 Gert-Jan Wijnant1
Gert-Jan Wijnant1 Franck Dumetz2
Franck Dumetz2 Laura Dirkx3
Laura Dirkx3 Dimitri Bulté3
Dimitri Bulté3 Bart Cuypers4
Bart Cuypers4 Katrien Van Bocxlaer5
Katrien Van Bocxlaer5 Sarah Hendrickx3*
Sarah Hendrickx3*- 1Department of Cellular and Molecular Pharmacology, Louvain Drug Research Institute, Université Catholique de Louvain, Brussels, Belgium
- 2Institute for Genome Sciences, University of Maryland, Baltimore, MD, United States
- 3Laboratory of Microbiology, Parasitology and Hygiene (LMPH), Department of Biomedical Sciences, University of Antwerp, Antwerp, Belgium
- 4Adrem Data Lab, Department of Computer Science, University of Antwerp, Antwerp, Belgium
- 5Department of Biology, York Biomedical Research Institute, University of York, York, United Kingdom
Leishmaniasis is a tropical infectious disease caused by the protozoan Leishmania parasite. The disease is transmitted by female sand flies and, depending on the infecting parasite species, causes either cutaneous (stigmatizing skin lesions), mucocutaneous (destruction of mucous membranes of nose, mouth and throat) or visceral disease (a potentially fatal infection of liver, spleen and bone marrow). Although more than 1 million new cases occur annually, chemotherapeutic options are limited and their efficacy is jeopardized by increasing treatment failure rates and growing drug resistance. To delay the emergence of resistance to existing and new drugs, elucidating the currently unknown causes of variable drug efficacy (related to parasite susceptibility, host immunity and drug pharmacokinetics) and improved use of genotypic and phenotypic tools to define, measure and monitor resistance in the field are critical. This review highlights recent progress in our understanding of drug action and resistance in Leishmania, ongoing challenges (including setbacks related to the COVID-19 pandemic) and provides an overview of possible strategies to tackle this public health challenge.
1 Introduction: Ongoing Treatment Challenges in Leishmaniasis
Leishmaniasis is a neglected tropical disease (NTD) caused by the obligate intracellular protozoan parasite Leishmania (1). The disease currently affects more than 1 million people per year, mostly disadvantaged populations living in East Africa, South East Asia, the Middle East (“Old World” leishmaniasis) and the Americas (“New World” leishmaniasis) (2). Transmission can occur from human to human (anthroponotic) or from an animal host reservoir to human (zoonotic) via the bite of haematophagous female sand flies. When the Leishmania-infected insect takes a blood meal, extracellular “promastigote” parasites are released into the skin of the mammalian host, where they are taken up by macrophages and other phagocytic immune cells and transform into intracellular “amastigotes”, which replicate and can disseminate to other tissues. If not asymptomatic (3), leishmaniasis presents itself as one of the following clinical forms: (i) cutaneous leishmaniasis (CL), causing stigmatizing skin lesions and sometimes life-long scars; (ii) mucocutaneous leishmaniasis (MCL), destroying the mucosa of the mouth, nose and throat; (iii) visceral leishmaniasis (VL) or kala-azar, a potentially fatal infection of spleen, liver and bone marrow and (iv) post kala-azar dermal leishmaniasis (PKDL), often causing skin papules or nodules all over the body after the apparent cure from VL. These manifestations are related to specific host factors and the causative parasite species, of which at least 20 are known to cause human disease (4).
In the absence of a protective human vaccine and the presence of various challenges related to vector control, the management of leishmaniasis relies heavily on prompt and effective diagnosis and treatment. Unfortunately, only a handful of antileishmanial drugs are available, which all suffer from limitations related to either severe side effects, high cost, invasive administration routes requiring prolonged hospitalization or unacceptably long treatment durations. Furthermore, their efficacy is increasingly jeopardized by rising rates of drug resistance (DR) and treatment failure (TF). While TF is increasingly common in almost all forms of leishmaniasis around the world, DR has been particularly described in the context of VL in South East Asia. Here, the pentavalent antimonials (SbV), which served as first-line treatment, were made redundant by the emergence of acquired DR in the 1980s (5). These were replaced in the early 2000s by more recently developed drugs, such as oral miltefosine (MF), in the local kala-azar elimination programme (KAEP), aiming to reduce VL incidence to less than 1 case per 10,000 people at the block level (6). However, after less than two decades of using MF to treat VL and PKDL, the number of post-treatment relapses has steadily increased and parasite strains with increased drug tolerance, and recently, even confirmed DR, have been detected in the field. Furthermore, the fact that MF (and other current drugs) can seemly cure VL, but not prevent a subpopulation of surviving parasites from establishing a new, dermal complication in the form of PKDL in up to 20% of patients (7), could also be interpreted as a special form of TF. This form could become increasingly important in the future, given the recent surge in PKDL in India and its role as an infection reservoir for VL transmission (8). In clinical practice, no resistance has yet been observed for the repurposed antifungal amphotericin B (AmB) and the repurposed aminoglycoside antibiotic paromomycin (PM), but AmB- and PM-resistant Leishmania strains have already been generated relatively easily under laboratory conditions. Thus, surveillance of emerging drug resistance seems warranted to safeguard the efficacy of current and new drugs, in particular in South East Asia and other leishmaniasis-endemic regions where anthroponotic transmission of resistant parasites could occur (e.g. L. donovani VL and PKDL in East Africa, L. tropica CL in the Middle East). This review provides an overview of the current state of knowledge on the causes of TF and DR in leishmaniasis and potential solutions to tackle this public health challenge (Table 1).

Table 1 Outstanding challenges and possible solutions related to drug resistance and other causes of treatment failure in leishmaniasis.
2 Defining “Drug Resistance” in Leishmaniasis
In general terms, DR in leishmaniasis can be defined as the decrease or absence of activity of a specific agent against a previously susceptible population of Leishmania parasites through the acquisition of molecular resistance mechanisms. This can lead to reduced or a lack of clinical efficacy even at the highest tolerated doses. Importantly, while DR can be a fundamental determinant in TF, these terms should not be used interchangeably. Many factors other than intrinsic parasite susceptibility play a role in the outcome of leishmaniasis treatment, most importantly host immunity, drug pharmacokinetics and posology [5]. Inversely, when viable parasites survive upon TF they can be subsequently exposed to lingering, subtherapeutic drug levels after treatment, thereby increasing the risk of DR emergence.
Formally detecting and defining antileishmanial DR based on the results of genotypic and phenotypic assays in the lab, however, remains a challenge. Indeed, while many mechanisms of drug action and resistance have been described for Leishmania, validated molecular resistance markers to enable genotypic testing are still lacking. Instead, surveillance of DR relies on culture-based phenotypic testing, but standardization of quality-controlled laboratory protocols, assays and endpoints remain poor. Furthermore, interpretation of drug susceptibility results should rely on a well-defined “breakpoint”, or a threshold value that can help distinguish “susceptible” (S) from “resistant” (R) parasites. Such an approach is standard for most antimicrobials used to treat major bacterial and some fungal infections (by the European Committee on Antimicrobial Susceptibility Testing EUCAST in Europe and the Clinical and Laboratory Standards Institute CLSI in the USA), as well as for antimalarials (by the WorldWide Antimalarial Resistance Network WWARN), but does not exist in the field of leishmaniasis, or for any obligate intracellular pathogen in general, for that matter.
Some recommendations to streamline antileishmanial drug susceptibility testing have been made for the VL-causing parasites L. donovani and L. infantum (9, 10), but remain to be widely implemented. As a metric of drug activity, Maes and colleagues have proposed using the 50% inhibitory concentration (IC50). This is the drug concentration that reduces the parasite burden by 50% compared to the untreated control, as measured ideally in intracellular amastigotes. However, several factors, including parasite infectivity and the type of host cell are known to influence intracellular drug susceptibility. A recent study indicated that Swiss primary peritoneal mouse exudate cells are better than cell lines in supporting infection and intracellular parasite multiplication and should therefore be considered as the first option of choice whenever possible (11, 12). By calculating the IC50 ratio (IC50 for a clinical VL isolate divided by the IC50 for the drug-susceptible laboratory reference strain L. donovani MHOM/ET/67/L82), “breakpoint estimates” for different standard drugs were suggested. For example, for MF, an isolate with unknown susceptibility would be classified as “S” if the IC50 ratio is > 10, but as “R” if this value is < 25. To truly validate the proposed “breakpoints”, a large drug susceptibility dataset would be required of hundreds to thousands of isolates from confirmed TF cases, non-responders and pre-treatment isolates from cured patients from all endemic areas over the world, measured via standardized methodology. Even then, modern breakpoint setting by EUCAST/CLSI considers not only microbiological, but also clinical, pharmacokinetic and PK/PD data, much of which is currently lacking for antileishmanial drugs. Similar principles could also be applied to the many different types of Leishmania parasites causing CL and MCL, although the huge intrinsic variations in drug susceptibility between species, as well as the well-known inter-strain variability, complicates defining DR in this context even more (7, 9, 10).
3 Causes of Drug Resistance in Leishmaniasis
Many factors, either parasite-, host- or drug-related, can impact antileishmanial drug efficacy and lead to TF and/or DR. This chapter will summarize and highlight the most important ones.
3.1 Parasite-Related Factors
First of all, the inherent in vitro drug susceptibility variations between the more than 20 clinically relevant Leishmania species are caused by biochemical and molecular species differences. Although pharmacokinetics and host immune responses also play a role in the infected host, these species-specific differences generally already result in a different in vivo drug efficacy. As the Leishmania parasite is known for its remarkable genomic plasticity, it can easily undergo genetic mutations allowing its survival under drug pressure. The acquisition of such resistance mechanisms can be associated with either (i) decreased drug uptake, (ii) increased drug efflux or sequestration, (iii) enzymatic drug inactivation, (iv) improved cellular mechanisms to deal with drug-induced stress of cell damage, and/or (v) changes in the expression, abundance or drug binding affinity of the primary therapeutic target (13) (Figure 1). These changes, however, might impact not only the parasite’s drug susceptibility but also the parasite’s fitness, which is defined as its virulence and propensity to spread in the environment (14). Here, we describe the current state of the scientific knowledge on these phenomena for the currently used antileishmanial reference drugs: SbV, MF, AmB and PM.
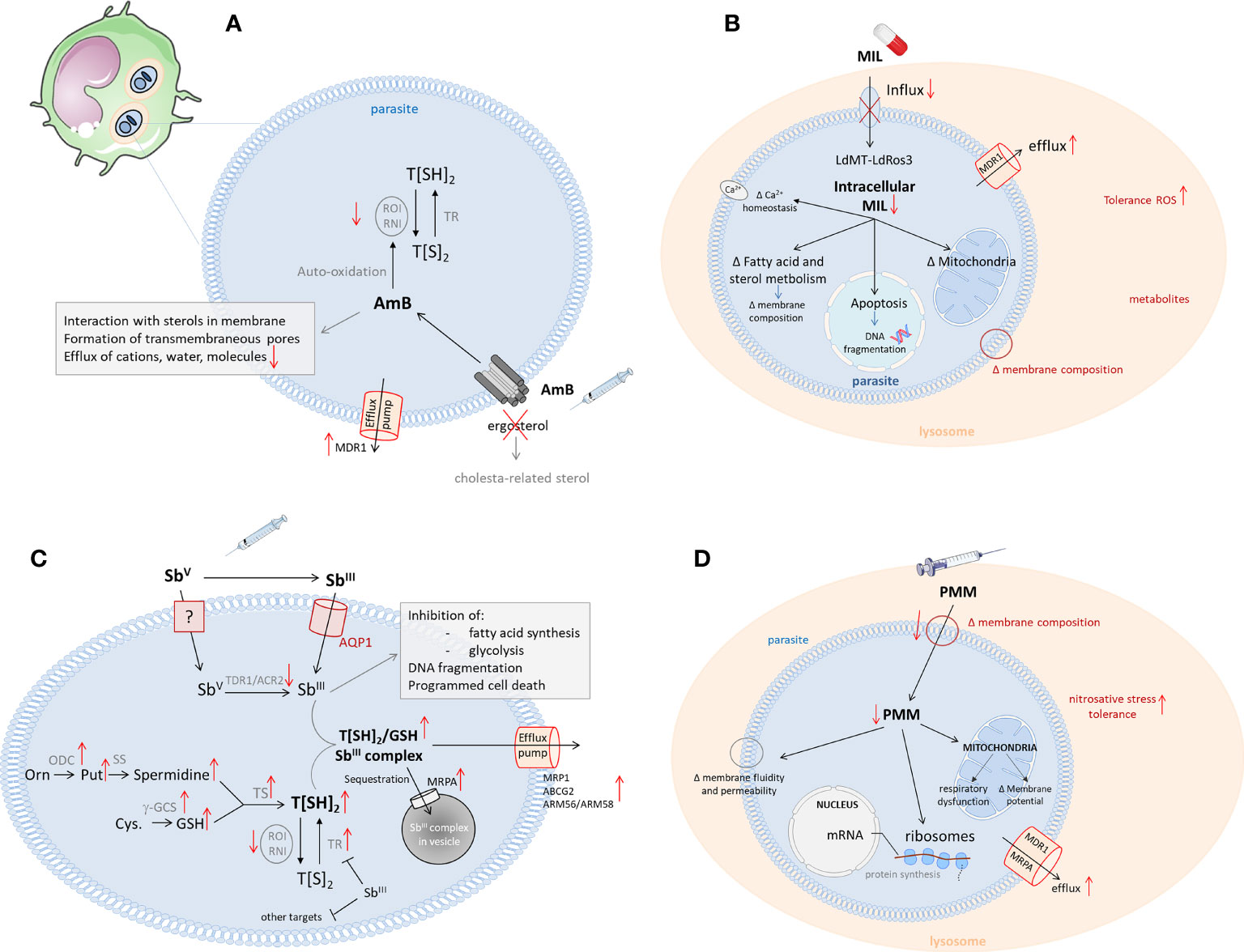
Figure 1 Schematic overview of the mechanisms of action and resistance of the antileishmanial reference drugs (13). Left corner: infected host cell (most commonly, macrophages) with the intracellular Leishmania amastigotes in phagolysosomes. (A) The mechanism of action of Amphotericin (B) Leishmania resistance mechanisms are shown in red. (AmB, Amphotericin B; Cys, cystein; g-GCS, gamma-glutamylcystein synthetase; GSH, glutathione; Orn, ornithine; ODC, ornithine decarboxylase; Put, putrescine; RNI, reactive nitrogen intermediates; ROI, reactive oxygen intermediates; SS, spermidine synthase; TR, trypanothione reductase; T[SH2], trypanothione; (B) The mechanisms of action for miltefosine in Leishmania. Possible resistance mechanisms are shown in red. (LdMT, Leishmania donovani miltefosine transporter; LdROS3, subunit of the LdMT transporter; MIL, miltefosine; MDR, multidrug resistance transporter 1); (C) The mechanism of action of antimonials. Mechanisms leading to Sb-resistance are depicted in red. (ACR, Arsenate reductase; AQP, aquaglyceroporine; Cys, cysteine; g-GCS, gammaglutamylcysteine synthetase; GSH, glutathione; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, reduced nicotinamide adenine dinucleotide phosphate; Orn, ornithine; ODC, ornithine decarboxylase; Put, putrescine; RNI, reactive nitrogen intermediates; ROI, reactive oxygen intermediates; SS, spermidine synthase; SbIII, trivalent antimony; SbV, pentavalent antimony; TDR1, Thiol-dependent reductase 1; TR, trypanothione reductase; T[SH2], trypanothione); (D) The mechanism of action of paromomycin. Possible resistance mechanisms in Leishmania are shown in red. (PMM, paromomycin; MDR, multidrug resistance transporter 1; MRPA, multidrug resistance-associated protein A).
3.1.1 Pentavalent Antimonials
The pentavalent antimonials (SbV) are considered the first-line drugs for leishmaniasis in most parts of the world since their introduction in the 1940s (15). Sodium stibogluconate (SSG) and meglumine antimoniate (MA) are the two available formulations and a standard treatment comprises systematic treatment with 20 mg/kg SbV over 28-30 days (15). Although widely used, SbV are toxic and can induce serious, sometimes life-threatening side effects such as cardiotoxicity, pancreatitis and nephrotoxicity (15). For VL in East Africa, Sb treatment shows an efficacy of 93.9% against infections with L. donovani (16, 17), while efficacies of up to 97% are observed against L. infantum in other regions (16). Despite its proven efficacy in other parts of the world, in South East Asia and especially in the region of Bihar (India) and Nepal (18, 19), the use of SbV for the treatment of VL has been abandoned due to increasing DR in L. donovani (20, 21). The widespread SbV resistance in this area has been linked to the high level of contamination of local freshwater with arsenic, an atom showing comparable properties as Sb and is often present in bedrock (22). An animal study confirmed that parasites isolated from L. donovani-infected hamsters that drank water spiked with arsenic showed a decreased SbV susceptibility. Nevertheless, underdosing, short treatment regimens and the use of expired medication in VL patients have also been implied to play a role in TF and DR in this setting (23). The SbV-resistant parasite phenotype could be linked to enhanced infectivity and lower sensitivity to oxidative stress (24–26). In contrast, so far no resistance has been observed for L. infantum isolates, although parasites isolated upon treatment relapse did show reduced drug sensitivity (27–29).
For CL the efficacy of SbV treatment is not only species-dependent, but also region-dependent. The World Health Organisation (WHO) recommends both intralesional and systemic SbV treatment (15). A recent systemic review by Brito et al., evaluating intralesional Sb treatment, reported an overall efficacy of 75% against Old World CL and 77% against New World CL (30). For systemic treatment of more complex CL, overall good cure rates are reported for L. major and L. aethiopica (75% to 98% and 69% to 85%, respectively), but poor efficacy against L. tropica in the Old World (31–35). In the New World, Sb is commonly used as a systemic treatment with cure rates varying between 77% and 90%, depending on the infecting species (36). This species-dependent efficacy is supported by studies in different Latin-American countries where TF rates of 30.4%, 24.5%, 8.3% and 8.9% have been reported in patients infected with L. braziliensis, L. peruviana, L. guyanensis and L. panamensis, respectively (37–39). Furthermore, susceptibility analysis of New World clinical CL isolates revealed an overall lower susceptibility of L. braziliensis compared to L. panamensis, L. amazonensis and L. guyanensis (40–42).
Although SbV has been used for over 80 years, its exact molecular targets still remain elusive. Nevertheless, it is widely accepted that SbV acts as a pro-drug requiring biological reduction to its more active trivalent SbIII form (43). The mechanism of SbV reduction remains controversial and may include both macrophage- and parasite-specific thiol compounds (non-enzymatic) and parasitic reductases (enzymatic), such as thiol-dependent reductase (TDR1) and Leishmania arsenate reductase homologues (LmACR2) (13, 44, 45). Inside the parasite, SbIII can interfere with the biosynthesis of macromolecules and is known to target the redox metabolism of the parasite by inhibiting trypanothione reductase (TR), which will reduce the intracellular trypanothione levels and increase susceptibility to oxidative stress (13, 46–48). However, when submitted to high levels of SbV, most parasites will increase the gene copy number of the multidrug resistance-associated protein 1 (MRP1), causing DR through increased drug efflux (49–52). Other efflux transporters, such as ABCG2 and ARM56/ARM58, have also been implicated, as well as deletion of the Aquaporin 1 (AQP1) pump, responsible for inward drug uptake (53–55) (Figure 1C). In general, CL species have a higher expression of AQP1 compared to VL species, making them more susceptible to SbV. The species-specific sensitivity of CL strains is therefore mostly attributed to post-transcriptional regulation of AQP1 (56).
