 Alexandra Cassano
Alexandra Cassano Anita S. Chong
Anita S. Chong Maria-Luisa Alegre
Maria-Luisa Alegre- 1Department of Medicine, University of Chicago, Chicago, IL, United States
- 2Department of Surgery, University of Chicago, Chicago, IL, United States
CD4+ Foxp3+ regulatory T cells (Tregs) are indispensable for preventing autoimmunity, and they play a role in cancer and transplantation settings by restraining immune responses. In this review, we describe evidence for the importance of Tregs in the induction versus maintenance of transplantation tolerance, discussing insights into mechanisms of Treg control of the alloimmune response. Further, we address the therapeutic potential of Tregs as a clinical intervention after transplantation, highlighting engineered CAR-Tregs as well as expansion of donor and host Tregs.
1. Introduction
Regulatory T cells (Tregs) are required to prevent autoimmunity and have a suppressive role on effector T cells (1, 2). The transcription factor forkhead box P3 (Foxp3) is expressed by Tregs and is indispensable for the Treg suppressive phenotype (3–6), with mutations in Foxp3 resulting in Immunodysregulation Polyendocrinopathy Enteropathy X-linked (IPEX) syndrome (7–9), although recent evidence indicates that microbiota-peripherally induced Tregs (pTregs) are able to retain some functionality in the absence of Foxp3 expression (10). Tregs control the T cell response to self-antigen, suppressing auto-reactive T cells and protecting from catastrophic autoimmunity (11–14). Regulatory CD8+ T cells have been characterized, such as CD8+ Qa-1-restricted suppressor cells, which have also been implicated in self-tolerance and allograft acceptance (15, 16), and CD8+ Foxp3+ regulatory cells, which have been described in both cancer and transplantation contexts (17–22). However, these regulatory subsets are less characterized, and this review article will focus on the role of CD4+ Foxp3+ Tregs in transplantation tolerance.
1.1. Treg subsets
Regulatory T cells are a heterogenous population and are often classified based on their origin. Tregs that develop in the thymus, termed tTregs (thymic Tregs) previously referred to as nTregs (natural Tregs), constitutively express Foxp3 and arise from either CD25+ or Foxp3low thymic progenitors during negative or positive selection, respectively (23–25). Tregs are often classified as CD3+ CD4+ Foxp3+ CD25high CD127low cells, since Foxp3 and CD127 expression are inversely correlated (26). Thymically derived tTregs make up the majority of the population of Tregs and are indispensable for controlling autoimmunity since they are reactive for self-antigens (2, 5, 24, 27).
Tregs can also develop from CD4+ conventional T cells (Tconvs) in the periphery in response to non-self-antigens. These Tregs are termed pTregs and are generated upon strong TCR activation in a tolerogenic environment, where the presence of IL-2, TGFβ and retinoic acid favors pTreg differentiation (24, 27–29). Induced Tregs generated in vitro, termed iTregs, are functionally similar to pTregs and require comparable conditions for generation (24, 27–29).
1.2. Phenotypic stability of Tregs
Continued expression of Foxp3 is required for the maintenance of the murine Treg phenotype and suppressive function (30), and both tTregs and pTregs adopt a memory T cell inflammatory phenotype upon cessation of Foxp3 expression (31). Inflammatory environments such as infection or autoimmunity can impair Treg stability, exacerbating disease progression. For instance, corneal inflammation following herpes simplex virus type 1 (HSV-1) infection led to a loss of Foxp3 expression in Tregs, which became pathogenic, producing IFNγ and driving stromal keratitis (32). In an autoimmune context, nTregs lost their Foxp3 expression and suppressive ability following co-culture with synovial fibroblasts from collagen-induced arthritic mice (33). On the other hand, certain inflammatory stimuli can enhance Treg stability, with type I interferon (IFN) augmenting Foxp3 acetylation and improving transplant survival (34).
2. Mechanisms of Treg suppression
Tregs can constrain immune responses through a variety of mechanisms that are contact-dependent and contact-independent. This review will focus on the role of Tregs in suppressing Tconvs, as Tconvs are known drivers of transplant rejection (35–37), although Tregs are also capable of mediating suppression of B cells, NK cells and NKT cells (38–40). Treg suppression mechanisms can be broadly categorized into three types: contact-dependent interactions with antigen-presenting cells (APCs), contact-dependent interactions with T cells, and contact-independent perturbations of the surrounding environment that alter cell metabolism or the cytokine milieu (Figure 1). Tconv proximity to Tregs following transplantation is promoted by Treg-produced chemokines; Foxp3 can bind to the promoter region of CCL3 and CCL4, leading to their increased production after TCR stimulation and the recruitment of CCR5+ Tconvs. CCL3-deficient Tregs are significantly less protective of islet allograft rejection (41), suggesting that this contributes to the ability of Treg suppression of graft-reactive Tconvs.

Figure 1. Mechanisms of Treg suppression. (A) Cell-contact-dependent interactions with T cells. Tregs can mediate suppression via transferring cAMP through gap junctions, membrane bound TGFβ, or the release of cytotoxic Granzyme A and Granzyme B. (B) Cell-contact-dependent interactions with APCs. Tregs express CTLA-4 which outcompetes CD28 on T cells for binding to CD80/CD86 and can mediate trogocytosis of CD80/86 as well as induce DC production of immunosuppressive IDO. LAG-3 binding to MHC-II on APCS inhibits their activation and function, impeding the ability of APCs to activate T cells. (C) Cell-contact-independent environmental perturbations. Tregs secrete immunosuppressive cytokines IL-10, IL-35 and TGFβ. Constitutive expression of the high affinity IL-2 receptor allows Tregs to outcompete T cells for IL-2 acting as a cytokine sink. Tregs increase environmental levels of immunosuppressive adenosine via CD39 and CD73 metabolism of ATP and AMP.
2.1. Contact-dependent suppression mechanisms
One of the major methods of contact-dependent Treg-mediated suppression of Tconv proliferation and activation is inducing the upregulation of cell surface inhibitory receptors on Tconvs, which typically occurs indirectly by inhibiting APC activation. For instance, Tregs constitutively express the inhibitory receptor cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) (42). CTLA-4 can outcompete the binding of the activating co-receptor CD28 to their shared ligands, CD80 and CD86, and also downregulate APC expression of CD80 and CD86 via trogocytosis, leaving the APCs less able to activate Tconvs (43–46). Importantly, the interaction between CTLA-4 on Tregs and CD80 and CD86 promotes dendritic cell (DC) expression of indoleamine-2,3-dioxygenase (IDO), a tolerogenic enzyme involved in tryptophan metabolism (47). The expression of IDO on DCs has been shown to inhibit T cell activation and proliferation (48–50), and overexpression of IDO on DCs prolonged murine intestinal allograft survival and expanded Tregs (51). In the same vein, Tregs express lymphocyte activation gene-3 (LAG-3) that binds to MHC-II and contributes to their suppressive activity, possibly by inhibiting DC activation, although this mechanism is less well understood (52–54).
Tregs can also act more directly on target cells themselves, driving apoptosis or suppression of target cells in a contact-dependent manner. Tregs are able to induce cell death through their expression of Granzyme A and Granzyme B (55–59). Tregs are also capable of inducing target cell death through activation of the TNF-related apoptosis inducing ligand (TRAIL)/Death Receptor 5 (DR5) pathway. This mechanism has been implicated in allograft survival as DR5-blocking antibodies prevented skin allograft survival in mice (60). Tregs can also express membrane-bound TGFβ on their cell surface and this may be an avenue of cell-cell contact-mediated Tconv suppression (61). Tregs are also able to enact metabolic changes in target cells to suppress proliferation and activation. For instance, Tconvs exhibit an increase in cyclic adenosine monophosphate (cAMP) and cAMP response element modulator (CREM) expression during cell contact-dependent Treg suppression via transfer of cAMP through gap junctions and this increased cAMP is necessary for inhibition of proliferation and IL-2 expression in target effector T cells (62, 63).
2.2. Contact-independent suppression mechanisms
Contact-independent mechanisms of suppression rely on Tregs driving a tolerogenic environment, therefore still requiring close proximity of Tregs to their target cells. For instance, Tregs can suppress Tconvs through secretion of the immunosuppressive cytokines IL-10 (64–68), IL-35 (69–72), and TGFβ (73, 74). Tregs also change the environmental cytokine concentration by acting as an “IL-2 sink” and outcompeting nearby Tconvs for IL-2 through expression of CD25, the high affinity IL-2 receptor α chain (64, 75, 76). Tregs also express the ectonucleotidases CD73 and CD39, which confer suppressive ability to Tregs by mediating the conversion of ATP and ADP to AMP then further to adenosine, thus modulating the extracellular adenosine content (77–80). Increased extracellular adenosine promotes a tolerogenic environment, impairing inflammatory cytokine production by DCs, while ATP has been shown to act as a pro-inflammatory signal (78–81). Tregs are therefore able to mediate bystander regulation in an antigen-agnostic manner; pre-treatment of recipient mice with donor alloantigen and anti-CD4 antibody generated Tregs that were not only sufficient to prevent the rejection of donor-matched skin allografts, but were also capable of preventing rejection of a 3rd party graft if the Tregs were reactivated with the tolerizing alloantigen prior to transplantation (82). Another mechanism through which Tregs are capable of promoting tolerance is through recruitment of other tolerogenic cells. Tregs have been found to recruit mast cells to skin allografts through production of IL-9, and this recruitment was indispensable for long-term tolerance in a costimulation blockade (αCD154) + donor-splenocyte transfer (CoB + DST)-mediated murine allograft tolerance model (83).
Foxp3+ Tregs can also secrete small extracellular vesicles (sEVs) that have Nrp1 in their membrane. Nrp1 in Treg-derived sEV promoted skin transplant tolerance and mice treated with Nrp1-KO Treg-derived sEV had decreased graft survival. Nrp1 expression in Tregs is associated with increased suppressive function and is necessary for phenotypic stability. Nrp1+ Treg-derived sEVs transferred to graft recipients resulted in significantly improved graft survival compared to Nrp1− sEVs or no sEVs. The presence of Nrp1+ sEVs skewed macrophages to a more tolerogenic phenotype in the graft highlighting the role of Tregs in promoting an environment permissive of allograft survival (84).
3. The fate of Tregs after transplantation
3.1. Treg proliferation and accumulation in the graft after transplantation
It has been well established than an increased ratio of Tregs:Tconvs is observed in tolerance, and that this high ratio is likely necessary for tolerance to occur/be maintained. After transplantation of an F1 Balb/c x B6 murine heart expressing the fusion protein 2W:OVA, endogenous donor-reactive Tregs (identified as 2W:I-Ab tetramer-binding Foxp3+ CD4+ T cells) accumulated in the spleen at comparable rates in both rejection and CoB + DST-mediated tolerance (85). In this model, during CoB + DST-mediated tolerance, endogenous donor-reactive Tconvs (identified as 2W:I-Ab tetramer-binding Foxp3− CD4+ T cells) proliferated less than they did in untreated hosts during acute rejection (85). This unequal expansion of Tregs and Tconvs led to a higher percentage of donor-reactive Tregs in both the spleen and graft (85). Treg accumulation in tolerant grafts and draining lymph nodes (dLNs) was also observed in αCD154+ rapamycin-treated mice that received rat islet xenografts (86). Corneal allograft recipient mice treated with αCD154 also exhibited increased graft-infiltrating Tregs and increased Treg to Tconv ratio in the spleen (87). Treg expansion was also observed in both the graft and dLNs of mice with spontaneously tolerant kidney allografts at both days 7 and 14 post-transplantation, along with an increase in splenic Treg proportions at day 14, possibly indicating initial Treg expansion in the graft followed by migration to the spleen (88).
A high Treg:Tconv ratio has been shown to be important in the maintenance of allograft tolerance. Indeed, transferring high numbers of alloreactive Tconvs to tolerant cardiac allograft recipient mice broke tolerance whereas low numbers did not (89). Similarly, in tolerant mice, transferring low numbers of alloreactive Tconvs in combination with αPD-L1 and αCD25 to partially deplete Tregs caused a loss of tolerance, suggesting that overwhelming Tregs can break established tolerance (89). In CoB + DST-treated tolerant mice, there was an enrichment of Foxp3 expression as well as an increased Foxp3+ Treg frequency in the allograft compared to that in the periphery or to rejecting allografts and syngeneic grafts (90–92). However, during Listeria monocytogenes (Lm) infection-mediated rejection, the high percentage of Foxp3+ cells in the CD4+ T cell compartment in the graft was reduced (91, 92). After resolution of Lm infection, the frequency of Foxp3+ Tregs in the graft returned to higher levels, accompanying a return of tolerance (92). Furthermore, after the infection was cleared, accumulation of Tregs reoccurred in a second donor-matched allograft, but αCD25-mediated depletion of Tregs at the time of second transplantation prevented acceptance of this new graft, indicating a role for Tregs in the restoration of tolerance (91).
3.2. Treg recruitment to the graft
As described above, Treg proximity to target cells is required for suppression. Therefore, Tregs must be recruited to the allograft to successfully constrain the Tconv-mediated rejection of the graft. DCs, particularly plasmacytoid DCs (pDCs), play a major role in the recruitment of Tregs to the graft and the induction of pTregs in the graft. During tolerance, pDCs picked up alloantigen in a cardiac allograft, migrated to peripheral LNs where they were necessary for the induction of Tregs and for tolerance (93). It has also been observed in mice that DBA/2 kidneys transplanted into B6 hosts are spontaneously accepted, while the reverse combination (B6 kidney transplanted into DBA/2 host) leads to rejection. Immunohistochemistry of the spontaneously accepted allografts showed Foxp3+-rich aggregates as well as the presence of SiglecHhigh pDCs. In vitro, pDCs from accepted kidneys induced more Foxp3 expression in T cells after co-culture than did syngeneic pDCs, and these Tregs were highly suppressive of sensitized T cells. Adoptive transfer of Tregs induced by culture with allogeneic pDCs was also capable of prolonging donor-matched cardiac allograft survival (94). Non-transplanted mice receiving allogeneic liver pDCs had increased percentage and counts of Tregs in the liver, spleen, and LNs. Furthermore, the presence of pDCs in the graft promoted Treg accumulation, protecting against graft injury and rejection, and depletion of pDCs from donor livers prior to transplantation led to acute rejection of the graft (95). Thus, pDCs seem central to tolerance and upstream of Treg accumulation.
Additionally, CCR4-mediated Treg recruitment to the graft was required for costimulation blockade mediated tolerance induction (90). CCR4 expression is regulated in part by Foxp3 expression and has been shown to promote Treg infiltration in the tumor microenvironment (96). CCL22 is a ligand for CCR4 and is produced by DCs and macrophages, highlighting a possible mechanism for DC-mediated Treg recruitment to the graft (90, 97, 98). Another potential pathway through which crosstalk between pDCs and Tregs can promote Treg differentiation and accumulation in allografts is through expression IDO, which has been shown to promote differentiation of Tregs in models of autoimmunity (99–101). The interaction between CTLA-4 on Tregs and CD80/CD86 on DCs in turn promotes IDO expression by DCs (47). Soluble CTLA-4 in the form of the fusion protein CTLA4-Ig has similarly been shown to induce long term graft survival in cardiac allograft recipient mice in an IDO-dependent manner (102–104). Patients who received CTLA4-Ig treatment after kidney transplantation had increased IDO+ cells in the periphery and increased Tregs in graft biopsies compared to cyclosporine-treated patients (105).
Although DCs have been shown to play a major role in Treg induction and trafficking to the graft, they are not the only innate immune cell type that can both promote rejection and be implicated in tolerance. For instance, monocytes, and neutrophils have also been shown to play a role in Treg recruitment. Recipient Gr-1+ innate immune cells were shown to play a role in promoting Treg trafficking to skin grafts in a human skin graft to mouse xenogeneic model using transfer of radiolabeled Tregs (106). Furthermore, macrophages may also play a role in Treg-mediated prolongation of graft survival; erythropoietin was capable of mediating prolongation of murine kidney allograft survival (BALB/c into B6) in a myeloid cell-dependent manner (107). Erythropoietin receptor signaling on myeloid cells drove a shift to a protective phenotype in macrophages, which in turn induced expansion of donor-reactive Tregs (107).
