 Xu Wang
Xu Wang Chunshu Rong1‡
Chunshu Rong1‡ Dexi Zhao
Dexi Zhao Jinhua Li
Jinhua Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Toxicol. , 27 January 2025
Sec. Neurotoxicology
Volume 7 - 2025 | https://doi.org/10.3389/ftox.2025.1543374
IAA is a by-product of the water disinfection process and has been found to be neurotoxic. However, the role and mechanism of IAA neurotoxicity remain unclear. In this review, we comprehensively discuss the neurotoxic effects and mechanisms of IAA from the molecular level, cellular level and neurological manifestations. At the molecular level, IAA causes neurotoxicity by reducing mitochondrial membrane potential, aggravating oxidative stress and DNA damage. At the cellular level, IAA causes neurotoxicity by inducing BBB disruption, neuroinflammation, and apoptosis. In neurological manifestations, IAA can lead to neurotransmitter disorders, neurodevelopment dysfunction, and even neurodegenerative diseases. Taken together, our review provides insights into the mechanisms of IAA neurotoxicity that will contribute to future studies of IAA neurotoxicity and its protective strategies.
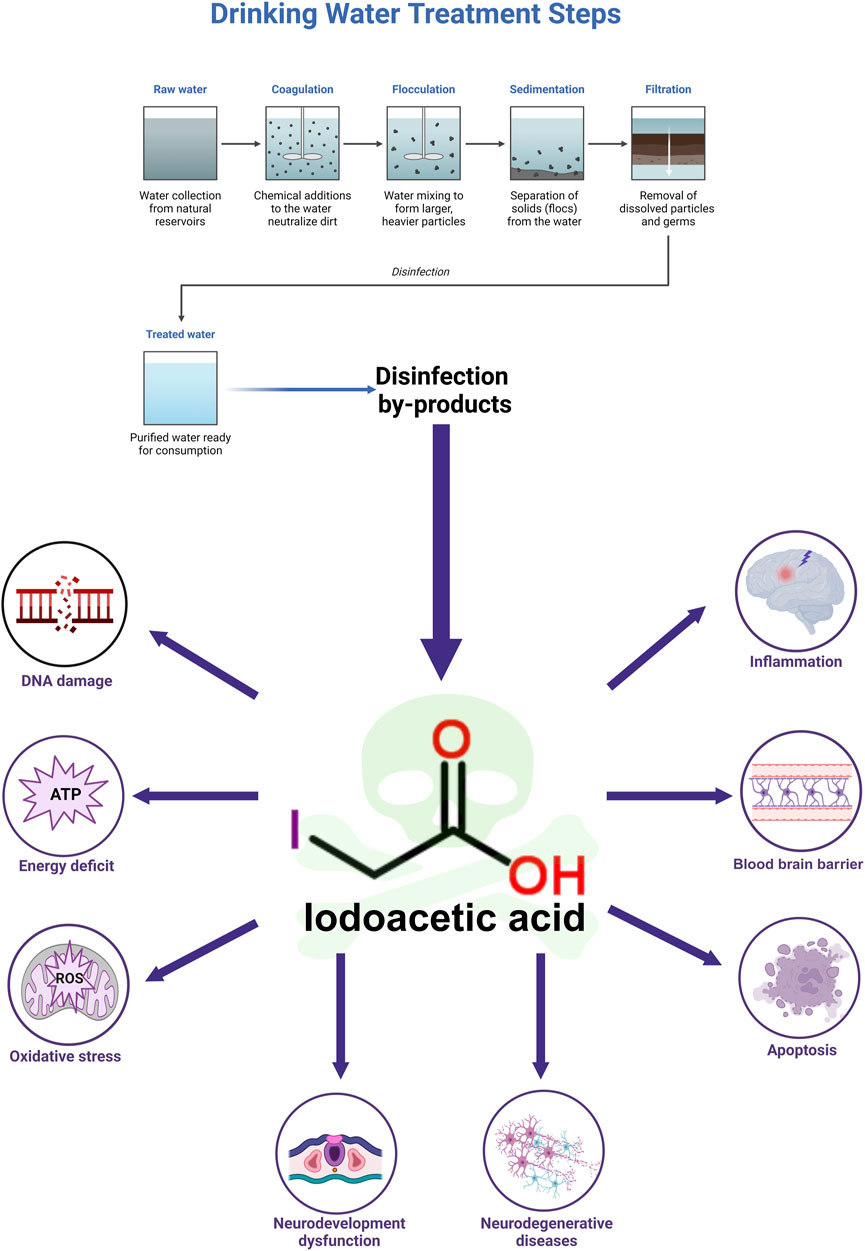
Disinfection plays a crucial role in safeguarding public health, serving as a method that effectively eliminates bacteria, viruses, and various pathogenic microorganisms from water, thus helping to prevent the transmission of waterborne illnesses. However, this essential process can result in the creation of disinfection by-products (DBPs), one of which is iodoacetic acid (IAA). IAA is a by-product that forms as a result of the reaction between disinfectants and organic materials, as well as iodine ions present in water (MacKeown et al., 2022). A significant compound produced during this oxidation reaction is hypoiodous acid, which interacts vigorously with organic substances in the environment, ultimately resulting in the synthesis of IAA. Moreover, when chlorinated tap water interacts with salt that has been fortified with potassium iodide, the production of IAA may occur (Becalski et al., 2006). Additionally, conditions such as saltwater intrusion (Dong et al., 2019), hydraulic fracturing (Harkness et al., 2015), and the release of X-ray contrast agents contribute to the occurrence of IAA in drinking water across the globe (Li et al., 2020).
The maximum concentration of IAA identified in water samples from 23 cities across North America reached 1.7 μg/L (Richardson et al., 2008). The globally highest concentration of IAA in drinking water has yet to be established. At present, the most significant level of IAA reported in drinking water was found to be 2.18 μg/L in Shanghai, China (Wei et al., 2013). Compared to other DBPs, IAA exhibits increased cytotoxicity and genotoxicity (Wagner and Plewa, 2017). In addition, IAA disrupts intestinal flora and metabolites to exert toxicity (Sha et al., 2022). While a reference limit for IAA in drinking water has been established in China at 0.02 mg/L (Shi, 2023), it is still not regulated in either the USA or Europe (Pérez-Albaladejo et al., 2023; Yu et al., 2024). Plasma and urinary levels of IAA were measured at 1.89 ± 0.29 and 5.92 ± 1.45 ng/mL, respectively, following an oral intake of 6 mg/kg of IAA after 6 h (Yu et al., 2024). Nevertheless, there have been no investigations assessing IAA concentrations in brain tissue.
IAA is capable of traversing the blood-brain barrier (BBB), leading to neurotoxic effects (Yang et al., 2021). The phenomena of IAA mutagenesis, carcinogenesis, and teratogenesis have been thoroughly investigated (Gonsioroski et al., 2020; Zeng et al., 2021). Nonetheless, research focusing on the neurotoxic impacts and underlying mechanisms of IAA remains limited, and a comprehensive review addressing its neurotoxicity is absent. This paper represents the first review of IAA’s neurotoxicity and its associated mechanisms, establishing a groundwork for future studies in this domain.
IAA, which inhibits glyceraldehyde-3-phosphate dehydrogenase (GAPDH), may decrease ATP levels within cells (Zhou et al., 2015). The compound can penetrate brain tissue by crossing the BBB and can lead to the destruction of nerve cells (Yang et al., 2021). IAA reduces viability of nerve cells. IAA is neurotoxic and can reduce cell viability, increase the release of lactate dehydrogenase (LDH), and even cause cell death in nerve cells (Yang et al., 2021; Chen et al., 2015; Sperling et al., 2003; Chen et al., 2017a; Lapchak et al., 2007). Compared to other tissues, brain tissue demands a higher supply of oxygen and energy (Wang et al., 2022). The mechanism by which IAA causes neurotoxicity is described below. Details of included studies and results of IAA shown in Table 1.
