-4) blocked of their synthesis pathway has been suggested as key to anti-leishmaniasis immunotherapy (42).This passive adoptive transfer of mAbs induces the synthesis of protective Th1 cytokines such as IFN- 𝛾 and TNF (83). As a result, patients with VL with anergic T cells were found have their functional response restored after treatment with anti-IL10 mAbs.
In another experiment on L. major challenged Balb/c mice treated with an infusion of anti-IL-4 and anti-IL-10 antibodies, nearly 85% of the treated group were cured of Leishmania infections (34). In addition, mice were challenged and injected intraperitoneally with anti-IL-10 and anti-IL-2 monoclonal antibodies at different time points, the group which received both antibodies together had early limited growth of the parasite in the spleen and controlled disease onset. At the initial phase of VL, the IL-2 cytokine is used as an IL-10 suppressor while inducing the secretion of the Th1 (IFN-γ) active immune response.
Furthermore, L. donovani-infected wild-type mice were treated with a single dose of anti-IL-10R mAbs and daily low doses of antimonials to assess the therapeutic impact of the antibody by blocking the IL-10 site of action. This experiment led to rapid L. donovani infection recovery and showed a significantly enhanced efficacy of the drug and monoclonal antibody with an over 10-fold dose-sparing effect and the length of the treatment period was shortened (84). In another separate study, BALB/c mice infected with L. donovani received a single dose of anti-IL-10R mAbs (0.5 mg) and this resulted in a 63% liver parasite burden reduction. Furthermore, when administered at a lower dose (0.1 mg), the anti-IL-10 mAbs enhanced the effect of antimonials, which had also been given at a suboptimal dose (50 mg/kg) and nearly 72% of liver parasites were killed.
The same result was found in L. donovani-infected BALB/c mice treated with a suboptimal single dose (0.1 mg) of an anti-IL-10R mAbs and low-dose amphotericin B (2 mg/kg total dose). The combination therapy reduced the hepatic parasite load by 76%, compared with anti-IL-10R mAbs alone (85, 86). Moreover, to understand whether the therapeutic effect was due to blocking the synthesis pathway of Th2 cytokine or the cytokine itself, there have been several studies in animal models. For example, a study blocked IL-10 production through the administration of a human monoclonal antibody (anti-hIL-10) to promote a Th1 response in CL patients infected with L. amazonensis, and a patient showed a reduction in IL-10, IL-4, and TNF-α levels (83).This cytokine reduction was found to be potentially protective for localized cutaneous leishmaniasis. Another experiment on the therapeutic efficacy of anti-IL-10R was conducted in a C57BL/6 mouse model infected with L. donovani. The results indicated that blocking the IL-10 receptor can reduce the parasite burden in mice and be an alternative to chemotherapy (Table 1). Hence, the direct role of IL-10 in the pathology of VL is supported by studies showing that IL-10 blockage could enhance IFN-γ synthesis.
More recently, phase 1 trials have revealed that IL-10 blockage has an anti-parasitic effect in human VL, indicating that neutralization of IL-10 results in a notable reduction in the parasite burden in splenic aspirate cells (4). In another trial, susceptible BALB/c mice strain infected with L. major were given a single dose of anti-IL-4 antibodies to investigate the role of anti-IL-4 mAbs against infections. Within 4 days, anti-IL-4 significantly changed the in vivo cytokine expression from a Th2 to a Th1 pattern. The treatment resulted in healing responses in the mice, demonstrating that IL-4 is necessary for Th cells to differentiate into Th2 cells in BALB/c mice leishmaniasis. Furthermore, research reveals that IL-4 severely degrades the Th1 immune response (87, 88). Since Th2/Th1 differentiation occurs during the first 2 weeks of infection, anti-IL-4 needs might be more effective if given during that week of infection. In general, this therapy against immunosuppressive cytokines is a justifiable and interesting alternative treatment of leishmaniasis (Table 1).
Moreover, a phase I study of anti-IL-10 mAbs alone and in combination with AmBisome has recently been proposed for the human trial, thus leading to possible ex vivo supportive findings. This combination is estimated to induce synergistic effects that will control VL immunopathology, overcoming the threat of drug resistance and possibly achieving a chemotherapeutic dose-sparing effect that results in better efficacy (4). Notably, proving that IL-10 neutralization has therapeutic value as a proof of concept will pave the way for additional approaches aimed at inhibiting IL-10 and other immunosuppressive components.