3.3. Phenotype of graft-infiltrating Tregs
Tregs that are recruited to the graft during transplantation tolerance exhibit a tolerogenic phenotype. Tregs accumulating in tolerant grafts expressed higher levels of TGFβ, IL-10, Cxcr3, and Blimp1 transcripts compared to splenic Tregs from both transplanted and naïve mice (88), as well as increased expression of Neuropilin-1 (Nrp1) and CD73 compared to Tregs in rejecting grafts (85). The majority of Tregs in tolerant grafts were PD-1+, but upon Lm infection the percentage of PD-1+ Tregs was reduced, which may account for part of the eroded tolerance observed after clearance of Lm infection (92). Similarly, liver Tregs induced by portal vein injection of allogeneic pDCs exhibited increased PD-1, Tim3, CD40l, and CD69 expression (95).
Inflammation due to tissue damage or infection poses a danger to graft survival due to the destabilizing effects of inflammation on Tregs. In a corneal transplant model, adoptively transferred Nrp1− pTregs were susceptible to conversion to IL-17- and IFNγ-producing exTregs in dry eye disease hosts but not control hosts. Furthermore, corneal transplant hosts with dry eye disease exhibited a decreased frequency of Tregs in the dLNs and impaired Treg suppressive capacity (108). Endothelial cells exposed to inflammatory conditions upregulated ICAM-1, PD-L1 and exposure to these endothelial cells led to an inhibition of Treg differentiation (109). Since some infections are capable of precipitating rejection of previously stably accepted allografts (110–112), one potential mechanism for this may be the destabilization of Tregs due to the inflammatory response to infection. Better understanding of the stability of Tregs after transplantation, at steady state and after infections would help improve clinical outcomes and offers new avenues for research.
4. Tregs as a prognostic tool in human transplantation
The different fate of Tregs in tolerance and rejection has prompted for the frequency and phenotype of Tregs in human transplant recipients to be used as a prognostic tool. In kidney allograft recipients, early after transplantation (1 and 3 months post-transplantation), the number of circulating Tregs and Foxp3 expression were associated with improved allograft survival and function both at the time of sampling and over the course of 5 years (113). In clinical trials to induce transient mixed chimerism and sustained graft acceptance in recipients of combined kidney and bone marrow transplants from haploidentical donors, recipients who remained tolerant to their graft as long as 5 years after immunosuppression cessation exhibited an expansion of pre-existing donor-specific Tregs at 6 months post-transplantation compared to pre-transplantation, whereas this same population was reduced at the same time point in a recipient that was not tolerant (114, 115). These Tregs expressed higher levels of Foxp3 and performed better in in vitro suppression assays after restimulation with donor B cells, compared to restimulation with 3rd party B cells (115). In transplant patients, Tregs in peripheral blood had increased surface and intracellular Notch-1 expression compared to Tconvs (116). Despite this, Notch-1low Tregs from human peripheral blood were more suppressive than Notch-1high Tregs, revealing an avenue of further research to understand the role of Notch-1 expression and signaling in Tregs (116). In kidney allograft recipients, a higher level of Foxp3 mRNA in cells from urine samples not only correlated with better graft function, but Foxp3 mRNA levels also predicted the reversibility of a T cell-mediated rejection event (117, 118). This is particularly promising as it demonstrates non-invasive methods of harnessing the prognostic potential of Tregs in transplantation success.
5. Interventions that modulate Treg suppressive ability
5.1. Post-transcriptional modifications
There are diverse factors that can impact the suppressive ability of Tregs in a transplantation context, one of which is post-transcriptional modifications that affect Treg differentiation, function, and stability. For instance, N6-methyladenosine mRNA modification was involved in Treg differentiation and function, and was regulated by “reader” and “writer” proteins including methyltransferase like 14 (METTL14), Wilms tumor 1-associating protein (WTAP), and methyltransferase like 3, all of which were necessary for Treg function (119–122). The ability of these proteins to modulate Treg functional ability is important in tolerance. Mice with METTL14-deficient Tregs rapidly rejected allografts and had lower levels of IL-10 and TGFβ (121). Similarly, expression of WTAP in peripheral blood samples from tolerant kidney allograft recipients was correlated with a higher Treg percentage in the peripheral blood and WTAP overexpression in naïve CD4+ T cells resulted in Foxo1 upregulation, promoting Treg differentiation and suppressive ability as well as improving allograft survival in mice (122). Tregs expressed increased miR-146a, a microRNA that targets TRAF6 and IRAK1 to negatively regulate NF-κB activation during rejection, and miR-146a-deficient Tregs had improved expansion but impaired function, although IFNγ blockade in miR-146a-deficient mice rescued Treg function and led to prolonged allograft survival (123). Another important post-transcriptional modification impacting Tregs is the acetylation of Foxp3 downstream of IFNβ-mediated activation of pSTAT1 (34). This acetylation promotes Treg function and stability and promotes long term allograft survival (34, 124).
5.2. Metabolic changes
Cellular metabolism plays a significant role in T cell function, and there is an increasingly appreciated role for metabolic changes in the suppressive function of Tregs, particularly in the context of anti-tumor immunity and in lupus. For example, in a mouse model of lupus, excess iron promoted the differentiation of pathogenic T follicular helper (Tfh) cells and increased production of pro-inflammatory cytokines. Moreover, increased transferrin receptor (CD71) expression was correlated with worse disease outcome, whereas blocking CD71 led to increased Treg differentiation and IL-10 production (125, 126).
The effects of metabolic changes on Tregs have been especially characterized in tumor settings due to the numerous metabolic alterations in the tumor microenvironment. Tregs exhibit a different metabolic profile from Tconvs, and Treg function is reduced upon high glucose uptake but enhanced with lactic acid uptake and metabolism (127, 128). Fatty acid oxidation and oxidative phosphorylation pathways are highly active in suppressive proliferating Tregs (128). Reduction in Tconv glycolytic activity via mTOR inhibition results in a more Treg-like metabolic profile, and this mTOR inhibition-mediated Tconv functional shift also prevented the induction of graft-versus-host disease (GvHD) (129). The distinct metabolic profile of Tregs suggests a promising avenue of therapeutic targeting in transplantation, as modulating glycolysis, oxidative phosphorylation, and fatty acid oxidation pathways could promote Treg proliferation and suppression without triggering Tconv activation.
5.3. Therapeutic interventions that affect Treg function
Treg phenotype and suppressive ability can also be affected via different therapeutic interventions (Figures 2A–C). In addition to inhibition the proliferation of Tconvs, CoB (αCD154 treatment) in transplant hosts was shown to augment Treg suppressive ability (130). In diabetic B6 Foxp3DTR mice transplanted with BALB/c islets, after normoglycemia was achieved with αCD154 treatment, Tregs from αCD154-treated tolerant mice suppressed B6 Tconvs better than Tregs from naïve mice when cultured with donor-matched BALB/c splenocytes compared to culture with third-party C3H splenocytes, indicating a donor-specific effect (131).
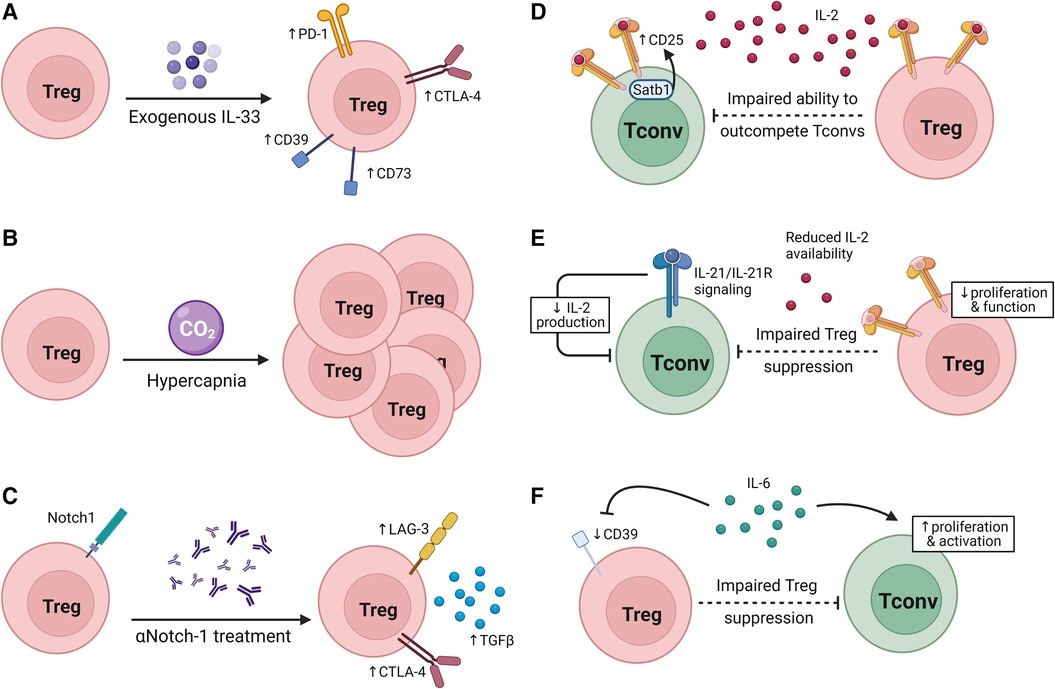
Figure 2. Modulating Treg suppressive ability. (A–C) Interventions that modulate Treg suppressive capacity in transplantation. (A) IL-33 treatment increases inhibitory receptor and CD39/CD73 expression on Tregs. (B) Hypercapnia promotes Treg expansion. (C) Neutralization of Notch-1 signaling increases inhibitory receptor expression and TGFβ production. (D–F) Factors that alter the susceptibility of Tconvs to Treg suppression. (D) Satb1 expression promotes CD25 expression on Tconvs, reducing Tregs’ ability to outcompete Tconvs for IL-2. (E) IL-21 signaling in Tconvs reduces IL-2 production, impairing the ability of Tregs to suppress Tconvs without impeding Tconv survival and function. (F) IL-6 signaling on Tregs can drive a reduction in CD39 expression by Tregs while IL-6 signaling drives Tconv activation and proliferation.
In line with the observation in both mice and humans that Notch-1low Tregs were more suppressive than Notch-1high Tregs in transplant recipients, Notch-1 blockade treatment prolonged allograft survival in a Treg-dependent manner (116). Furthermore, αNotch-1-treated cardiac allograft recipient mice not only had an increased proportion of Tregs in the spleen and graft, but splenic Tregs from these mice also expressed more LAG3, CTLA-4, Ki67, produced more TGFβ, and performed better in an in vitro Tconv suppression assay. Notch-1-deficient (N1c-KO) Tregs also outperformed wildtype (WT) Tregs in an in vitro suppression assay, and transfer of N1c-KO Tregs and WT CD4+ Tconvs improved graft survival in RAG-KO allograft recipients compared to WT CD4+ Tconvs and Tregs. Notch-1 inhibition in a humanized skin graft model reduced overall T cell infiltration to the graft, and increased the proportion of Tregs in the graft (116). These data emphasize the promise of using clinical observations of Treg phenotype post-transplantation to reveal targetable mechanisms to promote tolerance.
It has been shown in cancer, allergy, and lung injury that IL-33, an alarmin expressed at higher levels during inflammation (132, 133), promotes tissue repair and a more suppressive phenotype in ST2+ Tregs (134–138). However, IL-33 in the contexts of transplantation and GVHD has pleiotropic effects; there is evidence of IL-33 promoting Treg expansion, but IL-33 also has been shown to act as a costimulatory molecule for alloreactive Tconvs (138–142). In a GVHD model, IL-33/ST2 signaling in CD4+ cells promoted expansion and led to upregulation of Tbet while inhibiting IL-10 and Foxp3 expression which led to an exacerbation of GVHD severity (142, 143). Conversely, there is also evidence of IL-33 promoting ST2+ Treg expansion and therefore having a protective effect against GVHD (139). For instance, in a murine cardiac allograft model, IL-33 treatment not only prolonged graft survival but induced Foxp3+ Treg expansion in both the graft and spleen (140, 144). Treatment with exogenous IL-33 in skin allograft recipient mice caused a transcriptomic skewing towards an anti-inflammatory/tolerogenic phenotype, leading to an expansion of Foxp3+ cells in spleen, LNs, and periphery within the CD4+ population. These Tregs expanded after IL-33 treatment and expressed higher levels of the co-inhibitory receptors CTLA-4 and PD-1 as well as of CD39 and CD73 which are both involved in adenosine metabolism. Furthermore, IL-33 treatment led to long term graft survival in a skin allograft model (145). The complexities of the impacts of IL-33 on both Tregs and Tconvs in transplantation tolerance and rejection are yet to be fully understood.
Intermittent exposure to CO2 leads to hypercapnia, and subsequent prolongation of skin allograft survival. Mice with hypercapnia exhibited an increased percentage of Tregs in the CD4+ T cell compartment of both LNs and spleen as well as significantly decreased serum levels of pro-inflammatory cytokines IFNγ, TNFα, and IL-6. This may be due to increased numbers of iTregs since intermittent CO2 exposure-treated naïve T cells differentiated into Tregs at a higher rate than untreated cells (146). Interestingly, in the tumor environment, hypoxia has been shown to lead to increased extracellular adenosine and increased cAMP through adenosine receptor signaling, potentially indicating a mechanism through which hypercapnia may allow Tregs to suppress Tconvs and prolong graft survival (147, 148).
In a corneal allograft model, treatment with TIGIT-Fc, which significantly increased the mean graft survival time, led to increased Helios+ cells and also increased Foxp3+ cells compared to the PBS-treated control group. TIGIT-Fc treatment led to better suppression in a Treg suppression assay. The increased number and suppressive ability of Tregs after TIGIT-Fc treatment correlated with better graft survival (149).
Thus, several interventions can modulate Treg suppressive function in vivo, contributing to improved allograft survival. Whether some of these interventions can be combined or whether the maximal donor-specific suppression by Tregs is achieved by a single treatment remains to be investigated.
5.4. Clinical interventions that affect Treg function
Clinical treatment regimens for transplant recipients can be broadly divided into induction therapies and maintenance therapies. Induction therapies such as basiliximab and anti-thymocyte globulin (ATG) are often used in combination with longer term maintenance immunosuppression. The current standards of care for maintenance immunosuppression can be grouped into calcineurin inhibitors (CNI) such as cyclosporine and tacrolimus, mTOR inhibitors such as rapamycin/sirolimus, anti-proliferative therapies such as mycophenolate mofetil (MMF), JAK inhibitors, and co-stimulation blockade therapies such as belatacept. Unfortunately, many of these treatments are limited due to their depleting, destabilizing or otherwise inhibiting effects on Tregs (150–154).
Induction therapies are important for preventing acute rejection soon after transplantation (155). Rabbit anti-thymocyte globulin (rATG) is used to deplete lymphocytes and, along with basiliximab, a non-depleting IL-2R agonist, are commonly used induction agents (155). Basiliximab has been shown to lead to a decrease not only in Treg frequency, but also functional capacity. For instance, in vitro, basiliximab added to T cell culture from healthy donors led to a significant decrease in Treg frequency but not in the overall CD4+ or CD8+ T cell populations (156). Basiliximab suppressed Treg proliferation and IL-10 production in vitro, both functions which are known to be impacted by IL-2 signaling (156). Crucially, in a study of pediatric heart transplant recipients, the absolute number of CD4+Foxp3+CD25+ Tregs was significantly reduced compared to both pre-transplant levels and to patients who did not receive basiliximab (156). Kidney transplant recipients treated with basiliximab induction therapy had decreased circulating Tregs at both 1 and 2 weeks post-transplantation compared to patients who did not receive any induction therapy (151). A significant reduction in circulating Tregs in kidney recipients treated with basiliximab in combination with CNI + MMF + steroid therapy compared to patients who received only triple immunosuppression has been observed as early as 1 day post-transplantation and as far as 6 weeks post-transplantation (157). Basiliximab treatment also decreased the expression of CD25 on Tregs (157). In contrast to the effects of basiliximab, lymphocyte depletion via rATG may allow for the expansion of functional Tregs. In vitro, human peripheral blood mononuclear cells (PBMCs) cultured with rATG exhibited a conversion of CD4+CD25− T cells to CD4+CD25+ Tregs that were functionally suppressive (158). In both basiliximab and rATG induction therapies, Treg absolute number has been observed to decrease, but crucially the Treg frequency in rATG-treated recipients actually increased due to the decrease in Tconvs also observed in rATG induction therapy (154, 159, 160). Furthermore, rATG treatment has been shown to promote expansion of peripheral Tregs which likely contributed to the faster kinetics of Treg reconstitution in rATG-treated patients over basiliximab-treated patients (160–162). In a comparative study of islet allograft transplant recipients receiving either αCD25 or rATG induction therapies, ATG recipients maintained a stable frequency of CD25+CD4+ T cells, whereas the frequency of these Tregs decreased significantly in αCD25 induction therapy recipients (163). Of note, both groups had similar rates of insulin independence 1 year after islet transplantation (163), though the functional status of allospecific Tconvs was not compared.
