 Erin Taylor Kelly1†
Erin Taylor Kelly1† Lindsey K. Mack1†
Lindsey K. Mack1† Melina Campos2
Melina Campos2 Crystal Grippin3
Crystal Grippin3 Tse-Yu Chen4
Tse-Yu Chen4 Ana L. Romero-Weaver4
Ana L. Romero-Weaver4 Kyle J. Kosinski4Katherine K. Brisco1
Kyle J. Kosinski4Katherine K. Brisco1 Travis C. Collier4Eva A. Buckner4
Travis C. Collier4Eva A. Buckner4 Lindsay P. Campbell4Anthony J. Cornel1
Lindsay P. Campbell4Anthony J. Cornel1 Gregory C. Lanzaro2
Gregory C. Lanzaro2 Rodrigo Rosario-Cruz6Kirk Smith5
Rodrigo Rosario-Cruz6Kirk Smith5 Geoffrey M. Attardo1‡
Geoffrey M. Attardo1‡ Yoosook Lee4*‡
Yoosook Lee4*‡- 1Department of Entomology and Nematology, University of California – Davis, Davis, CA, United States
- 2Vector Genetics Laboratory, Department of Pathology, Microbiology, and Immunology, School of Veterinary Medicine, University of California – Davis, Davis, CA, United States
- 3Laboratory Division, Delta Vector Control District, Visalia, CA, United States
- 4Florida Medical Entomology Laboratory, Department of Entomology and Nematology, Institute of Food and Agricultural Sciences, University of Florida, Vero Beach, FL, United States
- 5Maricopa County Environmental Services Department, Phoenix, AZ, United States
- 6BioSA Research Lab, Natural Sciences College, Autonomous Guerrero State University, Campus “El Shalako” Las Petaquillas, Guerrero, Mexico
Established populations of Aedes aegypti, a mosquito vector of multiple major arthropod-borne viruses, were first found in three California (CA) cities in 2013. From 2013 to April 2021, Ae. aegypti thwarted almost all control efforts to stop its spread and expanded its range to 308 cities, including Exeter, in 22 counties in CA. Population genomic analyses have suggested that multiple genetically distinct Ae. aegypti populations were introduced into CA. However Ae. aegypti collected for the first time in 2014 in Exeter, appeared to be different from three major genetic clusters found elsewhere in CA. Due to intense control efforts by the Delta Vector Control District (DVCD), Ae. aegypti was thought to have been eliminated from Exeter in 2015. Unfortunately, it was recollected in 2018. It was not clear if the reemergence of Ae. aegypti in Exeter was derived from the bottlenecked remnants of the original 2014 Exeter population or from an independent invasion from a different population derived from surrounding areas. The goal of this work was to determine which of these scenarios occurred (recovery after bottleneck or reintroduction after elimination) and if elimination and reintroduction occurred to identify the origin of the invading population using a population genomic approach. Our results support the reintroduction after elimination hypothesis. The source of reintroduction, however, was unexpectedly from the southern CA cluster rather than from other two geographically closer central CA genetic clusters. We also conducted a knockdown resistance mutation profile, which showed Exeter 2014 had the lowest level of resistant alleles compared to the other populations, could have contributed towards DVCD’s ability to locally eliminate Ae. aegypti in 2014.
Introduction
Aedes aegypti serves as a major vector of four human disease-causing viruses, including yellow fever, dengue, chikungunya, and Zika viruses, posing a major threat to public health. Records indicate this species became established in the southeastern United States of America between the 15-18th centuries (1) and then spread throughout the east coast and southern states (2). California (CA) had remained free from Ae. aegypti until the summer of 2013 (3, 4) when the species were first detected in Menlo Park, Clovis, and Madera (4). Since then, this species has expanded its range to 308 cities in 22 counties, as of April, 2021 (5).
Attempts to locally eliminate and even control this highly invasive species has proven to be extremely challenging. For example, the Consolidated Mosquito Abatement District (CMAD) implemented labor intensive integrated vector control management in the city of Clovis where Ae. aegypti were first detected in 2013. Their efforts involved extensive public education, thorough property inspections, sanitation, insecticide treatment at larval sources, and residual barrier spraying with pyrethroids. Despite these efforts, Ae. aegypti successfully overwintered and continued to persist in Clovis (6).
Delta Vector Control District (DVCD), which covers northern Tulare County, just south of the area covered by CMAD, first detected Ae. aegypti in Exeter in August of 2014. Following detection, the DVCD initiated an intensive control campaign involving thorough, routine property inspections and barrier applications, public education, breeding site treatment or removal, as well as hand and truck-mounted adulticide fogging through October. DVCD encountered significant pushback from the public due to the regular adulticide applications, mandatory property inspections, and confiscation of container habitats. However, they reported that residents followed instructions and were able to control mosquito breeding. Exeter remained free of Ae. aegypti from the beginning of 2015 through the summer of 2017. DVCD detected Ae. aegypti again in 2017 in the neighboring cities of Visalia and Farmersville and, in 2018, at multiple sites in Exeter. In response to this detection, the district attempted to mount a response similar to 2014. Despite frequent, mandatory property inspections, breeding site elimination and sanitation, and barrier spraying, the infestation persisted.
Population genetic/genomic analyses suggest that multiple genetically distinct Ae. aegypti populations were introduced in CA (7, 8). The Ae. aegypti populations in California could be largely grouped into three major genetic clusters (8). One cluster includes samples from Fresno, Madera, and Menlo Park in Central CA (8). Another cluster includes samples from Clovis in central CA adjacent to Fresno. The third cluster includes all southern CA samples, as well as 2014 Exeter samples (8). Further clustering revealed that 2014 Exeter samples are similar to an Ae. aegypti population from Florida rather than the populations from southern CA. However, it is important to note that the data from Lee et al. (8) does not provide definitive evidence that Exeter Ae. aegypti were introduced from Florida because the cluster analysis included only Florida samples for comparison. Aedes aegypti from southeastern states such as Louisiana and Texas are genetically similar to Florida populations based on microsatellite analysis (3).
The genetic dissimilarity of 2014 Exeter to other California Ae. aegypti provided an opportunity to investigate the hypothesis that (1) the Exeter mosquito population went through a severe bottleneck due to intensive insecticide spraying and control followed by a recovery after a couple of years (bottleneck and recovery) or (2) DVCD successfully eliminated its initial introduction but later new mosquitoes migrated or were reintroduced to Exeter from other cities (local extinction and reintroduction). If bottleneck and recovery occurred, then we expected 2018 Exeter samples to have similar genetic profile as 2014 Exeter samples. If localized extinction and reintroduction occurred, then we expected 2018 Exeter samples to have a similar genetic profile to any of the other three CA genetic clusters. To investigate this hypothesis, we report the population genomic analysis of 243 Ae. aegypti from California, Arizona, Florida, and Mexico in this paper.
