
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Syst. Neurosci. , 13 April 2012
Volume 6 - 2012 | https://doi.org/10.3389/fnsys.2012.00023
This article is part of the Research Topic Histamine in the Brain View all 11 articles
 Yevgenij Yanovsky1† Jeffrey M. Zigman2† Anna Kernder1† Alisa Bein1 Ichiro Sakata2‡ Sherri Osborne-Lawrence2 Helmut L. Haas1 Olga A. Sergeeva1*
Yevgenij Yanovsky1† Jeffrey M. Zigman2† Anna Kernder1† Alisa Bein1 Ichiro Sakata2‡ Sherri Osborne-Lawrence2 Helmut L. Haas1 Olga A. Sergeeva1*The histaminergic neurons in the tuberomamillary nucleus (TMN) of the posterior hypothalamus are involved in the control of arousal. These neurons are sensitive to hypercapnia as has been shown in experiments examining c-Fos expression, a marker for increased neuronal activity. We investigated the mechanisms through which TMN neurons respond to changes in extracellular levels of acid/CO2. Recordings in rat brain slices revealed that acidification within the physiological range (pH from 7.4 to 7.0), as well as ammonium chloride (5 mM), excite histaminergic neurons. This excitation is significantly reduced by antagonists of type I metabotropic glutamate receptors and abolished by benzamil, an antagonist of acid-sensing ion channels (ASICs) and Na+/Ca2+ exchanger, or by ouabain which blocks Na+/K+ ATPase. We detected variable combinations of 4 known types of ASICs in single TMN neurons, and observed activation of ASICs in single dissociated TMN neurons only at pH lower than 7.0. Thus, glutamate, which is known to be released by glial cells and orexinergic neurons, amplifies the acid/CO2-induced activation of TMN neurons. This amplification demands the coordinated function of metabotropic glutamate receptors, Na+/Ca2+ exchanger and Na+/K+ ATPase. We also developed a novel HDC-Cre transgenic reporter mouse line in which histaminergic TMN neurons can be visualized. In contrast to the rat, the mouse histaminergic neurons lacked the pH 7.0-induced excitation and displayed only a minimal response to the mGluR I agonist DHPG (0.5 μM). On the other hand, ammonium-induced excitation was similar in mouse and rat. These results are relevant for the understanding of the neuronal mechanisms controlling acid/CO2-induced arousal in hepatic encephalopathy and obstructive sleep apnoea. Moreover, the new HDC-Cre mouse model will be a useful tool for studying the physiological and pathophysiological roles of the histaminergic system.
The mammalian brain depends on a constant supply of glucose and oxygen. Lack of either one of them rapidly leads to failure of neuronal function. Extracellular levels of CO2 change in response to alterations of blood oxygen levels and are fundamental physicochemical signals controlling breathing and wakefulness. However, the mechanisms connecting changes in extracellular CO2 to arousal are unclear.
Arousal-inducing systems are modulated, either directly or indirectly, in response to hypercapnia or acidification. For instance, serotonin neurons from the dorsal raphe respond to CO2 and genetic deletion of serotonin neurons abolishes the ventilatory response to hypercapnia (Corcoran et al., 2009; Buchanan and Richerson, 2010). Similarly, previous studies have shown that the orexin (hypocretin) neurons in the perifornical area of the posterior hypothalamus are highly sensitive to minor changes in the extracellular pH (Williams et al., 2007), displaying a 100% increase in firing rate in response to a 0.1 unit acidic shift. Furthermore, in wild type but not in orexin-deficient mice augmented ventilation is observed during the 2 h following a brief hypercapnia episode (Terada et al., 2008), implicating the orexin system in the ventilatory response.
The tuberomamillary nucleus (TMN) in the posterior hypothalamus contains the wake promoting histamine system, which provides a complementary and synergistic control of wakefulness together with the orexin neurons (Anaclet et al., 2009). The ventrolateral TMN group of neurons shows increased c-Fos expression indicating increased neuronal activity (Johnson et al., 2005) in response to hypercapnia and consistent with the increased arousal response. It is unclear however, whether CO2 or protons can directly affect TMN neurons or this is the consequence of a system response. In particular, it is possible that histaminergic neurons are excited indirectly via other arousal systems since they receive excitatory afferents from orexinergic neurons (Eriksson et al., 2001) in addition to inputs from other arousal centers of the brain (Haas and Panula, 2003; Haas et al., 2008).
Acid-induced excitation is a hallmark of chemosensing neurons (Williams et al., 2007) and contrasts with the majority of brain neurons which respond to acidification with a hyperpolarization. The cellular mechanisms underlying acid-induced excitation are diverse. In addition to direct activation of acid-sensitive ion channels (ASICs), acidification or hypercapnia may cause ATP-release from astrocytes. In brainstem, ATP-activation of respiratory neurons through P2Y1 receptors is a core event in the fundamental reflex controlling breathing (Gourine et al., 2010). In addition, some acid-sensing mechanisms can be activated by ammonium (Pidoplichko and Dani, 2006) and hyperammonemia leads to intracellular acidification (Kelly and Rose, 2010). In hepatic encephalopathy with ammonia as a main pathogenic factor, brain histamine concentration is increased (Lozeva et al., 2003).
This study investigates the role of the histaminergic system in acid/CO2-sensing and characterizes the mechanisms involved. We focus in detail on the ventrolateral part of the TMN since it is located close to the brain surface and may contact the cerebrospinal fluid. The expression of possible effector systems in the ventrolateral and the medial subdivisions of TMN are compared. The action of pH 7.0 is also compared with that of ammonium chloride at 5 mM. Despite similarity of time course and magnitude, we show that different mechanisms are involved in the effects of protons and ammonia. We identify glutamate acting through mGluR I as a mediator of acid-induced excitation in rats but not in mice.
Housing of rats, mice, and all animal procedures were in accordance with the Animal Protection Law of the Federal Republic of Germany and European Union: EEC (86/609/EEC) directives. They obtained approval of the UTSW Medical Center Institutional Animal Care and Use Committee (Dallas, TX, USA). All efforts were made to reduce the number of animals used. Recordings from histaminergic neurons in male Wistar rat (22–26 day old) or mouse hypothalamic slices were performed as previously described (Chepkova et al., 2012; Yanovsky et al., 2011). Cell-attached recording was used to register action potential currents (Perkins, 2006). Visual identification of histaminergic neurons was confirmed at the end of experiments by application of the histamine 3 (H3) receptor agonist R-α-methylhistamine (0.2 μM) (Yanovsky et al., 2012) (in mM): KCl 130, NaCl 10, MgCl2 2, CaCl2 0.25, glucose 5, Hepes 5, Mg-ATP 5, EGTA 10, Na-GTP 0.3, pH 7.3 adjusted with 1 M KOH. Temperature was held at 33 ± 0.5° if not mentioned otherwise. For gramicidin-perforated patch-clamp recording, (Akaike, 1996) pipettes (4.5–6.0 MΩ) were tip-filled with (in mM): K-gluconate 138, KCl 11, Hepes 10, MgCl2 2, pH 7.3, and back-filled with the same solution containing 50 μg/ml of gramicidin (Sigma). Gramicidin was dissolved in dimethyl sulfoxide (5 mg/0.1 ml), and then was diluted to the final concentration with recording solution. Gramicidin-perforated patch experiments were started when series resistance was below 100 MΩ. After identification of neurons properties, the holding potential was set to −50 mV. Voltage signals and synaptic currents were filtered at 3 kHz with a four-pole Bessel filter, sampled at 10 kHz using pCLAMP9 software (Axon Instruments, Union City, CA, USA), and stored on a PC for off-line analysis. Cells were discarded if their capacitance transients changed during recordings by more than 10%. Solutions of different pH were applied for 7 min through the bath perfusion system. They were obtained by changing NaHCO3 concentrations in the basal recording solution according to the protocols of Jarolimek et al. (1990). Briefly, to achieve pH 6.8 saturated with carbogen and heated to 33.5°C the control solution was supplemented with 5 mM NaHCO3; for pH 7.0 ± 0.05 with 9 mM NaHCO3, for pH 7.2 ± 0.05 with 14 mM NaHCO3. Given variations of pH are averages of measurements done on different experimental days. The control recording solution (ACSF: artificial cerebrospinal fluid, pH 7.4) was composed of (in mM): NaCl 125, KCl 3.7, CaCl2 1.0, MgCl2 1.0, NaH2PO4 1.3, NaHCO3 25, D-glucose 10.
The TMN was dissected from posterior hypothalamic slices (450 μm thick) and incubation with papain in crude form (0.3–0.5 mg/ml) for 20–30 min at 37°C. After rinsing the tissue was placed in a small volume of recording solution with the following composition (in mM): NaCl 150, KCl 3.7, CaCl2 2.0, MgCl2 2.0, HEPES 10, glucose 10 (pH 7.4). Cells were separated by gentle pipetting and placed in the recording chamber. TMN neurons were recorded and identified as previously described (Sergeeva et al., 2010; Yanovsky et al., 2012). Briefly, patch electrodes were sterilized by autoclaving and filled with the electrode solution for whole-cell recordings (in mM): KCl 130, NaCl 10, MgCl2 2, CaCl2 0.25, glucose 5, Hepes 5, Mg-ATP 5, EGTA 10, Na-GTP 0.3, pH 7.3 adjusted with 1 M KOH. The cells were voltage-clamped by an EPC-9 amplifier. The holding potential was –50 mV. An acutely isolated cell was lifted into the major chute of the application system, where it was continuously perfused with the sterile control bath solution. Substances were applied through a glass capillary (application tube), 0.08 mm in diameter. All solutions flowed continuously, gravity-driven, at the same speed and lateral movements of the capillaries exposed a cell either to control- or test-solutions. Solutions with different pH were prepared by the addition of different amounts of HCl to the recording solution (see above). In some experiments solutions at pH < 6.5 contained 10 mM MES [2-(N-morpholino) ethanesulfonic acid, Sigma] instead of HEPES. As the obtained pH50 did not differ between the two buffers data were pooled. To evaluate the proton sensitivity of the cells the mean of the peak current amplitude was fitted with a dose-response curve using the equation:
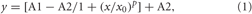
where A1 and A2 are the bottom and top asymptote, x0 is the half-activation between A1 and A2 and p is Hill's slope.
Amplification of cDNAs encoding for histidine decarboxylase was performed as previously described (Sergeeva et al., 2002, 2005, 2010). The expected size of the amplification product was 457 bp. For the first amplification round of the ASIC family we used following degenerate primers Dglo: 5′-ccRatRaacaRBcccatctg-3′ and Dgup: 5′-agatYcacagYcagKMKgaRcc-3′. In the second amplification round the primers varied for each ASIC-channel: ASIC 1 up: 5′-cctagtggagaaagaccaggaata-3′ was used in combination with Dglo (size of PCR product: 276 bp.), ASIC 2 lo: 5′-ccttttctgccagtagaccga-3′ was taken with Dgup (321 bp), ASIC 3 up: 5′-gggtgactgcaataccgcat-3′ with ASIC 3 lo: 5′-tcattcgacagccacacttc-3′ (164 bp) and ASIC 4 up: 5′-gaacagcggctaacttatctgc-3′ with ASIC 4 lo: 5′-catgggccctcagagccc-3′ (263 bp). For the analysis of ASIC1 splice variants (ASIC1a and ASIC1b) amplification was done with the following primers: ASIC1up: 5′-tccctgggcctgctgctg-3′ and ASIC1lo: 5′-gtggccgcccatcttg-3′ (size of PCR product for the ASIC 1b: 324 bp and for the ASIC 1a: 360 bp). Digestion of ASIC1a with the restriction endonuclease Eco RI (Invitrogen) confirmed presence of both splice variants in cDNA derived from the trigeminal ganglion: undigested amplimer of 324 bp size (ASIC1b) was seen together with ASIC1a restricted cDNA fragments of 210 bp und 150 bp size. Thin-walled PCR tubes contained a mixture of first strand cDNA template (1–1.5 μl), 10× PCR buffer, 10 pM each of sense and antisense primer, 200 μM of each dNTP and 2.5 units Taq polymerase. The final reaction volume was adjusted to 10 μl with nuclease-free water (Promega, Mannheim, Germany). (MgCl2) was 2.5 mM. Amplifications were performed on a thermal cycler (Perkin Elmer, GeneAmp 9600, Weiterstadt, Germany). In each round, 35 cycles of the following thermal programs were used: denaturation at 94°C for 48 s, annealing at 50–53°C for 48 s, and extension at 72°C for 1 min. For the second amplification round, 1.1 μl of the product of the first PCR was used as a template. The Taq enzyme, PCR buffer, Mg2+ solution, and four dNTPs were all purchased from Qiagen (Erkrath, Germany). All oligonucleotides were synthesized by MWG-Biotech (Ebersberg, Germany). Products were visualized by staining with ethidium bromide and analyzed by electrophoresis in 2% agarose gels. Randomly selected PCR products were purified in water (PCR purification kit, Qiagen, Erkrath, Germany) and sequenced on an automatic sequencing machine (ABI, model 377, Weiterstadt, Germany). Comparison with corresponding GENBANK sequences revealed no mismatches (ASIC 1: NM_024154.2, ASIC 2: NM_012892.2, ASIC 3: NM_173135.1, ASIC 4: NM_022234.2).
The TMN was dissected from hypothalamic slices and total cellular mRNA was isolated using an mRNA isolation kit (Pharmacia Biotech) according to the manufacturer's protocol. Total mRNA was eluted from the matrix with 30 μl of RNase-free water. For the reverse-transcription 8 μl of eluted mRNA was added to 7 μl of reagents mixture prepared according to the protocol of the “first strand cDNA synthesis kit” (Pharmacia Biotech). After incubation for 1 h at 37°C the reverse transcription reaction was stopped by freezing at –20°C. The reverse-transcription reactions were not normalized to contain the equivalent amounts of total mRNA. The PCR was performed in a PE Biosystems GeneAmp 5700 sequence detection system using the SYBR green master mix kit. Each reaction contained 2.5 μl of the 10xSYBR green buffer, 200 nM dATP, dGTP, and dCTP and 400 nM dUTP, 2 mM MgCl2, 0.25 units of uracil N-glycosylase, 0.625 units of Amplitaq Gold DNA polymerase, 10 pM forward and reverse primers, 5 μl of 1:4 diluted cDNA, and water to 25 μl. All reactions were normalized to β-actin expression. Primers for the β-actin, peripherin and mGluR I cDNA amplification were published previously (Chepkova et al., 2009; Yanovsky et al., 2011). Primers for the HDC—cDNA amplification (Sergeeva et al., 2002) and for the ASIC-cDNAs were the same as those used in the second round of single-cell RT-PCR. Primers for the histamine N-methyl-transferase were: HNMT up: 5′-ttcttggcacaaggagaca-3′and HNMT lo: 5′-gctaagagaccatggaaaaat-3′. The reactions were performed in tubes with MicroAmp optical caps. The reactions were incubated at 50°C for 2 min to activate uracil N′-glycosylase and then for 10 min at 95°C to inactivate the uracil N′-glycosylase and activate the Amplitaq Gold polymerase followed by 40 cycles of 15 s at 95°C, 1 min at 60°C. The PCR reactions were subjected to a heat dissociation protocol (PE Biosystems 5700 software). Following the final cycle of the PCR, the reactions were heat-denaturated over a 35°C temperature gradient at 0.03°C/s from 60°C to 95°C. Each PCR product showed a single peak in the denaturation curves. Standard curves for real-time PCR protocols with all primer-pairs obtained with sequential dilutions of one cDNA sample (till 1:64) were found optimal (linear regression coefficients were > 0.95). Semiquantitative analysis of target gene expression relative to the β-actin endogenous control was performed according to the “2−ΔΔCt”(ΔFold) method as described previously (Chepkova et al., 2009).
We generated several lines of transgenic mice that express cre recombinase (Cre) within histidine decarboxylase-expressing cells. These animals were made by first using various ET-cloning “recombineering” technologies (Lee et al., 2001; Muyrers et al., 2001); to construct an HDC-Cre transgene-containing bacterial artificial chromosome (BAC). The original HDC BAC (RP24-141N14), which was purchased from BACPAC Resources Center at Children's Hospital Oakland Research Institute, spanned the entire coding region of HDC and furthermore contained approximately 92.42 kb sequence upstream of the HDC start codon and approximately 13.91 kb sequence downstream of the HDC stop codon. This BAC was transformed into EL250 cells by electroporation. EL250 cells were provided by N. Copeland; they contain heat-inducible recE and recT recombinases for homologous recombination and arabinose-inducible Flp-recombinase for site-specific recombination at frt sites (Lee et al., 2001). Next, a DNA fragment containing the coding sequence of Cre recombinase followed by an SV40 polyadenylation (polyA) signal and a kanamycin resistance gene flanked by frt sites (FKF) was inserted into the HDC BAC, at the translational start site of HDC, by ET-cloning. The construction of the Cre-polyA-FKF cassette was described previously (Dhillon et al., 2006). This insertion resulted in the removal of 2194 bp of the HDC gene (this included the latter portion of exon 1 and the first portion of exon 2—which encodes the first 47 amino acids of histidine decarboxylase—and the intervening intron 1). Finally, the kanamycin resistance gene was removed by arabinose induction of Flp-recombinase, and the Cre recombinase coding region was sequenced to ensure that no mutations had been introduced. The Cre-modified HDC BAC was submitted to the UTSW Medical Center Transgenic Core Facility for microinjection into pronuclei of fertilized one-cell stage embryos of C57Bl6/J mice. We were successful in generating multiple potential HDC-Cre founder mice, of which one was shown to contain the expected Cre expression and activity within the CNS. Oligonucleotide primers used to confirm the genotype of mice harboring the HDC-Cre transgenes were as follows: M249: 5′-CAGGGAGTGCACAGCACAGACAA-3′ and M247: 5′-TGCGAACCTCATCACTCGTTGCAT-3′. The mice used in this study were on a pure C57BL6/J genetic background. Animals were housed under 12 h of light/12 h of dark per day in a temperature-controlled environment. They were fed standard chow diet (details) and had free access to water.
For histological validation of Transgene expression, HDC-Cre mice were bred to Rosa26-lox-STOP-lox-lacZ reporter mice [B6.129S4-Gt(ROSA)26–Sortm1Sor/J; stock#003474; The Jackson Laboratory, Bar Harbor, Maine], in which a transcriptional stop cassette is removed only in the presence of Cre activity, allowing for visualization of cells with Cre activity by assaying for β-galactosidase or for lacZ-IR, as below. Mice derived from the HDC-Cre X Rosa26-lox-STOP-lox-lacZ crosses were deeply anesthetized with an intraperitoneal injection of chloral hydrate (500 mg/kg) and subsequently perfused transcardially with diethylpyrocarbonate (DEPC)-treated 0.9% saline followed by 10% neutral buffered formalin. Brains were removed, stored in the same fixative for 4–6 h at 4°C, and then immersed in 20% sucrose in DEPC-treated phosphate buffered saline (PBS), pH 7.0 at 4°C overnight. Brains were sectioned coronally, using a sliding microtome, into five equal series at a 25 μm thickness. Brain sections were permeabilized and blocked in 3% normal goat serum/0.3% Triton X-100 for 1 h, and then incubated for 48 h at 4°C in 1:5000 guinea pig anti-HDC antiserum (Catalog number: B-GP 265-1, Lot: HS4061; ALPCO Diagnostics, Salem, NH). After washing in PBS, sections were incubated in Alexa Fluor 594 goat anti-guinea pig IgG (Invitrogen, Carlsbad, CA; 1:300) for 1 h at room temperature, followed by more PBS washes. For lacZ staining, sections were subsequently incubated for 24 h at 4°C in 1:10000 chicken anti-lacZ antiserum (Catalog number: ab9361, Lot: 713063, Abcam, Cambridge, MA). After washing in PBS, sections were incubated in Alexa Fluor 488 goat anti-chicken IgG (Invitrogen, Carlsbad, CA; 1:300) for 1 h at room temperature, followed by more PBS washes. Finally, the sections were mounted in Fluoromount G (Electron Microscopy Sciences, Hatfield, PA) and viewed using microscopy (Axioskop 2, Carl Zeiss). Images were taken using both the red filter and green filter at each level, and were placed side-by-side. All the cells at each level with either HDC-immunoreactivity alone, lacZ-immunoreactivity alone, or both were counted. An imaging editing software program, Adobe PhotoShop 7.0 (San Jose, CA) was used to adjust contrast, brightness, and color of the photomicrographs, and to merge the images taken using the red and green filters.
To be able to visualize TMN neurons in slices we crossed the HDC-Cre mouse to the Tomato reporter mouse B6J/N. Cg-Gt(ROSA) 26Sortm14(CAG−tdTomato) (Jackson Laboratory, #007908) yielding the HDC/Tmt mice. Offspring (both genders) carrying both transgenes were used for the slice recordings. Histamine immunostainings were done according to the previously published protocols (Yanovsky et al., 2011). Wilde type littermates of HDC-Cre breeders were used as a control in electrophysiological and molecular biological experiments (comparative gene expression analysis).
Experiments were conducted and analyzed with commercially available software (TIDA for Windows, HEKA, Lambrecht, Germany) and with Clampex 9.0. All graphs were assembled in GraphPad Prism (GraphPad Software, Inc., San Diego, CA, USA) and Photoshop.
Data are presented in text as the mean ± SEM. Statistical analysis was done with the non-parametrical Mann-Whitney U-test (real-time PCR) or unpaired Student t-test (comparison of magnitude of excitation between groups in cell-attached firing rate recordings). Comparison with the baseline activity in each individual experiment was done using paired Student t-test. Differences were considered statistically significant if p < 0.05.
As it was shown previously that only the ventrolateral TMN of the rat shows increased c-Fos expression in response to systemic hypercapnia (Johnson et al., 2005), we performed now patch-clamp recordings from ventrolateral TMN, which corresponds to the E2 group (Figure 1A). Acidification of the bath solution for 7 min increased the firing rate of most rat TMN neurons recorded in cell-attached mode (23 out of 28 neurons, 82%). Unresponsive neurons were not further investigated. On average pH 7.0-induced excitation amounted to 166 ± 15% of control firing rate (n = 23 at 33°C). The mean firing rate in these experiments under control conditions was 1.14 ± 0.05 Hz (n = 28). The excitatory response did not show a significant temperature-dependence: at room temperature it reached 157 ± 40% of control (n = 6). The period of high initial excitation in response to the acidification was followed by a reduction of activity with subsequent suppression of activity below control level in the beginning of the washout period (Figure 1B). The response to pH 7.0 in perforated patch whole-cell voltage-clamp recordings (Vh = –50 mV) perfectly matched the time course of responses seen in the cell-attached configuration (Figure 1C). The initial inward current of 22 ± 7 pA (starting after 1.9 ± 0.3 min, maximum after 4.6 ± 0.6 min) was followed by an outward current of 8.5 ± 1.4 pA (0.9 ± 0.1 min after beginning the washout) (Figure 1C3). Note a slight and delayed decrease in membrane resistance measured in parallel experiments (180 ± 10 MΩ versus 190 ± 4 MΩ in control, n = 4, p > 0.05), which did not correspond with respect to time to the fast rising phase of the inward current (Figure 1C4). Moreover, no robust membrane depolarisation was observed during the first phase of the response (n = 5, Figure 1C2), whereas the second phase was characterized by reduced firing frequency, outward current and membrane hyperpolarization. The magnitude of facilitation was dependent on the degree of acidification. The firing rate measured in cell-attached configuration was changed by 17 ± 2% for each 0.1 pH unit (Figure 2A).
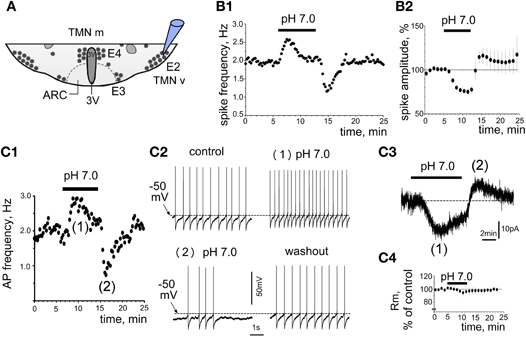
Figure 1. Proton-induced increase in TMN neuron firing. (A) Schematic presentation of the coronal hypothalamic slice indicates the TMN groups (E2 corresponds to ventrolateral TMN, whereas medial TMN contains the E3, E4 groups). 3V, third ventricle; ARC, arcuate nucleus. Electrode points to the recording site E2. (B1,2) Firing of TMN neuron recorded in cell-attached mode in control at pH 7.4 and at pH 7.0. (B1) Time course of firing-frequency in representative neuron. Every point on the histogram represents the frequency averaged for 15 s of recording. (B2) Change in the amplitude of action potential currents during acidification (each point: average recorded during 1 min in 6 neurons). (C1) Current clamp recording from one TMN neuron illustrates the action potential (AP) frequency in response to low pH. Each point is the average of 15 s recording. (C2) fragments of voltage trace recordings in the same neuron. Note no deflection of the control membrane potential ∼–50 mV during the first phase of the response and a slight hyperpolarization during the second phase. (C3) Biphasic current (voltage-clamp mode) recorded from the same neuron as in (C1,2). Note that phase (1) and phase (2) correspond timely to the excitation and inhibition phase in (C1). (C4) membrane resistance (average of 4 neurons) measured from current deflections in response to brief voltage steps (–10 mV for 10 ms from holding potential at 10 s intervals) in experiments like the one shown in (C3).
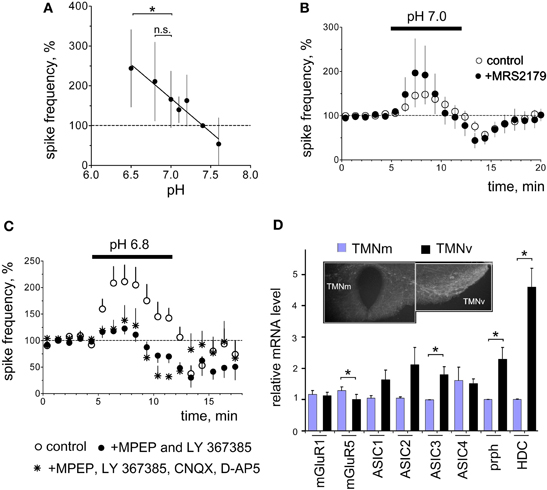
Figure 2. Functional identification and expression analysis of acid-sensors in TMN. (A) Acid-induced excitation depends on the degree of acidification. Each data point is an average of 4–23 neurons. The difference in excitation between pH 6.5 and 7.0 but not between pH 6.8 and 7.0 is significant (*p < 0.05). (B) The P2Y1 receptor antagonist MRS2179 (n = 5) does not prevent pH 7.0-excitation. Control diagram represents the average of 23 neurons. (C) Facilitation of firing in control and in the presence of the metabotropic GluR (mGluR) antagonists MPEP (mGluR5, 10 μM) and LY367385 (mGluR1, 20 μM) or mGluR antagonists combined with inotropic glutamate receptor antagonists CNQX (10 μM, AMPA) and D-AP5 (50 μM, NMDA). (D) mRNA levels normalized to the minimal expression level for mGluR1, mGluR5, ASIC (1–4), peripherin (prph) and histidine decarboxylase (HDC) detected with real-time RT-PCR (eight rats aged from 23 to 27 days). Hypothalamic slice shows regions dissected for the analysis. TMN neurons stained with the rabbit-anti-histamine antibody are visualized with the secondary donkey-anti-rabbit antibody conjugated to Alexa fluor 488. *p < 0.05 Mann–Whitney.
Previous studies have shown that glial cells respond to acid/CO2 by the release of ATP, which excites respiratory neurons in brain stem through the P2Y1 receptor (Gourine et al., 2010). We have shown that ATP/ADP excites TMN neurons through P2X (Vorobjev et al., 2003) and P2Y (Sergeeva et al., 2006) receptors. We also found that activation of P2Y1 receptors in hypothalamic slices containing TMN causes release of glutamate which amplifies the excitatory response through type I metabotropic glutamate receptors (mGluR I). Acid-induced excitation of TMN neurons in the present study was not affected by the P2Y1 receptor antagonist MRS 2179 (90 μM, n = 5; Figure 2B). The metabotropic glutamate receptor type I (mGluR I) antagonists MPEP (10 μM, mGluR5 antagonist) combined with LY 367385 (20 μM, mGluR1 antagonist) (Figure 2C) significantly reduced acid-induced excitation (p < 0.05). In the presence of mGluR I antagonists the excitation amounted to 123 ± 10% (n = 6) of baseline activity versus 211 ± 31% (n = 10) in the parallel control experiments. The AMPA and NMDA receptor antagonists CNQX (10 μM) and D-AP5 (50 μM) neither affected proton-induced excitation (199 ± 30%, n = 3) nor increased the block of excitation by metabotropic glutamate receptor antagonists when all glutamate receptor antagonists were applied together (138 ± 28% of control, n = 4, Figure 2C). Thus proton-induced excitation is largely (∼70%) mediated by metabotropic glutamate receptors.
Electrophysiological experiments revealed that mGluR mediate most of the acid-induced excitation of TMNv. Therefore, we investigated differential expression of these receptors with respect to functional diversity between TMNv and TMNm (Figure 2D). Semiquantitative RT-PCR was performed from mRNA isolated from two parts of TMN as shown in Figure 2D. The lateral part contained largely the E2 dense group of TMN neurons, whereas in the medial part only a minority of cells were histaminergic (groups E3 and E4, see Figure 1A). Therefore, histaminergic markers, such as histidine decarboxylase (HDC) or peripherin (prph) were expressed significantly higher in the ventrolateral part compared to the medial TMN (Figure 2D). Expression of mGluR1 did not differ between ventrolateral and medial TMN, whereas mGluR5 expression in the ventrolateral TMN represented 79% of that in the medial TMN (p < 0.05).
We then investigated the expression of classical acid-sensors, such as acid-sensing ion channels (ASICs) which others found expressed in the majority if not in all central neurons (Lingueglia, 2007; Grunder and Chen, 2010). We found the expression of ASIC3 significantly higher in ventrolateral TMN compared to the medial subdivision (Figure 2D). Interestingy, this member of the ASIC family responds to acidic pH with a fast inactivating followed by a sustained sodium current and is sensitive to physiological changes in proton concentration (Waldmann et al., 1997). Expression of ASIC3 in the hypothalamus was shown recently (Meng et al., 2009). Members of ASIC 1 and ASIC2 subfamilies are activated by much lower pH values (pH50 ∼ 4.1–6.2) except for ASIC 1a (Lingueglia, 2007), which also responds to ammonium (Pidoplichko and Dani, 2006).
Solutions with pH ranging from 7.0 to 4.0 were applied for 2 s at 30 s intervals in 17 acutely isolated TMN neurons. In none of the investigated neurons did pH 7.0 evoke measurable currents, whereas pH 6.5 evoked currents of 37 ± 8 pA in 82% of neurons (Figure 3B). Half-maximal current amplitude was evoked by pH 5.8± 0.1. In 12 neurons a solution with pH 6.0 was applied alone or in the presence of the ASIC blocker benzamil hydrochloride (bzm). Bzm 10 μM suppressed the peak current amplitude to 59± 8% of control (n = 4) without effects on plateau current. Bzm 20 μM suppressed peak current to 30± 6% and plateau to 57± 11% of control (n = 12)(see Figure 3). The reversal potential of the acid-induced current was determined in 3 TMN neurons. Currents in response to acidic pH were inward at negative membrane potentials and became outward at potentials more positive than +64 ± 4 mV (the predicted reversal potential for the sodium ion under our recording conditions is +63.4 mV). Previous studies have shown that ASICs are mainly permeable for sodium ions (Bassilana et al., 1997; Waldmann et al., 1997). Single-cell RT-PCR analysis was performed from 32 TMN neurons. We found variable expression of all four known ASICs in histaminergic cells: thus ASIC1 transcripts were detected in 71%, ASIC2 in 56%, ASIC3 in 63% and ASIC 4 in 47% of TMN neurons (n = 32, Figures 3C,D). Restriction analysis of PCR fragments containing the splice region of ASIC1 showed that only the ASIC1a subtype is expressed in TMN. The whole TMN region was analyzed from four rats. This result was confirmed by sequencing. Thus, similar to dopaminergic neurons of the midbrain (Pidoplichko and Dani, 2006) TMN neurons express ammonium-sensitive ASIC1a.
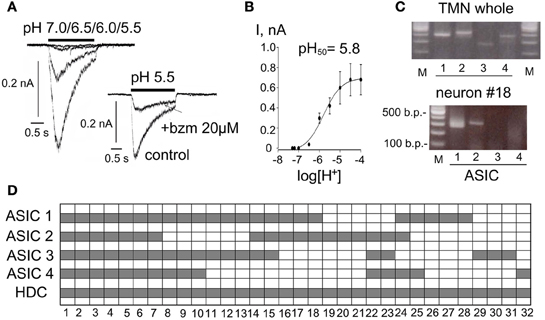
Figure 3. Expression and function of ASIC (acid sensing ion channels) in TMN neurons. (A) Acid-induced currents in two representative neurons. Neurons are moved to the test solution with indicated pH for 2 s from the control ACSF (pH 7.4). Benzamil hydrochloride (bzm) abolishes the fast inactivating phase of the response and reduces the non-desensitizing component. (B) Averaged current-pH histogram represents data obtained from 17 neurons. Data points are fitted with the logistic equation. (C) Photographs of gels illustrate RT-PCR analysis of ASIC (1–4) expression in whole TMN and in one representative neuron. (D) Floating bar histograms show co-expression of 4 ASICs in 32 TMN neurons positive for histidine decarboxylase (HDC).
To test the participation of ASICs in the acid-induced excitation of TMN, we applied pH 7.0 in the presence of bzm (20 μM, n = 3) and found that the firing increase was virtually abolished (Figure 4A). This can be explained by the simultaneous antagonism toward both: ASICs and Na+/Ca2+ exchange, which works in a reverse (depolarizing) mode downstream to mGluR activation. In addition, bzm blocks Na+/H+ exchange, preventing intracellular acidification during exposure to acid.
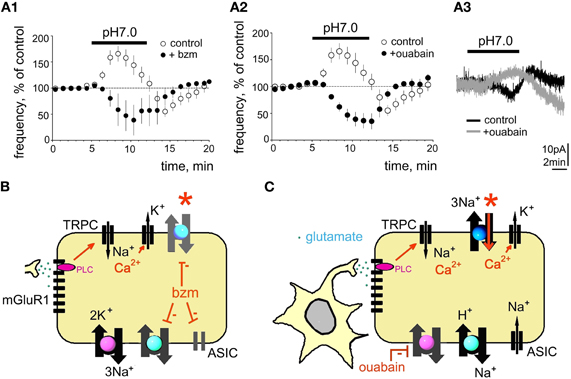
Figure 4. Benzamil or ouabain abolish acid-induced excitation. (A1) Benzamil (bzm, 20 μM) blocks acid-induced excitation. (A2) Time course of firing frequency in control and in ouabain (0.5 μM). (A3) gray trace shows acid-induced outward current recorded in ouabain (perforated patch-clamp mode) superimposed to the black (control) response recorded from the same neuron. (B,C) Models explaining action of bzm or ouabain. Bzm inhibits ASIC, Na+/H+ and Na+/Ca2+ exchangers. Ouabain (C) blocks Na+/K+ ATPase, which leads to the compensatory sodium removal by Na+/H+ and Na+/Ca2+ exchangers, which is accompanied by a rise of intracellular Ca2+ and calcium-activated potassium conductance. Red stars point to block of the direct mode of Na+/Ca2+ exchange in both experimental sessions.
Despite ubiquitous expression of ASICs in TMN neurons, the following findings argued against a massive ion flow through such membrane channels in response to pH 7.0 and favor the idea that the sodium gradient is reduced during the excitation phase of the response, possibly through inhibition of Na+/K+ ATPase by protons, as described previously for cardiomyocytes (Fuller et al., 2003): (1) lack of a pronounced membrane depolarisation during increased neuronal firing frequency; (2) decreased amplitude of action potential currents during the excitation phase; (3) no significant changes in Rin seen in perforated patch configuration. Fuller et al. (2003) demonstrated that Na+/K+ATPase subtypes expressed in brain and heart are inhibited by a proton-activated factor during the initial phase of sodium influx in response to ischemia, which leads to membrane depolarisation. Increased intracellular sodium concentration turns on the Na+/Ca2+ exchange in the reverse mode leading to the extrusion of sodium, intracellular acidification and rise of calcium (Fuller et al., 2003). Therefore, the second, inhibitory phase of the response to acidification recruits calcium-activated potassium channels acting synergistically with the reverse electrogenic Na+/Ca2+ exchange (3 sodium ions are exchanged for 1 calcium ion) to produce a hyperpolarization. Upon return to the control ACSF (pH 7.4) the Na+/K+ ATPase regains its functionality and contributes to the hyperpolarization. This scenario, schematically depicted in Figure 4, could explain the biophysical characteristics of the first (excitatory) and second (inhibitory) phase of the TMN response to the mild (physiological) acidification. Indeed, Na+/Ca2+ exchange and Na+/K+ ATPase were found to be essential for the first excitatory phase of the acid-induced excitation.
The Na+/K+ ATPase blocker ouabain (0.5 μM) increased the firing frequency of TMN neurons to 214 ± 20% of control (p < 0.0001; n = 8). In the presence of ouabain acidification of the extracellular solution produced neither excitation (Figure 4B) nor the rebound inhibition upon return to the control medium. When pH 7.0-induced currents were recorded in the presence of ouabain (perforated patch-clamp), outward currents were seen instead of inward currents in control experiments (Figure 4B). This can be explained by the activation of the reverse mode of Na+/Ca2+ exchange which leads, together with calcium-activated potassium currents to hyperpolarization.
As ASIC1a responds to ammonium (Pidoplichko and Dani, 2006) we compared the pharmacology of ammonium-induced excitation with the acid-induced excitation in the following experiments. In cell-attached recordings NH4Cl evoked a transient increase in firing rate to 177 ± 14% of control (n = 16). In some neurons an inhibition similar to that seen in experiments with pH 7.0 was present at the beginning of the washout period (Figure 5). In contrast to the acidification-induced increase in firing rate, the excitation by ammonium was not prevented by mGluR I-antagonists (n = 4), by benzamil hydrochloride (n = 4) or by ouabain (n = 3, Figure 5).
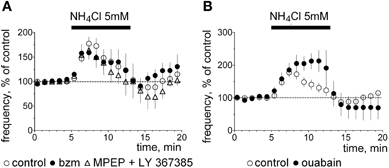
Figure 5. Ammonium-induced excitation. (A) Averaged time course diagrams show normalized firing frequency (cell-attached voltage-clamp mode) in control experiments and in the presence of indicated blockers, which abolish pH 7.0-induced excitation. (B) Ouabain does not block ammonium-induced excitation.
Ammonium can be transported instead of K+ by the Na+/K+ ATPase (Kelly and Rose, 2010) or interact with KCC2 (potassium/chloride co-transporter) (Liu et al., 2003). Thus mechanisms of action of ammonium and of protons are different in TMN neurons.
Our experiments in rats showed that metabotropic glutamate receptors of type I, NCX and Na+/K+ ATPase are the major players in proton-induced excitation of TMN, but not in response to ammonia. In order to identify the role of the histaminergic system in the response to hypercapnia or/and hyperammonemia it would be important to delete the major sensors only in histidine-decarboxylase (HDC) expressing neurons and see the behavioral response according to the previously published protocols (Johnson et al., 2005). For this purpose an HDC-Cre mouse was generated. A schematic diagram showing derivation of the HDC-Cre mouse is shown in Figure 6. In initial experiments a LacZ-reporter mouse was used for the visualization of HDC-Cre transgene in TMN. Results of this analysis are summarized in Table 1 and Figure 6.
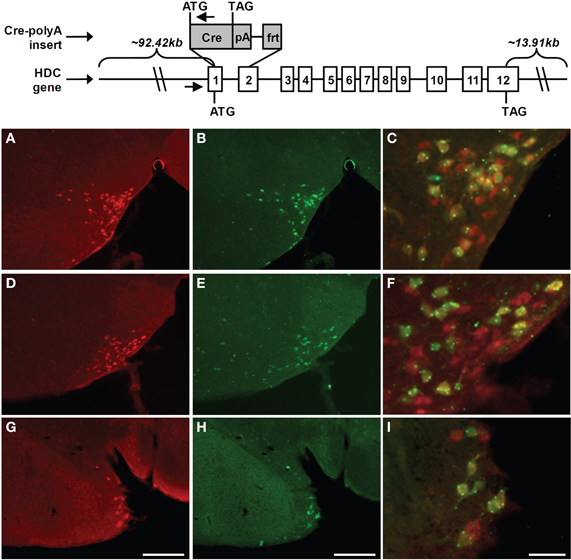
Figure 6. Schematic diagram of the derivation of HDC-Cre transgenic mice and immunohistochemical analysis of transgene expression in TMN. Mice expressing Cre recombinase under the control of the HDC promoter were generated by inserting a Cre recombinase expression cassette into a BAC genomic clone containing HDC. In particular, a 2194-bp region of HDC was replaced by the coding sequence of Cre recombinase followed by an SV40 polyadenylation signal (pA) and an frt site. HDC's exons (denoted by numbered rectangles), and the start codons (ATG) and stop codons (TAG) for HDC and Cre are all indicated. The amounts of mouse chromosomal sequence present in the BAC either upstream of the HDC start codon or downstream of the HDC stop codon also are indicated. Small arrows denote the locations of oligonucleotide primers used for genotyping. Expression of Cre activity within the tuberomamillary nucleus of the HDC-Cre transgenic mouse line was examined in coronal brain sections of Rosa26-lox-STOP-lox-lacZ reporter mice harboring the HDC-Cre transgene. (A,D,G) Rostrocaudally arranged brain sections [(A) Bregma −2.30 mm, (D) Bregma −2.70 mm, (G) Bregma −2.92 mm)] processed for HDC-immunoreactivity (red). (B,E,H) The same sections processed for lacZ-immunoreactivity (green). (C,F,I) Merged HDC (red) and lacZ signals (green), within cells containing co-localized HDC- and lacZ-immunoreactivity, appearing as yellow (yellow dots and green dots reflect highly localized collections of lacZ). Scale bars = 200 μM in G and H (applies to A, B, D, E, G, H); 50 μM in I (applies to C, F, I).
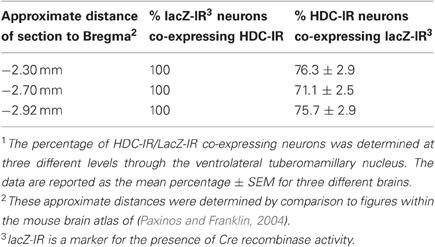
Table 1. Co-expression of Cre recombinase activity and HDC within the ventrolateral tuberomamillary nucleus of HDC-Cre transgenic line1.
For the electrophysiological recordings another reporter mouse was used which allowed us to visualize living TMN neurons in the slices. The co-localization pattern of transgene and histamine in the HDC/Tmt-mouse was similar qualitatively and quantitatively to that seen in the LacZ-reporter mouse. Thus, in the ventrolateral TMN (our recording site) all Tmt+ neurons contained histamine (Figure 7A). Among 1101 histaminergic neurons counted in similar fields as shown in Figure 7A (21 fields in slices obtained from five mice were investigated) 298 neurons were negative for the Tmt-protein (25.7 ± 2.8% of total in each mouse).
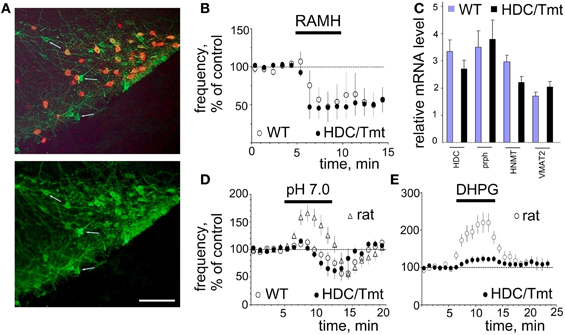
Figure 7. Mouse TMN neurons resemble those of the rat but hardly respond to pH 7.0 and DHPG. (A) Histamine (AF488) co-localization with Tmt protein (expressed in HDC-Cre positive neurons). Note, that virtually all red cells are histamine-positive, but some histamine+ cells do not express HDC-Cre (few cells are indicated by arrows). Scale bar 100 μm. (B) The H3 receptor agonist R-(α)-methyl histamine (RAMH, 2 μM) similarly decreases firing rate of mouse WT (n = 5) and Tmt+ (n = 6) TMN neurons. (C) Relative levels of mRNAs encoding for the markers of histaminergic activity in TMN region of 4 WT and 6 HDC/Tmt mice. HDC, histidine decarboxylase; prph, peripherin; HNMT, histamine N-methyl-transferase; VMAT2, vesicular monoamine transporter 2. (D) Acid-induced excitation is hardly seen in mouse WT (n = 6) or Tmt+ (n = 6) TMN neurons. Data points are superimposed to the time course diagram of acid-induced excitation from rats (n = 23). (E) The metabotropic glutamate receptor agonist DHPG (0.5 μM) excites rat TMN neurons to a significantly larger extent (n = 5) than mouse TMN neurons (n = 5).
The ventrolateral TMN (E2 group) in the mouse is not as compact as in the rat. Moreover, spontaneously firing cells in this region are not always histaminergic. Among 10 recorded cells in wild type (WT) mice (n = 3) only five reduced their firing activity in response to the histamine receptor 3 agonist R-α-methylhistamine (RAMH, 2 μM) and were considered histaminergic neurons (Figure 7). All red (Tmt+) TMN neurons from HDC/Tmt mice responded to RAMH (n = 6) with an inhibition of firing not different from that seen in TMN neurons of WT mice (Figure 7B). Orexin A (100 nM) excited mouse TMN (Tmt+) neurons to 255 ± 63% of control frequency (n = 4, not shown) similar to our previous results in the rat (Eriksson et al., 2001).
Relative mRNA levels encoding for HDC, peripherin, histamine N-methyl-transferase (HNMT) and vesicular monoamine transporter (VMAT2) in the TMN region were found similar between WT and transgenic HDC/Tmt mice (Figure 7C). In contrast to the rat neither WT nor Tmt+ mouse TMN neurons were excited by pH 7.0 (Figure 7D). Another difference was the significantly smaller excitation by the mGluR I agonist DHPG in mice (to 125 ± 12% of control, n = 5) compared to rats (to 220 ± 26% of control (n = 5), p < 0.05).
There was no difference between ammonium-induced excitation between rat and mouse TMN neurons: 177 ± 14 (n = 16) and 240 ± 42% (n = 6) of control, respectively (Figure 8). Thus the HDC-Cre mouse offers a good model for exploring the role of the histaminergic system in hepatic encephalopathy, characterized by the hyperammonemia and increased histamine level in the brain (Lozeva et al., 2003).
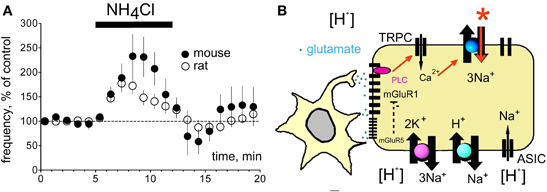
Figure 8. Ammonium and proton-sensing in histaminergic TMN neurons. (A) Excitation by ammonium chloride (5 mM) is not significantly different between mouse and rat. None of the effectors depicted in (B) participates in ammonium-sensing. (B) Proton sensing cascade in TMN neuron, see discussion.
The rat histaminergic neurons in the TMN respond to acidification in the physiological range (pH 7.0) with an excitation resembling that of chemosensors in the brainstem. A similar effect has also been described for the neighboring orexin/hypocretin-containing neurons (Williams et al., 2007) which express glutamatergic markers (Torrealba et al., 2003). Optogenetic stimulation of their axons results in glutamatergic currents in some TMN neurons (Schöne et al., 2011), which may add to the acid-response of TMN neurons. In principle, ATP release from glia in response to acid/CO2 could have excited TMN neurons, however antagonists at purinergic receptors failed to affect the excitation observed here.
We describe an unusually high sensitivity to glutamate of type I metabotropic receptors in TMN neurons. We applied the metabotropic glutamate receptor agonist DHPG at 0.5 μM, much below the concentrations used in previous studies (50–200 μM, Bonsi et al., 2005; Hartmann et al., 2008; Chepkova et al., 2009). Mice lack the pH 7.0-induced excitation and show much smaller responses to DHPG compared to the rat.
Our previous single-cell RT-PCR analysis of mGluR I receptor expression in the rat TMN showed that almost all histaminergic neurons express mGluR1 but only 78% of them are mGluR5-positive (Sergeeva et al., 2006). The rat ventrolateral but not the medial TMN is activated by hypercapnia (Johnson et al., 2005). We show now a lower level of mGluR5 transcripts in this TMN part than in the non-responsive medial TMN. In striatal cholinergic neurons mGluR5 receptors mediate an increased desensitization rate of mGluR1 receptors (Bonsi et al., 2005), thus antagonizing the mGluR1-response. Although different effector systems are involved in mGluR I signaling in striatum and in TMN, such antagonistic interactions between mGluR5 and mGluR1 may provide an explanation for the lacking pH 7.0- mediated excitation in the medial TMN with its higher expression level of mGluR5. Thus our current model explaining the proton-induced excitation of TMN (Figure 8B) depicts mGluR1 as a primary chain. The mGluR1 activates (through PLC) non-selective cation channels, most likely of the TRPC-type, which increase intracellular Ca2+. This is removed by the electrogenic Na+/Ca2+ exchanger (NCX) leading to depolarisation and increased firing. We have previously described NCX as the major effector for several excitatory neurotransmitters in TMN (Eriksson et al., 2004; Parmentier et al., 2009; Yanovsky et al., 2011). Block of ASIC, Na+/H+ and Na+/Ca2+ exchangers by benzamil (20 μM) also abolished the pH 7.0-induced excitation (Figure 4A), indicating that the direct mode of Na+/Ca2+ exchange is essential for the maintenance of low intracellular calcium level and for the neuronal excitation in response to pH 7.0. TMN neurons express variable types and splice variants of Na+/Ca2+ exchangers (NCX) (Sergeeva et al., 2003, 2004), their functional diversity remains to be elucidated. We have shown previously that serotonin, orexins/hypocretins, the dopamine receptor 2 agonist quinpirole and thyrotropin releasing hormone (TRH) excite TMN neurons by activation of NCX (Eriksson et al., 2004; Parmentier et al., 2009; Yanovsky et al., 2011). These transmitters are coupled to phospholipase C, like glutamate through type I metabotropic receptors (Wang et al., 2007), which are involved in acid-induced activation of TMN (see above). In our previous studies (Parmentier et al., 2009; Yanovsky et al., 2011) we found that non-selective cation channels (most likely of TRPC type) make the major contribution and are likely activated by the phospholipase C. Intracellular calcium rise allows sodium influx through the electrogenic (depolarizing) NCX. We did not observe pronounced depolarization in response to pH 7.0 in the present study most likely due to the dissipation of sodium gradient and subsequent reversal of Na+/Ca2+ exchange.
Benzamil but not metabotropic glutamate receptor antagonists blocks proton-induced excitation entirely. Furthermore, acid-induced excitation does not disappear in TMN neurons in slices at room temperature, where NCX function is dramatically reduced (Eriksson et al., 2001; Parmentier et al., 2009). Therefore, we assume contributions from ASICs in pH 7.0-excitation, which is seen only in slices where a larger fraction of dendrites and axons is preserved than in the acutely isolated cells. Such a strategically important location of ASICs may have no impact on somatic membrane conductances but can influence local sodium concentrations and action potential generation. Dissipation of the sodium gradient after blockade of Na+/K+ ATPase may also abolish or reduce ASIC-currents. ASIC1a and ASIC3 are most likely candidates as they are highly sensitive to small, physiological changes in pH (Waldmann et al., 1997; Lingueglia, 2007). Na+/K+ ATPase, Na+/H+ exchanger and the direct mode of Na+/Ca2+ exchange play supportive roles for the pH 7.0-induced excitation maintaining sodium and calcium homeostasis. Impairment of their function abolishes the excitatory response, most likely by turning on the reverse mode of Na+/Ca2+ exchange, which leads to accumulation of intracellular calcium and recruits calcium-activated potassium channels. The inhibition of Na+/K+ ATPase by protons (Fuller et al., 2003) can contribute to the observed excitation of TMN. A block of this pump by ouabain (0.5 μM) causes an acidification and an increase in firing rate comparable to that achieved by pH 7.0. The effects of ouabain and pH 7.0 are not additive: acidification in the presence of ouabain does not produce an excitatory response, indicating that preservation of the sodium gradient under basal conditions is necessary for the excitatory response.
The mechanisms of proton- and ammonium-evoked excitation of the histaminergic neurons are different: blockers of the excitation by protons (ouabain, benzamil hydrochloride, antagonists of mGluRI) did not influence ammonium (5 mM)-induced excitation. Moreover, ammonium-induced excitation did not differ between rat and mouse, whereas proton-induced excitation was only observed in rat, with the large DHPG (0.5 μM)-responses. As ammonium, similar to extracellular protons, causes intracellular acidification (Kelly and Rose, 2010) we believe that the increase in intracellular protons plays a role for both, acid- and ammonium-sensing. We demonstrate now that blockade of proton entry into the cell (Na+/H+ exchanger) eliminates acid-induced excitation, but does not affect ammonium-induced excitation. Kelly and Rose (2010) have shown that ammonium enters hippocampal neurons through the Na+/K+ ATPase, leading to intracellular acidification. We failed to block ammonium-induced excitation with the Na+/K+ ATPase antagonist ouabain.
The novel HDC-Cre mouse presented here will provide unique possibilities to investigate physiological functions of the histaminergic system in normal and pathological brain. In particular, the role of the histaminergic system can be studied now under conditions of hyperammonemia, where it is poorly understood. In hepatic encephalopathy brain concentrations of histamine are elevated (Lozeva et al., 2003). The resulting changes in the histaminergic system are unknown; it could be protective or deleterious. Our recent experiments in histamine-deficient mice (HDC−/−) indicated that histamine may play a protective role against damaging effects of ammonia on hippocampal long-term potentiation (Chepkova et al., 2012). Thus, ammonium but not proton-induced excitation can be studied in transgenic mice which allow manipulating receptors and signaling cascades selectively in histaminergic neurons. This mouse will shed more light on the role of the histaminergic system in health and disease, for instance on obstructive sleep apnea and hepatic encephalopathy.
In conclusion, acidification excites the histaminergic neurons which are prominent keepers of the waking state. Physiological and pathophysiological changes of pH are thus bound to influence many functions of the hypothalamus including sleep-waking, energy homeostasis and the release of hormones.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Deutsche Forschungsgemeinschaft (DFG) SE 1767/1(Heisenberg stipend to Olga A. Sergeeva) and SFB 575 C3 and C8 (to Olga A. Sergeeva and Helmut L. Haas). We wish to acknowledge Dr. Robert E. Hammer, director of the UTSW Transgenic Core Facility, for his assistance with the generation of the HDC-Cre mouse line, as well as Joel K. Elmquist for his technical support. This work was funded from the NIH (K08DK068069-01A2 to Jeffrey M. Zigman) and an International Research Alliance with The Novo Nordisk Foundation Center for Basic Metabolic Research (to Jeffrey M. Zigman). We acknowledge Forschungskomission of Heinrich-Heine University Düsseldorf for the stipend to Anna Kernder.
Akaike, N. (1996). Gramicidin perforated patch recording and intracellular chloride activity in excitable cells. Prog. Biophys. Mol. Biol. 65, 251–264.
Anaclet, C., Parmentier, R., Ouk, K., Guidon, G., Buda, C., Sastre, J. P., Akaoka, H., Sergeeva, O. A., Yanagisawa, M., Ohtsu, H., Franco, P., Haas, H. L., and Lin, J. S. (2009). Orexin/hypocretin and histamine: distinct roles in the control of wakefulness demonstrated using knock-out mouse models. J. Neurosci. 29, 14423–14438.
Bassilana, F., Champigny, G., Waldmann, R., de Weille, J. R., Heurteaux, C., and Lazdunski, M. (1997). The acid-sensitive ionic channel subunit ASIC and the mammalian degenerin MDEG form a heteromultimeric H+-gated Na+ channel with novel properties. J. Biol. Chem. 272, 28819–28822.
Bonsi, P., Cuomo, D., De Persis, C., Centonze, D., Bernardi, G., Calabresi, P., and Pisani, A. (2005). Modulatory action of metabotropic glutamate receptor (mGluR) 5 on mGluR1 function in striatal cholinergic interneurons. Neuropharmacology 49 (Suppl. 1), 104–113.
Buchanan, G. F., and Richerson, G. B. (2010). Central serotonin neurons are required for arousal to CO2. Proc. Natl. Acad. Sci. U.S.A. 107, 16354–16359.
Chepkova, A., Yanovsky, E., Parmentier, R., Ohtsu, H., Haas, H. L., Lin, J. S., and Sergeeva, O. A. (2012). Histamine receptor expression, hippocampal plasticity and ammonia in histidine decarboxylase knockout mice. Cell. Mol. Neurobiol. 32, 17–25.
Chepkova, A. N., Fleischer, W., Kazmierczak, T., Doreulee, N., Haas, H. L., and Sergeeva, O. A. (2009). Developmental alterations of DHPG-induced long-term depression of corticostriatal synaptic transmission: switch from NMDA receptor-dependent towards CB1 receptor-dependent plasticity. Pflugers Arch. 459, 131–141.
Corcoran, A. E., Hodges, M. R., Wu, Y., Wang, W., Wylie, C. J., Deneris, E. S., and Richerson, G. B. (2009). Medullary serotonin neurons and central CO2 chemoreception. Respir. Physiol. Neurobiol. 168, 49–58.
Dhillon, H., Zigman, J. M., Ye, C., Lee, C. E., McGovern, R. A., Tang, V., Kenny, C. D., Christiansen, L. M., White, R. D., Edelstein, E. A., Coppari, R., Balthasar, N., Cowley, M. A., Chua, S. Jr., Elmquist, J. K., and Lowell, B. B. (2006). Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191–203.
Eriksson, K. S., Sergeeva, O. A., Selbach, O., and Haas, H. L. (2004). Orexin (hypocretin)/dynorphin neurons control GABAergic inputs to tuberomammillary neurons. Eur. J. Neurosci. 19, 1278–1284.
Eriksson, K. S., Stevens, D. R., and Haas, H. L. (2001). Serotonin excites tuberomammillary neurons by activation of Na(+)/Ca(2+)-exchange. Neuropharmacology 40, 345–351.
Fuller, W., Parmar, V., Eaton, P., Bell, J. R., and Shattock, M. J. (2003). Cardiac ischemia causes inhibition of the Na/K ATPase by a labile cytosolic compound whose production is linked to oxidant stress. Cardiovasc. Res. 57, 1044–1051.
Gourine, A. V., Kasymov, V., Marina, N., Tang, F., Figueiredo, M. F., Lane, S., Teschemacher, A. G., Spyer, K. M., Deisseroth, K., and Kasparov, S. (2010). Astrocytes control breathing through pH-dependent release of ATP. Science 329, 571–575.
Grunder, S., and Chen, X. (2010). Structure, function, and pharmacology of acid-sensing ion channels (ASICs): focus on ASIC1a. Int. J. Physiol. Pathophysiol. Pharmacol. 2, 73–94.
Haas, H., and Panula, P. (2003). The role of histamine and the tuberomamillary nucleus in the nervous system. Nat. Rev. Neurosci. 4, 121–130.
Haas, H. L., Sergeeva, O. A., and Selbach, O. (2008). Histamine in the nervous system. Physiol. Rev. 88, 1183–1241.
Hartmann, J., Dragicevic, E., Adelsberger, H., Henning, H. A., Sumser, M., Abramowitz, J., Blum, R., Dietrich, A., Freichel, M., Flockerzi, V., Birnbaumer, L., and Konnerth, A. (2008). TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 59, 392–398.
Jarolimek, W., Misgeld, U., and Lux, H. D. (1990). Neurons sensitive to pH in slices of the rat ventral medulla oblongata. Pflugers Arch. 416, 247–253.
Johnson, P. L., Moratalla, R., Lightman, S. L., and Lowry, C. A. (2005). Are tuberomammillary histaminergic neurons involved in CO2-mediated arousal? Exp. Neurol. 193, 228–233.
Kelly, T., and Rose, C. R. (2010). Ammonium influx pathways into astrocytes and neurones of hippocampal slices. J. Neurochem. 115, 1123–1136.
Lee, E. C., Yu, D., Martinez de, V., Tessarollo, L., Swing, D. A., Court, D. L., Jenkins, N. A., and Copeland, N. G. (2001). A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73, 56–65.
Lingueglia, E. (2007). Acid-sensing ion channels in sensory perception. J. Biol. Chem. 282, 17325–17329.
Liu, X., Titz, S., Lewen, A., and Misgeld, U. (2003). KCC2 mediates NH4+ uptake in cultured rat brain neurons. J. Neurophysiol. 90, 2785–2790.
Lozeva, V., Tuomisto, L., Tarhanen, J., and Butterworth, R. F. (2003). Increased concentrations of histamine and its metabolite, tele-methylhistamine and down-regulation of histamine H3 receptor sites in autopsied brain tissue from cirrhotic patients who died in hepatic coma. J. Hepatol. 39, 522–527.
Meng, Q. Y., Wang, W., Chen, X. N., Xu, T. L., and Zhou, J. N. (2009). Distribution of acid-sensing ion channel 3 in the rat hypothalamus. Neuroscience 159, 1126–1134.
Muyrers, J. P., Zhang, Y., and Stewart, A. F. (2001). Techniques: recombinogenic engineering–new options for cloning and manipulating DNA. Trends Biochem. Sci. 26, 325–331.
Parmentier, R., Kolbaev, S., Klyuch, B. P., Vandael, D., Lin, J. S., Selbach, O., Haas, H. L., and Sergeeva, O. A. (2009). Excitation of histaminergic tuberomamillary neurons by thyrotropin-releasing hormone. J. Neurosci. 29, 4471–4483.
Paxinos, G., and Franklin, K. B. J. (2004). The Mouse Brain in Stereotaxic Coordinates: Compact, Second Edn. San Diego, CA: Academic Press.
Perkins, K. L. (2006). Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J. Neurosci. Methods 154, 1–18.
Pidoplichko, V. I., and Dani, J. A. (2006). Acid-sensitive ionic channels in midbrain dopamine neurons are sensitive to ammonium, which may contribute to hyperammonemia damage. Proc. Natl. Acad. Sci. U.S.A. 103, 11376–11380.
Schöne, C., Apergis-Schoute, J., and Burdakov, D. (2011). Optogenetic Probing of Synaptic Transmission Between Orexin/Hypocretin and Histamine Neurons. SFN meeting, Washington, DC:Abstract.
Sergeeva, O. A., Amberger, B. T., Eriksson, K. S., Scherer, A., and Haas, H. L. (2003). Co-ordinated expression of 5-HT2C receptors with the NCX1 Na+/Ca2+ exchanger in histaminergic neurones. J. Neurochem. 87, 657–664.
Sergeeva, O. A., Amberger, B. T., Vorobjev, V. S., Eriksson, K. S., and Haas, H. L. (2004). AMPA receptor properties and coexpression with sodium-calcium exchangers in rat hypothalamic neurons. Eur. J. Neurosci. 19, 957–965.
Sergeeva, O. A., Andreeva, N., Garret, M., Scherer, A., and Haas, H. L. (2005). Pharmacological properties of GABAA receptors in rat hypothalamic neurons expressing the epsilon-subunit. J. Neurosci. 25, 88–95.
Sergeeva, O. A., Eriksson, K. S., Sharonova, I. N., Vorobjev, V. S., and Haas, H. L. (2002). GABA(A) receptor heterogeneity in histaminergic neurons. Eur. J. Neurosci. 16, 1472–1482.
Sergeeva, O. A., Kletke, O., Kragler, A., Poppek, A., Fleischer, W., Schubring, S. R., Gorg, B., Haas, H. L., Zhu, X. R., Lubbert, H., Gisselmann, G., and Hatt, H. (2010). Fragrant dioxane derivatives identify β1-subunit-containing GABAA receptors. J. Biol. Chem. 285, 23985–23993.
Sergeeva, O. A., Klyuch, B. P., Fleischer, W., Eriksson, K. S., Korotkova, T. M., Siebler, M., and Haas, H. L. (2006). P2Y receptor-mediated excitation in the posterior hypothalamus. Eur. J. Neurosci. 24, 1413–1426.
Terada, J., Nakamura, A., Zhang, W., Yanagisawa, M., Kuriyama, T., Fukuda, Y., and Kuwaki, T. (2008). Ventilatory long-term facilitation in mice can be observed during both sleep and wake periods and depends on orexin. J. Appl. Physiol. 104, 499–507.
Torrealba, F., Yanagisawa, M., and Saper, C. B. (2003). Colocalization of orexin a and glutamate immunoreactivity in axon terminals in the tuberomammillary nucleus in rats. Neuroscience 119, 1033–1044.
Vorobjev, V. S., Sharonova, I. N., Haas, H. L., and Sergeeva, O. A. (2003). Expression and function of P2X purinoceptors in rat histaminergic neurons. Br. J. Pharmacol. 138, 1013–1019.
Waldmann, R., Bassilana, F., de Weille, J., Champigny, G., Heurteaux, C., and Lazdunski, M. (1997). Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J. Biol. Chem. 272, 20975–20978.
Wang, M., Bianchi, R., Chuang, S. C., Zhao, W., and Wong, R. K. (2007). Group I metabotropic glutamate receptor-dependent TRPC channel trafficking in hippocampal neurons. J. Neurochem. 101, 411–421.
Williams, R. H., Jensen, L. T., Verkhratsky, A., Fugger, L., and Burdakov, D. (2007). Control of hypothalamic orexin neurons by acid and CO2. Proc. Natl. Acad. Sci. U.S.A. 104, 10685–10690.
Yanovsky, Y., Li, S., Klyuch, B. P., Yao, Q., Blandina, P., Passani, M. B., Lin, J. S., Haas, H. L., and Sergeeva, O. A. (2011). L-Dopa activates histaminergic neurons. J. Physiol. 589, 1349–1366.
Keywords: mGluR, histamine, HDC-Cre, waking, ASIC
Citation: Yanovsky Y, Zigman JM, Kernder A, Bein A, Sakata I, Osborne-Lawrence S, Haas HL and Sergeeva OA (2012) Proton- and ammonium-sensing by histaminergic neurons controlling wakefulness. Front. Syst. Neurosci. 6:23. doi: 10.3389/fnsys.2012.00023
Received: 16 January 2012; Accepted: 21 March 2012;
Published online: 13 April 2012.
Edited by:
Jian-Sheng Lin, INSERM and Claude Bernard University, FranceReviewed by:
Ulrich Misgeld, Institut für Physiologie und Pathophysiologie, GermanyCopyright: © 2012 Yanovsky, Zigman, Kernder, Bein, Sakata, Osborne-Lawrence, Haas and Sergeeva. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Olga A. Sergeeva, Medical Faculty, Molecular Neurophysiology, Heinrich-Heine University, D-40204, Duesseldorf, Germany. e-mail:b2xnYS5zZXJnZWV2YUB1bmktZHVlc3NlbGRvcmYuZGU=
† These authors contributed equally to this work.
‡Present Address: Division of Life Sciences, Graduate School of Science and Engineering, Saitama University, Saitama, Japan.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.