CNIs such as cyclosporine (CsA) and tacrolimus are widely utilized immunosuppressive therapies in transplant recipients. These treatments block TCR-mediated activation of T cells and also block activation-induced cell death of T cells, which impairs the ability of co-stimulation blockade to induce tolerance (164–168). Additionally, CNIs have been shown to reduce peripheral Treg frequency in kidney transplant recipients (150, 169) which could limit the efficacy of these treatments. Importantly, clinical data on the effect of CNIs on Tregs are not all in agreement; for instance, in a study of kidney transplant recipients using either rapamycin or CsA as maintenance immunosuppression, no difference in circulating or graft-infiltrating Tregs was observed (168).
Belatacept, a CTLA4-Ig treatment, inhibits CD28 interaction with CD80/CD86, which is a critical pathway not only for Tconv activation but also for Treg homeostasis (170). Wildtype B6 mice administered four doses of CTLA4-Ig had half of the total Tregs by both percentage and absolute number than did their untreated counterparts (171). However, the effects of CTLA4-Ig on Tregs can be mixed. Cardiac allograft recipient mice that received 2 doses of CTLA4-Ig exhibited graft-reactive splenic Treg expansion at similar levels as αCD154 + DST-treated mice, but recipients treated with twice weekly CTLA4-Ig for 4 weeks showed a decrease in the percent of donor-specific Tregs (85), suggesting that more transient blockade of CD28 may preserve Treg viability. Indeed, it has been shown that the graft-protective effects of CTLA4-Ig are dependent on Tregs only at low doses, but not at high doses (172). Furthermore, CTLA4-Ig administration in murine recipients of a fully mismatched cardiac allograft prolonged graft survival but treatment in recipients of an MHC-II-mismatched allograft precipitated rejection (171). This could be due to the number of alloreactive Tconvs being smaller in a partially mismatched model, whereas in a fully-mismatched model the higher number of alloreactive Tconvs allows for CTLA4-Ig to have a greater effect on Tconvs than Tregs (171). Since CTLA4-Ig treatment may have negative impacts on Tregs, it may be beneficial to combine costimulation blockade therapies with Treg expanding interventions. For instance, IFNβ, which has been shown to promote Treg induction, synergized with CTLA4-Ig to prolong cardiac allograft survival in a murine model (34). Similarly, the addition of IL-2/αIL-2 complexes to CTLA4-Ig treatment in a murine cardiac allograft model reversed the observed decrease in Treg frequency observed with CTLA4-Ig and prolonged graft survival over CTLA4-Ig alone (173). These data illustrate the potential promise of combining Treg promoting therapies with more traditional immunosuppression. Belatacept has been approved for use in kidney transplant recipients and the BENEFIT Phase III study compared two belatacept regimens with CsA treatment (174). Interestingly, although belatacept treatment was associated with an increase in acute rejection episodes, by 1 year post-transplantation belatacept-treated recipients had similar patient and graft survival as CsA-treated recipients and by 7 years post-transplantation, belatacept-treated recipients had higher patient and graft survival as well as graft function than did CsA-treated patients (174, 175). Of note, in mixed lymphocyte reactions using Tregs from healthy volunteers, belatacept treatment inhibited both proliferation and Treg generation (153) highlighting the potential drawbacks of belatacept as a treatment option.
Inhibiting the JAK/STAT pathway is also a target pathway of interest to develop treatment regimens without the use of CNIs. A JAK3 inhibitor, CP-690,550, was efficacious in prolonging graft survival in mouse and monkey transplant recipients (176, 177). Alloactivated Tconvs from the peripheral blood of healthy individuals exhibited impaired proliferation when treated with CP-690,550 both in the presence and absence of Tregs (178). Furthermore, Tregs from human kidney transplant recipients treatment with CP-690,550 maintained functional suppressive abilities (178, 179). Despite the preservation of Treg functional capacity, CP-690,550 treatment led to a decrease in the percent of CD4+CD25high T cells from their pre-treatment baseline (179).
Unlike the therapies discussed above, mTOR inhibitors such as rapamycin (also known as sirolimus) may in fact be permissive of Treg function, expansion, and survival. Rapamycin binds to the FK506-binding protein-12 (FKBP12) but, unlike CsA or tacrolimus, rapamycin does not act as a CNI but rather inhibits mTOR (165, 168, 180, 181). Thus, rather than blocking TCR-mediated activation, rapamycin blocks CD4+ T cell progression through the cell cycle (165, 181). In vitro evidence has shown that rapamycin exposure leads to an expansion of functionally suppressive CD25hiCD4+ T cells, which are capable of preventing islet allograft rejection when adoptively transferred into recipient mice (165), and are more suppressive than Tregs cultured without rapamycin (182). Furthermore, rapamycin treatment in rats resulted in an increased ratio of CD25+CD4+ to CD25−CD4+ T cells (181). Rapamycin treatment also promoted the stability of phenotypic stability in an inflammatory context (183). In clinical contexts, rapamycin treatment in kidney transplant recipients did not cause a reduction in circulating Tregs (150, 169). Additionally, when human CD4+CD25+ Tregs were cultured with rapamycin, they outperformed Tregs cultured with CsA in an in vitro suppression assay, and more importantly were able to suppress on-going T cell responses in a mixed lymphocyte reaction (184). The benefits of rapamycin can also be observed in combination with other therapies. In a Phase II study, kidney recipients receiving a combination of rATG, belatacept, and sirolimus (rapamycin) not only had no incidence of acute rejection at 1 year post transplantation, but also had significantly higher frequencies of Tregs in the periphery than patients treated with combination therapies of rATG/belatacept/MMF, rATG/tacrolimus/MMF, or basiliximab/belatacept/MMF/steroids (185). Tregs from patients treated with rATG/belatacept/sirolimus also were highly suppressive in an in vitro suppression assay (185). Kidney transplant recipients treated with rapamycin did not experience a decrease in peripheral or kidney-infiltrating Treg frequencies (168).
It is important to note that existing research on the effects of post-transplantation therapies on Tregs, and the impact of potentially inhibited Tregs on graft survival and function are not yet fully understood. A more complete understanding of the impact of commonly used post-transplantation therapies is necessary for creating treatment regimens that preserve or promote Treg frequency and functionality. Accounting for Treg survival in post-transplantation treatment could be beneficial in promoting long-term graft survival and function, especially with the eventual goal of immunosuppression-free donor-specific tolerance.
5.5. Susceptibility of Tconvs to suppression and Tconv hypofunction
It is also conceivable that Tregs play a role in programming the Tconv hyporesponsiveness to alloantigen that can occur following tolerance induction (186), although this would need to be formally tested. In the tumor microenvironment, IL-10-producing and IL-35-producing Tregs together drive Blimp1 expression and an exhausted phenotype in CD8+ tumor-infiltrating T cells (187). In the transplantation setting, there is a correlation between hyporesponsiveness of alloreactive T cells and the presence of Tregs. For instance, in renal transplant patients, impaired production of IFNγ by PBMCs in response to donor-specific stimulation was associated with increased numbers of Tregs (188). An association between the presence of Tregs and more exhausted alloreactive T cells was also observed in a liver transplant model. Mice given pDC-depleted liver allografts not only had significantly decreased numbers and percentages of Tregs in both the graft and LNs, but also had fewer graft-infiltrating CD4+ and CD8+ T cells expressing the exhaustion markers Tim-3, PD-1, and CTLA-4. There were also increased levels of granzyme B, perforin and PD-L1 in pDC-depleted grafts, and CD4+ T cells from these livers produced significantly higher levels of IL-2 and TNFα. CD4+ and CD8+ T cells from recipients of pDC-depleted allografts had increased proliferation and in vitro pro-inflammatory cytokine production (IL-6, TNFα, IFNγ) compared to T cells from pDC-replete graft recipients (95). Whether this association between Tregs, pDCs and hyporesponsive or exhausted alloreactive T cells in transplantation tolerance is causal remains to be addressed.
Transplantation tolerance is not solely reliant on the suppressive ability of Tregs but also on the susceptibility of Tconvs to Treg suppression (Figures 2D–F). We reported that Tconvs deficient for the transcription factor Satb1 had impaired ability to upregulate CD25 upon TCR stimulation, allowing Tregs to outcompete them for IL-2 and leaving Satb1-deficient Tconvs more susceptible than control Tconvs to Treg suppression. Moreover, in a murine cardiac allograft model, mice with Satb1-deficient Tconvs and WT Tregs had significantly improved graft survival than mice with WT Tconvs and Tregs (189). Conversely, IL-21 inhibits the ability of Tregs to suppress Tconvs along the same axis. Indeed, IL-21/IL-21R signaling on Tconvs inhibited IL-2 production by Tconvs, impairing Treg proliferation and homeostasis while leaving Tconvs largely unaffected due to the ability of IL-21 to act in a similar manner as IL-2 on Tconvs (190, 191). However, the impact of IL-21 on the ability of Tregs to suppress Tconvs remains to be shown in transplantation settings.
Another axis through which Treg suppressive ability may be modulated in transplantation tolerance is IL-6 and TNFα signaling through PKB/c-Akt. It has been shown in multiple sclerosis, rheumatoid arthritis, psoriasis, and juvenile idiopathic arthritis that IL-6 and TNFα activate PKB/c-Akt in Tconvs and impair Treg suppression of Tconvs, permitting Tconv proliferation and activation. IL-6 receptor blockade or PKB inhibition both restored the suppressive capacity of Tregs (32, 192–198). Mechanistically, IL-6 may impair Treg suppression as a consequence of downregulating CD39 expression by Tregs (32, 192–198). This pathway has also been implicated in transplantation. IL-6 deficiency or its blockade in mouse models and human allograft recipients was accompanied by Treg expansion and prolonged graft survival (199–203). Improved suppressive capacity has also been observed in Tregs from IL-6-deficient allograft recipients, though the mechanism for this has not yet been defined (201). Since IL-6 is produced by DCs in response to damage associated with transplantation and in response to antigens (199, 204, 205), further understanding of the mechanisms conferring Tconv resistance to Treg suppression in transplantation is warranted.
6. Therapeutic potential of Tregs
6.1. Preclinical data
6.1.1. Donor-specific Tregs
Given the association between Tregs and better graft outcomes or responsiveness to immunosuppressive therapies, Tregs have been considered as a possible cell therapy to improve clinical transplant fate. One consideration for therapeutic harnessing of Tregs in clinical settings is choosing which Tregs to use. Polyclonal Tregs are more easily expanded or isolated, but donor-specific Tregs are likely more effective. In a murine islet allograft model, after recipient pre-conditioning by T cell depletion, transfer of fewer donor-reactive than polyclonal Tregs achieved indefinite graft survival (206). Transfer of iTregs stimulated in vitro with alloantigen prevented both acute and chronic rejection in a mouse model of bone marrow transplantation and cardiac or skin transplantation (207). Additionally, Tregs retrovirally transduced to express TCRs specific for donor peptide presented on host MHC-II were able to induce long-term allograft tolerance and protect against cellular infiltration of heart allograft (208). In vitro expanded donor-specific Tregs were functionally suppressive in vivo and trafficked to allografts where they were able to induce donor-specific tolerance better than polyclonal Tregs (82, 209, 210). The ability of alloreactive iTregs to promote donor-specific tolerance was augmented by the generation of these iTregs in the presence of vitamin C, which promoted lineage stability (211). This is an important finding since one major drawback of in vitro expanded Tregs is the potential for a loss of Foxp3 expression and Treg functionality (212).
Mouse data indicating more protection from donor-specific Tregs have been recapitulated with human cells. Human Tregs with direct allospecificity could be enriched and expanded and were more protective in a human skin xenotransplantation model (213). Furthermore, purified, expanded alloantigen-specific human Tregs more potently inhibited donor-reactive Tconvs in vitro than did expanded polyclonal Tregs (214). The findings that antigen-specific Tregs are more effective in tolerance induction are a further reason to pursue chimeric antigen receptor (CAR)-Treg therapies in which the antigen specificity of the Tregs can be manipulated.
6.1.2. Expansion of endogenous Tregs
As an alternative to transferring Tregs expanded in vitro, some groups have focused on expanding endogenous Tregs in vivo, using various approaches to deliver IL-2. To prevent low dose IL-2 treatment from expanding Tconvs, engineering approaches have been attempted. In a cardiac allograft model, transfer of Tregs expressing an engineered IL-2 receptor that binds exclusively to an engineered version of IL-2 led to indefinite heart graft survival after treatment with tacrolimus and engineered IL-2, and survival persisted after cessation of tacrolimus treatment (215). Other approaches to IL-2-mediated expansion of endogenous Tregs are also of interest. A controlled release microparticle system delivering TGFβ, IL-2, and rapamycin led to donor-specific tolerance characterized by indefinite graft survival, spontaneous acceptance of a second graft but rejection of a third-party graft, in a rat hindlimb major mismatched model. This system led to an increase in Treg frequency and a decrease in IFNγ+ Th1 cells when compared to untreated rats. Moreover, these Tregs were more effective at suppressing donor-reactive Tconvs than those from untreated rats (216). In a murine cardiac allograft model, low dose IL-2 administration, in combination with co-stimulation blockade, increased the frequency of Tregs in the graft, protecting against fibrosis and prolonging graft survival (217). Treatment of cardiac allograft recipient mice with IL-2/anti-IL-2 complexes in combination with CTLA4-Ig rescued the decrease in Tregs that usually accompanies CTLA4-Ig treatment because of its ability to block CD28 signals that are important for Treg survival, and significantly prolonged graft survival and health (173).
6.1.3. Donor-derived Tregs
It is well appreciated that recipient Tregs play an integral role in transplantation tolerance. However, the contribution of donor-derived Tregs, either transferred as passenger cells within the graft or adoptively transferred as therapy, is less well understood. Adoptive transfer of donor-derived nTregs prolonged murine cardiac allograft survival more effectively than recipient Tregs (218). Donor Tregs present in the graft itself were protective as evidenced by Treg depletion from the graft peri- and post-transplantation resulting in much faster rejection and more autoantibody response (218). Allogeneic donor-derived Tregs were able to inhibit in vitro IFNγ production by both CD8+ and CD4+ T cells more effectively than syngeneic or 3rd party Tregs. This inhibition was TGFβ- and IL-10-dependent and could be reversed by addition of IL-2 to the culture. Furthermore, in vivo adoptive transfer of donor-derived Tregs prolonged skin allograft survival and induced donor-specific tolerance (219).