Materials and Methods
Sample Collection
Genome data was obtained from deposited sequences available for specimens originating from earlier collections in Clovis, Fresno, Madera, Menlo Park, 2014 Exeter, East Los Angeles, San Diego (CA), Vero Beach, and Key West, Florida (FL) - NCBI BioProject PRJNA385349 (8, 9). Further samples from St. Augustine (FL), Naples (FL), St. Lucie (FL), Arizona, and Mexico were collected as adults using BG Sentinel traps and the remaining samples as eggs in ovicups, which were then reared to the adult stage in the laboratory and stored in >70% alcohol prior to DNA extraction. The global positioning system (GPS) coordinates from where each site specimens originated from as well as the number of samples sequenced and genotyped are provided in Table 1. The sample locations map is provided in Figure 1.
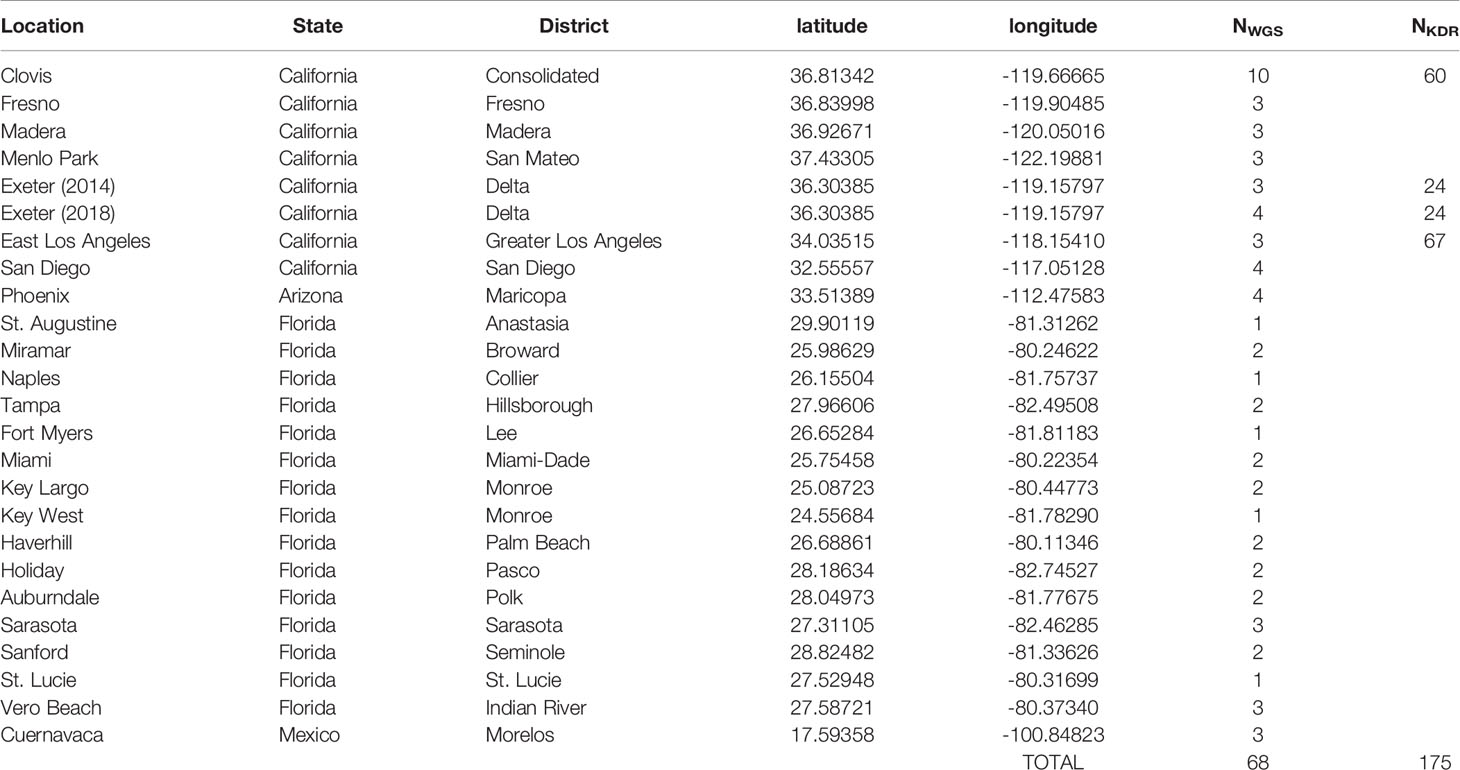
Table 1 Sample collection data. Number of Ae. aegypti sequenced are provided in NWGS column. NKDR denotes the number of samples genotyped for kdr and population specific SNPs using iPLEX assay.
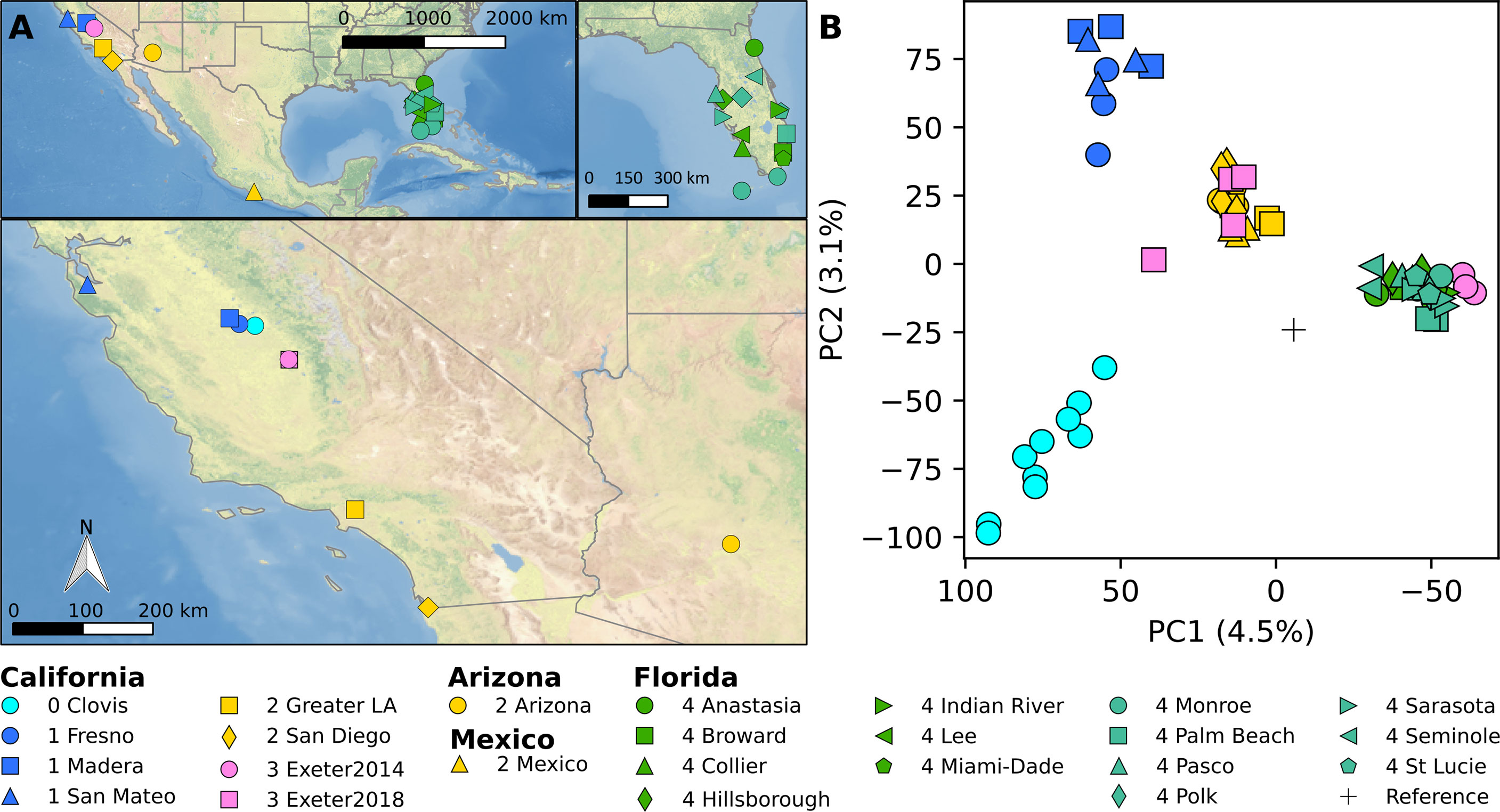
Figure 1 (A) Map of sample location. ClearnTOPO2, a public domain dataset, was used as a basemap. (B) PCA analysis based on genome-wide biallelic SNPs.
