
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Syst. Neurosci., 26 May 2022
Volume 16 - 2022 | https://doi.org/10.3389/fnsys.2022.867323
This article is part of the Research TopicTraumatic Brain Injury As a Systems Neuroscience Problem - Volume IIView all 5 articles
 Jeffery A. Boychuk1,2†‡
Jeffery A. Boychuk1,2†‡ Corwin R. Butler1,3‡
Corwin R. Butler1,3‡ Katalin Cs. Smith1,4
Katalin Cs. Smith1,4 Miklos B. Halmos5
Miklos B. Halmos5 Bret N. Smith1,4,6,7*
Bret N. Smith1,4,6,7*Type A GABA receptors (GABAARs) are pentameric combinations of protein subunits that give rise to tonic (ITonicGABA) and phasic (i.e., synaptic; ISynapticGABA) forms of inhibitory GABAAR signaling in the central nervous system. Remodeling and regulation of GABAAR protein subunits are implicated in a wide variety of healthy and injury-dependent states, including epilepsy. The present study undertook a detailed analysis of GABAAR signaling using whole-cell patch clamp recordings from mouse dentate granule cells (DGCs) in coronal slices containing dorsal hippocampus at 1–2 or 8–13 weeks after a focal, controlled cortical impact (CCI) or sham brain injury. Zolpidem, a benzodiazepine-like positive modulator of GABAARs, was used to test for changes in GABAAR signaling of DGCs due to its selectivity for α1 subunit-containing GABAARs. Electric charge transfer and statistical percent change were analyzed in order to directly compare tonic and phasic GABAAR signaling and to account for zolpidem’s ability to modify multiple parameters of GABAAR kinetics. We observed that baseline ITonicGABA is preserved at both time-points tested in DGCs ipsilateral to injury (Ipsi-DGCs) compared to DGCs contralateral to injury (Contra-DGCs) or after sham injury (Sham-DGCs). Interestingly, application of zolpidem resulted in modulation of ITonicGABA across groups, with Ipsi-DGCs exhibiting the greatest responsiveness to zolpidem. We also report that the combination of CCI and acute application of zolpidem profoundly augments the proportion of GABAAR charge transfer mediated by tonic vs. synaptic currents at both time-points tested, whereas gene expression of GABAAR α1, α2, α3, and γ2 subunits is unchanged at 8–13 weeks post-injury. Overall, this work highlights the shift toward elevated influence of tonic inhibition in Ipsi-DGCs, the impact of zolpidem on all components of inhibitory control of DGCs, and the sustained nature of these changes in inhibitory tone after CCI injury.
- Dentate granule cells (DGCs) ipsilateral to CCI brain injury exhibit a preserved GABAAR-mediated tonic current at 1–2 and 8–13 weeks post-injury.
- DGCs exhibit an unexpected zolpidem-sensitive tonic GABAAR current that is markedly enhanced by brain injury (≥84% of controls 1–2 weeks post-injury and ≥75% of controls 8–13 weeks post-injury).
- DGC’s receive more charge transfer from tonic GABAARs in comparison to synaptic GABAARs; the ratio of charge transfer is numerically (non-significantly) increased by CCI injury relative to controls (≥103% at 1–2 weeks post-injury and ≥64% at 8–13 weeks post-injury) whereas it is profoundly enhanced by the combination of CCI injury and acute application of zolpidem (≥381% at 1–2 weeks post-injury and ≥251% at 8–13 weeks post-injury).
- Gene expression of GABAAR α1, α2, α3, and γ2 subunits is unchanged in DG ipsilateral to injury, relative to DG contralateral to injury, 8–13 weeks post-injury.
Neurochemical inhibition of principal cells within the hippocampus is primarily mediated by type A GABA receptors (GABAARs), that are formed by combinations of α1–6, β1–4, γ1–3, δ, ε, θ, and π subunits (Lüddens and Wisden, 1991; Laurie et al., 1992; Macdonald and Olsen, 1994; Barnard et al., 1998; Whiting et al., 1999). These subunit combinations vary by cell type and result in distinct kinetic profiles and patterns of responses to pharmacological agents (Draguhn et al., 1990; Verdoorn et al., 1990; Davies et al., 1997). Hippocampal dentate granule cells (DGCs) are believed to predominantly express α1βxγ2 and α4βxδ GABAAR subunit combinations; these combinations make well-established contributions to synaptic (i.e., ISynapticGABA) and tonic (i.e., ITonicGABA) GABAAR signaling, respectively (Chang et al., 1996; Kapur and Macdonald, 1999; Mody, 2001; Brown et al., 2002; Stell et al., 2003; Wei et al., 2003; Farrant and Nusser, 2005; Mtchedlishvili and Kapur, 2006; Glykys et al., 2008).
Brain insults including repeated seizures, cerebral ischemia, or brain injury may change the expression or regulation of GABAAR subunits to alter functional GABAAR currents in DGCs (Gibbs et al., 1997; Brooks-Kayal et al., 1998; Redecker et al., 2002; Leroy et al., 2004; Peng et al., 2004; Sun et al., 2007; Zhang et al., 2007; Kharlamov et al., 2008; Zhan and Nadler, 2009; Pavlov et al., 2011; Gupta et al., 2012; Raible et al., 2012, 2015; Drexel et al., 2015). These changes have often been associated with the coincident loss of local GABAergic interneuron populations following traumatic brain injury (TBI) (Lowenstein et al., 1992; Zhu et al., 1997; Butler et al., 2016, 2017; Frankowski et al., 2019) and subsequent posttraumatic epileptogenesis. However, not all changes in inhibitory tone follow this pattern. Importantly, while synaptic inhibition of DGCs appears reduced after TBI in mouse models of posttraumatic epilepsy, presumably due to loss of inhibitory interneurons, the baseline tonic inhibition of DGCs after brain injury is maintained, even after spontaneous recurrent seizures have developed (Cossart et al., 2001; Pavlov et al., 2011; Boychuk et al., 2016; Butler et al., 2016; Becerra et al., 2021). Additionally, recordings of DGCs from mice with acquired epilepsy after experimental brain injury such as controlled cortical impact (CCI), or after pilocarpine-induced status epilepticus, often require efforts to “unmask” recurrent excitation due to the selective preservation/remodeling of inhibitory circuits (Winokur et al., 2004; Hunt et al., 2009, 2010, 2011). These observations suggest underlying compensatory mechanisms following brain injury to maintain inhibitory tone of DGCs despite a large percentage of local GABAergic interneurons being lost following brain insults.
We have previously reported a preservation of the tonic GABA current in DGCs after CCI that parallels a loss of responsiveness to THIP (Gaboxadol), a GABAAR “super-agonist” that exhibits preferential action (i.e., selectivity) for receptor pentamers containing the δ subunit along with a4 and a6 subunits (Saarelainen et al., 2008; Chandra et al., 2010; Boychuk et al., 2016; Butler et al., 2016). These findings using CCI have recently been replicated and expanded upon (Becerra et al., 2021) and suggest remodeling or altered regulation of GABAAR subunits, chloride transport, GABAAR signaling, and/or other mechanisms. In this study, we focused on the contribution of α1 containing GABAARs to inhibitory tone of DGCs following TBI injury using the benzodiazepine-like GABAAR augmenting agent zolpidem, a potent hypnotic and somnolescent with selectivity for α1 containing GABAARs and direct clinical implications (Kapur and Macdonald, 1996, 1999; Hollrigel and Soltesz, 1997; Brooks-Kayal et al., 1998; Defazio and Hablitz, 1999; Perrais and Ropert, 1999; Nusser and Mody, 2002; Cohen et al., 2003; Lindquist and Birnir, 2006; Leppä et al., 2011).
Using whole-cell patch-clamp recordings from DGCs in vitro, we tested the hypothesis that tonic GABAAR signaling is spared in DGCs located ipsilateral to CCI injury (Ipsi-DGCs), since this sparing has previously been observed and parallels a loss of responsiveness to THIP (Boychuk et al., 2016; Butler et al., 2016; Becerra et al., 2021). We also hypothesized that zolpidem sensitivity in GABAAR signaling would be reduced after CCI, since this compound is considered a modifier of synaptic α1 containing GABAARs and phasic signaling to DGCs is corrupted after brain injury. Outcomes of these studies have implications for treatments to alter DGC excitability after brain injury and consequent posttraumatic epileptogenesis.
Six- to eight-week-old adult male CD-1 mice (Invigro-Harlan) were housed under a normal 14/10 h light/dark cycle and given food and water ad libitum. All animals were acclimated to the University of Kentucky vivarium for at least 1 week prior to experimentation. Male mice were tested in this study because cycling female hormones, e.g., estrogen and progesterone, robustly alter GABAAR signaling and require dedicated examination of ovariectomized (OVX) females and OVX-treated females given select hormone replacement and/or pharmacological modification of key biochemical steps in ovarian hormone pathways. This biology has been studied by our labs (Littlejohn and Boychuk, 2021) and others (Twyman and Macdonald, 1992; Fáncsik et al., 2000; Wohlfarth et al., 2002; Griffiths and Lovick, 2005; Maguire and Mody, 2007; Abramian et al., 2014; Carver et al., 2014). All procedures were approved by the University of Kentucky Animal Care and Use Committee (Assurance #A3336-01) and adhered to NIH guidelines for the care and use of laboratory animals.
Mice were administered a unilateral, focal contusion injury using CCI as previously reported (Scheff et al., 1997; Hunt et al., 2009, 2010, 2011, 2012). Briefly, animals were anesthetized using 2% isoflurane and placed in a stereotaxic frame. A midline incision was made to reveal the skull and a 5 mm craniotomy was performed lateral to the sagittal suture and centered between bregma and lambda. The skull cap was removed without damage to the underlying dura. A computer controlled, pneumatically driven impactor fitted with a 3 mm stainless-steel beveled tip (TBI-0310; Precision Systems and Instrumentation, Fairfax Station, VA, United States) was used to compress the cortex without breaking the dura mater. Impact parameters were set to 1.0 mm depth (hard stop), 3.5 m/s velocity, and 500 ms duration. For sham-injured controls, craniotomy was performed but no impact was administered to the brain. The incision was sutured and the animals were allowed to recover. All mice given CCI survived and remained otherwise healthy until experimentation. These parameters result in a well-characterized injury that predisposes mice to development of posttraumatic epilepsy by 8–12 weeks post-injury (Hunt et al., 2009, 2010, 2011, 2012).
Mice were anesthetized by isoflurane inhalation to effect (lack of tail pinch response) and decapitated while anesthetized. The brain was then rapidly removed and immediately immersed in ice-cold (0–4°C), oxygenated (95% O2–5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mM) 124 NaCl, 3 KCl, 26 NaHCO3, 1.4 NaH2PO4, 11 glucose, 1.3 CaCl2, 1.3 MgCl2; 1.0 kynurenic acid (KYN; Sigma-Aldrich, St. Louis, MO, United States); pH = 7.2–7.4, with an osmolality of 290–305 mOsmol/kg H2O. The brain was blocked, mounted on a sectioning stage, and 350 μm slices were cut in the coronal plane with a vibrating microtome (Vibratome Series 1000; Technical Products International, St. Louis, MO, United States) to ensure consistency of transverse slices from the dorsal one-third of the hippocampus. Each hippocampus was isolated from surrounding brain areas, making sure to completely remove entorhinal cortex, and slice order was maintained so that the location relative to the injury within each hippocampus was known. The slices were then transferred to a holding chamber where they were superfused with warmed (32–34°C) ACSF.
After an equilibration period of at least 1 h, slices were transferred to a recording chamber on an upright, fixed-stage microscope equipped with infrared, differential interference contrast optics (IR-DIC; Olympus BX51WI, Melville, NY, United States), where they were continually superfused with warmed (32–34°C) ACSF. The ACSF always contained the glutamate receptor antagonist KYN (1 mM) and was identical to that used in the dissection and recovery, except when drugs [e.g., bicuculline methiodide (BIC) or zolpidem] were added, as described. Whole-cell patch-clamp recordings were performed from visualized DGCs. Recording pipettes were pulled (P-87, Sutter Instruments; Novato, CA, United States) from borosilicate glass capillaries with 1.65 mm outer diameter and 0.45 mm wall thickness (King Precision Glass, Claremont, CA, United States). Open tip resistance was 2–5 MΩ, seal resistance was 1–5 GΩ, series resistance was ≤20 MΩ (mean = 8.89 ± 0.60 MΩ, n = 79), uncompensated. Recordings were discontinued and not analyzed if series resistance varied by more than 25% during the recording. Recording pipettes were filled with (in mM) 140 Cs-gluconate, 1 NaCl, 5 EGTA, 10 HEPES, 1 MgCl2, 1 CaCl2, 3 CsOH, 2 ATP; pH = 7.15–7.30. A minimum of 5 min following establishment of whole-cell configuration was used in order to allow equilibration of the intracellular and recording pipette contents. Neural activity was recorded using Axon Instruments Axopatch 200B and MultiClamp 700B patch-clamp amplifiers (Molecular Devices, Sunnyvale, CA, United States), acquired at 10–20 kHz and low-pass filtered at 5 kHz using Digidata 1320A and 1322A digitizers and pClamp 10.3 software (Molecular Devices). Synaptic currents were analyzed off-line on a PC-style computer with pCLAMP programs (Molecular Devices) or MiniAnalysis 6.0.3 (Synaptosoft, Decatur, GA, United States). A value of 3× the root mean squared noise level for a given recording was used as the detection limit for synaptic currents. Spontaneous inhibitory postsynaptic currents (sIPSCs) and tonic GABAA currents (ITonicGABA) were recorded in voltage-clamp mode with a voltage command of 0 mV. Added to the ACSF for specific experiments were the following: zolpidem (N,N,6-Trimethyl-2-(4-methylphenyl)imidazo[1,2-a]pyridine-3-acetamide; 1 μM; Tocris Bioscience, Minneapolis, MN, United States) and BIC (30 μM; Tocris Bioscience, Minneapolis, MN, United States). Zolpidem and BIC were dissolved in ACSF. We did not test DGC responsiveness to the compound flumazenil, and/or its possible blocking effect on any observed zolpidem responses, due to the temporal constrains of the long-term whole-cell patch clamp recordings used in this study as well as possible changes in flumazenil-responsiveness following epileptogenic insults (Leroy et al., 2004). Unless otherwise stated, neurons were recorded for 5 min under baseline conditions, 10 min during zolpidem application, and then for 5 min during application of BIC. Tonic GABAAR currents were measured as the mean difference in holding current (during baseline or zolpidem) relative to BIC. Charge transfer for tonic and synaptic GABAAR currents was sampled by integrating the amplitude of these electrical signals (i.e., converting to Amperes/second (i.e., picoCoulombs, pC) during a standardized 60 s bin (i.e., units of pC/60). Synaptic charge transfer was sampled by determining the charge transfer of the mean sIPSC during this 60 s bin (to estimate Amperes/second) and then multiplying this value by the total number of sIPSC events that occurred within it. Tonic charge transfer was sampled as the mean tonic GABAAR current amplitude that occurred during this standardized 60 s bin (to estimate Amperes/second) and then multiplied by the bin duration (60 s). The ratio of charge transfer was calculated as (tonic GABAAR/synaptic GABAAR) to create values that were positive integers.
Isolated dentate gyrus (DG) was harvested from each hemisphere and processed according to previously described methodology (Hideo et al., 2009; Boychuk et al., 2016). The tissue was visualized with a dissecting microscope (SMZ800; Nikon, Melville, NY, United States) and was submerged in ice-cold (0–4°C) ACSF containing kynurenic acid for all steps of dissection. The dorsal half of each isolated DG was selected for transcript analysis because the greatest proportion of cell signaling changes in the ipsilateral DG (e.g., mossy fiber sprouting and interneuron loss) are observed in this region after CCI (Hunt et al., 2009, 2010, 2011, 2012). Resulting isolated, dorsal halves of DG ipsilateral and contralateral to CCI or sham-injury were treated as individual samples. Each sample was immediately homogenized in ice-cold 500 μL of TRIzol (Sigma-Aldrich). Chloroform (100–250 μL) was added and tubes were vortexed for 15 s and then maintained at 4°C for 20 min and subsequently centrifuged at 12,000 rpm for 15 min at 4°C. The pellet was discarded and the RNA supernatant was transferred into fresh 1.5 mL centrifuge tubes, mixed with 500 μL of ice-cold propanol, incubated at room temperature for 10 min, and centrifuged at 12,000 rpm for 10 min at 4°C. Propanol was decanted and RNA was washed by re-suspension in 500 μL 75% ethanol followed by centrifugation at 7500–12,000 rpm for 10 min at 4°C. The wash step was repeated, the ethanol decanted, and RNA samples were air-dried for 10–20 min. RNA was then re-dissolved in 20 μL ddH20. Spectrophotometry (NanoDrop, Fisher Scientific, Wilmington, DE, United States) was used to determine mRNA concentration and purity.
Quantitative real-time polymerase chain reaction (qRT-PCR) was performed as previously described (Boychuk et al., 2016). Samples of mRNA (2 μg) were used to create cDNA using SuperScript II reverse transcriptase kit (Invitrogen, Carlsbad, CA, United States) using a Mastercycler (Eppendorf, Hauppauge, NY, United States). All qRT-PCR reactions were run in triplicate in 96-well optical grade plates using a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, United States). Each run consisted of 1 cycle of 50°C for 2 min, then 1 cycle of 95°C for 10 min, and 40 cycles of 95°C for 15 s, and 60°C for 1 min. Total volume for each run was 20 μL containing 10 μL of TaqMan 2× PCR Master Mix (Applied Biosystems), 1 μL of primer (Applied Biosystems) and a combination of cDNA and ddH2O that provided a cDNA concentration of 50 ng. All reactions used forward and reverse primer concentrations of 100 nM. Probe concentrations for all reactions were 50 nM. Primer and TaqMan probe sets were purchased from Applied Biosystems. The sequences for each were generated from the listed references within GenBank: α1: Mm00439046_m1; α2: Mm00433435_m1; α3: Mm01294271_m1; γ2: Mm00433489_m1; and β-actin: Mm00607939_s1. No-template and no-RT controls were run for each plate and results discarded if false positives were present. Delta cycle thresholds were used for statistical analysis (two-tail T-tests) of gene expression of GABAAR subunits and these data are presented as relative change using the 2–ΔΔCT method (Wood and Giroux, 2003).
Repeated Measures 2-Way Analysis of Variance (RM 2-Way ANOVA) was used to test differences for all electrophysiology data followed by protected post hoc Tukey’s Multiple Comparisons to test between group differences and protected post hoc Sidak’s Multiple Comparisons to test within-group differences pre/post zolpidem. The outcome of main effects determined whether post hoc testing was performed (Wei et al., 2012). Percent relative change was calculated using the formula: [C = 100 × (Post − Pre/Pre)].
Synaptic and tonic GABAAR-mediated currents were assessed in DGCs at 1–2 and 8–13 weeks following CCI injury (Figure 1). Recordings were made from DGCs ipsilateral to CCI injury (i.e., Ipsi-DGCs) and contralateral to injury (i.e., Contra-DGCs) or after sham injury (i.e., Sham-DGCs). Sample sizes for 1–2 weeks post-surgery were the following: Sham-DGCs, n = 13 cells (8 mice); Contra-DGCs, n = 13 cells (9 mice); Ipsi-DGCs, n = 14 cells (9 mice). Sample sizes for 8–13 weeks post-surgery were the following: Sham-DGCs, n = 10 cells (8 mice); Contra-DGCs, n = 15 cells (8 mice); Ipsi-DGCs, n = 15 cells (9 mice); the one exception is that sample size for tonic GABAAR current for Ipsi-DGCs was 14 (rather than 15) as one recording lacked sufficient duration of bicuculline application. A total of 1–3 cells were recorded per mouse and we assessed the potential that the recordings from any given mouse may have unduly biased the data (i.e., “nesting effect”) and found no such issues.
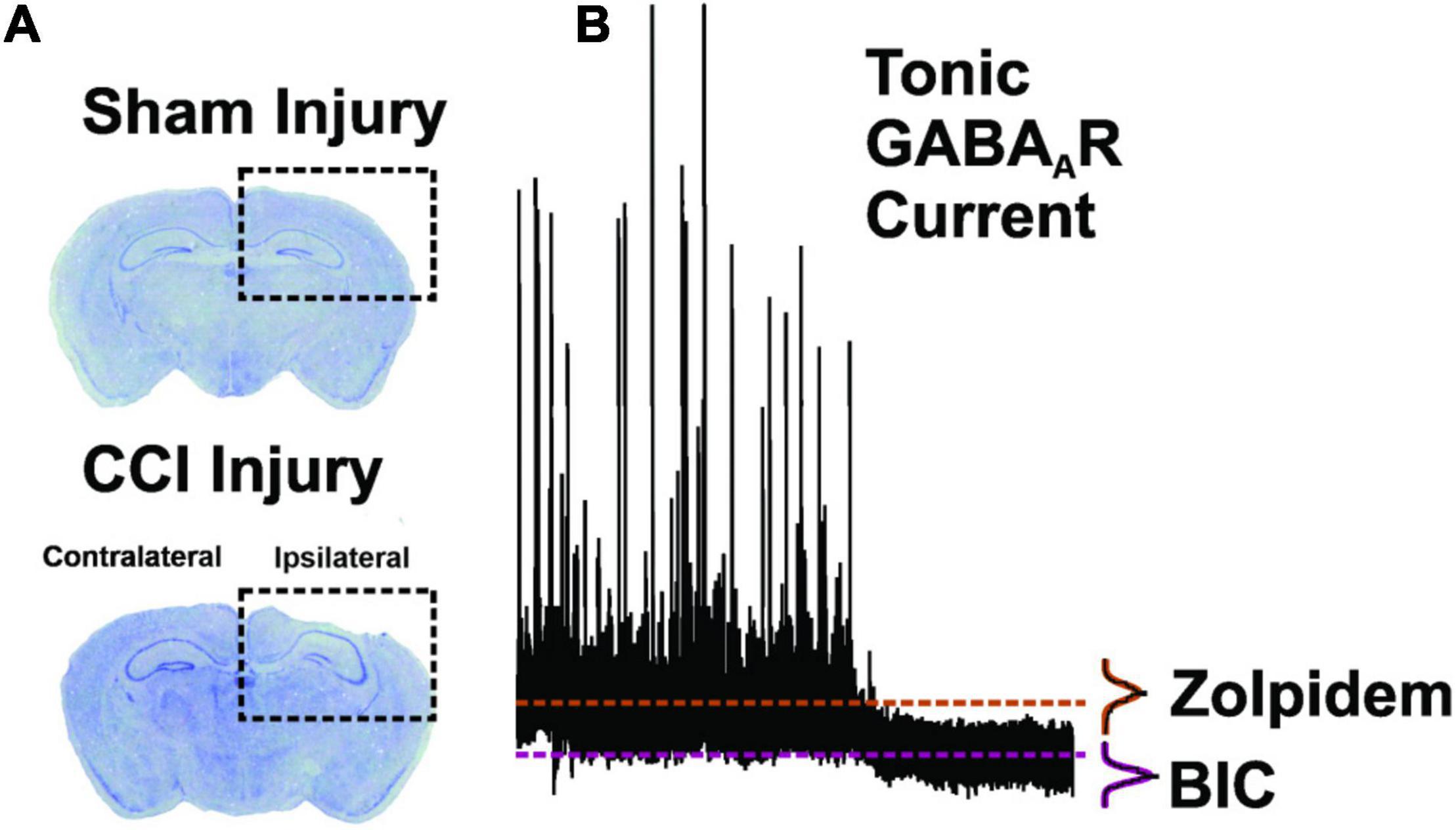
Figure 1. Methodology for brain injury and tonic GABAAR measurement. (A) Coronal brain sections of dorsal hippocampus stained with cresyl violet. In comparison to sham-injury (top panel), CCI-injury (bottom panel; 1.0 mm depth) resulted in cortical cavitation and distortion while sparing portions of the dentate gyrus (Hunt et al., 2009, 2010, 2012; Butler et al., 2015). (B) Representative neurophysiological recording from a DGC in order to depict measurement of the Tonic GABAAR at baseline and in the presence of zolpidem (1 μM) and the GABAAR antagonist, bicuculline methiodide (BIC; 30 μM). Mean current values during BIC application are subtracted from mean current values during zolpidem application to quantify the zolpidem-selective tonic GABAAR current (dashed lines). All points histograms, and subsequent Gaussian fits, are provided for each section of trace to demonstrate the population data from which mean values are derived.
ITonicGABA was evaluated in DGCs (Figure 1) because we have previously observed its sparing (and/or compensatory remodeling) in this cell type after CCI, findings that have recently been replicated (Boychuk et al., 2016; Butler et al., 2016; Becerra et al., 2021). This sparing of the baseline ITonicGABA in Ipsi-DGCs is surprising because it occurs coincident with robust alterations to phasic GABAAR (ISynapticGABA) signaling as attributed to loss of select groups of GABA-expressing cells within the hippocampus (Pavlov et al., 2011). Further, we have previously found that Ipsi-DGCs exhibit an evoked ITonicGABA that is less responsive to THIP, a GABAAR superagonist that predominantly targets GABAARs containing δ subunits along with α4 and α6 subunits (Saarelainen et al., 2008; Chandra et al., 2010; Boychuk et al., 2016; Butler et al., 2016; Becerra et al., 2021). Few, if any, reports exist for the effects of zolpidem on ITonicGABA and ISynapticGABA in DGCs after brain injury and the use of charge transfer provides helpful information regarding their relative contributions to GABAAR signaling (Bai et al., 2001).
For amplitude of baseline and zolpidem evoked ITonicGABA (Figures 2A,B and Table 1), a significant main effect of group was detected [2-Way RM ANOVA; F(2,37) = 14.37, p < 0.0001]. Baseline ITonicGABA amplitude of Ipsi-DGCs was comparable to Contra-DGCs (Tukey, p > 0.99) and Sham-DGCs (Tukey, p = 0.88). The zolpidem evoked ITonicGABA in Ipsi-DGCs (Figures 2A,B and Table 1), was significantly greater than in Contra-DGCs (Tukey, p < 0.0001), and Sham-DGCs (Tukey, p < 0.0001). Comparison of the amplitude of zolpidem evoked ITonicGABA between Sham- and Contra-DGCs revealed no significant differences before zolpidem (Tukey, p = 0.91) or after zolpidem (Tukey, p = 0.69). A significant main effect of zolpidem was also detected [2-Way RM ANOVA; F(1,37) = 78.99, p < 0.0001]. Zolpidem application evoked a change in the holding current in DGCs from all experimental groups thereby indicating a responsiveness of DGCs from all experimental groups to this compound. Amplitude of ITonicGABA was increased by zolpidem treatment in Ipsi-DGCs (211% change; Sidak, p < 0.0001), Contra-DGCs (72% change; Sidak, p = 0.010), and in Sham-DGCs (68% change; Sidak, p = 0.031). A significant interaction between main effects was also detected [2-Way RM ANOVA; F(2,37) = 14.64, p < 0.0001].

Figure 2. GABA receptor signaling in DGCs 1–2 weeks post-CCI before and after zolpidem application. Comparisons were made between DGCs from sham animals (i.e., Sham; n = 13 cells), DGCs contralateral to CCI injury (i.e., CCI-Contra; n = 13 cells), and DGCs ipsilateral to CCI injury (i.e., CCI-Ipsi; n = 14 cells). (A) Example traces showing the effect of zolpidem and bicuculline in DGCs recorded 1–2 weeks after CCI injury. (B) Baseline tonic GABAAR current is spared in CCI-Ipsi DGCs, whereas this current is profoundly augmented by zolpidem (85% increase vs. controls) in these cells after brain injury. (C–E) CCI-Ipsi DGCs also exhibit decreased sIPSC frequency and increased sIPSC decay time and rise time. Zolpidem augmented sIPSC decay time in all groups. (F) sIPSC amplitude was unaffected by zolpidem in any group at 1–2 weeks post-CCI injury. The symbol “*” denotes a significant within-group change between pre/post zolpidem. The symbol “^” denotes a significant change between the pre-zolpidem CCI-Ipsi group compared to both pre-zolpidem Sham- and CCI-Contra groups. The symbol “#” denotes a significant change between the post-zolpidem CCI-Ipsi group compared to both post-zolpidem Sham- and CCI-Contra groups. The symbol “+” denotes a significant difference between the CCI-Ipsi group compared to the CCI-Contra group (indicated as pre-zolpidem or post-zolpidem, as appropriate). Significance set as p ≤ 0.05; standard statistical codes were used to indicate level of significance (e.g., *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).

Table 1. ITonic amplitude for dentate granule cells (DGCs) from Sham controls, contralateral (Contra), or ipsilateral (Ipsi) to CCI injury.
Reduced sIPSC frequency in Ipsi-DGCs after CCI injury (Figure 2C and Table 2) has been previously reported (Hunt et al., 2011; Boychuk et al., 2016; Butler et al., 2016; Zhu et al., 2019; Becerra et al., 2021), presumably due to selective loss of hippocampal GABA-expressing neurons (Lowenstein et al., 1992; Zhu et al., 1997; Pavlov et al., 2011; Butler et al., 2016, 2017; Frankowski et al., 2019). Zolpidem application in a mouse model of epilepsy was less effective in augmenting GABA potency in isolated DGCs (Brooks-Kayal et al., 1998), but it is unclear if zolpidem application augments spontaneous inhibitory inputs onto DGCs in slices after brain injury. For sIPSC frequency, a significant main effect of group was detected [2-Way RM ANOVA; F(2,37) = 24.64, p < 0.0001]. Baseline sIPSC frequency (Figure 2C and Table 2) was significantly lower in Ipsi-DGCs in comparison to Contra-DGCs (Tukey, p < 0.0001) and Sham-DGCs (Tukey, p < 0.0001), similar to previous reports (Hunt et al., 2011; Boychuk et al., 2016; Butler et al., 2016; Becerra et al., 2021). A significant main effect of zolpidem was also detected [2-Way RM ANOVA; F(1,37) = 13.2, p = 0.0008]. In the presence of zolpidem, sIPSC frequency in Ipsi-DGCs remained reduced relative to the other groups of Contra-DGCs (Tukey, p < 0.0001 vs. Ipsi-DGCs), and Sham-DGCs (Tukey, p < 0.0001 vs. Ipsi-DGCs). No significant differences in sIPSC frequency were observed between Sham- and Contra-DGCs before (Tukey, p = 0.32) or after zolpidem (Tukey, p = 0.18). A significant main effect of and zolpidem was also detected [2-Way RM ANOVA; F(1,37) = 13.2, p = 0.0008]. However, there were no significant within-group differences in sIPSC frequency with zolpidem treatment. Instead, during post hoc testing, statistical trends (i.e., p < 0.10) were observed for changes in sIPSC frequency due to zolpidem treatment in some cases: Ipsi-DGCs (21% change; Sidak, p = 0.082), Contra-DGCs (9% change; Sidak, p = 0.059) and Sham-DGCs (7% change; Sidak, p = 0.32). The interaction between main effects was non-significant [2-Way RM ANOVA; F(2,37) = 0.1939, p = 0.82].
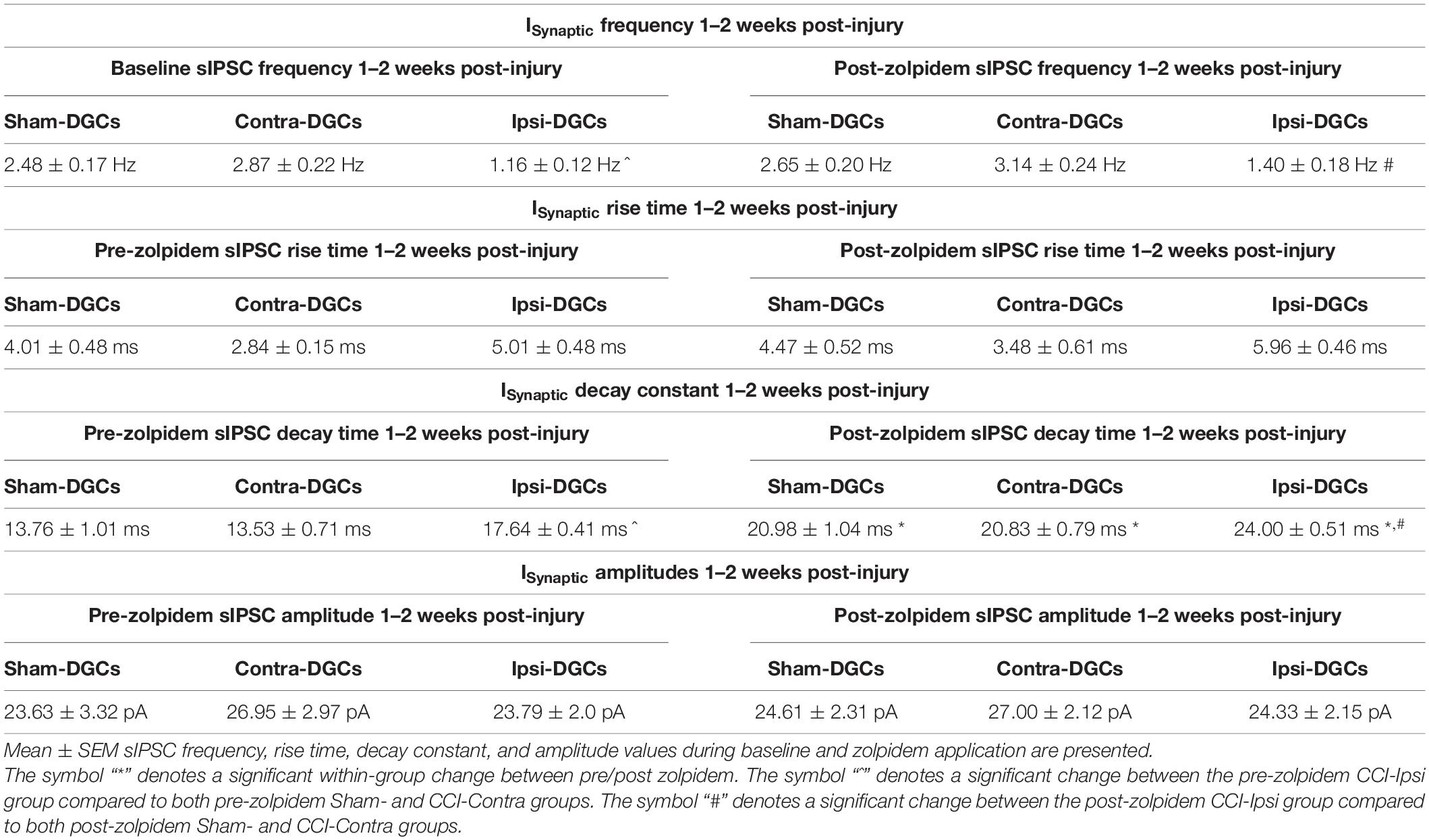
Table 2. ISynaptic frequency, kinetics, and amplitude properties 1–2 weeks post-injury.
Spontaneous inhibitory postsynaptic current decay time (Figure 2D and Table 2) was evaluated here because increased sIPSC decay times in Ipsi-DGCs have been reported previously (Hunt et al., 2011; Boychuk et al., 2016; Butler et al., 2016; Becerra et al., 2021), and zolpidem may increase sIPSC decay time by acting at the benzodiazepine binding site, predominantly through augmentation of α1 subunit-containing GABAARs (Hiu et al., 2016). For sIPSC decay time, a significant main effect of group was detected [2-Way RM ANOVA; F(2,37) = 8.14, p = 0.0012]. Baseline sIPSC decay time (Figure 2D and Table 2) was significantly greater in Ipsi-DGCs in comparison to Contra-DGCs (30% difference; Tukey, p = 0.0008) and Sham-DGCs (28% difference; Tukey, p = 0.0016). In the presence of zolpidem, sIPSC decay time was significantly greater in Ipsi-DGCs in comparison to Contra-DGCs (15% change; Tukey, p = 0.012) and Sham-DGCs (14% change; Tukey, p = 0.018). The comparison of sIPSC decay time between Sham- and Contra-DGCs revealed no significant differences before (Tukey, p = 0.98) or after zolpidem (Tukey, p = 0.99). A significant main effect of zolpidem was detected [2-Way RM ANOVA; F(1,37) = 589.9, p < 0.0001]. Importantly, sIPSC decay time was increased between baseline and zolpidem treatment in Ipsi-DGCs (36% change; Sidak, p < 0.0001), Contra-DGCs (54% change; Sidak, p < 0.0001) and Sham-DGCs (10% change; Sidak, p < 0.0001). However, The interaction between main effects was non-significant [2-Way RM ANOVA; F(2,37) = 1.14, p = 0.33].
Increased sIPSC rise times (Figure 2E and Table 2) in Ipsi-DGCs have been reported previously and is thought to be related to altered functional GABAAR’s expressed by Ipsi-DGCs after CCI injury (Brooks-Kayal et al., 1998, 2001; Boychuk et al., 2016; Butler et al., 2016; Becerra et al., 2021). For sIPSC rise time, a significant main effect of effect of group was detected [2-Way RM ANOVA; F(2,37) = 7.33, p = 0.0021]. Baseline sIPSC rise time (Figure 2E and Table 2) was significantly greater in Ipsi-DGCs compared to Contra-DGCs (76% greater; Tukey, p = 0.0045) whereas this effect was not detected relative to Sham-DGCs (24% greater; Tukey, p = 0.32). In the presence of zolpidem, sIPSC rise time remained significantly greater in Ipsi-DGCs in comparison to Contra-DGCs (71% greater; Tukey, p = 0.0010) whereas only a statistical trend (i.e., p < 0.10) was observed between Ipsi-DGCs relative to Sham-DGCs (33%; Tukey, p = 0.071). The comparison of sIPSC rise time between Sham- and Contra-DGCs revealed no significant differences before zolpidem (Tukey, p = 0.18) or after zolpidem (Tukey, p = 0.31). A significant main effect of zolpidem was also detected [2-Way RM ANOVA; F(1,37) = 9.51, p = 0.0038]. Interestingly, sIPSC rise time was only increased between baseline and zolpidem treatment in Ipsi-DGCs (19% change; Sidak, p = 0.041) whereas there was no significant effect in Contra-DGCs (23% change; Sidak, p = 0.27) and Sham-DGCs (10% change; Sidak, p = 0.62). The interaction between main effects was non-significant [2-Way RM ANOVA; F(2,37) = 0.50, p = 0.61]. Overall, this suggests that zolpidem application in the slice circuit was only partially capable of augmenting the activation of synaptic GABAARs of DGCs.
For sIPSC amplitude (Figure 2F and Table 2), there were no significant main effects for group [2-Way RM ANOVA; F(2,37) = 0.48, p = 0.62], zolpidem [2-Way RM ANOVA; F(1,37) = 0.34, p = 0.56], or the interaction between main effects [2-Way RM ANOVA; F(2,37) = 0.090, p = 0.91]. sIPSC amplitudes at baseline, and in the presence of zolpidem, are provided in Table 2 and are similar to previous reports (Brooks-Kayal et al., 1998, 2001; Boychuk et al., 2016; Butler et al., 2016; Becerra et al., 2021). Interestingly, zolpidem failed to significantly increase sIPSC amplitude under these experimental conditions; similar findings have been noted in other cortical cell types (Hiu et al., 2016).
De novo development of excitatory synaptic inputs onto surviving GABAergic interneurons after CCI injury may be a compensatory mechanism for loss of synaptic inhibition onto Ipsi-DGCs (Hunt et al., 2011; Boychuk et al., 2016; Butler et al., 2016, 2017; Becerra et al., 2021), but we and others have also reported previously that ITonicGABA is unaltered several weeks after CCI injury (Boychuk et al., 2016; Butler et al., 2016, 2017; Becerra et al., 2021). For amplitude of baseline ITonicGABA (Figure 3A and Table 1), there was no significant main effect of group [2-Way RM ANOVA; F(2,36) = 2.06, p = 0.14]. A significant main effect of zolpidem was detected for amplitude of ITonicGABA [2-Way RM ANOVA; F(1,36) = 14.59, p = 0.0005]. The application of zolpidem evoked a change in the holding current (Figures 3A,B and Table 1) in Ipsi-DGCs (45% change; Sidak, p < 0.0001), whereas this effect was not detected in Contra-DGCs (15% change; Sidak, p = 0.82) and Sham-DGCs (28% change; Sidak, p = 0.61), demonstrating that zolpidem application augments tonic inhibition prevalently in Ipsi-DGCs following CCI injury. The interaction between main effects was significant [2-Way RM ANOVA; F(2,36) = 4.90, p = 0.013].
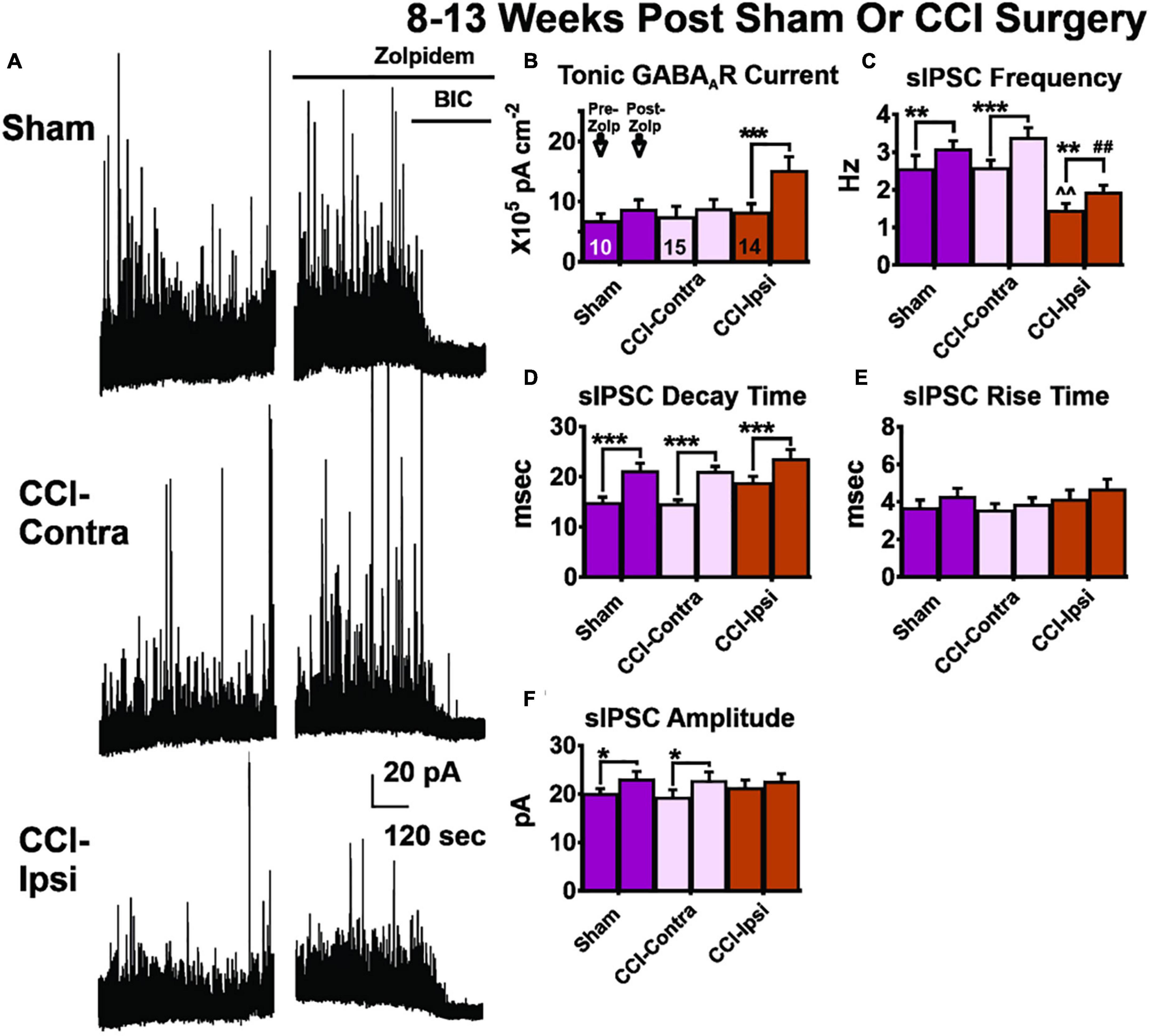
Figure 3. GABA receptor signaling in DGCs 8–13 weeks post-CCI before and after zolpidem application. Comparisons were made between DGCs from sham animals (i.e., Sham; n = 10 cells), DGCs contralateral to CCI injury (i.e., CCI-Contra; n = 14 cells for tonic GABAAR current; n = 15 cells for sIPSC parameters) and DGCs ipsilateral to CCI injury (i.e., CCI-Ipsi; n = 14 cells). (A) Example traces showing the effect of zolpidem and bicuculline in DGCs recorded 8–13 weeks after CCI injury. (B) Baseline tonic GABAAR current is spared in CCI-Ipsi DGCs whereas in the presence of zolpidem this current is 75% larger than in Sham-DGCs and 78% larger than in Contra-DGCs. (C–E) CCI-Ipsi DGCs also exhibit decreased sIPSC frequency and increased sIPSC decay time and rise time. Zolpidem augmented sIPSC decay time in all groups. (F) Zolpidem effect on sIPSC amplitude. The change in sIPSC amplitudes from baseline to zolpidem application was significantly increased in Sham-DGCs (15% change; Sidak, p = 0.035) and was significantly increased in Contra-DGCs (18% change; Sidak, p = 0.035) whereas no significant change was detected in Ipsi-DGCs (6% change; Sidak, p = 0.40). The symbol “*” denotes a significant within-group change between pre/post zolpidem. The symbol “^” denotes a significant change between the pre-zolpidem CCI-Ipsi group compared to both pre-zolpidem Sham and CCI-Contra groups. The symbol “#” denotes a significant change between the post-zolpidem CCI-Ipsi group compared to both post-zolpidem Sham and CCI-Contra groups. Significance set as p ≤ 0.05; standard statistical codes were used to indicate level of significance (e.g., *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
We previously reported that sIPSC frequency in Ipsi-DGCs remains reduced 8–13 weeks after injury, despite de novo development of excitatory synaptic inputs onto surviving GABAergic interneurons following CCI injury (Hunt et al., 2011; Boychuk et al., 2016; Butler et al., 2016, 2017). It is possible zolpidem could augment inhibitory input and we tested this possibility similarly to our measurements 1–2 weeks after CCI injury. For sIPSC frequency (Figure 3C and Table 3), a significant main effect of group was detected [2-Way RM ANOVA; F(2,37) = 9.72, p = 0.0004]. Baseline sIPSC frequency was significantly reduced in Ipsi-DGCs (Figure 3C and Table 3) compared to Contra-DGCs (43% change; Tukey, p = 0.0032) and Sham-DGCs (43% change; Tukey, p = 0.0078), similar to previous reports (Hunt et al., 2011; Boychuk et al., 2016; Butler et al., 2016; Zhu et al., 2019; Becerra et al., 2021). sIPSC frequency was still significantly reduced following zolpidem application in DGCs ipsilateral to CCI compared to DGCs contralateral to CCI (42% change; Tukey, p < 0.0001) and DGCs from Sham animals (37% change; Tukey, p = 0.0054). No differences in sIPSC frequency between Sham- and Contra-DGCs were observed before (Tukey, p > 0.99) or after zolpidem (Tukey, p = 0.74). A significant main effect of zolpidem was also detected [2-Way RM ANOVA; F(1,37) = 52.68, p < 0.0001]. Zolpidem treatment significantly increased sIPSC frequency in all three groups (Figure 3C and Table 3): Ipsi-DGCs (33% change; Sidak, p = 0.0029), Contra-DGCs (32% change; Sidak, p < 0.0001) and Sham-DGCs (20% change; Sidak, p = 0.0084) all exhibited significant increases in sIPSC frequency by zolpidem. The interaction between main effects was non-significant [2-Way RM ANOVA; F(2,37) = 1.75, p = 0.19]. These results demonstrate that zolpidem application equivalently impacts sIPSC frequency across experimental groups.
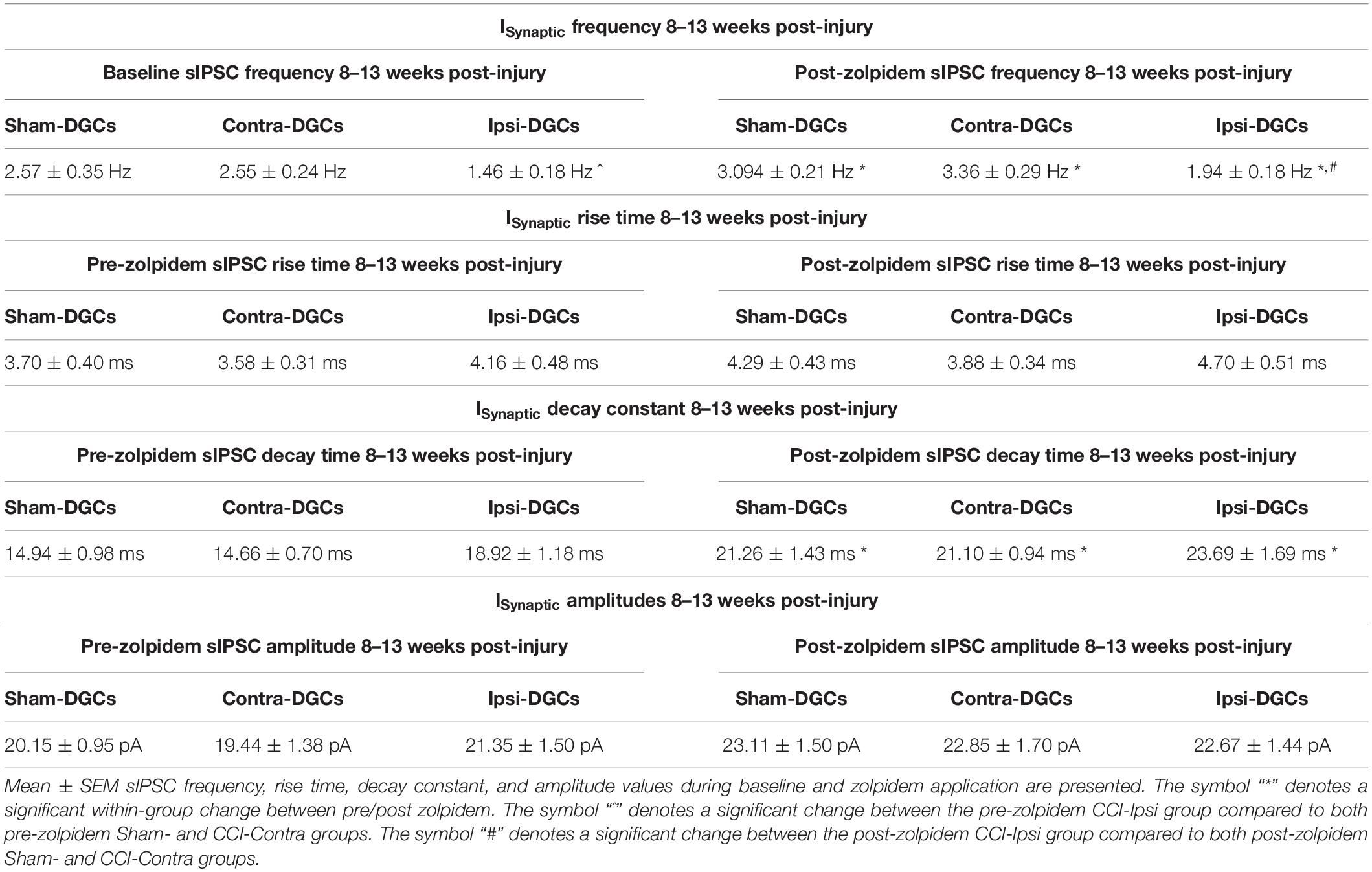
Table 3. ISynaptic frequency, kinetics, and amplitude properties 8–13 weeks post-injury.
For sIPSC decay time (Figure 3D and Table 3), only a statistical trend (i.e., p < 0.10) was observed for the overall effect of group [2-Way RM ANOVA; F(2,37) = 3.04, p = 0.060]. An overall effect of zolpidem was detected [2-Way RM ANOVA; F(1,37) = 143.8, p < 0.0001], wherein sIPSC decay time of Ipsi-DGCs (25% change; Sidak, p < 0.0001), Contra-DGCs (44% change; Sidak, p < 0.0001), and Sham-DGCs (42% change; Sidak, p < 0.0001) were significantly augmented by zolpidem in all groups. The interaction between main effects was non-significant [2-Way RM ANOVA; F(2,37) = 1.36, p = 0.27].
For sIPSC rise time (Figure 3E and Table 3), no overall effect was detected for group [2-Way RM ANOVA; F(2,37) = 0.97, p = 0.39]. An overall effect of zolpidem was detected [2-Way RM ANOVA; F(1,37) = 4.62, p = 0.038], however, no significant post hoc relationships were detected for zolpidem during multiple comparison testing: Ipsi-DGCs (13% change; Sidak, p = 0.36), Contra-DGCs (8% change; Sidak, p = 0.80), or Sham-DGCs animals (16% change; Sidak, p = 0.45). The interaction between main effects was non-significant [2-Way RM ANOVA; F(2,37) = 0.18, p = 0.83].
For sIPSC amplitude (Figure 3F and Table 3), no main effect was detected for group [2-Way RM ANOVA; F(2,37) = 0.11, p = 0.90]. An overall main effect was detected for zolpidem treatment on sIPSC amplitude [2-Way RM ANOVA; F(1,37) = 20.26, p < 0.001]. The change in sIPSC amplitudes, from baseline to zolpidem application (Figure 3E and Table 3), was significantly increased in Sham-DGCs (15% change; Sidak, p = 0.035) and Contra-DGCs (18% change; Sidak, p = 0.035) whereas no significant change was detected in Ipsi-DGCs (6% change; Sidak, p = 0.40). No significant interaction was detected between main effects [2-Way RM ANOVA; F(2,37) = 1.42, p = 0.25].
Ipsi-DGCs exhibit several differences from uninjured control conditions (Sham and contralateral to injury) such as kinetic properties of GABAAR sIPSCs as well as a profound reduction in sIPSC frequency and increase in zolpidem augmentation of their tonic GABAAR current (as assessed by % change). To better understand the overall effect of these relationships we calculated charge transfer of these signaling modalities for ISynapticGABA and ITonicGABA. Charge transfer is an “area under the curve”-type measurement of overall current over a matching duration of time that allows direct comparison of the contribution of tonic and synaptic GABAAR signaling and provides a separate way to analyze zolpidem augmentation of these signaling modalities since zolpidem affects many kinetic parameters simultaneously. Importantly, the ratio of charge transfer (ITonicGABA/ISynapticGABA) provides a means to test whether zolpidem augmentation predominantly occurs in tonic or phasic GABAAR signaling, as data here indicate that zolpidem profoundly augments ITonicGABA in Ipsi-DGCs whereas zolpidem augmentation mainly impacts ISynapticGABA in DGCs from uninjured control conditions.
For tonic charge transfer at 1–2 weeks post-CCI (Figure 4A and Table 4), a main effect of group was detected [2-Way RM ANOVA; F(2,37) = 14.37, p < 0.0001]. At baseline conditions, Ipsi-DGCs exhibit tonic charge transfer that is not different than Contra-DGCs (Tukey, p > 0.99) or Sham-DGCs (Tukey, p = 0.88). Markedly, in the presence of zolpidem, Ipsi-DGCs exhibit tonic charge transfer that is significantly greater (84%) than Contra-DGCs (Tukey, p < 0.0001) and is significantly greater (108%) than Sham-DGCs (Tukey, p < 0.0001). There was no difference in tonic charge transfer between Sham- and Contra-DGCs before zolpidem (Tukey, p = 0.91) or after zolpidem (Tukey, p = 0.69). A main effect of zolpidem was detected [2-Way RM ANOVA; F(1,37) = 78.99, p < 0.0001]. Zolpidem-mediated ITonicGABA charge transfer was greater in Ipsi-DGCs (211%; Sidak, p < 0.0001), Contra-DGCs (72%; Sidak, p = 0.010), and Sham-DGCs (68%; Sidak, p = 0.031) than their respective baseline values. A significant interaction between main effects was detected [2-Way RM ANOVA; F(2,37) = 14.64, p < 0.0001]. These results demonstrate a significant influence of zolpidem on inhibitory tone mediated by ITonicGABA in Ipsi-DGCs at an early time following CCI injury.
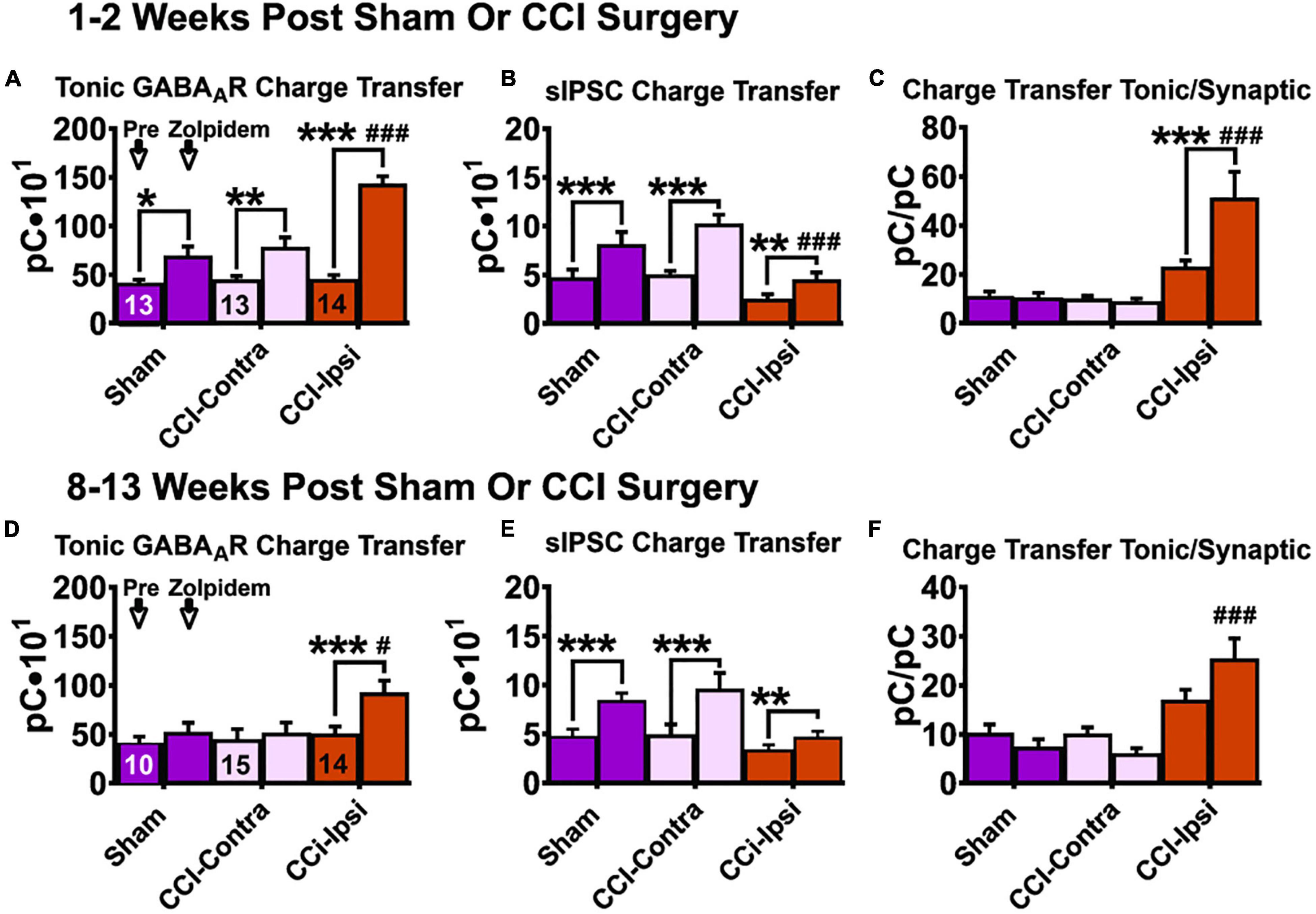
Figure 4. Charge transfer of tonic and synaptic GABAAR signaling in DGCs following CCI injury at baseline and after zolpidem application at 1–2 weeks and 8–13 weeks post-injury. Comparisons were made between DGCs from sham animals (i.e., Sham; n = 10 cells), DGCs contralateral to CCI injury (i.e., CCI-Contra; n = 15 cells) and DGCs ipsilateral to CCI injury (i.e., CCI-Ipsi; n = 14 cells). Tonic charge transfer is significantly augmented by zolpidem in DGCs from ipsilateral hemisphere of injured animals at 1–2 weeks (A) and 8–13 weeks (D) post-CCI. Synaptic charge transfer is significantly augmented by zolpidem in DGCs from all groups 1–2 weeks after injury (B) whereas this augmentation was less impactful in DGCs ipsilateral to CCI 8–13 weeks after injury (E). Ratio of baseline charge transfer (tonic/synaptic) is elevated in DGCs ipsilateral to CCI at 1–2 weeks (C) and at 8–13 weeks (F) after injury, whereas in the presence of zolpidem this ratio is larger at both 1–2 weeks post injury (382% larger than in Sham-DGCs and 499% larger than in Contra-DGCs) and 8–13 weeks post-injury (251% larger than in Sham-DGCs and 315% larger than in Contra-DGCs). The symbol “*” denotes a significant within-group change between pre/post zolpidem. The symbol “#” denotes a significant change between the post-zolpidem CCI-Ipsi group compared to both post-zolpidem Sham and CCI-Contra groups. Significance set as p ≤ 0.05; standard statistical codes were used to indicate level of significance (e.g., *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
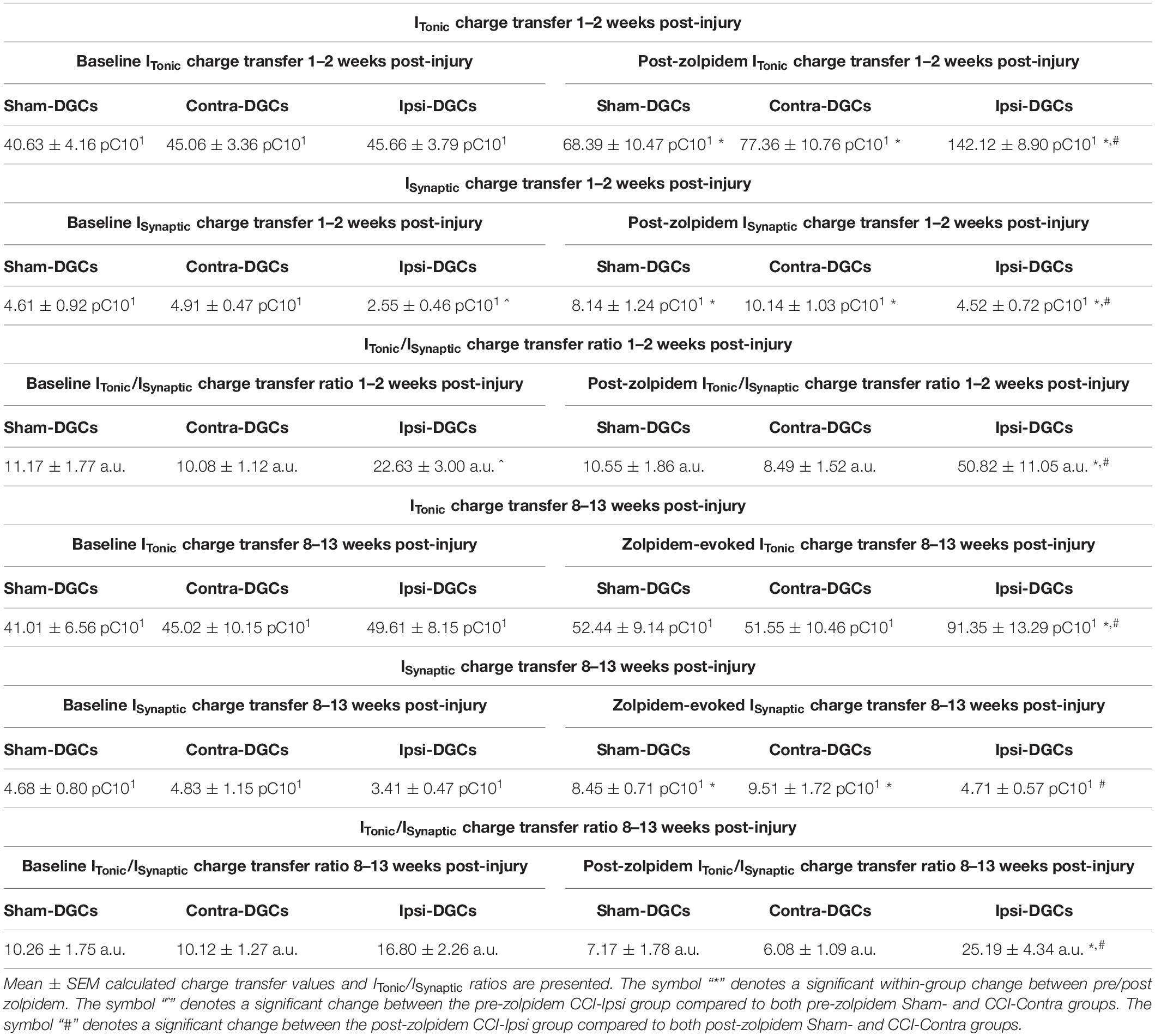
Table 4. ITonic, ISynaptic charge transfer, and ITonic/ISynaptic charge transfer ratio.
For synaptic charge transfer at 1–2 weeks post-CCI (Figure 4B and Table 4), a significant main effect of group was detected [2-Way RM ANOVA; F(2,37) = 6.73, p = 0.0032]. At baseline conditions, Ipsi-DGCs exhibit a synaptic charge transfer that is non-significantly reduced compared to Contra-DGCs (48% change; Tukey, p = 0.12) or Sham-DGCs (45% change; Tukey, p = 0.20). Markedly, in the presence of zolpidem, Ipsi-DGCs exhibit a synaptic charge transfer that is significantly decreased relative to Contra-DGCs (55% change; Tukey, p < 0.0001) and Sham-DGCs (44%; Tukey, p = 0.0088). The comparison of synaptic charge transfer between Sham- and Contra-DGCs revealed no significant differences before (Tukey, p = 0.97) or after zolpidem (Tukey, p = 0.23). A significant main effect of zolpidem was detected [2-Way RM ANOVA; F(1,37) = 129.8, p < 0.0001]. Zolpidem treatment significantly increased synaptic charge transfer in Ipsi-DGCs (78% change; Sidak, p = 0.0019), Contra-DGCs (106% change; Sidak, p < 0.0001), and Sham-DGCs (77% change; Sidak, p < 0.0001). The interaction between main effects was significant [2-Way RM ANOVA; F(2,37) = 9.041, p = 0.0006]. The application of zolpidem was therefore less effective in augmenting synaptic inhibition in Ipsi-DGCs, which is consistent with results from isolated DGCs taken from a rodent model of temporal lobe epilepsy (Brooks-Kayal et al., 1998). This is also despite the zolpidem-induced increase in sIPSC frequency observed in all groups (Figure 2).
For the ratio of charge transfer (ITonicGABA/ISynapticGABA) at 1–2 weeks post-CCI (Figure 4C and Table 4) a significant main effect of group was detected [2-Way RM ANOVA; F(2,37) = 14.97, p < 0.0001]. At baseline conditions, Ipsi-DGCs exhibit a charge transfer ratio that is non-significantly greater than Contra-DGCs (124% change; p = 0.19) and Sham-DGCs (103% change; Tukey, p = 0.25). In the presence of zolpidem, Ipsi-DGCs exhibit a ratio of charge transfer that is significantly greater than Contra-DGCs (499% change; Tukey, p < 0.0001) and Sham-DGCs (382% change; Tukey, p < 0.0001). The comparison of charge transfer ratio between Sham- and Contra-DGCs revealed no significant differences before (Tukey, p = 0.99) or after zolpidem (Tukey, p = 0.96). A main effect of zolpidem was detected [2-Way RM ANOVA; F(1,37) = 5.86, p = 0.021]. Zolpidem treatment significantly increased the charge transfer ratio in Ipsi-DGCs (125%; Sidak, p = 0.0001) whereas no significant change was detected in Contra-DGCs (−16% change; Sidak, p = 0.99) and Sham-DGCs (−6% change; Sidak, p > 0.99). No interaction of main effects was detected [2-Way RM ANOVA; F(2,37) = 7.65, p = 0.0017]. These results demonstrate that while tonic/synaptic GABAergic input is numerically imbalanced toward tonic GABAergic input early after CCI injury, the application of zolpidem amplifies this shift dramatically despite being a compound that is canonically characterized as selective for synaptic GABAergic input.
For the tonic charge transfer ratio at 8–13 weeks post-CCI (Figure 4D and Table 4), a significant main effect of group was detected [2-Way RM ANOVA; F(2,36) = 14.59, p = 0.0005]. At baseline conditions, Ipsi-DGCs exhibit tonic charge transfer that is not different than Contra-DGCs (Tukey, p = 0.94) and not different than Sham-DGCs (Tukey, p = 0.84); percent change not reported here since all groups are too similar. Markedly, in the presence of zolpidem, Ipsi-DGCs exhibit tonic charge transfer that is significantly greater than Contra-DGCs (77% change; Tukey, p = 0.015) and Sham-DGCs (74%; Tukey, p = 0.040). The comparison of tonic charge transfer between Sham- and Contra-DGCs revealed no significant differences before zolpidem (Tukey, p = 0.96) or after zolpidem (Tukey, p > 0.99). A significant main effect of zolpidem was also detected [2-Way RM ANOVA; F(1,36) = 2.064, p = 0.14]. Zolpidem treatment significantly increased tonic charge transfer in Ipsi-DGCs (84% change; Sidak, p < 0.0001), but non-significantly increased tonic charge transfer in Contra-DGCs (15% change; Sidak, p = 0.82) and Sham-DGCs (28% change; Sidak, p = 0.61). A significant interaction of main effects was detected [2-Way RM ANOVA; F(2,36) = 4.90, p = 0.013]. These results further emphasize the contribution of zolpidem to tonic inhibition of Ipsi-DGCs and that this effect is sustained after injury.
For synaptic charge transfer at 8–13 weeks post-CCI (Figure 4E and Table 4), there was no main effect of group [2-Way RM ANOVA; F(2,37) = 2.94, p = 0.066]. A significant main effect of zolpidem was detected [2-Way RM ANOVA; F(1,37) = 67.32, p < 0.0001]. Zolpidem treatment significantly increased synaptic charge transfer in Contra-DGCs (97% change; Sidak, p < 0.0001) and Sham-DGCs (81% change; Sidak, p < 0.0001), but was less impactful for Ipsi-DGCs (38%; Sidak, p = 0.0019). A significant interaction of the main effects was detected [2-Way RM ANOVA; F(2,37) = 7.40, p = 0.0020]. These results show that zolpidem application elevates synaptic input across groups similarly to earlier time points after CCI injury albeit with noticeable differences in the amount of augmentation of synaptic charge transfer in Ipsi-DGCs.
To better understand the balance of tonic and phasic GABAergic control in DGCs at later time points after CCI injury we compared the charge transfer ratio from these inhibitory sources (Figure 4F and Table 4). For charge transfer ratio at 8–13 weeks post-CCI, a significant main effect of group was detected [2-Way RM ANOVA; F(2,36) = 14.44, p < 0.0001]. At baseline, Ipsi-DGCs exhibit a ratio of charge transfer that is non-significantly greater than both Contra-DGCs (66% change; Tukey, p = 0.11) and Sham-DGCs (64% change; Tukey, p = 0.18). Markedly, in the presence of zolpidem, Ipsi-DGCs exhibit a charge transfer ratio that is significantly greater than Contra-DGCs (315% change; Tukey, p < 0.0001) and significantly greater than Sham-DGCs (251% change; Tukey, p < 0.0001). The comparison of charge transfer ratio between Sham and Contralateral DGCs revealed no significant differences before (Tukey, p = 0.99) or after zolpidem (Tukey, p = 0.95). The main effect of zolpidem was not significant [2-Way RM ANOVA; F(1,36) = 0.063, p = 0.80]. The interaction of main effects was significant [2-Way RM ANOVA; F(2,36) = 6.13, p = 0.0051]. Taken together with our observations 1–2 weeks after CCI injury, these results indicate that the relative balance of tonic/synaptic GABAergic input in Ipsi-DGCs remains shifted toward reliance on tonic inhibition at time points that correspond to numerous hippocampal circuit changes following injury. Additionally, application of zolpidem appears to further amplify this shift toward tonic inhibition, similar to earlier time points.
The shift in influence of tonic vs. synaptic inhibition following CCI injury could be due to alterations in subunit expression of GABAARs in Ipsi-DGCs. To test this possibility, we assessed mRNA expression in the dorsal half of the isolated dentate gyrus and analyzed it for α1, α2, α3, and γ2 GABAAR subunit mRNA expression using samples from Contra- and Ipsi-DG at the 8–13 week time-point. No significant changes in mRNA expression of GABAAR α1, α2, α3, and γ2 subunit expression 8–13 weeks after CCI injury were detected (Figures 5A–D, respectively), suggesting that shifts in the functional influence of tonic vs. synaptic GABAergic input are not likely due to underlying changes in composition of GABAARs in Ipsi-DGCs.
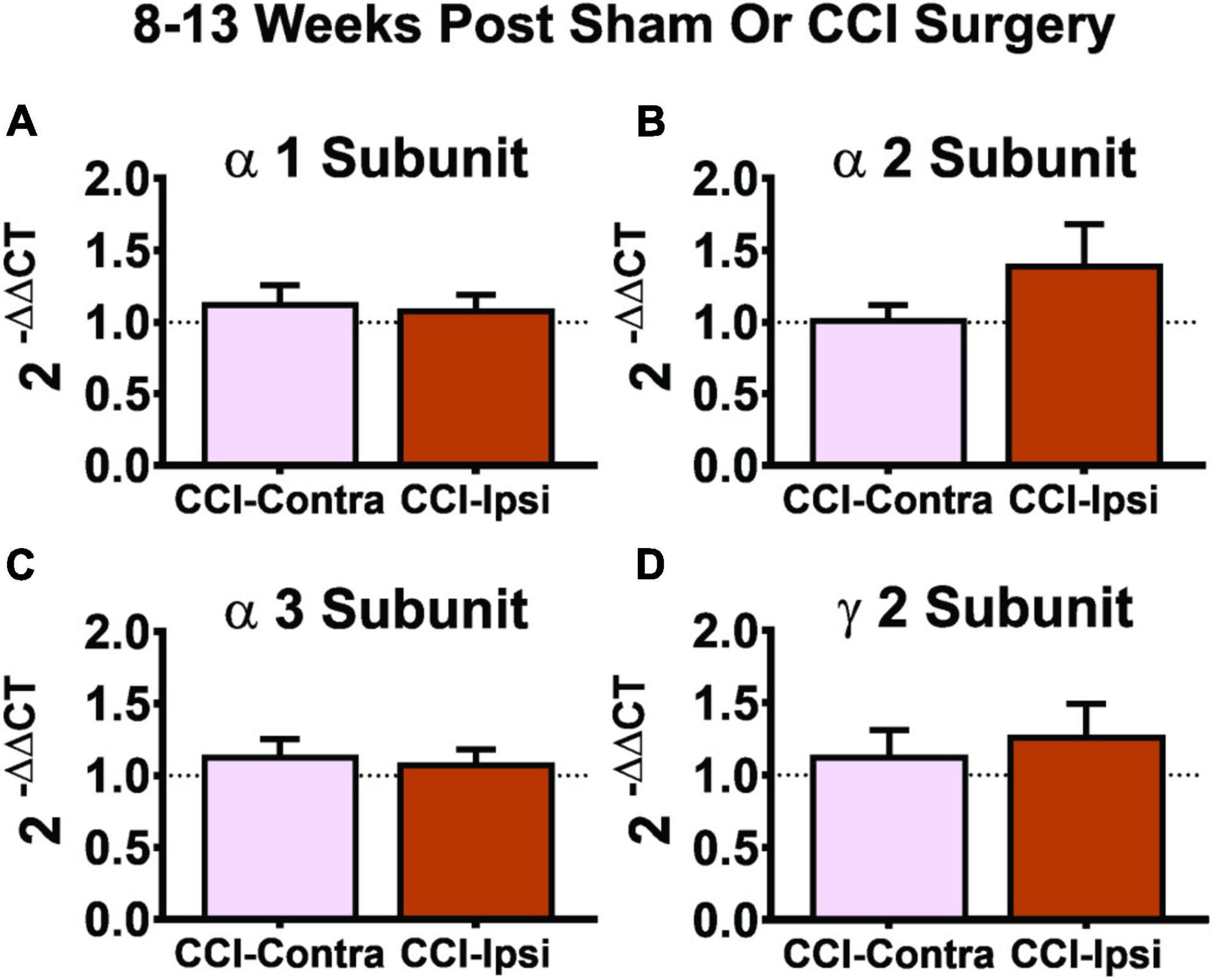
Figure 5. Gene expression of GABAAR subunits 8–13 weeks post-CCI. Comparisons of GABAAR subunits were made between the DG from the contralateral hemisphere (i.e., CCI-Contra) and ipsilateral hemisphere (i.e., CCI-Ipsi) relative to CCI injury. No effect was detected for the α1 subunit (A: CCI-Contra DG, n = 17; CCI-Ipsi DG, n = 19), α2 subunit (B: CCI-Contra DG, n = 8; CCI-Ipsi DG, n = 8), α3 subunit (C: CCI-Contra DG, n = 14; CCI-Ipsi DG, n = 16), or γ2 subunit (D: CCI-Contra DG, n = 9; CCI-Ipsi DG, n = 10). Delta cycle thresholds were used for statistical analysis (two-tailed T-tests) and data are presented as relative change using the 2–ΔΔCT method (no change indicated by dashed line). Significance set as p ≤ 0.05.
The present study provides several key findings. First, tonic GABAAR signaling is spared in DGCs from the ipsilateral hemisphere of CCI-injured animals, an effect that occurs early after injury and persists for at least several weeks (Hunt et al., 2011; Boychuk et al., 2016; Butler et al., 2016, 2017; Zhu et al., 2019). Second, DGCs exhibit a zolpidem-sensitive ITonicGABA that is markedly enhanced after CCI injury, which is surprising because zolpidem is traditionally considered to be a modifier of synaptic GABAAR signaling. Third, the proportion of charge transfer between baseline ITonicGABA and ISynapticGABA signaling (in DGCs) is non-significantly increased by CCI injury and profoundly enhanced by the combination of CCI injury and zolpidem. Fourth, GABAAR α1, α2, α3, and γ2 subunit gene expression is unchanged in Ipsi-DG relative to its contralateral counterpart at 8–13 weeks post-injury. These findings demonstrate the mechanistic disruption of GABAAR-dependent inhibition of DGCs at a cellular level following TBI. The results also demonstrate that zolpidem has surprising and powerful augmenting effects on ITonicGABA, and the proportion of charge transfer between tonic and synaptic GABAAR signaling in these cells, in a manner that is supra-additive with brain injury. These findings are consistent with a previous report that noted it is possible for GABAAR-modifying compounds such as midazolam and propofol to have differential responses of ITonicGABA and ISynapticGABA charge transfer in CA1 pyramidal neurons (Bai et al., 2001). Limitations of this study include how these cellular changes translate to post-injury hippocampus-dependent comorbidities such as posttraumatic epilepsy and hippocampal-dependent memory impairment and the potential for injury model-specific changes in GABAergic inhibition that we discuss in more detail below.
Altered GABAergic signaling is a common observation following brain insults, including TBI (Brooks-Kayal et al., 1998; Mtchedlishvili et al., 2010; Hunt et al., 2011; Pavlov et al., 2011; Raible et al., 2012, 2015; Boychuk et al., 2016; Butler et al., 2016, 2017; Frankowski et al., 2019; Zhu et al., 2019), and many treatment strategies attempt to rectify inhibitory control in disease through pharmaceutical targeting of the GABAAR’s, but with mixed results. Variation in how these different models of brain injury impact the components of inhibitory control (e.g., loss of GABAergic interneuron populations, changes in synaptic vs. extrasynaptic inhibition of target neurons, and excitation of surviving GABAergic interneurons) in the hippocampus is a potential source of these inconsistent outcomes. The CCI model has demonstrated reliable utility in understanding how severe TBI impacts hippocampal function, in particular because it closely reflects posttraumatic epilepsy outcomes in several key patient populations (Hunt et al., 2009, 2013), exhibits consistent loss of GABAergic interneuron populations (Lowenstein et al., 1992; Frankowski et al., 2019), and results in altered synaptic and tonic inhibition of DGCs in the hippocampus verified across multiple labs (Brooks-Kayal et al., 1998; Mtchedlishvili et al., 2010; Hunt et al., 2011; Pavlov et al., 2011; Raible et al., 2012, 2015; Boychuk et al., 2016; Butler et al., 2016, 2017; Frankowski et al., 2019; Zhu et al., 2019).
Inhibitory tone of DGCs in the hippocampus is directed by synaptic (phasic) and extrasynaptic (tonic) inhibition mediated by GABAARs (Chang et al., 1996; Mody, 2001; Brown et al., 2002; Stell et al., 2003; Wei et al., 2003; Semyanov et al., 2004; Farrant and Nusser, 2005; Mtchedlishvili and Kapur, 2006; Glykys et al., 2008). The mechanism(s) that alter GABAAR expression and function may be different depending on the injury model used, however, the work using CCI injury to model severe TBI has consistently demonstrated altered GABAAR function in DGCs ipsilateral to injury across multiple labs, highlighting the relevance of this dysfunction in TBI outcomes (Brooks-Kayal et al., 1998; Mtchedlishvili et al., 2010; Hunt et al., 2011; Pavlov et al., 2011; Raible et al., 2012, 2015; Boychuk et al., 2016; Butler et al., 2016, 2017; Frankowski et al., 2019; Zhu et al., 2019). One of the notable aspects of the present study, and our previous work describing δ subunit containing GABAAR’s, is that our observations of altered GABAAR function in DGCs ipsilateral to CCI injury were consistent at both early (1–2 weeks post-injury) and late (8–13 weeks post-injury) timepoints (Boychuk et al., 2016). This is particularly interesting given that many circuit alterations associated with posttraumatic epileptogenesis occur during the period between these timepoints, such as mossy fiber sprouting (Hunt et al., 2009, 2010; Butler et al., 2015) and de novo innervation and excitability changes of surviving GABAergic interneurons (Hunt et al., 2011; Butler et al., 2017), that parallel the change in inhibitory tone after CCI injury. These observations imply that while excitatory axon sprouting onto surviving interneurons may reflect a compensation for the loss of synaptic GABAergic input onto DGCs in the ipsilateral hippocampus, the preservation of tonic GABAAR signaling in DGCs also remains a critical mechanism to maintain hippocampal excitability and function.
In addition to molecular expression changes, multiple cell signaling mechanisms may affect GABAAR function in the DGCs ipsilateral to CCI injury (Butler et al., 2016; Becerra et al., 2021), and GABA transporters, chloride co-transporters and post-transcriptional mechanisms such as receptor trafficking and phosphorylation also contribute to ITonicGABA function (Van Den Pol et al., 1996; Mody, 2001; Stell et al., 2003; Mody and Pearce, 2004; Rivera et al., 2004; Farrant and Nusser, 2005; Glykys and Mody, 2007; Glykys et al., 2008; Clarkson et al., 2010). Further, it is unclear whether such signaling processes occur exclusively within the injured DGCs or in combination with the reactive glia and other cells within the local injured environment. GABA-uptake mechanisms by astrocytes may serve important roles in these processes (Bai et al., 2001; Nusser and Mody, 2002; Semyanov et al., 2003; Clarkson et al., 2010). Recent work demonstrating improved behavioral outcomes following CCI injury after eliminating reactive microglia (Henry et al., 2020), as well as work showing the ability of overactive cell signaling cascades in microglia (e.g., mechanistic target of rapamycin, mTOR) to drive neuronal injury (Erlich et al., 2007) suggest important roles for non-neuronal cells in the underlying mechanisms that alter GABAAR function after TBI.
Although zolpidem has a 10-fold greater selectivity for α1 subunit containing GABAAR’s, this drug also interacts with GABAAR’s containing α2, α3 and α5 subunits (Hiu et al., 2016). Hippocampal DGCs express α1βxγ2 and α4βxδ GABAAR subunit combinations; other combinations are possible, and this is an active area of research. We have previously found that DGCs do not differ between contralateral and ipsilateral hemispheres of CCI-injured animals in their response to L655,708, a compound that preferentially agonizes α5 subunit containing GABAARs (Boychuk et al., 2016). We and others have also demonstrated a markedly reduced response to THIP or THDOC, agonists that are selective for GABAARs containing the δ subunit in Ipsi-DGCs, after CCI relative to control DGCs (Boychuk et al., 2016; Butler et al., 2016; Becerra et al., 2021). Despite our observed functional changes in GABAAR signaling, gene expression analyses here and previously (Boychuk et al., 2016) were unable to detect any expression changes in GABAAR α1–5, δ, or γ2 subunits ipsilateral to CCI at 8–13 weeks after injury. Immunohistochemical testing suggests a reduction in GABAAR δ and GABABR β2 subunit immunoreactivity in DG about 2 weeks after CCI (Becerra et al., 2021), and western blot analyses detect a reduction in the α1 and γ2 GABAAR subunit protein expression 7 and 112 days after severe CCI (2.0 mm injury depth), whereas δ, β2 and β3 GABAAR subunits remained unchanged (Raible et al., 2015), and α1 GABAAR subunit mRNA expression is also reduced after severe CCI injury (Raible et al., 2015). Given that TBI results in a loss and/or disruption of synaptic GABAAR signaling, as indicated in our studies as a reduction in sIPSC frequency, but GABAAR mRNA and protein expression changes in this study and others have yielded few differences after CCI injury, an intriguing possibility is that post-synaptic populations of GABAARs (ISynapticGABA) can become orphaned and may thus be maintained after injury as extra-synaptic populations of GABAARs that could be zolpidem-sensitive and contribute to ITonicGABA. Mice with genetic deletion of the δ subunit exhibit a corresponding increase in γ2 subunit expression; thus a time-dependent inter-play between these subunits remains possible after CCI (Korpi et al., 2002). This line of inquiry requires further ultrastructural testing of GABAARs that mediate ISynapticGABA and ITonicGABA to more fully understand their location relative to the post-synaptic density (PSD), ITonicGABA’s dependence on GABA spill-over from the cleft, as well as time-course observation of PSD remodeling of GABAergic synapses during the evolution of the presynaptic injury caused by TBI. These findings suggest that subunit expression changes may develop in DGCs after severe CCI injury, but their contribution to the functional changes in GABAAR mediated responses reported here and elsewhere remains unclear.
The use of zolpidem to modulate GABAAR function highlights the importance of tonic inhibition of DGCs, especially in the hemisphere ipsilateral to injury. This is particularly interesting in light of recent work that implantation of progenitor GABAergic interneurons into the hippocampus following CCI injury is sufficient to restore synaptic inhibition and rescue behavioral impairments, including memory dysfunction, and reduce seizure susceptibility in these mice (Zhu et al., 2019). In particular, the influence of transplanted GABAergic interneurons on inhibitory synaptic input to DGCs suggests that these new GABAergic interneurons may restore the balance of synaptic and tonic inhibitory tone of the ipsilateral DGCs in a manner that is sufficient to correct the detrimental behavioral outcomes in these injured mice. It is unclear if these implanted interneurons integrate into the hippocampal circuitry similarly to those that migrate and integrate during neonatal development or if they receive de novo synaptic inputs similar to surviving GABAergic interneurons, as has been reported in this injury model and another model of epilepsy (Halabisky et al., 2010; Hunt et al., 2011; Butler et al., 2017). Regardless, a consistent theme in this study, and the literature for CCI, is the imbalance of inhibitory tone that develops in DGCs ipsilateral to CCI injury, as evidenced by an elevated reliance on tonic GABAAR-mediated inhibition. This tonic/synaptic GABAAR imbalance is observed early after injury, persists into timepoints when further axon reorganization and behavioral co-morbidities such as seizures develop in this model (Hunt et al., 2009, 2011, 2012, 2013; Halabisky et al., 2010; Butler et al., 2017). De novo excitatory synapse formation onto surviving GABAergic interneurons and other endogenous compensation mechanisms that develop during this time appear insufficient to modify this imbalance in inhibitory tone after injury. Further work is needed to understand if there is a way to enhance particular endogenous mechanisms to adjust this inhibitory balance or if exogenous methods, such as interneuron transplant, are the most effective strategies for improving the outcomes following TBI. The precise mechanism(s) by which DGC tonic GABAAR inhibition is spared and/or maintained after CCI is also critical to understand how to treat this imbalance and possibly learn how to better compensate for the loss of synaptic GABAAR signaling, which remains susceptible to damage after brain injury.
This study highlights that GABAergic inhibition of DGCs in the ipsilateral hemisphere after CCI injury is biased toward tonic inhibition vs. synaptic inhibition (per the respective charge transfers of these modalities at both 1–2 and 8–13 weeks post-injury). The comprehensive view of cellular inhibition in this study provides several key insights into altered inhibitory control after TBI, but there remain limitations in our understanding of the underpinning mechanisms and downstream consequences of these cellular changes. The observation that application of zolpidem, a drug believed to preferentially target GABAAR’s associated with synaptic inhibition, further shifts this bias toward tonic inhibition raises some intriguing perspectives on our views of synaptic reorganization after brain injury, particularly as it highlights a potential influence of this drug in extrasynaptic transmission. Two diverging views regarding the role of more volume-based transmission include concepts that are either neuronal- or glial-centric. Neuronal-centric processes that have been postulated for these phenomena include changes in GABAAR subunits: reorganization, activity, trafficking and/or their localization, in a manner that may depend on phosphorylation of GABAAR subunits as well as changes in GABAB receptor function. Glial-centric views may involve potential changes in synapse maintenance (Parkhurst et al., 2013; Rice et al., 2017), altered GABA transporter function (Yi and Hazell, 2006; Clarkson et al., 2010), and the need to reset “reactive” gene expression in glial cells after brain injury (Henry et al., 2020). Together, these diverging views demonstrate the great need to better understand how inhibitory synapses form, are maintained, and functionally contribute to phasic and tonic inhibition of target neurons in an integrative fashion involving many cell types and cell signals. Advancement in these research areas remains key to targeted intervention of disrupted excitation:inhibition balance found in a number of neurological diseases including posttraumatic epilepsy.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the University of Kentucky Animal Care and Use Committee.
All authors contributed to the conception and design of the work. JB, CB, KS, and MH performed the acquisition and analysis. All authors contributed to interpretation of the data, drafting and revision of the manuscript, and approved the final version to be published.
This work was supported by the Department of Defense (USAMRMC) grant W81XWH-11-1-0502 (BS), NINDS grant R01 NS092552 (BS), NINDS grant R01 NS114651 (JB), and a Fellowship from the American Epilepsy Society and Lennox and Lombroso Trust for Epilepsy Research and Training (JB). Funding sources had no involvement in study design, collection, analysis, and interpretation of data, writing of the report, or in the decision to submit the article for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
GABA, gamma-aminobutyric acid; ITonicGABA, tonic GABA current; GABAAR, type A GABA receptor; CCI, controlled cortical impact; DG, dentate gyrus; TBI, traumatic brain injury; DGC, dentate granule cell; THIP, 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-3-ol hydrochloride; L655,708, 11,12,13,13a-tetrahydro-7-methoxy-9-oxo-9H-imidazo[1,5-a]pyrrolo[2,1-c][1,4]benzodiazepine-1-carboxylic acid, ethyl ester; qRT-PCR, quantitative real-time polymerase chain reaction; ACSF, artificial cerebrospinal fluid; sIPSCs, spontaneous inhibitory postsynaptic currents.
Abramian, A. M., Comenencia-Ortiz, E., Modgil, A., Vien, T. N., Nakamura, Y., Moore, Y. E., et al. (2014). Neurosteroids promote phosphorylation and membrane insertion of extrasynaptic GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 111, 7132–7137. doi: 10.1073/pnas.1403285111
Bai, D., Zhu, G., Pennefather, P., Jackson, M. F., Macdonald, J. F., and Orser, B. A. (2001). Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by γ-aminobutyric acidA receptors in hippocampal neurons. Mol. Pharmacol. 59, 814–824. doi: 10.1124/mol.59.4.814
Barnard, E., Skolnick, P., Olsen, R., Mohler, H., Sieghart, W., Biggio, G., et al. (1998). International Union of Pharmacology. XV. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol. Rev. 50, 291–314.
Becerra, A. P., Logsdon, A. F., Banks, W. A., and Ransom, C. B. (2021). Traumatic brain injury broadly affects GABAergic signaling in dentate gyrus granule cells. eNeuro 8:ENEURO.0055-20.2021. doi: 10.1523/ENEURO.0055-20.2021
Boychuk, J. A., Butler, C. R., Halmos, K. C., and Smith, B. N. (2016). Enduring changes in tonic GABAA receptor signaling in dentate granule cells after controlled cortical impact brain injury in mice. Exp. Neurol. 277, 178–189. doi: 10.1016/j.expneurol.2016.01.005
Brooks-Kayal, A. R., Shumate, M. D., Jin, H., Rikhter, T. Y., and Coulter, D. A. (1998). Selective changes in single cell GABAA receptor subunit expression and function in temporal lobe epilepsy. Nat. Med. 4, 1166–1172. doi: 10.1038/2661
Brooks-Kayal, A. R., Shumate, M. D., Jin, H., Rikhter, T. Y., Kelly, M. E., and Coulter, D. A. (2001). γ−Aminobutyric acidA receptor subunit expression predicts functional changes in hippocampal dentate granule cells during postnatal development. J. Neurochem. 77, 1266–1278. doi: 10.1046/j.1471-4159.2001.00329.x
Brown, N., Kerby, J., Bonnert, T., Whiting, P., and Wafford, K. (2002). Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br. J. Pharmacol. 136, 965–974. doi: 10.1038/sj.bjp.0704795
Butler, C. R., Boychuk, J. A., and Smith, B. N. (2015). Effects of rapamycin treatment on neurogenesis and synaptic reorganization in the dentate gyrus after controlled cortical impact injury in mice. Front. Syst. Neurosci. 9:163. doi: 10.3389/fnsys.2015.00163
Butler, C. R., Boychuk, J. A., and Smith, B. N. (2016). Differential effects of rapamycin treatment on tonic and phasic GABAergic inhibition in dentate granule cells after focal brain injury in mice. Exp. Neurol. 280, 30–40. doi: 10.1016/j.expneurol.2016.03.022
Butler, C. R., Boychuk, J. A., and Smith, B. N. (2017). Brain injury-induced synaptic reorganization in hilar inhibitory neurons is differentially suppressed by rapamycin. eNeuro 4:ENEURO.0134-17.2017. doi: 10.1523/ENEURO.0134-17.2017
Carver, C. M., Wu, X., Gangisetty, O., and Reddy, D. S. (2014). Perimenstrual-like hormonal regulation of extrasynaptic δ-containing GABAA receptors mediating tonic inhibition and neurosteroid sensitivity. J. Neurosci. 34, 14181–14197. doi: 10.1523/JNEUROSCI.0596-14.2014
Chandra, D., Halonen, L. M., Linden, A.-M., Procaccini, C., Hellsten, K., Homanics, G. E., et al. (2010). Prototypic GABAA receptor agonist muscimol acts preferentially through forebrain high-affinity binding sites. Neuropsychopharmacology 35, 999–1007. doi: 10.1038/npp.2009.203
Chang, Y., Wang, R., Barot, S., and Weiss, D. S. (1996). Stoichiometry of a recombinant GABAA receptor. J. Neurosci. 16, 5415–5424.
Clarkson, A. N., Huang, B. S., Macisaac, S. E., Mody, I., and Carmichael, S. T. (2010). Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 468, 305–309. doi: 10.1038/nature09511
Cohen, A. S., Lin, D. D., Quirk, G. L., and Coulter, D. A. (2003). Dentate granule cell GABAA receptors in epileptic hippocampus: enhanced synaptic efficacy and altered pharmacology. Eur. J. Neurosci. 17, 1607–1616. doi: 10.1046/j.1460-9568.2003.02597.x
Cossart, R., Dinocourt, C., Hirsch, J. C., Merchan-Perez, A., De Felipe, J., Ben-Ari, Y., et al. (2001). Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat. Neurosci. 4, 52–62. doi: 10.1038/82900
Davies, P. A., Hanna, M. C., Hales, T. G., and Kirkness, E. F. (1997). Insensitivity to anaesthetic agents conferred by a class of GABAA receptor subunit. Nature 385, 820–823. doi: 10.1038/385820a0
Defazio, R. A., and Hablitz, J. J. (1999). Reduction of zolpidem sensitivity in a freeze lesion model of neocortical dysgenesis. J. Neurophysiol. 81, 404–407. doi: 10.1152/jn.1999.81.1.404
Draguhn, A., Verdorn, T. A., Ewert, M., Seeburg, P. H., and Sakmann, B. (1990). Functional and molecular distinction between recombinant rat GABAA receptor subtypes by Zn 2+. Neuron 5, 781–788. doi: 10.1016/0896-6273(90)90337-f
Drexel, M., Puhakka, N., Kirchmair, E., Hörtnagl, H., Pitkänen, A., and Sperk, G. (2015). Expression of GABA receptor subunits in the hippocampus and thalamus after experimental traumatic brain injury. Neuropharmacology 88, 122–133. doi: 10.1016/j.neuropharm.2014.08.023
Erlich, S., Alexandrovich, A., Shohami, E., and Pinkas-Kramarski, R. (2007). Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol. Disease 26, 86–93. doi: 10.1016/j.nbd.2006.12.003
Fáncsik, A., Linn, D. M., and Tasker, J. G. (2000). Neurosteroid modulation of GABA IPSCs is phosphorylation dependent. J. Neurosci. 20, 3067–3075. doi: 10.1523/JNEUROSCI.20-09-03067.2000
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci. 6, 215–229. doi: 10.1038/nrn1625
Frankowski, J. C., Kim, Y. J., and Hunt, R. F. (2019). Selective vulnerability of hippocampal interneurons to graded traumatic brain injury. Neurobiol. Disease 129, 208–216. doi: 10.1016/j.nbd.2018.07.022
Gibbs, J. W., Shumate, M. D., and Coulter, D. A. (1997). Differential epilepsy-associated alterations in postsynaptic GABAA receptor function in dentate granule and CA1 neurons. J. Neurophysiol. 77, 1924–1938. doi: 10.1152/jn.1997.77.4.1924
Glykys, J., Mann, E. O., and Mody, I. (2008). Which GABAA receptor subunits are necessary for tonic inhibition in the hippocampus? J. Neurosci. 28, 1421–1426. doi: 10.1523/jneurosci.4751-07.2008
Glykys, J., and Mody, I. (2007). Activation of GABAA receptors: views from outside the synaptic cleft. Neuron 56, 763–770. doi: 10.1016/j.neuron.2007.11.002
Griffiths, J., and Lovick, T. (2005). GABAergic neurones in the rat periaqueductal grey matter express α4, β1 and δ GABAA receptor subunits: plasticity of expression during the estrous cycle. Neuroscience 136, 457–466. doi: 10.1016/j.neuroscience.2005.08.013
Gupta, A., Elgammal, F. S., Proddutur, A., Shah, S., and Santhakumar, V. (2012). Decrease in tonic inhibition contributes to increase in dentate semilunar granule cell excitability after brain injury. J. Neurosci. 32, 2523–2537. doi: 10.1523/JNEUROSCI.4141-11.2012
Halabisky, B., Parada, I., Buckmaster, P., and Prince, D. (2010). Excitatory input onto hilar somatostatin interneurons is increased in a chronic model of epilepsy. J. Neurophysiol. 104, 2214–2223. doi: 10.1152/jn.00147.2010
Henry, R. J., Ritzel, R. M., Barrett, J. P., Doran, S. J., Jiao, Y., Leach, J. B., et al. (2020). Microglial depletion with CSF1R inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J. Neurosci. 40, 2960–2974. doi: 10.1523/JNEUROSCI.2402-19.2020
Hideo, H., Keiko, T., Nobuyuki, Y., and Tsuyoshi, M. (2009). Dissection of hippocampal dentate gyrus from adult mouse. J. Vis. Exp. 33:e1543. doi: 10.3791/1543
Hiu, T., Farzampour, Z., Paz, J. T., Wang, E. H. J., Badgely, C., Olson, A., et al. (2016). Enhanced phasic GABA inhibition during the repair phase of stroke: a novel therapeutic target. Brain 139, 468–480. doi: 10.1093/brain/awv360
Hollrigel, G. S., and Soltesz, I. (1997). Slow kinetics of miniature IPSCs during early postnatal development in granule cells of the dentate gyrus. J. Neurosci. 17, 5119–5128. doi: 10.1523/JNEUROSCI.17-13-05119.1997
Hunt, R. F., Boychuk, J. A., and Smith, B. N. (2013). Neural circuit mechanisms of post-traumatic epilepsy. Front. Cell Neurosci. 7:89. doi: 10.3389/fncel.2013.00089
Hunt, R. F., Haselhorst, L. A., Schoch, K. M., Bach, E. C., Rios-Pilier, J., Scheff, S. W., et al. (2012). Posttraumatic mossy fiber sprouting is related to the degree of cortical damage in three mouse strains. Epilepsy Res. 99, 167–170. doi: 10.1016/j.eplepsyres.2011.10.011
Hunt, R. F., Scheff, S. W., and Smith, B. N. (2009). Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp. Neurol. 215, 243–252. doi: 10.1016/j.expneurol.2008.10.005
Hunt, R. F., Scheff, S. W., and Smith, B. N. (2010). Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. J. Neurophysiol. 103, 1490–1500. doi: 10.1152/jn.00957.2009
Hunt, R. F., Scheff, S. W., and Smith, B. N. (2011). Synaptic reorganization of inhibitory hilar interneuron circuitry after traumatic brain injury in mice. J. Neurosci. 31, 6880–6890. doi: 10.1523/JNEUROSCI.0032-11.2011
Kapur, J., and Macdonald, R. L. (1996). Pharmacological properties of gamma-aminobutyric acidA receptors from acutely dissociated rat dentate granule cells. Mol. Pharmacol. 50, 458–466.
Kapur, J., and Macdonald, R. L. (1999). Postnatal development of hippocampal dentate granule cell Gamma-aminobutyric acidA receptor pharmacological properties. Mol. Pharmacol. 55, 444–452.
Kharlamov, E. A., Downey, K. L., Jukkola, P. I., Grayson, D. R., and Kelly, K. M. (2008). Expression of GABAA receptor α 1 subunit mRNA and protein in rat neocortex following photothrombotic infarction. Brain Res. 1210, 29–38. doi: 10.1016/j.brainres.2008.02.070
Korpi, E., Mihalek, R., Sinkkonen, S., Hauer, B., Hevers, W., Homanics, G., et al. (2002). Altered receptor subtypes in the forebrain of GABAA receptor δ subunit-deficient mice: recruitment of γ2 subunits. Neuroscience 109, 733–743. doi: 10.1016/s0306-4522(01)00527-9
Laurie, D., Wisden, W., and Seeburg, P. (1992). The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci. 12, 4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992
Leppä, E., Linden, A.-M., Rabe, H., Vekovischeva, O. Y., Wulff, P., Lüddens, H., et al. (2011). Actions of two GABAA receptor benzodiazepine-site ligands that are mediated via non-γ2-dependent modulation. Eur. J. Pharmacol. 666, 111–121. doi: 10.1016/j.ejphar.2011.05.011
Leroy, C., Poisbeau, P., Keller, A., and Nehlig, A. (2004). Pharmacological plasticity of GABAA receptors at dentate gyrus synapses in a rat model of temporal lobe epilepsy. J. Physiol. 557, 473–487. doi: 10.1113/jphysiol.2003.059246
Lindquist, C. E., and Birnir, B. (2006). Graded response to GABA by native extrasynaptic GABAA receptors. J. Neurochem. 97, 1349–1356. doi: 10.1111/j.1471-4159.2006.03811.x
Littlejohn, E. L., and Boychuk, C. R. (2021). Protein Kinase C-dependent effects of neurosteroids on synaptic GABAA receptor inhibition require the δ-Subunit. Front. Physiol. 12:742838. doi: 10.3389/fphys.2021.742838
Lowenstein, D. H., Thomas, M. J., Smith, D. H., and Mcintosh, T. K. (1992). Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 12, 4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992
Lüddens, H., and Wisden, W. (1991). Function and pharmacology of multiple GABAA receptor subunits. Trends Pharmacol. Sci. 12, 49–51. doi: 10.1016/0165-6147(91)90495-e
Macdonald, R. L., and Olsen, R. W. (1994). GABAA receptor channels. Annu. Rev. Neurosci. 17, 569–602.
Maguire, J., and Mody, I. (2007). Neurosteroid synthesis-mediated regulation of GABAA receptors: relevance to the ovarian cycle and stress. J. Neurosci. 27, 2155–2162. doi: 10.1523/JNEUROSCI.4945-06.2007
Mody, I. (2001). Distinguishing between GABAA receptors responsible for tonic and phasic conductances. Neurochem. Res. 26, 907–913. doi: 10.1023/a:1012376215967
Mody, I., and Pearce, R. A. (2004). Diversity of inhibitory neurotransmission through GABAA receptors. Trends Neurosci. 27, 569–575. doi: 10.1016/j.tins.2004.07.002
Mtchedlishvili, Z., and Kapur, J. (2006). High-affinity, slowly desensitizing GABAA receptors mediate tonic inhibition in hippocampal dentate granule cells. Mol. Pharmacol. 69, 564–575. doi: 10.1124/mol.105.016683
Mtchedlishvili, Z., Lepsveridze, E., Xu, H., Kharlamov, E. A., Lu, B., and Kelly, K. M. (2010). Increase of GABAA receptor-mediated tonic inhibition in dentate granule cells after traumatic brain injury. Neurobiol. Disease 38, 464–475. doi: 10.1016/j.nbd.2010.03.012
Nusser, Z., and Mody, I. (2002). Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J. Neurophysiol. 87, 2624–2628. doi: 10.1152/jn.2002.87.5.2624
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. III, Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Pavlov, I., Huusko, N., Drexel, M., Kirchmair, E., Sperk, G., Pitkänen, A., et al. (2011). Progressive loss of phasic, but not tonic, GABAA receptor-mediated inhibition in dentate granule cells in a model of post-traumatic epilepsy in rats. Neuroscience 194, 208–219. doi: 10.1016/j.neuroscience.2011.07.074
Peng, Z., Huang, C. S., Stell, B. M., Mody, I., and Houser, C. R. (2004). Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J. Neurosci. 24, 8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004
Perrais, D., and Ropert, N. (1999). Effect of zolpidem on miniature IPSCs and occupancy of postsynaptic GABAA receptors in central synapses. J. Neurosci. 19, 578–588. doi: 10.1523/JNEUROSCI.19-02-00578.1999
Raible, D. J., Frey, L. C., Cruz Del Angel, Y., Russek, S. J., and Brooks-Kayal, A. R. (2012). GABAA receptor regulation after experimental traumatic brain injury. J. Neurotrauma 29, 2548–2554.
Raible, D. J., Frey, L. C., Del Angel, Y. C., Carlsen, J., Hund, D., Russek, S. J., et al. (2015). JAK/STAT pathway regulation of GABAA receptor expression after differing severities of experimental TBI. Exp. Neurol. 271, 445–456. doi: 10.1016/j.expneurol.2015.07.001
Redecker, C., Wang, W., Fritschy, J.-M., and Witte, O. W. (2002). Widespread and long-lasting alterations in GABAA-receptor subtypes after focal cortical infarcts in rats: mediation by NMDA-dependent processes. J. Cereb. Blood Flow Metab. 22, 1463–1475. doi: 10.1097/01.WCB.0000034149.72481.BD
Rice, R. A., Pham, J., Lee, R. J., Najafi, A. R., West, B. L., and Green, K. N. (2017). Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia 65, 931–944. doi: 10.1002/glia.23135
Rivera, C., Voipio, J., Thomas-Crusells, J., Li, H., Emri, Z., Sipilä, S., et al. (2004). Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J. Neurosci. 24, 4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004
Saarelainen, K. S., Ranna, M., Rabe, H., Sinkkonen, S. T., Möykkynen, T., Uusi-Oukari, M., et al. (2008). Enhanced behavioral sensitivity to the competitive GABA agonist, gaboxadol, in transgenic mice over-expressing hippocampal extrasynaptic α6β GABAA receptors. J. Neurochem. 105, 338–350. doi: 10.1111/j.1471-4159.2007.05136.x
Scheff, S. W., Baldwin, S. A., Brown, R. W., and Kraemer, P. J. (1997). Morris water maze deficits in rats following traumatic brain injury: lateral controlled cortical impact. J. Neurotrauma 14, 615–627. doi: 10.1089/neu.1997.14.615
Semyanov, A., Walker, M. C., and Kullmann, D. M. (2003). GABA uptake regulates cortical excitability via cell type–specific tonic inhibition. Nat. Neurosci. 6, 484–490. doi: 10.1038/nn1043
Semyanov, A., Walker, M. C., Kullmann, D. M., and Silver, R. A. (2004). Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends Neurosci. 27, 262–269. doi: 10.1016/j.tins.2004.03.005
Stell, B. M., Brickley, S. G., Tang, C., Farrant, M., and Mody, I. (2003). Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δ subunit-containing GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 100, 14439–14444. doi: 10.1073/pnas.2435457100
Sun, C., Mtchedlishvili, Z., Erisir, A., and Kapur, J. (2007). Diminished neurosteroid sensitivity of synaptic inhibition and altered location of the α4 subunit of GABAA receptors in an animal model of epilepsy. J. Neurosci. 27, 12641–12650. doi: 10.1523/JNEUROSCI.4141-07.2007
Twyman, R., and Macdonald, R. (1992). Neurosteroid regulation of GABAA receptor single-channel kinetic properties of mouse spinal cord neurons in culture. J. Physiol. 456, 215–245. doi: 10.1113/jphysiol.1992.sp019334
Van Den Pol, A. N., Obrietan, K., and Chen, G. (1996). Excitatory actions of GABA after neuronal trauma. J. Neurosci. 16, 4283–4292. doi: 10.1523/JNEUROSCI.16-13-04283.1996
Verdoorn, T. A., Draguhn, A., Ymer, S., Seeburg, P. H., and Sakmann, B. (1990). Functional properties of recombinant rat GABAA receptors depend upon subunit composition. Neuron 4, 919–928. doi: 10.1016/0896-6273(90)90145-6
Wei, J., Carroll, R. J., Harden, K. K., and Wu, G. (2012). Comparisons of treatment means when factors do not interact in two-factorial studies. Amino Acids 42, 2031–2035. doi: 10.1007/s00726-011-0924-0
Wei, W., Zhang, N., Peng, Z., Houser, C. R., and Mody, I. (2003). Perisynaptic localization of delta subunit-containing GABA(A) receptors and their activation by GABA spillover in the mouse dentate gyrus. J. Neurosci. 23, 10650–10661. doi: 10.1523/JNEUROSCI.23-33-10650.2003
Whiting, P. J., Bonnert, T. P., Mckernan, R. M., Farrar, S., Bourdelles, B. L., Heavens, R. P., et al. (1999). Molecular and functional diversity of the expanding GABA-A receptor gene family. Ann. N. Y. Acad. Sci. 868, 645–653. doi: 10.1111/j.1749-6632.1999.tb11341.x
Winokur, R. S., Kubal, T., Liu, D., Davis, S. F., and Smith, B. N. (2004). Recurrent excitation in the dentate gyrus of a murine model of temporal lobe epilepsy. Epilepsy Res. 58, 93–105. doi: 10.1016/j.eplepsyres.2004.01.002
Wohlfarth, K. M., Bianchi, M. T., and Macdonald, R. L. (2002). Enhanced neurosteroid potentiation of ternary GABAA receptors containing the δ subunit. J. Neurosci. 22, 1541–1549. doi: 10.1523/JNEUROSCI.22-05-01541.2002
Wood, C. E., and Giroux, D. (2003). Central nervous system prostaglandin endoperoxide synthase-1 and− 2 responses to oestradiol and cerebral hypoperfusion in late-gestation fetal sheep. J. Physiol. 549, 573–581. doi: 10.1113/jphysiol.2002.038398
Yi, J.-H., and Hazell, A. S. (2006). Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 48, 394–403. doi: 10.1016/j.neuint.2005.12.001
Zhan, R.-Z., and Nadler, J. V. (2009). Enhanced tonic GABA current in normotopic and hilar ectopic dentate granule cells after pilocarpine-induced status epilepticus. J. Neurophysiol. 102, 670–681. doi: 10.1152/jn.00147.2009
Zhang, N., Wei, W., Mody, I., and Houser, C. R. (2007). Altered localization of GABA(A) receptor subunits on dentate granule cell dendrites influences tonic and phasic inhibition in a mouse model of epilepsy. J. Neurosci. 27, 7520–7531. doi: 10.1523/JNEUROSCI.1555-07.2007
Zhu, B., Eom, J., and Hunt, R. F. (2019). Transplanted interneurons improve memory precision after traumatic brain injury. Nat. Communi. 10:5156. doi: 10.1038/s41467-019-13170-w
Keywords: zolpidem, benzodiazepine, GABAA receptors, tonic GABA current, controlled cortical impact (CCI), traumatic brain injury, posttraumatic epilepsy
Citation: Boychuk JA, Butler CR, Smith KC, Halmos MB and Smith BN (2022) Zolpidem Profoundly Augments Spared Tonic GABAAR Signaling in Dentate Granule Cells Ipsilateral to Controlled Cortical Impact Brain Injury in Mice. Front. Syst. Neurosci. 16:867323. doi: 10.3389/fnsys.2022.867323
Received: 31 January 2022; Accepted: 05 May 2022;
Published: 26 May 2022.
Edited by:
Akiva Cohen, University of Pennsylvania, United StatesReviewed by:
Esa R. Korpi, University of Helsinki, FinlandCopyright © 2022 Boychuk, Butler, Smith, Halmos and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bret N. Smith, YnJldC5zbWl0aEBjb2xvc3RhdGUuZWR1
†ORCID: Jeffery A. Boychuk, orcid.org/0000-0002-7003-1553
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.