6.1.4. Treg allorecognition pathways
An important consideration in the use of transferred alloreactive Tregs to promote transplant survival is whether they should recognize alloantigen directly or indirectly. Direct allorecognition refers to a T cell recognizing donor MHC expressed on donor parenchymal cells of the graft or on donor APCs (220). Conversely indirect allorecognition occurs when a T cell recognizes alloantigen that has been processed and presented by host APCs (peptides from donor MHC or from any protein with polymorphisms distinct between the donor and host) (220). Tconvs activated by direct allorecognition contribute more to early acute graft rejection since donor APCs are short lived, whereas effector T cells with indirect allospecificity have been shown to drive both acute and chronic rejection (220). Whether it would be more desirable to use direct or indirect Tregs depends on their specificity and mechanism of Tconv suppression. Direct allorecognition for antigens expressed constitutively on the graft, such as donor MHC Class I, would allow Tregs to localize to the graft after their transfer, whether they are transferred early or late after transplantation. In contrast, direct allorecognition for alloantigens expressed more transiently in the graft, such as donor MHC Class II, may not be as useful if MHC Class II-expressing donor APCs migrate out of the graft or die shortly after transplantation. If a major mechanism of Treg suppression is bystander, via secretion of suppressor cytokines or engagement of co-inhibitors on Tconvs such that they can suppress all Tconvs that reach the graft, then direct specificity for constitutively expressed MHC Class I may be optimal. A potential problem in sensitized hosts is if the target of Treg recognition (say donor MHC Class I) is also the target of existing alloantibodies that may coat the protein and prevent Treg access and Treg activation. If a major mechanism of Treg suppression is via inactivating APCs that are also presenting antigens to Tconvs, then indirect Tregs may be optimal, especially for peptides derived from donor MHC Class I that may be presented by host APCs throughout the life of the graft. For ex vivo generation of Tregs, stimulation with donor APCs expands Tregs with direct allospecificity, whereas Tregs with indirect allospecificity can be expanded in vitro by culture with either APCs that co-express host MHC Class II and donor MHC such that host MHC Class II can present peptides from donor MHC, or with host APCs pulsed with alloantigen (207, 221, 222). It has been demonstrated in murine models of both skin and cardiac allografts that while Tregs with direct allospecificity, expanded ex vivo with donor APCs are sufficient to prevent acute graft rejection, they are not enough to prevent chronic rejection from occurring (207, 208, 222). Adoptive transfer of Tregs that recognize alloantigen through both the direct and indirect antigen recognition pathways expanded ex vivo with F1 APCs, however, are capable of also efficiently inhibiting chronic graft rejection in an antigen-dependent manner (207, 208, 222). This is an important consideration when weighing methods of expanding Tregs for adoptive transfer therapies in the clinic since it seems that Tregs with indirect allospecificity may be better poised to promote long-term graft survival.
6.1.5. CAR-Tregs
There is growing interest in the use of CAR-Tregs, Tregs engineered to express an extracellular binding domain (most often an antibody variable domain or a TCR), frequently specific for a donor-expressed HLA class I, since these molecules are only expressed in the graft and remain constitutively expressed. CAR-Tregs bypass concerns about limited precursor frequency of endogenous donor-specific Tregs.
Since mismatched HLA is an appealing target for CAR-Treg therapies, multiple groups have designed CAR Tregs with a surface antibody directed to HLA-A2 (A2-CAR). Infusion of A2-CAR Tregs to mice transplanted with a cardiac allograft transgenic for HLA-A2 improved graft survival over that in untreated mice and in polyclonal Treg-infused recipients and A2-CAR Treg infusion in combination with rapamycin treatment improved survival over rapamycin alone (223). The efficacy of donor-specific CAR Tregs has also been demonstrated in unsensitized skin allografted mice, leading to prolonged graft survival and decreased humoral response (224). However, this effect was not observed in previously sensitized mice, indicating that sensitization-induced alloantibodies may mask the target of CAR-Tregs and reduce the efficacy of this treatment (224). The use of these A2-CAR Tregs has been shown to be effective in skin xenograft and humanized mouse models, in which they suppressed alloreactivity and prolonged graft survival (225–228). A2-CAR Tregs demonstrated improved suppressive abilities over both polyclonal Tregs and Tregs bearing an irrelevant CAR, demonstrating the promise of this therapy (223, 225–228). However, although donor-reactive CAR Tregs have shown an ability to improve allograft survival, few CAR Tregs were found at later time points in a murine model, showing a lack of CAR-Treg persistence and a gradual loss of immunosuppressive control (223). Additionally, persistent signaling in CAR-Tregs recognizing ubiquitously expressed antigens, such as HLA Class I antigens, appeared to cause an exhausted phenotype and impaired suppressive functionality (229, 230). This could present a significant issue for the use of CAR-Tregs in transplantation since the currently developed A2-CAR Tregs would persistently engage their activating antigen. However, culture with rapamycin and vitamin C has been shown to restore CAR-Treg function after tonic signaling-induced dysfunction (230). These findings, in conjunction with the effects of mTOR inhibition described above, reveal a potential method of addressing the shortcomings of CAR-Treg therapy while avoiding adverse effects on endogenous Tregs.
6.2. Clinical data
There have also been attempts at expanding endogenous recipient Tregs ex vivo and reinfuse them for therapeutic benefit. For instance, human Tregs have been successfully purified from peripheral blood and expanded ex vivo then reinfused, showing feasibility of this approach, although more work on efficacy is necessary (231, 232). In human liver allograft recipients, Tregs from liver perfusate were capable of suppressing proliferation and IFNγ production by both donor and recipient T cells, and early after transplantation donor liver-derived Tregs could be found in the peripheral blood of patients, suggesting that early suppression of direct allorecognition response via donor Tregs may be useful to promote tolerance (233).
The multi-center Phase I/IIA ONE study is the largest clinical trial investigating the use of Tregs for the induction of transplantation tolerance to-date (234–236). The ONE study investigated several types of cell therapies, testing the efficacy and safety of polyclonal Tregs, donor-reactive Tregs, tolerogenic DCs, and regulatory macrophages (235–237). As the effects of tolerogenic DCs and macrophages is outside the scope of this review, we will focus on describing the results as they pertain to Treg cell therapy compared to conventional immunosuppressive treatments. Importantly, the ONE study found that the use of immune cell therapy for transplant recipients was overall both safe and feasible (235), which opens the door for future studies of these therapies. Transferred autologous Tregs were either expanded ex vivo with donor antigen (donor-antigen reactive Tregs, darTregs) or with TCR stimulation and co-stimulation (polyclonal Tregs) (235, 236). Monitoring of cell therapy recipients showed that even in darTreg recipients, there was not a loss of Foxp3 demethylated regions specific to Tregs which indicated Treg phenotypic stability, a crucial finding for the use of Tregs as a therapy (235). Despite the exciting findings of this study, further research is needed to fully investigate the efficacy of Treg transfer therapies. The ONE study is currently being followed up with a Phase IIB study termed the TWO study aiming to determine the efficacy of using ex vivo expanded autologous Tregs to minimize the need for immunosuppression (238, 239).
Although most CAR-Treg data have been confined to murine studies, and findings on the use of CAR-Tregs in the clinic have yet to be published, the promise of CAR-Tregs is being evaluated in active clinical trials. The STEADFAST study is an active Phase I/II clinical trial assessing the safety and efficacy of autologous HLA-A2 CAR-Tregs in kidney transplant recipients (240). Similarly, the LIBERATE study is an active Phase I/II clinical trial investigating the use of autologous HLA-A2 CAR-Tregs in liver transplant recipients (241). These active trials highlight the importance of continuing basic research for a more complete understanding of the role of Tregs in transplant tolerance and in Tconv hypofunction. Additionally, although HLA-A2 mismatch is an appealing target due to its prevalence, identifying CAR targets other than HLA-A2 should be pursued for use in patients with HLA-A2-negative grafts.
7. Discussion and limitations
Tregs play a crucial role in transplantation tolerance due to their ability both to suppress Tconvs which are known drivers of transplant rejection, and to drive a more tolerogenic milieu. Tregs suppress Tconvs through direct cell-cell contact with either T cells or APCs, as well as through contact-independent mechanisms that alter the environment. After transplantation, Tregs migrate to and accumulate in the allograft, as they are recruited by DCs. Surprisingly, Tregs accumulate in similar numbers in both tolerant and rejecting allografts, but the ratio of Tregs:Tconvs is significantly higher in tolerant grafts due to the lack of Tconv expansion in tolerance compared to rejection. Despite the comparable accumulation in the graft, Tregs from tolerant allografts exhibit a more tolerogenic phenotype, with increased expression of co-inhibitory receptors and anti-inflammatory cytokines. These observations translate to the clinical setting, where Tregs can be used as a prognostic tool. Higher frequencies of Tregs have been correlated with improved graft function and survival and better responsiveness to immunosuppressive therapy in cases of rejection episodes (113, 115, 117, 118). However, the ability of human Tconvs to transiently upregulate Foxp3 during activation without acquiring a regulatory phenotype (242–245), a phenomenon that does not occur in mouse Tconvs, can blur phenotypic distinctions between Tconvs and Tregs such that some of these human studies should be interpreted cautiously. Although the mechanisms of Treg suppression have been well studied, the factors that can challenge or augment Treg stability and suppressive ability in humans could still be better understood, to allow development of therapies that can prevent rejection and promote the suppressive ability of Tregs to enable long-term allograft acceptance or even donor-specific tolerance.
The promise of targeting Tregs to promote long term allograft stability has generated interest in using Tregs in clinical settings. However, there are currently multiple limitations constraining the therapeutic use of Tregs. Expansion of endogenous Tregs via IL-2 administration carries the risk of expanding alloreactive Tconvs that upregulate CD25 upon alloantigen recognition and needs therefore to be performed carefully to only target Tregs. This concern is highlighted by the results of a recent clinical trial in which low dose IL-2 treatment not only induced an expansion of circulating Tregs but also precipitated an IFNγ-driven inflammatory response that primed liver allografts for rejection (246). Furthermore, although circulating polyclonal Tregs were expanded by low dose IL-2 treatment, neither circulating donor-specific nor graft Tregs were preferentially expanded (246). The lack of targeted donor-specific Treg expansion in response to currently available IL-2 treatments is a major limitation in the feasibility of these interventions. However, there are exciting steps being taken to address the drawbacks of IL-2-mediated Treg expansion. Engineered “ortho IL-2” which activates only an engineered IL-2 receptor transduced to be present on Tregs has been shown in murine models to induce Treg expansion without impacting Tconvs (215, 247). The use of this ortho IL-2/IL-2R system also promoted donor-specific tolerance in a murine cardiac allograft model (215, 247). In these studies, allografts were tolerated even after tacrolimus treatment was stopped (215) which indicates a promising avenue of harnessing Tregs to induce lasting donor-specific tolerance without relying on lifelong immunosuppression.
The use of transferred Tregs in a therapeutic context is also limited by the potential loss of the Treg phenotype. This limitation impacts all types of Treg therapies. Expansion of Tregs by repeated in vitro stimulation has been shown to inhibit Treg stability and functionality (248, 249). Autologous donor-reactive Tregs adoptively transferred to lymphodepleted cardiac allografted cynomolgus monkeys were unable to prolong graft survival, and were found to have downregulated expression of Foxp3, Helios, CTLA-4, and Ki67 after transfusion despite in vitro demonstration of suppressive ability (250). This may be addressed by culturing Tregs for adoptive transfer in the presence of rapamycin which, along with minimizing the number of repeated stimulations, promotes Treg persistence and suppressive function (182). In humans, transferred Tregs exhibited a decrease in donor reactivity after transfusion (232). Genetic manipulation of Tregs may be an effective approach to promoting survival and stability in transferred Tregs. For instance, forced expression of Foxp3 in HLA-A2 CAR-Tregs improved Treg stability under inflammatory conditions without compromising suppressive function (251). Foxp3 is not the only possible target, as JNK1-deficient Tregs had improved survival as well as increased Lag-3 expression and IL-10 production compared to wildtype Tregs in a murine islet allograft model (252). A more complete understanding of the mechanisms of Treg function and stability has the potential to reveal genetic targets to improve Treg stability, survival, and long-term function.
An additional concern in Treg therapy, specifically in the ex vivo expansion of Tregs, is that attaining sufficient numbers of usable donor-specific Tregs is difficult, and some patients may not be able to produce an infusible Treg product (232). For this reason, the feasibility of Treg adoptive transfer therapy may be reliant on the ability to use allogeneic “off the shelf” Tregs. The obvious drawback of this approach is that these Tregs may be rapidly eliminated by alloreactive T cells. The logical solution of eliminating MHC expression from these third-party Tregs has the caveat of rendering them vulnerable to deletion by host NK cells activated by missing-self MHC (253–255). Various strategies have been attempted to protect edited cells from NK-mediated deletion including overexpression of the “don't-eat-me signal” CD47 (256), retention of a single HLA-C allele (257), or expression of HLA-E (258). Further progress in this field is likely needed before allogeneic Tregs can be safely and effectively harnessed to promote transplant tolerance.
In previously sensitized hosts, donor-specific CAR Tregs lost their ability to prolong graft survival and protect against production of donor specific antibodies, potentially due to anti-HLA antibodies blocking HLA on the graft and thus preventing CAR-Treg binding and activation, or to the reduced sensitivity of memory T cells to Treg-mediated suppression (224). The lack of efficacy in sensitized recipients is a particularly important consideration for clinical efficacy of CAR-Tregs since sensitization, due to pregnancy prior to transplantation, to transfusions, heterologous immunity, or prior transplantation, impacts a substantial proportion of transplant candidates. Over 10% of candidates on the U.S. waiting list have previously received transplants and up to 30% of candidates on the kidney transplant waiting list are considered highly sensitized (259, 260).
Another limitation is that Tregs may not induce true donor-specific tolerance, but merely prolongation of graft survival. One study of murine skin allograft recipients, in which endogenous Tregs were expanded in vivo using IL-2/anti-IL-2 complexes, found that although grafts were accepted long term, the induction of donor-specific tolerance did not occur, as 2nd donor-matched grafts were rejected (261). The absence of donor-specific tolerance implies reduced quality or quantity of redundant mechanisms maintaining acceptance of the first graft, such that transplant recipients may have to remain on permanent immunosuppressive therapies with possible consequent side effects and reduced quality of life. Thus, novel therapies may need to combine approaches to both augment the function of donor-specific Tregs and also tolerize conventional naïve and memory donor-reactive T and B cells to achieve universal and durable donor-specific transplantation tolerance.
In summary, some key aspects about Tregs in transplantation have emerged. First, is the ratio of Tregs:Tconvs in an allograft. This is associated with better graft outcome, relies on the expansion, recruitment, and induction of Tregs (85–89) and may be facilitated by DCs (93, 94, 99–101). Indeed, Foxp3 regulates both CCR4 and CCL3 to modulate Treg recruitment to the allograft and Treg proximity to Tconvs (41, 90, 96) and CCR4-mediated recruitment to the graft is driven by CCL22 produced by DCs and macrophages (90, 96). Second, is the phenotype of Tregs present in the graft as it is critical for Treg suppression of graft-infiltrating Tconvs. Increased expression of the inhibitory receptor PD-1 and tolerogenic cytokines TGFβ and IL-10 have been observed in Tregs in tolerance (85, 88, 92, 95). CTLA-4 expression on Tregs is important for both stripping CD80/CD86 from APCs via trogocytosis (43–46) and for triggering IDO expression by DCs which promotes Treg expansion and inhibits Tconv survival (47, 51). The role of Tregs as an IL-2 sink may be a key factor for Tconv suppression in allograft tolerance and down-regulation of Satb1 in Tconvs observed during tolerance leads to lower CD25 expression by Tconvs and better IL-2 competition by Tregs (189). Third, the therapeutic potential of Treg transfer is evident but the optimal way to harness the product to improve transplant outcomes is not yet clear. Donor-specific Tregs have greater efficacy in promoting graft survival, but polyclonal Tregs are easier to expand ex vivo (206–210, 213, 214). Furthermore, the use of both Tregs with direct and indirect allospecificity, expanded with F1 APCs, is more effective at promoting both acute and chronic graft survival than the use of Tregs with only direct allospecificity (207, 208, 221, 222). The use of CAR-Tregs allows their targeting to the graft and CAR-Tregs targeting HLA-A2 have been developed and tested in mouse models, and are currently in clinical trials (225–228, 240, 241). The efficacy of Treg transfer as a therapeutic is dependent on long-term stability, which may be improved by culture with rapamycin and vitamin C (230), on efficacy of suppression of memory alloreactive T cells, and on still reaching their antigenic target in sensitized hosts. Finally, as discussed above, many currently used immunosuppression therapies, including belatacept, basiliximab, and calcineurin inhibitors, deplete, destabilize, or otherwise inhibit Tregs. Interestingly, rapamycin seems much more permissive of Treg function and survival than other clinically used immunosuppressants. Thus, the impact of immunosuppressants on Tregs (endogenous or transferred) needs to be considered post-transplantation to allow Tregs the best opportunity to promote graft survival and function.
Author contributions
ACa wrote the manuscript, ACh and M-LA edited it. All authors contributed to the article and approved the submitted version.