Immune checkpoints are host cellular surface receptors with a stimulatory or inhibitory role that play a vital activity in regulating the effector function of T cells (89). For the development of functional immunity against foreign antigens, this checkpoint facilitates multidirectional cell-to-cell interactions, frequently among T cells, MQs, DCs, monocytes, and neutrophils (11). Different checkpoint receptors have been discovered with distinct roles that should be investigated in the context of intracellular infections such as Leishmania (89). For instance, the stimulatory arm of immune checkpoints triggers the inflammatory response and might lead to the destruction of the host cell, resulting in the establishment of disease (11, 17). Conversely, the use of immune checkpoint inhibitors in an HDT approach provides promising potential for the treatment of cancer and autoimmune and infectious diseases including leishmaniasis (17, 89). This treatment approach involves the downregulation of Th2 cytokine expression while inducing the activity of APCs and synthesis of defensive Th1 cytokine subsets (IFN-γ, TNF-α, and IL-12) acts to collapse intracellular amastigotes and halt disease progression (2). Of the immune checkpoints, programmed cell death protein 1 (PD-1; CD279) and its ligands, PD-L1 (B7-H1) and PD-L2 (B7-DC), are members of the B7/CD28 family and are a vital immune checkpoint axis that is frequently investigated in leishmaniasis. During the host-parasite interaction period, PD-1 to PD-L1 or PD-L2 induces phosphorylation of tyrosine residues in the PD-1 intracellular domain that recruits tyrosine phosphatases 2 (SHP2) (90). This dephosphorylation pathway produces crucial bioproducts that are used for parasite survival and proliferation, resulting in the progression of pathology (91). The subsequent effect of this signaling cascade is the inhibition and exhaustion of protective T cells (92). Thus, the neutralization and inhibition of such targeted immune checkpoint molecules using specific anti-bodies and inhibitory molecules looks to be a vital HDT modality to limit clinical disease advancement (89). Several individual experimental studies reported that the administration of antibodies that block PD-1/PD-L1 interaction in patients infected by different Leishmania species has been shown to increase the survival of CD8+ T cells, restore their function, and result in a notable reduction of parasite load after treatment (90, 93).
Additionally, a recent randomized controlled trial study that tested the therapeutic potential of anti-PD-1 and anti-PD-L1 mAbs against a non-healing L. amazonensis infection in BALB/c mice reported that the treatments significantly increased IFN-γ-producing CD4+ and CD8+ T cells, respectively. Thus, compared with infection controls, mice treated with anti-PD-1 and anti-PD-L1 demonstrated a significant reduction in lesion size and parasite load. The basic protection mechanism was established through the suppression of parasite-favorable IL-4 and TGF-β cytokine production in cells (94). This study concluded that treatment of leishmaniasis using anti-PD-1 has the potential to mobilize and build a strong T cell environment that activates MQs for intracellular parasite destruction, lowering the parasite load burden of the infected host (Table 1).
Another study in mice infected with L. donovani using cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) demonstrated that CTLA-4 blockade decreased parasite burden in both the liver and spleen and was associated with increased synthesis of IFN-γ major cytokines. Furthermore, CTLA-4 blockade has been shown to increase the efficiency of chemotherapy in L. donovani-infected mice (95, 96). Thus, these studies clearly show the therapeutic potential of immune checkpoint molecules for HDT against VL and other forms of leishmaniasis (11).
Host-directed immunotherapy has attracted attention for the treatment of leishmaniasis to save patients from frequent relapses and adverse effects of chemotherapy. In this review, the application of cytokine immunotherapy (IFN- γ, IL-12, and GM-CSF), cellular immunotherapy (dendritic cells and mesenchymal stem cells), mAbs immunotherapy (anti-IL-10 and anti-IL-4), and immune checkpoint inhibitor molecules against animal/human leishmaniasis infections were found to result in a significant reduction in parasite burden and/or complete cure of the Leishmania infection.
Treatments that enhance immune responses to fight against diseases are of significant clinical interest. While much progress in leishmaniasis treatment has been made over the past years, the field still has a limited understanding of the immune mechanism underlying human leishmaniasis. One of the major problems in translating discoveries from disease models into human treatment is the risk that potential treatment strategies will fail to give similar responses in humans as in the models (4). An additional central issue in immunotherapy is related to safety, cost, toxicity, and specificity. Since the production of some agents may be quite expensive and may cause severe sensitization/allergic reactions, this leads to low treatment efficacy. Additionally, there is a lack of clear practical precision on the adequate therapeutic dose, length of treatment duration, pharmacokinetics/dynamics, and route of administration for humans. This needs extensive prospective trial work with suitable animal models (77). Therefore, the problems and side effects associated with the use of cytokine/cellular therapy have to be addressed properly before its clinical application.
This review shows that HDT with pro-inflammatory cytokines, immune cells, and monoclonal antibodies has been found to show significant therapeutic potential, resulting in substantial parasite reduction and/or complete cure of all forms of leishmaniasis. In particular, cytokine and monoclonal antibody immunotherapy have promising efficacy. This review suggests the future deployment of immunotherapy as an alternative to conventional chemotherapy for the appropriate management of leishmaniasis. However, it needs extensive pre-clinical and clinical trials using local animal models which can reflect the precise nature of the human immune profile against leishmaniasis to select the most protective HDT candidates.