Genome Sequencing
Protocols for sequenced specimens available in the NCBI Bioproject database are described in Lee et al. (8) and Schmidt et al. (9). For the other specimens, DNA was extracted from individual mosquitoes using a magnetic-bead based DNA extraction protocol described in Chen et al. (10). DNA concentrations for each sample were measured using the Qubit dsDNA HS Assay Kit (Life Technologies) on a Qubit instrument (Life Technologies). A genomic DNA library was constructed for each individual mosquito with the QIAseq FX DNA Library UDI kit (Qiagen, Valencia, CA) using 20 ng DNA. Enzymatic fragmentation was conducted at 32°C for 11 minutes followed by 65°C for 30 minutes. Adapter ligation was conducted at 20°C for 2 hours. PCR amplification of the constructed library was carried out for 8 cycles [(98°C for 20 seconds, 60°C for 30 seconds, 72°C for 30 second) x 8]s. Library cleanup was done using PCRClean DX (Aline Biosciences, Woburn, MA). Library concentrations were measured using Qubit (Life Technologies). Libraries were sequenced as 150 bp paired-end reads using a NovaSeq instrument (Illumina) at the University of Florida Interdisciplinary Center for Biotechnology Research (ICBR) Nextgen DNA Sequencing Core.
SNP Genotyping
Eggs were collected from the field in Clovis and Greater LA and reared in the lab under existing protocols (11). Adult collections from Exeter were obtained from Delta Vector Control District. Sixty individuals from Clovis, 67 individuals from Greater LA, 24 individuals from Exeter 2014 and 24 individuals from Exeter 2018 were collected (Table 1, NKDR). DNA was extracted using the Zymo Quick-DNA/RNA Mini Prep Kit (#D7001) using the protocol for Solid Tissue. DNA quality and quantity were determined using a Qubit instrument (Life Technologies) and approximately 4 ng/µL was extracted from each individual.
DNA was submitted to the UC Davis Veterinary Genetics Laboratory for SNP genotyping using the iPLEX MassARRAY analysis following the protocol described in Lee et al. (12). Thirty-seven SNPs were selected from the published whole genome sequences (8), 29 of which were chosen due to their association with specific genetic clusters and eight within the Voltage Gated Sodium Channel gene. Data for 5 knockdown resistance (kdr) SNPs (F1534C, V410L, S723T, I915K, and V1016I) were included from (13). A full list of SNPs and primers can be found in Table S1. Populations were clustered using a Principal Component Analysis (PCA) performed in R v4.0.5, removing individuals with no calls for any SNPs.
Genome Sequence Data Analysis
Raw reads were trimmed using fastp (14) version 0.20.1. Trimmed reads were mapped to the Ae13CLOV028MT (Genbank ID: MH348176) first using BWA-MEM (15) version 0.7.15 following recommendation from Schmidt et al. (16) to minimize the impact of mitochondrial reads mapping to the nuclear genome due to presence of pseudogenes (17). Unmapped and mate-is-unmapped reads from mitogenome mapping were filtered using sambamba, converted to fastq files using samtools version 1.12, and mapped to AaegL5 reference genome (18) using BWA-MEM (15) version 0.7.15. Mapping statistics were calculated using Qualimap version 2.2 (19) (Table S2). Joint variant calling using all samples was done using Freebayes (20) version 1.0.1 with standard filters and population priors disabled.
The repeat regions were soft-masked in the AaegL5 reference genome and SNPs in these regions were excluded from analysis. Only biallelic SNPs with a minimum of 6X coverager were used for further analysis. A missing data threshold of 10% was used to filter SNPs. Hudson FST (21) and Principal Component Analysis (PCA) analyses was done in Python version 3.6.6 using the scikit-allel module version 1.2.0 (22). A phylogenetic tree based on the SNPs was constructed using the neighbor-joining algorithm as implemented in PHYLIP (23) version 3.696. Bayesian clustering method implemented in ADMIXTURE v1.3.0 (24) was used to estimate ancestry components for each individual. For this analysis, a total of 10 iterations were performed for values of K clusters from 1 to 10 with no prior population assignment. The results for each K were compiled using the online version of CLUMPAK and plotted in R. Windowed population genetic statistics such as FST and nucleotide diversity (π) were calculated using scikit-allel library with a 1Mbp window size and half-step overlapping windows.
Results
Thirty-eight Ae. aegypti genomes were sequenced, originating from Florida (N=24), Arizona (N=4), Exeter 2018 (N=4), and Clovis (N=6). These genomes were analyzed together with 30 sequenced genomes used in other publications (N=68) (Table 1, NWGS) (8, 9). Each sample was sequenced with a mean nuclear genome coverage of 11.9X per sample (Table S2; range = 8.3-37.9X; std = 5.0).
Principal component analysis (PCA) of genome-wide biallelic SNP genotypes reveal four major genetic clusters: (1) Clovis 2013 (cyan in Figure 1), (2) Fresno, Madera and Menlo Park (blue in Figure 1), (3) southern CA, Arizona, Mexico (yellow in Figure 1), and (4) Florida (green and sage in Figure 1). The Ae. aegypti population from Exeter 2014 cluster with Florida populations while 2018 Exeter samples mostly cluster together with southern CA samples. One sample from the 2018 Exeter group occupying the intermediate genetic space between southern CA and two other central populations appears to have genotypes intermediate between southern CA and Clovis populations (Figure 2).