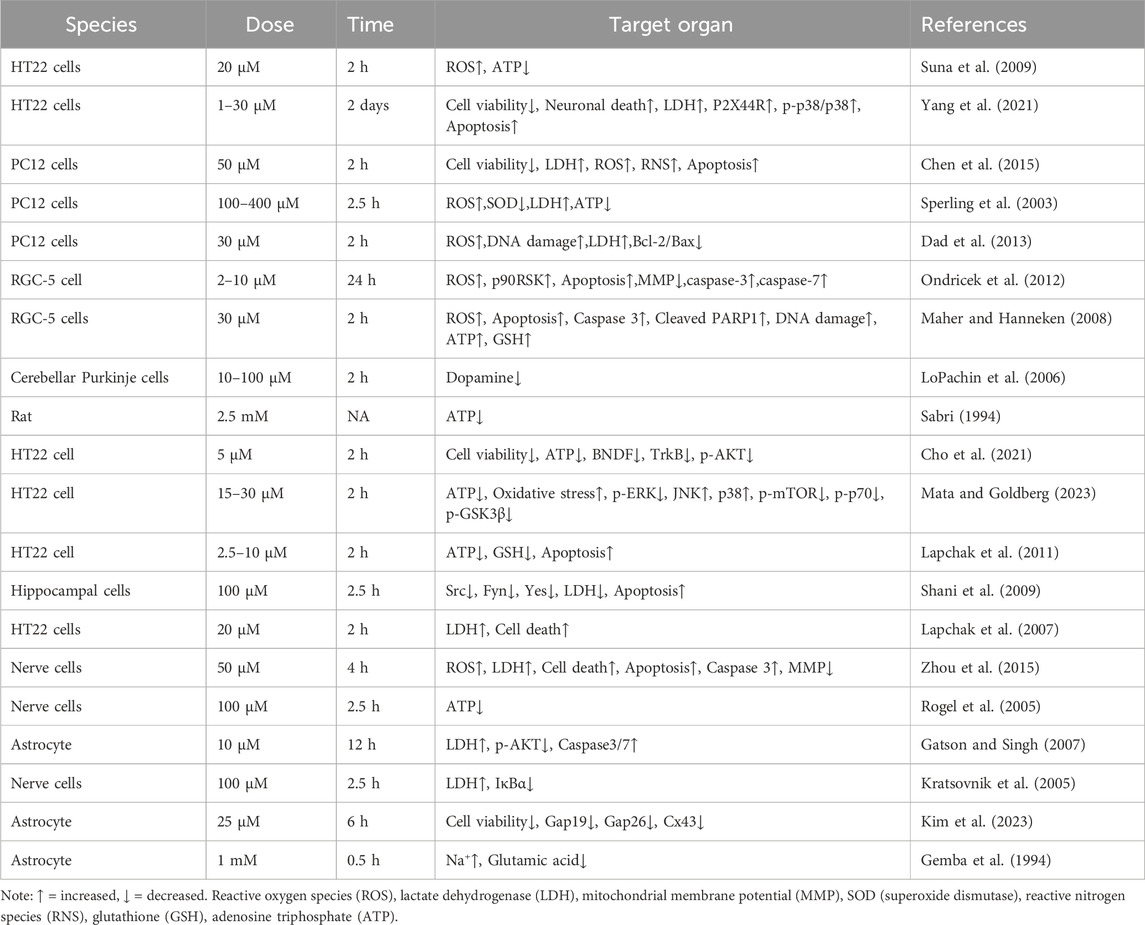
Table 1. Summarizing neurotoxicity of IAA.
GAPDH catalyzes the fifth step in glycolysis (Butera et al., 2018), the conversion of glyceraldehyde 3-phosphate into 1,3-biphosphoglycerate, a reaction that generates NADH, which is a “high energy” compound that subsequently generates ATP. GAPDH is best known for this reaction and is often used as a “housekeeping” gene or loading control in expression analyses, belying its complex role in growth and survival. GAPDH contains two domains for NAD+ binding and the catalytic domain. At the junction, cysteine 149 is a site for post-translational modification and is required for multiple GAPDH functions. While GAPDH is generally localized to the cytoplasm, it’s sub-cellular localization can change, particularly when modified by nitrosylation, oxidation and phosphorylation (Sirover, 2014).
GAPDH serves as an important enzyme within the glycolytic pathway, thereby inhibiting its function will hinder the glycolytic process, which in turn impacts the synthesis of ATP (Zhu et al., 2021). The glycolytic pathway produces pyruvate, which subsequently enters the mitochondria to take part in the citric acid cycle, generating substantial ATP. Inhibition of GAPDH impacts the ensuing metabolic pathways, leading to decreased ATP synthesis. Additionally, IAA entry into the brain hampers glycolysis, consequently lowering ATP levels in nerve cells (Sperling et al., 2003; Suna et al., 2009; Maher and Hanneken, 2008; Sabri, 1994; Rogel et al., 2005). Mitochondria function as the cell’s energy production sites, generating ATP via oxidative phosphorylation. Nerve cells have very high energy requirements, and any decrease in ATP synthesis may result in compromised function and potentially lead to cell death. Moreover, a lack of ATP diminishes the sodium-potassium pump’s activity, leading to the buildup of sodium inside the cell and a decline in potassium levels outside the cell, which reduces the membrane potential. Conversely, a reduction in the potential of the mitochondrial membrane indicates a lowered electrochemical gradient across the inner and outer mitochondrial membranes, subsequently impacting ATP production and causing an inadequate energy supply for the cell (Liang et al., 2007). The specific mechanism of IAA caused molecular level neurotoxicity is shown in Figure 1.
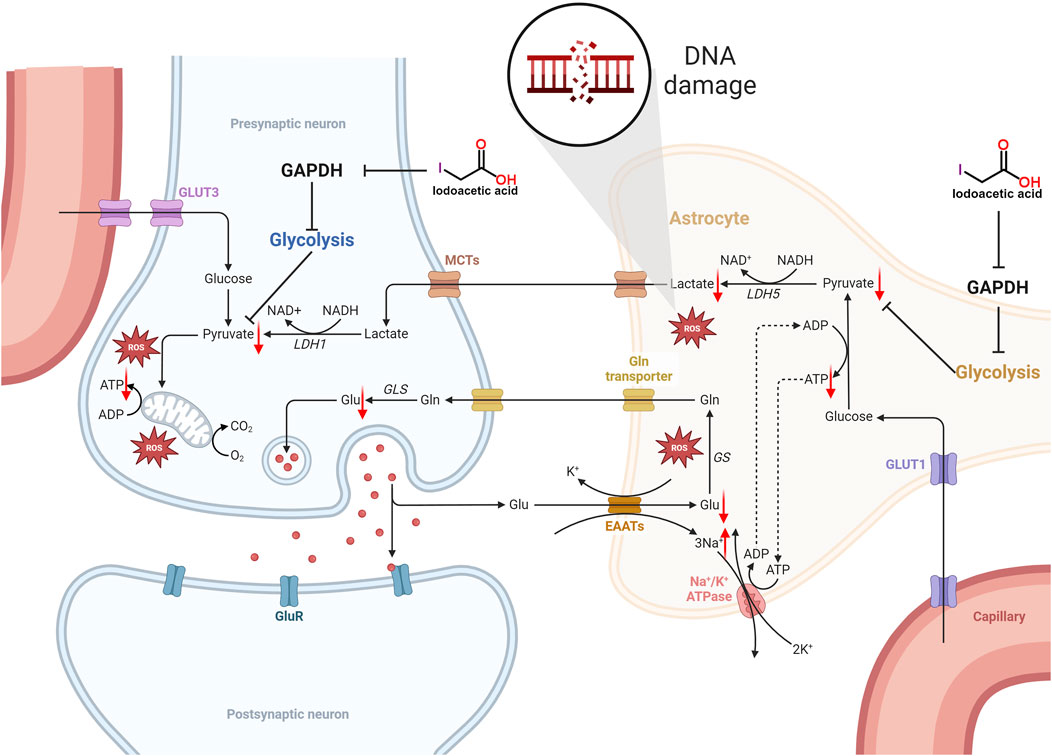
Figure 1. Schematic illustration of the molecular level neurotoxicity by IAA. IAA inhibited glycolysis by inhibiting GAPDH expression. The level of pyruvate is reduced, not enough to provide sufficient substrate for the tricarboxylic acid (TCA) cycle, resulting in an insufficient supply of ATP to nerve cells. In addition, ATP deficiency reduces the activity of the sodium-potassium pump, resulting in the accumulation of intracellular sodium and the decrease of extracellular potassium, which decreases the cell membrane potential. The decrease in mitochondrial membrane potential means that the electrochemical gradient between the inner and outer membrane of the mitochondria is reduced, which in turn affects ATP synthesis, and further leads to insufficient cell energy supply. In addition, lactate produced by glial cells can provide energy metabolism for neurons. Glycolysis is inhibited and lactate production is reduced. The imbalance of REDOX balance leads to the release and accumulation of excess ROS. Eventually it leads to DNA damage.
