 Zeinab Chahine
Zeinab Chahine Karine G. Le Roch
Karine G. Le Roch
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Syst. Biol. , 16 September 2022
Sec. Translational Systems Biology and In Silico Trials
Volume 2 - 2022 | https://doi.org/10.3389/fsysb.2022.940321
This article is part of the Research Topic Systems Biology, Women in Science 2021/22: Translational Systems Biology and In Silico Trials View all 5 articles
The human malaria parasite, Plasmodium falciparum, is a unicellular protozoan responsible for over half a million deaths annually. With a complex life cycle alternating between human and invertebrate hosts, this apicomplexan is notoriously adept at evading host immune responses and developing resistance to all clinically administered treatments. Advances in omics-based technologies, increased sensitivity of sequencing platforms and enhanced CRISPR based gene editing tools, have given researchers access to more in-depth and untapped information about this enigmatic micro-organism, a feat thought to be infeasible in the past decade. Here we discuss some of the most important scientific achievements made over the past few years with a focus on novel technologies and platforms that set the stage for subsequent discoveries. We also describe some of the systems-based methods applied to uncover gaps of knowledge left through single-omics applications with the hope that we will soon be able to overcome the spread of this life-threatening disease.
The Plasmodium parasite is transmitted to human hosts by the female Anopheles mosquito. This pathogen caused over 600,000 deaths worldwide in 2020, mostly affecting children under the age of five in malaria endemic countries (WHO, 2021). It is also important to note the global economic impact of malaria, in which roughly $4.3 billion dollars were spent worldwide in the efforts to treat and control the spread of disease in the year 2016 alone (WHO, 2021). Effective antimalarials and sensitive diagnostic kits play important roles in the efforts to monitor its spread and quell potential outbreaks. Most notably, the use of artemisinin-based combination therapies (ACTs) has had a prominent effect upon treatment since its approval by the WHO in 2006 (Dondorp et al., 2009). However, like several other antimalarial therapeutics discovered, evidence of resistance to artemisinin has been on the rise, with earliest cases of resistance observed as early as 2009 (Dondorp et al., 2009; Haakenstad et al., 2019). ACTs are the last line of defense against the most virulent species causing human malaria, P. falciparum (White et al., 2014; Noreen et al., 2021). Furthermore, over the past 2 years, control efforts have been radically hindered with the onset of the Covid-19 pandemic that resulted in drastic logistical impediments and financial redistricting of global funds and resources to combat the disease. This led to the sudden rise in death from 558,000 in the year 2019 to 627,000 in 2020 (an increase of 12%) (WHO, 2021). Moreover, with the lack of a highly protective vaccine, it is imperative to gain better insight into the parasite to identify novel therapeutic strategies and provide alternative diagnostic tools to prevent the spread of disease. In this review we provide a brief overview on some of the most important discoveries made in Plasmodium research with an emphasis on the innovative technologies used to advance our understanding of the parasite through a multi-omics, systems -based lens of study. In contrast to past decades of directed, reductionist biology research that take on a one-sided view of specific proteins or pathways, systems biology encompasses a multifaced vantage point of study. In doing so, we can identify how individual components fit together into the global scheme of a cell’s structure, behavioral responses and development. In this review, we bring attention to each of the major omics-based branches of study that serve as an important element of systems biology. In discussing the history of Plasmodium parasite research, we highlight some of the groundbreaking contributions brought forth with emerging innovations in technologies. We shed light on some of the major benefits and limitations of these technologies and how, through application of a systems-based approach, some of the more complex characteristics of the parasite life cycle can be understood and potentially overcome.
P. knowelsi is a human infecting parasite but is listed among the non-human infecting parasites. The Plasmodium species belong to the SAR supergroup of pathogens which includes Stramenopiles, Alveolates and Rhizaria. They can be categorized by their major hosts, infecting human (P. vivax, P. malariae, P. ovale, and P. falciparum), non-human primates (P. reichenowi, P. cynomolgi and P. knowlesi which infect chimpanzees and macaques, respectively), rodents (P. berghei, P. chabaudi, and P. yoelii) birds (P. gallinaceum) and even reptiles (P. mexicanum), each of which hold particular interest in the scientific community as they serve as important model systems (Pacheco et al., 2013; Bohme et al., 2018; van de Straat et al., 2022). Here we will primarily focus on human infective parasites. Infection begins through the inoculation of sporozoites into human hosts. Here, mature sporozoites will enter the blood stream where they circulate until they reach the liver within 30–60 min after first inoculation. Inside the liver, parasites initiate a series of differentiation and replication events to generate thousands of merozoites. It should be noted that the liver stages of the Plasmodium life cycle can be unique to each species. For example, in P. vivax and P. ovale, a dormant stage hypnozoite can persist for months or even years in the liver before being reactivated and released into the bloodstream (Mueller et al., 2007; Merrick, 2021; Mehra et al., 2022). After 6–10 days (sometimes longer depending on the species), the merozoites leave the liver to invade red blood cells where they initiate the asexual intraerythrocytic developmental cycle (IDC). The asexual cycle consists of a remarkable series of morphological changes, from the relatively dormant ring stage to the highly dynamic and rapidly metabolizing trophozoite stage or to the dividing schizont stage. During this trophozoite stage the parasite decondenses its chromatin to allow intensive transcription and cellular activity leading to parasite replication during schizogony. The asexual cycle can last 24, 48 or 72 h—depending on the species, and undergo asymmetric rounds of mitosis to produce 16 to 64 new infective merozoites that are then released to infect new red blood cells (White et al., 2014). This phase of the parasite life cycle is symptomatic and can cause febrile waves, headache, vomiting, anemia, coma, and eventually death. The IDC is repeated if the favored environmental conditions are met. Should the pathogen experience any nutrient deprivation, changes in temperature or stress signaling, it can shift from the IDC to the sexual stages of development. During sexual development, parasites differentiate into male and female gametocytes (Chawla et al., 2021). This stage involves five morphologically unique steps that may last 2–3 weeks to maturation. Once mature, gametocytes circulate the mammalian host circulatory systems awaiting uptake by a new Anopheles host. Inside mosquito gut lumen, increased Ca2+ levels, drop in pH and temperature levels along with other signaling cues such as xanthurenic acid, trigger the differentiation of the haploid gametocytes into gametes. A female gametocyte develops into a macro-gamete through a series of cytoplasmic reorganizing events and remains immotile in the midgut. The male gametocyte, on the other hand, morphs into a micro-gamete. The micro-gamete nucleus undergoes three rounds of replication to form 6–8 nuclei and undergoes a process of ex-flagellation. This cluster-like multinucleated cytoplasmic body subsequently breaks apart into individual male gametes that are directed to the macro-gamete. In the mosquito midgut male and female gametes fuse to form a diploid zygote that will subsequently develop into a motile ookinete. The ookinete travels and penetrates the inner walls of the mosquito gut to undergo encystation. Once encystation occurs, the ookinete (now called an oocyst) continues to develop in the epithelium of the midgut forming sporozoites. The mature sporozoites will then erupt from the oocyst and migrate to the salivary glands to infect a new human host (Figure 1) (Chawla et al., 2021; Dvorin and Goldberg, 2022). This elaborate life cycle is controlled by complex networks of proteins that are tightly regulated at the transcriptional and translational levels.
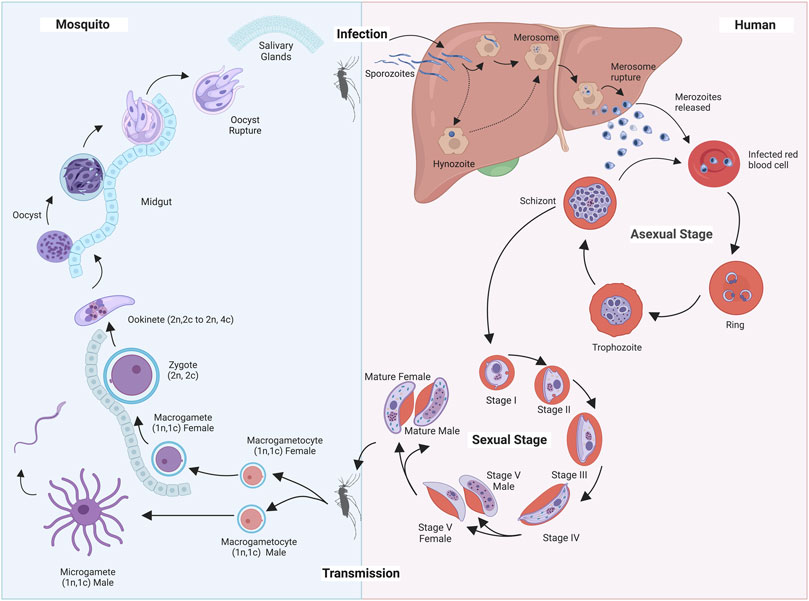
FIGURE 1. Plasmodium Life Cycle. Illustration depicting Plasmodium parasite life cycle alternating from an invertebrate Anopheles (Left) host to a mammalian human host (Right).
Sequencing of the Plasmodium falciparum genome using the Sanger sequencing technology began in 1997. It was completed and published in 2002 (Gardner et al., 1998; Gardner et al., 2002a; Gardner et al., 2002b). Sequencing, assembly, and annotation of the P. falciparum genome was amongst the greatest achievements made in the field. Accessibility to the genome helped shed light on the remarkably unique nature of the pathogen that set the groundwork for future scientific breakthroughs (Carlton et al., 2005; Florent et al., 2010; Mu et al., 2010; Ponts et al., 2011). P. falciparum has a characteristically high AT rich genome content (up to 80.6%). Its genome is roughly 23 Megabases (mb) in size organized into 14 chromosomes with an assessed 5,300 protein coding genes (52.3% of its genome). P. falciparum also has a 6 Kilobases (kb) mitochondrial genome encoding solely three proteins (Cox1, Cox3, and Cytb) along with a 35 kb apicoplast organelle genome which encodes 30 proteins (Jongwutiwes et al., 2005; Vaidya and Mather, 2009; Evers et al., 2021). Not long after gaining access to the genome, malariologists set forth to catalog the entire transcriptomic (Bozdech et al., 2003; Le Roch et al., 2003) and proteomic (Florens et al., 2002; Cooper and Carucci, 2004; Hall et al., 2005; Acharya et al., 2011) repertoire of P. falciparum along with another Plasmodium spp. With the genome characterized, identification of several important genetic markers for resistance genes such as the chloroquine resistance transporter gene (pfcrt) were established (Warhurst, 2001).
In 2005, Illumina (formally known as Solexa) sequencing technologies pioneered the development of next generation sequencing (NGS) through platforms such as the Genome Analyzer. NGS platforms gave researchers a more convenient and cost-effective means of high throughput sequencing techniques (Wadman, 2008; Wheeler et al., 2008; Bright et al., 2012). NGS technology was applied to the P. falciparum genome (Zhou et al., 2006; Taylor et al., 2013; Rao et al., 2016; Nag et al., 2017) and subsequently to the P. vivax genome (Carlton, 2003; Feng et al., 2003; Carlton et al., 2008), the latter of which was drastically hampered by the inability to be successfully cultured in vitro (Dharia et al., 2010; Gunalan et al., 2020; Noulin et al., 1912-2012). The ultra-high throughput sequencing of targeted DNA or RNA gave headways to the characterization of several novel genes responsible for drug resistance. The most prominent example lies in the mutation found in Pfkelch (Pf3d7_1343700) to be associated with dihydroartemisinin (DHA) resistance (Su et al., 2019). Similar associations were made in later studies that exposed the importance of polymorphic markers such as Single Nucleotide Polymorphisms (SNPs), indels and microsatellite mutations in parasite drug resistance adaptations (Gorobets et al., 2017).
First and second-generation sequencing platforms can provide remarkable depth and precision at the nucleotide level. Yet, these technologies have drawbacks when resolving regions of higher complexity or stretches of repetitive DNA. This is mainly due to the relatively short reading contigs to which these platforms are constrained (roughly 150-300bp). Genome segments consisting of Structural variants (SVs), Copy Number Variations (CNV), as well as segments of frequent recombination, are often beyond the sequencing capabilities of these platforms. Instead, third generation sequencing platforms that use single molecule sequencing technologies without any genome amplification, such as Pac-Bio (van Dijk et al., 2018) along with Oxford Nanopore Technologies (ONT) (Schatz, 2017), have helped researchers’ piece together these unresolved regions. PacBio technology uses labeled dNTPs that release fluorescent signals activated by a laser as they are added directly to the DNA templates. These signals are recorded by a camera system in real-time through flow cells equipped with Zero mode waveguides (ZMVs). Alternatively, ONT measures characteristic current disruptions of molecules passing through nanopores inserted in an electrical resistance membrane. DsDNA with bound polymerase or helicase enzymes, move across the membrane producing characteristic disruptions in the electrical currents of the membrane during sequencing (Xiao and Zhou, 2020). These sequencing technologies were used to analyze the genomic diversity of drug resistance genes (Runtuwene et al., 2018), as well as the diversity of genes involved in antigenic variation (var genes). There are roughly 60 var genes in the P. falciparum genome that can encode for PfEMP1, a protein exported on the surface of the infected erythrocytes that mediates the binding of infected erythrocytes to vascular endothelial cells. This binding allows the parasite to sequester in the microvasculature of the organs to avoid spleen clearance. One var gene of interest is the gene coding for VAR2CSA protein which plays a critical role in pathogenesis of placental malaria in pregnant women (Kim, 2012; Dara et al., 2017). P. falciparum-infected cells expressing VAR2CSA bind to the chondroitin sulfate A (CSA) expressed at the surface of placental tissues. Vaccines designed against VAR2CSA have been shown to be successful in preclinical studies. var genes are extremely polymorphic in field isolates and can code for proteins of up to 340 kDas, making sequencing a challenge. PacBio technology was successfully applied to characterize the extent of the genetic diversity across Plasmodium clinical specimens (Benavente et al., 2018; Bryant et al., 2018; Zhang et al., 2021). It was also recently applied to piece together the regions of high sequence repetition to resolve the P. knowlesi genome and annotate the SICAvar genes, an antigenic variant gene family similar to P. falciparum var genes. In this study the PacBio platforms were applied alongside High-throughput Chromosome Conformation Capture (Hi-C), which will be discussed later, to reorient and assemble the scaffoldings of misassembled repeat regions of the genome (Lapp et al., 2018).