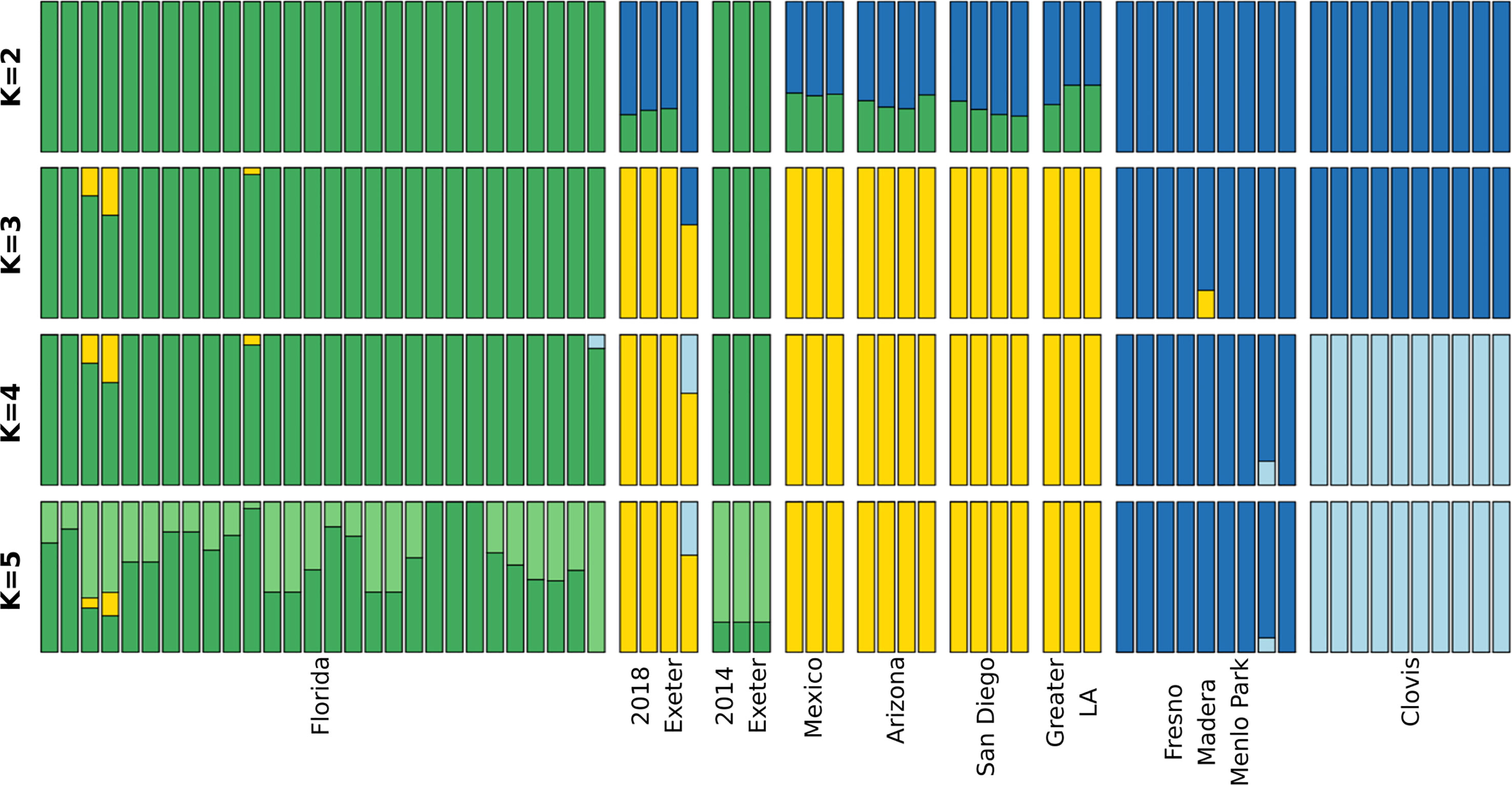
Figure 2 Admixture plot of individual Ae. aegypti mosquitoes. Each column represents an individual with probability of each cluster represented by different colors. The total probabilities add up to 1. Individuals with high likelihood of belonging to one cluster would appear as a single color column while individuals with mixed ancestry would have multiple colors in each column.
Consistent with PCA results, Admixture analysis of genome-wide SNPs clusters 2014 Exeter with Florida mosquitoes while 2018 Exeter clusters together with southern CA, Mexico and Arizona mosquitoes (Figure 2). One Exeter 2018 mosquito appears to have a signature of an admixture with the Clovis cluster. This particular sample corresponds to the outlier individual in the PCA that occupied a space between the southern CA and central CA clusters (Figure 1). The likelihood values increase continually with higher K (Figure S1) indicating that further substructure may be present beyond the K=5 clusters presented in Figure 2.
Exeter 2014 and 2018 were the most distant population pairs (FST = 0.152; Figure 3 and Table S3). Consistent with Admixture results, East Los Angeles, Arizona, Mexico, and Florida form a closely related group with FST < 0.05 (Table S3). Fresno, Madera, and Menlo Park also showed no differentiation with FST = 0 (Figures 2, 3 and Table S3). The closest population pair with Exeter 2014 was Florida (FST = 0.055) while the closest population pair with Exeter 2018 was East Los Angeles (FST = 0.038).
