


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Syst. Neurosci. , 03 May 2012
Volume 6 - 2012 | https://doi.org/10.3389/fnsys.2012.00032
This article is part of the Research Topic Histamine in the Brain View all 11 articles
Multiple sclerosis (MS) is a chronic inflammatory, neurodegenerative disease of the CNS whose pathogenesis remains largely unknown, and available therapies are rarely successful in reversing neurological deficits or stopping disease progression. Ongoing studies on MS and the widely used murine model of experimental autoimmune encephalomyelitis (EAE) are focused on the many components of this complex and heterogeneous neurodegenerative disease in the hope of providing a mechanism-based characterization of MS that will afford successful strategies to limit and repair the neuronal damage. Recently, histamine has been postulated to have a key regulatory role in EAE and MS pathogenesis. Histamine is a mediator of inflammation and immune responses, exerting its many actions through four G protein-coupled receptors (H1,2,3,4R) that signal through distinct intracellular pathways and have different therapeutic potentials as they vary in expression, isoform distribution, signaling properties, and function. Immune cells involved in MS/EAE, including dendritic cells (DCs) and T lymphocytes, express H1R, H2R and H4R, and histamine may have varying and counteracting effects on a particular cell type, depending on the receptor subtypes being activated. Here, we review evidence of the complex and controversial role of histamine in the pathogenesis of MS and EAE and evaluate the therapeutic potential of histaminergic ligands in the treatment of autoimmune diseases.
Multiple sclerosis (MS) is the most common, non-traumatic cause of neurological disability among young adults in Western Europe and North America. The current hypothesis asserts that MS is triggered by environmental factors in individuals with complex genetic-risk profiles. As with other autoimmune diseases, MS shows moderate polygenic inheritability and may be caused or exacerbated by environmental exposure such as viral infections or vitamin D deficiency (Correale et al., 2009). It is characterized by clinical and genetic heterogeneity and with individuals with MHC class II complex genes being most susceptible (Ballerini et al., 2004; Gourraud et al., 2011). Out of the MHC locus a plethora of genes have been described as minor contributors to genetic risk, among which are those encoding for IL-2 receptors and IFN-γ (Blankenhorn et al., 2011).
MS is characterized by inflammation of the central nervous system (CNS) in which focal lymphocytic infiltrations lead to damage of myelin and axons associated with neurological dysfunction. Initially, the inflammatory response is transient and remyelination occurs, but this is not durable and over time widespread microglial activation ensues along with extensive and chronic neurodegeneration.
The observation that histamine may be implicated in MS dates back to the early 1980s when Tuomisto et al. (1983) showed that patients with remitting or progressive disease have histamine levels about 60% higher than controls. Another clinical study, though, did not show elevated concentrations of histamine and its metabolite methylhistamine in MS patients when compared, in this case, with individuals affected by other neurological diseases (Rozniecki et al., 1995). More recently, gene-microarray analysis has shown that histamine H1 receptor (H1R) expression is upregulated in MS lesions (Lock et al., 2002), and epidemiological studies suggest a protective effect of brain penetrating H1R antagonists (Alonso et al., 2006). Furthermore, in a small pilot study, a cohort of MS patients treated with an H1R antagonist showed signs of neurological amelioration (Logothetis et al., 2005). The H1R has long been associated with inflammatory responses and for decades, the antiallergic and antiinflammatory activities of H1R antagonists have been used in therapy. Possible explanations of the therapeutic potential of H1R antagonists come from preclinical results in experimental allergic encephalomyelitis (EAE) in mice.
Pathological features of MS are studied in three main animal models: toxic induction of disease, viral induction, and several types of EAE. Cuprizone and lysolecithine, for example, are used to investigate demyelination and remyelination in the CNS (reviewed in Woodruff and Franklin, 1999; Kipp et al., 2009), and to investigate how viral infections can induce CNS autoimmunity; Theiler's murine encephalomyelitis virus (TMEV) inoculated intracerebrally is a currently used experimental protocol (Olson et al., 2001). EAE may be actively induced by immunization of susceptible mouse strains with myelin proteins or myelin protein immunodominant peptides in the presence of complete Freund's adjuvant (CFA), or may be passively induced by transfer of myelin specific T cells. EAE, in most animal models, is mainly driven by MHC class II restricted, autoimmune CD4+ T cells and the clinical course depends on the immunization protocol, as not all combinations of genetic background, antigen, and adjuvant induce EAE. Classically, EAE immunized animals develop an ascending flaccid paralysis from tail to head with variable disease score; manifestation of clinical symptoms may be relapsing remitting, monophasic, or chronic. In active disease pathogenesis of EAE, two main steps are usually described: priming of myelin-reactive T cells and CNS invasion through the blood brain barrier (BBB). Once cells invade the CNS, local, and infiltrated antigen presenting cells (APCs) will present myelin peptides for full activation. Many different types of cells contribute to the development of the disease: APCs, mainly dendritic cells (DCs), B cells, microglia, macrophages, and astrocytes, although astrocytes have an unclear function in EAE development (Volterra and Meldolesi, 2005).
For many years the principal paradigm of EAE pathogenesis has been centered on IFN-γ producing T cells (Th1). These cells differentiate upon exposure to IL-12 and IFN-γ, are characterized by transcription factors T-bet, STAT1, STAT4 and are found as infiltrates in CNS lesions. With time this paradigm has been partially revised and a recently described T cell subpopulation has been shown to have a central role in disease pathology: Th17 cells that differentiate in the presence of IL-6 and TGF-β and need RORγt as a transcription factor (Gutcher and Becher, 2007). Th17 cells have been extensively studied in several autoimmune diseases and there is wide agreement on the instability of the phenotype that may switch between Th1 and Th17. This plasticity derives from epigenetic factors with Th17-derived Th1 cells promoting autoimmune diseases (Annunziato et al., 2007; Mukasa et al., 2010). Altogether, the studies on CD4+ T cells involved in EAE do not ascribe a particular role to one specific T cell population, and T helper cells may undergo alterations. Furthermore, recent observations suggest that EAE can elicit both Th1 and Th2 immune responses in the same subjects, i.e., elements of the Th2 cell-mediated allergic response are associated with autoimmune demyelination (Pedotti et al., 2003).
In conclusion, EAE has provided mechanistic insights into the complex pathogenesis of MS and has proven to be a good model for the preclinical testing of new diagnostic or treatment modalities. In addition, it is considered a well-suited model because of its histopathological and immunological similarities to MS (Schreiner et al., 2009). Consequently, the large majority of studies looking for a possible implication of histamine in autoimmune diseases have been performed using the murine EAE experimental model.
Histamine has long been known to be a major promoter of allergic inflammatory conditions and gastric acid secretion. Synthesized by histidine decarboxylase (HDC) from histidine, histamine was later described as a neurotransmitter in the CNS that regulates several physiological processes and homeostatic functions including cognition, arousal, circadian, and feeding rhythms (Haas et al., 2008). These effects are mediated through four distinct G protein-coupled receptors (H1R, H2R, H3R, and H4R) with very low sequence homology. The H1R couples to Gq proteins, leading to phospholipase C activation and calcium mobilization (Bakker et al., 2002). The H2R activates Gαs and increases camp formation, whereas the H3R mediates its function through Gαi/o, inhibits cAMP synthesis (Bakker et al., 2002), activates MAP kinases and the AKT/GSK3β axis (Bongers et al., 2007; Mariottini et al., 2009).
Inflammatory responses consequent to histamine release have long been thought to be mediated by the H1R, and antihistamines commonly used to treat allergies are H1R antagonists. The discovery of a fourth histamine receptor (H4R) and its expression on virtually all inflammatory and immune cells, though, has prompted a reassessment of the role of histamine in inflammatory and immune disorders and widened the spectrum of potential therapeutic interventions (Thurmond et al., 2008). The H4R is coupled to Gαi/o proteins (Liu et al., 2001) and to the β-arrestin pathway (Rosethorne and Charlton, 2011), and signals via intracellular increases of calcium. Its functions include mediation of calcium mobilization, shape change, actin polymerization (Barnard et al., 2008) chemotaxis of mast cells, and eosinophils (Buckland et al., 2003), and up-regulation of adhesion molecules (Buckland et al., 2003).
All histamine receptors are expressed on the complement of cells involved in autoimmune diseases, with the exception of the H3R that is normally not expressed by hematopoietic cells, but is mostly confined to the CNS (Passani et al., 2011b). Histamine participates in the development and progression of EAE as it controls accessibility to the site of inflammation by modulating vasopermeability and adhesion molecule expression, chemotaxis, and the cytokine profile of DCs and T lymphocytes, the main players in autoimmune diseases.
The regulatory functions of histamine relevant to the onset and progression of neuroinflammatory diseases and in particular EAE, are being studied in genetically modified mice lacking histaminergic receptors, and with relatively selective agonists and antagonists. Hence, the contribution of each histamine receptor in autoimmune diseases has been identified. Histamine plays a complex role with varying and counteracting effects, depending on the receptor subtypes being activated and the targeted tissue (see Table 1). In vitro experiments have shown that activation of H1R and H2R on DCs modulates cytokine and chemokine production and their ability to drive CD4+ T-cell differentiation to the Th2 phenotype. On the other hand, H4R activation modulates chemotaxis (reviewed in Schneider et al., 2010). Depending on the receptor engaged on polarized T cells, histamine can promote Th1 responses through H1R and down-regulate both Th1 and Th2 responses through H2R (Jutel et al., 2001). Similarly to DCs, H1R and H4R activation on CD4+ T cells induces chemotaxis in vitro, whereas H1R and H2R modulate cytokine production (Schneider et al., 2010).
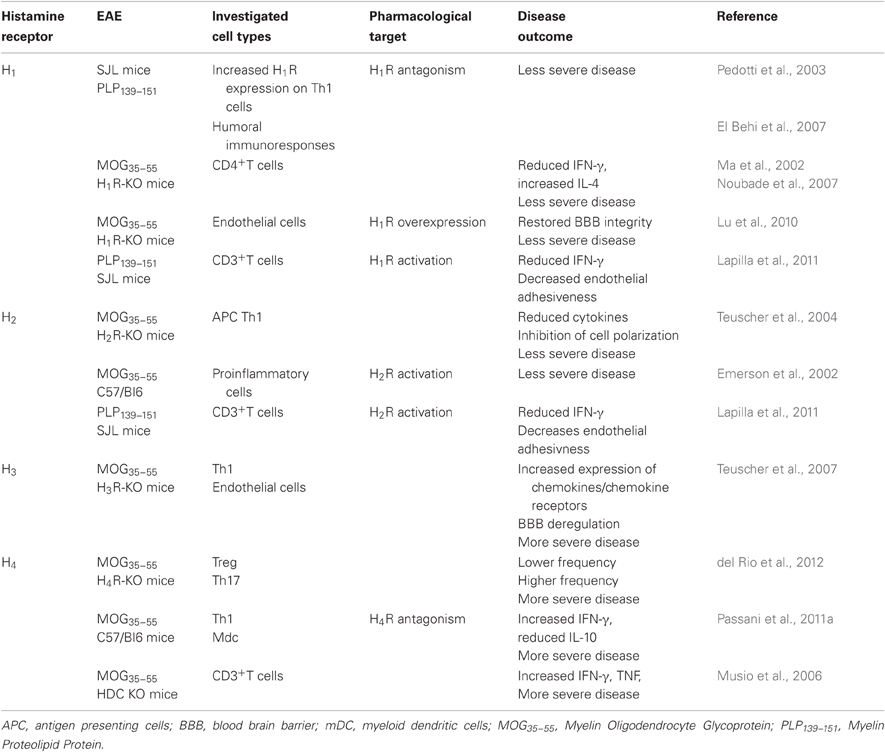
Table 1. Histamine receptors and EAE.
Mast cells are generally thought to be the major sources of histamine and can themselves be modulated by histamine as they express H1R, H2R, and H4R. There is ample correlative and direct evidence that supports mast cell involvement in amplifying the severity of both MS and EAE. Mast cell-deficient W/W(v) mice exhibit significantly less severe disease than wild type littermates in both progressive (Sayed et al., 2011) and relapsing-remitting (Secor et al., 2000) models of EAE.
Susceptibility to EAE requires expression of Hrh1, the gene encoding the H1R (Ma et al., 2002). The H1R is expressed on Th1 cells in EAE mice brain lesions (Pedotti et al., 2003), where its presence is necessary for full encephalitogenic expression (Noubade et al., 2007). Furthermore, expression of the H1R is up-regulated on encephalitogenic PLP139–151 specific Th1 compared to Th2 cell lines (Pedotti et al., 2003). Not surprisingly, specific pharmacological targeting of H1R results in amelioration of EAE (Pedotti et al., 2003; El Behi et al., 2007) and H1R-deficient (H1R-KO) mice exhibit a significant delay in the onset of EAE and a reduction in the severity of the clinical signs compared with WT mice (Ma et al., 2002; Table 1). In fact, CD4+ T-cells from H1R-KO mice produce significantly less IFN-γ and more IL-4 (that induces differentiation of naive CD4+ T cells to Th2 cells) in in vitro assays compared to wild-type controls, indicating that H1R signaling in CD4+ T cells plays a central role in regulating pathogenic T-cell responses (Ma et al., 2002).
The H2R also seems to partially regulate encephalitogenic Th1-cell responses and EAE susceptibility, as H2R-KO mice develop a less severe disease than wild-type littermates during the acute, early phase (Teuscher et al., 2004). The failure of H2R-KO mice to generate encephalitogenic Th1 effector cell responses is attributed to H2R-mediated regulation of cytokine production by DCs that affects T-cell-polarizing activity. In conclusion, H1R and H2R seem to have a pro-inflammatory role and disease-promoting effect, but the story is not quite as simple as that, as H1R or H2R activation may also play an important role in limiting autoimmune responses.
Adhesion of T cells to the inflamed microcirculation precedes their penetration in the brain parenchyma, following breakdown of BBB integrity. H1Rs are expressed on endothelial cells where they increase vascular permeability (Owen et al., 1980); however, functional expression of the H1R on endothelial cells does not restore EAE susceptibility in H1R-KO mice, rather, overexpression of the H1R further suppresses the residual symptoms shown by H1R-KO mice, suggesting that endothelial H1R signaling is needed to maintain cerebrovascular integrity (Lu et al., 2010). Pedotti and colleagues (Lapilla et al., 2011) have demonstrated that histamine activating H1R and H2R, inhibits in vitro the proliferation of mouse CD3+ T cells reactive against PLP139–151, and their adhesiveness to the inflamed endothelium. Also, treatment with an H2R agonist reduces the clinical signs of murine EAE (Emerson et al., 2002; Table 1). As pointed out by the authors (Pedotti et al., 2003; Lapilla et al., 2011) methodological differences may account for the discrepancies observed in in vivo and ex vivo experiments, such as the immunization protocols adopted. Also, ex vivo experiments may not necessarily recapitulate the integrated action of signals relevant to EAE and components of an integrated system maybe lost. On the other hand, genetically modified mice may carry alterations of systems other than the targeted ones, and activation of vicarious mechanisms may hinder the effects related to the deleted gene(s). In fact, the complete lack of HDC and histamine synthesis in HDC-KO mice exacerbates EAE and increases the production of proinflammatory cytokines (Musio et al., 2006). This phenotype does not seem to summarize the lack of signaling at histamine receptors observed in H1R- and H2R-deficient mice, rather a reduced effect of histamine at H3R and H4R (see below).
As mentioned earlier, H3R are normally not expressed by hematopoietic cells, but are mostly confined to the CNS where they limit histamine synthesis and release (Arrang et al., 1983), as well as regulate other neurotransmitters' release (reviewed in Passani and Blandina, 2011). It was recently shown that deletion of the H3R leads to more severe EAE, an effect associated with altered BBB permeability and an unexpected increased expression of chemokines/chemokine receptors that promote entry into the CNS on peripheral T cells that do not themselves express H3R (Teuscher et al., 2007). The authors suggest that neuronal H3R may serve as a central control of cerebrovascular tone, and negatively regulate susceptibility to neuroinflammatory diseases. Their proposed mechanism states that in H3R-KO mice the absence of a presynaptic inhibition would lead to increased release of neurotransmitters and postsynaptic activity that would exert neurogenic control of BBB permeability and T cell chemokine profile.
The distribution of the H4R on immune cells and its primary role in inflammatory functions has made it a very attractive target for the treatment of asthma and autoimmune diseases (Bhatt et al., 2010). Recent evidence has also shown the topological and functional localization of the H4R in the CNS of both humans and rodents (Connelly et al., 2009; Strakhova et al., 2009). Immunohistochemical detection revealed that H4Rs are expressed on the soma of sensory neurons with intense staining of small and medium diameter neurons, as well as lamina I-II of the lumbar spinal cord, where the immunoreactivity pattern suggests localization with terminals of primary afferent neurons (Connelly et al., 2009). These findings widen the range of therapeutic potentials of compounds targeting the H4R, as antagonists might relieve itching by decreasing not only inflammation, but also the urge to scratch. The H4R is detected on hematopoietic progenitor cells (Petit-Bertron et al., 2009), and its activation before exposure to growth factors leads to a drastic decrease in the percentage of cycling cells (Schneider et al., 2011). The H4R expression is dynamic as it is up-regulated during the differentiation from human monocytes to DCs (Gutzmer et al., 2005). In addition, receptor levels may change with the progression of pathophysiological responses, e.g., inflammatory stimuli can up-regulate the expression of H4R in monocytes (Dijkstra et al., 2007). The anti-inflammatory effect of H4R antagonists in asthma and pruritus is consolidated (reviewed in Thurmond et al., 2008; Zampeli and Tiligada, 2009). The use of selective antagonists demonstrated a pro-inflammatory role for this receptor in several paradigms and suggested a facilitating action on autoimmune diseases (Jadidi-Niaragh and Mirshafiey, 2010). Furthermore H4R are expressed on Th17 cells where their activation increases IL-17 production (Mommert et al., 2012). Quite surprisingly though, mice with a disrupted H4R gene develop more severe EAE together with increased neuroinflammatory signs and increased BBB permeability compared to wild type mice (del Rio et al., 2012). In this paradigm, H4R-KO mice have a lower frequency of infiltrating Treg cells that possess disease suppressive activity, more precisely during CNS invasion at day 10 post immunization (10 dpi), and a higher proportion of inflammatory Th17. Preliminary data in our laboratory are corroborating these findings (Table 1), as H4R antagonists such as JNJ7777120 administered daily for eight days to EAE mice at the onset of clinical signs exacerbate the clinical and neuropathological signs of the disease (Passani et al., 2011a). In our model, a decreased release of regulatory cytokines such as IL-10 is accompanied with augmented production of IFN-γ in MOG35–55-specific T cells during the acute phase of the disease (18–20 dpi), suggesting a more complex role of H4R not only on regulatory mechanisms of the immune response but also on T effector cells.
In conclusion, the use of different inflammatory and immune models is producing conflicting results about the role of the H4R in allergic and immune responses. For example, recent data show that the activation, and not antagonism, of H4R leads to reduced pro-inflammatory capacity of a subpopulation of DCs found in inflamed tissues in atopic dermatitis (Gschwandtner et al., 2011). Also, in a murine model of allergic asthma, the administration of H4R agonists mitigated airway hyperreactivity and inflammation with a suggested direct effect on T regulatory cell recruitment (Morgan et al., 2007). It is clear that in the context of the conflicting activities of the H4R that depend on its activation on different hematopoietic cells, additional research is needed to clarify whether H4R agonists can yield promising drugs in the treatment of autoimmune diseases.
Histamine receptors play multiple roles in immune reactions and autoimmune disorders. Strategies aimed at interfering with the histamine axis may have relevance in the therapy of autoimmune diseases of the CNS as histamine may determine, through different receptor activation pathways, a shift in T helper cell subpopulation, may influence migration of lymphocytes and myeloid cells during CNS invasion, interfere with antigen presentation at the immune synapse level and finally, determine variations in normal neuronal functions. It will be of paramount importance to define the temporal sequence of histamine receptor activation during disease initiation in peripheral tissues and during CNS invasion. Hopefully, this will help the scientific community to put the sometimes confusing and contradictory observations reviewed here into better focus and provide a perspective for evaluating potential therapeutic interventions using histaminergic compounds.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by Federazione Italiana Sclerosi Multipla (2010/R/27); COST BM0806; Universita' di Firenze funds, PRIN 2007 (519MIUR068).
APCs, antigen presenting cells; BBB, blood brain barrier; DCs, dendritic cells; EAE, experimental autoimmune encephalomyelitis; HDC, histidine decarboxylase; MOG35–55, Myelin Oligodendrocyte Glycoprotein; MS, multiple sclerosis; PLP139–151, myelin proteolipid protein.
Alonso, A., Jick, S. S., and Hernán, M. A. (2006). Allergy, histamine 1 receptor blockers, and the risk of multiple sclerosis. Neurology 66, 572–575.
Annunziato, F., Cosmi, L., Santarlasci, V., Maggi, L., Liotta, F., Mazzinghi, B., Parente, E., Filì, L., Ferri, S., Frosali, F., Giudici, F., Romagnani, P., Parronchi, P., Tonelli, F., Maggi, E., and Romagnani, S. (2007). Phenotypic and functional features of human Th17 cells. J. Exp. Med. 204, 1849–1861.
Arrang, J. M., Garbarg, M., and Schwartz, J. C. (1983). Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptors. Nature 302, 832–837.
Bakker, R. A., Timmerman, H., and Leurs, R. (2002). Histamine receptors: specific ligands, receptor biochemistry, and signal transduction. Clin. Allergy Immunol. 17, 27–64.
Ballerini, C., Guerini, F. R., Rombolà, G., Rosati, E., Massacesi, L., Ferrante, P., Caputo, D., Talamanca, L. F., Naldi, P., Liguori, M., Alizadeh, M., Momigliano-Richiardi, P., and D'Alfonso, S. (2004). HLA-multiple sclerosis association in continental Italy and correlation with disease prevalence in Europe. J. Neuroimmunol. 150, 178–185.
Barnard, R., Barnard, A., Salmon, G., Liu, W., and Sreckovic, S. (2008). Histamine-induced actin polymerization in human eosinophils: an imaging approach for histamine H4 receptor. Cytometry 73, 299–304.
Bhatt, H. G., Agrawal, Y. K., Raval, H. G., Manna, K., and Desai, P. R. (2010). Histamine H4 receptor: a novel therapeutic target for immune and allergic responses. Mini Rev. Med. Chem. 10, 1293–1308.
Blankenhorn, E. P., Butterfield, R., Case, L. K., Wall, E. H., Rio, R. D., Diehl, S. A., Krementsov, D. N., Saligrama, N., and Teuscher, C. (2011). Genetics of experimental allergic encephalomyelitis supports the role of T helper cells in multiple sclerosis pathogenesis. Ann. Neurol. 70, 887–896.
Bongers, G., Sallmen, T., Passani, M. B., Mariottini, C., Wendelin, D., Lozada, A., Marle, A. V., Navis, M., Blandina, P., Bakker, R. A., Panula, P., and Leurs, R. (2007). Akt/GSK-3b axis as a new signaling pathway of the histamine H3 receptor. J. Neurochem. 103, 248–258.
Buckland, K. F., Austin, N., Jackson, A., and Inder, T. (2003). Histamine induces cytoskeletal changes in human eosinophils via the histamine H4 receptor. Br. J. Pharmacol. 140, 1117–1126.
Connelly, W. M., Shenton, F. C., Lethbridge, N., Leurs, R., Waldvogel, H. J., Faull, R. L. M., Lees, G., and Chazot, P. L. (2009). The histamine H4 receptor is functionally expressed on neurons in the mammalian CNS. Br. J. Pharmacol. 1 57, 55–63.
Correale, J., Ysrraelit, M. C., and Mi, M. I. G. (2009). Immunomodulatory effects of Vitamin D in multiple sclerosis. Brain 132, 1146–1160.
del Rio, R., Noubade, R., Saligrama, N., Wall, E. H., Krementsov, D. N., Poynter, M. E., Zachary, J. F., Thurmond, R. L., and Teuscher, C. (2012). Histamine H4 receptor optimizes T tegulatory cell frequency and facilitates anti-inflammatory responses within the central nervous system. J. Immunol. 188, 541–547.
Dijkstra, D., Leurs, R., Chazot, P. L., Shenton, F. C., Stark, H., Werfel, T., and Gutzmer, R. (2007). Histamine downregulates monocyte CCL2 production through the histamine H4 receptor. J. Allergy Clin. Immunol. 120, 300–307.
El Behi, M., Zéphir, H., Lefranc, D., Dutoit, V., Dussart, P., Devos, P., Dessaint, J. P., Vermersch, P., and Prin, L. (2007). Changes in self-reactive IgG antibody repertoire after treatment of experimental autoimmune encephalomyelitis with antiallergic drugs. J. Neuroimmunol. 182, 80–88.
Emerson, M. R., Orentas, D. M., Lynch, S. G., and LeVine, S. M. (2002). Activation of histamine H2 receptors ameliorates experimental allergic encephalomyelitis. Neuroreport 13, 1407–1410.
Gourraud, P. A., Mcelroy, J. P., Caillier, S. J., Johnson, B. A., Santaniello, A., Hauser, S. L., and Oksenberg, J. R. (2011). Aggregation of multiple sclerosis genetic risk variants in multiple and single case families. Ann. Neurol. 69, 65–74.
Gschwandtner, R., Schäkel, K., Werfel, T., and Gutzmer, R. (2011). Histamine H(4) receptor activation on human slan-dendritic cells down-regulates their pro-inflammatory capacity. Immunology 132, 49–56.
Gutcher, I., and Becher, B. (2007). APC-derived cytokines and T cell polarization in autoimmune inflammation. J. Clin. Invest. 117, 1119–1127.
Gutzmer, R., Diestel, C., Mommert, S., Köther, B., Stark, H., Wittmann, M., and Werfel, T. (2005). Histamine H4 receptor stimulation suppresses IL-12p70 production and mediates chemotaxis in human monocyte-derived dendritic cells. J. Immunol. 174, 5224–5232.
Haas, H. L., Sergeeva, O. A., and Selbach, O. (2008). Histamine in the nervous system. Physiol. Rev. 88, 1183–1241.
Jadidi-Niaragh, F., and Mirshafiey, A. (2010). Histamine and histamine receptors in pathogenesis and treatment of multiple sclerosis. Neuropharmacology 59, 180–189.
Jutel, M., Watanabe, T., Klunker, S., Akdis, M., Thomet, O. A., Malolepszy, J., Zak-Nejmark, T., Koga, R., Kobayashi, T., Blaser, K., and Akdis, C. A. (2001). Histamine regulates T-cell and antibody responses by differential expression of H1 and H2 receptors. Nature 413, 420–425.
Kipp, M., Clarner, T., Dang, J., Copray, S., and Beyer, C. (2009). The cuprizone animal model: new insights into an old story. Acta Neuropathol. 118, 723–736.
Lapilla, M., Gallo, B., Martinello, M., Procaccini, C., Costanza, M., Musio, S., Rossi, B., Angiari, S., Farina, C., Steinman, L., Matarese, G., Constantin, G., and Pedotti, R. (2011). Histamine regulates autoreactive T cell activation and adhesiveness in inflamed brain microcirculation. J. Leukoc. Biol. 89, 259–267.
Liu, C., Ma, X-J., Wilson, S., Hofstra, C., Blevitt, J., Pyati, J., Li, X., Chai, W., Carruthers, N., and Lovenberg, T. (2001). Cloning and pharmacological characterization of a fourth histamine receptor (H4) expressed in bone marrow. Mol. Pharmacol. 59, 420–426.
Lock, C., Hermans, G., Pedotti, R., Brendolan, A., Schadt, E., Garren, H., Langer-Gould, A., Strober, S., Cannella, B., Allard, J., Klonowski, P., Austin, A., Lad, N., Kaminski, N., Galli, S. J., Oksenberg, J. R., Raine, C. S., Heller, R., and Steinman, L. (2002). Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 8, 500–508.
Logothetis, L., Mylonas, I. A., Baloyannis, S., Pashalidou, M., Orologas, A., Zafeiropoulos, A., Kosta, V., and Theoharides, T. C. (2005). A pilot, open label, clinical trial using hydroxyzine in multiple sclerosis. Int. J. Immunopathol. Pharmacol. 18, 771–778.
Lu, C., Diehl, S. A., Noubade, R., Ledoux, J., Nelson, M. T., Spach, K., Zachary, J. F., Blankenhorn, E. P., and Teuscher, C. (2010). Endothelial histamine H1 receptor signaling reduces blood-brain barrier permeability and susceptibility to autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 107, 18967–18972.
Ma, R. Z., Gao, J., Meeker, N. D., Fillmore, P. D., Tung, K. S., Watanabe, T., Zachary, J. F., Offner, H., Blankenhorn, E. P., and Teuscher, C. (2002). Identification of Bphs, an autoimmune disease locus, as histamine receptor H1. Science 297, 620–623.
Mariottini, C., Scartabelli, T., Bongers, G., Attucci, S., Nosi, D., Leurs, R., Chiarugi, A., Blandina, P., Pellegrini-Giampietro, D. E., and Passani, M. B. (2009). Activation of the histaminergic H3 receptor phosphorylates the Akt/GSK-3beta pathway incultured cortical neurons and protects against neurotoxic insults. J. Neurochem. 110, 1469–1478.
Mommert, S., Gschwandtner, M., Koether, B., Gutzmer, R., and Werfel, T. (2012). Human memory Th17 cells express a functional histamine H4 receptor. Am. J. Pathol. 180, 177–185.
Morgan, R. K., Mcallister, B., Cross, L., Green, D. S., Kornfeld, H., Center, D. M., and Cruikshank, W. W. (2007). Histamine 4 receptor activation induces recruitment of FoxP3+ T cells and inhibits allergic asthma in a murine model. J. Immunol. 178, 8081–8089.
Mukasa, R., Balasubramani, A., Lee, Y. K., Whitley, S. K., Weaver, B. T., Shibata, Y., Crawford, G. E., Hatton, R. D., and Weaver, C. T. (2010). Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity 32, 616–627.
Musio, S., Gallo, B., Scabeni, S., Lapilla, M., Poliani, P. L., Matarese, G., Ohtsu, H., Galli, S. J., Mantegazza, R., Steinman, L., and Pedotti, R. (2006). A key regulatory role for histamine in experimental autoimmune encephalomyelitis: disease exacerbation in histidine decarboxylase-deficient mice. J. Immunol. 176, 17–26.
Noubade, R., Milligan, G., Zachary, J. F., Blankenhorn, E. P., Rio, R. D., Rincon, M., and Teuscher, C. (2007). Histamine receptor H1 is required for TCR-mediated p38 MAPK activation and optimal IFN-gamma production in mice. J. Clin. Invest. 117, 3507–3518.
Olson, J. K., Croxford, J. L., Calenoff, M. A., Canto, M. C. D., and Miller, S. D. (2001). A virus-induced molecular mimicry model of multiple sclerosis. J. Clin. Invest. 108, 311–318.
Owen, D. A., Poy, E., Woodward, D. F., and Daniel, D. (1980). Evaluation of the role of Histamine H1- and H2-receptors in cutaneous inflammation in the guinea-pig produced by histamine and mast cell degranulation. Br. J. Pharmacol. 69, 615–623.
Passani, M. B., Aldinucci, A., Masini, E., Blandina, P., Bani, D., Chazot, P. L., and Ballerini, C. (2011a). “Antagonism of the histaminergic H4 receptor exacerbates clinical and pathological signs of EAE,” 256.12/AA14 42nd Meeting of the Society for Neuroscience, Washington, DC, USA.
Passani, M. B., Blandina, P., and Torrealba, F. (2011b). The histamine H3 receptor and eating behavior. J. Pharmacol. Exp. Ther. 336, 24–29.
Passani, M. B., and Blandina, P. (2011). Histamine receptors in the CNS as targets for therapeutic intervention. Trends Pharmacol. Sci. 32, 242–249.
Pedotti, R., Devoss, J. J., Youssef, S., Mitchell, D., Wedemeyer, J., Madanat, R., Garren, H., Fontoura, P., Tsai, M., Galli, S. J., Sobel, R. A., and Steinman, L. (2003). Multiple elements of the allergic arm of the immune response modulate autoimmune demyelination. Proc. Natl. Acad. Sci. U.S.A. 100, 1867–1872.
Petit-Bertron, A. F., Machavoine, F., Defresne, M. P., Gillard, M., Chatelain, P., Mistry, P., Schneider, E., and Dy, M. (2009). H4 histamine receptors mediate cell cycle arrest in growth factor-induced murine and human hematopoietic progenitor cells. PLoS One 4:e6504. doi: 10.1371/journal.pone.0006504
Rosethorne, E. M., and Charlton, S. J. (2011). Agonist-biased signaling at the histamine H4 receptor: JNJ7777120 recruits β-arrestin without activating G proteins. Mol. Pharmacol. 79, 749–757.
Rozniecki, J. J., Hauser, S. L., Stein, M., Lincoln, R., and Theoharides, T. C. (1995). Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Ann. Neurol. 37, 63–66.
Sayed, B. A., Walker, M. E., and Brown, M. A. (2011). Cutting edge: mast cells regulate disease severity in a relapsing-remitting model of multiple sclerosis. J. Immunol. 186, 3294–3298.
Schneider, E., Bertron, A. F., and Dy, M. (2011). Modulation of hematopoiesis through histamine receptor signaling. Front. Biosci. 3, 467–473.
Schneider, E., Leite-De-Moraes, M., and Dy, M. (2010). Histamine, immune cells and autoimmunity. Adv. Exp. Med. Biol. 709, 81–94.
Schreiner, B., Heppner, F. L., and Becher, B. (2009). Modeling multiple sclerosis in laboratory animals. Semin. Immunopathol. 31, 479–495.
Secor, V. H., Secor, W. E., Gutekunst, C. A., and Brown, M. A. (2000). Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J. Exp. Med. 191, 813–822.
Strakhova, M. I., Nikkel, A. L., Manelli, A. M., Hsieh, G. C., Esbenshade, T. A., Brioni, J. D., and Bitner, R. S. (2009). Localization of histamine H4 receptors in the central nervous system of human and rat. Brain Res. 1250, 41–48.
Teuscher, C., Poynter, M. E., Offner, H., Zamora, A., Watanabe, T., Fillmore, P. D., Zachary, J. F., and Blankenhorn, E. P. (2004). Attenuation of Th1 effector cell responses and susceptibility to experimental allergic encephalomyelitis in histamine H2 receptor knockout mice is due to dysregulation of cytokine production by antigen-presenting cells. Am. J. Pathol. 164, 883–892.
Teuscher, C., Subramanian, M., Noubade, R., Gao, J. F., Offner, H., Zachary, J. F., and Blankenhorn, E. P. (2007). Central histamine H3 receptor signaling negatively regulates susceptibility to autoimmune inflammatory disease of the CNS. Proc. Natl. Acad. Sci. U.S.A. 104, 10146–10151.
Thurmond, R. L., Gelfand, E. W., and Dunford, P. J. (2008). The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat. Rev. Drug Discov. 7, 41–53.
Tuomisto, L., Kilpeläinen, H., and Riekkinen, P. (1983). Histamine and histamine-N-methyltransferase in the CSF of patients with multiple sclerosis. Agents Actions 13, 255–257.
Volterra, A., and Meldolesi, J. (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 6, 626–640.
Woodruff, R. H., and Franklin, R. J. (1999). Demyelination and remyelination of the caudal cerebellar peduncle of adult rats following stereotaxic injections of lysolecithin, ethidium bromide, and complement/anti-galactocerebroside: a comparative study. Glia 25, 216–228.
Keywords: multiple sclerosis, EAE, autoimmune diseases, H1 receptor, H2 receptor, H4 receptor
Citation: Passani MB and Ballerini C (2012) Histamine and neuroinflammation: insights from murine experimental autoimmune encephalomyelitis. Front. Syst. Neurosci. 6:32. doi: 10.3389/fnsys.2012.00032
Received: 02 February 2012; Accepted: 14 April 2012;
Published online: 03 May 2012.
Edited by:
Pertti Panula, University of Helsinki, FinlandCopyright: © 2012 Passani and Ballerini. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Maria B. Passani, Dipartimento di Farmacologia Preclinica e Clinica, Universita' di Firenze, Viale Pieraccini 6, 50139 Firenze, Italy. e-mail:YmVhdHJpY2UucGFzc2FuaUB1bmlmaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.