Conflict of interest
The author M-LA declared that they were an editorial board member of Frontiers at the time of submission. This had no impact on the peer review process and the final decision.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. (1996) 184(2):387–96. doi: 10.1084/jem.184.2.387
2. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995) 155(3):1151–64. doi: 10.4049/jimmunol.155.3.1151
3. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. (2001) 27(1):68–73. doi: 10.1038/83784
4. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
5. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4(4):330–6. doi: 10.1038/ni904
6. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor Foxp3. Immunity. (2005) 22(3):329–41. doi: 10.1016/j.immuni.2005.01.016
7. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27(1):20–1. doi: 10.1038/83713
8. Park JH, Lee KH, Jeon B, Ochs HD, Lee JS, Gee HY, et al. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: a systematic review. Autoimmun Rev. (2020) 19(6):102526. doi: 10.1016/j.autrev.2020.102526
9. Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol. (2012) 3:211. doi: 10.3389/fimmu.2012.00211
10. van der Veeken J, Campbell C, Pritykin Y, Schizas M, Verter J, Hu W, et al. Genetic tracing reveals transcription factor Foxp3-dependent and Foxp3-independent functionality of peripherally induced Treg cells. Immunity. (2022) 55(7):1173–1184.e7. doi: 10.1016/j.immuni.2022.05.010
11. Chinen T, Volchkov PY, Chervonsky AV, Rudensky AY. A critical role for regulatory T cell–mediated control of inflammation in the absence of commensal microbiota. J Exp Med. (2010) 207(11):2323–30. doi: 10.1084/jem.20101235
12. Kim J, Lahl K, Hori S, Loddenkemper C, Chaudhry A, deRoos P, et al. Cutting edge: depletion of Foxp3+ cells leads to induction of autoimmunity by specific ablation of regulatory T cells in genetically targeted mice. J Immunol. (2009) 183(12):7631–4. doi: 10.4049/jimmunol.0804308
13. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. (2007) 8(2):191–7. doi: 10.1038/ni1428
14. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, et al. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. (2007) 204(1):57–63. doi: 10.1084/jem.20061852
15. Choi JY, Eskandari SK, Cai S, Sulkaj I, Assaker JP, Allos H, et al. Regulatory CD8 T cells that recognize qa-1 expressed by CD4 T-helper cells inhibit rejection of heart allografts. Proc Natl Acad Sci. (2020) 117(11):6042–6. doi: 10.1073/pnas.1918950117
16. Kim HJ, Cantor H. Regulation of self-tolerance by qa-1-restricted CD8+regulatory T cells. Semin Immunol. (2011) 23(6):446–52. doi: 10.1016/j.smim.2011.06.001
17. Chaput N, Louafi S, Bardier A, Charlotte F, Vaillant JC, Ménégaux F, et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut. (2009) 58(4):520–9. doi: 10.1136/gut.2008.158824
18. Niederlova V, Tsyklauri O, Chadimova T, Stepanek O. CD8+ Tregs revisited: a heterogeneous population with different phenotypes and properties. Eur J Immunol. (2021) 51(3):512–30. doi: 10.1002/eji.202048614
19. Bézie S, Anegon I, Guillonneau C. Advances on CD8+ Treg cells and their potential in transplantation. Transplantation. (2018) 102(9):1467. doi: 10.1097/TP.0000000000002258
20. Li XL, Ménoret S, Bezie S, Caron L, Chabannes D, Hill M, et al. Mechanism and localization of CD8 regulatory T cells in a heart transplant model of tolerance. J Immunol. (2010) 185(2):823–33. doi: 10.4049/jimmunol.1000120
21. Picarda E, Bézie S, Usero L, Ossart J, Besnard M, Halim H, et al. Cross-reactive donor-specific CD8+ Tregs efficiently prevent transplant rejection. Cell Rep. (2019) 29(13):4245–4255.e6. doi: 10.1016/j.celrep.2019.11.106
22. Picarda E, Bézie S, Venturi V, Echasserieau K, Mérieau E, Delhumeau A, et al. MHC-derived allopeptide activates TCR-biased CD8+ Tregs and suppresses organ rejection. J Clin Invest. (2014) 124(6):2497–512. doi: 10.1172/JCI71533
23. Liston A, Nutsch KM, Farr AG, Lund JM, Rasmussen JP, Koni PA, et al. Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc Natl Acad Sci. (2008) 105(33):11903–8. doi: 10.1073/pnas.0801506105
24. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. (2013) 14(4):307–8. doi: 10.1038/ni.2554
25. Owen DL, Mahmud SA, Sjaastad LE, Williams JB, Spanier JA, Simeonov DR, et al. Thymic regulatory T cells arise via two distinct developmental programs. Nat Immunol. (2019) 20(2):195–205. doi: 10.1038/s41590-018-0289-6
26. Liu W, Putnam AL, Xu-yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 Expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. (2006) 203(7):1701–11. doi: 10.1084/jem.20060772
27. Lee HM, Bautista JL, Hsieh CS. Chapter 2: thymic and peripheral differentiation of regulatory T cells. In: Alexander R, Shimon S, editors. Advances in immunology. Cambridge Massachusetts: Academic Press (2011). p. 25–71.
28. Plitas G, Rudensky AY. Regulatory T cells: differentiation and function. Cancer Immunol Res. (2016) 4(9):721–5. doi: 10.1158/2326-6066.CIR-16-0193
29. Shevach EM, Thornton AM. Ttregs, pTregs, and iTregs: similarities and differences. Immunol Rev. (2014) 259(1):88–102. doi: 10.1111/imr.12160
30. Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. (2007) 8(3):277–84. doi: 10.1038/ni1437
31. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. (2009) 10(9):1000–7. doi: 10.1038/ni.1774
32. Bhela S, Varanasi SK, Jaggi U, Sloan SS, Rajasagi NK, Rouse BT. The plasticity and stability of regulatory T cells during viral-induced inflammatory lesions. J Immunol Baltim Md. (2017) 199(4):1342–52. doi: 10.4049/jimmunol.1700520
33. Yang S, Zhang X, Chen J, Dang J, Liang R, Zeng D, et al. Induced, but not natural, regulatory T cells retain phenotype and function following exposure to inflamed synovial fibroblasts. Sci Adv. (2020) 6(44):eabb0606. doi: 10.1126/sciadv.abb0606
34. Fueyo-González F, McGinty M, Ningoo M, Anderson L, Cantarelli C, Angeletti A, et al. Interferon-β acts directly on T cells to prolong allograft survival by enhancing regulatory T cell induction through Foxp3 acetylation. Immunity. (2022) 55(3):459–474.e7. doi: 10.1016/j.immuni.2022.01.011
35. Allen KJ, Mifsud NA, Williamson R, Bertolino P, Hardikar W. Cell-mediated rejection results in allograft loss after liver cell transplantation. Liver Transpl. (2008) 14(5):688–94. doi: 10.1002/lt.21443
36. Pietra BA, Wiseman A, Bolwerk A, Rizeq M, Gill RG. CD4 T cell–mediated cardiac allograft rejection requires donor but not host MHC class II. J Clin Invest. (2000) 106(8):1003–10. doi: 10.1172/JCI10467
37. Jones ND, Turvey SE, Van Maurik A, Hara M, Kingsley CI, Smith CH, et al. Differential susceptibility of heart, skin, and islet allografts to T cell-mediated rejection. J Immunol. (2001) 166(4):2824–30. doi: 10.4049/jimmunol.166.4.2824
38. Azuma T, Takahashi T, Kunisato A, Kitamura T, Hirai H. Human CD4+ CD25+ regulatory T cells suppress NKT cell functions. Cancer Res. (2003) 63(15):4516–20.12907625
39. Ghiringhelli F, Ménard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor–β–dependent manner. J Exp Med. (2005) 202(8):1075–85. doi: 10.1084/jem.20051511
40. Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting edge: direct suppression of B cells by CD4+CD25+ regulatory T Cells1. J Immunol. (2005) 175(7):4180–3. doi: 10.4049/jimmunol.175.7.4180
41. Patterson SJ, Pesenacker AM, Wang AY, Gillies J, Mojibian M, Morishita K, et al. T regulatory cell chemokine production mediates pathogenic T cell attraction and suppression. J Clin Invest. (2016) 126(3):1039–51. doi: 10.1172/JCI83987
42. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by Cd25+Cd4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte–associated antigen 4. J Exp Med. (2000) 192(2):303–10. doi: 10.1084/jem.192.2.303
43. Oderup C, Cederbom L, Makowska A, Cilio CM, Ivars F. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology. (2006) 118(2):240–9. doi: 10.1111/j.1365-2567.2006.02362.x
44. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. (2008) 322(5899):271–5. doi: 10.1126/science.1160062
45. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell extrinsic function of CTLA-4. Science. (2011) 332(6029):600–3. doi: 10.1126/science.1202947
46. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci. (2021) 118(30):e2023739118. doi: 10.1073/pnas.2023739118
47. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. (2003) 4(12):1206–12. doi: 10.1038/ni1003
48. Mellor AL, Sivakumar J, Chandler P, Smith K, Molina H, Mao D, et al. Prevention of T cell–driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat Immunol. (2001) 2(1):64–8. doi: 10.1038/83183
49. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. (1998) 281(5380):1191–3. doi: 10.1126/science.281.5380.1191
50. Hwu P, Du MX, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol. (2000) 164(7):3596–9. doi: 10.4049/jimmunol.164.7.3596
51. Xie FT, Cao JS, Zhao J, Yu Y, Qi F, Dai XC. IDO expressing dendritic cells suppress allograft rejection of small bowel transplantation in mice by expansion of Foxp3+ regulatory T cells. Transpl Immunol. (2015) 33(2):69–77. doi: 10.1016/j.trim.2015.05.003
52. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity. (2004) 21(4):503–13. doi: 10.1016/j.immuni.2004.08.010
53. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II1. J Immunol. (2008) 180(9):5916–26. doi: 10.4049/jimmunol.180.9.5916
54. Do JS, Visperas A, Sanogo YO, Bechtel JJ, Dvorina N, Kim S, et al. An IL-27/Lag3 axis enhances Foxp3+ regulatory T cell–suppressive function and therapeutic efficacy. Mucosal Immunol. (2016) 9(1):137–45. doi: 10.1038/mi.2015.45
55. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. (2005) 174(4):1783–6. doi: 10.4049/jimmunol.174.4.1783
56. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. (2007) 27(4):635–46. doi: 10.1016/j.immuni.2007.08.014
57. Loebbermann J, Thornton H, Durant L, Sparwasser T, Webster KE, Sprent J, et al. Regulatory T cells expressing granzyme B play a critical role in controlling lung inflammation during acute viral infection. Mucosal Immunol. (2012) 5(2):161–72. doi: 10.1038/mi.2011.62
58. Velaga S, Ukena SN, Dringenberg U, Alter C, Pardo J, Kershaw O, et al. Granzyme A is required for regulatory T-cell mediated prevention of gastrointestinal graft-versus-host disease. PLoS One. (2015) 10(4):e0124927. doi: 10.1371/journal.pone.0124927
59. Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. (2004) 104(9):2840–8. doi: 10.1182/blood-2004-03-0859
60. Ren X, Ye F, Jiang Z, Chu Y, Xiong S, Wang Y. Involvement of cellular death in TRAIL/DR5-dependent suppression induced by CD4+CD25+ regulatory T cells. Cell Death Differ. (2007) 14(12):2076–84. doi: 10.1038/sj.cdd.4402220
61. Nakamura K, Kitani A, Strober W. Cell contact–dependent immunosuppression by Cd4+Cd25+ regulatory T cells is mediated by cell surface–bound transforming growth factor β. J Exp Med. (2001) 194(5):629–44. doi: 10.1084/jem.194.5.629
62. Bopp T, Becker C, Klein M, Klein-Heßling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell–mediated suppression. J Exp Med. (2007) 204(6):1303–10. doi: 10.1084/jem.20062129
63. Bodor J, Fehervari Z, Diamond B, Sakaguchi S. ICER/CREM-mediated transcriptional attenuation of IL-2 and its role in suppression by regulatory T cells. Eur J Immunol. (2007) 37(4):884–95. doi: 10.1002/eji.200636510
64. Barthlott T, Moncrieffe H, Veldhoen M, Atkins CJ, Christensen J, O’Garra A, et al. CD25+CD4+ T cells compete with naive CD4+ T cells for IL-2 and exploit it for the induction of IL-10 production. Int Immunol. (2005) 17(3):279–88. doi: 10.1093/intimm/dxh207
65. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. (2011) 34(4):566–78. doi: 10.1016/j.immuni.2011.03.018
66. Stewart CA, Metheny H, Iida N, Smith L, Hanson M, Steinhagen F, et al. Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J Clin Invest. (2013) 123(11):4859–74. doi: 10.1172/JCI65180
67. Yu L, Yang F, Zhang F, Guo D, Li L, Wang X, et al. CD69 enhances immunosuppressive function of regulatory T-cells and attenuates colitis by prompting IL-10 production. Cell Death Dis. (2018) 9(9):1–14. doi: 10.1038/s41419-018-0927-9
68. Tsuji-Takayama K, Suzuki M, Yamamoto M, Harashima A, Okochi A, Otani T, et al. The production of IL-10 by human regulatory T cells is enhanced by IL-2 through a STAT5-responsive intronic enhancer in the IL-10 locus. J Immunol. (2008) 181(6):3897–905. doi: 10.4049/jimmunol.181.6.3897
69. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450(7169):566–9. doi: 10.1038/nature06306
70. Collison LW, Pillai MR, Chaturvedi V, Vignali DAA. Regulatory T cell suppression is potentiated by target T cells in a cell contact, IL-35- and IL-10-dependent manner. J Immunol. (2009) 182(10):6121–8. doi: 10.4049/jimmunol.0803646
71. Niedbala W, Wei XQ, Cai B, Hueber AJ, Leung BP, McInnes IB, et al. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol. (2007) 37(11):3021–9. doi: 10.1002/eji.200737810
72. Wei X, Zhang J, Gu Q, Huang M, Zhang W, Guo J, et al. Reciprocal expression of IL-35 and IL-10 defines two distinct effector Treg subsets that are required for maintenance of immune tolerance. Cell Rep. (2017) 21(7):1853–69. doi: 10.1016/j.celrep.2017.10.090
73. Levings MK, Sangregorio R, Sartirana C, Moschin AL, Battaglia M, Orban PC, et al. Human CD25+CD4+ T suppressor cell clones produce transforming growth factor β, but not interleukin 10, and are distinct from type 1 T regulatory cells. J Exp Med. (2002) 196(10):1335–46. doi: 10.1084/jem.20021139
74. Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, et al. TGF-β1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol. (2004) 172(2):834–42. doi: 10.4049/jimmunol.172.2.834
75. Brandenburg S, Takahashi T, de la Rosa M, Janke M, Karsten G, Muzzulini T, et al. IL-2 induces in vivo suppression by CD4+CD25+Foxp3+ regulatory T cells. Eur J Immunol. (2008) 38(6):1643–53. doi: 10.1002/eji.200737791
76. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation–mediated apoptosis of effector CD4+ T cells. Nat Immunol. (2007) 8(12):1353–62. doi: 10.1038/ni1536
77. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. (2013) 19(6):355–67. doi: 10.1016/j.molmed.2013.03.005
78. Ernst PB, Garrison JC, Thompson LF. Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol. (2010) 185(4):1993–8. doi: 10.4049/jimmunol.1000108
79. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204(6):1257–65. doi: 10.1084/jem.20062512
80. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells*. J Biol Chem. (2010) 285(10):7176–86. doi: 10.1074/jbc.M109.047423
81. Di Virgilio F, Vuerich M. Purinergic signaling in the immune system. Auton Neurosci. (2015) 191:117–23. doi: 10.1016/j.autneu.2015.04.011
82. Karim M, Feng G, Wood KJ, Bushell AR. CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen-specific reactivation in vivo is critical for bystander regulation. Blood. (2005) 105(12):4871–7. doi: 10.1182/blood-2004-10-3888
83. Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. (2006) 442(7106):997–1002. doi: 10.1038/nature05010
84. Campos-Mora M, De Solminihac J, Rojas C, Padilla C, Kurte M, Pacheco R, et al. Neuropilin-1 is present on Foxp3+ T regulatory cell-derived small extracellular vesicles and mediates immunity against skin transplantation. J Extracell Vesicles. (2022) 11(6):e12237. doi: 10.1002/jev2.12237
85. Young JS, Yin D, Vannier AGL, Alegre ML, Chong AS. Equal expansion of endogenous transplant-specific regulatory T cell and recruitment into the allograft during rejection and tolerance. Front Immunol. (2018) 9:1385. doi: 10.3389/fimmu.2018.01385
86. Muller YD, Mai G, Morel P, Serre-Beinier V, Gonelle-Gispert C, Yung GP, et al. Anti-CD154 mAb and rapamycin induce T regulatory cell mediated tolerance in rat-to-mouse islet transplantation. PLoS One. (2010) 5(4):e10352. doi: 10.1371/journal.pone.0010352
87. Tan X, Zeng H, Jie Y, Zhang Y, Xu Q, Pan Z. CD154 blockade modulates the ratio of Treg to Th1 cells and prolongs the survival of allogeneic corneal grafts in mice. Exp Ther Med. (2014) 7(4):827–34. doi: 10.3892/etm.2014.1527
88. Hu M, Wang C, Zhang GY, Saito M, Wang YM, Fernandez MA, et al. Infiltrating Foxp3+ regulatory T cells from spontaneously tolerant kidney allografts demonstrate donor-specific tolerance. Am J Transplant. (2013) 13(11):2819–30. doi: 10.1111/ajt.12445
89. Miller ML, Daniels MD, Wang T, Wang Y, Xu J, Yin D, et al. Tracking of TCR-transgenic T cells reveals that multiple mechanisms maintain cardiac transplant tolerance in mice. Am J Transplant. (2016) 16(10):2854–64. doi: 10.1111/ajt.13814
90. Lee I, Wang L, Wells AD, Dorf ME, Ozkaynak E, Hancock WW. Recruitment of Foxp3+ T regulatory cells mediating allograft tolerance depends on the CCR4 chemokine receptor. J Exp Med. (2005) 201(7):1037–44. doi: 10.1084/jem.20041709
91. Miller ML, Daniels MD, Wang T, Chen J, Young J, Xu J, et al. Spontaneous restoration of transplantation tolerance after acute rejection. Nat Commun. (2015) 6(1):7566. doi: 10.1038/ncomms8566
92. Young JS, Daniels MD, Miller ML, Wang T, Zhong R, Yin D, et al. Erosion of transplantation tolerance after infection. Am J Transplant. (2016) 17(1):81–90. doi: 10.1111/ajt.13910
93. Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat Immunol. (2006) 7(6):652–62. doi: 10.1038/ni1333
94. Oh NA, O’Shea T, Ndishabandi DK, Yuan Q, Hong S, Gans J, et al. Plasmacytoid dendritic cell driven induction of Tregs is strain-specific and correlates with spontaneous acceptance of kidney allografts. Transplantation. (2020) 104(1):39–53. doi: 10.1097/TP.0000000000002867
95. Nakano R, Yoshida O, Kimura S, Nakao T, Yokota S, Ono Y, et al. Donor plasmacytoid dendritic cells modulate effector and regulatory T cell responses in mouse spontaneous liver transplant tolerance. Am J Transplant. (2021) 21(6):2040–55. doi: 10.1111/ajt.16412
96. Sarkar T, Dhar S, Chakraborty D, Pati S, Bose S, Panda AK, et al. FOXP3/HAT1 axis controls Treg infiltration in the tumor microenvironment by inducing CCR4 expression in breast cancer. Front Immunol. (2022) 13:740588. doi: 10.3389/fimmu.2022.740588
97. Rapp M, Wintergerst MWM, Kunz WG, Vetter VK, Knott MML, Lisowski D, et al. CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. J Exp Med. (2019) 216(5):1170–81. doi: 10.1084/jem.20170277
98. Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol. (2015) 27(1):11–20. doi: 10.1093/intimm/dxu079
99. Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO activates regulatory T cells and blocks their conversion into TH17-like T cells. J Immunol Baltim Md. (2009) 183(4):2475–83. doi: 10.4049/jimmunol.0900986
100. Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol Baltim Md. (2010) 185(10):5953–61. doi: 10.4049/jimmunol.1001628
101. Lippens C, Duraes FV, Dubrot J, Brighouse D, Lacroix M, Irla M, et al. IDO-orchestrated crosstalk between pDCs and Tregs inhibits autoimmunity. J Autoimmun. (2016) 75:39–49. doi: 10.1016/j.jaut.2016.07.004
102. Mellor AL, Munn DH. Ido expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. (2004) 4(10):762–74. doi: 10.1038/nri1457
103. Puccetti P, Grohmann U. IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-κB activation. Nat Rev Immunol. (2007) 7(10):817–23. doi: 10.1038/nri2163
104. Sucher R, Fischler K, Oberhuber R, Kronberger I, Margreiter C, Ollinger R, et al. IDO and regulatory T cell support are critical for cytotoxic T lymphocyte-associated ag-4 ig-mediated long-term solid organ allograft survival. J Immunol. (2012) 188(1):37–46. doi: 10.4049/jimmunol.1002777
105. Furuzawa-Carballeda J, Lima G, Uribe-Uribe N, Avila-Casado C, Mancilla E, Morales-Buenrostro LE, et al. High levels of IDO-expressing CD16+ peripheral cells, and Tregs in graft biopsies from kidney transplant recipients under belatacept treatment. Transplant Proc. (2010) 42(9):3489–96. doi: 10.1016/j.transproceed.2010.08.037
106. Jacob J, Nadkarni S, Volpe A, Peng Q, Tung SL, Hannen RF, et al. Spatiotemporal in vivo tracking of polyclonal human regulatory T cells (Tregs) reveals a role for innate immune cells in Treg transplant recruitment. Mol Ther Methods Clin Dev. (2021) 20:324–36. doi: 10.1016/j.omtm.2020.12.003
107. Horwitz JK, Bin S, Fairchild RL, Karen SK, Yi Z, Zhang W, et al. Linking erythropoietin to regulatory T-cell-dependent allograft survival through myeloid cells. JCI Insight. (2022) 7(10):e158856. doi: 10.1172/jci.insight.158856
108. Hua J, Inomata T, Chen Y, Foulsham W, Stevenson W, Shiang T, et al. Pathological conversion of regulatory T cells is associated with loss of allotolerance. Sci Rep. (2018) 8(1):7059. doi: 10.1038/s41598-018-25384-x
109. Cross AR, Lion J, Poussin K, Glotz D, Mooney N. Inflammation determines the capacity of allogenic endothelial cells to regulate human Treg expansion. Front Immunol. (2021) 12:666531. doi: 10.3389/fimmu.2021.666531
110. Higdon LE, Tan JC, Maltzman JS. Infection, rejection, and the connection. Transplantation. (2023) 107(3):584. doi: 10.1097/TP.0000000000004297
111. Lopez C, Simmons RL, Mauer SM, Najarian JS, Good RA. Association of renal allograft rejection with virus infections. Am J Med. (1974) 56(3):280–9. doi: 10.1016/0002-9343(74)90609-3
112. Wang T, Ahmed EB, Chen L, Xu J, Tao J, Wang CR, et al. Infection with the intracellular bacterium, Listeria monocytogenes, overrides established tolerance in a mouse cardiac allograft model. Am J Transplant. (2010) 10(7):1524–33. doi: 10.1111/j.1600-6143.2010.03066.x
113. Krajewska M, Kościelska-Kasprzak K, Kamińska D, Żabińska M, Myszka-Kozłowska M, Gomułkiewicz A, et al. Kidney transplant outcome is associated with regulatory T cell population and gene expression early after transplantation. J Immunol Res. (2019) 2019:e7452019. doi: 10.1155/2019/7452019
114. National Institute of Allergy and Infectious Diseases (NIAID). Renal allograft tolerance through mixed chimerism. clinicaltrials.gov (2015). Report No.: study/NCT00801632. Available at: https://clinicaltrials.gov/ct2/show/study/NCT00801632 (Accessed March 7, 2023).
115. Savage TM, Shonts BA, Obradovic A, Dewolf S, Lau S, Zuber J, et al. Early expansion of donor-specific Tregs in tolerant kidney transplant recipients. JCI Insight. (2018) 3(22):e124086. doi: 10.1172/jci.insight.124086
116. Magee CN, Murakami N, Borges TJ, Shimizu T, Safa K, Ohori S, et al. Notch-1 inhibition promotes immune regulation in transplantation via regulatory T cell–dependent mechanisms. Circulation. (2019) 140(10):846–63. doi: 10.1161/CIRCULATIONAHA.119.040563
117. Luan D, Dadhania DM, Ding R, Muthukumar T, Lubetzky M, Lee JR, et al. FOXP3 mRNA profile prognostic of acute T cell–mediated rejection and human kidney allograft survival. Transplantation. (2021) 105(8):1825. doi: 10.1097/TP.0000000000003478
118. Muthukumar T, Dadhania D, Ding R, Snopkowski C, Naqvi R, Lee JB, et al. Messenger RNA for FOXP3 in the urine of renal-allograft recipients. N Engl J Med. (2005) 353(22):2342–51. doi: 10.1056/NEJMoa051907
119. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. (2014) 24(2):177–89. doi: 10.1038/cr.2014.3
120. Tong J, Cao G, Zhang T, Sefik E, Amezcua Vesely MC, Broughton JP, et al. M6a mRNA methylation sustains Treg suppressive functions. Cell Res. (2018) 28(2):253–6. doi: 10.1038/cr.2018.7
121. Liu Y, Yuan Y, Zhou Z, Cui Y, Teng Y, Huang H, et al. Mettl14-mediated m6A modification enhances the function of Foxp3+ regulatory T cells and promotes allograft acceptance. Front Immunol. (2022) 13:1022015. doi: 10.3389/fimmu.2022.1022015
122. Wang Z, Qi Y, Feng Y, Xu H, Wang J, Zhang L, et al. The N6-methyladenosine writer WTAP contributes to the induction of immune tolerance post kidney transplantation by targeting regulatory T cells. Lab Invest. (2022) 102(11):1268–79. doi: 10.1038/s41374-022-00811-w
123. Lu J, Wang W, Li P, Wang X, Gao C, Zhang B, et al. MiR-146a regulates regulatory T cells to suppress heart transplant rejection in mice. Cell Death Discov. (2021) 7(1):1–12. doi: 10.1038/s41420-021-00534-9
124. Tao R, de Zoeten EF, Özkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. (2007) 13(11):1299–307. doi: 10.1038/nm1652
125. Voss K, Sewell AE, Krystofiak ES, Gibson-Corley KN, Young AC, Basham JH, et al. Elevated transferrin receptor impairs T cell metabolism and function in systemic lupus erythematosus. Sci Immunol. (2023) 8(79):eabq0178. doi: 10.1126/sciimmunol.abq0178
126. Gao X, Song Y, Wu J, Lu S, Min X, Liu L, et al. Iron-dependent epigenetic modulation promotes pathogenic T cell differentiation in lupus. J Clin Invest. (2022) 132(9):e152345. doi: 10.1172/JCI152345
127. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature. (2021) 591(7851):645–51. doi: 10.1038/s41586-020-03045-2
128. Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D, et al. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J Hematol OncolJ Hematol Oncol. (2022) 15(1):104. doi: 10.1186/s13045-022-01322-3
129. Nguyen HD, Chatterjee S, Haarberg KMK, Wu Y, Bastian D, Heinrichs J, et al. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J Clin Invest. (2016) 126(4):1337–52. doi: 10.1172/JCI82587
130. Jarvinen LZ, Blazar BR, Adeyi OA, Strom TB, Noelle RJ. CD154 on the surface of CD4+CD25+ regulatory T cells contributes to skin transplant tolerance. Transplantation. (2003) 76(9):1375. doi: 10.1097/01.TP.0000093462.16309.73
131. Lee SJ, Kim HJ, Byun NR, Park CG. Donor-specific regulatory T cell-mediated immune tolerance in an intrahepatic murine allogeneic islet transplantation model with short-term anti-CD154 mAb single treatment. Cell Transplant. (2020) 29:0963689720913876. doi: 10.1177/0963689720913876
132. Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. (2014) 31:31–7. doi: 10.1016/j.coi.2014.09.004
133. Chan BCL, Lam CWK, Tam LS, Wong CK. IL33: roles in allergic inflammation and therapeutic perspectives. Front Immunol. (2019) 10:364. doi: 10.3389/fimmu.2019.00364
134. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. (2014) 513(7519):564–8. doi: 10.1038/nature13577
135. Pastille E, Wasmer MH, Adamczyk A, Vu VP, Mager LF, Phuong NNT, et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. (2019) 12(4):990–1003. doi: 10.1038/s41385-019-0176-y
136. Faustino LD, Griffith JW, Rahimi RA, Nepal K, Hamilos DL, Cho JL, et al. Interleukin-33 activates regulatory T cells to suppress innate γδ T cell responses in the lung. Nat Immunol. (2020) 21(11):1371–83. doi: 10.1038/s41590-020-0785-3
137. Liu Q, Dwyer GK, Zhao Y, Li H, Mathews LR, Chakka AB, et al. IL-33–mediated IL-13 secretion by ST2+ Tregs controls inflammation after lung injury. JCI Insight. (2019) 4(6):e123919. doi: 10.1172/jci.insight.123919
138. Bou Saba J, Turnquist HR. The reparative roles of IL-33. Transplantation. (2023) 107(5):1069. doi: 10.1097/TP.0000000000004447
139. Matta BM, Reichenbach DK, Zhang X, Mathews L, Koehn BH, Dwyer GK, et al. Peri-alloHCT IL-33 administration expands recipient T-regulatory cells that protect mice against acute GVHD. Blood. (2016) 128(3):427–39. doi: 10.1182/blood-2015-12-684142
140. Brunner SM, Schiechl G, Falk W, Schlitt HJ, Geissler EK, Fichtner-Feigl S. Interleukin-33 prolongs allograft survival during chronic cardiac rejection. Transpl Int. (2011) 24(10):1027–39. doi: 10.1111/j.1432-2277.2011.01306.x
141. Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 is an unconventional alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol. (2014) 193(8):4010–20. doi: 10.4049/jimmunol.1400481
142. Dwyer GK, Mathews LR, Villegas JA, Lucas A, de Peredo AG, Blazar BR, et al. IL-33 acts as a costimulatory signal to generate alloreactive Th1 cells in graft-versus-host disease. J Clin Invest. (2022) 132(12):e150927. doi: 10.1172/JCI150927
143. Reichenbach DK, Schwarze V, Matta BM, Tkachev V, Lieberknecht E, Liu Q, et al. The IL-33/ST2 axis augments effector T-cell responses during acute GVHD. Blood. (2015) 125(20):3183–92. doi: 10.1182/blood-2014-10-606830
144. Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, et al. IL-33 Expands suppressive CD11b+ gr-1int and regulatory T cells, including ST2l+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol. (2011) 187(9):4598–610. doi: 10.4049/jimmunol.1100519
145. Kawai K, Uchiyama M, Hester J, Issa F. IL-33 drives the production of mouse regulatory T cells with enhanced in vivo suppressive activity in skin transplantation. Am J Transplant. (2021) 21(3):978–92. doi: 10.1111/ajt.16266
146. Tzeng YS, Peng YJ, Tang SE, Huang KL, Chu SJ, Wu SY, et al. Intermittent exposure of hypercapnia suppresses allograft rejection via induction of Treg differentiation and inhibition of neutrophil accumulation. Biomedicines. (2022) 10(4):836. doi: 10.3390/biomedicines10040836
147. Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. (2004) 22(1):657–82. doi: 10.1146/annurev.immunol.22.012703.104731