Oxidative stress arises from an imbalanced redox state, characterized by an excess of ROS or malfunctioning of the antioxidant systems (Ping et al., 2024). The brain is particularly sensitive to ROS due to its high requirement for oxygen and the prevalence of lipid cells that are prone to peroxide damage. Research has indicated that oxidative stress is a significant contributor to the shared pathology seen in neurodegenerative disorders. One notable aspect of oxidative stress is the heightened production of ROS, which correlates with mitochondrial dysfunction (Marchetti et al., 2006). Exposure to IAA has been shown to elevate oxidative stress levels within nerve cells. Specifically, IAA led to increased concentrations of ROS and reactive nitrogen species (RNS), along with a reduction in GSH and SOD levels in these cells (Zhou et al., 2015; Chen et al., 2015; Sperling et al., 2003; Suna et al., 2009; Maher and Hanneken, 2008). Also, IAA induces oxidative stress in germ cells via the GRP78/IRE1/XBP1s and cGAS/STING/NF-kappa B signaling pathways (Mou et al., 2024). Nonetheless, the specific molecular pathways through which IAA triggers oxidative stress in nerve cells are still not fully understood. The oxidative stress agents produced by IAA have the potential to inflict damage on DNA, resulting in compromised cellular function and potentially leading to cell death (Marchetti et al., 2006).
IAA was found to increase intracellular ROS and gamma-H2AX levels in a dose-dependent manner (Jiao et al., 2021). Heightened oxidative harm to DNA detected in both nuclear and mitochondrial DNA (mtDNA) obtained from the postmortem brain areas of individuals suffering from neurodegenerative disorders indicates cytogenetic injury associated with these diseases (Coppedè and Migliore, 2015). The DNA damage resulting from oxidative stress induced by IAA was observed. In retinal ganglion cells RGC-5 exposed to IAA, there was an elevation in the levels of cleaved Poly(ADP-ribose) polymerase 1 (PARP1) and p90 ribosomal s6 kinase (RSK) protein, which exacerbated the DNA damage (Maher and Hanneken, 2008). PARP1 and the PAR play a significant role in the repair of DNA damage, and a rise in the expression of cleaved PARP1 suggests a higher level of DNA damage (Ping et al., 2024). IAA additionally contributes to significant DNA damage as an inhibitor of GAPDH. Furthermore, GAPDH interacts with the DNA repair protein PARP1 (Nakajima et al., 2015). PARP1 plays a crucial role in repairing single strand DNA breaks as well as double strand DNA breaks (Ping et al., 2024). PARP1 also has the ability to ADP-ribosylate and deactivate GAPDH, which results in a reduction of GAPDH levels and may, in certain situations, lead to cell death (Du et al., 2003). Thus, IAA causes DNA damage by increasing PARP1 activity (Maher and Hanneken, 2008). The buildup of DNA damage and the malfunctioning of DNA repair mechanisms play a role in the development of various neurodegenerative disorders. Pathways that respond to DNA damage create the necessary environment to initiate cellular senescence and/or apoptosis when the extent of DNA damage is excessive. Although double-strand DNA breaks are infrequent occurrences, they represent the most lethal type of DNA damage. In neuronal cells, these double-strand DNA breaks prove especially harmful due to their diminished ability to repair DNA in comparison to proliferating cells (Merlo et al., 2016). Therefore, DNA damage is closely associated with neurodegeneration.
mtDNA, the genetic material found within this cellular energy factory, is an essential component of cellular life processes (Vazquez-Villasenor et al., 2021; Christie and Beekman, 2017). Mitochondria play a crucial role in energy conversion, and the integrity of their function is essential for maintaining normal cellular operations and energy metabolism. IAA-induced oxidative stress triggers excessive activation of mitochondrial complex I in response to persistent DNA damage. These altered responses to continuous oxidative damage may result in further oxidative stress, contributing to neuronal dysfunction, and ultimately, neurodegeneration (Vazquez-Villasenor et al., 2021). When mtDNA is damaged, cells initiate a series of complex repair mechanisms to address the issue. Among these mechanisms, PARP1 plays a crucial role. PARP1 is an enzyme located in both the nucleus and mitochondria, capable of sensing DNA damage and responding rapidly. Following the occurrence of DNA damage, PARP1 is swiftly activated and commences its essential enzymatic activity—catalyzing PARylation (Han et al., 2019). This process involves the transfer of ADP ribose units to proteins, resulting in the formation of poly(ADP-ribose) (PAR) chains. These chains not only facilitate the repair of damaged DNA but also function as signaling molecules that recruit additional repair proteins to the site of damage, thereby initiating a complex network of DNA damage repair (Smith et al., 2019). However, IAA activates cleaved PARP1 (89 kDa), which promotes nerve cell death rather than repair (Maher and Hanneken, 2008). The 116 kDa form refers to the full-length variant of PARP1, representing the original protein’s molecular weight without any cleavage or modifications (Ping et al., 2024). Full-length PARP1 (116 kDa) possesses complete catalytic activity in cells and is involved in various biological processes, including DNA repair and transcriptional regulation. Thus, IAA promotes mtDNA damage rather than DNA repair.
IAA inhibits GAPDH and reduces ATP to perturb Na+, K+ ions in cells (Gemba et al., 1994). Furthermore, IAA induced a decrease in mitochondrial membrane potential of nerve cells (Zhou et al., 2015; Ondricek et al., 2012). The reduction in the potential of the mitochondrial membrane due to disturbances in energy metabolism can significantly impact cellular functions. This decline in mitochondrial membrane potential shows a strong correlation with neurodegenerative disorders. It is essential to keep cellular energy metabolism balance to ensure the stability of mitochondrial membrane potential and preserve the proper functioning of neural cells. A decline in mitochondrial membrane potential results in dysfunction of the electron transport chain located in the mitochondria, which heightens the generation of free radicals and reactive oxygen species ROS. The potential difference across the cell membrane, known as the cell membrane potential, is primarily upheld by ion pumps and channels located within the membrane. This potential is essential for processes such as nerve conduction and intercellular signaling. An imbalance in energy metabolism may lead to decreased ATP production, subsequently affecting the function of sodium-potassium pumps embedded in the cell membrane. These pumps play a crucial role in regulating the concentrations of sodium and potassium ions both inside and outside the cell, and rely on ATP for the energy necessary to operate. When there is a lack of ATP, the functionality of the sodium-potassium pump diminishes, causing sodium ions to accumulate within the cell while extracellular potassium levels drop, ultimately resulting in a decline in the cell membrane potential (Bartolome et al., 2013; Zugno et al., 2003). The decrease in membrane potential further leads to a decrease in ATP production (Wang et al., 2003; Elmi et al., 2001). IAA inhibits GAPDH and reduces ATP to perturb Na+, K+ ions in cells (Gemba et al., 1994). This is the main reason for the disturbance of cell membrane potential caused by IAA.
Neuroinflammation is closely linked to the neurotransmitter system and plays a crucial role in the onset and progression of neurodegenerative disorders (Davis et al., 2023). Following treatment with IAA, there was an upregulation in the expression levels of Purinergic Receptor P2X4 (P2X4R) (Yang et al., 2021). Recent research has indicated that the heightened neurotoxicity linked to the overexpression of P2X4R plays a significant role in the pathological mechanisms of neurodegenerative conditions, including injury, inflammation, Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis (Ma et al., 2020). IAA also affects the integrity of the BBB through the P2X4R/p38 signaling pathway. Furthermore, the expression of IκBα was found to be reduced in neurons that were treated with IAA (Kratsovnik et al., 2005). The degraded IκBα activates both subunits of NF-κB, transitioning them from an inactive state to an active one. This process facilitates their movement from the cytoplasm into the nucleus, where they attach to specific genes associated with inflammation, leading to the initiation of transcription for inflammatory cytokines and the induction of inflammation.
The BBB serves as a protective shield that stops harmful and toxic substances from accessing and damaging brain tissue, thereby fulfilling a crucial function (Wang et al., 2024). Harm to the BBB allows more harmful substances and immune cells to infiltrate the brain, which further worsens nerve cell injury. Following treatment with IAA, the levels of the tight junction-associated protein Connexin43 (Cx43) were reduced (Kim et al., 2023). Cx43 is crucial in the central nervous system (CNS). It is the most prevalent connexin found in the CNS, primarily located in astrocytes, and it exists as gap junctions and hemichannels, facilitating both intercellular and intracellular communication (Zhan et al., 2023). CX43 is a crucial protein involved in the BBB (Zhang et al., 2023). The BBB is formed by brain capillary endothelial cells, pericytes, and the pseudopodia of glial cells, functioning to allow the passage of nutrients while preventing harmful substances in the blood from entering the brain (Wang et al., 2024). CX43 plays a significant role in maintaining the integrity and function of the BBB (Chen et al., 2017b). Studies have shown that with aging, the expression level of CX43 in BBB-related cell subpopulations decreases significantly (Wilson et al., 2008), indicating a close relationship between CX43 expression and the integrity of the BBB (Zhan et al., 2023). Furthermore, CX43 directly interacts with PARP1, a NAD + -consuming enzyme. Enhanced activity of PARP1 leads to excessive consumption of NAD+, which may adversely affect the integrity of the BBB (Zhan et al., 2023). The specific mechanism of IAA caused BBB disruption is shown in Figure 2.