3.1.2 Miltefosine
From 2002 to 2012, miltefosine (MF) was used in South East Asia to replace SbV in the frame of the KAEP (57) and nowadays MF is still used to treat PKDL. The treatment regimen generally consists of a 28-day oral schedule with 25, 50 or 100 mg/kg/day (for children < 12 years, patients < 25kg and patients > 25 kg) (15). The main adverse effects of this drug are gastrointestinal disturbances, including nausea, vomiting and diarrhoea (15). Upon its initial use, MF showed a treatment efficacy of more than 94%. Unfortunately, the number of relapses in South East Asia has steadily increased after only one decade of use (58, 59). By 2012, 7% of the Indian VL patients relapsed within 6 months after MF monotherapy (60), while in Nepal the relapse rate in 2013 was 20% within 12 months (61). However, several studies (62–64) have demonstrated that in vitro MF susceptibility between L. donovani parasites isolated from cured and relapsed patients remained comparable. This indicates that TF probably was not linked to the parasite’s intrinsic MF sensitivity. However, parasites isolated upon MF TF did show an increase in drug tolerability, infectivity and resistance to oxidative stress (63–66). Although initial cure rates in PKDL were over 96%, nowadays 15% to 20% of treated patients relapse months to years after initial cure (67–69). The rapid emergence of TF in South East Asia has been linked to its initial over-the-counter availability that enabled patients to shorten the recommended, highly expensive treatment courses. Since 2008, MF provision has been therefore restricted to the public sector in India, where directly observed treatment (DOT) and the use of combination regimens are recommended (70). For the treatment of VL in East Africa, MF is mainly used in combination with other drugs due to the unsatisfactory efficacy of monotherapy (36). MF is not used to treat VL in Latin America due to a poor response to treatment (71). The reason for this is likely two-fold (i) positive response to MF treatment was correlated with the presence of a genetically stable MF sensitivity locus (MSL) present in the genomes of all sequenced L. infantum and L. donovani isolates from the Old World, but not in New World L. infantum (72, 73). The absence of this MSL in New World L. infantum strains increases TF more than 9-fold (72). In addition (ii) Dorlo et al. have demonstrated subtherapeutic plasma concentrations of MF in children treated using linearly as opposed to allometricly scaled drug doses, which may explain the observed poor treatment success rates.
In 2014, the use of MF in Latin America was approved for the treatment of CL caused by L. braziliensis, L. panamensis and L. guyanensis, though with variable success (36, 74). In Colombia, for example, an efficacy of 50% was reported for L. braziliensis CL, while a treatment failure of 8.92% was reported for L. panamensis infections (38). Inter-species differences were observed with L. braziliensis showing the lowest drug susceptibility, followed by L. panamensis, whereas L. guyanensis showed high susceptibility (41, 75).
The exact mode of action of MF is not well understood and several potential mechanisms have been described, suggesting different targets within the parasite. A well-studied mechanism is an interference with the parasite’s lipid metabolism (76). MF was shown to decrease the amount of phosphocholine in the membrane by inhibiting the cytidine-5-diphosphocholine pathway while increasing the proportion of phosphatidylethanolamine (PE) through the stimulation of cytidine triphosphate:PE cytidylyltransferase and inhibition of PE-N- methyltransferase (77). MF also induces apoptosis-like death, which is linked to the inhibition of the cytochrome-c oxidase in the mitochondria of the parasite (78–82) (Figure 1B). The drug also exhibits several immunomodulatory properties that promote a pro-inflammatory Th1 response in the host (76). It induces IFN-γ, IL-12 and TNF-α secretion and increases the responsiveness of macrophages to IFN-γ by inducing the upregulation of the IFN-γ receptor (76, 83, 84). Recently, MF has also been shown to disrupt intracellular Ca2+ homeostasis by activating plasma membrane Ca2+ channels, which could contribute to apoptosis-like cell death (85). Although phenotypic resistance in clinical isolates is still rare, parasites isolated from MF relapse patients often show increased tolerability to the drug (63–66, 86). Despite the low number of resistant clinical isolates, many studies have generated MF resistant parasites in vitro, leading to the discovery of mutations in two genes, ROS3 and MT, encoding the Leishmania miltefosine transporter complex (87–89). This transporter complex, which is involved in the translocation of phospholipids from the outer to the inner leaflet of the parasite’s membrane, is responsible for the uptake of MF in the parasite (89–91). In 2012, a first study reported the isolation of an MF-resistant clinical L. infantum isolate from a French HIV patient with a single nucleotide polymorphism (SNP) in the MT gene (92) and in 2017 clinical MF resistance was also described for the first time for L. donovani in India (93). The overall spread and abundance of these MF-resistant Leishmania strains remain limited to date, probably because mutations in the MF transporter appear to cause a severe loss of fitness and infectivity for the parasite (94–96). However, a reduced expression and copy number of the MT and/or ROS3 genes in L. donovani parasites with a higher tolerance to MF did not impact parasite fitness (63, 91), and an increased fitness was reported for an experimentally selected MF resistant L. major strain (97), which supports the hypothesis that its impact on fitness is species-specific. Even with the low abundance of clinical resistance and the switch to liposomal amphotericin B (L-AmB) as the first-line treatment option in 2014, the emergence of MF resistant parasites in South East Asia still poses a potential threat to the success of the KAEP. Nevertheless, MF remains an essential leishmaniasis treatment to manage specific clinical presentations (HIV/VL coinfections, some pediatric CL and MCL) and as a general partner drug in combination therapies (15, 74, 98, 99).
3.1.3 Amphotericin B and Its Lipid Formulations
Amphotericin B (AmB) is a polyene antibiotic that is isolated from Streptomyces nodosus and has been used as an antifungal agent for over 70 years (13, 100). AmB was introduced as an antileishmanial drug in the 1960s and its liposomal formulation is currently the recommended first-line treatment for VL in India and the standard treatment in Europe and the USA (101). The drug shows excellent leishmanicidal activity against a broad range of Leishmania species (40, 102–105), but also induces some severe side effects, including myocarditis and nephrotoxicity (15). To minimize the adverse effects of the original deoxycholate salt formulation (e.g. Fungizone®), various types of lipid-associated formulations of AmB with improved safety profiles were developed, including lipid complexes (e.g. Abelcet®), colloidal dispersions (e.g. Amphocil®) and liposomal formulations (e.g. AmBisome® and Fungisome®) (106). Liposomal AmB (L-AmB) has been predominantly used in leishmaniasis, although effective doses vary greatly depending on the geographical region. In South East Asia (L. donovani), a single intravenous dose of 10 mg/kg was shown to efficiently cure >95% of VL patients, with reported relapse rates of 2.4% (100, 107, 108). However, trials in East Africa using similar dosing regimens had to be terminated due to poor efficacy, with cure rates ranging from 40 to 58% (109). In this region higher doses of 20-35 mg/kg are required to obtain cure rates of 87-92%, with 7-10% relapse (110, 111). L-AmB has also shown promising results for the treatment of PKDL with cure rates of 89% and higher (112, 113).
Data regarding the efficacy of L-AmB for Old World CL are scarce with no large randomized trials and variable outcomes. Retrospective studies showed a good efficacy of L-AmB against L. major, L. tropica and L. aethiopica, but poor response against CL caused by L. infantum (114, 115). However, a recent study by Ubals et al. showed a cure rate of 100% in L. major CL (116). Although the infecting species or strain may affect the effectiveness of L-AmB in these studies, other factors such as immunosuppressive conditions are more likely to have influenced the treatment outcome (115). In the New World, several trials with L-AmB for the treatment of CL caused by L. braziliensis have shown cure rates ranging from 90% to 100% (114, 115, 117, 118), while a poor response was observed against L. amazonensis and L. shawi (119). In Brazil, L-AmB also showed cure rates ranging from 88% to 100% for MCL (120, 121). Despite its good efficacy and generally good safety profile, the widespread use of L-AmB is limited mainly due to its high cost (up to 18 $ for a single dose) (122).
Resistance emergence against AmB has been considered to be a low risk as, despite its long-term use as an antifungal agent, AmB resistance in fungi is relatively uncommon. Nevertheless, in addition to some reports of AmB unresponsiveness in immunocompromised patients in Europe, some cases of TF with AmB have already been reported in India (123–126). Although AmB resistance markers have yet to be identified, several studies already proposed potential biomarkers which generally relate to the interaction of AmB with the parasite’s membrane sterols (127, 128). AmB has an amphipathic structure that binds to ergosterol and episterol in the parasite’s membrane (13, 101). While its hydrophobic surface interacts with membrane lipids, the hydrophilic part will generate a pore that increases the permeability of the membrane, resulting in cell death (Figure 1A). Several clinical and laboratory-selected AmB resistant L. donovani and L. mexicana lines display alterations in their membrane sterol composition (127–129). As a result of changes in the (ergo) sterol biosynthesis pathway, they have replaced ergosterol and other related sterols with alternative, cholesterol-associated sterols, resulting in a reduced affinity to AmB. The absence of ergosterol also increases membrane fluidity, which further contributes to the decreased binding of AmB to the membrane (128). Next to reduced binding, an increased AmB efflux was also reported in resistant strains, due to upregulation of the MDR1 efflux pump. These AmB resistant parasites were also more tolerant to oxidative stress (128).
3.1.4 Paromomycin
Paromomycin (PM) is a broad-spectrum aminoglycosidic aminocyclitol antibiotic with activity against a wide range of bacteria and protozoa, including Leishmania (130). It is the cheapest of all available antileishmanial drugs and has a good safety profile with only relatively mild adverse effects, such as pain at the injection site and ototoxicity (15). For the treatment of VL caused by L. donovani, a large phase IV trial in South East Asia indicated cure rates >94% using a systemic dose of 11mg/kg for 21 days (131–134). However, a similar dosing regimen was not sufficient to cure VL patients in East Africa and higher doses of 20 mg/kg or longer treatment periods of 28 days were needed to obtain cure rates up to 84.3% (134–136). The use of PM for the treatment of VL caused by L. infantum in the Mediterranean region and South America, respectively, has not yet been documented, but in vitro susceptibility assays using clinical and reference strains showed a similar PM activity against all strains of the L. donovani complex (40, 105, 137).
Systemic use of PM for CL treatment is not common, with only a few studies in the New World indicating poor efficacy of 59% at low doses in Belize, but excellent efficacy with cure rates >90% at higher doses in Brazil (138, 139). For the treatment of CL, PM has been more commonly used in topical formulations. A meta-analysis of 14 randomized control trials revealed that topical PM was effective against both Old World and New World CL and achieved cure rates similar to those obtained with intralesional SbV treatment (140). Two recent Phase III trials in Tunisia and Panama reported cure rates of 82% and 79% against CL caused by L. major and L. panamensis, respectively, whereas an efficacy of 77.5% was reported against L. braziliensis in Bolivia (141–143). Furthermore, clinical isolates of L. braziliensis, L. amazonensis and L. aethiopica were all highly susceptible to PM in vitro, in contrast to L. guyanensis which showed higher tolerability towards the drug (40, 144–146).
While the mechanisms of action and resistance of PM are well-understood in bacteria, its exact effects in Leishmania remain unclear (130, 132, 133). The drug has been proposed to interfere with the parasite’s lipid metabolism and membrane fluidity (leading to changes in membrane permeability), ribosomes (affecting translation and protein synthesis) and energy metabolism (by dysregulating the mitochondrial activity) (147–149). As PM has not been extensively used for the treatment of leishmaniasis, resistant clinical isolates have not yet been reported. However, several in vitro and in vivo laboratory studies have demonstrated a rapid selection of PM resistant parasites, indicating that the development of clinical resistance is not unlikely (137, 150, 151). Furthermore, it was shown that PM resistance was well tolerated in the sand fly vector, which increases its potential to be spread among the population (152). Research on laboratory selected strains already proposed several resistance mechanisms, such as increased membrane fluidity, impacting binding and uptake of the drug, an elevated drug efflux through overexpression of ABC transporters and increased tolerance to nitrosative stress (151, 153) (Figure 1D). However, no genetic resistance markers have been identified yet. Recently, Hendrickx et al. identified 11 short nucleotide variations and copy number alterations in 39 genes that were correlated with PM resistance (154). These identified genes were involved in transcription/translation processes, virulence, mitochondrial functioning and cell signalling and underline the probably multifactorial origin of PM resistance.
3.2 Host-Related Factors
In addition to these parasite-specific factors, host-related factors also determine drug efficacy. In this context, we will focus on the role of the antileishmanial immune response and the impact of immunodeficiency. Nevertheless, other patient-specific or demographic factors related to the host, such as geographic region (discussed in the previous section), age, gender, weight, nutrition status, socioeconomic factors, the severity of disease and treatment adherence have also been directly or indirectly associated with treatment outcomes in the past (38, 155–158).
3.2.1 Immune Response
As Leishmania is an obligate intracellular parasite residing within immune cells, leishmaniasis involves a complex interplay between the host and parasite and is classified as an immune-mediated disease (159). Various types of immune responses are observed during infections with different Leishmania spp. (159), which may contribute to differences in drug efficacy and disease progression. Differences in treatment response between individuals are common for most drugs and are observed for the antileishmanials as well (160). The efficacy of drugs generally relies on effective immune response, as demonstrated by the decreased efficacy of SbV in immunocompromised individuals (161). Intracellular parasites can induce modifications to the immune system or alter the infected host cell characteristics, all of which render parasites less responsive to treatment. For example, SSG TF has been linked to immune suppression, caused by the upregulation of the anti-inflammatory cytokine IL-10 that is produced by regulatory T cells (Tregs) (162–164). MF has been described to enhance monocytic function, such as inducing phagocytosis and increasing oxidative burst. In addition, the drug increases the number of CD4+ T cells, which is associated with the necessary Th1 response to control infection (165). Furthermore, the inherent virulence of the infecting isolate can define the type of immunological response in the host (166, 167). It is known that Leishmania parasites produce virulence factors, such as glycoprotein-63 (GP63), to trick the immune system into inducing a weaker response. GP63 proteolytically degrades specific signalling proteins that attract and activate certain immune cells (168). Finally, Leishmania can release exosomes (extracellular vesicles) that carry bioactive molecules to interfere with host cell function, to modulate the parasite-macrophage interaction and to overcome host protective immune mechanisms (169).
Host immune responses and genetic factors do not only play a crucial role in determining drug efficacy, but also in the progression of disease pathology. Recently, the role of cytokines and host genetics in the susceptibility or resistance to leishmaniasis has gained more interest (170). Various cytokines (IFN-γ, IL-2, IL-12, and TNF-α) play an important role during protection, while some other cytokines (e.g. IL-10, IL-6, IL-17, TGF-β) are associated with disease progression (171). Mice with different genetic backgrounds, for example, are known to show different immune responses to leishmaniasis infection, e.g. BALB/c and C57BL/6 mice are susceptible, while SV/129 mice are considered resistant (172). A recent study in Brazil also found an impairment of the production of Th1 cytokines and a high rate of treatment failure in Leishmania skin test-negative CL patients (173).
3.2.2 HIV and Other Immunosuppressive Conditions
As treatment efficacy in leishmaniasis in part depends on an effective host immune response, immunosuppressed patients are typically more difficult to cure, in particular those co-infected with human immunodeficiency virus (HIV). HIV/VL co-infection is associated with higher initial failure rates due to immune exhaustion and chronic immunostimulation. HIV infection decreases the number of CD4+ T cells, which enhances VL disease progression. Eventually, immunosenescence can be detected, characterized by the exhaustion of immune resources and the presence of senescent CD4+ and CD8+ T cells, which together with the chronic immunostimulation induced by Leishmania, enhances the multiplication of HIV and stimulates progression to acquired immunodeficiency syndrome (AIDS) (174). Such patients suffer from increased drug toxicity and more frequent relapses posing an ideal reservoir for the development of DR, especially in anthroponotic leishmaniasis settings such as East Africa and South East Asia (175). A recent systematic review on the treatment of HIV/VL co-infections reported L-AmB to be the most commonly used drug with an overall cure rate of 68% (176). For SbV low efficacy, pronounced side effects and increased mortality rates are observed (177). Data on MF mainly report low efficacy on L. infantum in Southern Europe (178) with cure rates of 64% and treatment relapse in all patients (179). Also in Ethiopia, although it was considered safer, MF was less effective than SSG (180). Furthermore, for HIV/VL co-infected patients, secondary prophylaxis to prevent relapse is crucial. A recent example in Ethiopian HIV co-infected VL patients, where pentamidine was used as secondary prophylaxis, the relapse-free survival rate at 2 years was only 58.3% (181). As treatment monitoring in these patients is particularly important, a recent study used transcriptomics of peripheral blood to evaluate the immunological responses related to relapse. An enrichment of pathways consistent with disease remission was observed in successfully cured HIV/VL co-infected patients, while these were completely absent in TF cases. Subsequently, a 4-gene signature was identified as able to discriminate treatment success at 4 weeks with a sensitivity of 84% and specificity of 85% (182).
The knowledge on the impact of non-HIV immunosuppressive conditions (such as organ transplantations, rheumatology, haematology, and oncology) on leishmaniasis is more fragmented and mostly based on individual case reports (161). The available data seem to suggest that, compared to HIV/VL co-infected patients, treatment efficacy is higher, although still lower than in immunocompetent patients (183). Also here, relatively high rates of potentially life-threatening toxicity were reported for SbV (184).
3.3 Drug-Related Factors
Drug efficacy, TF and the emergence of DR in leishmaniasis also depend on several drug-related factors providing Leishmania with the opportunity to survive treatment. Here, we will only focus on the role of subtherapeutic drug exposure due to poor pharmacokinetics or pharmaceutical quality of the medication.
3.3.1 Drug Exposure and Pharmacokinetics
Drug pharmacokinetics (PK) describes aspects of drug absorption, distribution, metabolism and excretion (ADME) in the body of the host over time. After administration to the patient, an antileishmanial drug must reach the target tissue, being the viscera (VL), the skin (CL, PKDL) or the mucosal membranes (MCL), to allow uptake into the infected host cells, and exert its activity. In terms of drug delivery to such target sites, there has been an increased interest in so-called “sanctuary sites”, which can be cells or tissues where the pathogen can survive and escape treatment or immune response. Known examples of such sanctuary sites in other NTDs are adipose tissue for Trypanosoma cruzi (185), hepatocytes for Plasmodium vivax (186), stem cells for Mycobacterium tuberculosis (187) and primary skeletal muscle cells for Toxoplasma gondii (188). Besides surviving inside different macrophages populations in various tissues, Leishmania can also infect fibroblasts (189), keratinocytes (190), hepatocytes (191) and stem cells (192). Recently, intracellular parasite survival was demonstrated in different immune cell types in granulomatous lesions, which might shield the pathogen from drug exposure and lead to the adaptation of intracellular amastigotes into a reversible, quiescent stage with limited metabolic activity and replication (193, 194), potentially altering drug susceptibility. The likelihood of Leishmania surviving chemotherapy due to inadequate drug penetration and/or a switch to a ‘drug-tolerant phenotype’ at the aforementioned infection sites (195) is an important concern because drug tolerance is presumed to be a precursor of “classic” genotypic DR in many bacterial infections (196).
While many inherent PK properties of a drug (e.g., protein-binding, plasma exposure, volume of distribution, clearance, metabolism) affect its bioavailability at the intracellular site of infection required for antileishmanial drug action, a particularly important one in the context of the risk for DR emergence is the elimination half-life. For example, the plasma half-life for MF and L-AmB both exceed 5 days (100, 133), causing them to linger in the body at subtherapeutic concentrations for weeks after treatment, a major concern when viable “persister” parasites may remain present in the host. Still, most drug dosing regimens in leishmaniasis are empirical and based on maximally tolerated doses, rather than rationally designed to minimize the risk of toxicity, relapse or DR emergence. Clinical PK studies are therefore extremely important to explain the underlying reasons for variable treatment outcomes and improve dosing strategies. Subtherapeutic drug exposure due to underdosing has already been linked to TF in leishmaniasis, in particular in paediatrics. Age-related differences in PK lead to subtherapeutic MF concentrations in children with VL (< 12 years) and more pronounced TF compared to the adult population treated with the same dose (61, 197). This age-effect was corroborated in another PK study with MF in Columbian CL patients where 30 children (age 2-12 years old) and 30 adults (18-60 years old) were compared (198). MF dose regimen optimization, using allometric rather than linear dosing extrapolations, was proposed as a way to normalise the metabolic differences between adults and children (199). Also in Ethiopian HIV/VL co-infected patients receiving antiviral therapy, plasma exposure for MF and AmB was significantly lower than in immunocompetent VL patients, potentially contributing to the higher TF and relapse rates in this population (200). Although PK alone could not explain the geographical variability in drug efficacy, another study recently demonstrated differences in bioavailability, absorption rates and plasma drug exposure following intramuscular PM administration in East African and Indian VL patients as well (201).