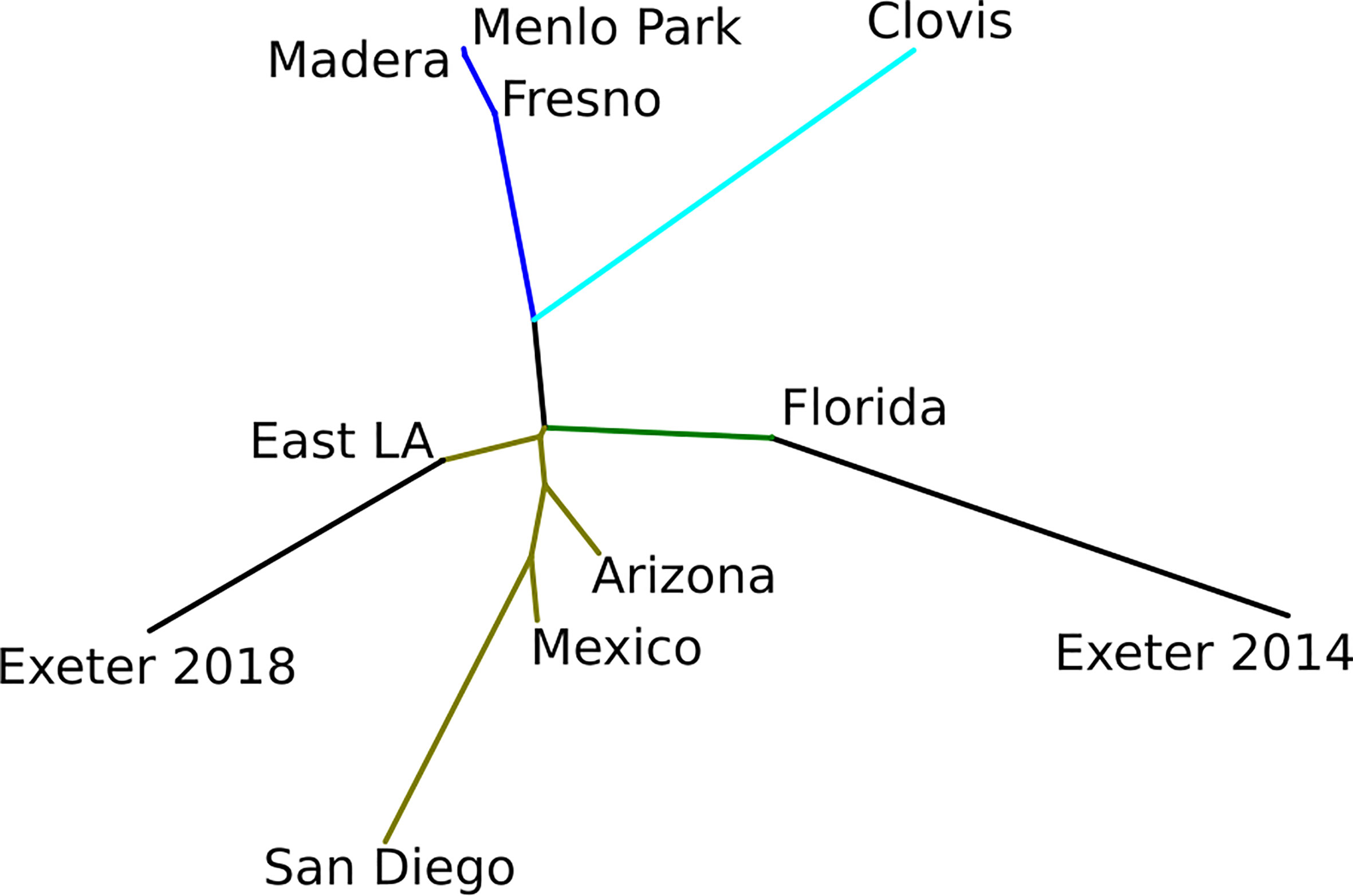
Figure 3 Neighbor-joining tree based on pairwise FST distance.
Based on published genome sequences (8), we selected a set of biallelic SNPs most informative for separating individuals into each of the major three genetic clusters found in CA. We designed a multiplex SNP genotyping assay using the iPLEX MassARRAY system to screen 37 SNPs simultaneously for their genetic background (Table S1). Thirty-one of these SNPs differentiate Ae. aegypti into each of the four genetic clusters present in California and 6 of these SNPs are associated with permethrin resistance. We genotyped 2018 Clovis, Greater Los Angeles, Exeter 2014, and Exeter 2018 samples using this new multiplex SNP assay. Our PCA analysis of SNP genotypes (Figure 4) shows Clovis and Greater Los Angeles forming separate genetic clusters, although the boundary is not as clear as genome-wide SNP data shown in Figure 1. Consistent with genome-wide SNP data, Exeter 2018 has large overlap with southern CA samples with two samples potentially having mixed ancestry from the southern CA and Clovis clusters. Exeter 2014 Ae. aegypti clearly clustered separately from the Clovis 2018, Exeter 2018, and Greater Los Angeles Ae. aegypti mosquitoes.
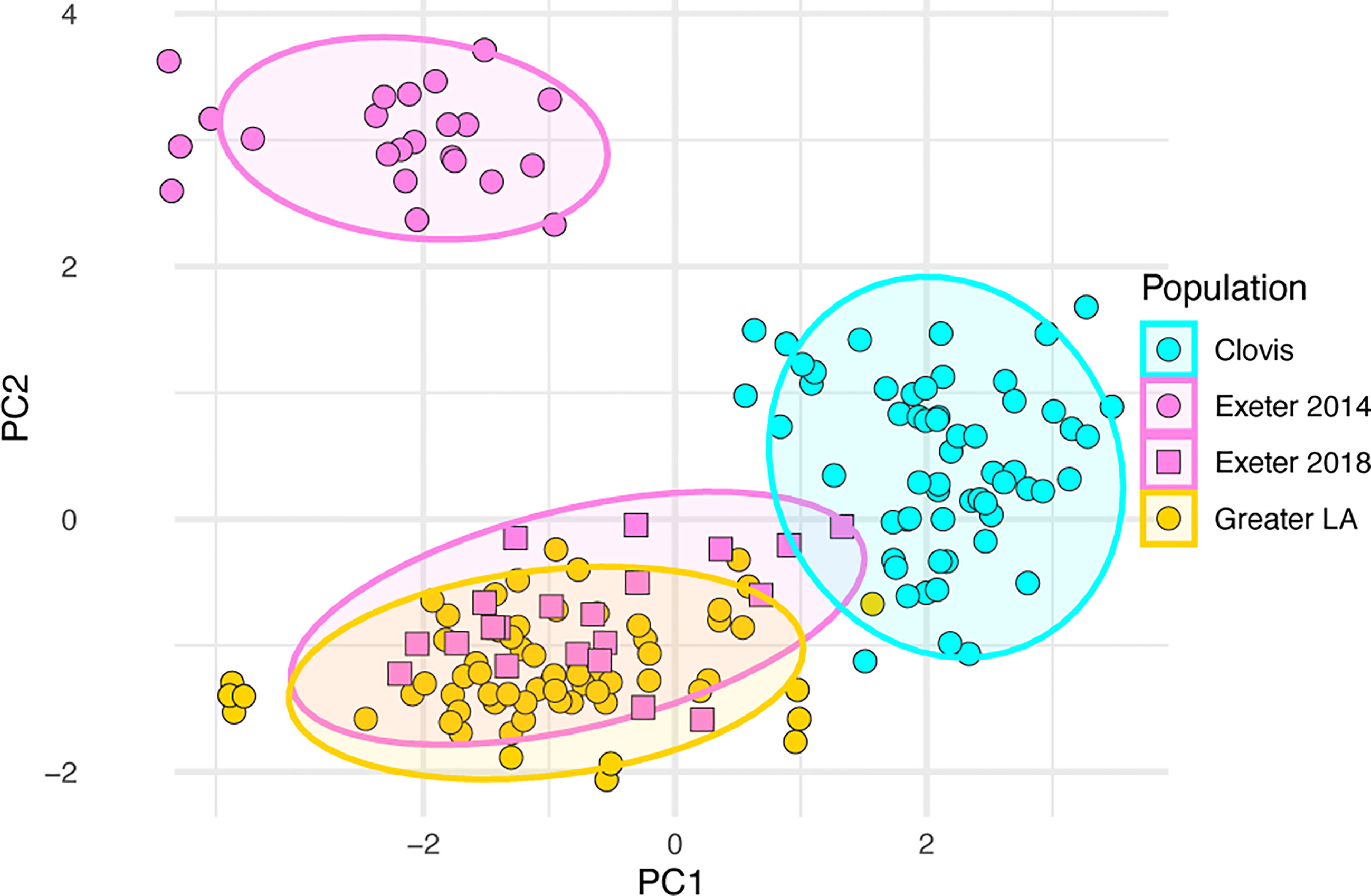
Figure 4 Principal component analysis based on 37 SNPs in Ae. aegypti from Central and Southern California using the iPLEX MASSarray genotyping.