One drawback of current sequencing platforms is a result of bulk sample preparation. Oftentimes, major genetic variants are lost or obscured by dominant genotypes within a population. Single-cell genomic sequencing (sc-seq) is a relatively novel technology that can capture these missed genetic variations. The sc-seq protocol begins with preparation of isolated single cells of interest, either through fluorescence activated cell sorting (FACS) or microfluidic cell sorting instruments where they are tagged with unique molecular identifiers (UMIs). These identifiers serve as barcodes allowing the identification of individual cells within the sequenced population. Single-cell instruments allow for both genomic and transcriptomic sequencing (Tang et al., 2009; Bai et al., 2021). Single-cell genomics was used to generate multiple P. vivax genomes from patients sampled during sequential febrile episodes. These genome sequences revealed hundreds of de novo mutations and variations that may otherwise be lost through conventional sequencing technologies (Dia et al., 2021). Single-cell genome technology was also applied to characterize parasite relatedness and identify distinct haplotypes during co-infection (Nkhoma et al., 2020; Siegel and Rayner, 2020).
Growing access to Plasmodium sequence information helped widen the scope of knowledge in the field of parasitology (Jayakumar and Sakakibara, 2019). Over subsequent years, the advancements of sequencing platforms will be further refined and will open the door new avenues of genetic information along with the discovery of novel drug resistance genes. The previous decade had celebrated the capabilities of these technologies and raced to collect a formerly untapped source of data. Today, genome sequencing may set an important backbone of information, but this information serves little without the means to derive accurate connections or logical sequence networks. Systems-based applications may offer an appropriate means to decode some of the complex genomic information by applying multiple platforms concurrently to fill in gaps of knowledge that sequencing alone cannot resolve (Figure 2).
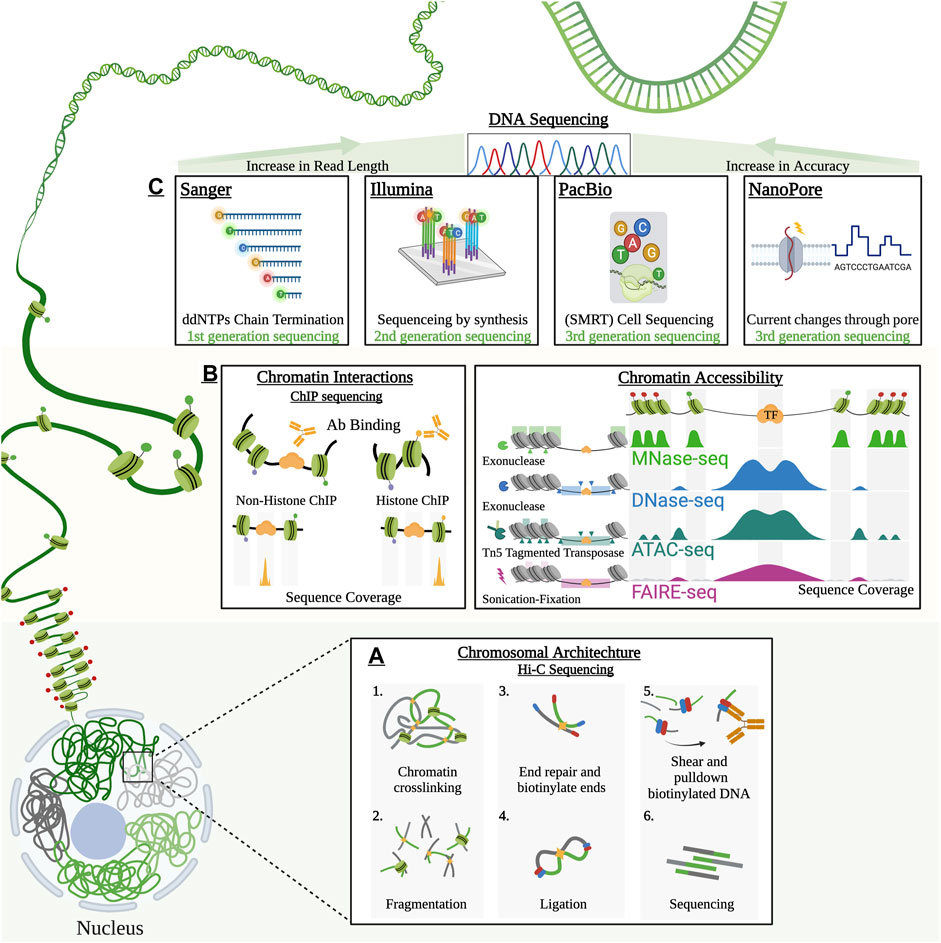
FIGURE 2. Schematic representation of hierarchical chromatin organization along with the technologies applied for their characterization. The hierarchical chromatin landscape can be broken down to three major compartments. (A) Hi-C provide 3D spatial architectural landscapes of chromosomes within the cell. These chromatin architectural analyses uncover important chromosomal territories, topologically associating domain (TAD) and chromatin contacts required for gene regulation. (B) Chromosomal landscape is divided into territories of heterochromatin and euchromatin. The chromatin is regulated by histone modifying enzymes to open (euchromatin) or close (heterochromatin) chromatin boundaries. Tools such as ChIP-sequencing, MNase, FAIRE-seq, DNase-seq, ATAC-seq serve as important means to uncover important PTMS and gene regulators. (C) DNA sequencing platforms provide high-throughput WGS capable of identifying genomic variations such as SNPs and CNVs. Over the past decade these sequencing have evolved drastically in the depth of sequencing capabilities.
Gene regulation is a major foundation of cell development, differentiation, and adaptation. A cell’s gene expression pattern is dynamically dependent on internal and environmental stimuli. Understanding how the human malaria parasite regulates its transcription profile will open the door to novel therapeutic strategies. As previously stated, one of the first efforts to characterize the full Plasmodium transcriptome repertoire was followed shortly after publication of the P. falciparum genome sequence (Bozdech et al., 2003; Le Roch et al., 2003). Researchers developed specific P. falciparum DNA microarrays, either through Affymetrix technology or DNA spotted microarrays, and applied this technology to examine the expression profiles throughout different time points of the parasite life cycle including the gametocyte and sporozoite stages (Le Roch et al., 2003). Microarrays were also used to characterize the transcriptome of P. berghei, P. yoelii and P. vivax across the different stages of development (Bozdech et al., 2008; Tarun et al., 2008; Zhou et al., 2008; Hoo et al., 2016). Such studies helped in understanding genes involved in cell cycle development including mechanisms of transcriptional control and drug resistance.
The advent of NGS platforms brought new opportunities and applications for high-throughput transcriptome studies. RNA sequencing (RNA-seq) results provided details on alternate splicing events (Otto et al., 2010), identified lowly expressed transcripts at single-base level resolution, and even characterized overlapping, sense/anti-sense, and non-coding transcripts. With advances in NGS platforms and development of targeted RNA-seq came a boom in comparative transcriptomic expression studies. Global transcriptome patterns and the mechanisms regulating post-transcriptional gene expression profiles became more apparent (Bunnik et al., 2016; Lee et al., 2018a; Chappell et al., 2020). Sequencing of polysome-associated mRNA (Poly-seq) (Lacsina et al., 2011; Bunnik et al., 2013) or ribosome foot-printing (Ribo-seq) (Caro et al., 2014) identified key regulatory mechanisms involved in translational regulation. Some of these regulatory mechanisms were found to be involved in upstream open reading frames that control translational repression of major virulence factors. Other works even shed light on the mechanisms of alternative splicing events in the parasite (Sorber et al., 2011). Today, advances in NGS technologies give researchers the opportunity to study parasite development at a systems-wide level by examining the interaction between several omics’-based technologies simultaneously (Liu et al., 2019). Tools such as RNA immunoprecipitation (RIP-seq) or enhanced cross-linking and immunoprecipitation (eCLIP-seq), characterize the interactions between RNAs and their associated proteins. These tools are being applied to uncover major mechanisms regulating gene expression at the post-transcriptional level. For example, a major mechanism recently found to be involved in regulating the atypical biogenesis of the mitoribosome in Plasmodium parasites were identified through eCLIP sequencing (Bunnik et al., 2016; Hollin et al., 2022). These technologies will undoubtedly lead to the discovery of novel key regulators of parasite development in the future.
With increasing specificity and resolution of new sequencing technologies along with improvement in RNA extraction protocols, researchers were able to undertake more systems-based approaches to understanding the dynamic nature of the pathogen’s transcriptome. From the study of transcriptional variation within parasite field isolates to single-cell transcriptomic analyses within a population, these studies helped gain new knowledge on the pathogen’s behavioral responses to different environmental conditions. As an example, parasite transcriptomes extracted from patients during the dry or rainy season were found to have distinctly extended circulation cycles during the dry season when mosquito populations were low. This unique behavioral shift results in increased parasite clearance to levels below host immune activation to enable the parasite to survive until the end of the dry season (Andrade et al., 2020; Thomson-Luque et al., 2021). Another example of the power of NGS technology is the ability to undertake dual transcriptomic-sequencing (Dual-RNA sequencing) of both the host cell and pathogen simultaneously. These methods can be applied to monitor the unique temporal responses and cellular interplay during infection. Studies offered a more comprehensive analysis of host-pathogen interactions in infected patients with uncomplicated or severe malaria (Westermann et al., 2012). Researchers identified hundreds of distinct expression profiles between patients of severe malaria over those with uncomplicated malaria associated with coma, hyperlactatemia, and thrombocytopenia symptoms. These studies also revealed positive correlations between human neutrophil granule protein (GBP) expression with onset of severe malaria symptoms (Lee et al., 2018b). More recent studies have applied this technology to identify the human mucosal immunity gene, mucin-13. MUC13 acts as an innate immune response mechanism against parasite infection similar to what was observed in defense mechanisms against several forms of cancer and bacterial infections (LaMonte et al., 2019). Dual-RNA sequencing has yet to find solid and widespread application in the field of Plasmodium research, but this system wide approach can be a promising tool to be implemented into future exploration of the parasite mechanism of pathogenesis in human patients.
One of the more advanced technologies currently available for transcriptomic analysis is through single-cell RNA sequencing (scRNA-seq). This technology can be applied in developmental or precision medicine studies to detect discrete differences in gene expression patterns among individual cells within a population. This level of detection allows the identification of cell-to-cell variations that are often masked by classical population “bulk” RNA-seq. It shows major promise in the study of drug therapies to distinguish effective treatment options or even identify gene expression profiles between sensitive and drug resistant isolates. scRNA-seq experiment has been successfully used in Plasmodium to better understand the role of AP2-G, a master regulator on sexual commitment and development (Rawat et al., 2021). These platforms were also applied to uncover previously hidden transcriptional signatures in more than 500 individual parasites in both rodent and human malaria infected cells comprising asexual and sexual life-cycle stages. The data suggested that gene expression during parasite development was not as continuous as commonly thought. In focusing attention on the transcriptional shifts and variations that did not proceed along expected incremental paths during development, researchers bring doubt on the notion of the cascading nature of the transcription initiation asexual cycles (Reid et al., 2018). These findings can help researchers characterize novel transcriptional signatures and differential expression patterns that may potentially expose previously overlooked therapeutic targets. More recently, researchers at the Wellcome Sanger Institute generated the first malaria single-cell gene expression profile in P. berghei. Over 1780 individual parasites were profiled to define a high-resolution transcriptional atlas of the entire P. berghei life cycle. This atlas was later used to precisely define developmental stages of single cells from three different human malaria parasite species (P. knowlesi, P. malariae and P. falciparum), including parasites isolated directly from infected individuals (Howick et al., 2019). Other studies focused on understanding P. vivax blood-stage parasite expression patterns (Sa et al., 2020) and cultivated our knowledge on the transcriptome across the mosquito midgut to the salivary glands in P. falciparum (Real et al., 2021). Very recently, single-cell analyses performed on both parasites and host transcripts in individual hepatocytes were able to capture host- and state-dependent transcriptional signatures throughout the course of liver infection (Mancio-Silva et al., 2022). The authors identified replicative and non-replicative parasites that share features with sexual transmissive forms. They also discovered that infection suppresses transcripts of key hepatocyte genes that control anti-parasite innate immune responses. These works have provided a comprehensive view of genes expressed throughout all parasitic stages of several malaria parasite species. They also revealed insights into the biology of malaria parasites including the P. vivax dormancy and transmission stages and even provided a reference dataset for understanding host-parasite interactions in clinical samples.
The field of transcriptomics research has made remarkable headways over the years to provide a more rounded understanding of the unicellular pathogen that genomic studies alone simply cannot uncover. What was previously denoted as redundant transcriptional sequences or simple “noise” was later recognized as a major focal point of organismal study. From transcription initiation, post-transcriptional regulation and eventually post-translational regulatory functions, the parasite transcript can give insight on all aspects of the parasite behavior and development. Refinement of extraction protocols and new systems-based, multi-omics approaches along with the progressively lower costs of NGS technologies are changing the landscape of the field of parasitology (Figure 3).