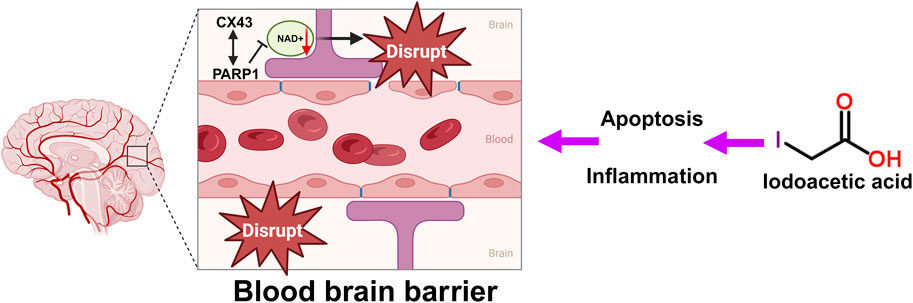
Figure 2. Schematic illustration of the BBB disruption, oxidative stress and DNA damage by IAA. DNA damage caused by IAA-mediated inflammation and oxidative stress disrupts the tight junctions and gap junctions of the BBB. CX43 directly interacts with PARP1. Excessive consumption of NAD+, which affects the integrity of the BBB.
A reduction in the potential of the mitochondrial membrane serves as a critical indicator of apoptosis. As the mitochondrial membrane potential drops, this triggers the opening of the mitochondrial permeability transition pore, resulting in the release of cytochrome C and various other apoptotic factors from the mitochondria into the cytoplasm. This event activates the subsequent apoptotic signaling pathways, ultimately causing cell apoptosis (Rehfeldt et al., 2021). Reduced ATP levels, diminished membrane potential, oxidative stress, heightened DNA damage, and lowered neurotransmitter levels in nerve cells treated with IAA contribute to apoptosis. The molecular mechanisms behind IAA-induced neuronal apoptosis involve the upregulation of mitogen-activated Protein Kinase (MAPK) and JNK, where pMAPK can enhance the expression of TNF, consequently triggering cell apoptosis (Lapchak et al., 2011; Rehfeldt et al., 2021). Furthermore, following the treatment of RGC-5 cells with IAA, there was an elevation in the levels of cysteinyl aspartate specific proteinase-3 (Caspase-3) and Caspase-7 proteins (Ondricek et al., 2012; Gatson and Singh, 2007). Caspase-3 play crucial roles in the apoptosis pathway, working alongside Caspase-7 (Eslami et al., 2020) to facilitate this process. Additionally, treatment with IAA led to an upregulation of cleaved PARP1 expression in RGC-5 cells (Maher and Hanneken, 2008). The generation of cleaved PARP1 is also driven by Caspase-3, which cleaves PARP, leading to its activation, and subsequently triggers other proteases associated with apoptosis to conclude the apoptotic process (Ping et al., 2024). A decrease in the Bcl-2/Bax ratio was observed in rat pheochromocytoma cells (PC12), that were treated with IAA (Chen et al., 2017a). Bcl-2 and Bax modulate the opening of channels induced by the apoptotic activator Bax dimer on the mitochondrial membrane, enhancing permeability by managing the mitochondrial membrane’s permeability. A reduction in the Bcl-2/Bax ratio signifies an increase in apoptosis.
Furthermore, following IAA treatment, the levels of p-AKT, p-mTOR, p-p70, and p-GSK3β were found to be decreased (Cho et al., 2021; Mata and Goldberg, 2023). Akt plays a crucial role in the growth of cells. It serves as a key modulator of cell survival by directly suppressing proteins that promote apoptosis (Hiromura et al., 2004). p-mTOR is an essential molecule that regulates growth by detecting and interacting with various nutritional and environmental elements, such as growth factors, energy availability, cellular stress, and amino acids. It integrates these signals to stimulate cell growth through the phosphorylation of substrates, thereby promoting anabolism or suppressing catabolism (Yao et al., 2016). p70 is essential in regulating the cell cycle, growth, and survival. One of the primary mechanisms for managing cell survival, proliferation, and metabolism is the PI3K/mTOR signaling pathway (Gao et al., 2003). GSK-3β has the ability to modulate Bax, which subsequently influences mitochondrial permeability and the release of cytochrome C, playing a role in the regulation of apoptosis (Xu et al., 2023). The decline in mitochondrial membrane potential over an extended period, alongside energy shortages and cellular apoptosis, is associated with the development of various neurodegenerative disorders, including Alzheimer’s and Parkinson’s diseases. In these conditions, both mitochondrial dysfunction and apoptosis represent widespread pathological characteristics (Rehfeldt et al., 2021). The specific mechanism of IAA neurotoxicity is shown in Figure 3.
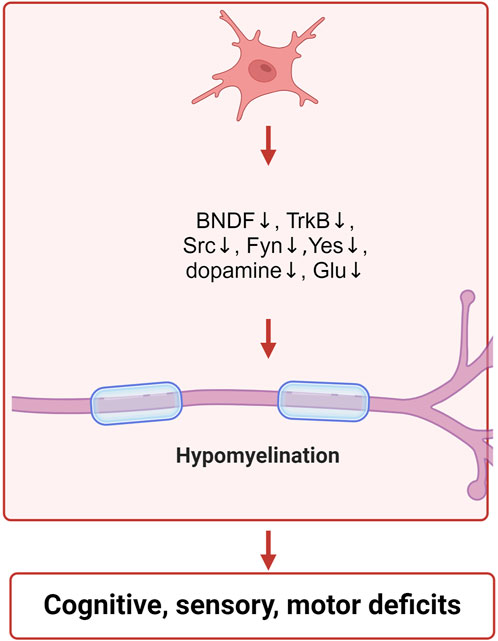
Figure 3. Schematic illustration of the neurotoxic mechanisms of IAA inclued neurotransmitter disorders and neurodevelopment. IAA inhibits the expression of BNDF, TrkB, Src, Fyn, Yes, and reduces the levels of dopamine and glutamate, leading to neurodevelopmental abnormalities.
Neurons depend on the energy supplied by mitochondria to sustain regular neurotransmitter release. A reduction in mitochondrial membrane potential results in an inadequate energy supply, impacting the production and release of neurotransmitters, and consequently influencing the transmission of nerve signals (Sun et al., 2016). IAA inhibits dopamine levels in cerebellar Purkinje cells (LoPachin et al., 2006). One of the key pathological alterations observed in Parkinson’s disease is the loss of dopaminergic neurons located in the substantia nigra of the midbrain. This neuronal death results in a reduction of dopamine levels within the striatum, which serves as the brain’s center for motor control, ultimately causing impairment in the striatal regulation of motor functions (Salamon et al., 2020). Moreover, ATP serves not only as an energy source for tissue cells but also acts as a neurotransmitter that has excitatory properties (Edwards and Gibb, 1993). Glutamic acid (Glu) is recognized broadly as a molecule involved in metabolism, yet it also functions as a neurotransmitter (Franco et al., 2021). IAA enters the brain to inhibit glycolysis and reduce the levels of ATP and Glu in nerve cells (Sperling et al., 2003; Suna et al., 2009; Maher and Hanneken, 2008; Sabri, 1994; Rogel et al., 2005; Gemba et al., 1994). ATP synthase in mitochondria plays a crucial role in generating cellular ATP; ATP synthase accomplishes this by harnessing the mitochondrial membrane potential created through the ongoing oxidation of particular cellular metabolites (Ebanks and Chakrabarti, 2022). The process of releasing, binding, reabsorbing, and breaking down glutamate in the brain plays a significant role in both physiology and pathophysiology. Notably, alterations in glutamatergic neurotransmission become more pronounced during neurodegenerative disorders, correlating with cognitive deficits that manifest as challenges in memory, learning, attention, and decision-making (Castillo-Vazquez et al., 2024). Intracellular molecules known as neurotransmitters are essential for amplifying, transmitting, and converting signals, significantly impacting information transfer within the nervous system. These compounds influence various functions such as mood, cognition, memory, learning, and motor activity. Consequently, an imbalance in the homeostasis of neurotransmitters is increasingly linked to numerous neurological and neurodegenerative disorders (Teleanu et al., 2022).