We calculated the windowed FST, nucleotide diversity (π), and change in π between 2018 and 2014 Exeter samples (Figure 5) to identify any hotspots of divergence. Any FST greater than 0.1 (red line in Figure 5A) indicates high genetic distance in the corresponding genomic region between two years. The genomic divergence between the two years appears to be genome-wide except in limited locations such as in the immediate vicinity of the centromeres. Overall elevated π was observed near the centromeres of Chromosome 1 and 3 (Figure 5B). While 2018 Exeter samples had higher π in Chromosome 1, 2014 Exeter samples had higher π in Chromosome 3 (Figure 5C).
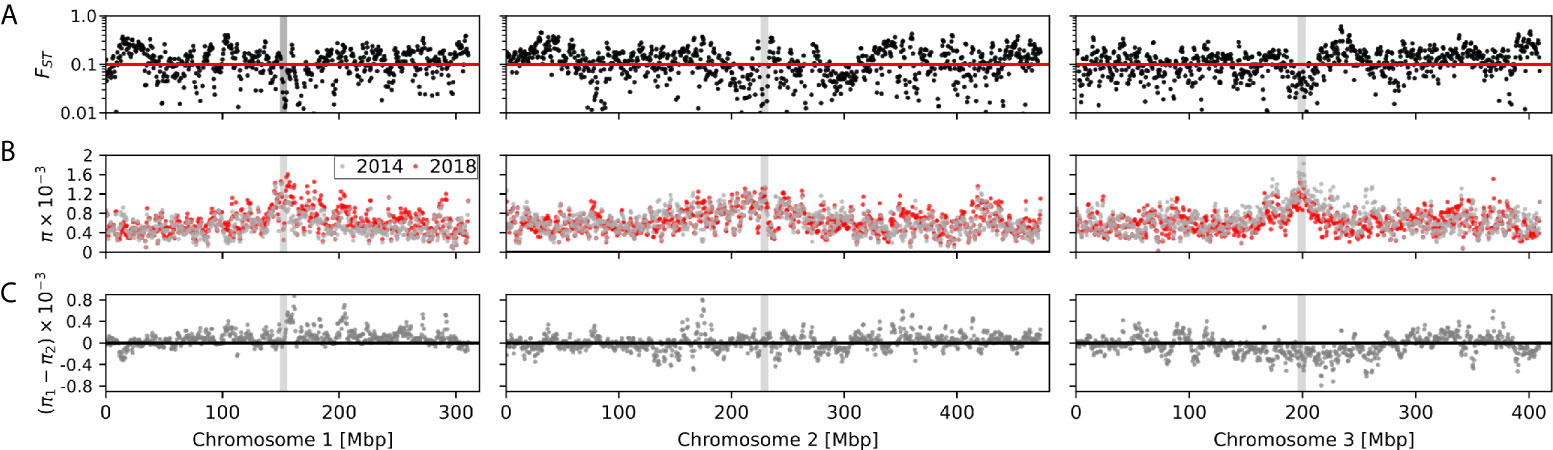
Figure 5 (A) Windowed FST, (B) nucleotide diversity (π), and (C) change in nucleotide diversity (π2018 - π2014) between 2014 and 2018 Exeter samples. The gray bar in the middle of each chromosome indicates location of the centromere.
We also analyzed 6 SNPs in the voltage-gated sodium channel gene, typically known as knockdown resistance gene (kdr) in mosquito literature (AAEL023266) located on Chromosome 3 using the method described in Mack et al. (13). Five of these 6 SNPs were previously used in Mack et al. (13). The SNP Q1853R (3:315931672) is a new addition to this study as it was only detected in the mosquitoes derived from the Exeter 2014 population. None of the other Californian populations possess this SNP and it appears to have disappeared with the eradication of that population. More than 60% of Exeter 2014 and 2018 Ae. aegypti had a V1016 resistant allele, while >90% of Clovis mosquitoes had this resistant allele present. Other nonsynonymous mutations in the kdr gene also occurred at the highest frequencies in Clovis. Except for allele F1534C, which was almost fixed in Clovis, alleles V410L, S723T and I915K were at much lower frequencies in Exeter 2014, 2018 and Greater LA Ae. aegypti (Table 2).
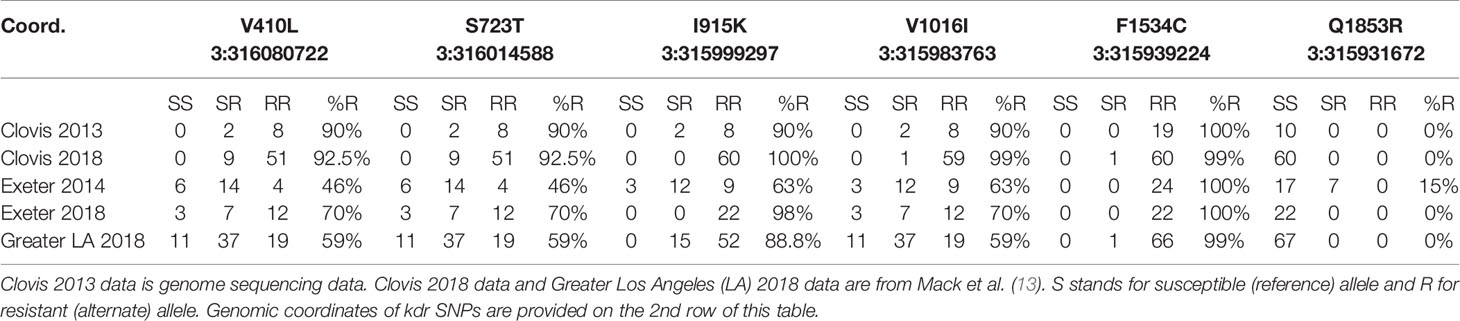
Table 2 Knockdown resistance (kdr) mutation genotypes per population.
Discussion
Our results strongly suggest that control actions taken by Delta Vector Control District personnel eliminated the local Ae. aegypti population in Exeter in 2014-2015. Aedes aegypti reinvaded Exeter in 2018 originating from different location/s than what originally invaded in 2014 and became established and are still present. We were able to confirm elimination and reinvasion in Exeter by comparative genetic analyses on 2014 and 2018 Exeter individuals. Had the Exeter population experienced a severe reduction due to DVCD control efforts, followed by 3-4 years of recovery before detection again, then the 2018 Exeter individuals would be expected to be genetically similar to those from Exeter 2014. However, the 2018 Exeter samples form a genetic cluster separate from that of the 2014 Exeter cluster. Genetic differentiation between 2014 and 2018 Exeter samples appears genome-wide with the majority of genomic regions exceeding FST > 0.1 (Figure 5). This pattern is in contrast to the level of genetic differentiation we observed between Clovis 2013 and 2016 (8). Such a drastic change in genome structure would be extremely unlikely to develop within a few years simply by genetic drift or natural selection, adding additional evidence toward the local elimination and reintroduction hypothesis for Ae. aegypti population in Exeter, CA.