148. Vignali PDA, DePeaux K, Watson MJ, Ye C, Ford BR, Lontos K, et al. Hypoxia drives CD39-dependent suppressor function in exhausted T cells to limit antitumor immunity. Nat Immunol. (2022) 24(2):267–79. doi: 10.1038/s41590-022-01379-9
149. Li S, Zhang P, Li A, Bao J, Pan Z, Jie Y. TIGIT-Fc prolongs corneal allograft survival in mice by upregulating TIGIT/CD226 expression and the proportion of Helios+Foxp3+ Treg cells. Transplantation. (2022) 107(2):372–381. doi: 10.1097/TP.0000000000004257
150. Segundo DS, Ruiz JC, Izquierdo M, Fernández-Fresnedo G, Gómez-Alamillo C, Merino R, et al. Calcineurin inhibitors, but not rapamycin, reduce percentages of CD4+CD25+FOXP3+ regulatory T cells in renal transplant recipients. Transplantation. (2006) 82(4):550. doi: 10.1097/01.tp.0000229473.95202.50
151. Zhao T, Yang C, Xue Y, Qiu YY, Hu L, Qiu Y, et al. Impact of basiliximab on the proportion of regulatory T cells and their subsets early after renal transplantation: a preliminary report. Transplant Proc. (2012) 44(1):175–8. doi: 10.1016/j.transproceed.2011.11.026
152. Cortés-Hernández A, Alvarez-Salazar E, Arteaga-Cruz S, Alberu-Gómez J, Soldevila G. Ex vivo expansion of regulatory T cells from long-term belatacept-treated kidney transplant patients restores their phenotype and suppressive function but not their FOXP3 TSDR demethylation status. Cell Immunol. (2020) 348:104044. doi: 10.1016/j.cellimm.2020.104044
153. Levitsky J, Miller J, Huang X, Chandrasekaran D, Chen L, Mathew JM. Inhibitory effects of belatacept on allospecific regulatory T cell generation in humans. Transplantation. (2013) 96(8):689–96. doi: 10.1097/TP.0b013e31829f1607
154. Bluestone JA, Liu W, Yabu JM, Laszik ZG, Putnam A, Belingheri M, et al. The effect of costimulatory and interleukin 2 receptor blockade on regulatory T cells in renal transplantation. Am J Transplant. (2008) 8(10):2086–96. doi: 10.1111/j.1600-6143.2008.02377.x
155. Hardinger KL, Brennan DC, Klein CL. Selection of induction therapy in kidney transplantation. Transpl Int. (2013) 26(7):662–72. doi: 10.1111/tri.12043
156. López-Abente J, Martínez-Bonet M, Bernaldo-de-Quirós E, Camino M, Gil N, Panadero E, et al. Basiliximab impairs regulatory T cell (TREG) function and could affect the short-term graft acceptance in children with heart transplantation. Sci Rep. (2021) 11(1):827. doi: 10.1038/s41598-020-80567-9
157. Vondran FWR, Timrott K, Tross J, Kollrich S, Schwarz A, Lehner F, et al. Impact of basiliximab on regulatory T-cells early after kidney transplantation: down-regulation of CD25 by receptor modulation. Transpl Int. (2010) 23(5):514–23. doi: 10.1111/j.1432-2277.2009.01013.x
158. Feng X, Kajigaya S, Solomou EE, Keyvanfar K, Xu X, Raghavachari N, et al. Rabbit ATG but not horse ATG promotes expansion of functional CD4+CD25highFOXP3+regulatory T cells in vitro. Blood. (2008) 111(7):3675–83. doi: 10.1182/blood-2008-01-130146
159. Krystufkova E, Sekerkova A, Striz I, Brabcova I, Girmanova E, Viklicky O. Regulatory T cells in kidney transplant recipients: the effect of induction immunosuppression therapy. Nephrol Dial Transplant. (2012) 27(6):2576–82. doi: 10.1093/ndt/gfr693
160. Bouvy AP, Klepper M, Kho MML, Boer K, Betjes MGH, Weimar W, et al. The impact of induction therapy on the homeostasis and function of regulatory T cells in kidney transplant patients. Nephrol Dial Transplant. (2014) 29(8):1587–97. doi: 10.1093/ndt/gfu079
161. Gurkan S, Luan Y, Dhillon N, Allam SR, Montague T, Bromberg JS, et al. Immune reconstitution following rabbit antithymocyte globulin. Am J Transplant. (2010) 10(9):2132–41. doi: 10.1111/j.1600-6143.2010.03210.x
162. Lopez M, Clarkson MR, Albin M, Sayegh MH, Najafian N. A novel mechanism of action for anti-thymocyte globulin: induction of CD4: +: CD25: +: foxp3: +: regulatory T cells. J Am Soc Nephrol. (2006) 17(10):2844. doi: 10.1681/ASN.2006050422
163. Toso C, Edgar R, Pawlick R, Emamaullee J, Merani S, Dinyari P, et al. Effect of different induction strategies on effector, regulatory and memory lymphocyte sub-populations in clinical islet transplantation. Transpl Int. (2009) 22(2):182–91. doi: 10.1111/j.1432-2277.2008.00746.x
164. Gao W, Lu Y, El Essawy B, Oukka M, Kuchroo VK, Strom TB. Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. Am J Transplant. (2007) 7(7):1722–32. doi: 10.1111/j.1600-6143.2007.01842.x
165. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+regulatory T cells. Blood. (2005) 105(12):4743–8. doi: 10.1182/blood-2004-10-3932
166. Azzi JR, Sayegh MH, Mallat SG. Calcineurin inhibitors: 40 years later, can’t live without. J Immunol. (2013) 191(12):5785–91. doi: 10.4049/jimmunol.1390055
167. Wiederrecht G, Lam E, Hung S, Martin M, Sigal N. The mechanism of action of FK-506 and cyclosporin A. Ann N Y Acad Sci. (1993) 696(1):9–19. doi: 10.1111/j.1749-6632.1993.tb17137.x
168. Libetta C, Esposito P, Gregorini M, Margiotta E, Martinelli C, Borettaz I, et al. Sirolimus vs cyclosporine after induction with basiliximab does not promote regulatory T cell expansion in de novo kidney transplantation: results from a single-center randomized trial. Transpl Immunol. (2015) 33(2):117–24. doi: 10.1016/j.trim.2015.07.005
169. Segundo DS, Ruiz JC, Fernández-Fresnedo G, Izquierdo M, Gómez-Alamillo C, Cacho E, et al. Calcineurin inhibitors affect circulating regulatory T cells in stable renal transplant recipients. Transplant Proc. (2006) 38(8):2391–3. doi: 10.1016/j.transproceed.2006.08.081
170. Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, et al. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells 1. J Immunol. (2003) 171(7):3348–52. doi: 10.4049/jimmunol.171.7.3348
171. Riella LV, Liu T, Yang J, Chock S, Shimizu T, Mfarrej B, et al. Deleterious effect of CTLA4-ig on a Treg-dependent transplant model. Am J Transplant. (2012) 12(4):846–55. doi: 10.1111/j.1600-6143.2011.03929.x
172. Schwarz C, Unger L, Mahr B, Aumayr K, Regele H, Farkas AM, et al. The immunosuppressive effect of CTLA4 immunoglobulin is dependent on regulatory T cells at low but not high doses. Am J Transplant. (2016) 16(12):3404–15. doi: 10.1111/ajt.13872
173. Schwarz C, Mahr B, Muckenhuber M, Weijler AM, Unger LW, Pilat N, et al. In vivo Treg expansion under costimulation blockade targets early rejection and improves long-term outcome. Am J Transplant. (2021) 21(11):3765–74. doi: 10.1111/ajt.16724
174. Vincenti F, Charpentier B, Vanrenterghem Y, Rostaing L, Bresnahan B, Darji P, et al. A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study). Am J Transplant. (2010) 10(3):535–46. doi: 10.1111/j.1600-6143.2009.03005.x
175. Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, et al. Belatacept and long-term outcomes in kidney transplantation. N Engl J Med. (2016) 374(4):333–43. doi: 10.1056/NEJMoa1506027
176. Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. (2003) 302(5646):875–8. doi: 10.1126/science.1087061
177. Kudlacz E, Perry B, Sawyer P, Conklyn M, McCurdy S, Brissette W, et al. The novel JAK-3 inhibitor CP-690550 is a potent immunosuppressive agent in various murine models. Am J Transplant. (2004) 4(1):51–7. doi: 10.1046/j.1600-6143.2003.00281.x
178. Sewgobind VDKD, Quaedackers ME, Laan LJWVD, Kraaijeveld R, Korevaar SS, Chan G, et al. The jak inhibitor CP-690,550 preserves the function of CD4+CD25brightFoxP3+ regulatory T cells and inhibits effector T cells. Am J Transplant. (2010) 10(8):1785–95. doi: 10.1111/j.1600-6143.2010.03200.x
179. van Gurp EAFJ, Schoordijk-Verschoor W, Klepper M, Korevaar SS, Chan G, Weimar W, et al. The effect of the JAK inhibitor CP-690,550 on peripheral immune parameters in stable kidney allograft patients. Transplantation. (2009) 87(1):79. doi: 10.1097/TP.0b013e31818bbea7
180. Valmori D, Tosello V, Souleimanian NE, Godefroy E, Scotto L, Wang Y, et al. Rapamycin-mediated enrichment of T cells with regulatory activity in stimulated CD4+ T cell cultures is not due to the selective expansion of naturally occurring regulatory T cells but to the induction of regulatory functions in conventional CD4+ T Cells1. J Immunol. (2006) 177(2):944–9. doi: 10.4049/jimmunol.177.2.944
181. Tian L, Lu L, Yuan Z, Lamb JR, Tam PKH. Acceleration of apoptosis in CD4+CD8+ thymocytes by rapamycin accompanied by increased CD4+CD25+ T cells in the periphery1. Transplantation. (2004) 77(2):183. doi: 10.1097/01.TP.0000101005.44661.3E
182. Wu D, Wong MQ, Vent-Schmidt J, Boardman DA, Steiner TS, Levings MK. A method for expansion and retroviral transduction of mouse regulatory T cells. J Immunol Methods. (2021) 488:112931. doi: 10.1016/j.jim.2020.112931
183. Yurchenko E, Shio MT, Huang TC, Martins MDS, Szyf M, Levings MK, et al. Inflammation-driven reprogramming of CD4+Foxp3+ regulatory T cells into pathogenic Th1/Th17 T effectors is abrogated by mTOR inhibition in vivo. PLoS One. (2012) 7(4):e35572. doi: 10.1371/journal.pone.0035572
184. Coenen JJA, Koenen HJPM, van Rijssen E, Hilbrands LB, Joosten I. Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood. (2006) 107(3):1018–23. doi: 10.1182/blood-2005-07-3032
185. Bestard O, Cassis L, Cruzado JM, Torras J, Franquesa M, Gil-Vernet S, et al. Costimulatory blockade with mTor inhibition abrogates effector T-cell responses allowing regulatory T-cell survival in renal transplantation. Transpl Int. (2011) 24(5):451–60. doi: 10.1111/j.1432-2277.2011.01223.x
186. Miller ML, McIntosh CM, Wang Y, Chen L, Wang P, Lei YM, et al. Resilience of T cell-intrinsic dysfunction in transplantation tolerance. Proc Natl Acad Sci. (2019) 116(47):23682–90. doi: 10.1073/pnas.1910298116
187. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. (2019) 20(6):724–35. doi: 10.1038/s41590-019-0346-9
188. Bestard O, Cruzado JM, Mestre M, Caldés A, Bas J, Carrera M, et al. Achieving donor-specific hyporesponsiveness is associated with FOXP3+ regulatory T cell recruitment in human renal allograft infiltrates. J Immunol. (2007) 179(7):4901–9. doi: 10.4049/jimmunol.179.7.4901
189. Gupta PK, Allocco JB, Fraipont JM, McKeague ML, Wang P, Andrade MS, et al. Reduced Satb1 expression predisposes CD4+ T conventional cells to Treg suppression and promotes transplant survival. Proc Natl Acad Sci. (2022) 119(40):e2205062119. doi: 10.1073/pnas.2205062119
190. Clough LE, Wang CJ, Schmidt EM, Booth G, Hou TZ, Ryan GA, et al. Release from regulatory T cell-mediated suppression during the onset of tissue-specific autoimmunity is associated with elevated IL-211. J Immunol. (2008) 180(8):5393–401. doi: 10.4049/jimmunol.180.8.5393
191. Attridge K, Wang CJ, Wardzinski L, Kenefeck R, Chamberlain JL, Manzotti C, et al. IL-21 inhibits T cell IL-2 production and impairs Treg homeostasis. Blood. (2012) 119(20):4656–64. doi: 10.1182/blood-2011-10-388546
192. Trinschek B, Lüssi F, Haas J, Wildemann B, Zipp F, Wiendl H, et al. Kinetics of IL-6 production defines T effector cell responsiveness to regulatory T cells in multiple sclerosis. PLoS One. (2013) 8(10):e77634. doi: 10.1371/journal.pone.0077634
193. Wehrens EJ, Vastert SJ, Mijnheer G, Meerding J, Klein M, Wulffraat NM, et al. Brief report: anti-tumor necrosis factor α targets protein kinase B/c-akt-induced resistance of effector cells to suppression in juvenile idiopathic arthritis: anti-TNFα targets resistance of effector cells to suppression. Arthritis Rheum. (2013) 65(12):3279–84. doi: 10.1002/art.38132
194. Wehrens EJ, Mijnheer G, Duurland CL, Klein M, Meerding J, van Loosdregt J, et al. Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c-akt hyperactivation in effector cells. Blood. (2011) 118(13):3538–48. doi: 10.1182/blood-2010-12-328187
195. Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD. IL-6 signaling in psoriasis prevents immune suppression by regulatory T Cells1. J Immunol. (2009) 183(5):3170–6. doi: 10.4049/jimmunol.0803721
196. Thiolat A, Semerano L, Pers YM, Biton J, Lemeiter D, Portales P, et al. Interleukin-6 receptor blockade enhances CD39+ regulatory T cell development in rheumatoid arthritis and in experimental arthritis. Arthritis Rheumatol. (2014) 66(2):273–83. doi: 10.1002/art.38246
197. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. (2003) 299(5609):1033–6. doi: 10.1126/science.1078231
198. Chen X, Howard OMZ, Oppenheim JJ. Pertussis toxin by inducing IL-6 promotes the generation of IL-17-producing CD4 Cells1. J Immunol. (2007) 178(10):6123–9. doi: 10.4049/jimmunol.178.10.6123
199. Chen L, Ahmed E, Wang T, Wang Y, Ochando J, Chong AS, et al. TLR signals promote IL-6/IL-17-dependent transplant Rejection1. J Immunol. (2009) 182(10):6217–25. doi: 10.4049/jimmunol.0803842
200. Jordan S, Ge S, Ammerman N, Toyoda M, Huang E, Peng A, et al. Clazakizumab (Anti-IL-6) induces Foxp3+ Tregs in highly HLA sensitized patients receiving HLAi kidney transplantation (NCT03380962). Transplantation. (2020) 104(S3):S105. doi: 10.1097/01.tp.0000698800.17823.68
201. Zhao X, Boenisch O, Yeung M, Mfarrej B, Yang S, Turka LA, et al. Critical role of proinflammatory cytokine IL-6 in allograft rejection and tolerance. Am J Transplant. (2012) 12(1):90–101. doi: 10.1111/j.1600-6143.2011.03770.x
202. Granofszky N, Farkas AM, Muckenhuber M, Mahr B, Unger L, Maschke S, et al. Anti-interleukin-6 promotes allogeneic bone marrow engraftment and prolonged graft survival in an irradiation-free murine transplant model. Front Immunol. (2017) 8:821. doi: 10.3389/fimmu.2017.00821
203. Kodati S, Chauhan SK, Chen Y, Emami-Naeini P, Omoto M, Dohlman TH, et al. Interleukin-6 neutralization prolongs corneal allograft survival. Curr Trends Immunol. (2018) 19:105–13. PMID: 30906117
204. Perona-Wright G, Jenkins SJ, O’Connor RA, Zienkiewicz D, McSorley HJ, Maizels RM, et al. A pivotal role for CD40-mediated IL-6 production by dendritic cells during IL-17 induction in Vivo1. J Immunol. (2009) 182(5):2808–15. doi: 10.4049/jimmunol.0803553
205. Liang Y, Christopher K, Finn PW, Colson YL, Perkins DL. Graft produced interleukin-6 functions as a danger signal and promotes rejection after transplantation. Transplantation. (2007) 84(6):771. doi: 10.1097/01.tp.0000281384.24333.0b
206. Lee K, Nguyen V, Lee KM, Kang SM, Tang Q. Attenuation of donor-reactive T cells allows effective control of allograft rejection using regulatory T cell therapy. Am J Transplant. (2014) 14(1):27–38. doi: 10.1111/ajt.12509
207. Joffre O, Santolaria T, Calise D, Saati TA, Hudrisier D, Romagnoli P, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med. (2008) 14(1):88–92. doi: 10.1038/nm1688