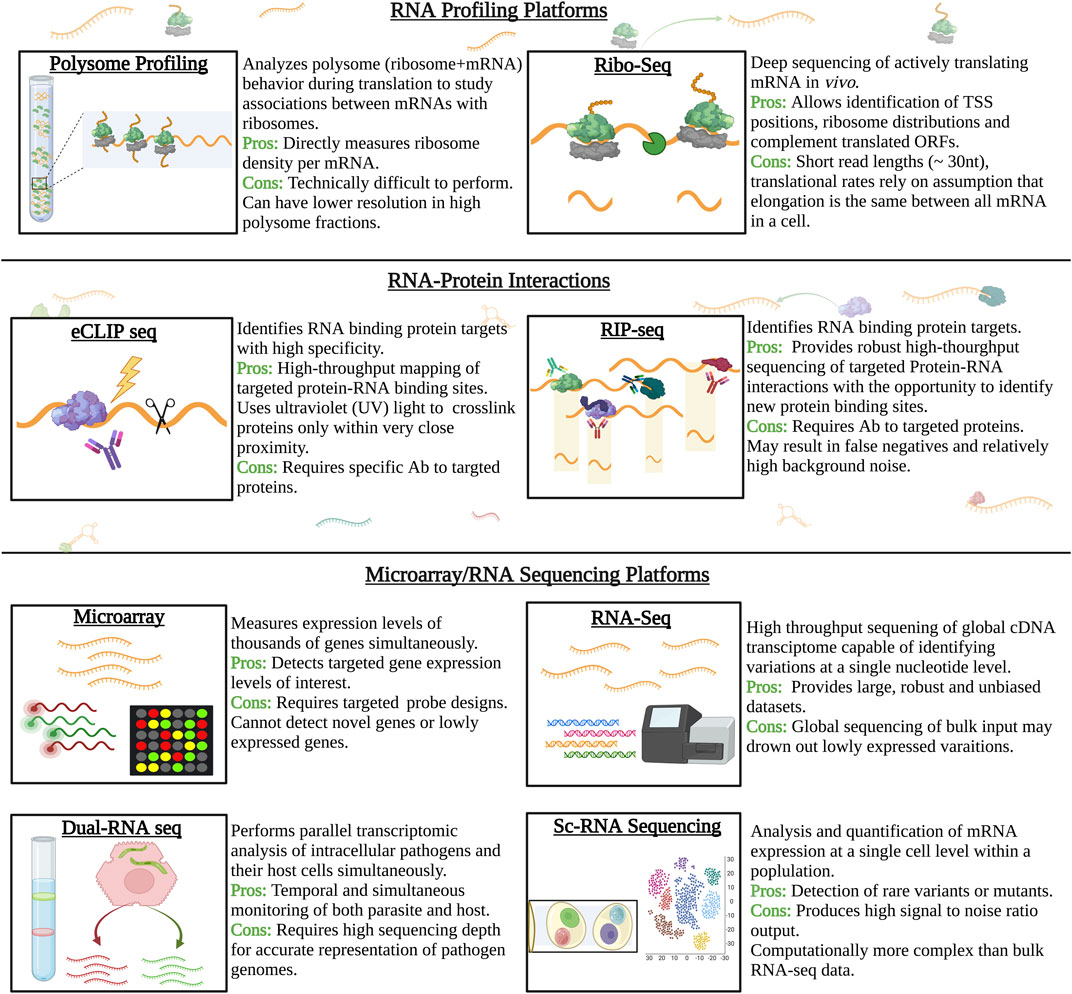
FIGURE 3. Schematic illustration of major tools and technologies used to characterize a cell’s transcriptome. Gene expression is a major factor of cell development, regulation, and response to stimuli. Thus, adaptations in tools and technologies have provided new opportunities to study a cell transcriptome at all stages pre and post transcriptional levels. Unique tools have now allowed for the capture of actively transcribing genes by targeting RNA-ribosome interactions. Other platforms seek to decipher RNA-protein interactions. RNA sequencing platforms have also shown great improvement overtime.
There has been growing interest in the field of epigenetics and the 3D architectural framework of the plasmodial genome that establishes the parasites’ genomic regulatory patterns. The spatial layout and architectural organization of the genome have been found to play important roles in the modulation of major biological processes during cell division. Evidence has revealed the dynamic nature of the chromatin during the important transitory stages of the Plasmodium developmental cycles (Batugedara et al., 2017; Davis et al., 2021). Among chromatin conformational regulatory features and moderators are nucleosome positionings, histone post-translational modifications (PTMs), DNA methylation and transcription activating or repressing factors (TFs). These features control the heterochromatin and euchromatin arrangements of the genome leading to regulation of gene expression throughout the parasite’s life cycle (Rivera et al., 2014; Hollin and Le Roch, 2020; Hollin et al., 2021). Micrococcal nuclease sequencing (MNase-seq) (Kaplan et al., 2009; Ponts et al., 2010a; Kensche et al., 2016), Formaldehyde-Assisted Isolation of Regulatory Elements sequencing (FAIRE-seq) (Giresi et al., 2007; Ponts et al., 2010a; Ponts et al., 2010b), chromatin immunoprecipitation sequencing (ChIP-seq) (Ponts et al., 2010b), methylome profiling methods such as Bisulfite sequencing (BS-seq) (Lister et al., 2009) as well as Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) (Toenhake et al., 2018) have all been applied to profile chromatin landscapes within the parasite to characterize some important epigenetic landscapes including the positioning of histone modification, the identification of transcriptional start sites (TSS) regions, some enhancer-like elements, and mechanism regulating the gene expression of clonally variant gene families (CVGs), including eba, phist, var and clag genes (Ruiz et al., 2018; Toenhake et al., 2018).
Earliest insight on the role of chromatin architecture on parasite development arose through fluorescence in situ hybridization (FISH), a technique only applicable to known gene target sites (Yokota et al., 1995; Walczak et al., 2018). More recently, the development of chromatin conformation capture technologies, including Hi-C (Dekker et al., 2002), allowed the identification of chromatin contact loops, topologically associating domains (TADs) and cis- and trans-elements at the genome-wide level (Dostie et al., 2006; Simonis et al., 2006; Zhao et al., 2006; Lieberman-Aiden et al., 2009; van de Werken et al., 2012). When applied to Plasmodium, Hi-C technology allowed researchers to gain a macro-level representation of the chromatin interactions during important developmental stages of the parasite life cycles in an “all-vs-all” approach. Surprisingly, loops and TADs were not identified. Instead, the chromatin was shown to adopt an open structure and increased chromosomal interactions during stages of high transcriptional activity and revealed patterns of heterochromatin and virulence gene clusters present. Altogether these findings revealed a strong correlation between gene expression and genome organization and validate the importance of genome architecture during the parasite’s life cycle (Ay et al., 2014; Mumbach et al., 2016; Bunnik et al., 2018).
Scientists have long established the importance of nucleosome positioning on gene expression levels by regulating access of transcription factors and other DNA binding proteins to the DNA. These nucleosomes encapsulate approximately 147 DNA base pairs wrapped around a histone octamer consisting of two H2A, two H2B, two H3, and two H4. One widely used technique used to reveal the role of histone PTMs on chromatin remodeling and gene expression in Plasmodium research is through chromatin-immunoprecipitation sequencing (ChIP-seq) (Solomon et al., 1988; Johnson et al., 2007). The identification of the genome-wide distribution of the H3K9me3 histone represents a classic example of well-studied repressive markers mainly associated with telomere ends and repressed genes like the var genes. Other histone markers including H3K4me3, H3K9ac and H4K8ac represent some substantiated players of the epigenetic pathway that lead to gene activation throughout the IDC cycle (Salcedo-Amaya et al., 2009; Ay et al., 2015; Saraf et al., 2016; Bunnik et al., 2018). Most recent work surveyed the regulatory roles and the network of apicomplexan AP2 (ApiAP2) heterochromatin-associated transcription factors (PfAP2-HFs) to characterize eight previously unknown PfAP2-HFs. These PfAP2-HFs were shown to exhibit preferential enrichment within heterochromatic regions. Subsequent knockout of these PfAP2-HFs correlated with significant changes in var gene expression. These findings validated the role of the ApiAP2 family in gene repression and regulatory networks (Campbell et al., 2010; Shang et al., 2022). The importance of the ApiAP2 DNA-binding proteins as master regulators of sexual development in Plasmodium has been well established in the field (Pieszko et al., 2015; Josling et al., 2020). Other regulators such as AP2-I, on the other hand, was shown to adopt a more targeted role in its control of genes involved in merozoite invasion into red blood cells (RBCs) (Kafsack et al., 2014; Sinha et al., 2014; Santos et al., 2017). Recently, another important regulator of transcription, HDP1, was found to serve as a positive regulator and enhancer of key genes required for sexual differentiation (Campelo Morillo et al., 2022).
In higher eukaryotes, ChIP technology has been applied in congruence with other chromatin capture techniques to understand the broader implications of these pairwise chromatin contacts within individual parasites and between parasite-host cells (Zhao et al., 2006; Beagrie et al., 2017; Han et al., 2018; Quinodoz et al., 2018; Cook et al., 2020). These studies have even been assessed at the single-cell level to divulge these regulatory features and associations within cells in a population. Single-cell sequencing platforms are used in hand with tools such as ChIP and Hi-C. Although not yet widely available in the field of Plasmodium research, it is not difficult to envision that these technologies could soon be adapted to Plasmodium cultures. These applications may provide great promise in detection of discrete and complex transcriptional networks regulating the variant parasite development throughout its life cycle (Clark et al., 2016; Luo et al., 2020).
Like a cell’s transcriptome, a cell’s proteome is distinct within individual cell types and is temporally dynamic. The domain of proteomics explores quantitative gene expression levels to study the structure and function of proteins. In a systems biology facet, the branch of proteomics often fell behind genomics and transcriptomics interests. This was mainly a result of the lack of appropriate tools needed to capture distinct and fleeting protein interactions, phosphorylation, and other modifications associated with more elusive protein pathways. A major advent of genomic and RNA expression analyses came through the ability to amplify targeted genome fragments, giving researchers the ability to recognize, replicate and manipulate target segments. Proteins, on the other hand, cannot be amplified and thus rely solely on capture technology that is often-times lost in the background noise or simply too unstable to be properly collected and studied. Thus, researchers were limited to capturing only highly expressed and stable proteins with little understanding on the complex modulators and regulars associated with these networks. Earlier methods of protein identification focused simply on two-dimensional (2-D) gel electrophoresis with the purpose of cataloging presence or absence of targeted protein interactions. The advances of Mass-spectrometry (MS) platforms, in contrast, were capable of large-scale, high throughput analysis that allowed detecting the parasite proteomes with significantly higher accuracy and specificity, giving researchers to ability to capture and catalogue complex cascading protein networks at play during important cellular processes. The initial characterizations of P. falciparum and P. yoelii proteomes were first published in 2002 (Florens et al., 2002). Ultimately, the proteomes of P. berghei and P. chabaudi identified 4,500 conserved core Plasmodium genes that were categorized according to different expression stages during parasite development. These technologies have now been applied to record the proteomic profiles of Plasmodium parasites at all stages of development (Hall et al., 2005) including zygote, ookinete (Patra et al., 2008), sporozoite and the intra-hepatic parasite stages (Doolan et al., 2003). Advanced proteomic approaches have also supported the detection of parasite-encoded surface proteins of infected erythrocytes (PIESPs) to better understand mechanisms regulating pathogenesis and immunity (Florens et al., 2004).
Understanding the dynamics of protein interaction within an organism will provide a more detailed framework of complex regulatory networks at play during cell development and homeostasis (Ramaprasad et al., 2012). One early publication generated an extensive protein-interaction analysis using the yeast-two hybrid (Y2H) technology. This study detected 2,846 unique interactions, identified major protein associations, and predicted novel protein functions involved in chromatin modification, mRNA stability, ubiquitination, and host cell invasion (LaCount et al., 2005). In recent years, BioID (proximity-dependent biotin identification) has been a favored tool for unbiased proteomic profiling. These tools are also used for protein–protein interaction detection and subcellular proteome characterizations ex vivo (Kimmel et al., 2022). Proximity labeling methods have been used for proteomic profiling of targeted subcellular compartments in various parasitic unicellular organisms, including Plasmodium spp. Yet older techniques resulted in high false positive reads and was relatively unstable and untargeted. BioID, in comparison was successfully applied to uncover 19 previously unknown Inner Membrane Complex (IMC) proteins in sub-organellar compartments of Toxoplasma gondii (Chen et al., 2015). In Plasmodium, this technology was used in characterization of the interactome of Tail-anchored (TA) proteins and their association with Guided Entry of TA (GET) machinery. This machinery is involved in several cellular processes including vesicular trafficking and protein translocation (Kumar et al., 2021a). Similar approaches have been used to target compartmentalized proteins within the parasite’s organelles. Using photosensitized INA-Labeled protein 1 (PhIL1) as a bait protein, researchers successfully enriched for several established IMC marker proteins along with 12 known and six unknown IMC proteins expressing distinct apical localization within cells (Wichers et al., 2021). Although relatively new in the field of Plasmodium research, these methods can provide new insight on the parasite proteomic networks.
Additional mass spectrometry approaches including the cellular thermal shift assay coupled with mass spectrometry (MS-CETSA) are now being used to bolster our understanding of the mechanisms of action for known or novel drugs. Despite decades of research, the specific mechanisms of action and direct cellular and biochemical targets of most antimalarial drugs remained elusive. MS-CETSA defines direct interactions between small molecules and protein targets in cells. It involves two strategies that enable proteome-wide target screening: a melt curve and an isothermal dose-response (ITDR) approach. A recent study demonstrated the capabilities of CETSA to successfully detect the direct drug interactions of previously known antimalarial compounds including the PfDHFR-TS interaction with pyrimethamine (Dziekan et al., 2019; Dziekan et al., 2020). Such an approach is expected to gain widespread application in mechanistic and pharmacokinetic studies as it can greatly benefit drug and resistome based research.