In the above-mentioned PK studies in patients, extracellular drug concentrations are typically measured in the blood due to the ease of sample collection, but these are not necessarily representative of intracellular drug exposure in Leishmania-containing macrophages inside infected tissues. Quantifying drugs at the cellular and subcellular level at the infection site in vivo is technically challenging and tissue homogenates are often used instead, despite significant limitations of their own (202). In this regard, L-AmB is the best-studied antileishmanial drug in terms of PK and PK/PD in animal models. Following IV administration of L-AmB, AmB accumulation in the liver and spleen is lower in BALB/c mice infected with L. donovani than in those of uninfected animals, indicating the impact of pathophysiology and disease-induced organ enlargement on PK (203, 204). Furthermore, dose fractionation and PK/PD studies revealed the concentration-dependent in vivo activity of L-AmB in VL, suggesting higher, less frequent dosing maximizes clinical efficacy. In contrast, AmB accumulation was significantly higher in the CL skin lesions than in the uninfected control skin of mice infected with L. major. The severely inflamed state of the infected dermis had a profound effect on local drug accumulation and contributed to variable in vivo efficacy of L-AmB against Old and New World CL (205, 206). In addition, damage to the epidermal skin barrier and dermal inflammation in CL lesions can alter topical drug penetration and activity (207, 208). To study target site PK in CL and PKDL patients in the clinic, dermal microdialysis holds great potential as an alternative to invasive skin biopsies and following tissue homogenisation, as already shown in murine disease models (209) and could prove a tool to optimize clinical drug dosing regimens (210, 211).
3.3.2 Pharmaceutical Formulation and Quality
The use of poor pharmaceutical quality products can also result in subtherapeutic drug exposure and increased risks of TF and DR. Drug formulations lacking (counterfeit medication) or containing inferior levels of the active pharmaceutical ingredient (API) than indicated on the packaging, have been reported for both MF and SbV. LC-MS analysis of “Miltefos”, the counterfeit MF tablets sourced in Bangladesh at 10 and 50 mg, demonstrated the absence of the API (212). More recently, a counterfeit formulation of Glucantime® named “Gulucatime” was intercepted in Iran after physicians reported its lack of efficacy. The WHO later confirmed the circulation of this counterfeit formulation in both Iran and Pakistan (213). For AmBisome®, a single bilayer liposome containing AmB, the need for cold chain maintenance increases the risk of formulation instability and degradation if incorrectly stored, a concern in some low-resource settings. Furthermore, because of its relatively high price, alternative generic liposomal formulations with different lipid compositions have been developed, but their bioequivalence and pharmaceutical quality is not always known (214).
4 Overcoming Drug Resistance in Leishmaniasis
4.1 Improved Monitoring and Surveillance
4.1.1 Phenotypic Testing
Monitoring and surveillance of DR in leishmaniasis still relies primarily on phenotypic susceptibility testing following parasite isolation from clinical samples. Considering the bi-phasic life cycle of Leishmania, in vitro drug susceptibility testing can be performed on extracellular promastigotes (sand fly vector stage) or intracellular amastigotes (mammalian host stage). Despite being more labour-intensive, expensive and complex than the promastigote model (which is still routinely used for drug susceptibility testing in many hospital laboratories), the intracellular amastigote model remains the gold standard due to its increased biological relevance (10). For example, AmB and MF show comparable activity against promastigotes and intracellular amastigotes, but SbV is inactive against extracellular parasites, indicating the importance of host-cell mediated effects (215). Indeed, testing outcomes can depend not only on the choice of macrophage host cell type (216), but also on many other technical factors (including parasite inoculum, incubation time, culture medium and end points) (12). As already discussed earlier, standardization of these assays and defining clear susceptibility/resistance breakpoints remains a major challenge for the leishmaniasis research community.
Thus, to improve the phenotypic surveillance of DR in leishmaniasis, (i) correct Leishmania species identification to rationalize therapy choice, (ii) the use of standardized drug susceptibility assays on intracellular amastigotes, (iii) interpretation of the results based on well-defined susceptibility/resistance breakpoint criteria, all seem critical.
4.1.2 Genotypic Testing
Although the surveillance of DR should preferably rely on the identification of molecular resistance markers, the scarcity of knowledge on molecular and biochemical resistance mechanisms to both old and newer antileishmanial drugs hampers this approach. Nevertheless, over the last decade, whole-genome sequencing (WGS) has vastly increased our molecular understanding of how Leishmania can adapt to drug pressure (87, 88). More and more, specific parasite genomic variants can be linked to DR against certain anti-leishmanial drugs (see 3.1), holding tremendous potential for future DR detection and surveillance. WGS studies characterizing DR typically result in a list of genomic variants, such as SNPs, insertions or deletions (INDELs), and gene copy number variants present in drug-resistant strains but not, or less frequently, in drug-sensitive strains. These variants can be directly wheeled for DR surveillance. If it concerns a single or small set of variants, assays such as a PCR or line probe assay can be developed, being a relatively low-cost and straightforward solution (217). However, these assays need redesigning and validation every time new variants appear, and increased multiplexing quickly becomes expensive and complicated. WGS does not have these limitations and provides the richest information about a strain’s genetics and DR profile. For many years, the high costs and the complexity of data analysis blocked application of WGS for DR surveillance in remote or resource-poor settings. Recently, however, new and much cheaper sequencers, such as the Illumina Iseq 100 and the Oxford Nanopore MinION, have become available, with prices decreasing from the original ≥ 100K to 20K or as little as 1K US $. Furthermore, these new sequencers provide automated, user-friendly and computationally-efficient workflows, reducing the need for specialized bioinformaticians to run them. These systems have already been successfully used to detect DR in Mycobacterium tuberculosis (218) and could also be applied to Leishmania.
To enable successful genotypic DR surveillance for Leishmania (i) the number of (now affordable) sequencers in the clinical labs in endemic regions should be increased; (ii) more sequencing data of (potential) DR as well as reference sensitive strains should be generated, so that DR phenotypes can be linked to genomic variants; (iii) these DR genomic variants should be catalogued and (iv) these variants should be monitored with WGS in the field.
4.2 Better Use of Existing Drugs
4.2.1 Combination Therapy
Co-administration of multiple drugs can show some major advantages over monotherapy, including the potential for the synergistic or additive activity of the partner drugs, a lower risk for the emergence of DR and reduced drug doses, which in turn reduce treatment costs and the risk of adverse effects. Considering the rise of SbV and MF TF and DR in South East Asia (20), the variable efficacy of PM monotherapy for VL in East Africa (201), rising TF for intralesional SbV monotherapy for CL in Sri Lanka (219) and the poor overall patient adherence due to drug toxicity and long treatment durations, the application of multidrug regimens to treat the different forms of leishmaniasis and to combat TF and DR has increasingly been explored. Combined treatment of PM and MF, for example, is not only efficacious and safe, but has also been shown to delay the onset of experimental DR in L. infantum in vitro and in vivo (220).
The safety and efficacy of MF/L-AmB and PM/MF combinations were first tested in phase III clinical trials in India, after which the efficacy in field conditions was evaluated in India and Bangladesh in large studies between 2010 and 2015. The encouraging preliminary results led to a policy change in India, recommending PM/MF combination as second-line treatment (while single-dose L-AmB remained fist-line when cold chain maintenance can be guaranteed). At the 12-month follow-up, efficacy rates were as high as 91.5% and 98.6% for MF/L-AmB and PM/MF, respectively (221). Despite the highly variable efficacy of PM in Africa, the combined effect of both injectables PM and SSG administered for 17, instead of 30 days, demonstrated similar efficacy to SSG monotherapy. The efficacy and low mortality rate of this regimen remain evident even after the WHO recommendation in 2010 to treat VL in Africa accordingly. The treatment, however, is not suitable for all VL patients; increased mortality and reduced efficacy were observed in patients aged over 50 and HIV/VL co-infected patients, probably because of the lower drug exposure and antileishmanial immune responses reported in this population (200). A retrospective analysis with a co-infected population in Bihar, India that received L-AmB and MF combination therapy for 14 days was well-tolerated, safe and effective (222).
For CL and PKDL, the presence of parasites in the dermis offers the opportunity to combine either two systemic treatments or a local treatment (for small/few lesions) with a systemic agent (for numerous/large lesions) to reduce the potential of pathogen dissemination and post-treatment relapse. Currently, the Drugs for Neglected Diseases Initiative (DNDi) is evaluating the immunomodulator CpG-D35 as a combination therapy with standard antileishmanial drugs for the treatment of complicated CL and PKDL in late preclinical development stages. Furthermore, a combination of thermotherapy (hereby one application of radio frequency waves equivalent to 50°C is applied to the skin lesion for 30 seconds) and a standard daily dose of 2.5 mg/kg MF for 21 days was found to be effective and safe in patients with uncomplicated CL during phase II clinical trials in Peru and Columbia (223).
4.2.2 Increasing Compliance
Overall, most of the existing antileishmanial chemotherapeutics are patient-unfriendly, as they generally require either long-term drug administration and hospitalisation, cause cumbersome adverse effects, or are expensive, either directly through their high purchase cost or indirectly through the inability to work. All of these factors contribute to delayed treatment-seeking behaviour, low treatment adherence and high patient drop-out rates. Such phenomena may have played a role in the emergence of DR for MF in India. The oral drug was temporarily available over the counter at high prices in private pharmacies, which may have caused patients to start treatment without medical supervision, but terminate treatment earlier than the recommended 28-day course. Since 2008, its provision is restricted to the public sector and caregiver directly observed treatment (DOT) was advocated to increase compliance. Similar approaches may help to preserve the efficacy of new drugs once they reach the clinic (see 4.3).
New pharmaceutical formulations can also help to increase compliance and minimize the risk of drug misuse by shortening treatment duration or the frequency of administration. This is exemplified by the liposomal formulation of AmB, which is safer and more effective than the original AmB deoxycholate and can be given as a single intravenous injection to treat VL in South East Asia. Ample research describes novel micro- and nano-pharmaceutical formulations of standard antileishmanial agents in an attempt to overcome current treatment limitations, although none of these has yet reached the clinic (224). A promising example is an AmB-loaded PGLA (poly(lactide-co-glycolide acid) microparticle with prolonged release, which cured L. amazonensis CL with a single dose (225). Some recent therapeutic breakthroughs from other major infectious diseases remain unexplored. Long-acting injectable nanoformulations have been announced to soon “revolutionize” HIV care. Such formulations, containing an antiretroviral combination of cabotegravir and rilpivirin, recently received FDA and EMA approval in 2020, replacing the need for daily oral pill intake by a single monthly intramuscular injection. These particles are prepared via a novel “nanosuspension” technology, where pure nanosized drug crystals are stabilized by surfactants, resulting in a formulation suitable for intramuscular depot injection that significantly increases the half-life of the two active drugs (226). Similarly, a long-acting injectable formulation of atovaquone solid drug nanoparticles provided long-lived prophylaxis against Plasmodium berghei malaria in mice. These particles were created using a new “emulsion-templated freeze-drying” (ETFD) methodology, which allows a higher drug loading per particle in comparison to other preparations methods (227). Important concerns for evaluating the feasibility of developing such novel formulations for an NTD such as leishmaniasis are their potentially high cost, low thermal stability and difficulties during the scaling-up process.
4.3 Development of New Therapeutics
While chemotherapeutics directly target the Leishmania parasite, immunotherapeutics modulate the antileishmanial host immune response. Adjuvant therapeutics to cure leishmaniases, such as anti-parasitic peptides (228) or resistance reversal agents (229), are under investigation but have not resulted in new treatments to date.
4.3.1 Leishmania-Targeting Therapeutics
Over the past decade, significant progress has been made in the discovery of new drugs with novel mechanisms of action that remain active against Leishmania strains resistant to current agents. Prospective DR studies during the early stages of drug development can help to identify drug targets, evaluate the overall risk/benefit ratio and help in the selection of appropriate partner drugs for combination therapy. A high in vitro “frequency of resistance” for a certain drug, however, does not necessarily indicate a rapid loss of in vivo efficacy in the clinic (as pharmacokinetics and host immunity also play a role) and should not be used as a sole criterium to drop compounds from the R&D pipeline. Such a “DR-based” approach is a cornerstone of much of mainstream anti-infective drug development, but it has only very recently been applied to NTDs such as leishmaniasis (230). TCMDC-143345, a promising anti-VL compound of the GSK ‘Leishbox’, was shown to exhibit a longer time-to-resistance than MF and SbV in L. donovani promastigotes under sub-leishmanicidal drug pressure (10 versus 6 and 1 versus 4 selection rounds, respectively), which was maintained in intracellular amastigotes. Mutations in the L. donovani dynamin-1-like protein (LdoDLP1), a protein involved in mitochondrial fission, were associated with drug action and resistance. No cross-resistance with standard antileishmanial drugs could be observed (231). DNDi has several VL drug candidates in their portfolio, belonging to the oxaborole series (lead DNDI-6148 and backup DNDI-5421, thought to target the endonuclease Cleavage and Polyadenylation Specificity Factor 3 CPSF3) and aminopyrazole classes (DNDI-1044, DNDI-8012 and DNDI-5561, thought to target mitogen-activated protein kinases MAPK and cdc2-related kinases CRK) (232). For these highly active compounds, repeated in vivo drug exposure in the L. infantum Syrian hamster model and in vitro selection in both promastigotes and amastigotes did not lead to the emergence of DR. Furthermore, none of these DNDi compounds was found to be a substrate for common Leishmania drug efflux pumps such as ABC, MDR and MRP (233). Besides VL, many DNDi compounds appear to retain good activity in vitro against 6 Old and New World CL-causing Leishmania species and in vivo against L. major (234).
The pharmacokinetic/pharmacodynamic (PK/PD) relationships of novel antileishmanial drugs should also be examined before release into the clinic, alone or in combination therapy. For example, LXE408, a promising kinetoplastid-selective proteasome inhibitor discovered by Novartis is currently in phase 1 clinical trials and demonstrates oral bioavailability, an appropriate safety profile and excellent efficacy in mice infected with L. donovani. PK and PK/PD modelling suggest a dose of 85–190 mg/day for 10 days could cure VL patients in monotherapy, but combination therapy would be optimal (235). Little is currently known on how to design the best matching new drug combinations to treat leishmaniasis, although it seems pharmacodynamically plausible that partner drugs ideally have different targets, lack cross-resistance and show therapeutic synergy, additive effect or, in the very least, no antagonism. In malaria research, it is commonly assumed that based on such principles, the chance that a mutant arises that becomes resistant to both drugs simultaneously is much smaller than the chance that a parasite becomes resistant to the individual drugs alone (e.g. decreasing the risk for DR emergence from 1/109 to 1/1018) (236). From a PK point of view, drug-drug interactions should be avoided and drugs must be bioavailable at the site of infection at the same time to allow theoretical synergy. In tuberculosis and malaria combination therapy design, different drugs with similar half-lives are often “PK-matched” to keep continuous multiple drug pressure on the pathogen and prevent the emergence of DR, but this principle is increasingly under debate (237, 238).
4.3.2 Host-Directed Therapy
To overcome the notable limitations of chemotherapeutics, and especially the emergence of DR, the general interest in the application of host-directed therapies (HDTs) for the treatment of infectious diseases is steadily growing (239). HDT aims to interfere with host cell factors that are required by the pathogen for survival, to enhance the protective immune responses and/or to reduce excessive inflammation and balance the immune response at the site of infection. Such approaches can include cytokines, small molecule inhibitors, humanized monoclonal antibodies, and drugs directed against immune targets (240). Over the last decades, various approaches of immunotherapies have been developed and applied in the treatment of human leishmaniasis, both for VL and CL (241). In VL, combining SbV with the cytokine IL-12 helped recover animals infected with L. donovani (242). Other studies focused on promoting the production of nitric oxide (NO) (243) and reactive oxygen species (ROS) (244), improving antileishmanial immune responses and healing. For CL and MCL, topical simvastatin treatment enhanced host protection against L. major by increasing macrophage phagosome maturation and killing effector functions (245). Another study revealed that Imatinib, an anticancer drug, was useful for reducing the severity of skin lesions caused by L. amazonensis (246). Another agent currently undergoing clinical investigation for oncological treatment, the Heat shock protein 90 (HSP90)-inhibitor 17-AAG, was shown to kill intracellular L. amazonensis and reduce pro-inflammatory host responses that can cause tissue damage in CL and MCL (247, 248). Recently, the pathology induced by CD8+ T cells in CL was blocked using tofacitinib, an inhibitor of IL-15 signalling (249). Imidazoquinolines, agonists of the Toll-like receptor (TLR) 7 and 8, promote the host cell production of free radicals and inflammatory cytokines and assist macrophages in killing intracellular L. amazonensis (250). With the ever-emerging risk of DR and the considerably slower development of new antileishmanial drugs, redirecting our focus to the combination with drugs that modulate the host immune system could be promising in the future (251).
4.4 Preventing Transmission of Resistant Strains
Transmission of DR parasites in endemic regions can be prevented in several ways, i.e. by (i) the implementation of vector control strategies, (ii) control of reservoir hosts and by (iii) early diagnosis and prompt and correct treatment.
First, related to vector control, previous research has already indicated that most drug-resistant Leishmania strains can be successfully transmitted via the sand fly, potentially leading to the spread of primary resistant isolates in endemic regions (91, 94, 152, 252–254). Depending on previous drug exposure and the resulting impact on parasite fitness, transmission efficiency might even be altered. Vector population control can be achieved via various chemical or biological approaches reviewed elsewhere (255). Another concern here is climate change, more specifically global warming contributing to the expansion of the vector and potentially DR parasites to new geographical areas where anthroponotic transmission is possible (256).
A second approach relevant to zoonotic leishmaniasis to decrease transmission is to reduce the number of animal hosts living near the at-risk populations in leishmaniasis endemic areas. For L. infantum VL, for example, dogs are known primary reservoirs for human infection (257, 258). Although canine vaccination campaigns and insecticide-impregnated dog collars nowadays are frequently used to reduce the number of secondary infections, the only intervention that has shown real proven efficacy in reducing the number of human leishmaniasis cases so far are the large nation-wide dog culling campaigns in the 1950s in China (L. donovani) and the national eradication programmes of the great gerbil in the 1970-1980s in former USSR territories (L. major) (258–260). Treatment of infected animals will not result in total parasite clearance, making them predisposed to treatment relapse. Chemotherapeutic interventions with MF for example should therefore be avoided as they will have little effect on decreasing parasite transmission and will merely result in the emergence of drug-resistant Leishmania species (261).
Third, early diagnosis and treatment, possibly via active case detection, is not only very important to reduce morbidity and mortality, but also to block the transmission of resistant parasites in the community (262). Diagnosis of leishmaniasis can be challenging due to the wide spectrum of clinical symptoms (partly overlapping with those of tuberculosis, typhoid, and malaria), undermining the potential for prompt chemotherapy to control the disease. Diagnosis relies on either (i) parasitology (direct detection of Leishmania in tissue smears or culture), (ii) serology or (iii) molecular diagnostics (263). In VL, the rK39 rapid diagnostic test (that detects antibodies against the 39-amino acid repeat antigens encoded by a kinesin-related gene of the L. donovani complex) has been played a key role in the past decades to perform quick, cheap, equipment-free diagnosis in remote areas (264). For ongoing VL elimination programs in East Africa and South East Asia, active case detection and early diagnosis and treatment of PKDL cases also should be integrated, as these patients could carry L. donovani parasites in their skin and be infectious to sand flies (265, 266). However, convincing PKDL (and CL) patients to seek medical care can be challenging due to the social stigma and exclusion associated with these disfiguring skin conditions. For CL, Leishmania species diagnosis is done via laborious isoenzyme analysis or more modern molecular approaches, for example, Restriction Fragment Length Polymorphism (RFLP) genotyping (PCR) (267). An rK39-style rapid bedside test for CL species discrimination does not yet exist and would be of great clinical value in settings where multiple parasite species are endemic that may require different treatment, such as in the Middle East and South America. Overall, to improve diagnosis and treatment, increased education and awareness about leishmaniasis and its management are also needed. Regardless of the approach chosen, a lot of advocacy, political support and central coordination will be required, embedded in close collaborations between local governments, the pharmaceutical industry and non-governmental agencies (268, 269).