MT: Conceptualization, Writing – original draft, Writing – review & editing. BB: Writing – review & editing. DM: Writing – review & editing. AAA: Writing – review & editing. AM: Conceptualization, Writing – review & editing. NC: Conceptualization, Writing – review & editing. GG: Conceptualization, Supervision, Writing – review & editing. AA: Conceptualization, Supervision, Writing – review & editing. TE: Conceptualization, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AD-MSC: adipose tissue-mesenchymal stromal cell
anti-IL-4 mAbs: anti-interleukin-4 monoclonal antibody
anti-IL-10R: anti-interleukin-10 receptor
Balb/susceptible: albino mice
C57BL/6: resistant albino mice
CL: cutaneous leishmaniasis
DCs: dendritic cells
IFN-γ: interferon gamma
IL-10: interleukin-10
IL-12: interleukin-12
IL-1: interleukin-1
IL-4: interleukin-4
mAbs: monoclonal antibody
MHC class-II: major histocompatibility complex II
MQ: macrophage
MSC: mesenchymal stem cells
NK: natural killer cells
NO: Nitric oxide
RIFN-γ: recombinant interferon gamma
ROS: reactive oxygen species
TGF: transforming growth factor beta
Th1: T-helper 1
Th2: T-helper 2
TNF-α: tumor necrosis factor-alpha
VL: visceral leishmaniasis
anti-PD/PDL: anti-programmed cell death/ligand
GCSF: granulocyte colony-stimulating factor
CTLA-4: cytotoxic T-lymphocyte associated protein 4
1. Akbari M, Oryan A, Hatam G. Immunotherapy in treatment of leishmaniasis. Immunol Letters. (2021) 233:80–6. doi: 10.1016/j.imlet.2021.03.011
2. Ikeogu NM, Akaluka GN, Edechi CA, Salako ES, Onyilagha C, Barazandeh AF, et al. Leishmania immunity: advancing immunotherapy and vaccine development. Microorganisms. (2020) 8:1201. doi: 10.3390/microorganisms8081201
3. Akhoundi M, Kuhls K, Cannet A, Votýpka J, Marty P, Delaunay P, et al. A historical overview of the classification, evolution, and dispersion of leishmania parasites and sandflies. PLoS Negl Trop Diseases. (2016) 10:e0004349. doi: 10.1371/journal.pntd.0004349
4. Singh OP, Sundar S. Immunotherapy and targeted therapies in treatment of visceral leishmaniasis: current status and future prospects. Front Immunol. (2014) 5:296. doi: 10.3389/fimmu.2014.00296
5. Roatt BM, Aguiar-Soares RDDO, Coura-Vital W, Ker HG, Moreira NDD, Vitoriano-Souza J, et al. Immunotherapy and immunochemotherapy in visceral leishmaniasis: promising treatments for this neglected disease. Front Immunol. (2014) 5:272. doi: 10.3389/fimmu.2014.00272
6. Mcgwire BS, Satoskar A. Leishmaniasis: clinical syndromes and treatment. QJM: Int J Med. (2014) 107:7–14. doi: 10.1093/qjmed/hct116
7. Wamai RG, Kahn J, McGloin J, Ziaggi G. Visceral leishmaniasis: a global overview. J Global Health Sci. (2020) 2:3–22. doi: 10.35500/jghs.2020.2.e3
8. Tamiru HF, Mashalla YJ, Mohammed R, Tshweneagae GT. Cutaneous leishmaniasis a neglected tropical disease: community knowledge, attitude and practices in an endemic area, Northwest Ethiopia. BMC Infect diseases. (2019) 19:855. doi: 10.1186/s12879-019-4506-1
9. Oryan A, Akbari M. Worldwide risk factors in leishmaniasis. Asian Pacific J Trop Med. (2016) 9:925–32. doi: 10.1016/j.apjtm.2016.06.021
10. de Medeiros MDGF, da Silva AC, Citó AMDGL, Borges AR, de Lima SG, Lopes JAD, et al. In vitro antileishmanial activity and cytotoxicity of essential oil from Lippia sidoides Cham. Parasitol Int. (2011) 60:237–41. doi: 10.1016/j.parint.2011.03.004
11. Kumar R, Chauhan SB, Ng SS, Sundar S, Engwerda CR. Immune checkpoint targets for host-directed therapy to prevent and treat leishmaniasis. Front Immunol. (2017) 8:1492. doi: 10.3389/fimmu.2017.01492
12. Novais FO, Amorim CF, Scott P. Host-directed therapies for cutaneous leishmaniasis. Front Immunol. (2021) 12:660183. doi: 10.3389/fimmu.2021.660183
13. Gurung P, Kanneganti T-D. Innate immunity against Leishmania infections. Cell Microbiol. (2015) 17:1286–94. doi: 10.1111/cmi.12484