GAPDH is also crucial for the growth and differentiation of neurons. In mice, the inhibition of the GluA2-GAPDH interaction adversely affects neurodevelopment. The GluA2 subunit is essential for the development of cortical neurons due to its association with GAPDH (Lee et al., 2018). The expressions of brain-derived neurotrophic factor (BDNF), tyrosine receptor kinase B (TrkB), SRC proto-oncogene (Src), FYN proto-oncogene (Fyn) and YES proto-oncogene 1 (Yes) were decreased after IAA treatment (Cho et al., 2021; Shani et al., 2009; Gatson and Singh, 2007). BDNF aids in the maintenance of neurons and brain cells, fosters synaptic connections among neurons, and plays a crucial role in learning as well as the storage of long-term memories (Klein et al., 2011). The receptor for BDNF is TrkB, which is an essential component of the BDNF-TrkB signaling pathway. BDNF performs a distinctive biological role by interacting with its receptor, TrkB, which primarily contributes to neuronal development and is crucial for signal transduction. This pathway encompasses the swift modulation of excitatory and inhibitory synaptic transmission in neurons located in the hippocampus (Otani et al., 2017; Rantamäki et al., 2011). The SRC, YES, and FYN genes produce three related tyrosine protein kinases that are found in human neural tissues (Pyper and Bolen, 1989). The roles of SRC, YES, and FYN are significant in synaptic transmission and the processes underlying excitatory synaptic plasticity (Kalia and Salter, 2003). Src family kinases are involved in the regulation of signaling pathways that contribute to the formation of neuromuscular synapses (Stein et al., 1994). The non-receptor Src tyrosine kinase is a crucial element in intracellular signal transduction and is highly expressed within the nervous system (Zhao et al., 2003). Src has the ability to enhance the proliferation and dedifferentiation of Schwann cells, aiding in the repair of peripheral nerve injuries (Govindasamy et al., 2022). Fyn, a cytoplasmic tyrosine kinase, is essential for various biological processes. Within both the central and peripheral nervous systems, Fyn is pivotal for initiating myelination through the formation of myelinated glial cells, such as Schwann cells and oligodendrocytes (Torii et al., 2017). Furthermore, Fyn has been shown to facilitate the regulation of the growth of axons and dendrites (Liu et al., 2006). The Fyn protein, associated with myelin, plays a crucial role in starting the myelination process and is significant in the overall myelination mechanism (Biffiger et al., 2000). IAA treatment led to the inhibition of nerve cell growth and differentiation, which was closely associated with the suppression of neurodevelopmental proteins by IAA. The specific mechanism of IAA neurotoxicity is shown in Figure 3.
As most studies of IAA have focused on the fields of genotoxicity, carcinogenicity and reproductive toxicity, few studies have been conducted on neurotoxicity. And there has been no review of IAA neurotoxicity. For the first time, we review the neurotoxicity of IAA. IAA causes neurotoxicity through a variety of mechanisms, including oxidative stress, DNA damage, inflammation, BBB disruption, and apoptosis. Neurotoxic manifestations include neurodevelopmental disorders (abnormal neural differentiation and demyelination), neurotransmitter disorders, etc. The unavoidable limitation of this review is that most of the studies are based on cell experiments and there are few studies on neurotoxicity in mammals. In addition, there are no studies that have confirmed neurotoxicity caused by daily intake of IAA through drinking water. This may be related to the daily dose of IAA ingested. However, oxidative stress and inflammation in nerve cells caused by long-term low level of IAA intake are closely related to neurodegeneration. Future experimental studies should consider these factors. Our review provides insights into the mechanism of IAA neurotoxicity. The molecular mechanism of neurotoxicity induced by IAA is shown in Figure 4.
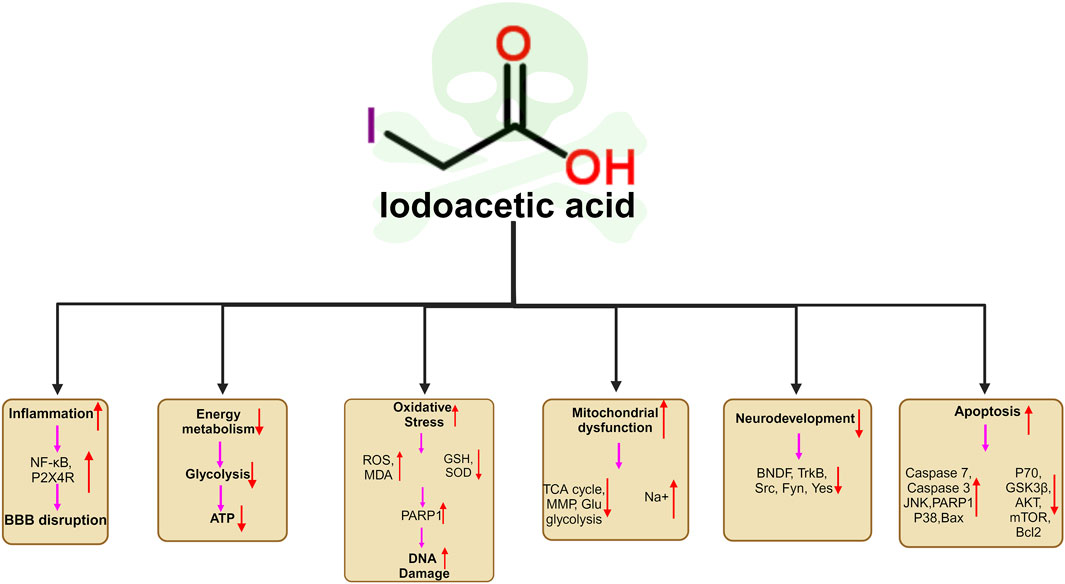
Figure 4. Schematic illustration of the neurotoxic mechanisms of IAA. (1) IAA promoted neuroinflammation by promoting the expression of NFκB, and P2X4R. (2) IAA caused Energy metabolism disorder. (3) The neurotoxicity of IAA caused oxidative stress and DNA damage by stimulating the increase of ROS and reducing the level of antioxidant SOD, GSH (4) IAA induced mitochondrial dysfunction and ATP production. (5) IAA neurotoxicity cause nerve cell developmental disorders, and abnormal differentiation by reducing the BNDF, TrkB,Src, Fyn, Yes expression. (6) IAA cause nerve cell apoptosis by reducing anti-apoptotic proteins P70, GSK3B, AKT, mTOR, Bcl2 expression and promoting apoptosis protein JNK, PARP1, Caspase3, Caspase7, Bax, MAPK, PARP1, Bax expression.
XW: Writing–original draft, Writing–review and editing. CR: Writing–original draft, Writing–review and editing. PN: Writing–original draft, Writing–review and editing. WL: Writing–original draft, Writing–review and editing. GW: Writing–original draft, Writing–review and editing. ZH: Writing–original draft, Writing–review and editing. XQ: Writing–original draft, Writing–review and editing. DZ: Writing–original draft, Writing–review and editing. JL: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the grants from the Science and Technology Department of Jilin Province (20240305055YY), and Graduate Innovation Fund of Jilin University (2024CX253).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
adenosine Triphosphate, ATP; brain-derived neurotrophic factor, BDNF; central nervous system, CNS; Connexin43, Cx43; cysteinyl aspartate specific proteinase-3, Caspase-3; disinfection by-products, DBPs; FYN proto-oncogene, Fyn; Glutamic acid, Glu; glutathione, GSH; glyceraldehyde-3-phosphate dehydrogenase, GAPDH; Iodoacetic acid, IAA; lactate dehydrogenase, LDH; mitogen-activated Protein Kinase, MAPK; mitochondrial membrane potential, MMP; pheochromocytoma cells, PC12; Poly(ADP-ribose) polymerase 1, PARP1; Purinergic Receptor P2X4, P2X4R; reactive nitrogen species, RNS; reactive oxygen species, ROS; retinal ganglion cells, RGC-5 cells; RSK, ribosomal s6 kinase; SRC proto-oncogene, Src; superoxide dismutase, SOD; tricarboxylic acid, TCA; tyrosine receptor kinase B, TrkB; FYN proto-oncogene: Fyn.