4.5 Drug Policy: Tackling Programmatic Problems
An important prerequisite to enabling fast and correct leishmaniasis treatment is the availability and accessibility to antileishmanial drugs. Drug access is influenced by numerous factors (268) and one of the most important factors when discussing NTDs is drug affordability. Several economic analyses in the past showed that drugs for NTDs should not cost more than US$50–60 per treatment to be an effective public health tool (268, 270). That is why the WHO still prioritizes benchmarking this price in price negotiations for antileishmanial drugs extensively used in control programs, such as MF, SbV and L-AmB (268). Drugs for NTDs are therefore often unaffordable unless low-income countries enrol in control programs and join hands with organizations like WHO and pharmaceutical companies to form public-private partnerships. These collaborations enable pharmaceutical companies to either provide drugs at non-profit prices, donate drugs or work out preferential pricing schemes, and WHO to coordinate and arrange drug distribution (268). This way, drugs can be made accessible to governments of low- to middle-income countries and large non-governmental organisations such as WHO, Pan American Health Organization (PAHO), Médecins Sans Frontières (MSF) and DNDi, which are tackling disease control in the field. However, negotiations on preferential prices of some drugs take years. For example, MF preferential pricing in India was only put in place in 2008, seven years after its registration and its introduction as first-line therapy in the KAEP (271). Before that, it was only available in private pharmacies at the cost of US$150–200 per treatment (58). The WHO also partnered up with Sanofi-aventis and reached an indefinite preferential, no profit price for Glucantime® (meglumine antimoniate) in Africa, the Balkan region and post-Soviet States. Prices of generic SSG are comparable to the price of Glucantime®, although the sole producer of generic SSG [Albert David (Calcutta, India)] has made no agreement about the sustainability of this price. Access to Pentostam® on the other hand is restricted, as GlaxoSmithKline (GSK) does not offer preferential prices and intends to discontinue the production of Pentostam®, which will certainly reduce access in some areas. In Sudan and Ethiopia, the WHO and AECID (Agencia Española de Cooperación Internacional Para el Desarrollo) have donated several drugs, including SbV, L-AmB and PM. In 2014, the WHO negotiated a large-scale donation of AmBisome® with Gilead for the control of VL in several countries in South East Asia and East Africa for a no-profit-no-loss price that is re-established yearly. PM, produced by Gland Pharma (India) is still the cheapest antileishmanial drug available, but its production is irregular and the drug is only registered (but not WHO recommended) for treatment of VL in India, Bangladesh, Nepal and African countries (268, 269, 272). However, over time preferential prices are steadily increasing and are often only valid when buying large drug batches at once, which is challenging for countries engaged in elimination programs decreasing their case numbers and drug needs (273).
Because of the low number of patients and the unlikely return on investment, some drugs have not been registered in the low-income countries to be used in control programmes. This lack of drug registration complicates and extends drug import as special import permissions are required. Inclusion of all SbV formulations (SSG and MA) in the national essential drug lists and agreements on the import of unregistered drugs (both on the authority level and customs level) and the establishment of an easily accessible stock could expedite the treatment of patients (268). Most drugs for leishmaniasis are produced by one single manufacturer, which has already caused issues for MF and PM (268). Ideally, manufacturers should predict yearly drug needs in close collaboration with WHO and local authorities to avoid financial damages due to overproduction, but still, be able to continuously meet market demands while maintaining a healthy buffer stock. The production of such evidence-based estimates would also provide a useful tool in the epidemiological monitoring of drug efficacy and drug resistance in surveillance systems (268). In addition, some antileishmanials are produced in ways not compliant with the WHO GMP (good manufacturing processes) standards. As centralized quality assurance and control measures are lacking, the distribution of counterfeit drugs and drugs with unacceptable toxicity profiles for Glucantime® and MF have already been reported in the past (212, 274) and quality issues have been reported for PM and generic SSG (268). The over-the-counter availability of antileishmanial drugs is common in many countries, facilitating counterfeit drug trafficking, drug misuse, suboptimal treatment and thus ultimately drug resistance. For most drugs, phase IV clinical studies on pharmacovigilance are lacking as well, indicating that some serious adverse events are not followed up and reported as such. Drug access is also influenced by drug distribution and storage, both having their own respective challenges. Drug distribution from manufacturers to the affected countries, but towards the peripheral health centres is often threatened by exposure to inadequate temperatures during shipment, at customs level and during subsequent storage and the time-sensitive supply of centres in remote areas. Drug manufacturers should be encouraged to invest in ensuring validated “cold chain” transport, which is particularly important for thermosensitive L-AmB formulations. In addition, drug stock should be properly managed to avoid the elimination of quickly expiring drugs (268). Distribution of drugs ideally arranged through official institutions would be one of the most efficient ways to guarantee their proper transport, distribution, but also drug quality.
A final policy challenge is balancing the need for treatment access in the most remote and neglected areas with the risk of widespread drug misuse and associated TF/DR. The basic concepts of Antimicrobial Stewardship programs to measure and improve the use of antibiotics are quite well described and endorsed by the WHO in the global fight against antimicrobial resistance. Implementing such types of policies (e.g. prospective audit and feedback, prescriber education, evidence-based treatment guidelines) for leishmaniasis is challenging in many endemic settings due to inadequate funding, lack of political commitment and/or fragility of health and regulatory systems. In any case, to be effective in tackling DR, such policies must be adapted to each context and focus on the local causes of suboptimal or incorrect antileishmanial drug use (275, 276).
4.6 Unexpected Setbacks: The SARS-CoV-2 Pandemic
Ever since the alarming spread of the SARS-CoV-2 virus, health systems were challenged with millions of deaths worldwide and urged nationwide lockdowns. In addition, the pandemic also had a big impact on numerous control programs for various neglected diseases. In India, for example, a nationwide lockdown was declared in March 2020 to control the COVID-19 pandemic, resulting in a nationwide pause in running VL control programs. Given the relatively fast epidemic growth rate of leishmaniasis, mathematical models predict a substantial re-emergence of the number of VL cases in high transmission areas that will considerably delay the achievement of the preset 2030 VL elimination targets (i.e. incidence < 1 VL case/10 000 people/year) (277, 278). Interruptions in early case detection (either passive or active) will likely lead to a build-up of undetected cases that need to be addressed as soon as control programs return to full strength. Nevertheless, the delay in addressing all cases will probably lead to increased transmission rates, emphasizing the importance of minimizing COVID-19 pandemic-related interruptions of NTD control programs and urging their swift restart (278). A similar situation can be found in other countries in Africa where MSF in Pakistan, for example, was forced to close its clinics for people with CL since the disease is not life-threatening. Hospitals refused patients for supportive treatment while patients often were clueless with little information about the COVID-19 pandemic reaching the poorer, rural communities. Even after treatment centres were reopened road blockages and problems with public transport still complicated CL treatment. In addition, co-infections of SARS-CoV-2 and VL have been reported, which are generally difficult to diagnose because of their aspecific clinical features. Although the possible relationships between VL and SARS-CoV-2 infection still require further investigation, it is likely that VL endemic settings, associated with an inadequate immune response, could have been in favour of the spread of SARS-CoV-2 (279). In contrast, there has been speculation on the ability of SARS-CoV-2 to reactivate latent, asymptomatic L. donovani infections as well, similar to HIV.
The pandemic did not only exert huge pressure on public health systems worldwide, but also severely impacted the global economy, leading to the rise of new threats as well. In the UK, the leading donor of the Global leishmaniasis response organization, recent budget cuts threaten the constant supply of chemotherapeutics to endemic regions, which will inevitably lead to shortages and increased mortality (272). Moreover, Bio-Rad Laboratories will discontinue the production of IT-Leish, which is the only rapid diagnostic test with a sensitivity that is high enough to detect VL in East Africa (272). Finally, a shortage of L-AmB, the first-line treatment for many patients with VL, can be expected as in India the recent outbreak of COVID-19-related mucormycosis has significantly increased the global demand for L-AmB. Half of the doses of L-AmB donated by Gilead to India at preferential prices for VL have already been diverted to respond to the urgent needs for mucormycosis (272), leaving patients with VL in the dark once more. Finally, the development of urgently needed new antileishmanial drugs has been delayed, as the recruitment of healthy volunteers by DNDi for the Phase 1 clinical trials of DNDI-6148 and DNDI-0690 was cancelled during the early days of the pandemic (280). Overall, due to setbacks in many leishmaniasis control campaigns and chemotherapy being the main pillar of disease control, the battle against DR and other causes of TF in leishmaniasis remains highly relevant.
5 Conclusions and Future Perspectives
Treatment failure is an increasingly worrying problem in the management of leishmaniasis, as chemotherapy remains central to disease control and the ongoing COVID-19 pandemic is undermining recent global progress towards its elimination. The exact causes of variations in antileishmanial drug efficacy are not fully understood, although host immunity, pharmacokinetics and the drug susceptibility, tolerance and resistance profile of the causative Leishmania parasite all play a role. In South Asia, acquired drug resistance against the pentavalent antimonials has enforced the implementation of other drugs such as miltefosine and liposomal amphotericin B. Here and in East Africa, the further emergence of resistance should be particularly closely monitored due to the high prevalence of HIV/VL co-infection and the potential for anthroponotic transmission of resistant parasites between patients. However, detection and surveillance of drug resistance in Leishmania is currently challenged by a lack of validated genotypic and phenotypic assays. The introduction of now cheaper and easier-to-use Whole Genome Sequencing methods to identify resistance-associated parasite mutations and formal guidelines to measure and interpret the results of antileishmanial susceptibility testing would be an important first step to address this issue. A decent number of new drugs with novel mechanisms of action and activity against isolates resistant to current agents are currently under (pre)clinical development. To optimize and safeguard their future efficacy, these compounds should ideally be combined with host-directed therapies or used in multi-drug regimens with carefully selected partner drugs. Particular concerns in this context include the short time-to-resistance for paromomycin in the lab and the risk of subtherapeutic exposure to miltefosine and liposomal amphotericin B in patients due to their long half-lives. Finally, strong political and programmatic commitments towards universal access to high-quality diagnostics and medicines remain essential to delay the emergence of resistance to new and current antileishmanial drugs.
Author Contributions
G-JW and SH conceptualized and designed the table of contents. G-JW, FD, LD, DB, BC, KVB, and SH wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
G-JW received funding in the past from the European Horizon’s 2020 Research and Innovation Program as part of the EuroLeish.Net Training Network under Marie Sklodowska-Curie grant agreement number 642609. KVB is supported by a fellowship awarded from the Research Council United Kingdom Grand Challenges Research Funder under grant agreement ‘A Global Network for Neglected Tropical Diseases’ grant number MR/P027989/1, the MRC CiC grant (number18014) and European Union’s Horizon 2020 (number 815622). SH was funded by the Fonds Wetenschappelijk Onderzoek Vlaanderen (FWO: project G051812N, G013118N and 12I0317N). LMPH is a partner of the Excellence Centre ‘Infla-Med’ (www.uantwerpen.be/infla-med).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Professor Simon Croft and Professor Guy Caljon for carefully revising the manuscript.
References
1. McGhee RB, Cosgrove WB. Biology and Physiology of the Lower Trypanosomatidae. Microbiol Rev (1980) 44(1):140–73. doi: 10.1128/mr.44.1.140-173.1980
2. WHO. Leishmaniasis (2021). Available at: https://www.who.int/health-topics/leishmaniasis#tab=tab_1.
3. Singh OP, Hasker E, Sacks D, Boelaert M, Sundar S. Asymptomatic Leishmania Infection: A New Challenge for Leishmania Control. Clin Infect Dis (2014) 58(10):1424–9. doi: 10.1093/cid/ciu102
4. Van der Auwera G, Dujardin JC. Species Typing in Dermal Leishmaniasis. Clin Microbiol Rev (2015) 28(2):265–94. doi: 10.1128/CMR.00104-14
5. Sereno D, Maia C, Aït-Oudhia K. Antimony Resistance and Environment: Elusive Links to Explore During Leishmania Life Cycle. Int J Parasitol Drugs Drug Resist (2012) 2:200–3. doi: 10.1016/j.ijpddr.2012.07.003
6. Kumar V, Mandal R, Das S, Kesari S, Dinesh DS, Pandey K, et al. Kala-Azar Elimination in a Highly-Endemic District of Bihar, India: A Success Story. PloS Negl Trop Dis (2020) 14(5):e0008254. doi: 10.1371/journal.pntd.0008254
7. Gedda MR, Singh B, Kumar D, Singh AK, Madhukar P, Upadhyay S, et al. Post Kala-Azar Dermal Leishmaniasis: A Threat to Elimination Program. PloS Negl Trop Dis (2020) 14(7):e0008221. doi: 10.1371/journal.pntd.0008221
8. Zijlstra EE, Kumar A, Sharma A, Rijal S, Mondal D, Routray S. Report of the Fifth Post-Kala-Azar Dermal Leishmaniasis Consortium Meeting, Colombo, Sri Lanka, 14–16 May 2018. Parasit Vectors (2020) 13(1):159. doi: 10.1186/s13071-020-04011-7
9. Croft SL, Yardley V, Kendrick H. Drug Sensitivity of Leishmania Species: Some Unresolved Problems. Trans R Soc Trop Med Hyg (2002) 96(Suppl 1):S127–9. doi: 10.1016/S0035-9203(02)90063-5
10. Hendrickx S, Guerin PJ, Caljon G, Croft SL, Maes L. Evaluating Drug Resistance in Visceral Leishmaniasis: The Challenges. Parasitology (2018) 145(4):453–63. doi: 10.1017/S0031182016002031
11. Van den Kerkhof M, Van Bockstal L, Gielis JF, Delputte P, Cos P, Maes L, et al. Impact of Primary Mouse Macrophage Cell Types on Leishmania Infection and In Vitro Drug Susceptibility. Parasitol Res (2018) 117(11):3601–12. doi: 10.1007/s00436-018-6059-4
12. Hendrickx S, Van Bockstal L, Caljon G, Maes L. In-Depth Comparison of Cell-Based Methodological Approaches to Determine Drug Susceptibility of Visceral Leishmania Isolates. PloS Negl Trop Dis (2019) 13(12):e0007885. doi: 10.1371/journal.pntd.0007885
13. Ponte-Sucre A, Gamarro F, Dujardin J-C, Barrett MP, López-Vélez R, García-Hernández R, et al. Drug Resistance and Treatment Failure in Leishmaniasis: A 21st Century Challenge. PloS Negl Trop Dis (2017) 11(12):e0006052. doi: 10.1371/journal.pntd.0006052
14. Natera S, Machuca C, Padrón-Nieves M, Romero A, Díaz E, Ponte-Sucre A. Leishmania Spp.: Proficiency of Drug-Resistant Parasites. Int J Antimicrob Agents (2007) 29(6):637–42. doi: 10.1016/j.ijantimicag.2007.01.004
15. Organization WH. Control of the Leishmaniasis: Report of a Meeting of the WHO Expert Committee on the Control of Leishmaniases, Geneva, 22-26 March 2010. Geneva: World Health Organization (2010) 2010(949).
16. Burza S, Croft SL, Boelaert M. Leishmaniasis. Lancet (2018) 392(10151):951–70. doi: 10.1016/S0140-6736(18)31204-2
17. Musa A, Khalil E, Hailu A, Olobo J, Balasegaram M, Omollo R, et al. Sodium Stibogluconate (SSG) & Paromomycin Combination Compared to SSG for Visceral Leishmaniasis in East Africa: A Randomised Controlled Trial. PloS Negl Trop Dis (2012) 6(6):e1674. doi: 10.1371/journal.pntd.0001674
18. Lira R, Sundar S, Makharia A, Kenney R, Gam A, Saraiva E, et al. Evidence That the High Incidence of Treatment Failures in Indian Kala-Azar is Due to the Emergence of Antimony-Resistant Strains of Leishmania Donovani. J Infect Dis (1999) 180(2):564–7. doi: 10.1086/314896
19. Rijal S, Yardley V, Chappuis F, Decuypere S, Khanal B, Singh R, et al. Antimonial Treatment of Visceral Leishmaniasis: Are Current In Vitro Susceptibility Assays Adequate for Prognosis of In Vivo Therapy Outcome? Microbes Infect (2007) 9(4):529–35. doi: 10.1016/j.micinf.2007.01.009
20. Rijal S, Chappuis F, Singh R, Bovier PA, Acharya P, Karki BM, et al. Treatment of Visceral Leishmaniasis in South-Eastern Nepal: Decreasing Efficacy of Sodium Stibogluconate and Need for a Policy to Limit Further Decline. Trans R Soc Trop Med Hyg (2003) 97(3):350–4. doi: 10.1016/S0035-9203(03)90167-2
21. Sundar S, More DK, Singh MK, Singh VP, Sharma S, Makharia A, et al. Failure of Pentavalent Antimony in Visceral Leishmaniasis in India: Report From the Center of the Indian Epidemic. Clin Infect Dis (2000) 31(4):1104–7. doi: 10.1086/318121
22. Chakraborti D, Rahman MM, Murrill M, Das R, Siddayya, Patil SG, et al. Environmental Arsenic Contamination and its Health Effects in a Historic Gold Mining Area of the Mangalur Greenstone Belt of Northeastern Karnataka, India. J Hazard Mater (2013) 262:1048–55. doi: 10.1016/j.jhazmat.2012.10.002
23. Perry MR, Prajapati VK, Menten J, Raab A, Feldmann J, Chakraborti D, et al. Arsenic Exposure and Outcomes of Antimonial Treatment in Visceral Leishmaniasis Patients in Bihar, India: A Retrospective Cohort Study. PloS Negl Trop Dis (2015) 9(3):e0003518. doi: 10.1371/journal.pntd.0003518
24. Ouakad M, Vanaerschot M, Rijal S, Sundar S, Speybroeck N, Kestens L, et al. Increased Metacyclogenesis of Antimony-Resistant Leishmania Donovani Clinical Lines. Parasitology (2011) 138(11):1392–9. doi: 10.1017/S0031182011001120
25. Vanaerschot M, De Doncker S, Rijal S, Maes L, Dujardin JC, Decuypere S. Antimonial Resistance in Leishmania Donovani is Associated With Increased In Vivo Parasite Burden. PloS One (2011) 6(8):e23120. doi: 10.1371/journal.pone.0023120
26. Vanaerschot M, Maes I, Ouakad M, Adaui V, Maes L, De Doncker S, et al. Linking In Vitro and In Vivo Survival of Clinical Leishmania Donovani Strains. PloS One (2010) 5(8):e12211. doi: 10.1371/journal.pone.0012211
27. de Moura TR, Santos ML, Braz JM, Santos LF, Aragão MT, de Oliveira FA, et al. Cross-Resistance of Leishmania Infantum Isolates to Nitric Oxide From Patients Refractory to Antimony Treatment, and Greater Tolerance to Antileishmanial Responses by Macrophages. Parasitol Res (2016) 115(2):713–21. doi: 10.1007/s00436-015-4793-4
28. Diotallevi A, Buffi G, Corbelli G, Ceccarelli M, Ortalli M, Varani S, et al. In Vitro Reduced Susceptibility to Pentavalent Antimonials of a Leishmania Infantum Isolate From a Human Cutaneous Leishmaniasis Case in Central Italy. Microorganisms (2021) 9(6). doi: 10.3390/microorganisms9061147
29. Eddaikra N, Ait-Oudhia K, Oury B, Farida M-M, Harrat Z, Denis S. Leishmania Antimony Resistance/ Susceptibility in Algerian Foci. Open J Trop Med (2017) 1. doi: 10.17352/ojtm.000005
30. Brito NC, Rabello A, Cota GF. Efficacy of Pentavalent Antimoniate Intralesional Infiltration Therapy for Cutaneous Leishmaniasis: A Systematic Review. PloS One (2017) 12(9):e0184777. doi: 10.1371/journal.pone.0184777
31. Firdous R, Yasinzai M, Ranja K. Efficacy of Glucantime in the Treatment of Old World Cutaneous Leishmaniasis. Int J Dermatol (2009) 48(7):758–62. doi: 10.1111/j.1365-4632.2009.04072.x
32. Firooz A, Khamesipour A, Ghoorchi MH, Nassiri-Kashani M, Eskandari SE, Khatami A, et al. Imiquimod in Combination With Meglumine Antimoniate for Cutaneous Leishmaniasis: A Randomized Assessor-Blind Controlled Trial. Arch Dermatol (2006) 142(12):1575–9. doi: 10.1001/archderm.142.12.1575
33. Monge-Maillo B, López-Vélez R. Therapeutic Options for Old World Cutaneous Leishmaniasis and New World Cutaneous and Mucocutaneous Leishmaniasis. Drugs (2013) 73(17):1889–920. doi: 10.1007/s40265-013-0132-1
34. van Griensven J, Gadisa E, Aseffa A, Hailu A, Beshah AM, Diro E. Treatment of Cutaneous Leishmaniasis Caused by Leishmania Aethiopica: A Systematic Review. PloS Negl Trop Dis (2016) 10(3):e0004495. doi: 10.1371/journal.pntd.0004495
35. Zerehsaz F, Salmanpour R, Handjani F, Ardehali S, Panjehshahin MR, Tabei SZ, et al. A Double-Blind Randomized Clinical Trial of a Topical Herbal Extract (Z-HE) vs. Systemic Meglumine Antimoniate for the Treatment of Cutaneous Leishmaniasis in Iran. Int J Dermatol (1999) 38(8):610–2. doi: 10.1046/j.1365-4362.1999.00727.x
36. Chakravarty J, Sundar S. Current and Emerging Medications for the Treatment of Leishmaniasis. Expert Opin Pharmacother (2019) 20(10):1251–65. doi: 10.1080/14656566.2019.1609940
37. Arevalo J, Ramirez L, Adaui V, Zimic M, Tulliano G, Miranda-Verástegui C, et al. Influence of Leishmania (Viannia) Species on the Response to Antimonial Treatment in Patients With American Tegumentary Leishmaniasis. J Infect Dis (2007) 195(12):1846–51. doi: 10.1086/518041
38. Castro MDM, Cossio A, Velasco C, Osorio L. Risk Factors for Therapeutic Failure to Meglumine Antimoniate and Miltefosine in Adults and Children With Cutaneous Leishmaniasis in Colombia: A Cohort Study. PloS Negl Trop Dis (2017) 11(4):e0005515. doi: 10.1371/journal.pntd.0005515
39. Llanos-Cuentas A, Tulliano G, Araujo-Castillo R, Miranda-Verastegui C, Santamaria-Castrellon G, Ramirez L, et al. Clinical and Parasite Species Risk Factors for Pentavalent Antimonial Treatment Failure in Cutaneous Leishmaniasis in Peru. Clin Infect Dis (2008) 46(2):223–31. doi: 10.1086/524042
40. de Morais-Teixeira E, Gallupo MK, Rodrigues LF, Romanha AJ, Rabello A. In Vitro Interaction Between Paromomycin Sulphate and Four Drugs With Leishmanicidal Activity Against Three New World Leishmania Species. J Antimicrob Chemother (2014) 69(1):150–4. doi: 10.1093/jac/dkt318
41. Fernández OL, Diaz-Toro Y, Ovalle C, Valderrama L, Muvdi S, Rodríguez I, et al. Miltefosine and Antimonial Drug Susceptibility of Leishmania Viannia Species and Populations in Regions of High Transmission in Colombia. PloS Negl Trop Dis (2014) 8(5):e2871.