14. Amare GA, Mekonnen GG, Kassa M, Addisu A, Kendie DA, Tegegne B, et al. First report of cutaneous leishmaniasis caused by Leishmania donovani in Ethiopia. Parasites Vectors. (2023) 16:457. doi: 10.1186/s13071-023-06057-9
15. Lypaczewski P, Chauhan Y, Paulini K, Thakur L, Chauhan S, Roy EI, et al. Emerging leishmania donovani lineages associated with cutaneous Leishmaniasis, Himachal Pradesh, India, 2023. Emerging Infect Dis J. (2024) 30:1–3. doi: 10.3201/eid3009.231595
16. Welay GM, Alene KA, Dachew BA. Visceral leishmaniasis treatment outcome and its determinants in northwest Ethiopia. Epidemiol Health. (2017) 39:2–6. doi: 10.4178/epih.e2017001
17. Faleiro RJ, Kumar R, Bunn PT, Singh N, Chauhan SB, Sheel M, et al. Combined immune therapy for the treatment of visceral leishmaniasis. PLoS Negl Trop Diseases. (2016) 10:e0004415. doi: 10.1371/journal.pntd.0004415
18. Rossi M, Fasel N. How to master the host immune system? Leishmania parasites have the solutions! Int Immunol. (2018) 30:103–11. doi: 10.1093/intimm/dxx075
19. Gonçalves AAM, Leite JC, Resende LA, Mariano RMDS, Silveira P, Melo-Júnior OADO, et al. An overview of immunotherapeutic approaches against canine visceral leishmaniasis: what has been tested on dogs and a new perspective on improving treatment efficacy. Front Cell Infection Microbiol. (2019) 9:427. doi: 10.3389/fcimb.2019.00427
20. Okwor I, Uzonna JE. Immunotherapy as a strategy for treatment of leishmaniasis: a review of the literature. Immunotherapy. (2009) 1:765–76. doi: 10.2217/imt.09.40
21. Pifer R, Benson A, Sturge CR, Yarovinsky F. UNC93B1 is essential for TLR11 activation and IL-12-dependent host resistance to Toxoplasma gondii. J Biol Chem. (2011) 286:3307–14. doi: 10.1074/jbc.M110.171025
22. Srivastava S, Shankar P, Mishra J, Singh S. Possibilities and challenges for developing a successful vaccine for leishmaniasis. Parasites Vectors. (2016) 9:277. doi: 10.1186/s13071-016-1553-y
23. Faria MS, Reis FCG, Lima APCA. Toll-like receptors in leishmania infections: guardians or promoters? J Parasitol Res. (2012) 2012:930257. doi: 10.1155/2012/930257
24. Chandra D, Naik S. Leishmania donovani infection down-regulates TLR2-stimulated IL-12p40 and activates IL-10 in cells of macrophage/monocytic lineage by modulating MAPK pathways through a contact-dependent mechanism. Clin Exp Immunol. (2008) 154:224–34. doi: 10.1111/j.1365-2249.2008.03741.x
25. Carvalho E, Carvalho L, Passos S, Schriefer A. Protective and pathologic immune responses in human tegumentary leishmaniasis. Front Immunol. (2012) 3. doi: 10.3389/fimmu.2012.00301
26. Varikuti S, Jha BK, Volpedo G, Ryan NM, Halsey G, Hamza OM, et al. Host-directed drug therapies for neglected tropical diseases caused by protozoan parasites. Front Microbiol. (2018) 9:2655. doi: 10.3389/fmicb.2018.02655
27. Lamotte S, Späth GF, Rachidi N, Prina E. The enemy within: Targeting host–parasite interaction for antileishmanial drug discovery. PLoS Negl Trop Diseases. (2017) 11:e0005480. doi: 10.1371/journal.pntd.0005480
28. Dayakar A, Chandrasekaran S, Kuchipudi SV, Kalangi SK. Cytokines: key determinants of resistance or disease progression in visceral leishmaniasis: opportunities for novel diagnostics and immunotherapy. Front Immunol. (2019) 10:670. doi: 10.3389/fimmu.2019.00670
29. Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+ CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. (2002) 420:502–7. doi: 10.1038/nature01152
30. Atia AM, Mumina A, Tayler-Smith K, Boulle P, Alcoba G, Elhag MS, et al. Sodium stibogluconate and paromomycin for treating visceral leishmaniasis under routine conditions in eastern Sudan. Trop Med Int Health. (2015) 20:1674–84. doi: 10.1111/tmi.2015.20.issue-12
31. Sundar S, Olliaro PL. Miltefosine in the treatment of leishmaniasis: clinical evidence for informed clinical risk management. Ther Clin Risk management. (2007) 3:733.