208. Tsang JYS, Tanriver Y, Jiang S, Xue SA, Ratnasothy K, Chen D, et al. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Invest. (2008) 118(11):3619–28. doi: 10.1172/JCI33185
209. Golshayan D, Jiang S, Tsang J, Garin MI, Mottet C, Lechler RI. In vitro–expanded donor alloantigen–specific CD4+CD25+ regulatory T cells promote experimental transplantation tolerance. Blood. (2007) 109(2):827–35. doi: 10.1182/blood-2006-05-025460
210. Brennan TV, Tang Q, Liu FC, Hoang V, Bi M, Bluestone JA, et al. Requirements for prolongation of allograft survival with regulatory T cell infusion in lymphosufficient hosts. J Surg Res. (2011) 169(1):e69–75. doi: 10.1016/j.jss.2011.03.021
211. Nikolouli E, Hardtke-Wolenski M, Hapke M, Beckstette M, Geffers R, Floess S, et al. Alloantigen-Induced regulatory T cells generated in presence of vitamin C display enhanced stability of Foxp3 expression and promote skin allograft acceptance. Front Immunol. (2017) 8:748. doi: 10.3389/fimmu.2017.00748
212. Koenecke C, Czeloth N, Bubke A, Schmitz S, Kissenpfennig A, Malissen B, et al. Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. Eur J Immunol. (2009) 39(11):3091–6. doi: 10.1002/eji.200939432
213. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med. (2011) 3(83):83ra42. doi: 10.1126/scitranslmed.3002076
214. Mathew JM, Voss JH, McEwen ST, Konieczna I, Chakraborty A, Huang X, et al. Generation and characterization of alloantigen-specific regulatory T cells for clinical transplant tolerance. Sci Rep. (2018) 8(1):1136. doi: 10.1038/s41598-018-19621-6
215. Hirai T, Lin PY, Ramos TL, Simonetta F, Su LL, Picton LK, et al. IL-2 receptor engineering enhances regulatory T cell function suppressed by calcineurin inhibitor. Am J Transplant. (2022) 22(12):3061–8. doi: 10.1111/ajt.17181
216. Fisher JD, Balmert SC, Zhang W, Schweizer R, Schnider JT, Komatsu C, et al. Treg-inducing microparticles promote donor-specific tolerance in experimental vascularized composite allotransplantation. Proc Natl Acad Sci. (2019) 116(51):25784–9. doi: 10.1073/pnas.1910701116
217. Ravichandran R, Itabashi Y, Fleming T, Bansal S, Bowen S, Poulson C, et al. Low-dose IL-2 prevents murine chronic cardiac allograft rejection: role for IL-2-induced T regulatory cells and exosomes with PD-L1 and CD73. Am J Transplant. (2022) 22(9):2180–94. doi: 10.1111/ajt.17101
218. Harper IG, Gjorgjimajkoska O, Siu JHY, Parmar J, Mulder A, Claas FHJ, et al. Prolongation of allograft survival by passenger donor regulatory T cells. Am J Transplant. (2019) 19(5):1371–9. doi: 10.1111/ajt.15212
219. Velásquez-Lopera MM, Eaton VL, Lerret NM, Correa LA, DeCresce RP, García LF, et al. Induction of transplantation tolerance by allogeneic donor-derived CD4+CD25+Foxp3+ regulatory T cells. Transpl Immunol. (2008) 19(2):127–35. doi: 10.1016/j.trim.2008.02.003
220. Siu JHY, Surendrakumar V, Richards JA, Pettigrew GJ. T cell allorecognition pathways in solid organ transplantation. Front Immunol. (2018) 9:2548. doi: 10.3389/fimmu.2018.02548
221. Veerapathran A, Pidala J, Beato F, Yu XZ, Anasetti C. Ex vivo expansion of human Tregs specific for alloantigens presented directly or indirectly. Blood. (2011) 118(20):5671–80. doi: 10.1182/blood-2011-02-337097
222. Takasato F, Morita R, Schichita T, Sekiya T, Morikawa Y, Kuroda T, et al. Prevention of allogeneic cardiac graft rejection by transfer of ex vivo expanded antigen-specific regulatory T-cells. PLoS One. (2014) 9(2):e87722. doi: 10.1371/journal.pone.0087722
223. Wagner JC, Ronin E, Ho P, Peng Y, Tang Q. Anti-HLA-A2-CAR Tregs prolong vascularized mouse heterotopic heart allograft survival. Am J Transplant. (2022) 22(9):2237–45. doi: 10.1111/ajt.17063
224. Sicard A, Lamarche C, Speck M, Wong M, Rosado-Sánchez I, Blois M, et al. Donor-specific chimeric antigen receptor Tregs limit rejection in naive but not sensitized allograft recipients. Am J Transplant. (2020) 20(6):1562–73. doi: 10.1111/ajt.15787
225. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MAA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. (2016) 17(4):931–43. doi: 10.1111/ajt.14185
226. Dawson NAJ, Lamarche C, Hoeppli RE, Bergqvist P, Fung VCW, McIver E, et al. Systematic testing and specificity mapping of alloantigen-specific chimeric antigen receptors in regulatory T cells. JCI Insight. (2019) 4(6):e123672. doi: 10.1172/jci.insight.123672
227. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant. (2016) 17(4):917–30. doi: 10.1111/ajt.14175
228. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126(4):1413–24. doi: 10.1172/JCI82771
229. Lamarche C, Ward-Hartstonge K, Mi T, Lin DTS, Huang Q, Brown A, et al. Tonic-signaling chimeric antigen receptors drive human regulatory T cell exhaustion. Proc Natl Acad Sci. (2023) 120(14):e2219086120. doi: 10.1073/pnas.2219086120
230. Lamarthée B, Marchal A, Charbonnier S, Blein T, Leon J, Martin E, et al. Transient mTOR inhibition rescues 4-1BB CAR-Tregs from tonic signal-induced dysfunction. Nat Commun. (2021) 12(1):6446. doi: 10.1038/s41467-021-26844-1
231. Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal regulatory T cell therapy for control of inflammation in kidney transplants. Am J Transplant. (2017) 17(11):2945–54. doi: 10.1111/ajt.14415
232. Tang Q, Leung J, Peng Y, Sanchez-Fueyo A, Lozano JJ, Lam A, et al. Selective decrease of donor-reactive Tregs after liver transplantation limits Treg therapy for promoting allograft tolerance in humans. Sci Transl Med. (2022) 14(669):eabo2628. doi: 10.1126/scitranslmed.abo2628
233. Demirkiran A, Bosma BM, Kok A, Baan CC, Metselaar HJ, IJzermans JNM, et al. Allosuppressive donor CD4+CD25+ regulatory T cells detach from the graft and circulate in recipients after liver transplantation. J Immunol. (2007) 178(10):6066–72. doi: 10.4049/jimmunol.178.10.6066
234. Guy’s and St Thomas’ NHS Foundation Trust. The ONE study: a unified approach to evaluating cellular immunotherapy in solid Organ transplantation. clinicaltrials.gov (2019). Report No.: NCT02129881. Available at: https://clinicaltrials.gov/ct2/show/NCT02129881 (Accessed June 13, 2023).
235. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory cell therapy in kidney transplantation (the ONE study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet. (2020) 395(10237):1627–39. doi: 10.1016/S0140-6736(20)30167-7
236. Lamarche C, Maltzman JS. The ONE study: one small step for patient care, a giant leap for cell therapy. Am J Kidney Dis. (2021) 77(2):297–9. doi: 10.1053/j.ajkd.2020.07.006
237. Alexander SI, Hu M, O’Connell PJ. One for all and all for one: the triumph of the one study. Transplantation. (2021) 105(2):273. doi: 10.1097/TP.0000000000003474
238. Brook MO, Hester J, Petchey W, Rombach I, Dutton S, Bottomley MJ, et al. Transplantation without overimmunosuppression (TWO) study protocol: a phase 2b randomised controlled single-centre trial of regulatory T cell therapy to facilitate immunosuppression reduction in living donor kidney transplant recipients. BMJ Open. (2022) 12(4):e061864. doi: 10.1136/bmjopen-2022-061864
239. Issa F. TWO study: Treg cell therapy trial v1.0. (2018). Available at: https://www.hra.nhs.uk/planning-and-improving-research/application-summaries/research-summaries/two-study-treg-cell-therapy-trial-v10/ (Accessed June 13, 2023).
240. Sangamo Therapeutics. Multicentre open-label single ascending dose dose-ranging phase I/IIa study to evaluate safety and tolerability of an autologous antigen-specific chimeric antigen receptor tregulatory cell therapy in living donor renal transplant recipients. clinicaltrials.gov (2023). Report No.: mu. Available at: https://clinicaltrials.gov/study/NCT04817774 (Accessed July 24, 2023).
241. Quell Therapeutics Limited. A single-arm, open-label, multi-center, phase I/II study evaluating the safety and clinical activity of QEL-001, an autologous CAR T regulatory cell treatment targeting HLA-A2, in HLA-A2/A28neg patients that have received an HLA-A2pos liver transplant. clinicaltrials.gov (2022). Report No.: NCT05234190. Available at: https://clinicaltrials.gov/study/NCT05234190 (Accessed July 24, 2023).
242. Morgan ME, van Bilsen JHM, Bakker AM, Heemskerk B, Schilham MW, Hartgers FC, et al. Expression of FOXP3 mRNA is not confined to CD4+CD25+ T regulatory cells in humans. Hum Immunol. (2005) 66(1):13–20. doi: 10.1016/j.humimm.2004.05.016
243. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3− T cells by T-cell receptor stimulation is transforming growth factor-β–dependent but does not confer a regulatory phenotype. Blood. (2007) 110(8):2983–90. doi: 10.1182/blood-2007-06-094656
244. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. (2007) 19(4):345–54. doi: 10.1093/intimm/dxm014
245. Pillai V, Ortega SB, Wang CK, Karandikar NJ. Transient regulatory T-cells: a state attained by all activated human T-cells. Clin Immunol. (2007) 123(1):18–29. doi: 10.1016/j.clim.2006.10.014
246. Lim TY, Perpiñán E, Londoño MC, Miquel R, Ruiz P, Kurt AS, et al. Low dose interleukin-2 selectively expands circulating regulatory T cells but fails to promote liver allograft tolerance in humans. J Hepatol. (2023) 78(1):153–64. doi: 10.1016/j.jhep.2022.08.035
247. Hirai T, Ramos TL, Lin PY, Simonetta F, Su LL, Picton LK, et al. Selective expansion of regulatory T cells using an orthogonal IL-2/IL-2 receptor system facilitates transplantation tolerance. J Clin Invest. (2021) 131(8):e139991. doi: 10.1172/JCI139991
248. Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, et al. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol. (2009) 39(4):1088–97. doi: 10.1002/eji.200838904
249. Chai JG, Coe D, Chen D, Simpson E, Dyson J, Scott D. In vitro expansion improves in vivo regulation by CD4+CD25+ regulatory T cells. J Immunol. (2008) 180(2):858–69. doi: 10.4049/jimmunol.180.2.858
250. Ezzelarab MB, Zhang H, Sasaki K, Lu L, Zahorchak AF, van der Windt DJ, et al. Ex vivo expanded donor alloreactive regulatory T cells lose immunoregulatory, proliferation, and antiapoptotic markers after infusion into ATG-lymphodepleted, nonhuman primate heart allograft recipients. Transplantation. (2021) 105(9):1965–79. doi: 10.1097/TP.0000000000003617
251. Henschel P, Landwehr-Kenzel S, Engels N, Schienke A, Kremer J, Riet T, et al. Supraphysiological FOXP3 expression in human CAR-Tregs results in improved stability, efficacy, and safety of CAR-Treg products for clinical application. J Autoimmun. (2023) 138:103057. doi: 10.1016/j.jaut.2023.103057
252. Tripathi D, Cheekatla SS, Paidipally P, Radhakrishnan RK, Welch E, Thandi RS, et al. c-Jun N-terminal kinase 1 defective CD4+CD25+FoxP3+ cells prolong islet allograft survival in diabetic mice. Sci Rep. (2018) 8(1):3310. doi: 10.1038/s41598-018-21477-9
253. Lu P, Chen J, He L, Ren J, Chen H, Rao L, et al. Generating hypoimmunogenic human embryonic stem cells by the disruption of Beta 2-microglobulin. Stem Cell Rev Rep. (2013) 9(6):806–13. doi: 10.1007/s12015-013-9457-0
254. Torikai H, Reik A, Soldner F, Warren EH, Yuen C, Zhou Y, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. (2013) 122(8):1341–9. doi: 10.1182/blood-2013-03-478255
255. Ichise H, Nagano S, Maeda T, Miyazaki M, Miyazaki Y, Kojima H, et al. NK cell alloreactivity against KIR-ligand-mismatched HLA-haploidentical tissue derived from HLA haplotype-homozygous iPSCs. Stem Cell Rep. (2017) 9(3):853–67. doi: 10.1016/j.stemcr.2017.07.020
256. Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol. (2019) 37(3):252–8. doi: 10.1038/s41587-019-0016-3
257. Xu H, Wang B, Ono M, Kagita A, Fujii K, Sasakawa N, et al. Targeted disruption of HLA genes via CRISPR-Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell. (2019) 24(4):566–578.e7. doi: 10.1016/j.stem.2019.02.005
258. Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol. (2017) 35(8):765–72. doi: 10.1038/nbt.3860
259. Organ Procurement and Transplantation Network (OPTN) and Scientific Registry of Transplant Recipients (SRTR). OPTN/SRTR 2021 annual data report. U.S. Department of Health and Human Services, Health Resources and Services Administration (2023). Available at: https://srtr.transplant.hrsa.gov/annual_reports/Default.aspx (Accessed April 18, 2023).
260. Jordan SC, Choi J, Vo A. Kidney transplantation in highly sensitized patients. Br Med Bull. (2015) 114(1):113–25. doi: 10.1093/bmb/ldv013
Keywords: Tregs (regulatory T cells), Foxp 3, transplantation, tolerance, alloimmunity, acute rejection (AR)
Citation: Cassano A, Chong AS and Alegre M-L (2023) Tregs in transplantation tolerance: role and therapeutic potential. Front. Transplant. 2:1217065. doi: 10.3389/frtra.2023.1217065
Received: 4 May 2023; Accepted: 14 August 2023;
Published: 30 August 2023.
Edited by:
Sheri M. Krams, Stanford University, United StatesReviewed by:
Everett Meyer, Stanford University, United StatesRobert Ian Lechler, King’s College London, United Kingdom
© 2023 Cassano, Chong and Alegre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria-Luisa Alegre bWFsZWdyZUBtaWR3YXkudWNoaWNhZ28uZWR1
Abbreviations ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATG, anti-thymocyte globulin; APC, antigen presenting cell; ATP, adenosine triphosphate; CAR, chimeric antigen receptor; CCL, chemokine ligand; CCR, chemokine receptor; CD, cluster of differentiation protein; CNI, calcineurin inhibitor(s); CoB, co-stimulation blockade; CsA, cyclosporine; CTLA-4, cytotoxic T-lymphocyte associated antigen-4; DC, dendritic cell; dLN, draining lymph node; DST, donor splenocyte transfusion; Foxp3, forkhead box P3; GvHD, graft versus host disease; HLA, human leukocyte antigen; ICAM-1, intercellular adhesion molecule 1; IDO, indoleamine-2,3-dioxygenase; IFNγ, interferon gamma; IL, interleukin; iTreg, induced Treg; LAG-3, lymphocyte activation gene 3; Lm, Listeria monocytogenes; LN, lymph node; METTL14, methyltransferase like 14; MHC-II, major histocompatibility complex class II; Nrp1, neuropilin 1; N1c-KO, Notch-1 conditional knock out; pDC, plasmacytoid dendritic cell; PD-1, programmed death protein 1; PD-L1, programmed death ligand 1; pTreg, peripherally induced Treg; sEV, small extracellular vesicle; TCR, T cell receptor; TGFβ, transforming growth factor beta; TIGIT, T cell immunoreceptor with Ig and ITIM domains; TIM-3, T cell immunoglobulin and mucin domain-containing protein 3; TNFα, tumor necrosis factor alpha; tTreg, thymically derived Treg; WT, wildtype; WTAP, Wilms tumor 1-associating protein.