PTMs present an additional hurdle to overcome when analyzing cell proteomes and protein features involved in major biological pathways. Protein phosphorylation, acetylation, methylation, glycosylation, and ubiquitylation, are some examples of PTMs that regulate parasite infection and disease progression (Rashidi et al., 2021). With over 60% of most eukaryotic organismal proteins capable of phosphorylation, phosphoproteomics research opens a significant window into the multifaceted and dynamic biochemical mechanisms at play during parasite development. The apicomplexan kinome is relatively distinct from most eukaryotes. It is characterized by the absence or loss of the tyrosine kinase (TK) protein families that are involved in cellular regulatory mechanisms. It also has a uniquely reduced STE group, that include homologs of the yeast sterile kinases which activate the Mitogen-activated protein kinases (MAP). They have instead adapted an expanded repertoire of CMGC Kinases involved in cell proliferation and development. These CMGC Kinases that include a group of Cyclin-dependent kinases (CDKs), MAP kinases, Glycogen synthase kinases (GSKs) and CDK-like kinases control cell proliferation and developmental processes in the pathogen (Khan et al., 2002; Gubbels et al., 2008; Francia and Striepen, 2014). Earliest studies applied in silico analyses report approximately 84 (Ward et al., 2004) or 99 (Anamika and Krupa, 2005) genes encoding protein kinases, including the discovery of 20 serine/threonine kinases (FIKK family) restricted to apicomplexans. These 20 kinases comprise the largest kinase family in P. falciparum with roughly 21 members identified to date. Through combined kinome-wide gene knockouts, reverse genetics approaches and global phosphoproteomics, these kinases have been confirmed to play a role in parasite viability, maintenance, invasion, and cyto-adherence (Solyakov et al., 2011). When analyzing the enriched peptides of P. falciparum schizonts or merozoites during invasion or egress using LC-MS/MS, researchers revealed over 5,000 specific phosphorylation sites (Treeck et al., 2011). Calcium-independent protein kinases (CDPKs), a family of kinases that are restricted to plants and alveolates, have been established as essential in the parasite life cycle. As an example, PfCDPK4 has been involved in gametogenesis. CDPK1 and CDPK5 have been shown to play a role in merozoite egress (Bansal et al., 2013; Govindasamy and Bhanot, 2020) while PfCDPK7 seems to be involved in signaling regulation of phospholipid biosynthesis (Maurya et al., 2022). Most recently, PfCDPK4 was also proven to serve as a central regulatory kinase in male gametocyte exflagellation, making PfCDPK4 critical for parasite transmission into the invertebrate vectors (Kumar et al., 2021b). Other studies were applied to determine 29 protein kinases and phosphatases involved in previously unknown sex-specific regulatory pathways of P. berghei parasites, many of which found to be gender specific. They also demonstrated that the mitogen-activated protein kinase 2 (MAP2) was involved in male gametocyte cell differentiation while the NIMA-related kinase (NEK4) was responsible for zygote-to-ookinete transformation, by regulating DNA replication that precedes meiosis (Khan et al., 2005; Reininger et al., 2005). Altogether these studies uncovered the role of kinases and their dynamics in regulating major parasite developmental cycles. They can also reveal parasite-host interactions at play and serve as a diagnostic tool. Using Phos-tag and magnetic TiO2 beads, comparison of the phosphoproteome of P. falciparum infected and un-infected RBC membrane extracts showed identifiable sickle cell traits along with notable associations with parasite infections, bringing renewed optimism in clinical and diagnostic research (Chauvet et al., 2021). Phosphoproteome capture technology has been refined to increase the specificity, stability, and spatial resolution across different conditions. Advancements in Immobilized metal ion affinity chromatography (IMAC), Phos-tag and magnetic TiO2 beads have been used over the years to refine the current phosphoproteome repertoire (Lindeberg et al., 1991; Lasonder et al., 2012). Understanding of phosphorylation events will reveal the cross-talks and complex networking pathways required for the parasite development.
Recently, researchers at the university of Cambridge have uncovered a means of subcellular localization of proteins and determinization of their spatial organization and relative compartmentalization in T. gondii parasites (Barylyuk et al., 2020). Through application of the hyperplexed localization of organelle proteins by isotope tagging (hyperLOPIT), they localized over 4,100 proteins. This was determined by identifying the distinctive distribution patterns formed post biochemical fractionation and nitrogen cavitation. It is likely that such technology will soon be adapted to Plasmodium to support our understanding of one of the deadliest protozoa parasites. While proteomics-based technologies and mass spectrometry tools have improved significantly over the past few years, these methods are still facing prominent limitations. One major example arises from protein sequestration protocols that require large volumes of parasite sample input that are often difficult to procure due to protein degradation. Another obstruction is a result of limited sensitivity of current mass spectrometry platforms that may yield false negative signals. Despite the growth in the protein capture technology, the field of proteomics leaves much to be discovered.
Persistent prevalence of drug resistance to antimalarials has sparked deeper interest in the global metabolic profiles of parasitic pathogens. The branch of metabolomics presents a uniquely sensitive perspective on small and transient metabolic fluxes such as oxidative stress responses or non-enzymatic processes that other omics-based datasets cannot offer. This makes it a powerful tool for identifying specific drug targets and resulting metabolic cascade consequences (Carolino and Winzeler, 2020) as many antimalarial mechanisms of action disrupt important metabolic pathways (Linares and Rodriguez, 2007). For example, it is suggested that Dihydroartemisinin (DHA) directly inhibits pyrimidine biosynthesis (Cobbold et al., 2016; Murithi et al., 2020). Categorization of the metabolomic changes of infected hosts has also shown some application in clinical diagnostics research. Studies done in cerebral malaria patients have demonstrated a significant increase in arachidonic acid levels, a precursor for inflammation responses and molecules associated with seizure symptoms (Leopold et al., 2019). In addition, P. falciparum infected patients with varying severities of disease progression, exhibited a rise in malaria-specific plasma lipoproteins as well as a decrease in plasma glycoproteins (Sengupta et al., 2016). Other studies have revealed significant metabolic changes in the activation of hypnozoites of P. vivax and P. ovale parasites in patient livers, which can serve to uncover new information on the distinct nutrient and metabolic requirements between Plasmodium species (Mueller et al., 2009; Sengupta et al., 2016).
Advances in the detection rate, sensitivity and scope of LC-MS/MS and NMR spectroscopy technologies has promoted significant headways in parasite global metabolic profile analyses and exposed notable characteristics of the apicomplexan host-parasite interactions throughout its growth. Parasite metabolomic and lipidomic profiles across the asexual cycle are spatially and temporally regulated. They are also highly dependent on environmental signals (Koyama et al., 2009). One study unveiled an estimated 100-fold increase in host glucose consumption in P. falciparum parasite infected erythrocytes relative to uninfected RBCs, shedding light on the striking metabolic activity by malaria-causing agents (Mehta et al., 2006). Characterizing the pathogen’s overall metabolomic and lipidomic profiles presents a significant achievement that sets a solid baseline from which more complex regulatory networks and mechanistic pathways can be inferred (Kafsack and Llinas, 2010). Overall, the patterns of these nutrient requirements and metabolomic changes can serve to identify new drug targets and help understand important pathways that have remained unidentified. For example, although Isopentenyl pyrophosphate (IPP) is a known essential product of the apicoplast organelle that is required for many downstream cellular processes, its complete metabolic pathway remains elusive. Recent knockout studies have discovered polyprenyl synthase (PPS) as an essential factor for this pathway that functions in the synthesis of the long chain polyprenols ultimately used for apicoplast organelle biogenesis. The PPS functions in the formation of farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), which form the synthase FPPS/GGPPS. FPPS/GGPPS is involved in the condensation of isoprenoids precursors into long chain polyprenyl-PPs through the condensation of DMAPP (dimethylallyl pyrophosphate) and IPP. Parasites subjected to PPS-KO were only rescued though IPP and long- (C50) chain prenol alcohols, bringing some clarity to the biological mechanisms involved in the biogenesis and function of the apicoplast and exposing a previously unknown drug target (Okada et al., 2022).
While metabolomic studies have been directed towards drug mechanisms of action and targeted clinical diagnostic investigations, profiling of the global metabolome across Plasmodium spp. at specific stages of the parasite life cycle has improved significantly (Tewari et al., 2021). However, to establish sound metabolic associations and understand how these changes may regulate the cell’s transcriptomes and proteomes, global metabolomic profiles across Plasmodium spp. in vitro and in vivo must be chronicled. Prolonged extraction protocols, inadequate sample preservation and inefficient enrichment methods remain a challenging facet in metabolomics acquisition that must be supplemented with transcriptomics and proteomics data sets.
A notable feature of P. falciparum is the configuration of its genome, with up to 80.6% composed of Adenine and Thymine bases. This unique aspect of the pathogen’s genome has, much to the dismay of researchers, presented an additional obstacle in genome editing efforts. Recently, researchers were able to identify essential genes through transposon-based mutagenesis. This functional genomic approach used a piggyBac system for the random integration of transposons into parasite genome sites (Zhang et al., 2018). Results were shared and served as a critical source of knowledge when other means of gene manipulation were more challenging. One important factor in genome editing tools is the reliance on the double stranded breakage (DSB) of target genes to induce homology repair mechanisms (HR) with introduction of the desired recombination fragment through donor DNA or nonhomologous end-joining (Orr-Weaver et al., 1981; Goonewardene et al., 1993; Rouet et al., 1994). However, Plasmodium is found to be deficient in canonical NHEJ and subsequently hindered attempts at gene editing. The first successful attempt at gene editing in P. falciparum was performed by Fidock et al. in 1997 (Fidock and Wellems, 1997). Shortly thereafter an episomal plasmid “single-site crossover” method was used to create dhfr resistant P. berghei and P. falciparum parasite lines. This work, along with several other ensuing successful publications set the framework for central advances in the study of these apicomplexans (Wu et al., 1996; Ghorbal et al., 2014; Lee et al., 2014). Different techniques were developed and modified with varying success. Some of which involved the homologous recombination through electroporation of circular or linear DNA, double strand break-based genome editing techniques, such as Zinc Finger Nucleases (ZFNs), and Plasmodium Artificial Chromosomes (PACs) (Iwanaga et al., 2010; Straimer et al., 2012).
Recent success was achieved using the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 gene editing tools to knock-out or knockdown essential and non-essential genes in P. falciparum (Bryant et al., 2020; Vieira et al., 2021). Improvements made in transfection protocols including the use of the 4D-Nucleofector X electroporator increased transfection efficiency rates by 3-4 logs in P. berghei (Janse et al., 2006). By combining these novel functional genomics tools with transcriptomics, epigenomics and proteomics studies, we have improved our ability to understand the core drivers of Plasmodium cell cycle progression and parasite fitness (Siciliano and Alano, 2015; Wang et al., 2022). Despite the success made in our abilities to alter this pathogen’s genome, CRISPR/Cas9 tools remain a component of targeted bottom-up research approaches. Many aspects of the Plasmodium genome have hindered capabilities for targeted genome-wide knockouts for malaria researchers and we remain reliant on the transposon-based genome-wide mutagenesis accomplishments of the previous decades. Ultimately, genome wide CRISPR/Cas9 screens across the Plasmodium genome will assess the contributions and roles of individual genes throughout its life cycle. These genome-wide screens are already performed in the human T. gondii parasite. The screens were used to identify previously uncharacterized conserved proteins and novel essential invasion factors (Wu et al., 2019). The accomplishments made on this Plasmodium relative gives foresight on the opportunities CRISPR/Cas9 tools can offer in malaria studies on a system-wide scale. Sequencing of the pathogen genome set an important framework from which most omics-based studies have originated but the ability to undergo functional genomic screens on the parasite can greatly support and solidify our research findings. The capability to genetically modify the malaria parasites gives researchers a key advantage: the objective to elucidate its biology and provide stronger weapons to fight malaria. Systems biology brings together a global understanding of the internal and external stimuli along with the multi-player interactions that have allowed the pathogen to thrive in its niche environments. It is through the use of global gene knockouts experiments followed by multi-omics studies including those designed for more targeted approach such as the kinome-wide gene knockouts (Solyakov et al., 2011), that we will be able to fully understand the function of each of the proteins in the parasite genome.
Malaria eradication is a key global health objective that requires contemporary efforts to combat the adaptive capabilities of the pathogen. Using systems-wide experimental approaches, researchers have now opened new avenues to tackle the spread of the disease. To help process these vast datasets, researchers have made significant achievements in bioinformatics tools and databases. These accomplishments have helped create complex computing platforms, machine learning algorithms and mathematical modeling systems dedicated to extrapolating meaningful information and pattern detection. These tools are necessary to uncover important networks and interactions that can oftentimes be hidden among the background “noise” of data outputs. Tools such as Altered Chromatin COnformation STatistics (ACCOST) is an example of a modeling algorithm used to better understand the many aspects of epigenetic features critical to regulate gene expression in the parasite (Cook et al., 2020). Many of the mathematical modeling tools currently available are generally applied as predictive models for potential host–pathogen, protein–protein interactions (HP-PPIs) or intra parasitic interactome models to better predict and uncover novel parasite vulnerabilities for potential therapeutic targets. Machine learning algorithms can detect regions of homology using sequence-based similarity approaches, domain or motif-based approaches or potential epitope target models. Machine learning software undergo a method of computational inference to predict HP-PPIs by first collecting directories of sequencing information to identify patterns of similarity or trends within the Plasmodium parasite pathways. As of recently, these algorithms have also been adapted to identify patterns within interspecies systems through data mining. In deriving shared intra- and interspecies sequences or domains of interaction, these modeling systems formulate inferred maps of homologous or orthologous interaction sites in different organisms. For example, a recent publication identified several HP-PPIs between P. falciparum and Helicobacter pylori within host systems through sequences and domain-based approaches of mathematical modeling systems (Krishnadev and Srinivasan, 2008; Lee et al., 2008; Tyagi et al., 2009; Mariano and Wuchty, 2017).
One powerful application of computer simulation is through structural based approaches that allow virtual screening to make predictions about the behavior of proteins and/or compounds. Such methodology can analyze in silico the physiochemical and electrostatic properties between host cell and pathogen surface proteins to detect components critical for pathogen host invasion and serve as attractive epitope target sites for vaccine discovery. Other machine learning platforms have been applied to spearhead the advances in drug discovery. As an example, researchers successfully trained supervised classifiers to screen and validate compounds from Traditional Chinese Medicine Databases (Maindola et al., 2015). This was achieved using the physicochemical properties of the compounds available from a confirmatory high throughput screening against the interaction between the conserved Apical Membrane Antigen (AMA1) and Rhoptry Neck (RON2) proteins. These two molecular components are known to be essential for successful invasion of erythrocyte by Plasmodium and were used to validate their findings. Another study was able to test the dosing regimens of primaquine, a cure for the treatment and relapse prevention of P. vivax parasites. Treatment entails multiple doses of the drug for parasite clearance and has a major safety risk of hemolysis, especially in patients with glucose-6-phosphate dehydrogenase deficiency (G6PDd). G6PD is an enzyme involved in protecting RBCs from oxidative stress. In this study, mathematical modeling systems analyzed the RBC clearance and production to hemoglobin and reticulocyte levels of young patients (believed to have highest G6PD enzyme activity) and older parents with G6PDd. The models were used to predict the optimal dosing pattern that would minimize hemolysis in patients. These powerful predictive tools give researchers an informed standing on predictive treatment regimens that can have a direct impact on future clinical studies (Watson et al., 2017).