Bartolome, F., Wu, H. C., Burchell, V. S., Preza, E., Wray, S., Mahoney, C. J., et al. (2013). Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 78, 57–64. doi:10.1016/j.neuron.2013.02.028
Becalski, A., Lau, B. P. Y., Schrader, T. J., Seaman, S. W., and Sun, W. F. (2006). Formation of iodoacetic acids during cooking: interaction of iodized table salt with chlorinated drinking water. Food Addit. Contam. 23, 957–962. doi:10.1080/02652030600838407
Biffiger, K., Bartsch, S., Montag, D., Aguzzi, A., Schachner, M., and Bartsch, U. (2000). Severe hypomyelination of the murine CNS in the absence of myelin-associated glycoprotein and Fyn tyrosine kinase. J. Neurosci. 20, 7430–7437. doi:10.1523/JNEUROSCI.20-19-07430.2000
Butera, G., Pacchiana, R., Mullappilly, N., Margiotta, M., Bruno, S., Conti, P., et al. (2018). Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Biochimica Biophysica Acta-Molecular Cell Res. 1865, 1914–1923. doi:10.1016/j.bbamcr.2018.10.005
Castillo-Vazquez, S. K., Massieu, L., Rincón-Heredia, R., García-delaTorre, P., Quiroz-Baez, R., Gomez-Verjan, J. C., et al. (2024). Glutamatergic neurotransmission in aging and neurodegenerative diseases: a potential target to improve cognitive impairment in aging. Archives Med. Res. 55, 103039. doi:10.1016/j.arcmed.2024.103039
Chen, H. Y., Tan, G. L., Cao, J., Zhang, G. X., Yi, P., Yu, P., et al. (2017a). Design, synthesis, and biological evaluation of novel tetramethylpyrazine derivatives as potential neuroprotective agents. Chem. and Pharm. Bull. 65, 56–65. doi:10.1248/cpb.c16-00699
Chen, H. Y., Xu, D. P., Tan, G. L., Cai, W., Zhang, G. X., Cui, W., et al. (2015). A potent multi-functional neuroprotective derivative of tetramethylpyrazine. J. Mol. Neurosci. 56, 977–987. doi:10.1007/s12031-015-0566-x
Chen, W., Feng, J. G., and Tong, W. S. (2017b). Phosphorylation of astrocytic connexin43 by ERK1/2 impairs blood-brain barrier in acute cerebral ischemia. Cell Biosci. 7, 43. doi:10.1186/s13578-017-0170-6
Cho, Y. J., Choi, S. H., Lee, R. M., Cho, H. S., Rhim, H., Kim, H. C., et al. (2021). Protective effects of gintonin on reactive oxygen species-induced HT22 cell damages: involvement of LPA1 receptor-BDNF-AKT signaling pathway. Molecules 26, 4138. doi:10.3390/molecules26144138
Christie, J. R., and Beekman, M. (2017). Selective sweeps of mitochondrial DNA can drive the evolution of uniparental inheritance. Evolution 71, 2090–2099. doi:10.1111/evo.13291
Coppedè, F., and Migliore, L. (2015). DNA damage in neurodegenerative diseases. Mutat. Res. 776, 84–97. doi:10.1016/j.mrfmmm.2014.11.010
Dad, A., Jeong, C. H., Pals, J. A., Wagner, E. D., and Plewa, M. J. (2013). Pyruvate remediation of cell stress and genotoxicity induced by haloacetic acid drinking water disinfection by-products. Environ. Mol. Mutagen. 54, 629–637. doi:10.1002/em.21795
Davis, S. E., Cirincione, A. B., Jimenez-Torres, A. C., and Zhu, J. (2023). The impact of neurotransmitters on the neurobiology of neurodegenerative diseases. Int. J. Mol. Sci. 24, 15340. doi:10.3390/ijms242015340
Dong, H. Y., Qjang, Z. M., and Richardson, S. D. (2019). Formation of iodinated disinfection byproducts (I-DBPs) in drinking water: emerging concerns and current issues. Accounts Chem. Res. 52, 896–905. doi:10.1021/acs.accounts.8b00641
Du, X. L., Matsumura, T., Edelstein, D., Rossetti, L., Zsengellér, Z., Szabó, C., et al. (2003). Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investigation 112, 1049–1057. doi:10.1172/jci200318127
Ebanks, B., and Chakrabarti, L. (2022). Mitochondrial ATP synthase is a target of oxidative stress in neurodegenerative diseases. Front. Mol. Biosci. 9, 854321. doi:10.3389/fmolb.2022.854321
Edwards, F. A., and Gibb, A. J. (1993). ATP - a Fast neurotransmitter. Febs Lett. 325, 86–89. doi:10.1016/0014-5793(93)81419-z
Elmi, A., Idahl, L., and Sehlin, J. (2001). Modulation of islet ATP content by inhibition or stimulation of the Na+/K+ pump. Eur. J. Pharmacol. 426, 139–143. doi:10.1016/s0014-2999(01)01214-6
Eslami, M., Alizadeh, L., Morteza-Zadeh, P., and Sayyah, M. (2020). The effect of Lipopolysaccharide (LPS) pretreatment on hippocampal apoptosis in traumatic rats. Neurological Res. 42, 91–98. doi:10.1080/01616412.2019.1709139
Franco, R., Rivas-Santisteban, R., Lillo, J., Camps, J., Navarro, G., and Reyes-Resina, I. (2021). 5-Hydroxytryptamine, glutamate, and ATP: much more than neurotransmitters. Front. Cell Dev. Biol. 9, 667815. doi:10.3389/fcell.2021.667815
Gao, N., Zhang, Z., Jiang, B. H., and Shi, X. L. (2003). Role of PI3K/AKT/mTOR signaling in the cell cycle progression of human prostate cancer. Biochem. Biophysical Res. Commun. 310, 1124–1132. doi:10.1016/j.bbrc.2003.09.132
Gatson, J. W., and Singh, M. (2007). Activation of a membrane-associated androgen receptor promotes cell death in primary cortical astrocytes. Endocrinology 148, 2458–2464. doi:10.1210/en.2006-1443
Gemba, T., Oshima, T., and Ninomiya, M. (1994). Glutamate efflux via the reversal of the sodium-dependent glutamate transporter caused by glycolytic inhibition in rat cultured astrocytes. Neuroscience 63, 789–795. doi:10.1016/0306-4522(94)90523-1
Gonsioroski, A., Meling, D. D., Gao, L. Y., Plewa, M. J., and Flaws, J. A. (2020). Iodoacetic acid inhibits follicle growth and alters expression of genes that regulate apoptosis, the cell cycle, estrogen receptors, and ovarian steroidogenesis in mouse ovarian follicles. Reprod. Toxicol. 91, 101–108. doi:10.1016/j.reprotox.2019.10.005
Govindasamy, N., Chok, K. C., Ng, P. Y., Koh, R. Y., and Chye, S. M. (2022). Melatonin induced Schwann cell proliferation and dedifferentiation through NF-ĸB, FAKDependent but src-independent pathways. Rep. Biochem. Mol. Biol. 11, 63–73. doi:10.52547/rbmb.11.1.63
Han, Y., Jin, F., Xie, Y., Liu, Y. K., Hu, S., Liu, X. D., et al. (2019). DNA-PKcs PARylation regulates DNA-PK kinase activity in the DNA damage response. Mol. Med. Rep. 20, 3609–3616. doi:10.3892/mmr.2019.10640
Harkness, J. S., Dwyer, G. S., Warner, N. R., Parker, K. M., Mitch, W. A., and Vengosh, A. (2015). Iodide, bromide, and ammonium in hydraulic fracturing and oil and gas wastewaters: environmental implications. Environ. Sci. and Technol. 49, 1955–1963. doi:10.1021/es504654n
Hiromura, M., Okada, F., Obata, T., Auguin, D., Shibata, T., Roumestand, C., et al. (2004). Inhibition of Akt kinase activity by a peptide spanning the betaA strand of the proto-oncogene TCL1. J. Biol. Chem. 279, 53407–53418. doi:10.1074/jbc.M403775200
Jiao, X. F., Gonsioroski, A., Flaws, J. A., and Qiao, H. Y. (2021). Iodoacetic acid disrupts mouse oocyte maturation by inducing oxidative stress and spindle abnormalities. Environ. Pollut. 268, 115601. doi:10.1016/j.envpol.2020.115601
Kalia, L. V., and Salter, M. W. (2003). Interactions between Src family protein tyrosine kinases and PSD-95. Neuropharmacology 45, 720–728. doi:10.1016/s0028-3908(03)00313-7
Kim, S., Kubelka, N. K., Laporte, H. M., Krishnamoorthy, V. R., and Singh, M. (2023). Estradiol and 3β-diol protect female cortical astrocytes by regulating connexin 43 Gap Junctions. Mol. Cell. Endocrinol. 578, 112045. doi:10.1016/j.mce.2023.112045