32. Mueller M, Ritmeijer K, Balasegaram M, Koummuki Y, Santana MR, Davidson R. Unresponsiveness to AmBisome in some Sudanese patients with kala-azar. Trans R Soc Trop Med Hygiene. (2007) 101:19–24. doi: 10.1016/j.trstmh.2006.02.005
33. Kurtzhals J, Hey A, Jardim A, Kemp M, Schaefer KU, Odera E, et al. Dichotomy of the human T cell response to Leishmania antigens. II. Absent or Th2-like response to gp63 and Thl-like response to lipophosphoglycan-associated protein in cells from cured visceral leishmaniasis patients. Clin Exp Immunol. (1994) 96:416–21. doi: 10.1111/j.1365-2249.1994.tb06044.x
34. Taslimi Y, Zahedifard F, Rafati S. Leishmaniasis and various immunotherapeutic approaches. Parasitology. (2018) 145:497–507. doi: 10.1017/S003118201600216X
35. Fernández OL, Diaz-Toro Y, Ovalle C, Valderrama L, Muvdi S, Rodríguez I, et al. Miltefosine and antimonial drug susceptibility of leishmania viannia species and populations in regions of high transmission in Colombia. PLoS Negl Trop Diseases. (2014) 8:e2871. doi: 10.1371/journal.pntd.0002871
36. Guerin PJ, Olliaro P, Sundar S, Boelaert M, Croft SL, Desjeux P, et al. Visceral leishmaniasis: current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect Diseases. (2002) 2:494–501. doi: 10.1016/S1473-3099(02)00347-X
37. Chakravarty J, Sundar S. Drug resistance in leishmaniasis. J Global Infect diseases. (2010) 2:167. doi: 10.4103/0974-777X.62887
38. Barrett MP, Croft SL. Management of trypanosomiasis and leishmaniasis. Br Med bulletin. (2012) 104:175–96. doi: 10.1093/bmb/lds031
39. Yousofi Darani H, Yousefi M, Safari M, Jafari R. Parasites and immunotherapy: with or against? J Parasitic Dis. (2016) 40:217–26.
40. Adriaensen W, Dorlo TPC, Vanham G, Kestens L, Kaye PM, van Griensven J. Immunomodulatory therapy of visceral leishmaniasis in human immunodeficiency virus-coinfected patients. Front Immunol. (2018) 8. doi: 10.3389/fimmu.2017.01943
41. Oldham RK, Dillman RO. Principles of cancer biotherapy. USA, University of California: Springer Science & Business Media (2009).
42. El-On J. Current status and perspectives of the immunotherapy of leishmaniasis. Israel Med Assoc journal: IMAJ. (2009) 11:623–8.
43. Zumla A, Rao M, Wallis RS, Kaufmann SH, Rustomjee R, Mwaba P, et al. Host-directed therapies for infectious diseases: current status, recent progress, and future prospects. Lancet Infect diseases. (2016) 16:e47–63. doi: 10.1016/S1473-3099(16)00078-5
44. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc London Ser B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
45. Wallis RS, O’Garra A, Sher A, Wack A. Host-directed immunotherapy of viral and bacterial infections: past, present and future. Nat Rev Immunol. (2022) 23:1–13. doi: 10.1038/s41577-022-00734-z
46. Murray HW. Effect of continuous administration of interferon-γ in experimental visceral leishmaniasis. J Infect Diseases. (1990) 161:992–4. doi: 10.1093/infdis/161.5.992
47. Murray HW, Moreira AL, Lu CM, DeVecchio JL, Matsuhashi M, Ma X, et al. Determinants of response to interleukin-10 receptor blockade immunotherapy in experimental visceral leishmaniasis. J Infect diseases. (2003) 188:458–64. doi: 10.1086/jid.2003.188.issue-3
48. Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNγ signaling—does it mean JAK–STAT? Cytokine & growth factor reviews. Cytokine and growth factor review. (2008) 19:383–94. doi: 10.1016/j.cytogfr.2008.08.004
49. Nathan CF, Hibbs JB Jr. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr Opin Immunol. (1991) 3:65–70. doi: 10.1016/0952-7915(91)90079-G
50. Carvalho EM, Bacellar O, Brownell C, Regis T, Coffman RL, Reed SG. Restoration of IFN-gamma production and lymphocyte proliferation in visceral leishmaniasis. J Immunol. (1994) 152:5949–56. doi: 10.4049/jimmunol.152.12.5949
51. Hart P, Whitty G, Piccoli D, Hamilton J. Control by IFN-gamma and PGE2 of TNF alpha and IL-1 production by human monocytes. Immunology. (1989) 66:376. doi: 10.1089/jir.1991.11.177
52. Malefyt RDW, Haanen J, Spits H, Roncarolo MG, Te Velde A, Figdor C, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. (1991) 174:915–24. doi: 10.1084/jem.174.4.915
53. Immunotherapy of murine visceral leishmaniasis with murine recombinant interferon-γ and MTP-PE encapsulated in liposomes. J Interferon Res. (1991) 11:177–85.