Other applications for these mathematical modeling systems have been used to uncover relationships between parasite dynamics and host erythrocyte age. Malaria infection has been found to impact a dispersed age range based on the species. P. vivax, for example, is associated with increasing levels of infection in reticulocytes and P. falciparum lines are more ubiquitous in the infection of RBC age. In this study, mathematical models tested and validated the notion that RBC selectivity was a significant determinant of infection outcome. These studies tested variations of HbA1c levels over time and exposed hematologic perturbations that could be applied to assess disease severity. Their finding’s showed parasites were efficiently invading younger erythrocytic cells and were able to cause severe anemia at relatively low levels of parasite burden (Malka et al., 2016; Foy et al., 2020).
Overall, advances in measurements and computation will help researchers better correlate important mechanisms of parasite cellular pathways. It may also spearhead the efforts in drug therapeutic studies. Machine learning programs have already begun to make headways in the realms of clinical and diagnostic research. They have been used to offer optimized predictive accuracy, risk-sensitive clinical decisions, and reliable recommended treatment regimens for compounds of some health concern. Access to greater multi-omics-based research provides additional resources to refine the mathematical modeling platforms and help future researchers understand the parasite pathophysiology.
With increased resistance to all clinically administered anti-malarial drugs and no fully protective vaccines available to date, it is wise to propose that future eradication efforts will require new approaches to better understand the pathogen and the environment in which it thrives. Systems biology can be applied to uncover complex hidden networks that are normally overlooked using a single-omic branch of study. Previous fundamental technologies were restricted, and encouraged linear and direct, cause-and-effect associations that were commonly reduced to a single or di-omic branch of research. Yet with innovation and advancements in modern day technologies researchers have unlocked previously untapped sources of information that now allow the study of biological mechanisms through innovative planes of view. Collectively, multi-omics technologies and mathematical modeling systems are increasingly being used to study, not only how Plasmodium parasites affect their hosts but also how the host environment affects the parasite. Altogether, these novel approaches can help lead to a systems wide understanding of host-parasite interactions necessary for new drug targets and vaccine development in the fight against malaria (Figure 4).
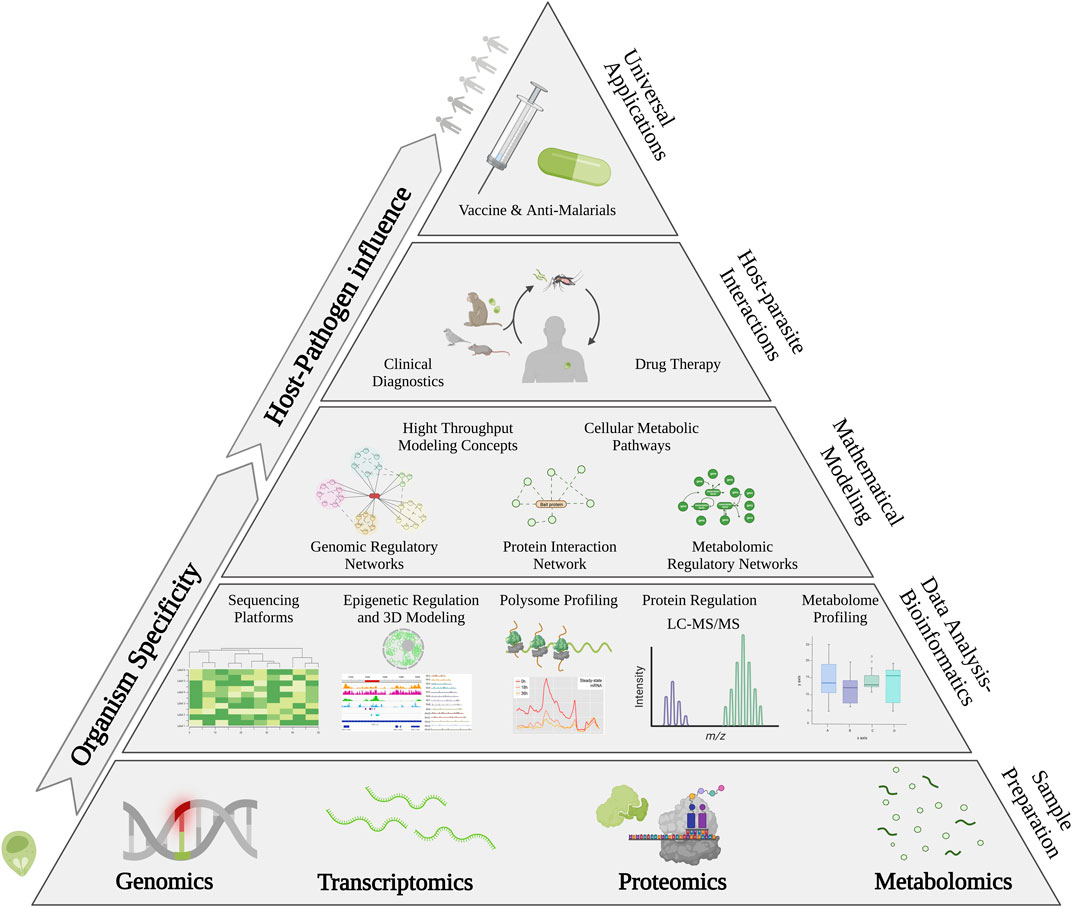
FIGURE 4. Systems-Wide Approach to Determine the Cell’s Functional Organization. Genomics, Transcriptomics, Proteomics, and Metabolomics based techniques are applied towards precision medicine against malaria. These multi-omics-based approaches are used to understand the intrinsic characteristics of the human host and Plasmodium involved in clinical malaria as well as the complex host-parasite interactions that occur. Complex mathematical models provide opportunities for precision based computational inferences to better uncover new potential drug targets. Ultimately, the integration of various layers of regulatory and structural and computational level analysis will help in developing precision medicine against malaria.
ZC and KL conceived and wrote the manuscript. All authors contributed too the article and approve the submitted version.
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (Grants R01 AI136511, R01 AI142743, R01AI138139, R21 AI142506-01 AND R21 A1154386 to KL) and the University of California, Riverside (NIFA-Hatch-225935 to KL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acharya, P., Pallavi, R., Chandran, S., Dandavate, V., Sayeed, S. K., Rochani, A., et al. (2011). Clinical proteomics of the neglected human malarial parasite Plasmodium vivax. PLoS One 6 (10), e26623. doi:10.1371/journal.pone.0026623
Anamika, N. S., and Krupa, A. (2005). A genomic perspective of protein kinases in Plasmodium falciparum. Proteins 58 (1), 180–189. doi:10.1002/prot.20278
Andrade, C. M., Fleckenstein, H., Thomson-Luque, R., Doumbo, S., Lima, N. F., Anderson, C., et al. (2020). Increased circulation time of Plasmodium falciparum underlies persistent asymptomatic infection in the dry season. Nat. Med. 26 (12), 1929–1940. doi:10.1038/s41591-020-1084-0
Ay, F., Bunnik, E. M., Varoquaux, N., Bol, S. M., Prudhomme, J., Vert, J. P., et al. (2014). Three-dimensional modeling of the P. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression. Genome Res. 24 (6), 974–988. doi:10.1101/gr.169417.113
Ay, F., Bunnik, E. M., Varoquaux, N., Vert, J. P., Noble, W. S., and Le Roch, K. G. (2015). Multiple dimensions of epigenetic gene regulation in the malaria parasite Plasmodium falciparum: Gene regulation via histone modifications, nucleosome positioning and nuclear architecture in P. Falciparum. Bioessays 37 (2), 182–194. doi:10.1002/bies.201400145
Bai, D., Peng, J., and Yi, C. (2021). Advances in single-cell multi-omics profiling. RSC Chem. Biol. 2 (2), 441–449. doi:10.1039/d0cb00163e
Bansal, A., Singh, S., More, K. R., Hans, D., Nangalia, K., Yogavel, M., et al. (2013). Characterization of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1) and its role in microneme secretion during erythrocyte invasion. J. Biol. Chem. 288 (3), 1590–1602. doi:10.1074/jbc.M112.411934
Barylyuk, K., Koreny, L., Ke, H., Butterworth, S., Crook, O. M., Lassadi, I., et al. (2020). A comprehensive subcellular atlas of the Toxoplasma proteome via hyperLOPIT provides spatial context for protein functions. Cell Host Microbe 28 (5), 752–766. doi:10.1016/j.chom.2020.09.011
Batugedara, G., Lu, X. M., Bunnik, E. M., and Le Roch, K. G. (2017). The role of chromatin structure in gene regulation of the human malaria parasite. Trends Parasitol. 33 (5), 364–377. doi:10.1016/j.pt.2016.12.004
Beagrie, R. A., Scialdone, A., Schueler, M., Kraemer, D. C. A., Chotalia, M., Xie, S. Q., et al. (2017). Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543 (7646), 519–524. doi:10.1038/nature21411
Benavente, E. D., de Sessions, P. F., Moon, R. W., Grainger, M., Holder, A. A., Blackman, M. J., et al. (2018). A reference genome and methylome for the Plasmodium knowlesi A1-H.1 line. Int. J. Parasitol. 48 (3-4), 191–196. doi:10.1016/j.ijpara.2017.09.008
Bohme, U., Otto, T. D., Cotton, J. A., Steinbiss, S., Sanders, M., Oyola, S. O., et al. (2018). Complete avian malaria parasite genomes reveal features associated with lineage-specific evolution in birds and mammals. Genome Res. 28 (4), 547–560. doi:10.1101/gr.218123.116
Bozdech, Z., Llinas, M., Pulliam, B. L., Wong, E. D., Zhu, J., and DeRisi, J. L. (2003). The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 1 (1), E5. doi:10.1371/journal.pbio.0000005
Bozdech, Z., Mok, S., Hu, G., Imwong, M., Jaidee, A., Russell, B., et al. (2008). The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc. Natl. Acad. Sci. U. S. A. 105 (42), 16290–16295. doi:10.1073/pnas.0807404105
Bright, A. T., Tewhey, R., Abeles, S., Chuquiyauri, R., Llanos-Cuentas, A., Ferreira, M. U., et al. (2012). Whole genome sequencing analysis of Plasmodium vivax using whole genome capture. BMC Genomics 13, 262. doi:10.1186/1471-2164-13-262
Bryant, J. M., Baumgarten, S., Dingli, F., Loew, D., Sinha, A., Claes, A., et al. (2020). Exploring the virulence gene interactome with CRISPR/dCas9 in the human malaria parasite. Mol. Syst. Biol. 16 (8), e9569. doi:10.15252/msb.20209569
Bryant, J. M., Baumgarten, S., Lorthiois, A., Scheidig-Benatar, C., Claes, A., and Scherf, A. (2018). De novo genome assembly of a Plasmodium falciparum NF54 clone using single-molecule real-time sequencing. Genome Announc. 6 (5), e01479. doi:10.1128/genomeA.01479-17
Bunnik, E. M., Batugedara, G., Saraf, A., Prudhomme, J., Florens, L., and Le Roch, K. G. (2016). The mRNA-bound proteome of the human malaria parasite Plasmodium falciparum. Genome Biol. 17 (1), 147. doi:10.1186/s13059-016-1014-0
Bunnik, E. M., Chung, D. W. D., Hamilton, M., Ponts, N., Saraf, A., Prudhomme, J., et al. (2013). Polysome profiling reveals translational control of gene expression in the human malaria parasite Plasmodium falciparum. Genome Biol. 14 (11), R128. doi:10.1186/gb-2013-14-11-r128
Bunnik, E. M., Cook, K. B., Varoquaux, N., Batugedara, G., Prudhomme, J., Cort, A., et al. (2018). Changes in genome organization of parasite-specific gene families during the Plasmodium transmission stages. Nat. Commun. 9 (1), 1910. doi:10.1038/s41467-018-04295-5
Campbell, T. L., De Silva, E. K., Olszewski, K. L., Elemento, O., and Llinas, M. (2010). Identification and genome-wide prediction of DNA binding specificities for the ApiAP2 family of regulators from the malaria parasite. PLoS Pathog. 6 (10), e1001165. doi:10.1371/journal.ppat.1001165
Campelo Morillo, R. A., Tong, X., Xie, W., Abel, S., Orchard, L. M., Daher, W., et al. (2022). The transcriptional regulator HDP1 controls expansion of the inner membrane complex during early sexual differentiation of malaria parasites. Nat. Microbiol. 7 (2), 289–299. doi:10.1038/s41564-021-01045-0
Carlton, J. M., Adams, J. H., Silva, J. C., Bidwell, S. L., Lorenzi, H., Caler, E., et al. (2008). Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455 (7214), 757–763. doi:10.1038/nature07327
Carlton, J., Silva, J., and Hall, N. (2005). The genome of model malaria parasites, and comparative genomics. Curr. Issues Mol. Biol. 7 (1), 23–37.