42. Rojas R, Valderrama L, Valderrama M, Varona MX, Ouellette M, Saravia NG. Resistance to Antimony and Treatment Failure in Human Leishmania (Viannia) Infection. J Infect Dis (2006) 193(10):1375–83. doi: 10.1086/503371
43. Haldar AK, Sen P, Roy S. Use of Antimony in the Treatment of Leishmaniasis: Current Status and Future Directions. Mol Biol Int (2011) 2011:571242. doi: 10.4061/2011/571242
44. Denton H, McGregor JC, Coombs GH. Reduction of Anti-Leishmanial Pentavalent Antimonial Drugs by a Parasite-Specific Thiol-Dependent Reductase, TDR1. Biochem J (2004) 381(Pt 2):405–12. doi: 10.1042/BJ20040283
45. Zhou Y, Messier N, Ouellette M, Rosen BP, Mukhopadhyay R. Leishmania Major LmACR2 is a Pentavalent Antimony Reductase That Confers Sensitivity to the Drug Pentostam. J Biol Chem (2004) 279(36):37445–51. doi: 10.1074/jbc.M404383200
46. Demicheli C, Frézard F, Lecouvey M, Garnier-Suillerot A. Antimony(V) Complex Formation With Adenine Nucleosides in Aqueous Solution. Biochim Biophys Acta (2002) 1570(3):192–8. doi: 10.1016/S0304-4165(02)00198-8
47. Ferreira Cdos S, Martins PS, Demicheli C, Brochu C, Ouellette M, Frézard F. Thiol-Induced Reduction of Antimony(V) Into Antimony(III): A Comparative Study With Trypanothione, Cysteinyl-Glycine, Cysteine and Glutathione. Biometals (2003) 16(3):441–6. doi: 10.1023/a:1022823605068
48. Yan S, Li F, Ding K, Sun H. Reduction of Pentavalent Antimony by Trypanothione and Formation of a Binary and Ternary Complex of Antimony(III) and Trypanothione. J Biol Inorg Chem (2003) 8(6):689–97. doi: 10.1007/s00775-003-0468-1
49. El Fadili K, Messier N, Leprohon P, Roy G, Guimond C, Trudel N, et al. Role of the ABC Transporter MRPA (PGPA) in Antimony Resistance in Leishmania Infantum Axenic and Intracellular Amastigotes. Antimicrob Agents Chemother (2005) 49(5):1988–93. doi: 10.1128/AAC.49.5.1988-1993.2005
50. Légaré D, Richard D, Mukhopadhyay R, Stierhof YD, Rosen BP, Haimeur A, et al. The Leishmania ATP-Binding Cassette Protein PGPA is an Intracellular Metal-Thiol Transporter ATPase. J Biol Chem (2001) 276(28):26301–7. doi: 10.1074/jbc.M102351200
51. Moreira DS, Monte Neto RL, Andrade JM, Santi AM, Reis PG, Frézard F, et al. Molecular Characterization of the MRPA Transporter and Antimony Uptake in Four New World Leishmania Spp. Susceptible and Resistant to Antimony. Int J Parasitol Drugs Drug Resist (2013) 3:143–53. doi: 10.1016/j.ijpddr.2013.08.001
52. Mukherjee B, Mukhopadhyay R, Bannerjee B, Chowdhury S, Mukherjee S, Naskar K, et al. Antimony-Resistant But Not Antimony-Sensitive Leishmania Donovani Up-Regulates Host IL-10 to Overexpress Multidrug-Resistant Protein 1. Proc Natl Acad Sci USA (2013) 110(7):E575–82. doi: 10.1073/pnas.1213839110
53. Maharjan M, Singh S, Chatterjee M, Madhubala R. Role of Aquaglyceroporin (AQP1) Gene and Drug Uptake in Antimony-Resistant Clinical Isolates of Leishmania Donovani. Am J Trop Med Hyg (2008) 79(1):69–75. doi: 10.4269/ajtmh.2008.79.69
54. Monte-Neto R, Laffitte MC, Leprohon P, Reis P, Frézard F, Ouellette M. Intrachromosomal Amplification, Locus Deletion and Point Mutation in the Aquaglyceroporin AQP1 Gene in Antimony Resistant Leishmania (Viannia) Guyanensis. PloS Negl Trop Dis (2015) 9(2):e0003476. doi: 10.1371/journal.pntd.0003476
55. Rugani JN, Gontijo CMF, Frézard F, Soares RP, Monte-Neto RLD. Antimony Resistance in Leishmania (Viannia) Braziliensis Clinical Isolates From Atypical Lesions Associates With Increased ARM56/ARM58 Transcripts and Reduced Drug Uptake. Mem Inst Oswaldo Cruz (2019) 114:e190111. doi: 10.1590/0074-02760190111
56. Mandal G, Mandal S, Sharma M, Charret KS, Papadopoulou B, Bhattacharjee H, et al. Species-Specific Antimonial Sensitivity in Leishmania is Driven by Post-Transcriptional Regulation of AQP1. PloS Negl Trop Dis (2015) 9(2):e0003500. doi: 10.1371/journal.pntd.0003500
57. Dhillon GP, Sharma SN, Nair B. Kala-Azar Elimination Programme in India. J Indian Med Assoc (2008) 106(10):664, 6–, 8.
58. Sundar S, Murray HW. Availability of Miltefosine for the Treatment of Kala-Azar in India. Bull World Health Organ (2005) 83(5):394–5.
59. Sundar S, Jha TK, Thakur CP, Engel J, Sindermann H, Fischer C, et al. Oral Miltefosine for Indian Visceral Leishmaniasis. N Engl J Med (2002) 347(22):1739–46. doi: 10.1056/NEJMoa021556
60. Sundar S, Singh A, Rai M, Prajapati VK, Singh AK, Ostyn B, et al. Efficacy of Miltefosine in the Treatment of Visceral Leishmaniasis in India After a Decade of Use. Clin Infect Dis (2012) 55(4):543–50. doi: 10.1093/cid/cis474
61. Rijal S, Ostyn B, Uranw S, Rai K, Bhattarai NR, Dorlo TP, et al. Increasing Failure of Miltefosine in the Treatment of Kala-Azar in Nepal and the Potential Role of Parasite Drug Resistance, Reinfection, or Noncompliance. Clin Infect Dis (2013) 56(11):1530–8. doi: 10.1093/cid/cit102
62. Prajapati VK, Sharma S, Rai M, Ostyn B, Salotra P, Vanaerschot M, et al. In Vitro Susceptibility of Leishmania Donovani to Miltefosine in Indian Visceral Leishmaniasis. Am J Trop Med Hyg (2013) 89(4):750–4. doi: 10.4269/ajtmh.13-0096
63. Deep DK, Singh R, Bhandari V, Verma A, Sharma V, Wajid S, et al. Increased Miltefosine Tolerance in Clinical Isolates of Leishmania Donovani is Associated With Reduced Drug Accumulation, Increased Infectivity and Resistance to Oxidative Stress. PloS Negl Trop Dis (2017) 11(6):e0005641. doi: 10.1371/journal.pntd.0005641
64. Bhandari V, Kulshrestha A, Deep DK, Stark O, Prajapati VK, Ramesh V, et al. Drug Susceptibility in Leishmania Isolates Following Miltefosine Treatment in Cases of Visceral Leishmaniasis and Post Kala-Azar Dermal Leishmaniasis. PloS Negl Trop Dis (2012) 6(5):e1657. doi: 10.1371/journal.pntd.0001657
65. Rai K, Cuypers B, Bhattarai NR, Uranw S, Berg M, Ostyn B, et al. Relapse After Treatment With Miltefosine for Visceral Leishmaniasis is Associated With Increased Infectivity of the Infecting Leishmania Donovani Strain. mBio (2013) 4(5):e00611–13. doi: 10.1128/mBio.00611-13
66. Das M, Saudagar P, Sundar S, Dubey VK. Miltefosine-Unresponsive Leishmania Donovani has a Greater Ability Than Miltefosine-Responsive L. Donovani to Resist Reactive Oxygen Species. FEBS J (2013) 280(19):4807–15. doi: 10.1111/febs.12449
67. Ramesh V, Singh R, Avishek K, Verma A, Deep DK, Verma S, et al. Decline in Clinical Efficacy of Oral Miltefosine in Treatment of Post Kala-Azar Dermal Leishmaniasis (PKDL) in India. PloS Negl Trop Dis (2015) 9(10):e0004093. doi: 10.1371/journal.pntd.0004093
68. Ramesh V, Katara GK, Verma S, Salotra P. Miltefosine as an Effective Choice in the Treatment of Post-Kala-Azar Dermal Leishmaniasis. Br J Dermatol (2011) 165(2):411–4. doi: 10.1111/j.1365-2133.2011.10402.x
69. Pijpers J, den Boer ML, Essink DR, Ritmeijer K. The Safety and Efficacy of Miltefosine in the Long-Term Treatment of Post-Kala-Azar Dermal Leishmaniasis in South Asia - A Review and Meta-Analysis. PloS Negl Trop Dis (2019) 13(2):e0007173. doi: 10.1371/journal.pntd.0007173
70. van Griensven J, Balasegaram M, Meheus F, Alvar J, Lynen L, Boelaert M. Combination Therapy for Visceral Leishmaniasis. Lancet Infect Dis (2010) 10(3):184–94. doi: 10.1016/S1473-3099(10)70011-6
71. Carnielli JBT, Monti-Rocha R, Costa DL, Molina Sesana A, Pansini LNN, Segatto M, et al. Natural Resistance of Leishmania Infantum to Miltefosine Contributes to the Low Efficacy in the Treatment of Visceral Leishmaniasis in Brazil. Am J Trop Med Hyg (2019) 101(4):789–94. doi: 10.4269/ajtmh.18-0949
72. Carnielli JBT, Crouch K, Forrester S, Silva VC, Carvalho SFG, Damasceno JD, et al. A Leishmania Infantum Genetic Marker Associated With Miltefosine Treatment Failure for Visceral Leishmaniasis. EBioMedicine (2018) 36:83–91. doi: 10.1016/j.ebiom.2018.09.029
73. Bhattacharya A, Ouellette M. New Insights With Miltefosine Unresponsiveness in Brazilian Leishmania Infantum Isolates. EBioMedicine (2018) 37:13–4. doi: 10.1016/j.ebiom.2018.10.016
74. Aronson N, Herwaldt BL, Libman M, Pearson R, Lopez-Velez R, Weina P, et al. Diagnosis and Treatment of Leishmaniasis: Clinical Practice Guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Am J Trop Med Hyg (2017) 96(1):24–45. doi: 10.4269/ajtmh.16-84256
75. Obonaga R, Fernández OL, Valderrama L, Rubiano LC, Castro Mdel M, Barrera MC, et al. Treatment Failure and Miltefosine Susceptibility in Dermal Leishmaniasis Caused by Leishmania Subgenus Viannia Species. Antimicrob Agents Chemother (2014) 58(1):144–52. doi: 10.1128/AAC.01023-13
76. Dorlo TP, Balasegaram M, Beijnen JH, de Vries PJ. Miltefosine: A Review of its Pharmacology and Therapeutic Efficacy in the Treatment of Leishmaniasis. J Antimicrob Chemother (2012) 67(11):2576–97. doi: 10.1093/jac/dks275
77. Rakotomanga M, Blanc S, Gaudin K, Chaminade P, Loiseau PM. Miltefosine Affects Lipid Metabolism in Leishmania Donovani Promastigotes. Antimicrob Agents Chemother (2007) 51(4):1425–30. doi: 10.1128/AAC.01123-06
78. Khademvatan S, Gharavi MJ, Rahim F, Saki J. Miltefosine-Induced Apoptotic Cell Death on Leishmania Major and L. Tropica Strains. Korean J Parasitol (2011) 49(1):17–23. doi: 10.3347/kjp.2011.49.1.17
79. Luque-Ortega JR, Rivas L. Miltefosine (Hexadecylphosphocholine) Inhibits Cytochrome C Oxidase in Leishmania Donovani Promastigotes. Antimicrob Agents Chemother (2007) 51(4):1327–32. doi: 10.1128/AAC.01415-06
80. Marinho Fde A, Gonçalves KC, Oliveira SS, Oliveira AC, Bellio M, d'Avila-Levy CM, et al. Miltefosine Induces Programmed Cell Death in Leishmania Amazonensis Promastigotes. Mem Inst Oswaldo Cruz (2011) 106(4):507–9. doi: 10.1590/s0074-02762011000400021
81. Paris C, Loiseau PM, Bories C, Bréard J. Miltefosine Induces Apoptosis-Like Death in Leishmania Donovani Promastigotes. Antimicrob Agents Chemother (2004) 48(3):852–9. doi: 10.1128/AAC.48.3.852-859.2004
82. Verma NK, Dey CS. Possible Mechanism of Miltefosine-Mediated Death of Leishmania Donovani. Antimicrob Agents Chemother (2004) 48(8):3010–5. doi: 10.1128/AAC.48.8.3010-3015.2004
83. Wadhone P, Maiti M, Agarwal R, Kamat V, Martin S, Saha B. Miltefosine Promotes IFN-Gamma-Dominated Anti-Leishmanial Immune Response. J Immunol (2009) 182(11):7146–54. doi: 10.4049/jimmunol.0803859
84. Zeisig R, Rudolf M, Eue I, Arndt D. Influence of Hexadecylphosphocholine on the Release of Tumor Necrosis Factor and Nitroxide From Peritoneal Macrophages In Vitro. J Cancer Res Clin Oncol (1995) 121(2):69–75. doi: 10.1007/BF01202215
85. Pinto-Martinez AK, Rodriguez-Durán J, Serrano-Martin X, Hernandez-Rodriguez V, Benaim G. Mechanism of Action of Miltefosine on Leishmania Donovani Involves the Impairment of Acidocalcisome Function and the Activation of the Sphingosine-Dependent Plasma Membrane Ca(2+) Channel. Antimicrob Agents Chemother (2018) 62(1). doi: 10.1128/AAC.01614-17
86. Khanra S, Sarraf NR, Das AK, Roy S, Manna M. Miltefosine Resistant Field Isolate From Indian Kala-Azar Patient Shows Similar Phenotype in Experimental Infection. Sci Rep (2017) 7(1):10330. doi: 10.1038/s41598-017-09720-1
87. Pérez-Victoria FJ, Sánchez-Cañete MP, Castanys S, Gamarro F. Phospholipid Translocation and Miltefosine Potency Require Both L. Donovani Miltefosine Transporter and the New Protein LdRos3 in Leishmania Parasites. J Biol Chem (2006) 281(33):23766–75. doi: 10.1074/jbc.M605214200
88. Pérez-Victoria FJ, Castanys S, Gamarro F. Leishmania Donovani Resistance to Miltefosine Involves a Defective Inward Translocation of the Drug. Antimicrob Agents Chemother (2003) 47(8):2397–403. doi: 10.1128/AAC.47.8.2397-2403.2003
89. Mondelaers A, Sanchez-Cañete MP, Hendrickx S, Eberhardt E, Garcia-Hernandez R, Lachaud L, et al. Genomic and Molecular Characterization of Miltefosine Resistance in Leishmania Infantum Strains With Either Natural or Acquired Resistance Through Experimental Selection of Intracellular Amastigotes. PloS One (2016) 11(4):e0154101. doi: 10.1371/journal.pone.0154101
90. Laffitte MC, Leprohon P, Légaré D, Ouellette M. Deep-Sequencing Revealing Mutation Dynamics in the Miltefosine Transporter Gene in Leishmania Infantum Selected for Miltefosine Resistance. Parasitol Res (2016) 115(10):3699–703. doi: 10.1007/s00436-016-5195-y
91. Hendrickx S, Van Bockstal L, Bulté D, Mondelaers A, Aslan H, Rivas L, et al. Phenotypic Adaptations of Leishmania Donovani to Recurrent Miltefosine Exposure and Impact on Sand Fly Infection. Parasit Vectors (2020) 13(1):96. doi: 10.1186/s13071-020-3972-z
92. Cojean S, Houze S, Haouchine D, Huteau F, Lariven S, Hubert V, et al. Leishmania Resistance to Miltefosine Associated With Genetic Marker. Emerg Infect Dis (2012) 18(4):704–6. doi: 10.3201/eid1804.110841
93. Srivastava S, Mishra J, Gupta AK, Singh A, Shankar P, Singh S. Laboratory Confirmed Miltefosine Resistant Cases of Visceral Leishmaniasis From India. Parasit Vectors (2017) 10(1):49. doi: 10.1186/s13071-017-1969-z
94. Van Bockstal L, Bulté D, Hendrickx S, Sadlova J, Volf P, Maes L, et al. Impact of Clinically Acquired Miltefosine Resistance by Leishmania Infantum on Mouse and Sand Fly Infection. Int J Parasitol Drugs Drug Resist (2020) 13:16–21. doi: 10.1016/j.ijpddr.2020.04.004
95. Eberhardt E, Bulté D, Van Bockstal L, Van den Kerkhof M, Cos P, Delputte P, et al. Miltefosine Enhances the Fitness of a non-Virulent Drug-Resistant Leishmania Infantum Strain. J Antimicrob Chemother (2019) 74(2):395–406. doi: 10.1093/jac/dky450
96. Bulté D, Van Bockstal L, Dirkx L, Van den Kerkhof M, De Trez C, Timmermans JP, et al. Miltefosine Enhances Infectivity of a Miltefosine-Resistant Leishmania Infantum Strain by Attenuating its Innate Immune Recognition. PloS Negl Trop Dis (2021) 15(7):e0009622. doi: 10.1371/journal.pntd.0009622
97. Turner KG, Vacchina P, Robles-Murguia M, Wadsworth M, McDowell MA, Morales MA. Fitness and Phenotypic Characterization of Miltefosine-Resistant Leishmania Major. PloS Negl Trop Dis (2015) 9(7):e0003948. doi: 10.1371/journal.pntd.0003948
98. Rubiano LC, Miranda MC, Muvdi Arenas S, Montero LM, Rodríguez-Barraquer I, Garcerant D, et al. Noninferiority of Miltefosine Versus Meglumine Antimoniate for Cutaneous Leishmaniasis in Children. J Infect Dis (2012) 205(4):684–92. doi: 10.1093/infdis/jir816
99. Berger BA, Cossio A, Saravia NG, Castro MDM, Prada S, Bartlett AH, et al. Cost-Effectiveness of Meglumine Antimoniate Versus Miltefosine Caregiver DOT for the Treatment of Pediatric Cutaneous Leishmaniasis. PloS Negl Trop Dis (2017) 11(4):e0005459. doi: 10.1371/journal.pntd.0005459