Klein, A. B., Williamson, R., Santini, M. A., Clemmensen, C., Ettrup, A., Rios, M., et al. (2011). Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int. J. Neuropsychopharmacol. 14, 347–353. doi:10.1017/s1461145710000738
Kratsovnik, E., Bromberg, Y., Sperling, O., and Zoref-Shani, E. (2005). Oxidative stress activates transcription factor NF-kB-mediated protective signaling in primary rat neuronal cultures. J. Mol. Neurosci. 26, 27–32. doi:10.1385/jmn:26:1:027
Lapchak, P. A., Maher, P., Schubert, D., and Zivin, J. A. (2007). Baicalein, an antioxidant 12/15-lipoxygenase inhibitor improves clinical rating scores following multiple infarct embolic strokes. Neuroscience 150, 585–591. doi:10.1016/j.neuroscience.2007.09.033
Lapchak, P. A., Schubert, D. R., and Maher, P. A. (2011). De-risking of stilbazulenyl nitrone (STAZN), a lipophilic nitrone to treat stroke using a unique panel of in vitro assays. Transl. Stroke Res. 2, 209–217. doi:10.1007/s12975-011-0071-7
Lee, F. H. F., Zhang, H. L., Jiang, A. L., Zai, C. C., and Liu, F. (2018). Specific alterations in astrocyte properties via the GluA2-GAPDH complex associated with multiple sclerosis. Sci. Rep. 8, 12856. doi:10.1038/s41598-018-31318-4
Li, J., Jiang, J., Pang, S. Y., Yang, Y., Sun, S. F., Wang, L. H., et al. (2020). Transformation of X-ray contrast media by conventional and advanced oxidation processes during water treatment: efficiency, oxidation intermediates, and formation of iodinated byproducts. Water Res. 185, 116234. doi:10.1016/j.watres.2020.116234
Liang, H. Y., Van Remmen, H., Frohlich, V., Lechleiter, J., Richardson, A., and Ran, Q. T. (2007). Gpx4 protects mitochondrial ATP generation against oxidative damage. Biochem. Biophysical Res. Commun. 356, 893–898. doi:10.1016/j.bbrc.2007.03.045
Liu, H., Nakazawa, T., Tezuka, T., and Yamamoto, T. (2006). Physical and functional interaction of Fyn tyrosine kinase with a brain-enriched Rho GTPase-activating protein TCGAP. J. Biol. Chem. 281, 23611–23619. doi:10.1074/jbc.M511205200
LoPachin, R. M., Barber, D. S., He, D. K., and Das, S. (2006). Acrylamide inhibits dopamine uptake in rat striatal synaptic vesicles. Toxicol. Sci. 89, 224–234. doi:10.1093/toxsci/kfj005
Ma, J. N., Gao, J. Z., Niu, M. Y., Zhang, X. N., Wang, J., and Xie, A. M. (2020). P2X4R overexpression upregulates interleukin-6 and exacerbates 6-OHDA-induced dopaminergic degeneration in a rat model of PD. Front. Aging Neurosci. 12, 580068. doi:10.3389/fnagi.2020.580068
MacKeown, H., von Gunten, U., and Criquet, J. (2022). Iodide sources in the aquatic environment and its fate during oxidative water treatment - a critical review. Water Res. 217, 118417. doi:10.1016/j.watres.2022.118417
Maher, P., and Hanneken, A. (2008). Flavonoids protect retinal ganglion cells from ischemia in vitro. Exp. Eye Res. 86, 366–374. doi:10.1016/j.exer.2007.11.009
Marchetti, M. A., Lee, W., Cowell, T. L., Wells, T. M., Weissbach, H., and Kantorow, M. (2006). Silencing of the methionine sulfoxide reductase A gene results in loss of mitochondrial membrane potential and increased ROS production in human lens cells. Exp. Eye Res. 83, 1281–1286. doi:10.1016/j.exer.2006.07.005
Mata, V. C., and Goldberg, J. (2023). Morin and isoquercitrin protect against ischemic neuronal injury by modulating signaling pathways and stimulating mitochondrial biogenesis. Nutr. Neurosci. 26, 796–806. doi:10.1080/1028415x.2022.2094855
Merlo, D., Mollinari, C., Racaniello, M., Garaci, E., and Cardinale, A. (2016). DNA double strand breaks: a common theme in neurodegenerative diseases. Curr. Alzheimer Res. 13, 1208–1218. doi:10.2174/1567205013666160401114915
Mou, L., Sun, D. G., Qu, J. Y., Tan, X. Y., Wang, S. L., Zeng, Q., et al. (2024). GRP78/IRE1 and cGAS/STING pathway crosstalk through CHOP facilitates iodoacetic acid-mediated testosterone decline. J. Hazard. Mater. 476, 135101. doi:10.1016/j.jhazmat.2024.135101
Nakajima, H., Kubo, T., Ihara, H., Hikida, T., Danjo, T., Nakatsuji, M., et al. (2015). Nuclear-translocated glyceraldehyde-3-phosphate dehydrogenase promotes poly(ADP-ribose) polymerase-1 activation during oxidative/nitrosative stress in stroke. J. Biol. Chem. 290, 14493–14503. doi:10.1074/jbc.M114.635607
Ondricek, A. J., Kashyap, A. K., Thamake, S. I., and Vishwanatha, J. K. (2012). A comparative study of phytoestrogen action in mitigating apoptosis induced by oxidative stress. Vivo 26, 765–775.
Otani, K., Okada, M., and Yamawaki, H. (2017). Diverse distribution of tyrosine receptor kinase B isoforms in rat multiple tissues. J. Veterinary Med. Sci. 79, 1516–1523. doi:10.1292/jvms.17-0257
Pérez-Albaladejo, E., Pinteño, R., Aznar-Luque, M. D., Casado, M., Postigo, C., and Porte, C. (2023). Genotoxicity and endocrine disruption potential of haloacetic acids in human placental and lung cells. Sci. Total Environ. 879, 162981. doi:10.1016/j.scitotenv.2023.162981
Ping, N., Zuo, K. Y., Cai, J. H., Rong, C. S., Yu, Z. Q., Zhang, X., et al. (2024). Apigenin protects against ischemic stroke by increasing DNA repair. Front. Pharmacol. 15, 1362301. doi:10.3389/fphar.2024.1362301
Pyper, J. M., and Bolen, J. B. (1989). Neuron-specific splicing of c-src rna in human-brain. J. Neurosci. Res. 24, 89–96. doi:10.1002/jnr.490240113
Rantamäki, T., Vesa, L., Antila, H., Di Lieto, A., Tammela, P., Schmitt, A., et al. (2011). Antidepressant drugs transactivate TrkB neurotrophin receptors in the adult rodent brain independently of BDNF and monoamine transporter blockade. Plos One 6, e20567. doi:10.1371/journal.pone.0020567
Rehfeldt, S. C. H., Laufer, S., and Goettert, M. I. (2021). A highly selective in vitro JNK3 inhibitor, FMU200, restores mitochondrial membrane potential and reduces oxidative stress and apoptosis in SH-SY5Y cells. Int. J. Mol. Sci. 22, 3701. doi:10.3390/ijms22073701
Richardson, S. D., Fasano, F., Ellington, J. J., Crumley, F. G., Buettner, K. M., Evans, J. J., et al. (2008). Occurrence and mammalian cell toxicity of iodinated disinfection byproducts in drinking water. Environ. Sci. and Technol. 42, 8330–8338. doi:10.1021/es801169k
Rogel, A., Bromberg, Y., Sperling, O., and Zoref-Shani, E. (2005). Phospholipase C is involved in the adenosine-activated signal transduction pathway conferring protection against iodoacetic acid-induced injury in primary rat neuronal cultures. Neurosci. Lett. 373, 218–221. doi:10.1016/j.neulet.2004.10.012
Sabri, M. I. (1994). Attenuation of glyceraldehyde-3-phosphate dehydrogenase-activity and atp levels in rat-brain synaptosomes by acrylamide. Neurochem. Res. 19, 1439–1444. doi:10.1007/bf00972473
Salamon, A., Zádori, D., Szpisjak, L., Klivényi, P., and Vécsei, L. (2020). Neuroprotection in Parkinson's disease: facts and hopes. J. Neural Transm. 127, 821–829. doi:10.1007/s00702-019-02115-8
Sha, Y. J., Wu, H., Guo, Y., Liu, X., Mo, Y., Yang, Q. Y., et al. (2022). Effects of iodoacetic acid drinking water disinfection byproduct on the gut microbiota and its metabolism in rats. J. Environ. Sci. 117, 91–104. doi:10.1016/j.jes.2022.02.048
Shani, V., Bromberg, Y., Sperling, O., and Zoref-Shani, E. (2009). Involvement of src tyrosine kinases (SFKs) and of focal adhesion kinase (FAK) in the injurious mechanism in rat primary neuronal cultures exposed to chemical ischemia. J. Mol. Neurosci. 37, 50–59. doi:10.1007/s12031-008-9113-3