Carlton, J. (2003). The Plasmodium vivax genome sequencing project. Trends Parasitol. 19 (5), 227–231. doi:10.1016/s1471-4922(03)00066-7
Caro, F., Ahyong, V., Betegon, M., and DeRisi, J. L. (2014). Genome-wide regulatory dynamics of translation in the Plasmodium falciparum asexual blood stages. Elife 3. doi:10.7554/eLife.04106
Carolino, K., and Winzeler, E. A. (2020). The antimalarial resistome - finding new drug targets and their modes of action. Curr. Opin. Microbiol. 57, 49–55. doi:10.1016/j.mib.2020.06.004
Chappell, L., Ross, P., Orchard, L., Russell, T. J., Otto, T. D., Berriman, M., et al. (2020). Refining the transcriptome of the human malaria parasite Plasmodium falciparum using amplification-free RNA-seq. BMC Genomics 21 (1), 395. doi:10.1186/s12864-020-06787-5
Chauvet, M., Chhuon, C., Lipecka, J., Dechavanne, S., Dechavanne, C., Lohezic, M., et al. (2021). Sickle cell trait modulates the proteome and phosphoproteome of Plasmodium falciparum-infected erythrocytes. Front. Cell. Infect. Microbiol. 11, 637604. doi:10.3389/fcimb.2021.637604
Chawla, J., Oberstaller, J., and Adams, J. H. (2021). Targeting gametocytes of the malaria parasite Plasmodium falciparum in a functional genomics era: Next steps. Pathogens 10 (3), 346. doi:10.3390/pathogens10030346
Chen, A. L., Kim, E. W., Toh, J. Y., Vashisht, A. A., Rashoff, A. Q., Van, C., et al. (2015). Novel components of the Toxoplasma inner membrane complex revealed by BioID. mBio 6 (1), e02357–14. doi:10.1128/mBio.02357-14
Clark, S. J., Lee, H. J., Smallwood, S. A., Kelsey, G., and Reik, W. (2016). Single cell epigenomics: Powerful new methods for understanding gene regulation and cell identity. Genome Biol. 17, 72. doi:10.1186/s13059-016-0944-x
Cobbold, S. A., Chua, H. H., Nijagal, B., Creek, D. J., Ralph, S. A., and McConville, M. J. (2016). Metabolic dysregulation induced in Plasmodium falciparum by dihydroartemisinin and other front-line antimalarial drugs. J. Infect. Dis. 213 (2), 276–286. doi:10.1093/infdis/jiv372
Cook, K. B., Hristov, B. H., Le Roch, K. G., Vert, J. P., and Noble, W. S. (2020). Measuring significant changes in chromatin conformation with ACCOST. Nucleic Acids Res. 48 (5), 2303–2311. doi:10.1093/nar/gkaa069
Cooper, R. A., and Carucci, D. J. (2004). Proteomic approaches to studying drug targets and resistance in Plasmodium. Curr. Drug Targets. Infect. Disord. 4 (1), 41–51. doi:10.2174/1568005043480989
Dara, A., Travassos, M. A., Adams, M., Schaffer DeRoo, S., Drabek, E. F., Agrawal, S., et al. (2017). A new method for sequencing the hypervariable Plasmodium falciparum gene var2csa from clinical samples. Malar. J. 16 (1), 343. doi:10.1186/s12936-017-1976-8
Davis, S. Z., Hollin, T., Lenz, T., and Le Roch, K. G. (2021). Three-dimensional chromatin in infectious disease-A role for gene regulation and pathogenicity? PLoS Pathog. 17 (2), e1009207. doi:10.1371/journal.ppat.1009207
Dekker, J., Rippe, K., Dekker, M., and Kleckner, N. (2002). Capturing chromosome conformation. Science 295 (5558), 1306–1311. doi:10.1126/science.1067799
Dharia, N. V., Bright, A. T., Westenberger, S. J., Barnes, S. W., Batalov, S., Kuhen, K., et al. (2010). Whole-genome sequencing and microarray analysis of ex vivo Plasmodium vivax reveal selective pressure on putative drug resistance genes. Proc. Natl. Acad. Sci. U. S. A. 107 (46), 20045–20050. doi:10.1073/pnas.1003776107
Dia, A., Jett, C., Trevino, S. G., Chu, C. S., Sriprawat, K., Anderson, T. J. C., et al. (2021). Single-genome sequencing reveals within-host evolution of human malaria parasites. Cell Host Microbe 29 (10), 1496–1506. doi:10.1016/j.chom.2021.08.009
Dondorp, A. M., Nosten, F., Yi, P., Das, D., Phyo, A. P., Tarning, J., et al. (2009). Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361 (5), 455–467. doi:10.1056/NEJMoa0808859
Doolan, D. L., Southwood, S., Freilich, D. A., Sidney, J., Graber, N. L., Shatney, L., et al. (2003). Identification of Plasmodium falciparum antigens by antigenic analysis of genomic and proteomic data. Proc. Natl. Acad. Sci. U. S. A. 100 (17), 9952–9957. doi:10.1073/pnas.1633254100
Dostie, J., Richmond, T. A., Arnaout, R. A., Selzer, R. R., Lee, W. L., Honan, T. A., et al. (2006). Chromosome conformation capture carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 16 (10), 1299–1309. doi:10.1101/gr.5571506
Dvorin, J. D., and Goldberg, D. E. (2022). Plasmodium egress across the parasite life cycle. Annu. Rev. Microbiol. doi:10.1146/annurev-micro-041320-020659
Dziekan, J. M., Wirjanata, G., Dai, L., Go, K. D., Yu, H., Lim, Y. T., et al. (2020). Cellular thermal shift assay for the identification of drug-target interactions in the Plasmodium falciparum proteome. Nat. Protoc. 15 (6), 1881–1921. doi:10.1038/s41596-020-0310-z
Dziekan, J. M., Yu, H., Chen, D., Dai, L., Wirjanata, G., Larsson, A., et al. (2019). Identifying purine nucleoside phosphorylase as the target of quinine using cellular thermal shift assay. Sci. Transl. Med. 11 (473), eaau3174. doi:10.1126/scitranslmed.aau3174
Evers, F., Cabrera-Orefice, A., Elurbe, D. M., Kea-Te Lindert, M., Boltryk, S. D., Voss, T. S., et al. (2021). Composition and stage dynamics of mitochondrial complexes in Plasmodium falciparum. Nat. Commun. 12 (1), 3820. doi:10.1038/s41467-021-23919-x
Feng, X., Carlton, J. M., Joy, D. A., Mu, J., Furuya, T., Suh, B. B., et al. (2003). Single-nucleotide polymorphisms and genome diversity in Plasmodium vivax. Proc. Natl. Acad. Sci. U. S. A. 100 (14), 8502–8507. doi:10.1073/pnas.1232502100
Fidock, D. A., and Wellems, T. E. (1997). Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc. Natl. Acad. Sci. U. S. A. 94 (20), 10931–10936. doi:10.1073/pnas.94.20.10931
Florens, L., Liu, X., Wang, Y., Yang, S., Schwartz, O., Peglar, M., et al. (2004). Proteomics approach reveals novel proteins on the surface of malaria-infected erythrocytes. Mol. Biochem. Parasitol. 135 (1), 1–11. doi:10.1016/j.molbiopara.2003.12.007
Florens, L., Washburn, M. P., Raine, J. D., Anthony, R. M., Grainger, M., Haynes, J. D., et al. (2002). A proteomic view of the Plasmodium falciparum life cycle. Nature 419 (6906), 520–526. doi:10.1038/nature01107
Florent, I., Marechal, E., Gascuel, O., and BreheLin, L. (2010). Bioinformatic strategies to provide functional clues to the unknown genes in Plasmodium falciparum genome. Parasite 17 (4), 273–283. doi:10.1051/parasite/2010174273
Foy, B. H., Goncalves, B. P., and Higgins, J. M. (2020). Unraveling disease pathophysiology with mathematical modeling. Annu. Rev. Pathol. 15, 371–394. doi:10.1146/annurev-pathmechdis-012419-032557
Francia, M. E., and Striepen, B. (2014). Cell division in apicomplexan parasites. Nat. Rev. Microbiol. 12 (2), 125–136. doi:10.1038/nrmicro3184
Gardner, M. J., Hall, N., Fung, E., White, O., Berriman, M., Hyman, R. W., et al. (2002). Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419 (6906), 498–511. doi:10.1038/nature01097
Gardner, M. J., Shallom, S. J., Carlton, J. M., Salzberg, S. L., Nene, V., Shoaibi, A., et al. (2002). Sequence of Plasmodium falciparum chromosomes 2, 10, 11 and 14. Nature 419 (6906), 531–534. doi:10.1038/nature01094
Gardner, M. J., Tettelin, H., Carucci, D. J., Cummings, L. M., Aravind, L., Koonin, E. V., et al. (1998). Chromosome 2 sequence of the human malaria parasite Plasmodium falciparum. Science 282 (5391), 1126–1132. doi:10.1126/science.282.5391.1126
Ghorbal, M., Gorman, M., Macpherson, C. R., Martins, R. M., Scherf, A., and Lopez-Rubio, J. J. (2014). Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat. Biotechnol. 32 (8), 819–821. doi:10.1038/nbt.2925
Giresi, P. G., Kim, J., McDaniell, R. M., Iyer, V. R., and Lieb, J. D. (2007). FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17 (6), 877–885. doi:10.1101/gr.5533506
Goonewardene, R., Daily, J., Kaslow, D., Sullivan, T. J., Duffy, P., CaRteR, R., et al. (1993). Transfection of the malaria parasite and expression of firefly luciferase. Proc. Natl. Acad. Sci. U. S. A. 90 (11), 5234–5236. doi:10.1073/pnas.90.11.5234
Gorobets, N. Y., Sedash, Y. V., Singh, B. K., and Rathi, B. (2017). An overview of currently available antimalarials. Curr. Top. Med. Chem. 17 (19), 2143–2157. doi:10.2174/1568026617666170130123520
Govindasamy, K., and Bhanot, P. (2020). Overlapping and distinct roles of CDPK family members in the pre-erythrocytic stages of the rodent malaria parasite, Plasmodium berghei. PLoS Pathog. 16 (8), e1008131. doi:10.1371/journal.ppat.1008131
Gubbels, M. J., White, M., and Szatanek, T. (2008). The cell cycle and Toxoplasma gondii cell division: tightly knit or loosely stitched? Int. J. Parasitol. 38 (12), 1343–1358. doi:10.1016/j.ijpara.2008.06.004
Gunalan, K., Rowley, E. H., and Miller, L. H. (2020). A way forward for culturing Plasmodium vivax. Trends Parasitol. 36 (6), 512–519. doi:10.1016/j.pt.2020.04.002
Haakenstad, A., Harle, A. C., Tsakalos, G., Micah, A. E., Tao, T., Anjomshoa, M., et al. (2019). Tracking spending on malaria by source in 106 countries, 2000-16: An economic modelling study. Lancet. Infect. Dis. 19 (7), 703–716. doi:10.1016/S1473-3099(19)30165-3
Hall, N., Karras, M., Raine, J. D., Carlton, J. M., Kooij, T. W. A., Berriman, M., et al. (2005). A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science 307 (5706), 82–86. doi:10.1126/science.1103717
Han, J., Zhang, Z., and Wang, K. (2018). 3C and 3C-based techniques: The powerful tools for spatial genome organization deciphering. Mol. Cytogenet. 11, 21. doi:10.1186/s13039-018-0368-2
Hollin, T., Abel, S., Falla, A., Pasaje, C. F. A., Bhatia, A., Hur, M., et al. (2022). Functional genomics of RAP proteins and their role in mitoribosome regulation in Plasmodium falciparum. Nat. Commun. 13 (1), 1275. doi:10.1038/s41467-022-28981-7
Hollin, T., Gupta, M., Lenz, T., and Le Roch, K. G. (2021). Dynamic chromatin structure and epigenetics control the fate of malaria parasites. Trends Genet. 37 (1), 73–85. doi:10.1016/j.tig.2020.09.003
Hollin, T., and Le Roch, K. G. (2020). From genes to transcripts, a tightly regulated journey in Plasmodium. Front. Cell. Infect. Microbiol. 10, 618454. doi:10.3389/fcimb.2020.618454
Hoo, R., Zhu, L., Amaladoss, A., Mok, S., Natalang, O., Lapp, S. A., et al. (2016). Integrated analysis of the Plasmodium species transcriptome. EBioMedicine 7, 255–266. doi:10.1016/j.ebiom.2016.04.011
Howick, V. M., Russell, A. J. C., Andrews, T., Heaton, H., Reid, A. J., Natarajan, K., et al. (2019). The Malaria Cell Atlas: Single parasite transcriptomes across the complete Plasmodium life cycle. Science 365 (6455), eaaw2619. doi:10.1126/science.aaw2619
Iwanaga, S., Khan, S. M., Kaneko, I., Christodoulou, Z., Newbold, C., Yuda, M., et al. (2010). Functional identification of the Plasmodium centromere and generation of a Plasmodium artificial chromosome. Cell Host Microbe 7 (3), 245–255. doi:10.1016/j.chom.2010.02.010
Janse, C. J., Franke-Fayard, B., and Waters, A. P. (2006). Selection by flow-sorting of genetically transformed, GFP-expressing blood stages of the rodent malaria parasite, Plasmodium berghei. Nat. Protoc. 1 (2), 614–623. doi:10.1038/nprot.2006.88
Jayakumar, V., and Sakakibara, Y. (2019). Comprehensive evaluation of non-hybrid genome assembly tools for third generation PacBio long-read sequence data. Brief. Bioinform. 20 (3), 866–876. doi:10.1093/bib/bbx147
Johnson, D. S., Mortazavi, A., Myers, R. M., and Wold, B. (2007). Genome-wide mapping of in vivo protein-DNA interactions. Science 316 (5830), 1497–1502. doi:10.1126/science.1141319
Jongwutiwes, S., Putaporntip, C., Iwasaki, T., Ferreira, M. U., Kanbara, H., and Hughes, A. L. (2005). Mitochondrial genome sequences support ancient population expansion in Plasmodium vivax. Mol. Biol. Evol. 22 (8), 1733–1739. doi:10.1093/molbev/msi168
Josling, G. A., Russell, T. J., Venezia, J., Orchard, L., van Biljon, R., Painter, H. J., et al. (2020). Dissecting the role of PfAP2-G in malaria gametocytogenesis. Nat. Commun. 11 (1), 1503. doi:10.1038/s41467-020-15026-0
Kafsack, B. F., and Llinas, M. (2010). Eating at the table of another: Metabolomics of host-parasite interactions. Cell Host Microbe 7 (2), 90–99. doi:10.1016/j.chom.2010.01.008
Kafsack, B. F., Rovira-Graells, N., Clark, T. G., Bancells, C., Crowley, V. M., Campino, S. G., et al. (2014). A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature 507 (7491), 248–252. doi:10.1038/nature12920
Kaplan, N., Moore, I. K., Fondufe-Mittendorf, Y., Gossett, A. J., Tillo, D., Field, Y., et al. (2009). The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458 (7236), 362–366. doi:10.1038/nature07667
Kensche, P. R., Hoeijmakers, W. A. M., Toenhake, C. G., Bras, M., Chappell, L., Berriman, M., et al. (2016). The nucleosome landscape of Plasmodium falciparum reveals chromatin architecture and dynamics of regulatory sequences. Nucleic Acids Res. 44 (5), 2110–2124. doi:10.1093/nar/gkv1214
Khan, F., Tang, J., Qin, C. L., and Kim, K. (2002). Cyclin-dependent kinase TPK2 is a critical cell cycle regulator in Toxoplasma gondii. Mol. Microbiol. 45 (2), 321–332. doi:10.1046/j.1365-2958.2002.03026.x
Khan, S. M., Franke-Fayard, B., Mair, G. R., Lasonder, E., Janse, C. J., Mann, M., et al. (2005). Proteome analysis of separated male and female gametocytes reveals novel sex-specific Plasmodium biology. Cell 121 (5), 675–687. doi:10.1016/j.cell.2005.03.027
Kim, K. (2012). Malaria var gene expression: Keeping up with the neighbors. Cell Host Microbe 11 (1), 1–2. doi:10.1016/j.chom.2012.01.002
Kimmel, J., Kehrer, J., Frischknecht, F., and Spielmann, T. (2022). Proximity-dependent biotinylation approaches to study apicomplexan biology. Mol. Microbiol. 117 (3), 553–568. doi:10.1111/mmi.14815
Koyama, F. C., Chakrabarti, D., and Garcia, C. R. (2009). Molecular machinery of signal transduction and cell cycle regulation in Plasmodium. Mol. Biochem. Parasitol. 165 (1), 1–7. doi:10.1016/j.molbiopara.2009.01.003
Krishnadev, O., and Srinivasan, N. (2008). A data integration approach to predict host-pathogen protein-protein interactions: Application to recognize protein interactions between human and a malarial parasite. Silico Biol. 8 (3-4), 235–250.