100. Sundar S, Chakravarty J. Liposomal Amphotericin B and Leishmaniasis: Dose and Response. J Glob Infect Dis (2010) 2(2):159–66. doi: 10.4103/0974-777X.62886
101. Roatt BM, Aguiar-Soares RD, Coura-Vital W, Ker HG, Moreira N, Vitoriano-Souza J, et al. Immunotherapy and Immunochemotherapy in Visceral Leishmaniasis: Promising Treatments for This Neglected Disease. Front Immunol (2014) 5:272. doi: 10.3389/fimmu.2014.00272
102. Escobar P, Matu S, Marques C, Croft SL. Sensitivities of Leishmania Species to Hexadecylphosphocholine (Miltefosine), ET-18-OCH(3) (Edelfosine) and Amphotericin B. Acta Trop (2002) 81(2):151–7. doi: 10.1016/S0001-706X(01)00197-8
103. Franco-Muñoz C, Manjarrés-Estremor M, Ovalle-Bracho C. Intraspecies Differences in Natural Susceptibility to Amphotericine B of Clinical Isolates of Leishmania Subgenus Viannia. PloS One (2018) 13(4):e0196247. doi: 10.1371/journal.pone.0196247
104. Lachaud L, Bourgeois N, Plourde M, Leprohon P, Bastien P, Ouellette M. Parasite Susceptibility to Amphotericin B in Failures of Treatment for Visceral Leishmaniasis in Patients Coinfected With HIV Type 1 and Leishmania Infantum. Clin Infect Dis (2009) 48(2):e16–22. doi: 10.1086/595710
105. Seifert K, Munday J, Syeda T, Croft SL. In Vitro Interactions Between Sitamaquine and Amphotericin B, Sodium Stibogluconate, Miltefosine, Paromomycin and Pentamidine Against Leishmania Donovani. J Antimicrob Chemother (2011) 66(4):850–4. doi: 10.1093/jac/dkq542
106. Adler-Moore JP, Gangneux J-P, Pappas PG. Comparison Between Liposomal Formulations of Amphotericin B. Med Mycol (2016) 54(3):223–31. doi: 10.1093/mmy/myv111
107. Burza S, Sinha PK, Mahajan R, Lima MA, Mitra G, Verma N, et al. Five-Year Field Results and Long-Term Effectiveness of 20 Mg/Kg Liposomal Amphotericin B (Ambisome) for Visceral Leishmaniasis in Bihar, India. PloS Negl Trop Dis (2014) 8(1):e2603. doi: 10.1371/journal.pntd.0002603
108. Mondal D, Alvar J, Hasnain MG, Hossain MS, Ghosh D, Huda MM, et al. Efficacy and Safety of Single-Dose Liposomal Amphotericin B for Visceral Leishmaniasis in a Rural Public Hospital in Bangladesh: A Feasibility Study. Lancet Glob Health (2014) 2(1):e51–7. doi: 10.1016/S2214-109X(13)70118-9
109. Khalil EAG, Weldegebreal T, Younis BM, Omollo R, Musa AM, Hailu W, et al. Safety and Efficacy of Single Dose Versus Multiple Doses of AmBisome® for Treatment of Visceral Leishmaniasis in Eastern Africa: A Randomised Trial. PloS Negl Trop Dis (2014) 8(1):e2613. doi: 10.1371/journal.pntd.0002613
110. Salih NA, van Griensven J, Chappuis F, Antierens A, Mumina A, Hammam O, et al. Liposomal Amphotericin B for Complicated Visceral Leishmaniasis (Kala-Azar) in Eastern Sudan: How Effective is Treatment for This Neglected Disease? Trop Med Int Health (2014) 19(2):146–52. doi: 10.1111/tmi.12238
111. Tamiru A, Tigabu B, Yifru S, Diro E, Hailu A. Safety and Efficacy of Liposomal Amphotericin B for Treatment of Complicated Visceral Leishmaniasis in Patients Without HIV, North-West Ethiopia. BMC Infect Diseases (2016) 16(1):548. doi: 10.1186/s12879-016-1746-1
112. Musa AM, Khalil EA, Mahgoub FA, Hamad S, Elkadaru AM, El Hassan AM. Efficacy of Liposomal Amphotericin B (AmBisome) in the Treatment of Persistent Post-Kala-Azar Dermal Leishmaniasis (PKDL). Ann Trop Med Parasitol (2005) 99(6):563–9. doi: 10.1179/136485905X514127
113. den Boer M, Das AK, Akhter F, Burza S, Ramesh V, Ahmed B-N, et al. Safety and Effectiveness of Short-Course AmBisome in the Treatment of Post–Kala-Azar Dermal Leishmaniasis: A Prospective Cohort Study in Bangladesh. Clin Infect Dis (2018) 67(5):667–75. doi: 10.1093/cid/ciy172
114. Wortmann G, Zapor M, Ressner R, Fraser S, Hartzell J, Pierson J, et al. Lipsosomal Amphotericin B for Treatment of Cutaneous Leishmaniasis. Am J Trop Med Hyg (2010) 83(5):1028–33. doi: 10.4269/ajtmh.2010.10-0171
115. Shirzadi MR. Lipsosomal Amphotericin B: A Review of its Properties, Function, and Use for Treatment of Cutaneous Leishmaniasis. Res Rep Trop Med (2019) 10:11–8. doi: 10.2147/RRTM.S200218
116. Ubals M, Bosch-Nicolau P, Sánchez-Montalvá A, Salvador F, Aparicio-Español G, Sulleiro E, et al. Treatment of Complex Cutaneous Leishmaniasis With Liposomal Amphotericin B. Pathogens (2021) 10(10). doi: 10.3390/pathogens10101253
117. Pedras MJ, Carvalho JP, Silva RED, Ramalho DB, Senna MCR, Moreira HSA, et al. Mucosal Leishmaniasis: The Experience of a Brazilian Referral Center. Rev Soc Bras Med Trop (2018) 51(3):318–23. doi: 10.1590/0037-8682-0478-2017
118. Solomon M, Baum S, Barzilai A, Scope A, Trau H, Schwartz E. Liposomal Amphotericin B in Comparison to Sodium Stibogluconate for Cutaneous Infection Due to Leishmania Braziliensis. J Am Acad Dermatol (2007) 56(4):612–6. doi: 10.1016/j.jaad.2006.06.044
119. Motta JO, Sampaio RN. A Pilot Study Comparing Low-Dose Liposomal Amphotericin B With N-Methyl Glucamine for the Treatment of American Cutaneous Leishmaniasis. J Eur Acad Dermatol Venereol (2012) 26(3):331–5. doi: 10.1111/j.1468-3083.2011.04070.x
120. Amato VS, Tuon FF, Camargo RA, Souza RM, Santos CR, Nicodemo AC. Can We Use a Lower Dose of Liposomal Amphotericin B for the Treatment of Mucosal American Leishmaniasis? Am J Trop Med Hyg (2011) 85(5):818–9. doi: 10.4269/ajtmh.2011.11-0287
121. Santos CR, Tuon FF, Cieslinski J, de Souza RM, Imamura R, Amato VS. Comparative Study on Liposomal Amphotericin B and Other Therapies in the Treatment of Mucosal Leishmaniasis: A 15-Year Retrospective Cohort Study. PloS One (2019) 14(6):e0218786. doi: 10.1371/journal.pone.0218786
122. Barrett MP, Croft SL. Management of Trypanosomiasis and Leishmaniasis. Br Med Bull (2012) 104(1):175–96. doi: 10.1093/bmb/lds031
123. Eichenberger A, Buechi AE, Neumayr A, Hatz C, Rauch A, Huguenot M, et al. A Severe Case of Visceral Leishmaniasis and Liposomal Amphotericin B Treatment Failure in an Immunosuppressed Patient 15 Years After Exposure. BMC Infect Diseases (2017) 17(1):81. doi: 10.1186/s12879-017-2192-4
124. Morizot G, Jouffroy R, Faye A, Chabert P, Belhouari K, Calin R, et al. Antimony to Cure Visceral Leishmaniasis Unresponsive to Liposomal Amphotericin B. PloS Negl Trop Dis (2016) 10(1):e0004304. doi: 10.1371/journal.pntd.0004304
125. Mueller M, Ritmeijer K, Balasegaram M, Koummuki Y, Santana MR, Davidson R. Unresponsiveness to AmBisome in Some Sudanese Patients With Kala-Azar. Trans R Soc Trop Med Hyg (2007) 101(1):19–24. doi: 10.1016/j.trstmh.2006.02.005
126. Srivastava P, Prajapati VK, Rai M, Sundar S. Unusual Case of Resistance to Amphotericin B in Visceral Leishmaniasis in a Region in India Where Leishmaniasis is Not Endemic. J Clin Microbiol (2011) 49(8):3088–91. doi: 10.1128/JCM.00173-11
127. Bansal R, Sen SS, Muthuswami R, Madhubala R. Stigmasterol as a Potential Biomarker for Amphotericin B Resistance in Leishmania Donovani. J Antimicrob Chemother (2020) 75(4):942–50. doi: 10.1093/jac/dkz515
128. Purkait B, Kumar A, Nandi N, Sardar AH, Das S, Kumar S, et al. Mechanism of Amphotericin B Resistance in Clinical Isolates of Leishmania Donovani. Antimicrob Agents Chemother (2012) 56(2):1031–41. doi: 10.1128/AAC.00030-11
129. Mwenechanya R, Kovářová J, Dickens NJ, Mudaliar M, Herzyk P, Vincent IM, et al. Sterol 14α-Demethylase Mutation Leads to Amphotericin B Resistance in Leishmania Mexicana. PloS Negl Trop Dis (2017) 11(6):e0005649. doi: 10.1371/journal.pntd.0005649
130. Davidson RN, den Boer M, Ritmeijer K. Paromomycin. Trans R Soc Trop Med Hyg (2009) 103(7):653–60. doi: 10.1016/j.trstmh.2008.09.008
131. Sinha PK, Jha TK, Thakur CP, Nath D, Mukherjee S, Aditya AK, et al. Phase 4 Pharmacovigilance Trial of Paromomycin Injection for the Treatment of Visceral Leishmaniasis in India. J Trop Med (2011) 2011:645203. doi: 10.1155/2011/645203
132. Sundar S, Chakravarty J. Paromomycin in the Treatment of Leishmaniasis. Expert Opin Investig Drugs (2008) 17(5):787–94. doi: 10.1517/13543784.17.5.787
133. Sundar S, Jha TK, Thakur CP, Sinha PK, Bhattacharya SK. Injectable Paromomycin for Visceral Leishmaniasis in India. N Engl J Med (2007) 356(25):2571–81. doi: 10.1056/NEJMoa066536
134. Pokharel P, Ghimire R, Lamichhane P. Efficacy and Safety of Paromomycin for Visceral Leishmaniasis: A Systematic Review. J Trop Med (2021) 2021:8629039. doi: 10.1155/2021/8629039
135. Musa AM, Younis B, Fadlalla A, Royce C, Balasegaram M, Wasunna M, et al. Paromomycin for the Treatment of Visceral Leishmaniasis in Sudan: A Randomized, Open-Label, Dose-Finding Study. PloS Negl Trop Dis (2010) 4(10):e855. doi: 10.1371/journal.pntd.0000855
136. Hailu A, Musa A, Wasunna M, Balasegaram M, Yifru S, Mengistu G, et al. Geographical Variation in the Response of Visceral Leishmaniasis to Paromomycin in East Africa: A Multicentre, Open-Label, Randomized Trial. PloS Negl Trop Dis (2010) 4(10):e709. doi: 10.1371/journal.pntd.0000709
137. Hendrickx S, Boulet G, Mondelaers A, Dujardin JC, Rijal S, Lachaud L, et al. Experimental Selection of Paromomycin and Miltefosine Resistance in Intracellular Amastigotes of Leishmania Donovani and L. Infantum. Parasitol Res (2014) 113(5):1875–81. doi: 10.1007/s00436-014-3835-7
138. Correia D, Macêdo VO, Carvalho EM, Barral A, Magalhães AV, de Abreu MV, et al. Comparative Study of Meglumine Antimoniate, Pentamidine Isethionate and Aminosidine Sulfate in the Treatment of Primary Skin Lesions Caused by Leishmania (Viannia) Braziliensis. Rev Soc Bras Med Trop (1996) 29(5):447–53. doi: 10.1590/S0037-86821996000500007
139. Hepburn NC, Tidman MJ, Hunter JA. Aminosidine (Paromomycin) Versus Sodium Stibogluconate for the Treatment of American Cutaneous Leishmaniasis. Trans R Soc Trop Med Hyg (1994) 88(6):700–3. doi: 10.1016/0035-9203(94)90237-2
140. Kim DH, Chung HJ, Bleys J, Ghohestani RF. Is Paromomycin an Effective and Safe Treatment Against Cutaneous Leishmaniasis? A Meta-Analysis of 14 Randomized Controlled Trials. PloS Negl Trop Dis (2009) 3(2):e381. doi: 10.1371/journal.pntd.0000381
141. Ben Salah A, Ben Messaoud N, Guedri E, Zaatour A, Ben Alaya N, Bettaieb J, et al. Topical Paromomycin With or Without Gentamicin for Cutaneous Leishmaniasis. N Engl J Med (2013) 368(6):524–32. doi: 10.1056/NEJMoa1202657
142. Sosa N, Pascale JM, Jiménez AI, Norwood JA, Kreishman-Detrick M, Weina PJ, et al. Topical Paromomycin for New World Cutaneous Leishmaniasis. PloS Negl Trop Dis (2019) 13(5):e0007253. doi: 10.1371/journal.pntd.0007253
143. Soto J, Soto P, Ajata A, Luque C, Tintaya C, Paz D, et al. Topical 15% Paromomycin-Aquaphilic for Bolivian Leishmania Braziliensiscutaneous Leishmaniasis: A Randomized, Placebo-Controlled Trial. Clin Infect Dis (2019) 68(5):844–9. doi: 10.1093/cid/ciy619
144. Coser EM, Ferreira BA, Branco N, Yamashiro-Kanashiro EH, Lindoso JAL, Coelho AC. Activity of Paromomycin Against Leishmania Amazonensis: Direct Correlation Between Susceptibility In Vitro and the Treatment Outcome In Vivo. Int J Parasitol Drugs Drug Resist (2020) 14:91–8. doi: 10.1016/j.ijpddr.2020.08.001
145. Coser EM, Ferreira BA, Yamashiro-Kanashiro EH, Lindoso JAL, Coelho AC. Susceptibility to Paromomycin in Clinical Isolates and Reference Strains of Leishmania Species Responsible for Tegumentary Leishmaniasis in Brazil. Acta Trop (2021) 215:105806. doi: 10.1016/j.actatropica.2020.105806
146. Utaile M, Kassahun A, Abebe T, Hailu A. Susceptibility of Clinical Isolates of Leishmania Aethiopica to Miltefosine, Paromomycin, Amphotericin B and Sodium Stibogluconate Using Amastigote-Macrophage In Vitro Model. Exp Parasitol (2013) 134(1):68–75. doi: 10.1016/j.exppara.2013.01.022
147. Maarouf M, Lawrence F, Brown S, Robert-Gero M. Biochemical Alterations in Paromomycin-Treated Leishmania Donovani Promastigotes. Parasitol Res (1997) 83(2):198–202. doi: 10.1006/excr.1997.3500
148. Maarouf M, de Kouchkovsky Y, Brown S, Petit PX, Robert-Gero M. In Vivo Interference of Paromomycin With Mitochondrial Activity of Leishmania. Exp Cell Res (1997) 232(2):339–48. doi: 10.1006/excr.1997.3500
149. Chawla B, Jhingran A, Panigrahi A, Stuart KD, Madhubala R. Paromomycin Affects Translation and Vesicle-Mediated Trafficking as Revealed by Proteomics of Paromomycin -Susceptible -Resistant Leishmania Donovani. PloS One (2011) 6(10):e26660. doi: 10.1371/journal.pone.0026660
150. Hendrickx S, Leemans A, Mondelaers A, Rijal S, Khanal B, Dujardin JC, et al. Comparative Fitness of a Parent Leishmania Donovani Clinical Isolate and its Experimentally Derived Paromomycin-Resistant Strain. PloS One (2015) 10(10):e0140139. doi: 10.1371/journal.pone.0140139
151. Jhingran A, Chawla B, Saxena S, Barrett MP, Madhubala R. Paromomycin: Uptake and Resistance in Leishmania Donovani. Mol Biochem Parasitol (2009) 164(2):111–7. doi: 10.1016/j.molbiopara.2008.12.007
152. Hendrickx S, Van Bockstal L, Aslan H, Sadlova J, Maes L, Volf P, et al. Transmission Potential of Paromomycin-Resistant Leishmania Infantum and Leishmania Donovani. J Antimicrob Chemother (2019) 75(4):951–7. doi: 10.1093/jac/dkz517
153. Bhandari V, Sundar S, Dujardin JC, Salotra P. Elucidation of Cellular Mechanisms Involved in Experimental Paromomycin Resistance in Leishmania Donovani. Antimicrob Agents Chemother (2014) 58(5):2580–5. doi: 10.1128/AAC.01574-13
154. Hendrickx S, Reis-Cunha JL, Forrester S, Jeffares DC, Caljon G. Experimental Selection of Paromomycin Resistance in Leishmania Donovani Amastigotes Induces Variable Genomic Polymorphisms. Microorganisms (2021) 9(8):1546. doi: 10.3390/microorganisms9081546
155. Valencia C, Arevalo J, Dujardin JC, Llanos-Cuentas A, Chappuis F, Zimic M. Prediction Score for Antimony Treatment Failure in Patients With Ulcerative Leishmaniasis Lesions. PloS Negl Trop Dis (2012) 6(6):e1656. doi: 10.1371/journal.pntd.0001656
156. Rijal S, Uranw S, Chappuis F, Picado A, Khanal B, Paudel IS, et al. Epidemiology of Leishmania Donovani Infection in High-Transmission Foci in Nepal. Trop Med Int Health (2010) 15(Suppl 2):21–8. doi: 10.1111/j.1365-3156.2010.02518.x
157. Jervis S, Chapman LAC, Dwivedi S, Karthick M, Das A, Le Rutte EA, et al. Variations in Visceral Leishmaniasis Burden, Mortality and the Pathway to Care Within Bihar, India. Parasit Vectors (2017) 10(1):601. doi: 10.1186/s13071-017-2530-9
158. Wondimeneh Y, Takele Y, Atnafu A, Ferede G, Muluye D. Trend Analysis of Visceral Leishmaniasis at Addis Zemen Health Center, Northwest Ethiopia. BioMed Res Int (2014) 2014:545393. doi: 10.1155/2014/545393
159. Aruleba RT, Carter KC, Brombacher F, Hurdayal R. Can We Harness Immune Responses to Improve Drug Treatment in Leishmaniasis? Microorganisms (2020) 8(7). doi: 10.3390/microorganisms8071069