Shi, X. M. (2023). Revision and prospect of the standards for drinking water quality gb5749-2022. Zhonghua yu fang yi xue za zhi Chin. J. Prev. Med. 57, 801–805. doi:10.3760/cma.j.cn112150-20221027-01039
Sirover, M. A. (2014). Structural analysis of glyceraldehyde-3-phosphate dehydrogenase functional diversity. Int. J. Biochem. and Cell Biol. 57, 20–26. doi:10.1016/j.biocel.2014.09.026
Smith, R., Lebeaupin, T., Juhász, S., Chapuis, C., D'Augustin, O., Dutertre, S., et al. (2019). Poly(ADP-ribose)-dependent chromatin unfolding facilitates the association of DNA-binding proteins with DNA at sites of damage. Nucleic Acids Res. 47, 11250–11267. doi:10.1093/nar/gkz820
Sperling, O., Bromberg, Y., Oelsner, H., and Zoref-Shani, E. (2003). Reactive oxygen species play an important role in iodoacetate-induced neurotoxicity in primary rat neuronal cultures and in differentiated PC12 cells. Neurosci. Lett. 351, 137–140. doi:10.1016/s0304-3940(03)00858-9
Stein, P. L., Vogel, H., and Soriano, P. (1994). Combined deficiencies of src, fyn, and yes tyrosine kinases in mutant mice. Genes and Dev. 8, 1999–2007. doi:10.1101/gad.8.17.1999
Sun, X. G., Shi, X., Lu, L. J., Jiang, Y. F., and Liu, B. (2016). Stimulus-dependent neuronal cell responses in SH-SY5Y neuroblastoma cells. Mol. Med. Rep. 13, 2215–2220. doi:10.3892/mmr.2016.4759
Suna, H., Arai, M., Tsubotani, Y., Hayashi, A., Setiawan, A., and Kobayashi, M. (2009). Dysideamine, a new sesquiterpene aminoquinone, protects hippocampal neuronal cells against iodoacetic acid-induced cell death. Bioorg. and Med. Chem. 17, 3968–3972. doi:10.1016/j.bmc.2009.04.025
Teleanu, R. I., Niculescu, A. G., Roza, E., Vladâcenco, O., Grumezescu, A. M., and Teleanu, D. M. (2022). Neurotransmitters-key factors in neurological and neurodegenerative disorders of the central nervous system. Int. J. Mol. Sci. 23, 5954. doi:10.3390/ijms23115954
Torii, T., Miyamoto, Y., Kawahara, K., Tanoue, A., Seki, Y., Morimoto, T., et al. (2017). Data supporting the role of Fyn in embryonic sciatic nerve fasciculation. Data Brief 11, 358–363. doi:10.1016/j.dib.2017.02.042
Vazquez-Villasenor, I., Garwood, C. J., Simpson, J. E., Heath, P. R., Mortiboys, H., and Wharton, S. B. (2021). Persistent DNA damage alters the neuronal transcriptome suggesting cell cycle dysregulation and altered mitochondrial function. Eur. J. Neurosci. 54, 6987–7005. doi:10.1111/ejn.15466
Wagner, E. D., and Plewa, M. J. (2017). CHO cell cytotoxicity and genotoxicity analyses of disinfection by-products: an updated review. J. Environ. Sci. 58, 64–76. doi:10.1016/j.jes.2017.04.021
Wang, X., Li, J. J., Zhao, D. X., and Li, J. H. (2022). |Therapeutic and preventive effects of apigenin in cerebral ischemia: a review. Food and Funct. 13, 11425–11437. doi:10.1039/d2fo02599j
Wang, X., Yu, Z. Q., Dong, F. X., Li, J. J., Niu, P., Ta, Q., et al. (2024). Clarifying the mechanism of apigenin against blood-brain barrier disruption in ischemic stroke using systems pharmacology. Mol. Divers. 28, 609–630. doi:10.1007/s11030-023-10607-9
Wang, X. Q., Xiao, A. Y., Sheline, C., Hyrc, K., Yang, A. Z., Goldberg, M. P., et al. (2003). Apoptotic insults impair Na+, K+-ATPase activity as a mechanism of neuronal death mediated by concurrent ATP deficiency and oxidant stress. J. Cell Sci. 116, 2099–2110. doi:10.1242/jcs.00420
Wei, X., Wang, S., Zheng, W. W., Wang, X., Liu, X. L., Jiang, S. H., et al. (2013). Drinking water disinfection byproduct iodoacetic acid induces tumorigenic transformation of NIH3T3 cells. Environ. Sci. and Technol. 47, 5913–5920. doi:10.1021/es304786b
Wilson, A. C., Clemente, L., Liu, T. B., Bowen, R. L., Meethal, S. V., and Atwood, C. S. (2008). Reproductive hormones regulate the selective permeability of the blood-brain barrier. Biochimica Biophysica Acta-Molecular Basis Dis. 1782, 401–407. doi:10.1016/j.bbadis.2008.02.011
Xu, L., Jiang, X. J., Wei, F., and Zhu, H. L. (2023). [Corrigendum] Leonurine protects cardiac function following acute myocardial infarction through anti‑apoptosis by the PI3K/AKT/GSK3β signaling pathway. Mol. Med. Rep. 27, 65. doi:10.3892/mmr.2023.12952
Yang, M. X., Feng, Y., Yan, S. C., Wu, Z. Y., Xiao, X., Sang, J. C., et al. (2021). Evans blue might produce pathologically activated neuroprotective effects via the inhibition of the P2X4R/p38 signaling pathway. Cell. Mol. Neurobiol. 41, 293–307. doi:10.1007/s10571-020-00852-z
Yao, H., Shang, Z. M., Wang, P. H., Li, S. X., Zhang, Q. Y., Tian, H. Q., et al. (2016). Protection of luteolin-7-O-glucoside against doxorubicin-induced injury through PTEN/akt and ERK pathway in H9c2 cells. Cardiovasc. Toxicol. 16, 101–110. doi:10.1007/s12012-015-9317-z
Yu, H. N., Wu, L. Y., Xuan, D. L., Peng, Q., Qu, W. D., and Zhou, Y. (2024). Development and validation of a GC-MS/MS method for the determination of iodoacetic acid in biological samples. Anal. Bioanal. Chem. 416, 3185–3194. doi:10.1007/s00216-024-05266-0
Zeng, L., Bi, Y. Q., Guo, P. F., Bi, Y. L., Wang, T. T., Dong, L., et al. (2021). Metabolic analysis of schizochytrium mutants with high DHA content achieved with ARTP mutagenesis combined with iodoacetic acid and dehydroepiandrosterone screening. Front. Bioeng. Biotechnol. 9, 738052. doi:10.3389/fbioe.2021.738052
Zhan, R., Meng, X., Tian, D. P., Xu, J., Cui, H. T., Yang, J. L., et al. (2023). NAD+ rescues aging-induced blood-brain barrier damage via the CX43-PARP1 axis. Neuron 111, 3634–3649.e7. doi:10.1016/j.neuron.2023.08.010
Zhang, Y., Liu, Y., Zhang, X. Y., Yong, V. W., and Xue, M. Z. (2023). Omarigliptin protects the integrity of the blood-brain barrier after intracerebral hemorrhage in mice. J. Inflamm. Res. 16, 2535–2548. doi:10.2147/jir.S411017
Zhao, Y. L., Takagawa, K., Oya, T., Yang, H. F., Gao, Z. Y., Kawaguchi, M., et al. (2003). Active Src expression is induced after rat peripheral nerve injury. Glia 42, 184–193. doi:10.1002/glia.10223
Zhou, X. H., Zhu, L. J., Wang, L., Guo, B. J., Zhang, G. X., Sun, Y. W., et al. (2015). Protective effect of edaravone in primary cerebellar granule neurons against iodoacetic acid-induced cell injury. Oxidative Med. Cell. Longev. 2015, 606981. doi:10.1155/2015/606981
Zhu, X. B., Jin, C. M., Pan, Q. R., and Hu, X. (2021). Determining the quantitative relationship between glycolysis and GAPDH in cancer cells exhibiting the Warburg effect. J. Biol. Chem. 296, 100369. doi:10.1016/j.jbc.2021.100369
Keywords: neurotoxicity, neurodegeneration, disinfection byproducts, iodoacetic acid, neuroinflammation
Citation: Wang X, Rong C, Niu P, Leng W, Wang G, He Z, Qi X, Zhao D and Li J (2025) The neurotoxicity of iodoacetic acid, a byproduct of drinking water disinfection. Front. Toxicol. 7:1543374. doi: 10.3389/ftox.2025.1543374
Received: 11 December 2024; Accepted: 10 January 2025;
Published: 27 January 2025.
Edited by:
Konrad Andrzej Szychowski, University of Information Technology and Management in Rzeszow, PolandReviewed by:
Jae-hyeon Park, National Institute on Aging (NIH), United StatesCopyright © 2025 Wang, Rong, Niu, Leng, Wang, He, Qi, Zhao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dexi Zhao, emR4MDJAMTYzLmNvbQ==; Jinhua Li, amluaHVhMUBqbHUuZWR1LmNu
†ORCID: Jinhua Li, orcid.org/0000-0003-0085-9168
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.