Kumar, S., Haile, M. T., Hoopmann, M. R., Tran, L. T., Michaels, S. A., Morrone, S. R., et al. (2021). Plasmodium falciparum calcium-dependent protein kinase 4 is critical for male gametogenesis and transmission to the mosquito vector. mBio 12 (6), e0257521. doi:10.1128/mBio.02575-21
Kumar, T., Maitra, S., Rahman, A., and Bhattacharjee, S. (2021). A conserved guided entry of tail-anchored pathway is involved in the trafficking of a subset of membrane proteins in Plasmodium falciparum. PLoS Pathog. 17 (11), e1009595. doi:10.1371/journal.ppat.1009595
LaCount, D. J., Vignali, M., Chettier, R., Phansalkar, A., Bell, R., Hesselberth, J. R., et al. (2005). A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 438 (7064), 103–107. doi:10.1038/nature04104
Lacsina, J. R., LaMonte, G., Nicchitta, C. V., and Chi, J. T. (2011). Polysome profiling of the malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 179 (1), 42–46. doi:10.1016/j.molbiopara.2011.05.003
LaMonte, G. M., Orjuela-Sanchez, P., Calla, J., Wang, L. T., Li, S., Swann, J., et al. (2019). Dual RNA-seq identifies human mucosal immunity protein Mucin-13 as a hallmark of Plasmodium exoerythrocytic infection. Nat. Commun. 10 (1), 488. doi:10.1038/s41467-019-08349-0
Lapp, S. A., Geraldo, J. A., Chien, J. T., AyF., , Pakala, S. B., BatuGedara, G., et al. (2018). PacBio assembly of a Plasmodium knowlesi genome sequence with Hi-C correction and manual annotation of the SICAvar gene family. Parasitology 145 (1), 71–84. doi:10.1017/S0031182017001329
Lasonder, E., Green, J. L., Camarda, G., Talabani, H., Holder, A. A., Langsley, G., et al. (2012). The Plasmodium falciparum schizont phosphoproteome reveals extensive phosphatidylinositol and cAMP-protein kinase A signaling. J. Proteome Res. 11 (11), 5323–5337. doi:10.1021/pr300557m
Le Roch, K. G., Zhou, Y., Blair, P. L., Grainger, M., Moch, J. K., Haynes, J. D., et al. (2003). Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301 (5639), 1503–1508. doi:10.1126/science.1087025
Lee, A. H., Symington, L. S., and Fidock, D. A. (2014). DNA repair mechanisms and their biological roles in the malaria parasite Plasmodium falciparum. Microbiol. Mol. Biol. Rev. 78 (3), 469–486. doi:10.1128/MMBR.00059-13
Lee, H. J., Georgiadou, A., Otto, T. D., Levin, M., Coin, L. J., Conway, D. J., et al. (2018). Transcriptomic studies of malaria: A paradigm for investigation of systemic host-pathogen interactions. Microbiol. Mol. Biol. Rev. 82 (2), e00071. doi:10.1128/MMBR.00071-17
Lee, H. J., Georgiadou, A., Walther, M., Nwakanma, D., Stewart, L. B., Levin, M., et al. (2018). Integrated pathogen load and dual transcriptome analysis of systemic host-pathogen interactions in severe malaria. Sci. Transl. Med. 10 (447), eaar3619. doi:10.1126/scitranslmed.aar3619
Lee, S. A., Chan, C. h., Tsai, C. H., Lai, J. M., Wang, F. S., Kao, C. Y., et al. (2008). Ortholog-based protein-protein interaction prediction and its application to inter-species interactions. BMC Bioinforma. 9 (12), S11. doi:10.1186/1471-2105-9-S12-S11
Leopold, S. J., Ghose, A., Allman, E. L., Kingston, H. W. F., Hossain, A., Dutta, A. K., et al. (2019). Identifying the components of acidosis in patients with severe Plasmodium falciparum malaria using metabolomics. J. Infect. Dis. 219 (11), 1766–1776. doi:10.1093/infdis/jiy727
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326 (5950), 289–293. doi:10.1126/science.1181369
Linares, G. E., and Rodriguez, J. B. (2007). Current status and progresses made in malaria chemotherapy. Curr. Med. Chem. 14 (3), 289–314. doi:10.2174/092986707779941096
Lindeberg, G., Bennich, H., and Engstrom, A. (1991). Purification of synthetic peptides. Immobilized metal ion affinity chromatography (IMAC). Int. J. Pept. Protein Res. 38 (3), 253–259. doi:10.1111/j.1399-3011.1991.tb01436.x
Lister, R., Pelizzola, M., Dowen, R. H., Hawkins, R. D., Hon, G., Tonti-Filippini, J., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462 (7271), 315–322. doi:10.1038/nature08514
Liu, M., Lu, B., Fan, Y., He, X., Shen, S., Jiang, C., et al. (2019). TRIBE uncovers the role of Dis3 in shaping the dynamic transcriptome in malaria parasites. Front. Cell Dev. Biol. 7, 264. doi:10.3389/fcell.2019.00264
Luo, G., Gao, Q., Zhang, S., and Yan, B. (2020). Probing infectious disease by single-cell RNA sequencing: Progresses and perspectives. Comput. Struct. Biotechnol. J. 18, 2962–2971. doi:10.1016/j.csbj.2020.10.016
Maindola, P., Jamal, S., and Grover, A. (2015). Cheminformatics based machine learning models for AMA1-RON2 abrogators for inhibiting Plasmodium falciparum erythrocyte invasion. Mol. Inf. 34 (10), 655–664. doi:10.1002/minf.201400139
Malka, R., Nathan, D. M., and Higgins, J. M. (2016). Mechanistic modeling of hemoglobin glycation and red blood cell kinetics enables personalized diabetes monitoring. Sci. Transl. Med. 8 (359), 359ra130. doi:10.1126/scitranslmed.aaf9304
Mancio-Silva, L., Gural, N., Real, E., Wadsworth, M. H., Butty, V. L., March, S., et al. (2022). A single-cell liver atlas of Plasmodium vivax infection. Cell Host Microbe 30, 1048–1060. doi:10.1016/j.chom.2022.03.034
Mariano, R., and Wuchty, S. (2017). Structure-based prediction of host-pathogen protein interactions. Curr. Opin. Struct. Biol. 44, 119–124. doi:10.1016/j.sbi.2017.02.007
Maurya, R., Tripathi, A., Kumar, M., Antil, N., Yamaryo-Botte, Y., Kumar, P., et al. (2022). PI4-kinase and PfCDPK7 signaling regulate phospholipid biosynthesis in Plasmodium falciparum. EMBO Rep. 23 (2), e54022. doi:10.15252/embr.202154022
Mehra, S., Stadler, E., Khoury, D., McCaw, J. M., and Flegg, J. A. (2022). Hypnozoite dynamics for Plasmodium vivax malaria: The epidemiological effects of radical cure. J. Theor. Biol. 537, 111014. doi:10.1016/j.jtbi.2022.111014
Mehta, M., Sonawat, H. M., and Sharma, S. (2006). Glycolysis in Plasmodium falciparum results in modulation of host enzyme activities. J. Vector Borne Dis. 43 (3), 95–103.
Merrick, C. J. (2021). Hypnozoites in Plasmodium: Do parasites parallel plants? Trends Parasitol. 37 (4), 273–282. doi:10.1016/j.pt.2020.11.001
Mu, J., Seydel, K. B., Bates, A., and Su, X. Z. (2010). Recent progress in functional genomic research in Plasmodium falciparum. Curr. Genomics 11 (4), 279–286. doi:10.2174/138920210791233081
Mueller, I., Galinski, M. R., Baird, J. K., Carlton, J. M., Kochar, D. K., Alonso, P. L., et al. (2009). Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet. Infect. Dis. 9 (9), 555–566. doi:10.1016/S1473-3099(09)70177-X
Mueller, I., Zimmerman, P. A., and Reeder, J. C. (2007). Plasmodium malariae and Plasmodium ovale--the "bashful" malaria parasites. Trends Parasitol. 23 (6), 278–283. doi:10.1016/j.pt.2007.04.009
Mumbach, M. R., Rubin, A. J., Flynn, R. A., Dai, C., Khavari, P. A., Greenleaf, W. J., et al. (2016). HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13 (11), 919–922. doi:10.1038/nmeth.3999
Murithi, J. M., Owen, E. S., Istvan, E. S., Lee, M. C. S., Ottilie, S., Chibale, K., et al. (2020). Combining stage specificity and metabolomic profiling to advance antimalarial drug discovery. Cell Chem. Biol. 27 (2), 158–171. doi:10.1016/j.chembiol.2019.11.009
Nag, S., Dalgaard, M. D., Kofoed, P. E., Ursing, J., Crespo, M., Andersen, L. O., et al. (2017). High throughput resistance profiling of Plasmodium falciparum infections based on custom dual indexing and Illumina next generation sequencing-technology. Sci. Rep. 7 (1), 2398. doi:10.1038/s41598-017-02724-x
Nkhoma, S. C., Trevino, S. G., Gorena, K. M., Nair, S., Khoswe, S., Jett, C., et al. (2020). Co-Transmission of related malaria parasite lineages shapes within-host parasite diversity. Cell Host Microbe 27 (1), 93–103. doi:10.1016/j.chom.2019.12.001
Noreen, N., Ullah, A., Salman, S. M., Mabkhot, Y., Alsayari, A., and Badshah, S. L. (2021). New insights into the spread of resistance to artemisinin and its analogues. J. Glob. Antimicrob. Resist. 27, 142–149. doi:10.1016/j.jgar.2021.09.001
Noulin, F., Borlon, C., Van Den Abbeele, J., D'Alessandro, U., and Erhart, A. (1912-20122013). 1912-2012: A century of research on Plasmodium vivax in vitro culture. Trends Parasitol. 29 (6), 286–294. doi:10.1016/j.pt.2013.03.012
Okada, M., Rajaram, K., Swift, R. P., Mixon, A., Maschek, J. A., Prigge, S. T., et al. (2022). Critical role for isoprenoids in apicoplast biogenesis by malaria parasites. Elife 11, e73208. doi:10.7554/eLife.73208
Orr-Weaver, T. L., Szostak, J. W., and Rothstein, R. J. (1981). Yeast transformation: A model system for the study of recombination. Proc. Natl. Acad. Sci. U. S. A. 78 (10), 6354–6358. doi:10.1073/pnas.78.10.6354
Otto, T. D., Wilinski, D., Assefa, S., Keane, T. M., Sarry, L. R., Bohme, U., et al. (2010). New insights into the blood-stage transcriptome of Plasmodium falciparum using RNA-Seq. Mol. Microbiol. 76 (1), 12–24. doi:10.1111/j.1365-2958.2009.07026.x
Pacheco, M. A., Cranfield, M., Cameron, K., and Escalante, A. A. (2013). Malarial parasite diversity in chimpanzees: The value of comparative approaches to ascertain the evolution of Plasmodium falciparum antigens. Malar. J. 12, 328. doi:10.1186/1475-2875-12-328
Patra, K. P., Johnson, J. R., Cantin, G. T., Yates, J. R., and Vinetz, J. M. (2008). Proteomic analysis of zygote and ookinete stages of the avian malaria parasite Plasmodium gallinaceum delineates the homologous proteomes of the lethal human malaria parasite Plasmodium falciparum. Proteomics 8 (12), 2492–2499. doi:10.1002/pmic.200700727
Pieszko, M., Weir, W., Goodhead, I., Kinnaird, J., and Shiels, B. (2015). ApiAP2 factors as candidate regulators of stochastic commitment to merozoite production in theileria annulata. PLoS Negl. Trop. Dis. 9 (8), e0003933. doi:10.1371/journal.pntd.0003933
Ponts, N., Harris, E. Y., Prudhomme, J., Wick, I., Eckhardt-Ludka, C., Hicks, G. R., et al. (2010). Nucleosome landscape and control of transcription in the human malaria parasite. Genome Res. 20 (2), 228–238. doi:10.1101/gr.101063.109
Ponts, N., Harris, E. Y., Lonardi, S., and Le Roch, K. G. (2011). Nucleosome occupancy at transcription start sites in the human malaria parasite: A hard-wired evolution of virulence? Infect. Genet. Evol. 11 (4), 716–724. doi:10.1016/j.meegid.2010.08.002
Ponts, N., Harris, E. Y., Prudhomme, J., Wick, I., Eckhardt-Ludka, C., Hicks, G. R., et al. (2010). Nucleosome landscape and control of transcription in the human malaria parasite. Genome Res. 20 (2), 228–238. doi:10.1101/gr.101063.109
Quinodoz, S. A., Ollikainen, N., Tabak, B., Palla, A., Schmidt, J. M., Detmar, E., et al. (2018). Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174 (3), 744–757. doi:10.1016/j.cell.2018.05.024
Ramaprasad, A., Pain, A., and Ravasi, T. (2012). Defining the protein interaction network of human malaria parasite Plasmodium falciparum. Genomics 99 (2), 69–75. doi:10.1016/j.ygeno.2011.11.006
Rao, P. N., Uplekar, S., Kayal, S., Mallick, P. K., Bandyopadhyay, N., Kale, S., et al. (2016). A method for amplicon deep sequencing of drug resistance genes in Plasmodium falciparum clinical isolates from India. J. Clin. Microbiol. 54 (6), 1500–1511. doi:10.1128/JCM.00235-16
Rashidi, S., Tuteja, R., Mansouri, R., Ali-Hassanzadeh, M., Shafiei, R., Ghani, E., et al. (2021). The main post-translational modifications and related regulatory pathways in the malaria parasite Plasmodium falciparum: An update. J. Proteomics 245, 104279. doi:10.1016/j.jprot.2021.104279
Rawat, M., Srivastava, A., Johri, S., Gupta, I., and Karmodiya, K. (2021). Single-cell RNA sequencing reveals cellular heterogeneity and stage transition under temperature stress in synchronized Plasmodium falciparum cells. Microbiol. Spectr. 9 (1), e0000821. doi:10.1128/Spectrum.00008-21
Real, E., Howick, V. M., Dahalan, F. A., Witmer, K., Cudini, J., Andradi-Brown, C., et al. (2021). A single-cell atlas of Plasmodium falciparum transmission through the mosquito. Nat. Commun. 12 (1), 3196. doi:10.1038/s41467-021-23434-z
Reid, A. J., Talman, A. M., Bennett, H. M., Gomes, A. R., Sanders, M. J., Illingworth, C. J. R., et al. (2018)., 7. Elife, e33105. doi:10.7554/eLife.33105Single-cell RNA-seq reveals hidden transcriptional variation in malaria parasitesElife.