54. Sundar S, Rosenkaimer F, Murray HW. Successful treatment of refractory visceral leishmaniasis in India using antimony plus interferon-ã. J Infect diseases. (1994) 170:659–62. doi: 10.1093/infdis/170.3.659
55. Murray HW. Effect of continuous administration of interferon-gamma in experimental visceral leishmaniasis. J Infect diseases. (1990) 161:992–4. doi: 10.1093/infdis/161.5.992
56. Abdi K. IL-12: the role of p40 versus p75. Scandinavian J Immunol. (2002) 56:1–11. doi: 10.1046/j.1365-3083.2002.01101.x
57. Sypek J, Chung CL, Mayor S, Subramanyam JM, Goldman SJ, Sieburth DS, et al. Resolution of cutaneous leishmaniasis: interleukin 12 initiates a protective T helper type 1 immune response. J Exp Med. (1993) 177:1797–802. doi: 10.1084/jem.177.6.1797
58. Roberts M. Current understandings on the immunology of leishmaniasis and recent developments in prevention and treatment. Br Med bulletin. (2005) 75:115–30. doi: 10.1093/bmb/ldl003
59. Murray HW, Hariprashad J, Coffman RL. Behavior of visceral Leishmania donovani in an experimentally induced T helper cell 2 (Th2)-associated response model. J Exp Med. (1997) 185:867–74. doi: 10.1084/jem.185.5.867
60. Heinzel FP, Rerko RM, Ahmed F, Pearlman E. Endogenous IL-12 is required for control of Th2 cytokine responses capable of exacerbating leishmaniasis in normally resistant mice. J Immunol (Baltimore Md: 1950). (1995) 155:730–9. doi: 10.4049/jimmunol.155.2.730
61. Wang Z-E, Reiner SL, Zheng S, Dalton DK, Locksley RM. CD4+ effector cells default to the Th2 pathway in interferon gamma-deficient mice infected with Leishmania major. J Exp Med. (1994) 179:1367–71. doi: 10.1084/jem.179.4.1367
62. Bacellar O, Brodskyn C, Guerreiro J, Barral-Netto M, Costa CH, Coffman RL, et al. Interleukin-12 restores interferon-γ production and cytotoxic responses in visceral leishmaniasis. J Infect Diseases. (1996) 173:1515–8. doi: 10.1093/infdis/173.6.1515
63. Heinzel F, Schoenhaut DS, Rerko R, Rosser L, Gately M. Recombinant interleukin 12 cures mice infected with Leishmania major. J Exp Med. (1993) 177:1505–9. doi: 10.1084/jem.177.5.1505
64. Chen BDM, Clark CR, Chou T-H. Granulocyte/macrophage colony-stimulating factor stimulates monocyte and tissue macrophage proliferation and enhances their responsiveness to macrophage colony-stimulating factor. Blood. (1988) 71:997–1002. doi: 10.1182/blood.V71.4.997.997
65. Murray HW, Cervia JS, Hariprashad J, Taylor AP, Stoeckle MY, Hockman H. Effect of granulocyte-macrophage colony-stimulating factor in experimental visceral leishmaniasis. J Clin Invest. (1995) 95:1183–92. doi: 10.1172/JCI117767
66. Hübel K, Dale DC, Liles WC. Therapeutic use of cytokines to modulate phagocyte function for the treatment of infectious diseases: current status of granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, macrophage colony-stimulating factor, and interferon-γ. J Infect Diseases. (2002) 185:1490–501. doi: 10.1086/340221
67. Ruef C, Coleman DL. Granulocyte-macrophage colony-stimulating factor: pleiotropic cytokine with potential clinical usefulness. Rev Infect Diseases. (1990) 12:41–62. doi: 10.1093/clinids/12.1.41
68. Lieschke GJ, Burgess AW. Granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor. New Engl J Med. (1992) 327:99–106. doi: 10.1056/NEJM199207093270207
69. Jones TC. Future uses of granulocyte-macrophage colony-stimulating factor (GM-CSF). Stem Cells. (1994) 12:229–40. doi: 10.1002/stem.5530120719
70. Tomiotto-Pellissier F, Bortoleti BTDS, Assolini JP, Gonçalves MD, Carloto ACM, Miranda-Sapla MM, et al. Macrophage polarization in leishmaniasis: broadening horizons. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.02529
71. Jones TC. The effects of rhGM-CSF on macrophage function. Eur J Cancer. (1993) 29:S10–S3. doi: 10.1016/0959-8049(93)90625-P