The Exeter 2018 cluster more closely aligns with the samples from the Southwestern USA and Mexico indicating that 2018 samples likely resulted from a reintroduction to Exeter. The most likely source of reinvading individuals would intuitively come from neighboring cities like Clovis and Fresno, which experience high abundances of Ae. aegypti in the middle to late summer. However, the genetic similarity of the Exeter 2018 to southern CA Ae. aegypti suggests that the founding Exeter 2018 mosquito/es were likely escapees traveling in a vehicle traveling from southern CA, demonstrating the propensity of Ae. aegypti for human-mediated dispersal across long distances. Our study is the first report of Ae. aegypti from a Central CA location sharing the majority of its genetic profile with Ae. aegypti from southern CA. Importantly, when Ae. aegypti were detected in 2018 in Exeter, they were also detected in several Visalia suburbs (unpublished data), and it remains to be learned whether the Ae. aegypti that are now present in Visalia and other towns neighboring Exeter are genetically similar or different to those from Exeter and whether they are migrants from the second Exeter invasion or from other neighboring central California locations. The success in eliminating the population detected in 2014 is likely due to the limited geographical scope of the population and the intensive control efforts that were implemented. However, the possibility of eliminating the new populations of Ae. aegypti in Exeter and suburbs of Visalia is unlikely as resources are stretched too thin to mount such an intense control effort across such an extensive area.
Southern CA Ae. aegypti had different frequencies of non-synonymous SNPs or mutations in the VGSC gene than in Central CA populations, including the ubiquitous V410L mutation that is known to confer resistance to pyrethroids (Table 2). If all six of the mutations in the VGSC gene included in this study confer some levels of resistance to pyrethroids (kdr resistant alleles) then Southern CA populations have a higher proportion of susceptible alleles relative to central CA populations which is consistent with a previous report (13). Although Exeter 2014 and 2018 samples had different overall genetic profiles, they both contained a greater number of pyrethroid susceptible kdr alleles than the Clovis population. Presence of higher frequencies of kdr susceptible alleles may have been a factor leading to elimination of Ae. aegypti in Exeter in 2014, while other neighboring central CA districts struggled to manage dispersal of their Ae. aegypti populations.
These findings confirm that implementation of intensive control efforts can eliminate Ae. aegypti locally. However, the replacement of the Exeter 2014 population with individuals related to the Southern CA population that had a higher proportion of pyrethroid resistance alleles adds to the difficulty of adulticide control that could be further exacerbated when they hybridize to CA Central Valley populations that have an even greater number of kdr alleles. Elimination strategies require intensive efforts and resources, including public consent and establishment of a strong public relations program. Anticipating challenges resulting from the dispersal of resistant populations throughout California will be critical to comprehensive and sustainable control strategies.
This is the first record of southern CA-like Ae. aegypti genetic cluster found north of Kern county [latitude 35.21N; (25)]. The results from our study suggest that the population of southern CA Ae. aegypti continues to expand its range northwards and will hybridize with the existing Clovis population, corroborating previous indications of this possibility presented in Lee et al. (25). Additional statewide surveillance on the geographic distribution of Ae. aegypti genetic clusters combined with socio-environmental parameters may inform the nature and potential mechanisms of Ae. aegypti dispersal pathways within CA. Our relatively low-cost SNP genotyping assay or a similar approach could be a cost-effective way to screen a larger number of samples than current whole genome sequencing approaches and would be a useful tool to elucidate genetic mixing, origin of introductions, and pyrethroid resistance status of local populations.
Data Availability Statement
The data presented in the study are deposited in the NCBI BioProject repository with accession number PRJNA725510.
Author Contributions
YL, GA, GL, and AC conceived experimental design. EK, LM, and GA conducted SNP genotyping analysis. AR-W, T-YC, and KK conducted genomic DNA library preparations for whole genome sequencing. YL, MC, and TC conducted genome data analysis. EK, LM, KB, CG, RR-C, KS, LC, and EB conducted sample collection or arranged sample acquisition and provided the sample metadata. All authors contributed to the article and approved the submitted version.
Funding
We acknowledge funding support from the Pacific Southwest Regional Center of Excellence for Vector-Borne Diseases funded by the U.S. Centers for Disease Control and Prevention (Cooperative Agreement 1U01CK000516), CDC grant NU50CK000420-04-04, the Southern IPM Center (Project S21-002) as part of USDA National Institute of Food and Agriculture Crop Protection and Pest Management Regional coordination Program (Agreement No. 2018-70006-28884), the USDA National Institute of Food and Agriculture (Hatch project 1025565), UF/IFAS Florida Medical Entomology Laboratory fellowship to T-YC, and Florida Department of Health (Contract CODQJ). The findings and conclusions in this article are those of the author(s) and do not necessarily represent the views of the funding agencies.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ms. Mir Bear-Johnson at the Delta Vector Control District for providing materials for genetic analysis and sharing a detailed account of the mosquito control activities in Exeter, CA. We also thank Drs. Heinig and Lucas and their team at Collier Mosquito Control District, Dr. Sarah Shafer-Graham at Hillsborough County Mosquito Management Services, Dr. Carl Boohene at Polk County Mosquito Control District, Drs. Rui-de Xue, Steven Peper, and their team from Anastasia Mosquito Control District, Dr. Roger Jacobsen and his team at St. Lucie Mosquito Control & Coastal Management Services, Drs. Wade Brennan and Katie Cassell from Sarasota County Mosquito Management Services, for providing mosquito specimens for genetic analysis. We thank Rishi Kondapaneni, Ashley Malcolm, Brian Vasquez, and Eric Zeng at University of Florida to facilitate Ae. aegypti sample acquisition. We also thank Dr. David Moraga Amador at University of Florida NextGen DNA Sequencing Core and his team for their outstanding sequencing service.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fitd.2021.703873/full#supplementary-material
Supplementary Table 1 | iPLEX SNP primers and assay information
Supplementary Table 2 | Whole genome sequence metadata.
Supplementary Table 3 | Pairwise FST values.
Supplementary Figure 1 | Log likelihood values of Admixture runs.
Supplementary Figure 2 | Admixture plots for higher K values.
References
1. Tabachnick WJ. The Evolutionary Relationships Among Arboviruses and the Evolutionary Relationships of Their Vectors Provides a Method for Understanding Vector-Host Interactions. J Med Entomol (1991) 28:297–8.
2. Crosby MC. The American Plague : The Untold Story of Yellow Fever, the Epidemic That Shaped Our History. New York: Berkley Books (2006).