160. Vanaerschot M, Dumetz F, Roy S, Ponte-Sucre A, Arevalo J, Dujardin JC. Treatment Failure in Leishmaniasis: Drug-Resistance or Another (Epi-) Phenotype? Expert Rev Anti Infect Ther (2014) 12(8):937–46. doi: 10.1586/14787210.2014.916614
161. van Griensven J, Carrillo E, Lopez-Velez R, Lynen L, Moreno J. Leishmaniasis in Immunosuppressed Individuals. Clin Microbiol Infect (2014) 20(4):286–99. doi: 10.1111/1469-0691.12556
162. Bourreau E, Prevot G, Gardon J, Pradinaud R, Launois P. High Intralesional Interleukin-10 Messenger RNA Expression in Localized Cutaneous Leishmaniasis is Associated With Unresponsiveness to Treatment. J Infect Dis (2001) 184(12):1628–30. doi: 10.1086/324665
163. Katara GK, Ansari NA, Verma S, Ramesh V, Salotra P. Foxp3 and IL-10 Expression Correlates With Parasite Burden in Lesional Tissues of Post Kala Azar Dermal Leishmaniasis (PKDL) Patients. PloS Negl Trop Dis (2011) 5(5):e1171. doi: 10.1371/journal.pntd.0001171
164. Mukhopadhyay R, Mukherjee S, Mukherjee B, Naskar K, Mondal D, Decuypere S, et al. Characterisation of Antimony-Resistant Leishmania Donovani Isolates: Biochemical and Biophysical Studies and Interaction With Host Cells. Int J Parasitol (2011) 41(13-14):1311–21. doi: 10.1016/j.ijpara.2011.07.013
165. Peixoto F, Nascimento MT, Costa R, Silva J, Renard G, Guimaraes LH, et al. Evaluation of the Ability of Miltefosine Associated With Topical GM-CSF in Modulating the Immune Response of Patients With Cutaneous Leishmaniasis. J Immunol Res (2020) 2020:2789859. doi: 10.1155/2020/2789859
166. McMahon-Pratt D, Alexander J. Does the Leishmania Major Paradigm of Pathogenesis and Protection Hold for New World Cutaneous Leishmaniases or the Visceral Disease? Immunol Rev (2004) 201:206–24. doi: 10.1111/j.0105-2896.2004.00190.x
167. Vanaerschot M, Huijben S, Van den Broeck F, Dujardin JC. Drug Resistance in Vectorborne Parasites: Multiple Actors and Scenarios for an Evolutionary Arms Race. FEMS Microbiol Rev (2014) 38(1):41–55. doi: 10.1111/1574-6976.12032
168. Antonia AL, Barnes AB, Martin AT, Wang L, Ko DC. Variation in Leishmania Chemokine Suppression Driven by Diversification of the GP63 Virulence Factor. PloS Negl Trop Dis (2021) 15(10):e0009224. doi: 10.1371/journal.pntd.0009224
169. Dong G, Filho AL, Olivier M. Modulation of Host-Pathogen Communication by Extracellular Vesicles (EVs) of the Protozoan Parasite Leishmania. Front Cell Infect Microbiol (2019) 9(100). doi: 10.3389/fcimb.2019.00100
170. Krayem I, Lipoldova M. Role of Host Genetics and Cytokines in Leishmania Infection. Cytokine (2021) 147:155244. doi: 10.1016/j.cyto.2020.155244
171. Samant M, Sahu U, Pandey SC, Khare P. Role of Cytokines in Experimental and Human Visceral Leishmaniasis. Front Cell Infect Microbiol (2021) 11:624009. doi: 10.3389/fcimb.2021.624009
172. Perez-Cabezas B, Cecilio P, Gaspar TB, Gartner F, Vasconcellos R, Cordeiro-da-Silva A. Understanding Resistance vs. Susceptibility in Visceral Leishmaniasis Using Mouse Models of Leishmania Infantum Infection. Front Cell Infect Microbiol (2019) 9:30. doi: 10.3389/fcimb.2019.00030
173. Carvalho AM, Guimaraes LH, Costa R, Saldanha MG, Prates I, Carvalho LP, et al. Impaired Th1 Response is Associated With Therapeutic Failure in Patients With Cutaneous Leishmaniasis Caused by Leishmania Braziliensis. J Infect Dis (2021) 223(3):527–35. doi: 10.1093/infdis/jiaa374
174. Lindoso JAL, Moreira CHV, Cunha MA, Queiroz IT. Visceral Leishmaniasis and HIV Coinfection: Current Perspectives. HIV AIDS (Auckl) (2018) 10:193–201. doi: 10.2147/HIV.S143929
175. Alvar J, Aparicio P, Aseffa A, Den Boer M, Canavate C, Dedet JP, et al. The Relationship Between Leishmaniasis and AIDS: The Second 10 Years. Clin Microbiol Rev (2008) 21(2):334–59. table of contents. doi: 10.1128/CMR.00061-07
176. Graepp-Fontoura I, Soeiro Barbosa D, Paes AMA, Santos FS, Santos Neto M, Fontoura VM, et al. Epidemiological, Clinical and Laboratory Aspects of Human Visceral Leishmaniasis (HVL) Associated With Human Immunodeficiency Virus (HIV) Coinfection: A Systematic Review. Parasitology (2018) 145(14):1801–18. doi: 10.1017/S003118201800080X
177. Diro E, Lynen L, Mohammed R, Boelaert M, Hailu A, van Griensven J. High Parasitological Failure Rate of Visceral Leishmaniasis to Sodium Stibogluconate Among HIV Co-Infected Adults in Ethiopia. PloS Negl Trop Dis (2014) 8(5):e2875. doi: 10.1371/journal.pntd.0002875
178. Troya J, Casquero A, Refoyo E, Fernandez-Guerrero ML, Gorgolas M. Long Term Failure of Miltefosine in the Treatment of Refractory Visceral Leishmaniasis in AIDS Patients. Scand J Infect Dis (2008) 40(1):78–80. doi: 10.1080/00365540701466215
179. Sindermann H, Engel KR, Fischer C, Bommer W. Miltefosine Compassionate Use P. Oral Miltefosine for Leishmaniasis in Immunocompromised Patients: Compassionate Use in 39 Patients With HIV Infection. Clin Infect Dis (2004) 39(10):1520–3. doi: 10.1086/425359
180. Ritmeijer K, Dejenie A, Assefa Y, Hundie TB, Mesure J, Boots G, et al. A Comparison of Miltefosine and Sodium Stibogluconate for Treatment of Visceral Leishmaniasis in an Ethiopian Population With High Prevalence of HIV Infection. Clin Infect Dis (2006) 43(3):357–64. doi: 10.1086/505217
181. Diro E, Ritmeijer K, Boelaert M, Alves F, Mohammed R, Abongomera C, et al. Long-Term Clinical Outcomes in Visceral Leishmaniasis/Human Immunodeficiency Virus-Coinfected Patients During and After Pentamidine Secondary Prophylaxis in Ethiopia: A Single-Arm Clinical Trial. Clin Infect Dis (2018) 66(3):444–51. doi: 10.1093/cid/cix807
182. Adriaensen W, Cuypers B, Cordero CF, Mengasha B, Blesson S, Cnops L, et al. Host Transcriptomic Signature as Alternative Test-of-Cure in Visceral Leishmaniasis Patients Co-Infected With HIV. EBioMedicine (2020) 55:102748. doi: 10.1016/j.ebiom.2020.102748
183. Antinori S, Cascio A, Parravicini C, Bianchi R, Corbellino M. Leishmaniasis Among Organ Transplant Recipients. Lancet Infect Dis (2008) 8(3):191–9. doi: 10.1016/S1473-3099(08)70043-4
184. Garcia-Cordoba F, Ortuno FJ, Segovia M, Gonzalez Diaz G. Fatal Visceral Leishmaniasis, With Massive Bone-Marrow Infection, in an Immunosuppressed But HIV-Negative Spanish Patient, After the Initiation of Treatment With Meglumine Antimoniate. Ann Trop Med Parasitol (2005) 99(2):125–30. doi: 10.1179/136485905X19810
185. Ferreira AV, Segatto M, Menezes Z, Macedo AM, Gelape C, de Oliveira Andrade L, et al. Evidence for Trypanosoma Cruzi in Adipose Tissue in Human Chronic Chagas Disease. Microbes Infect (2011) 13(12-13):1002–5. doi: 10.1016/j.micinf.2011.06.002
186. Shanks GD, White NJ. The Activation of Vivax Malaria Hypnozoites by Infectious Diseases. Lancet Infect Dis (2013) 13(10):900–6. doi: 10.1016/S1473-3099(13)70095-1
187. Beamer G, Major S, Das B, Campos-Neto A. Bone Marrow Mesenchymal Stem Cells Provide an Antibiotic-Protective Niche for Persistent Viable Mycobacterium Tuberculosis That Survive Antibiotic Treatment. Am J Pathol (2014) 184(12):3170–5. doi: 10.1016/j.ajpath.2014.08.024
188. Ferreira-da-Silva Mda F, Takacs AC, Barbosa HS, Gross U, Luder CG. Primary Skeletal Muscle Cells Trigger Spontaneous Toxoplasma Gondii Tachyzoite-to-Bradyzoite Conversion at Higher Rates Than Fibroblasts. Int J Med Microbiol (2009) 299(5):381–8. doi: 10.1016/j.ijmm.2008.10.002
189. Bogdan C, Donhauser N, Doring R, Rollinghoff M, Diefenbach A, Rittig MG. Fibroblasts as Host Cells in Latent Leishmaniosis. J Exp Med (2000) 191(12):2121–30. doi: 10.1084/jem.191.12.2121
190. Scorza BM, Wacker MA, Messingham K, Kim P, Klingelhutz A, Fairley J, et al. Differential Activation of Human Keratinocytes by Leishmania Species Causing Localized or Disseminated Disease. J Invest Dermatol (2017) 137(10):2149–56. doi: 10.1016/j.jid.2017.05.028
191. Gangneux JP, Lemenand O, Reinhard Y, Guiguen C, Guguen-Guillouzo C, Gripon P. In Vitro and Ex Vivo Permissivity of Hepatocytes for Leishmania Donovani. J Eukaryot Microbiol (2005) 52(6):489–91. doi: 10.1111/j.1550-7408.2005.00055.x
192. Carvalho-Gontijo R, Moreira DR, Resende M, Costa-Silva MF, Peruhype-Magalhaes V, Ribeiro CMF, et al. Infection of Hematopoietic Stem Cells by Leishmania Infantum Increases Erythropoiesis and Alters the Phenotypic and Functional Profiles of Progeny. Cell Immunol (2018) 326:77–85. doi: 10.1016/j.cellimm.2017.10.016
193. Jara M, Maes I, Imamura H, Domagalska MA, Dujardin JC, Arevalo J. Tracking of Quiescence in Leishmania by Quantifying the Expression of GFP in the Ribosomal DNA Locus. Sci Rep (2019) 9(1):18951. doi: 10.1038/s41598-019-55486-z
194. Kloehn J, Boughton BA, Saunders EC, O'Callaghan S, Binger KJ, McConville MJ. Identification of Metabolically Quiescent Leishmania Mexicana Parasites in Peripheral and Cured Dermal Granulomas Using Stable Isotope Tracing Imaging Mass Spectrometry. mBio (2021) 12(2). doi: 10.1128/mBio.00129-21
195. Lopes CS, Daifalla N, Das B, Dias da Silva V, Campos-Neto A. CD271+ Mesenchymal Stem Cells as a Possible Infectious Niche for Leishmania Infantum. PloS One (2016) 11(9):e0162927. doi: 10.1371/journal.pone.0162927
196. Windels EM, Michiels JE, BVd B, Fauvart M, Michiels J, Epstein S, et al. Antibiotics: Combatting Tolerance to Stop Resistance. mBio (2019) 10(5):e02095–19. doi: 10.1128/mBio.02095-19
197. Dorlo TP, Rijal S, Ostyn B, de Vries PJ, Singh R, Bhattarai N, et al. Failure of Miltefosine in Visceral Leishmaniasis is Associated With Low Drug Exposure. J Infect Dis (2014) 210(1):146–53. doi: 10.1093/infdis/jiu039
198. Castro MD, Gomez MA, Kip AE, Cossio A, Ortiz E, Navas A, et al. Pharmacokinetics of Miltefosine in Children and Adults With Cutaneous Leishmaniasis. Antimicrob Agents Chemother (2017) 61(3). doi: 10.1128/AAC.02198-16
199. Dorlo TP, Huitema AD, Beijnen JH, de Vries PJ. Optimal Dosing of Miltefosine in Children and Adults With Visceral Leishmaniasis. Antimicrob Agents Chemother (2012) 56(7):3864–72. doi: 10.1128/AAC.00292-12
200. Kip AE, Blesson S, Alves F, Wasunna M, Kimutai R, Menza P, et al. Low Antileishmanial Drug Exposure in HIV-Positive Visceral Leishmaniasis Patients on Antiretrovirals: An Ethiopian Cohort Study. J Antimicrob Chemother (2021) 76(5):1258–68. doi: 10.1093/jac/dkab013
201. Verrest L, Wasunna M, Kokwaro G, Aman R, Musa AM, Khalil EAG, et al. Geographical Variability in Paromomycin Pharmacokinetics Does Not Explain Efficacy Differences Between Eastern African and Indian Visceral Leishmaniasis Patients. Clin Pharmacokinet (2021) 60(11):1463–73. doi: 10.1007/s40262-021-01036-8
202. Mouton JW, Theuretzbacher U, Craig WA, Tulkens PM, Derendorf H, Cars O. Tissue Concentrations: Do We Ever Learn? J Antimicrob Chemother (2008) 61(2):235–7. doi: 10.1093/jac/dkm476
203. Voak AA, Harris A, Coteron-Lopez JM, Angulo-Barturen I, Ferrer-Bazaga S, Croft SL, et al. Pharmacokinetic / Pharmacodynamic Relationships of Liposomal Amphotericin B and Miltefosine in Experimental Visceral Leishmaniasis. PloS Negl Trop Dis (2021) 15(3):e0009013. doi: 10.1371/journal.pntd.0009013
204. Voak AA, Harris A, Qaiser Z, Croft SL, Seifert K. Pharmacodynamics and Biodistribution of Single-Dose Liposomal Amphotericin B at Different Stages of Experimental Visceral Leishmaniasis. Antimicrob Agents Chemother (2017) 61(9):e00497–17. doi: 10.1128/AAC.00497-17
205. Wijnant GJ, Van Bocxlaer K, Yardley V, Harris A, Murdan S, Croft SL. Relation Between Skin Pharmacokinetics and Efficacy in AmBisome Treatment of Murine Cutaneous Leishmaniasis. Antimicrob Agents Chemother (2018) 62(3):e02009–17. doi: 10.1128/AAC.02009-17
206. Wijnant GJ, Van Bocxlaer K, Fortes Francisco A, Yardley V, Harris A, Alavijeh M, et al. Local Skin Inflammation in Cutaneous Leishmaniasis as a Source of Variable Pharmacokinetics and Therapeutic Efficacy of Liposomal Amphotericin B. Antimicrob Agents Chemother (2018) 62(10):e00631–18. doi: 10.1128/AAC.00631-18
207. Van Bocxlaer K, Gaukel E, Hauser D, Park SH, Schock S, Yardley V, et al. Topical Treatment for Cutaneous Leishmaniasis: Dermato-Pharmacokinetic Lead Optimization of Benzoxaboroles. Antimicrob Agents Chemother (2018) 62(5):e02419–17. doi: 10.1128/AAC.02419-17
208. Van Bocxlaer K, Yardley V, Murdan S, Croft SL. Drug Permeation and Barrier Damage in Leishmania-Infected Mouse Skin. J Antimicrob Chemother (2016) 71(6):1578–85. doi: 10.1093/jac/dkw012
209. Wijnant G-J, Croft SL, Rdl F, Alavijeh M, Yardley V, Braillard S, et al. Pharmacokinetics and Pharmacodynamics of the Nitroimidazole DNDI-0690 in Mouse Models of Cutaneous Leishmaniasis. Antimicrob Agents Chemother (2019) 63(9):e00829–19. doi: 10.1128/AAC.00829-19
210. Roseboom IC, Rosing H, Beijnen JH, Dorlo TPC. Skin Tissue Sample Collection, Sample Homogenization, and Analyte Extraction Strategies for Liquid Chromatographic Mass Spectrometry Quantification of Pharmaceutical Compounds. J Pharm BioMed Anal (2020) 191:113590. doi: 10.1016/j.jpba.2020.113590
211. Van Bocxlaer K, Croft SL. Pharmacokinetics and Pharmacodynamics in the Treatment of Cutaneous Leishmaniasis - Challenges and Opportunities. RSC Med Chem (2021) 12(4):472–82. doi: 10.1039/D0MD00343C
212. Dorlo TP, Eggelte TA, de Vries PJ, Beijnen JH. Characterization and Identification of Suspected Counterfeit Miltefosine Capsules. Analyst (2012) 137(5):1265–74. doi: 10.1039/c2an15641e
213. Abtahi-Naeini B, Shahmoradi Z, Niknami E, Saffaei A. Gulucatime Versus Glucantime: A Serious Warning on Counterfeit Medicines. J Res Pharm Pract (2019) 8(4):228–9. doi: 10.4103/jrpp.JRPP_19_84
214. Dorlo TPC, Ostyn BA, Uranw S, Dujardin JC, Boelaert M. Treatment of Visceral Leishmaniasis: Pitfalls and Stewardship. Lancet Infect Dis (2016) 16(7):777–8. doi: 10.1016/S1473-3099(16)30091-3
215. Vermeersch M, da Luz RI, Toté K, Timmermans JP, Cos P, Maes L. In Vitro Susceptibilities of Leishmania Donovani Promastigote and Amastigote Stages to Antileishmanial Reference Drugs: Practical Relevance of Stage-Specific Differences. Antimicrob Agents Chemother (2009) 53(9):3855–9. doi: 10.1128/AAC.00548-09
216. Seifert K, Escobar P, Croft SL. In Vitro Activity of Anti-Leishmanial Drugs Against Leishmania Donovani is Host Cell Dependent. J Antimicrob Chemother (2010) 65(3):508–11. doi: 10.1093/jac/dkp500
217. World Health O. Global Antimicrobial Resistance Surveillance System (GLASS): Molecular Methods for Antimicrobial Resistance (AMR) Diagnostics to Enhance the Global Antimicrobial Resistance Surveillance System. Geneva: World Health Organization (2019).
218. Cervantes J, Yokobori N, Hong BY. Genetic Identification and Drug-Resistance Characterization of Mycobacterium Tuberculosis Using a Portable Sequencing Device. A Pilot Study. Antibiotics (Basel) (2020) 9(9). doi: 10.3390/antibiotics9090548
219. Silva H, Liyanage A, Deerasinghe T, Chandrasekara V, Chellappan K, Karunaweera ND. Treatment Failure to Sodium Stibogluconate in Cutaneous Leishmaniasis: A Challenge to Infection Control and Disease Elimination. PloS One (2021) 16(10):e0259009. doi: 10.1371/journal.pone.0259009
220. Hendrickx S, Van den Kerkhof M, Mabille D, Cos P, Delputte P, Maes L, et al. Combined Treatment of Miltefosine and Paromomycin Delays the Onset of Experimental Drug Resistance in Leishmania Infantum. PloS Negl Trop Dis (2017) 11(5):e0005620. doi: 10.1371/journal.pntd.0005620
221. Goyal V, Burza S, Pandey K, Singh SN, Singh RS, Strub-Wourgaft N, et al. Field Effectiveness of New Visceral Leishmaniasis Regimens After 1 Year Following Treatment Within Public Health Facilities in Bihar, India. PloS Negl Trop Dis (2019) 13(9):e0007726. doi: 10.1371/journal.pntd.0007726
222. Mahajan R, Das P, Isaakidis P, Sunyoto T, Sagili KD, Lima MA, et al. Combination Treatment for Visceral Leishmaniasis Patients Coinfected With Human Immunodeficiency Virus in India. Clin Infect Dis (2015) 61(8):1255–62. doi: 10.1093/cid/civ530
223. DNDi. Cutaneous Leishmaniasis. New CL Combination Therapies 2021. Available at: https://dndi.org/research-development/portfolio/new-cl-combos/.
224. Varma DM, Redding EA, Bachelder EM, Ainslie KM. Nano- and Microformulations to Advance Therapies for Visceral Leishmaniasis. ACS Biomater Sci Eng (2021) 7(5):1725–41. doi: 10.1021/acsbiomaterials.0c01132
225. Sousa-Batista AJ, Pacienza-Lima W, Ré MI, Rossi-Bergmann B. Novel and Safe Single-Dose Treatment of Cutaneous Leishmaniasis With Implantable Amphotericin B-Loaded Microparticles. Int J Parasitol Drugs Drug Resist (2019) 11:148–55. doi: 10.1016/j.ijpddr.2019.06.001
226. Thoueille P, Choong E, Cavassini M, Buclin T, Decosterd LA. Long-Acting Antiretrovirals: A New Era for the Management and Prevention of HIV Infection. J Antimicrob Chemother (2021) 77(2):290–302. doi: 10.1093/jac/dkab324
227. Bakshi RP, Tatham LM, Savage AC, Tripathi AK, Mlambo G, Ippolito MM, et al. Long-Acting Injectable Atovaquone Nanomedicines for Malaria Prophylaxis. Nat Commun (2018) 9(1):315. doi: 10.1038/s41467-017-02603-z
228. Robles-Loaiza AA, Pinos-Tamayo EA, Mendes B, Teixeira C, Alves C, Gomes P, et al. Peptides to Tackle Leishmaniasis: Current Status and Future Directions. Int J Mol Sci (2021) 22(9):4400. doi: 10.3390/ijms22094400
229. Manzano JI, Lecerf-Schmidt F, Lespinasse M-A, Di Pietro A, Castanys S, Boumendjel A, et al. Identification of Specific Reversal Agents for Leishmania ABCI4-Mediated Antimony Resistance by Flavonoid and Trolox Derivative Screening. J Antimicrob Chemother (2013) 69(3):664–72. doi: 10.1093/jac/dkt407
230. Hefnawy A, Berg M, Dujardin JC, De Muylder G. Exploiting Knowledge on Leishmania Drug Resistance to Support the Quest for New Drugs. Trends Parasitol (2017) 33(3):162–74. doi: 10.1016/j.pt.2016.11.003
231. Hefnawy A, Negreira G, Jara M, Cotton JA, Maes I, D’ Haenens E, et al. Genomic and Phenotypic Characterization of Experimentally Selected Resistant Leishmania Donovani Reveals a Role for Dynamin-1 Like Protein in the Mechanism of Resistance to a Novel Anti-Leishmanial Compound. bioRxiv (2021) 2021.01.05.425522. doi: 10.1101/2021.01.05.425522
232. Mowbray CE, Braillard S, Glossop PA, Whitlock GA, Jacobs RT, Speake J, et al. DNDI-6148: A Novel Benzoxaborole Preclinical Candidate for the Treatment of Visceral Leishmaniasis. J Med Chem (2021) 64(21):16159–76. doi: 10.1021/acs.jmedchem.1c01437
233. Van den Kerkhof M, Leprohon P, Mabille D, Hendrickx S, Tulloch LB, Wall RJ, et al. Identification of Resistance Determinants for a Promising Antileishmanial Oxaborole Series. Microorganisms (2021) 9(7):1408. doi: 10.3390/microorganisms9071408
234. Van Bocxlaer K, Caridha D, Black C, Vesely B, Leed S, Sciotti RJ, et al. Novel Benzoxaborole, Nitroimidazole and Aminopyrazoles With Activity Against Experimental Cutaneous Leishmaniasis. Int J Parasitol Drugs Drug Resist (2019) 11:129–38. doi: 10.1016/j.ijpddr.2019.02.002
235. Nagle A, Biggart A, Be C, Srinivas H, Hein A, Caridha D, et al. Discovery and Characterization of Clinical Candidate LXE408 as a Kinetoplastid-Selective Proteasome Inhibitor for the Treatment of Leishmaniases. J Med Chem (2020) 63(19):10773–81. doi: 10.1021/acs.jmedchem.0c00499
236. Hastings I. How Artemisinin-Containing Combination Therapies Slow the Spread of Antimalarial Drug Resistance. Trends Parasitol (2011) 27(2):67–72. doi: 10.1016/j.pt.2010.09.005
237. Hastings IM, Hodel EM. Pharmacological Considerations in the Design of Anti-Malarial Drug Combination Therapies – is Matching Half-Lives Enough? Malaria J (2014) 13(1):62. doi: 10.1186/1475-2875-13-62
238. Srivastava S, Sherman C, Meek C, Leff R, Gumbo T. Pharmacokinetic Mismatch Does Not Lead to Emergence of Isoniazid- or Rifampin-Resistant Mycobacterium Tuberculosis But to Better Antimicrobial Effect: A New Paradigm for Antituberculosis Drug Scheduling. Antimicrob Agents Chemother (2011) 55(11):5085–9. doi: 10.1128/AAC.00269-11
239. Zumla A, Rao M, Wallis RS, Kaufmann SH, Rustomjee R, Mwaba P, et al. Host-Directed Therapies for Infectious Diseases: Current Status, Recent Progress, and Future Prospects. Lancet Infect Dis (2016) 16(4):e47–63. doi: 10.1016/S1473-3099(16)00078-5
240. Martinez-Hernandez JE, Hammoud Z, de Sousa AM, Kramer F, Monte-Neto RLD, Maracaja-Coutinho V, et al. Network-Based Approaches Reveal Potential Therapeutic Targets for Host-Directed Antileishmanial Therapy Driving Drug Repurposing. Microbiol Spectr (2021) 9(2):e0101821. doi: 10.1128/Spectrum.01018-21
241. Novais FO, Amorim CF, Scott P. Host-Directed Therapies for Cutaneous Leishmaniasis. Front Immunol (2021) 12:660183. doi: 10.3389/fimmu.2021.660183
242. Murray HW, Montelibano C, Peterson R, Sypek JP. Interleukin-12 Regulates the Response to Chemotherapy in Experimental Visceral Leishmaniasis. J nfect Dis (2000) 182(5):1497–502. doi: 10.1086/315890
243. Saha P, Bhattacharjee S, Sarkar A, Manna A, Majumder S, Chatterjee M. Berberine Chloride Mediates its Anti-Leishmanial Activity via Differential Regulation of the Mitogen Activated Protein Kinase Pathway in Macrophages. PloS One (2011) 6(4):e18467. doi: 10.1371/journal.pone.0018467
244. Kyriazis ID, Koutsoni OS, Aligiannis N, Karampetsou K, Skaltsounis AL, Dotsika E. The Leishmanicidal Activity of Oleuropein is Selectively Regulated Through Inflammation- and Oxidative Stress-Related Genes. Parasit Vectors (2016) 9(1):441. doi: 10.1186/s13071-016-1701-4
245. Parihar SP, Hartley MA, Hurdayal R, Guler R, Brombacher F. Topical Simvastatin as Host-Directed Therapy Against Severity of Cutaneous Leishmaniasis in Mice. Sci Rep (2016) 6:33458. doi: 10.1038/srep33458
246. Wetzel DM, McMahon-Pratt D, Koleske AJ. The Abl and Arg Kinases Mediate Distinct Modes of Phagocytosis and are Required for Maximal Leishmania Infection. Mol Cell Biol (2012) 32(15):3176–86. doi: 10.1128/MCB.00086-12
247. Petersen A, Guedes CES, Versoza CL, Lima JGB, de Freitas LAR, Borges VM, et al. 17-AAG Kills Intracellular Leishmania Amazonensis While Reducing Inflammatory Responses in Infected Macrophages. PloS One (2012) 7(11):e49496. doi: 10.1371/journal.pone.0049496
248. Talaei S, Mellatyar H, Asadi A, Akbarzadeh A, Sheervalilou R, Zarghami N. Spotlight on 17-AAG as an Hsp90 Inhibitor for Molecular Targeted Cancer Treatment. Chem Biol Drug Des (2019) 93(5):760–86. doi: 10.1111/cbdd.13486
249. Novais FO, Nguyen BT, Scott P. Granzyme B Inhibition by Tofacitinib Blocks the Pathology Induced by CD8 T Cells in Cutaneous Leishmaniasis. J Invest Dermatol (2021) 141(3):575–85. doi: 10.1016/j.jid.2020.07.011
250. Kaushik D, Granato JT, Macedo GC, Dib PRB, Piplani S, Fung J, et al. Toll-Like Receptor-7/8 Agonist Kill Leishmania Amazonensis by Acting as Pro-Oxidant and Pro-Inflammatory Agent. J Pharm Pharmacol (2021) 73(9):1180–90. doi: 10.1093/jpp/rgab063
251. Okwor I, Uzonna JE. Immunotherapy as a Strategy for Treatment of Leishmaniasis: A Review of the Literature. Immunotherapy (2009) 1(5):765–76. doi: 10.2217/imt.09.40
252. Van Bockstal L, Hendrickx S, Maes L, Caljon G. Sand Fly Studies Predict Transmission Potential of Drug-Resistant Leishmania. Trends Parasitol (2020) 36(9):785–95. doi: 10.1016/j.pt.2020.06.006
253. Van Bockstal L, Sádlová J, Suau HA, Hendrickx S, Meneses C, Kamhawi S, et al. Impaired Development of a Miltefosine-Resistant Leishmania Infantum Strain in the Sand Fly Vectors Phlebotomus Perniciosus and Lutzomyia Longipalpis. Int J Parasitol Drugs Drug Resist (2019) 11:1–7. doi: 10.1016/j.ijpddr.2019.09.003
254. Seblova V, Oury B, Eddaikra N, Aït-Oudhia K, Pratlong F, Gazanion E, et al. Transmission Potential of Antimony-Resistant Leishmania Field Isolates. Antimicrob Agents Chemother (2014) 58(10):6273–6. doi: 10.1128/AAC.02406-13
255. Wilson AL, Courtenay O, Kelly-Hope LA, Scott TW, Takken W, Torr SJ, et al. The Importance of Vector Control for the Control and Elimination of Vector-Borne Diseases. PloS Negl Trop Dis (2020) 14(1):e0007831. doi: 10.1371/journal.pntd.0007831
256. Kholoud K, Denis S, Lahouari B, El Hidan MA, Souad B. Management of Leishmaniases in the Era of Climate Change in Morocco. Int J Environ Res Public Health (2018) 15(7). doi: 10.3390/ijerph15071542
257. Shimozako HJ, Wu J, Massad E. The Preventive Control of Zoonotic Visceral Leishmaniasis: Efficacy and Economic Evaluation. Comput Math Methods Med (2017) 2017:4797051260. doi: 10.1155/2017/4797051
258. Quinnell RJ, Courtenay O. Transmission, Reservoir Hosts and Control of Zoonotic Visceral Leishmaniasis. Parasitology (2009) 136(14):1915–34. doi: 10.1017/S0031182009991156
259. Zhi-Biao X. Present Situation of Visceral Leishmaniasis in China. Parasitol Today (1989) 5(7):224–8. doi: 10.1016/0169-4758(89)90276-7
260. Eliseev LN. Strategy of the Control With Great Gerbils in Epidemically Dangerous Regions of Zoonotic Leishmaniasis. In: V KV, editor. Ecology and Medical Value of Great Gerbils in the Fauna of the USSR Alma-Ata1981.
261. Gonçalves G, Campos MP, Gonçalves AS, Soares Medeiros LC, Figueiredo FB. Treatment of Canine Visceral Leishmaniasis With Milteforan™ Induces Leishmania Infantum Resistance to Miltefosine and Amphotericin B. bioRxiv (2021). 2021.04.08.438938. doi: 10.1101/2021.04.08.438938
262. Kumari D, Perveen S, Sharma R, Singh K. Advancement in Leishmaniasis Diagnosis and Therapeutics: An Update. Eur J Pharmacol (2021) 910:174436. doi: 10.1016/j.ejphar.2021.174436
263. Thakur S, Joshi J, Kaur S. Leishmaniasis Diagnosis: An Update on the Use of Parasitological, Immunological and Molecular Methods. J Parasit Dis (2020) 44(2):1–20. doi: 10.1007/s12639-020-01212-w
264. Hagos DG, Schallig H, Kiros YK, Abdulkadir M, Wolday D. Performance of Rapid Rk39 Tests for the Diagnosis of Visceral Leishmaniasis in Ethiopia: A Systematic Review and Meta-Analysis. BMC Infect Dis (2021) 21(1):1166. doi: 10.1186/s12879-021-06826-w
265. Ganguly S, Saha P, Chatterjee M, Roy S, Ghosh TK, Guha SK, et al. PKDL–A Silent Parasite Pool for Transmission of Leishmaniasis in Kala-Azar Endemic Areas of Malda District, West Bengal, India. PloS Negl Trop Dis (2015) 9(10):e0004138. doi: 10.1371/journal.pntd.0004138
266. Mondal D, Bern C, Ghosh D, Rashid M, Molina R, Chowdhury R, et al. Quantifying the Infectiousness of Post-Kala-Azar Dermal Leishmaniasis Toward Sand Flies. Clin Infect Dis (2019) 69(2):251–8. doi: 10.1093/cid/ciy891
267. de Vries HJ, Reedijk SH, Schallig HD. Cutaneous Leishmaniasis: Recent Developments in Diagnosis and Management. Am J Clin Dermatol (2015) 16(2):99–109. doi: 10.1007/s40257-015-0114-z
268. den Boer M, Argaw D, Jannin J, Alvar J. Leishmaniasis Impact and Treatment Access. Clin Microbiol Infect (2011) 17(10):1471–7. doi: 10.1111/j.1469-0691.2011.03635.x
269. Sunyoto T, Potet J, den Boer M, Ritmeijer K, Postigo JAR, Ravinetto R, et al. Exploring Global and Country-Level Barriers to an Effective Supply of Leishmaniasis Medicines and Diagnostics in Eastern Africa: A Qualitative Study. BMJ Open (2019) 9(5):e029141. doi: 10.1136/bmjopen-2019-029141
270. Moran M RA-L, Guzman J, Dias J, Garrison C. The New Landscape of Neglected Disease Drug Development. In: A Pharmacetical R&D Policy Project. London: London School of Economics and Political Science (2005).
271. Olliaro P, Sundar S. Anthropometrically Derived Dosing and Drug Costing Calculations for Treating Visceral Leishmaniasis in Bihar, India. Trop Med Int Health (2009) 14(1):88–92. doi: 10.1111/j.1365-3156.2008.02195.x
272. Dahl EH, Hamdan HM, Mabrouk L, Matendechero SH, Mengistie TB, Elhag MS, et al. Control of Visceral Leishmaniasis in East Africa: Fragile Progress, New Threats. BMJ Global Health (2021) 6(8):e006835. doi: 10.1136/bmjgh-2021-006835
273. Sunyoto T, Potet J, Boelaert M. Why Miltefosine-a Life-Saving Drug for Leishmaniasis-is Unavailable to People Who Need it the Most. BMJ Glob Health (2018) 3(3):e000709. doi: 10.1136/bmjgh-2018-000709
274. Alvar J, Croft S, Olliaro P. Chemotherapy in the Treatment and Control of Leishmaniasis. Adv Parasitol (2006) 61:223–74. doi: 10.1016/S0065-308X(05)61006-8
275. Kakkar AK, Shafiq N, Singh G, Ray P, Gautam V, Agarwal R, et al. Antimicrobial Stewardship Programs in Resource Constrained Environments: Understanding and Addressing the Need of the Systems. Front Public Health (2020) 8:140–. doi: 10.3389/fpubh.2020.00140
276. Organisation WH. Antimicrobial Stewardship Programmes in Health-Care Facilities in Low- and Middle-Income Countries - A WHO Practical Toolkit 2019. Available at: https://apps.who.int/iris/bitstream/handle/10665/329404/9789241515481-eng.pdf.
277. Le Rutte EA, Coffeng LE, Muñoz J, de Vlas SJ. Modelling the Impact of COVID-19-Related Programme Interruptions on Visceral Leishmaniasis in India. Trans R Soc Trop Med Hyg (2021) 115(3):229–35. doi: 10.1093/trstmh/trab012
278. Hollingsworth TD, Mwinzi P, Vasconcelos A, de Vlas SJ. Evaluating the Potential Impact of Interruptions to Neglected Tropical Disease Programmes Due to COVID-19. Trans R Soc Trop Med Hyg (2021) 115(3):201–4. doi: 10.1093/trstmh/trab023
279. Carvalho SFG, Vieira TM, Moura APV, Andrade MC. Should an Intersection Between Visceral Leishmaniasis Endemicity and the COVID-19 Pandemic be Considered? Med Hypotheses (2020) 144:110289–. doi: 10.1016/j.mehy.2020.110289
280. Initiative DfND. Status of DNDi Clinical Trials During the COVID-19 Pandemic 2021. Available at: https://dndi.org/research-and-development/clinical-trials/status-dndi-clinical-trials-during-covid19-pandemic/.
Keywords: leishmania, drug resistance, treatment failure, pharmacokinetics, surveillance, leishmaniasis, neglected tropical disease (NTD), antimicrobial therapy
Citation: Wijnant G-J, Dumetz F, Dirkx L, Bulté D, Cuypers B, Van Bocxlaer K and Hendrickx S (2022) Tackling Drug Resistance and Other Causes of Treatment Failure in Leishmaniasis. Front. Trop. Dis 3:837460. doi: 10.3389/fitd.2022.837460
Received: 16 December 2021; Accepted: 12 April 2022;
Published: 12 May 2022.
Edited by:
Megha Raj Banjara, Tribhuvan University, NepalReviewed by:
Denis Sereno, Institut de Recherche Pour le Développement (IRD), FrancePatricia Sampaio Tavares Veras, Gonçalo Moniz Institute (IGM), Brazil
Nahid Ali, Indian Institute of Chemical Biology (CSIR), India
Copyright © 2022 Wijnant, Dumetz, Dirkx, Bulté, Cuypers, Van Bocxlaer and Hendrickx. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah Hendrickx, c2FyYWguaGVuZHJpY2t4QHVhbnR3ZXJwZW4uYmU=