72. MaChado PR, Prates FV, Boaventura V, Lago T, Guimarães LH, Schriefer A, et al. A double-blind, randomized trial to evaluate Miltefosine and topical granulocyte macrophage colony-stimulating factor in the treatment of cutaneous leishmaniasis caused by Leishmania Braziliensis in Brazil. Clin Infect Diseases. (2021) 73:e2465–e9. doi: 10.1093/cid/ciaa1337
73. Yadagiri G, Singh A, Arora K, Mudavath SL. Immunotherapy and immunochemotherapy in combating visceral leishmaniasis. Front Med (Lausanne). (2023) 10:1096458. doi: 10.3389/fmed.2023.1096458
74. Moll H. Dendritic cells and host resistance to infection. Cell Microbiol. (2003) 5:493–500. doi: 10.1046/j.1462-5822.2003.00291.x
75. Costa-da-Silva AC, Nascimento DDO, Ferreira JR, Guimarães-Pinto K, Freire-de-Lima L, Morrot A, et al. Immune responses in leishmaniasis: an overview. Trop Med Infect Disease. (2022) 7:54. doi: 10.3390/tropicalmed7040054
76. Ghosh M, Pal C, Ray M, Maitra S, Mandal L, Bandyopadhyay S. Dendritic cell-based immunotherapy combined with antimony-based chemotherapy cures established murine visceral leishmaniasis. J Immunol (Baltimore Md: 1950). (2003) 170:5625–9. doi: 10.4049/jimmunol.170.11.5625
77. Navard SH, Rezvan H, Haddad MHF, Ali S, Nourian A, Eslaminejad MB, et al. Therapeutic effects of mesenchymal stem cells on cutaneous leishmaniasis lesions caused by Leishmania major. J Global antimicrobial resistance. (2020) 23:243–50. doi: 10.1016/j.jgar.2020.09.005
78. Di Ianni M, Del Papa B, De Ioanni M, Moretti L, Bonifacio E, Cecchini D, et al. Mesenchymal cells recruit and regulate T regulatory cells. Exp hematology. (2008) 36:309–18. doi: 10.1016/j.exphem.2007.11.007
79. Pereira JC, Ramos TD, Silva JD, de Mello MF, Pratti JES, da-Fonseca-Martins AM, et al. Effects of bone marrow mesenchymal stromal cell therapy in experimental cutaneous leishmaniasis in BALB/c mice induced by leishmania amazonensis. Front Immunol. (2017) 8:893. doi: 10.3389/fimmu.2017.00893
80. Ramos TD, Silva JD, da-Fonseca-Martins AM, da Silveira Pratti JE, Firmino-Cruz L, Maciel-Oliveira D, et al. Combined therapy with adipose tissue-derived mesenchymal stromal cells and meglumine antimoniate controls lesion development and parasite load in murine cutaneous leishmaniasis caused by Leishmania amazonensis. Stem Cell Res Ther. (2020) 11:374. doi: 10.1186/s13287-020-01889-z
81. Shaw TD, Krasnodembskaya AD, Schroeder GN, Zumla A, Maeurer M, O’Kane CM. Mesenchymal stromal cells: an antimicrobial and host-directed therapy for complex infectious diseases. Clin Microbiol Rev. (2021) 34:e00064–21. doi: 10.1128/CMR.00064-21
82. Murphy ML, Wille U, Villegas EN, Hunter CA, Farrell JP. IL-10 mediates susceptibility to Leishmania donovani infection. Eur J Immunol. (2001) 31:2848–56. doi: 10.1002/1521-4141(2001010)31:10<2848::AID-IMMU2848>3.0.CO;2-T
83. Castellano LR, Argiro L, Dessein H, Dessein A, da Silva MV, Correia D, et al. Potential use of interleukin-10 blockade as a therapeutic strategy in human cutaneous leishmaniasis. J Immunol Res. (2015) 2015:152741. doi: 10.1155/2015/152741
84. Murray HW. Interleukin 10 receptor blockade—pentavalent antimony treatment in experimental visceral leishmaniasis. Acta Tropica. (2005) 93:295–301. doi: 10.1016/j.actatropica.2004.11.008
85. Murray HW, Brooks EB, DeVecchio JL, Heinzel FP. Immunoenhancement combined with amphotericin B as treatment for experimental visceral leishmaniasis. Antimicrobial Agents Chemotherapy. (2003) 47:2513–7. doi: 10.1128/AAC.47.8.2513-2517.2003
86. Murray HW, Flanders KC, Donaldson DD, Sypek JP, Gotwals PJ, Liu J, et al. Antagonizing deactivating cytokines to enhance host defense and chemotherapy in experimental visceral leishmaniasis. Infection Immunity. (2005) 73:3903–11. doi: 10.1128/IAI.73.7.3903-3911.2005
87. Chatelain R, Varkila K, Coffman RL. IL-4 induces a Th2 response in Leishmania major-infected mice. J Immunol (Baltimore Md: 1950). (1992) 148:1182–7. doi: 10.4049/jimmunol.148.4.1182
88. Lehn M, Weiser WY, Engelhorn S, Gillis S, Remold HG. IL-4 inhibits H2O2 production and antileishmanial capacity of human cultured monocytes mediated by IFN-gamma. J Immunol (Baltimore Md: 1950). (1989) 143:3020–4. doi: 10.4049/jimmunol.143.9.3020
89. de Freitas ESR, von Stebut E. Unraveling the role of immune checkpoints in leishmaniasis. Front Immunol. (2021) 12:620144. doi: 10.3389/fimmu.2021.620144
90. Joshi T, Rodriguez S, Perovic V, Cockburn IA, Stäger S. B7-H1 blockade increases survival of dysfunctional CD8(+) T cells and confers protection against Leishmania donovani infections. PLoS Pathog. (2009) 5:e1000431. doi: 10.1371/journal.ppat.1000431
91. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci. (2001) 98:13866–71. doi: 10.1073/pnas.231486598
92. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR–induced stop signal. Nat Immunol. (2009) 10:1185–92. doi: 10.1038/ni.1790
93. Bankoti R, Stäger S. Differential regulation of the immune response in the spleen and liver of mice infected with Leishmania donovani. J Trop Med. (2012) 2012:639304. doi: 10.1155/2012/639304
94. da-Fonseca-Martins AM, Ramos TD, Pratti JES, Firmino-Cruz L, Gomes DCO, Soong L, et al. Immunotherapy using anti-PD-1 and anti-PD-L1 in Leishmania amazonensis-infected BALB/c mice reduce parasite load. Sci Rep. (2019) 9:20275. doi: 10.1038/s41598-019-56336-8
95. Murray HW, Brooks EB, DeVecchio JL, Heinzel FP. Immunoenhancement combined with amphotericin B as treatment for experimental visceral leishmaniasis. Antimicrob Agents Chemother. (2003) 47:2513–7. doi: 10.1128/AAC.47.8.2513-2517.2003
96. Zubairi S, Sanos SL, Hill S, Kaye PM. Immunotherapy with OX40L-Fc or anti-CTLA-4 enhances local tissue responses and killing of Leishmania donovani. Eur J Immunol. (2004) 34:1433–40. doi: 10.1002/eji.200324021
97. Hockertz S, Franke G, Paulini I, Lohmann-Matthes M-L. Immunotherapy of murine visceral leishmaniasis with murine recombinant interferon-γ and MTP-PE encapsulated in liposomes. J Interferon Res. (1991) 11:177–85. doi: 10.1089/jir.1991.11.177
98. Sundar S, Rosenkaimer F, Murray HW. Successful treatment of refractory visceral leishmaniasis in India using antimony plus Interferon-ã. J Infect Diseases. (1994) 170:659–62. doi: 10.1093/infdis/170.3.659
99. Do JL, Reed SG, Wick EA, Giordano M. Granulocyte-macrophage and macrophage colony-stimulating factors activate intramacrophage killing of Leishmania mexicana amazonensis. J Infect Diseases. (1990) 162:224–30. doi: 10.1093/infdis/162.1.224
100. Murray HW, Flanders KC, Donaldson DD, Sypek JP, Gotwals PJ, Liu J, et al. Antagonizing deactivating cytokines to enhance host defense and chemotherapy in experimental visceral leishmaniasis. Infection immunity. (2005) 73:3903–11. doi: 10.1128/IAI.73.7.3903-3911.2005
Keywords: leishmaniasis, host-directed immunotherapy, candidate agents, Gondar, Ethiopia, cytokine threapy, cellular threapy
Citation: Tamir M, Birke Teketelew B, Berta DM, Angelo AA, Terekegne AM, Cherie N, Mekonnen GG, Abere A and Eshetu T (2025) Review of host-directed immunotherapy and candidate agents against leishmaniasis. Front. Trop. Dis 6:1450908. doi: 10.3389/fitd.2025.1450908
Received: 18 June 2024; Accepted: 04 February 2025;
Published: 11 March 2025.
Edited by:
Adam P. Roberts, Liverpool School of Tropical Medicine, United KingdomReviewed by:
Elham Kazemirad, Tehran University of Medical Sciences, IranCopyright © 2025 Tamir, Birke Teketelew, Berta, Angelo, Terekegne, Cherie, Mekonnen, Abere and Eshetu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mebratu Tamir, bWVicmF0dXQzQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.