3. Gloria-Soria A, Brown JE, Kramer V, Hardstone Yoshimizu M, Powell JR. Origin of the Dengue Fever Mosquito, Aedes Aegypti, in California. PLoS Negl Trop Dis (2014) 8:e3029. doi: 10.1371/journal.pntd.0003029
4. Cornel AJ, Holeman J, Nieman CC, Lee Y, Smith C, Amorino M, et al. Surveillance, Insecticide Resistance and Control of an Invasive Aedes Aegypti (Diptera: Culicidae) Population in California. F1000Res (2016) 5:194. doi: 10.12688/f1000research.8107.2
5. CDPH. Aedes aegyptiAedes albopictusAedes Aegypti and Aedes albopictusMosquitoes in California by County/City (2021). California Department of Health. Available at: https://www.cdph.ca.gov/Programs/CID/DCDC/CDPH%20Document%20Library/AedesDistributionMap.pdf (Accessed Apri, 2021).
6. Metzger ME, Hardstone Yoshimizu M, Padgett KA, Hu R, Kramer VL. Detection and Establishment of Aedes Aegypti and Aedes Albopictus (Diptera: Culicidae) Mosquitoes in Californi-2015. J Med Entomol (2017) 54:533–43. doi: 10.1093/jme/tjw237
7. Pless E, Gloria-Soria A, Evans BR, Kramer V, Bolling BG, Tabachnick WJ, et al. Multiple Introductions of the Dengue Vector, Aedes Aegypti, Into California. PLoS Negl Trop Dis (2017) 11:e0005718. doi: 10.1371/journal.pntd.0005718
8. Lee Y, Schmidt H, Collier TC, Conner WR, Hanemaaijer MJ, Slatkin M, et al. Genome-wide Divergence Among Invasive Populations of Aedes Aegypti in California. BMC Genomics (2019a) 20:204. doi: 10.1186/s12864-019-5586-4
9. Schmidt H, Collier TC, Hanemaaijer MJ, Houston PD, Lee Y, Lanzaro GC. Abundance of Conserved CRISPR-Cas9 Target Sites Within the Highly Polymorphic Genomes of Anopheles and Aedes Mosquitoes. Nat Commun (2020) 11:1425. doi: 10.1038/s41467-020-15204-0
10. Chen T, Vorsino AE, Kosinski KJ, Romero-Weaver AL, Buckner EA, Chiu JC, et al. A Magnetic-Bead-Based Mosquito DNA Extraction Protocol for Next-Generation Sequencing. J Vis Exp (2021) 170:e62354. doi: 10.3791/62354
11. Clemons A, Mori A, Haugen M, Severson DW, Duman-Scheel M. Culturing and Egg Collection of Aedes Aegypti. Cold Spring Harb Protoc (2010) 2010:pdb prot5507. doi: 10.1101/pdb.prot5507
12. Lee Y, Weakley AM, Nieman CC, Malvick J, Lanzaro GC. A Multi-Detection Assay for Malaria Transmitting Mosquitoes. J Vis Exp (2015) 96:e52385. doi: 10.3791/52385
13. Mack LK, Kelly ET, Lee Y, Brisco KK, Shen KV, Zahid A, et al. Frequency of Sodium Channel Genotypes and Association With Pyrethrum Knockdown Time in Populations of Californian Aedes Aegypti. Parasit Vectors (2021) 14:141. doi: 10.1186/s13071-021-04627-3
14. Chen S, Zhou Y, Chen Y, Gu J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
15. Li H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs With BWA-MEM. arXiv (2013). doi: arXiv:1303.3997v2
16. Schmidt H, Hanemaaijer MJ, Cornel AJ, Lanzaro GC, Braack L, Lee Y. Complete Mitogenome Sequence of Aedes (Stegomyia) Aegypti Derived From Field Isolates From California and South Africa. Mitochondrial DNA Part B Resources (2018) 3:994–5. doi: 10.1080/23802359.2018.1495117
17. Hlaing T, Tun-Lin W, Somboon P, Socheat D, Setha T, Min S, et al. Mitochondrial Pseudogenes in the Nuclear Genome of Aedes Aegypti Mosquitoes: Implications for Past and Future Population Genetic Studies. BMC Genet (2009) 10:11. doi: 10.1186/1471-2156-10-11
18. Matthews BJ, Dudchenko O, Kingan SB, Koren S, Antoshechkin I, Crawford JE, et al. Improved Reference Genome of Aedes Aegypti Informs Arbovirus Vector Control. Nature (2018) 563:501–7. doi: 10.1038/s41586-018-0692-z
19. Okonechnikov K, Conesa A, Garcia-Alcalde F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics (2016) 32:292–4. doi: 10.1093/bioinformatics/btv566
20. Garrison E, Marth G. Haplotype-Based Variant Detection From Short-Read Sequencing. arXiv (2012).
21. Hudson RR, Slatkin M, Maddison WP. Estimation of Levels of Gene Flow From DNA Sequence Data. Genetics (1992) 132:583–9. doi: 10.1093/genetics/132.2.583
22. Miles A, Harding N. Scikit-Allel - Explore and Analyse Genetic Variation (2018). Available at: https://github.com/cggh/scikit-allel.
23. Felsenstein J. “Phylip (Phylogeny Inference Package)”. In: Seattle, 3.6 ed. Seattle, WA: Department of Genome Sciences, University of Washington (2005). Available at: http://evolution.genetics.washington.edu/phylip.html.
24. Alexander DH, Novembre J, Lange K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res (2009) 19:1655–64. doi: 10.1101/gr.094052.109
25. Lee Y, Schmidt H, Collier TC, Weakley AM, Gripkey H, Person K, et al. “Population Genomics of Aedes Aegypti in California”. In: Reisen WK, editor. Eighty-Senventh Annual Conference of the Mosuqito and Vector Control Association of California. Burlingame, CA: Mosquito and Vector Control Association of California (2019b). p. 23–9.
Keywords: mosquito, Aedes aegypti, California, population genomics, mosquito control, reintroduction assessment
Citation: Kelly ET, Mack LK, Campos M, Grippin C, Chen T-Y, Romero-Weaver AL, Kosinski KJ, Brisco KK, Collier TC, Buckner EA, Campbell LP, Cornel AJ, Lanzaro GC, Rosario-Cruz R, Smith K, Attardo GM and Lee Y (2021) Evidence of Local Extinction and Reintroduction of Aedes aegypti in Exeter, California. Front. Trop. Dis 2:703873. doi: 10.3389/fitd.2021.703873
Received: 30 April 2021; Accepted: 15 June 2021;
Published: 08 July 2021.
Edited by:
Andre Barretto Bruno Wilke, University of Miami, United StatesReviewed by:
JohnPaul Mutebi, Centers for Disease Control and Prevention (CDC), United StatesTom Schmidt, The University of Melbourne, Australia
Copyright © 2021 Kelly, Mack, Campos, Grippin, Chen, Romero-Weaver, Kosinski, Brisco, Collier, Buckner, Campbell, Cornel, Lanzaro, Rosario-Cruz, Smith, Attardo and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoosook Lee, eW9vc29vay5sZWVAdWZsLmVkdQ==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship