
 Kunal D. Chaniary2
Kunal D. Chaniary2- 1 Department of Neurology, Virginia Commonwealth University, Richmond, VA, USA
- 2 Department of Biomedical Engineering, Virginia Commonwealth University, Richmond, VA, USA
Classical rate-based pathway models are invaluable for conceptualizing direct/indirect basal ganglia pathways, but cannot account for many aspects of normal and abnormal motor control. To better understand the contribution of patterned basal ganglia signaling to normal and pathological motor control, we simultaneously recorded multi-neuronal and EMG activity in normal and dystonic rats. We used the jaundiced Gunn rat model of kernicterus as our experimental model of dystonia. Stainless steel head fixtures were implanted on the skulls and EMG wires were inserted into antagonistic hip muscles in nine dystonic and nine control rats. Under awake, head-restrained conditions, neuronal activity was collected from up to three microelectrodes inserted in the principal motor regions of the globus pallidus (GP), subthalamic nucleus, and entopeduncular nucleus (EP). In normal animals, most neurons discharged in regular or irregular patterns, without appreciable bursting. In contrast, in dystonic animals, neurons discharged in slow bursty or irregular, less bursty patterns. In normal rats, a subset of neurons showed brief discharge bursts coinciding with individual agonist or antagonist EMG bursts. In contrast, in dystonics, movement related discharges were characterized by more prolonged bursts which persist over multiple dystonic co-contraction epics. The pattern of movement related decreases in discharge activity however did not differ in dystonics compared to controls. In severely dystonic rats, exclusively, simultaneously recorded units often showed abnormally synchronized movement related pauses in GP and bursts in EP. In conclusion, our findings support that slow, abnormally patterned neuronal signaling is a fundamental pathophysiological feature of intrinsic basal ganglia nuclei in dystonia. Moreover, from our findings, we suggest that excessive movement related silencing of neuronal signaling in GP profoundly disinhibits EP and in turn contributes to sustained, unfocused dystonic muscle contractions.
Introduction
Dystonia is a devastating condition characterized by ineffective, twisting movements and contorted postures. The prevalence is estimated at 10.7 per 1000 people aged 50–89 years for secondary dystonias due to such causes as Parkinson’s disease (PD), cerebral palsy (CP), and strokes (Wenning et al., 2005). Despite being considered the third most common movement disorder (Kernich, 2003), the underlying pathophysiology remains poorly understood. Largely as a result, no current medical or surgical therapies were intentionally designed to treat dystonia.
According to longstanding basal ganglia circuitry models, the striatum is the major site for motor-related inputs into the basal ganglia and the globus pallidus internus (GPi) is the primary output nucleus (Albin et al., 1989; DeLong, 1990). Excitatory inputs to the striatum via the cortex are thought to inhibit GPi neurons along the “direct” monosynaptic pathway. In contrast, cortical activation of the striatum is considered to excite GPi neurons along the “indirect” GP externa (GPe)–subthalamus (STN) pathway by disinhibiting excitatory STN–GPi projections and by inhibiting inhibitory GPe–GPi projections. Ultimately, net reduced inhibitory output from GPi to the ventrolateral (VL) thalamus is thought to release thalamocortical activation. Although the classical basal ganglia circuitry models provide an invaluable framework for conceptualizing basal ganglia connections, these models have major limitations. Firstly, the models attribute the various hypo- and hyperkinetic movement disorders to opposing alterations in neuronal discharge rates (on the order of seconds to minutes). Yet, overall discharge rates in GPe (Bezard et al., 1999; Raz et al., 2000; Boraud et al., 2001), GPi (Bergman et al., 1994; Bezard et al., 1999; Wichmann and DeLong, 1999), and the thalamus (Pessiglione et al., 2005) are not clearly altered in primate models of PD. Secondly, while such models correctly predict that pallidotomy (GPi ablation) should ameliorate hypokinesia in PD patients (by disinhibiting thalamocortical activity), the models cannot likewise account for comparable surgical benefits of pallidotomy on medication-induced dyskinesias (excessive movements) in these patients (Lozano et al., 1995; Baron et al., 1996). Thirdly, in PD patients undergoing deep brain stimulator (DBS) surgery, intra-operative induction of dyskinesias was associated with alterations in phasic patterned neuronal activity in GPi without corresponding changes in the discharge rates (Lee et al., 2007).
Although many experts in the field are now suggesting that pathological alterations in discharge patterned activity hold the key to understanding the various movement disorders, to date, the abnormally patterned discharge activity has not been systematically investigated in animal models of dystonia. To date, the most extensive neurophysiological recording studies in animals have been conducted in dtsz hamsters, which exhibit paroxysmal dystonia only in response to prolonged stress (Loscher et al., 1989; Richter and Loscher, 1993; Bhatia, 2001). Besides this limitation of the model, the neuronal recordings have been collected under general anesthesia, thereby altering the neuronal signals and not reflecting actual dystonic movements. The tottering mutant mice model, which also has paroxysmal dystonia, as well as ataxia, has also been extensively investigated (Scholle et al., 2010), though the basal ganglia physiology has not been explored in this model. Nambu and colleagues (Chiken et al., 2008) have recorded in the basal ganglia in a mouse model of human DTY1 genetic dystonia without the use of sedation. These mice show prominent hyperkinesia with modest evidence of dystonia. Although abnormal neuronal burst and pause patterns were discovered in GP and entopeduncular nucleus (EP) in these animals, the investigators did not monitor for concurrent dystonia during the neuronal recordings.
Toward the present studies, we advanced techniques to record extracellular neuronal discharge activity from basal ganglia nuclei in dystonic rats without the requirement of ear bars and sedation (Chaniary et al., 2011) and developed objective EMG indicators of dystonia (Chaniary et al., 2008). For our animal model, we used the well-established jaundiced Gunn rat model which has clinical and pathological changes closely resembling human kernicterus (Byers et al., 1955; Perlstein, 1960; Volpe, 2008). Gunn rats are genetically deficient of UDP glucuronosyl transferase, the principal liver enzyme responsible for bilirubin clearance. Homozygous recessive (jj) pups appear normal apart from being jaundiced in the first weeks of life. Free unbound bilirubin blood levels are highest at 16–17 days of age when levels of blood albumin, which binds bilirubin and normally keeps it out of brain, are relatively low (Schutta and Johnson, 1969). We create our model by injecting 16-day-old jjs with sulfonamide (sulfa) to competitively displace bilirubin from blood albumin into the brain (Diamond and Schmid, 1966). The rats become prominently dystonic within minutes to hours after injection. Heterozygous non-jaundiced (Nj) rats have about 50% of the normal enzyme activity (Strebel and Odell, 1971) and remain phenotypically normal even after sulfa injection.
Materials and Methods
Animals and Behavioral Ratings
Nine jj dystonic and nine Nj control rats were used for the present studies. All jjs were injected with sulfonamide and Njs with saline at day 16 ± 24 h. Post-sulfa administration, the acute bilirubin toxicity often resulted in poor spontaneous hydration and food intake. All animals were carefully monitored each day for loss of body weight. As necessary, the animals were compensated with oral feedings of kitten milk formula and given subcutaneous injections of 5% dextrose in 0.45% NaCl. Animals were assessed clinically on a daily basis and a clinical score (CS) between 0 and 5 was assigned based on the severity of the movement disorder (0 – normal, 1 – slight limb dystonia and gait abnormality, 2 – mild limb dystonia and gait abnormality and impaired righting reflex, 3 – moderate limb dystonia and gait abnormality, with prolonged righting reflex, 4 – severe failure of ambulation, general lack of spontaneous movement with occasional bursts of hyperactivity and no righting reflex, and 5 – moribund, including seizures and agonal respiration; Shaia and Shapiro, 2002), and 0.5 was added to scores when midway between categories. All experiments were approved and monitored by the Virginia Commonwealth University IRB and performed in accordance with relevant regulatory guidelines.
Surgical and Stereotaxic Techniques
Surgeries were carried out with 2% general isoflurane anesthesia on day 45 ± 24 h. A manufactured stainless steel head holder was secured to the skull with two sets of four screws to thick portions of the parietal bones on each side of the skull. Next, one hindlimb was surgically exposed and fine Teflon coated (50 μm bare, 110 μm coated) stainless steel wires (A-M systems, Carlsborg, WA, USA) were directly implanted with the aid of a 30 gauge needle and sutured into antagonistic hip (gluteus superficialis, gluteus medius) muscle pairs. To assure correct placement of the wires, the wires were stimulated electrically (Grass Technologies, West Warwick, RI, USA) and the expected hindlimb responses verified. The EMG wires were tunneled together subcutaneously and passed through the opening over the skull and then soldered to a micro-circuit board. The board in turn was secured to a removable Teflon cap, which covered the head holder in-between experiments and was secured to the stereotaxic apparatus during the recording sessions. After the initial surgery, the animals were put back in their cages and allowed 24 h for recovery.
On the first day of recordings, the head holder was clamped into a manufactured positioning mechanism via tapered grooves in its walls (Chaniary et al., 2011). The positioner is mounted onto a standard sized, commercial large animal stereotaxic frame. The system contains three control knobs, which permit independent tilting of the head in three directions (yaw, pitch, and roll). A commercial dial test indicator is mounted onto a customized Plexiglas platform, which slides on the arms of the stereotactic frame. The dial test indicator apparatus was used to achieve flat skull positions along all three directions. The three positioner dials indicated the final placements in each direction. Subsequently, a burr hole was made with a surgical drill to expose the dura for targeting of the hemisphere contralateral to the study hindlimb.
Electrophysiological Recordings
A five-channel mini-xyz-manipulator (Thomas Recording GmbH, Giessen, Germany) was mounted onto a Kopf stereotactic arm and up to three quartz glass-insulated platinum/tungsten microelectrodes (100 μm shaft diam.) were individually passed into the brain using a concentric head configuration with 305 μm intra-electrode spacing targeting single nuclei. The globus pallidus (GP, rodent equivalent of GPe), subthalamic nucleus (STN), and (EP, equivalent of GPi) were identified by their characteristic neuronal firing patterns (illustrated in Figure 1). These neuronal discharge patterns are readily distinguished from those of surrounding structures in normal and dystonic rats. The optic tract, just below EP, was readily identified by its distinct response to light flashes and provided additional confirmation of the location of the microelectrodes. The posterolateral, presumed motor portions of GP, EP, and the STN were specifically targeted. Successful targeting of the intended motor regions was supported by distinguishing neurons whose discharge activity changed in relation to movement. To curtail damage, the number of microelectrode tracts was restricted in each animal.
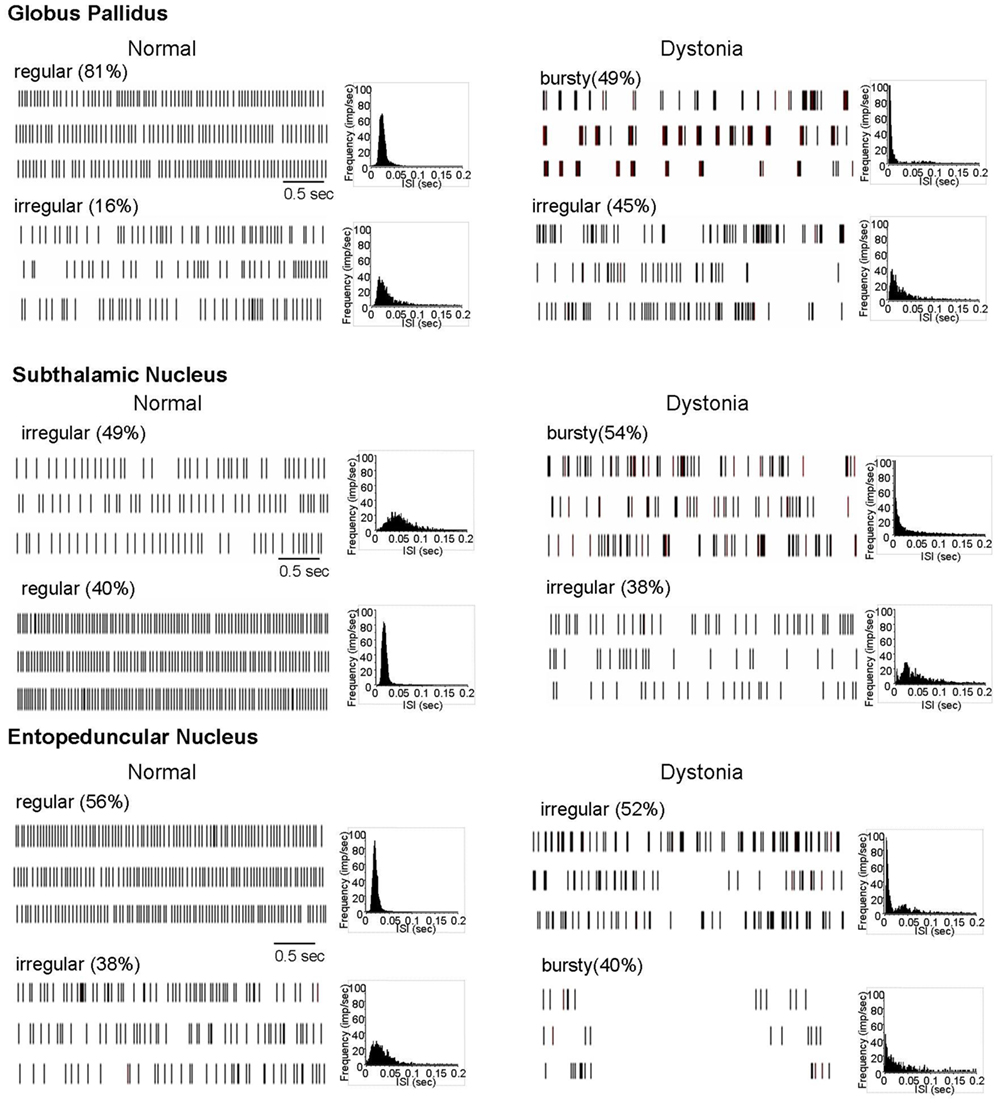
Figure 1. Extracellular neuronal recordings at rest. In normal animals, most neurons in the motor territories of GP, STN, and EP discharged in either a regular or irregular pattern, without appreciable bursting. In contrast, in dystonic animals, most neurons discharged in either a bursty or highly irregular pattern. Three examples of spike rasters for each of the principal patterns are shown, along with grouped ISI histograms.
During the recording sessions, the animal stood on the Plexiglas platform whose height was adjusted to position the animal to stand comfortably and freely move its limbs, with only the head-restrained. During collection of rest activity, towels were often placed on both sides of the animals’ bodies to comfort them and minimize spontaneous movement. Under these conditions, the rats only infrequently displayed overt dystonia. The animals were continuously monitored to assure that they were not moving during collection of rest activity. Towards investigating neuronal correlates of dystonia, neuronal and EMG activity were collected both during spontaneous movements and in response to light touch of the animal’s tail by the examiner. The motor activity varied widely from isolated brief to more prolonged movements of individual limbs to more exaggerated whole body movements and the intent of the movements did not appear to differ between dystonic versus normal animals.
Extracellularly recorded action potentials were monitored online using two digital storage oscilloscopes (HAMEG Instruments, Mainhausen, Germany) and collected at a sampling rate of 20 kHz. Quality of neuronal isolation was additionally monitored online on a desktop computer using Sort Client (Plexon Inc., Dallas, TX, USA). Neuronal and EMG activity were collected for a minimum of 120 s at rest in all 18 rats and with movement in 7 of the dystonics and 7 controls. The neuronal data were plotted on graph paper to establish the boundaries of recorded nuclei and transparencies of parasagittal sections from the Paxinos and Watson (1998) atlas were in turn superimposed on these tracks to estimate the precise location in the brain. EMG signals were amplified (×1000) and filtered (10 Hz–1 kHz) through a differential AC amplifier (A-M Systems, Carlsborg, WA, USA) and digitized through a NI-DAQ card (National Instruments Co., Austin, TX, USA) at a sampling frequency = 4 kHz. The EMG activity was continuously monitored using Sort Client.
Data Analysis and Statistics
Stored digital data were replayed off-line and individual units were sorted using Off-line Spike Sorter (Plexon Inc.) using a combination of manual (k-means clustering, contours and waveform crossing method) and automated sorting techniques (valley-seeking; Lewicki, 1998). This permitted excellent separation of waveforms collected from single or multiple electrodes into distinct clusters. Each separated cluster was carefully examined for loss of neuronal recording and for quality of neuronal isolation. Any obvious artifacts in the signal were removed. Neuronal units were included only if a unit displayed high quality of separation from background noise, the number of recorded potentials exceeded 800, and the activity was recorded for a minimum of 120 s. Also, neuronal units whose location could not be established from the plotted tracks were excluded from all further analysis. Neuronal epics with and without motor activity were considered separately. Neuronal analyses were largely carried out by importing the data into NeuroExplorer software (Nex Technologies, Littleton, MA, USA). For most analyses, neuronal spike data for each nuclei were pooled by treatment groups (jj sulfa, Nj saline).
To define burst periods in a spike train, we initially investigated the Poisson surprise method developed by Legendy and Salcman (1985). This method uses definable levels of “surprise” to determine the unlikeliness of finding burst interspike intervals (ISIs) in a random stream of ISIs, which are assumed to be Poisson-distributed. Despite its popularity, the Poisson surprise method proved to be unreliable for detecting bursts in dystonic rats. Figure 2 shows a representative, well-isolated unit from GP with a clear burst pattern in a dystonic Gunn rat (Figures 2A–C). Using the Poisson surprise method, at lower surprise values, several adjacent burst epics were wrongly grouped as a single burst, while at progressively higher surprise values, the method failed to detect clear bursts (Figure 2D). To overcome these shortcomings of the Poisson surprise method, the interval method was used in which the bursts were identified by individually defining the burst parameters within the algorithm (Figure 3). Based on visual inspection of spike trains in dystonic and control rats, various parameters were chosen and subsequently modified until they proved to be highly reliable in detecting and separating out individual bursts in the spike train. The following final parameters were chosen: max. interval to start burst = 6 ms, max. interval to end burst = 9 ms, min. interval between bursts = 20 ms, min. duration of burst = 5 ms, and min. number of spikes in a burst = 3. As can be seen for the representative unit, using these parameters, the interval method provides far superior burst detection compared to the Poisson surprise method in our animals (Figure 2E).
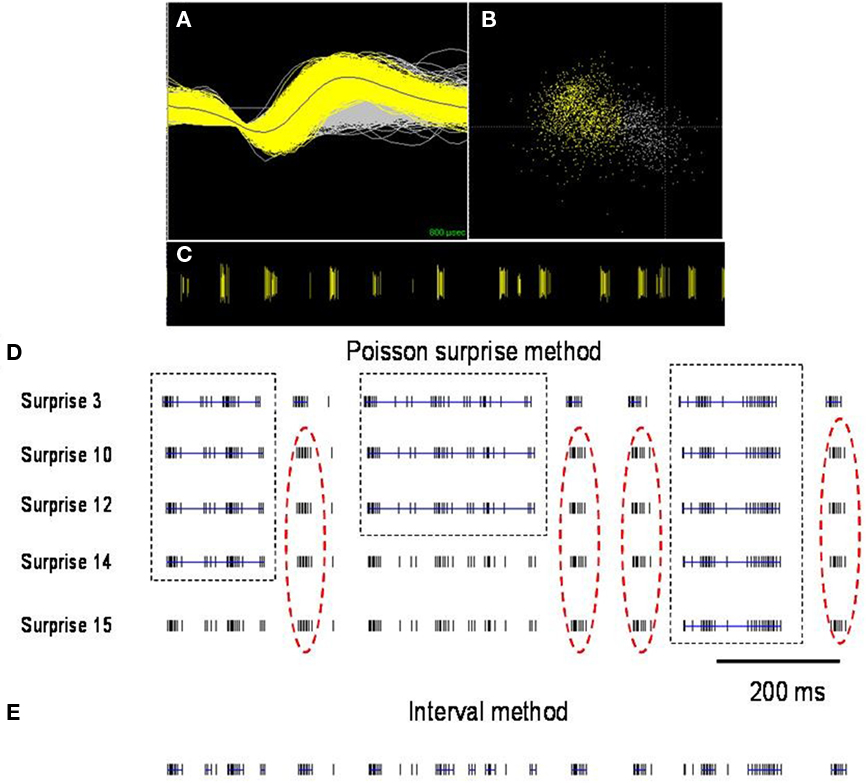
Figure 2. Comparison of burst detection using the Poisson surprise and interval methods. (A) Spike discharge activity (yellow) superimposed on background noise (gray) from a representative neuron in GP of a dystonic rat (B). PCA analysis highlights the neuronal cluster (yellow) from the background (gray). (C) Raster display highlights the representative distinct burst clusters. Each vertical line represents an action potential. (D) Using lower surprise values, the Poisson surprise method wrongly classifies adjacent burst periods as one single burst (dashed boxes) and with higher surprise values, obvious bursts are rejected by the algorithm (red dashed oval). (E) The interval method provides greater reliability for defining individual burst periods. Blue lines indicate bursts detected in the spike trains using the two illustrated burst detection methods.
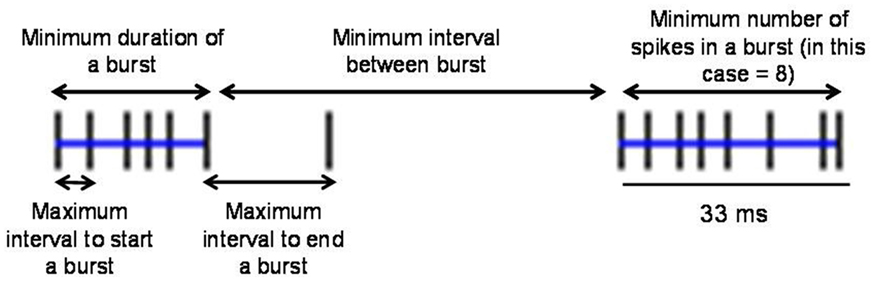
Figure 3. Explanation of parameters used in the interval method to define bursts. The interval method uses the five indicated definable parameters to delineate individual bursts. Blue lines indicate two bursts in the spike train.
Based on inspection of the spike rasters and the shape of the ISI histograms and the results of burst analyses, most neurons in GP, EP, and STN, with the exception of a few outliers, were classified as (1) regular, (2) irregular, or (3) bursty. More specifically, regular firing neurons were characterized by showing tonic discharge activity and symmetrical, sharply peaked, near normally distributed ISI histograms. Irregular firing neurons were characterized by showing tonic discharge activity interrupted by variable length pauses, with flattened, relatively dispersed ISI histograms, with longer pauses reflected by a tail in the ISI histogram. Bursty neurons were characterized by having grouped discharge activity reflected by a leftward peak in the ISI histogram, with significant burstiness using the interval method for burst detection.
Statistically significant relations between neuronal discharge activity and EMG movement epics were detected using peri-event time histogram (PETH) calculations. All segments of the spike train corresponding to EMG movement epics were cut and realigned at the onset of the EMG changes (time = 0) and the histograms plotted for 200 ms before and after the onset of EMG epics. Neurons were considered to show significant movement related responses if four consecutive 10 ms bins differed above or below the baseline mean firing rate (95% confidence limits, p < 0.001). Because we did not perform passive examinations and only hip flexor and extensor muscles were recorded, these relations however could not be established with certainty, nor can we exclude the possibility that non-responsive (or responsive) cells were related to different directional movements at the hip.
Statistical analysis was performed using JMP software package (SAS Institute Inc.). Mean ± SD of the firing rates were determined for each nucleus. Comparisons between the discharge rates, coefficient of variance of ISI’s, and burst parameters between groups were made using one-way ANOVA analysis. Multiple comparisons between groups were made using the Tukey–Kramer honestly significant difference (HSD) test. A probability value of <0.05 was considered to be statistically significant for comparisons between groups.
Results
Phenotype of Dystonic Gunn Rats
Within 24 h after sulfa injection, sulfa treated jaundiced animals developed a movement dystonia that closely resembles pure generalized dystonia in humans. While unrestrained (not formally assessed in the present study), the rats regularly showed mild to severe axial twisting and variable disruptive, prolonged hindlimb extensions with relatively milder forelimb. Of the nine dystonic animals used in this study, six had milder dystonia (CS: ∼2.0), and three had more severe dystonia (CS: ∼3.5). At rest, with their heads restrained for the neuronal and EMG recordings, even more severely dystonic animals showed no active signs of dystonia. With self-initiated or provoked movement while restrained, dystonic rats consistently showed variable severities of dystonic extensor posturing of single to multiple limbs mostly without overt truncal involvement. As illustrated by the recordings of antagonistic hip flexor and extensor EMGs (see Figures 4 and 5), the dystonic movements were heralded by brief (∼1 s) to more prolonged antagonistic muscle co-contractions. Refer to our previous publications for further EMG characterization and videos of the animals (Chaniary et al., 2008) and objective assessments of the dystonic gait (Chaniary et al., 2009).
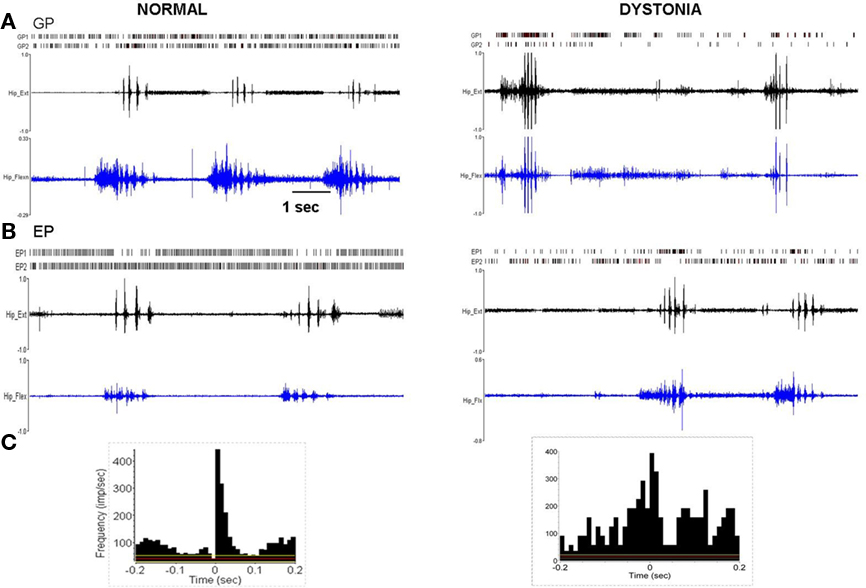
Figure 4. Multi-unit discharge activity during movement in normal and dystonic animals. (A) In normal rats, movement related increases in neuronal discharge activity (illustrated for GP, but also in EP and STN) were dominated by brief bursts which recurred in relation to individual agonist or antagonist EMG epics (p < 0.001). In dystonic rats, movement related increases in discharge were characterized by prolonged bursts mostly persisting over multiple dystonic co-contraction epics (p < 0.001). (B) The pattern of movement related decreases in neuronal discharge activity (illustrated for EP) was similar in normal and dystonic animals. Note that in the illustrated example for a dystonic rat, one neuron shows movement related reduced discharge activity (EP2), while the second neuron (EP1) shows coincident increased movement related discharge activity (a rare co-occurrence in our sample). (C) Peri-event histograms corresponding to the examples in Figure 4A confer significant movement related related increases in discharge activity. The histograms illustrate collective neuronal discharge activity (y-axis) 200 ms before and after the onset of voluntary movement over multiple movement epics (minimum 15). The voluntary movements were either self-initiated or followed a brief flexor withdrawal response in response to light touch of the animal’s tail by the examiner. Red lines indicate the mean firing rate and yellow lines indicate the 95% confidence intervals. Bin size is 10 ms.
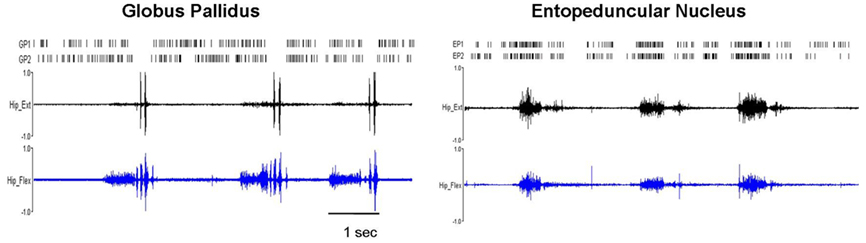
Figure 5. Pathologically synchronized signaling in severely dystonic animals. In more severely dystonic rats (n = 2), exclusively, simultaneously recorded units showed abnormally synchronized movement related pauses in GP (n = 5 pairs, p < 0.001) and bursts in EP (n = 4 pairs, p < 0.001). In distinction, synchronized discharges were never seen in the STN.
Major Alterations in Neuronal Discharge Activity in GP, STN, and EP in Dystonic Rats at Rest
Neuronal discharge rates in dystonic rats were markedly reduced in GP (62%, p < 0.0001) and EP (67%, p < 0.0001) and showed a trend toward a reduction in the STN (33%, p = 0.07). See Table 1 for details of discharge rates and patterns and numbers of animals and neurons.
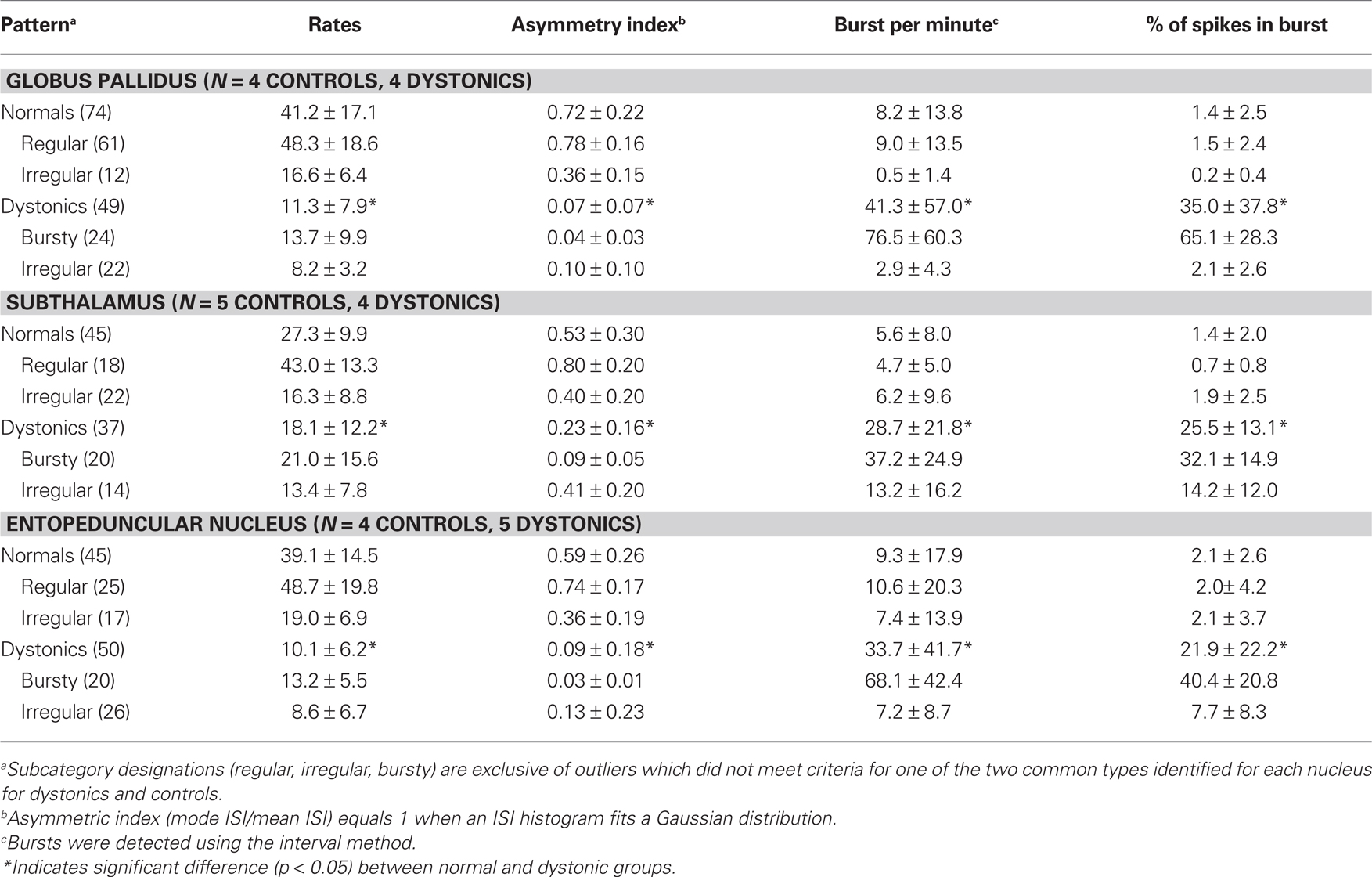
Table 1. Resting discharge rates and patterned activity in GP, STN, and EP (n = 9 dystonic, 9 controls).
In control animals, neuronal activity in GP, STN, and EP was dominated by (1) regular tonic and (2) irregular discharge patterns, with little burst activity (Figure 1). In contrast, in dystonic animals, the patterned activity was dominated by (1) highly irregular and (2) irregular burst activity. During prolonged recordings of up to 5 min, neurons never changed patterns. In distinction from PD and MPTP-treated parkinsonian animals (Hammond et al., 2007), the burst activity was non-oscillatory.
Major Alterations in Neuronal Discharge Activity in GP, STN, and EP in Dystonic Rats during Movement
During voluntary movement epics, hip responsive units in GP, STN, and EP showed phasic increases or decreases in discharge (both p < 0.001) in normal and dystonic rats. In GP, there was a significant shift toward more movement related decreases from increases in neuronal discharge activity in dystonic rats compared to controls. The recorded neuronal discharge activity did not change during externally triggered reflexive withdrawal movements. See Table 2 for details of the movement related neuronal responses, including numbers of animals and neuronal/movement relations.
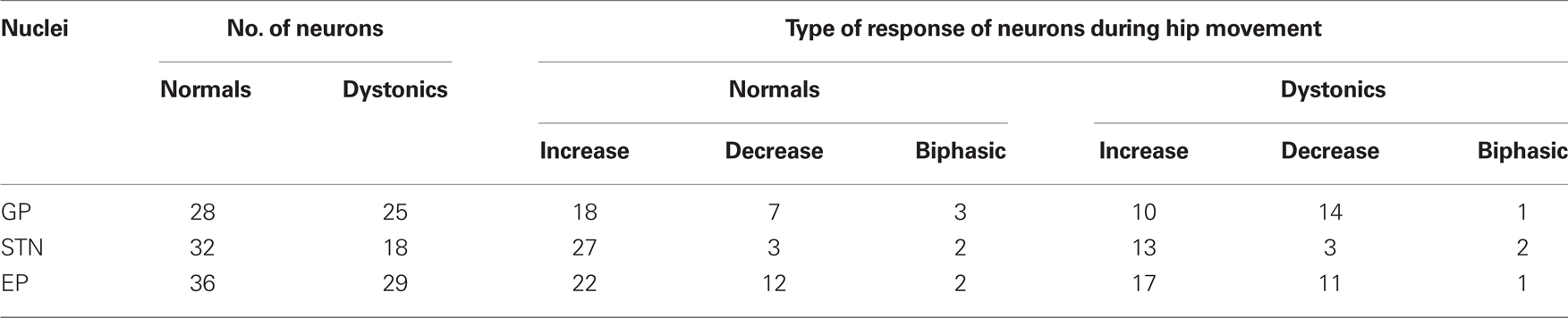
Table 2. Neuronal responses during hip movement (n = 7 dystonics, 7 controls).
In normal rats, movement related increases were characterized by brief discharge bursts coinciding with either individual agonist or antagonist EMG bursts. In contrast, in dystonic rats, neurons showed prolonged bursts persisting over multiple dystonic co-contraction epics (Figure 4). The pattern of movement related decreases in discharge activity however did not differ in dystonics compared to controls. Exclusively in the two severely dystonic rats that were assessed during movement, simultaneously recorded pairs or triplets of neurons separated by an appreciable distance (∼300 μm) regularly showed abnormally synchronized movement related discharge activity, without appreciable time lag between neurons (Figure 5). The synchronized movement related neuronal discharge activity was specifically confined to pauses in GPe (n = 5 pairs) and to bursts in EP (n = 4 pairs). Such synchronization was not observed in the STN.
Discussion
We describe for the first time to our knowledge findings from combined EMG and multi-neuronal recordings in the basal ganglia in unsedated rats with active dystonia. In both normal and dystonic Gunn rats, neurons in motor regions of the basal ganglia commonly increased or decreased discharge firing with movement. In normal rats, movement related increases in neuronal discharge activity were confined to short bursts, which coincided with individual agonist or antagonist EMG bursts. In distinction, in dystonic rats, increases in discharge activity were characterized by prolonged bursts which persisted over multiple co-contraction epics. Also, in more severely affected animals, pairs or triplets of neurons in GP and EP frequently discharged in synchrony. Moreover, with movement, synchronized alternations in discharge activity were limited in GP to practical silencing of discharge and in EP to prominent bursting. While our findings in part support traditional basal ganglia modeling of inhibitory influences of GPe on GPi along the indirect pathway, they provide novel insight into the contributions of precision multi-neuronal phasic basal ganglia signaling to normal and pathological motor control.
At rest, without clinical or EMG evidence for active dystonia, neuronal activity in the basal ganglia of dystonic rats was notable for prominently irregular burst discharge activity with markedly reduced discharge rates in GP and EP, and to a lesser degree in the STN. These background changes in discharge activity are for the most part comparable to reports from humans undergoing DBS surgery for various dystonias. For instance, the discharge rates in GPi in humans with dystonia were mostly reduced compared to rates in normal monkeys (Lenz et al., 1998; Vitek et al., 1999; Sanghera et al., 2003; Merello et al., 2004; Zhuang et al., 2004; Starr et al., 2005). In contrast to the tonic discharge activity in GPi in normal monkeys, the discharge pattern in dystonic patients was dominated by grouped burst discharges on a particularly silent background. In one investigation of GPe (Vitek et al., 1999), neuronal activity also showed abnormal burst activity, with reduced discharge rates compared to normal monkeys. In contrast, another group (Starr et al., 2005) did not find any definite abnormalities in the neuronal activity in GPe apart from occasional atypical oscillations. However, of potential significance, the motor portion of GPi is specifically targeted during these surgeries using different angular approaches, and therefore, there may be considerable differences between groups with respect to traversing motor versus non-motor territories of GPe. In a single investigation of the STN (Starr et al., 2005), the discharge pattern was dominated by irregular grouped discharges, without clear alterations in the discharge rates.
The presence of highly similar resting neuronal discharge rates and patterned activity in the basal ganglia in dystonic Gunn rats and in humans with various dystonias importantly supports the validity of the Gunn rat animal model. Further, as demonstrated in Gunn rats, patterned discharge activity in GPi (Vitek et al., 1999), STN (Zhuang et al., 2004), and the thalamus (Lenz et al., 1999; Zhuang et al., 2004) was found to correlate with dystonic EMG activity in humans. Moreover, Lenz et al. (1999) showed that the thalamic activity preceded correlated EMG activity. This observation supports that at least for the thalamus, the correlated activity in dystonia is not likely to be epiphenomena. Additionally, because human recording studies have not shown clear differences between various dystonias, our findings should have wide relevance for many forms of dystonia.
In severely dystonic animals, exclusively, two to three simultaneously recorded neurons in GP or EP separated by an appreciable distance (∼300 μm) were often observed to discharge largely in unison without appreciable time lags. From this finding, we hypothesize that in dystonia, abnormally synchronized basal ganglia signaling occurs in lieu of normally highly selective activation of intended and inactivation of unintended muscles during movement and in turn, is responsible for the hallmark overflow of contractions to unintended muscles. Further, we postulate that our findings of exaggerated and synchronized burst activation of neurons in EP which persist throughout repetitive muscle co-contractions contributes to prolonged co-contractions, another hallmark feature of this disorder. Additionally, from prior studies suggesting that inactivation of GP could cause various dystonias and our discovery of excessive and synchronized silencing of neuronal discharge in GP during dystonic movements, we postulate that the etiological source for dystonia in many human forms of dystonia is exaggerated and synchronized movement related silencing of neuronal discharge activity in GP. A surprising finding is the apparently less dramatic alterations in rates and patterned activity in the STN, along with the absence of clear prominent synchronization in dystonia. We reason that in dystonia, excessive and synchronized silencing of GP neurons could lead to a prominent and powerfully influential reduction in GABAergic inhibitory output from GP directly to the proximal dendrites and soma of EP neurons, and, in turn, largely induce prominent synchronized bursting of EP neurons.
Dopamine (DA) concentrations were found to be reduced in a mouse model of DYT1 dystonia, the most common genetic form of human dystonia (Balcioglu et al., 2007; Hewett et al., 2010). DA depletion can be expected to disinhibit DA D2 striatal GABAergic projection neurons and in turn suppress neuronal discharge activity in GPe. Further, in transgenic DTY1 mice, striatal DA D2 receptor function was reported to be impaired and associated with disinhibition of GABAergic transmission (Sciamanna et al., 2009). In humans, Dopa-responsive dystonia has been linked to specific genetic defects which lead to deficiencies in DA production (Ichinose et al., 1994). Using positive emission tomography (PET), Perlmutter et al. (1997a) showed that striatal DA D2 receptor labeling striatum was reduced in patients with focal idiopathic dystonia. These investigators (Perlmutter et al., 1997b) also demonstrated that transiently induced dystonia in MPTP-treated monkeys was temporally correlated with reductions in DA D2-like receptor binding. This background, along with our present findings, leads us to hypothesize that various pathologies along the indirect pathway similarly cause dystonia via exaggerated and synchronized silencing of GPe neurons. This postulate is in part supported by human reports of GP strokes producing dystonia (Ünchau et al., 2000).
Post-mortem studies from human newborns with kernicterus have shown prominent cell loss and gliosis in GPe, GPi, STN, substantia nigra pars reticulata, and less consistently in the cerebellum, hippocampus, and brain stem auditory and oculomotor nuclei (Jew and Sandquist, 1979; Ahdab-Barmada and Moossy, 1984). A number of groups have shown that the pathological changes in jaundiced Gunn rats closely approximate those of human kernicterus (Blanc and Johnson, 1959; Schutta and Johnson, 1967, 1969, 1971; Sturrock and Jew, 1978; Rose and Wisniewski, 1979). Considering the extent of the pathology in kernicterus, the pathogenesis of the dystonia in kernicterus has remained elusive. We initially considered two not necessarily exclusive possibilities: (1) that the dystonia in kernicterus results from a loss of motor-related signaling as a direct result of the loss of cells or (2) that it occurs due to abnormal signaling in surviving cells. The presence of largely movement-induced dystonia in Gunn rats that correlates with phasic changes in signaling of basal ganglia neurons is more supportive of the latter explanation. Studies in torsinA mutant mice have shown that torsinA, the principal protein product of the DYT1 gene is essential to maintain the normal neuronal nuclear membrane structure (Kim et al., 2010). Therefore, mechanisms other than DA dysfunction may play major contributory roles in the development of aberrant signaling in both primary and secondary forms of dystonia.
We acknowledge a number of limitations of the present studies. Firstly, although synchronization was only observed in more severely dystonic animals, we suggest that our small sampling size could have been expected to only uncover profound degrees of synchronization. Secondly, our limited sampling of multi-unit recordings in the STN in dystonic rats was inadequate to exclude comparable synchronization as demonstrated in GP and EP. Thirdly, because we did not perform passive examinations or more extensive EMG recordings over multiple joints, direct relations between the recorded neurons and hip muscle flexor or extensor contractions could not be reliably established. In turn, the temporal relations between the neuronal and EMG events could not be established. Lastly, although the clinical features appear to be highly similar between primary and secondary forms, the relative shortcomings of DBS and pallidotomy for most secondary dystonias (Holloway et al., 2006) infer limitations in generalizing our findings to other dystonias, in particular to primary forms.
In summary, we have presented novel findings from simultaneously recording from the basal ganglia, along with EMGs from antagonist muscle pairs in unsedated rodents with active dystonia. From our findings, we suggest that in dystonia, dramatic alterations in the underlying discharge activity of basal ganglia neurons, along with a profound loss of independent neuronal signaling, leads to highly pathological signaling during movement. Moreover, we propose that exaggerated and unfocused silencing of GPi neurons during movement results in profound, highly synchronized movement related burst activity in EP which contributes to the hallmark sustained co-contractions and spread to unintended muscles in dystonia. Why surgical approaches to the GPi do not generally improve such abnormalities in secondary dystonias to a similar degree as seen for primary dystonias remains to be determined. Our methodology could serve to lay the groundwork for pursing similar comparison studies in other animal models of dystonia.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by NIH/NINDS grant R01NS47151 to Steven M. Shapiro and a Thomas F. Kate Miller Jeffress Memorial Trust award to Mark S. Baron.
References
Ahdab-Barmada, M., and Moossy, J. (1984). The neuropathology of kernicterus in the premature neonate: diagnostic problems. J. Neuropathol. Exp. Neurol. 43, 45–56.
Albin, R. L., Young, A. B., and Penney, J. B. (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375.
Balcioglu, A., Kim, M. O., Sharma, N., Cha, J. H., Breakefield, X. O., and Standaert, D. G. (2007). Dopamine release is impaired in a mouse model of DYT1 dystonia. J. Neurochem. 102, 783–788.
Baron, M. S., Vitek, J. L., Bakay, R. A., Green, J., Kaneoke, Y., Hashimoto, T., Turner, R. S., Woodard, J. L., Cole, S. A., McDonald, W. M., and DeLong, M. R. (1996). Treatment of advanced Parkinson’s disease by posterior GPi pallidotomy, 1-year results of a pilot study. Ann. Neurol. 40, 355–366.
Bergman, H., Wichmann, T., Karmon, B., and DeLong, M. R. (1994). The primate subthalamic nucleus. II. neuronal activity in the MPTP model of parkinsonism. J. Neurophysiol. 72, 507–520.
Bezard, E., Boraud, T., Bioulac, B., and Gross, C. E. (1999). Involvement of the subthalamic nucleus in glutamatergic compensatory mechanisms. Eur. J. Neurosci. 11, 2167–2170.
Bhatia, K. P. (2001). Familial (idiopathic) paroxysmal dyskinesias: an update. Semin. Neurol. 21, 69–74.
Blanc, W. A., and Johnson, L. (1959). Studies on kernicterus; relationship with sulfonamide intoxication, report on kernicterus in rats with glucuronyl transferase deficiency and review of pathogenesis. J. Neuropathol. Exp. Neurol. 18, 165–187; discussion 187–189.
Boraud, T., Bezard, E., Bioulac, B., and Gross, C. E. (2001). Dopamine agonist-induced dyskinesias are correlated to both firing pattern and frequency alterations of pallidal neurones in the MPTP-treated monkey. Brain 124, 546–557.
Byers, R. K., Paine, R. S., and Crothers, B. (1955). Extrapyramidal cerebral palsy with hearing loss following erythroblastosis. Pediatrics 15, 248–254.
Chaniary, K., Baron, M., Rice, A., Wetzel, P., and Shapiro, S. (2008). Electromyographic characterization in an animal model of dystonia. Mov. Disord. 23, 1122–1129.
Chaniary, K. D., Baron, M. S., Rice, A. C., Wetzel, P. A., Ramakrishnan, V., and Shapiro, S. M. (2009). Quantification of gait in dystonic Gunn rats. J. Neurosci. Methods 180, 273–277.
Chaniary, K. D., Baron, M. S., Robinson, P., Rice, A. C., and Shapiro, S. M. (2011). A novel stereotaxic apparatus for neuronal recordings in awake head-restrained rats. J. Neurosci. Methods 198, 29–35.
Chiken, S., Shashidharan, P., and Nambu, A. (2008). Cortically evoked long-lasting inhibition of pallidal neurons in a transgenic mouse model of dystonia. J. Neurosci. 28, 13967–13977.
DeLong, M. R. (1990). Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13, 281–285.
Diamond, I., and Schmid, R. (1966). Experimental bilirubin encephalopathy. the mode of entry of bilirubin-14C into the central nervous system. J. Clin. Invest. 45, 678–689.
Hammond, C., Bergman, H., and Brown, P. (2007). Pathological synchronization in Parkinson’s disease: networks, models and treatments. Trends Neurosci. 30, 357–364.
Hewett, J., Johanson, P., Sharma, N., Standaert, D., and Balcioglu, A. (2010). Function of dopamine transporter is compromised in DYT1 transgenic animal model in vivo. J. Neurochem. 113, 228–235.
Holloway, K. L., Baron, M. S., Brown, R., Cifu, D. X., and Ramesh, V. (2006). Deep brain stimulation for dystonia: a meta-analysis. Neuromodulation 9, 253–261.
Ichinose, H., Ohye, T., Takahashi, E., Seki, N., Hori, T., Segawa, M., Nomura, Y., Endo, K., Tanaka, H., Tsuji, S., Fujita, K., and Nagatsu, T. (1994). Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat. Genet. 8, 236–242.
Jew, J. Y., and Sandquist, D. (1979). CNS changes in hyperbilirubinemia. Functional implications. Arch. Neurol. 36, 149–154.
Kim, C. E., Perez, A., Perkins, G., Ellisman, M. H., and Dauer, W. T. (2010). A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc. Natl. Acad. Sci. U.S.A. 107, 9861–9866.
Lee, J. I., Verhagen Metman, L., Ohara, S., Dougherty, P. M., Kim, J. H., and Lenz, F. A. (2007). Internal pallidal neuronal activity during mild drug-related dyskinesias in Parkinson’s disease: decreased firing rates and altered firing patterns. J. Neurophysiol. 97, 2627–2641.
Legendy, C. R., and Salcman, M. (1985). Bursts and recurrences of bursts in the spike trains of spontaneously active striate cortex neurons. J. Neurophysiol. 53, 926–939.
Lenz, F. A., Jaeger, C. J., Seike, M. S., Lin, Y. C., Reich, S. G., DeLong, M. R., and Vitek, J. L. (1999). Thalamic single neuron activity in patients with dystonia: dystonia-related activity and somatic sensory reorganization. J. Neurophysiol. 82, 2372–2392.
Lenz, F. A., Suarez, J. I., Metman, L. V., Reich, S. G., Karp, B. I., Hallett, M., Rowland, L. H., and Dougherty, P. M. (1998). Pallidal activity during dystonia: somatosensory reorganization and changes with severity. J. Neurol. Neurosurg. Psychiatr. 65, 767–770.
Lewicki, M. S. (1998). A review of methods for spike sorting: the detection and classification of neural action potentials. Network 9, R53–R78.
Loscher, W., Fisher, J. E. Jr., Schmidt, D., Fredow, G., Honack, D., and Iturrian, W. B. (1989). The sz mutant hamster: a genetic model of epilepsy or of paroxysmal dystonia? Mov. Disord. 4, 219–232.
Lozano, A. M., Lang, A. E., Galvez-Jimenez, N., Miyasaki, J., Duff, J., Hutchinson, W. D., and Dostrovsky, J. O. (1995). Effect of GPi pallidotomy on motor function in Parkinson’s disease. Lancet 346, 1383–1387.
Merello, M., Cerquetti, D., Cammarota, A., Tenca, E., Artes, C., Antico, J., Antico, J., and Leiguarda, R. (2004). Neuronal globus pallidus activity in patients with generalised dystonia. Mov. Disord. 19, 548–554.
Paxinos, G., and Watson, W. C. (1998). The Rat Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press.
Perlmutter, J. S., Stambuk, M. K., Markham, J., Black, K. J., McGee-Minnich, L., Jankovic, J., and Moerlein, S. M. (1997a). Decreased [18F]spiperone binding in putamen in idiopathic focal dystonia. J. Neurosci. 17, 843–850.
Perlmutter, J. S., Tempel, L. W., Black, K. J., Parkinson, D., and Todd, R. D. (1997b). MPTP induces dystonia and parkinsonism. Clues to the pathophysiology of dystonia. Neurology 49, 1432–1438.
Perlstein, M. A. (1960). The late clinical syndrome of posticteric encephalopathy. Pediatr. Clin. North Am. 7, 665–687.
Pessiglione, M., Guehl, D., Rolland, A. S., Francois, C., Hirsch, E. C., Feger, J., and Tremblay, L. (2005). Thalamic neuronal activity in dopamine-depleted primates: evidence for a loss of functional segregation within basal ganglia circuits. J. Neurosci. 25, 1523–1531.
Raz, A., Vaadia, E., and Bergman, H. (2000). Firing patterns and correlations of spontaneous discharge of pallidal neurons in the normal and the tremulous 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine vervet model of parkinsonism. J. Neurosci. 20, 8559–8571.
Richter, A., and Loscher, W. (1993). Alterations in pharmacological sensitivity of GABAergic but not dopaminergic and glutamatergic systems during ontogenesis in dystonic mutant hamsters. Eur. J. Pharmacol. 231, 111–119.
Rose, A. L., and Wisniewski, H. (1979). Acute bilirubin encephalopathy induced with sulfadimethoxine in Gunn rats. J. Neuropathol. Exp. Neurol. 38, 152–164.
Sanghera, M. K., Grossman, R. G., Kalhorn, C. G., Hamilton, W. J., Ondo, W. G., and Jankovic, J. (2003). Basal ganglia neuronal discharge in primary and secondary dystonia in patients undergoing pallidotomy. Neurosurgery 52, 1358–1370.
Scholle, H. C., Jinnah, H. A., Arnold, D., Biedermann, F. H., Faenger, B., Grassme, R., Hess, E. J., and Schumann, N. P. (2010). Kinematic and electromyographic tools for characterizing movement disorders in mice. Mov. Disord. 25,265–274.
Schutta, H. S., and Johnson, L. (1967). Bilirubin encephalopathy in the Gunn rat: a fine structure study of the cerebellar cortex. J. Neuropathol. Exp. Neurol. 26, 377–396.
Schutta, H. S., and Johnson, L. (1969). Clinical signs and morphologic abnormalities in Gunn rats treated with sulfadimethoxine. J. Pediatr. 75, 1070–1079.
Schutta, H. S., and Johnson, L. (1971). Electron microscopic observations on acute bilirubin encephalopathy in Gunn rats induced by sulfadimethoxine. Lab. Invest. 24, 82–89.
Sciamanna, G., Bonsi, P., Tassone, A., Cuomo, D., Tscherter, A., Viscomi, M. T., Martella, G., Sharma, N., Bernardi, G., Standaert, D. G., and Pisani, A. (2009). Impaired striatal D2 receptor function leads to enhanced GABA transmission in a mouse model of DYT1 dystonia. Neurobiol. Dis. 34, 133–145.
Shaia, W., and Shapiro, S. M. (2002). Immunhistochemical localization of calcium-binding proteins in the brainstem vestibular nuclei of the jaundiced Gunn rat. Hear. Res. 173, 82–90.
Starr, P. A., Rau, G. M., Davis, V., Marks, W. J. Jr, Ostrem, J. L., Simmons, D., Lindsey, N., and Turner, R. S. (2005). Spontaneous pallidal neuronal activity in human dystonia: comparison with Parkinson’s disease and normal macaque. J. Neurophysiol. 93, 3165–3176.
Strebel, L., and Odell, G. B. (1971). Bilirubin uridine disphosphoglucuronyltransferase in rat liver microsomes: genetic variation and maturation. Pediatr. Res. 5, 548–559.
Sturrock, R. R., and Jew, J. Y. (1978). A quantitative histological study of changes in neurons and glia in the Gunn rat brain. Neuropathol. Appl. Neurobiol. 4, 209–223.
Ünchau, A., Mathen, D., Cox, T., Quinn, N. P., Marsden, C. D., and Bhatia, K. P. (2000). Unilateral lesions of the globus pallidus: report of four patients presenting with focal or segmental dystonia. J. Neurol. Neurosurg. Psychiatr. 69, 494–498.
Vitek, J. L., Chockkan, V., Zhang, J. Y., Kaneoke, Y., Evatt, M., DeLong, M. R., Triche, S., Mewes, K., Hashimoto, T., and Bakay, R. A. (1999). Neuronal activity in the basal ganglia in patients with generalized dystonia and hemiballismus. Ann. Neurol. 46, 22–35.
Volpe, J. J. (2008) “Bilirubin and brain injury,” in Neurology of the Newborn, 5th Edn, ed. J. J. Volpe (Philadelphia, PA: W. B. Saunders), 619.
Wenning, G. K., Kiechl, S., Seppi, K., Muller, J., Hogl, B., Saletu, M., Rungger, G., Gasperi, A., Willeit, J., and Poewe, W. (2005). Prevalence of movement disorders in men and women aged 50–89 years (Bruneck study cohort): a population-based study. Lancet Neurol. 4, 815–820.
Keywords: dystonia, basal ganglia, globus pallidus, subthalamus, entopeduncular nucleus, microelectrode recording, rats
Citation: Baron MS, Chaniary KD, Rice AC and Shapiro SM (2011) Multi-neuronal recordings in the basal ganglia in normal and dystonic rats. Front. Syst. Neurosci. 5:67. doi: 10.3389/fnsys.2011.00067
Received: 23 April 2011; Accepted: 01 August 2011;
Published online: 31 August 2011.
Edited by:
Charles J. Wilson, University of Texas at San Antonio, USAReviewed by:
Atsushi Nambu, National Institute for Physiological Sciences, JapanHitoshi Kita, The University Tennessee Health Science Center, USA
Copyright: © 2011 Baron, Chaniary, Rice and Shapiro. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Mark S. Baron, Department of Neurology, Virginia Commonwealth University, 6th Floor Sanger Hall, Box 980599, Richmond, VA 23220, USA. e-mail:bWJhcm9uQG1jdmgtdmN1LmVkdQ==