Reininger, L., Billker, O., Tewari, R., Mukhopadhyay, A., Fennell, C., Dorin-Semblat, D., et al. (2005). A NIMA-related protein kinase is essential for completion of the sexual cycle of malaria parasites. J. Biol. Chem. 280 (36), 31957–31964. doi:10.1074/jbc.M504523200
Rivera, C., Gurard-Levin, Z. A., Almouzni, G., and Loyola, A. (2014). Histone lysine methylation and chromatin replication. Biochim. Biophys. Acta 1839 (12), 1433–1439. doi:10.1016/j.bbagrm.2014.03.009
Rouet, P., Smih, F., and Jasin, M. (1994). Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 14 (12), 8096–8106. doi:10.1128/mcb.14.12.8096
Ruiz, J. L., Tena, J. J., Bancells, C., Cortes, A., Gomez-Skarmeta, J. L., and Gomez-Diaz, E. (2018). Characterization of the accessible genome in the human malaria parasite Plasmodium falciparum. Nucleic Acids Res. 46 (18), 9414–9431. doi:10.1093/nar/gky643
Runtuwene, L. R., Tuda, J. S. B., Mongan, A. E., Makalowski, W., Frith, M. C., Imwong, M., et al. (2018). Nanopore sequencing of drug-resistance-associated genes in malaria parasites, Plasmodium falciparum. Sci. Rep. 8 (1), 8286. doi:10.1038/s41598-018-26334-3
Sa, J. M., Cannon, M. V., Caleon, R. L., Wellems, T. E., and Serre, D. (2020). Single-cell transcription analysis of Plasmodium vivax blood-stage parasites identifies stage- and species-specific profiles of expression. PLoS Biol. 18 (5), e3000711. doi:10.1371/journal.pbio.3000711
Salcedo-Amaya, A. M., van Driel, M. A., Alako, B. T., Trelle, M. B., van den Elzen, A. M. G., Cohen, A. M., et al. (2009). Dynamic histone H3 epigenome marking during the intraerythrocytic cycle of Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A. 106 (24), 9655–9660. doi:10.1073/pnas.0902515106
Santos, J. M., Josling, G., Ross, P., Joshi, P., Orchard, L., Campbell, T., et al. (2017). Red blood cell invasion by the malaria parasite is coordinated by the PfAP2-I transcription factor. Cell Host Microbe 21 (6), 731–741. doi:10.1016/j.chom.2017.05.006
Saraf, A., Cervantes, S., Bunnik, E. M., Ponts, N., Sardiu, M. E., Chung, D. W. D., et al. (2016). Dynamic and combinatorial landscape of histone modifications during the intraerythrocytic developmental cycle of the malaria parasite. J. Proteome Res. 15 (8), 2787–2801. doi:10.1021/acs.jproteome.6b00366
Schatz, M. C. (2017). Nanopore sequencing meets epigenetics. Nat. Methods 14 (4), 347–348. doi:10.1038/nmeth.4240
Sengupta, A., Ghosh, S., Das, B. K., Panda, A., Tripathy, R., Pied, S., et al. (2016). Host metabolic responses to Plasmodium falciparum infections evaluated by (1)H NMR metabolomics. Mol. Biosyst. 12 (11), 3324–3332. doi:10.1039/c6mb00362a
Shang, X., Wang, C., Fan, Y., Guo, G., Wang, F., Zhao, Y., et al. (2022). Genome-wide landscape of ApiAP2 transcription factors reveals a heterochromatin-associated regulatory network during Plasmodium falciparum blood-stage development. Nucleic Acids Res. 50 (6), 3413–3431. doi:10.1093/nar/gkac176
Siciliano, G., and Alano, P. (2015). Enlightening the malaria parasite life cycle: Bioluminescent Plasmodium in fundamental and applied research. Front. Microbiol. 6, 391. doi:10.3389/fmicb.2015.00391
Siegel, S. V., and Rayner, J. C. (2020). Single cell sequencing shines a light on malaria parasite relatedness in complex infections. Trends Parasitol. 36 (2), 83–85. doi:10.1016/j.pt.2019.12.007
Simonis, M., Klous, P., Splinter, E., Moshkin, Y., Willemsen, R., de Wit, E., et al. (2006). Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet. 38 (11), 1348–1354. doi:10.1038/ng1896
Sinha, A., Hughes, K. R., Modrzynska, K. K., Otto, T. D., Pfander, C., Dickens, N. J., et al. (2014). A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature 507 (7491), 253–257. doi:10.1038/nature12970
Solomon, M. J., Larsen, P. L., and Varshavsky, A. (1988). Mapping protein-DNA interactions in vivo with formaldehyde: Evidence that histone H4 is retained on a highly transcribed gene. Cell 53 (6), 937–947. doi:10.1016/s0092-8674(88)90469-2
Solyakov, L., Halbert, J., Alam, M. M., Semblat, J. P., Dorin-Semblat, D., Reininger, L., et al. (2011). Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun. 2, 565. doi:10.1038/ncomms1558
Sorber, K., Dimon, M. T., and DeRisi, J. L. (2011). RNA-Seq analysis of splicing in Plasmodium falciparum uncovers new splice junctions, alternative splicing and splicing of antisense transcripts. Nucleic Acids Res. 39 (9), 3820–3835. doi:10.1093/nar/gkq1223
Straimer, J., Lee, M. C. S., Lee, A. H., Zeitler, B., Williams, A. E., Pearl, J. R., et al. (2012). Site-specific genome editing in Plasmodium falciparum using engineered zinc-finger nucleases. Nat. Methods 9 (10), 993–998. doi:10.1038/nmeth.2143
Su, X. Z., Lane, K. D., Xia, L., Sa, J. M., and Wellems, T. E. (2019). Plasmodium genomics and genetics: New insights into malaria pathogenesis, drug resistance, epidemiology, and evolution. Clin. Microbiol. Rev. 32 (4), e00019. doi:10.1128/CMR.00019-19
Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., et al. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6 (5), 377–382. doi:10.1038/nmeth.1315
Tarun, A. S., Peng, X., Dumpit, R. F., Ogata, Y., Silva-Rivera, H., Camargo, N., et al. (2008). A combined transcriptome and proteome survey of malaria parasite liver stages. Proc. Natl. Acad. Sci. U. S. A. 105 (1), 305–310. doi:10.1073/pnas.0710780104
Taylor, S. M., Parobek, C. M., Aragam, N., Ngasala, B. E., Martensson, A., Meshnick, S. R., et al. (2013). Pooled deep sequencing of Plasmodium falciparum isolates: An efficient and scalable tool to quantify prevailing malaria drug-resistance genotypes. J. Infect. Dis. 208 (12), 1998–2006. doi:10.1093/infdis/jit392
Tewari, S. G., Rajaram, K., Swift, R. P., Kwan, B., Reifman, J., Prigge, S. T., et al. (2021). Inter-study and time-dependent variability of metabolite abundance in cultured red blood cells. Malar. J. 20 (1), 299. doi:10.1186/s12936-021-03780-5
Thomson-Luque, R., Votborg-Novel, L., Ndovie, W., Andrade, C. M., Niangaly, M., Attipa, C., et al. (2021). Plasmodium falciparum transcription in different clinical presentations of malaria associates with circulation time of infected erythrocytes. Nat. Commun. 12 (1), 4711. doi:10.1038/s41467-021-25062-z
Toenhake, C. G., Fraschka, S. A. K., Vijayabaskar, M. S., Westhead, D. R., van Heeringen, S. J., and Bartfai, R. (2018). Chromatin accessibility-based characterization of the gene regulatory network underlying Plasmodium falciparum blood-stage development. Cell Host Microbe 23 (4), 557–569. doi:10.1016/j.chom.2018.03.007
Treeck, M., Sanders, J. L., Elias, J. E., and Boothroyd, J. C. (2011). The phosphoproteomes of Plasmodium falciparum and Toxoplasma gondii reveal unusual adaptations within and beyond the parasites' boundaries. Cell Host Microbe 10 (4), 410–419. doi:10.1016/j.chom.2011.09.004
Tyagi, N., Krishnadev, O., and Srinivasan, N. (2009). Prediction of protein-protein interactions between Helicobacter pylori and a human host. Mol. Biosyst. 5 (12), 1630–1635. doi:10.1039/b906543c
Vaidya, A. B., and Mather, M. W. (2009). Mitochondrial evolution and functions in malaria parasites. Annu. Rev. Microbiol. 63, 249–267. doi:10.1146/annurev.micro.091208.073424
van de Straat, B., Sebayang, B., Grigg, M. J., Staunton, K., Garjito, T. A., Vythilingam, I., et al. (2022). Zoonotic malaria transmission and land use change in southeast asia: What is known about the vectors. Malar. J. 21 (1), 109. doi:10.1186/s12936-022-04129-2
van de Werken, H. J., Landan, G., Holwerda, S. J. B., Hoichman, M., Klous, P., Chachik, R., et al. (2012). Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat. Methods 9 (10), 969–972. doi:10.1038/nmeth.2173
van Dijk, E. L., Jaszczyszyn, Y., Naquin, D., and Thermes, C. (2018). The third revolution in sequencing technology. Trends Genet. 34 (9), 666–681. doi:10.1016/j.tig.2018.05.008
Vieira, T. B., Astro, T. P., and de Moraes Barros, R. R. (2021). Genetic manipulation of non-falciparum human malaria parasites. Front. Cell. Infect. Microbiol. 11, 680460. doi:10.3389/fcimb.2021.680460
Wadman, M. (2008). James Watson's genome sequenced at high speed. Nature 452 (7189), 788. doi:10.1038/452788b
Walczak, M., Ganesan, S. M., Niles, J. C., and Yeh, E. (2018). ATG8 is essential specifically for an autophagy-independent function in apicoplast biogenesis in blood-stage malaria parasites. mBio 9 (1), e02021. doi:10.1128/mBio.02021-17
Wang, S., Zeng, W., Zhao, W., Xiang, Z., Zhao, H., Yang, Q., et al. (2022). Comparison of in vitro transformation efficiency methods for Plasmodium falciparum. Mol. Biochem. Parasitol. 247, 111432. doi:10.1016/j.molbiopara.2021.111432
Ward, P., Equinet, L., Packer, J., and Doerig, C. (2004). Protein kinases of the human malaria parasite Plasmodium falciparum: The kinome of a divergent eukaryote. BMC Genomics 5, 79. doi:10.1186/1471-2164-5-79
Warhurst, D. (2001). New developments: Chloroquine-resistance in Plasmodium falciparum. Drug resist. updat. 4 (3), 141–144. doi:10.1054/drup.2001.0194
Watson, J., Taylor, W. R., Menard, D., Kheng, S., and White, N. J. (2017). Modelling primaquine-induced haemolysis in G6PD deficiency. Elife 6, e23061. doi:10.7554/eLife.23061
Westermann, A. J., Gorski, S. A., and Vogel, J. (2012). Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 10 (9), 618–630. doi:10.1038/nrmicro2852
Wheeler, D. A., Srinivasan, M., Egholm, M., Shen, Y., Chen, L., McGuire, A., et al. (2008). The complete genome of an individual by massively parallel DNA sequencing. Nature 452 (7189), 872–876. doi:10.1038/nature06884
White, N. J., Pukrittayakamee, S., Hien, T. T., Faiz, M. A., Mokuolu, O. A., and Dondorp, A. M. (2014). Lancet 383 (9918), 723–735. doi:10.1016/S0140-6736(13)60024-0
Wichers, J. S., Wunderlich, J., Heincke, D., Pazicky, S., Strauss, J., Schmitt, M., et al. (2021). Identification of novel inner membrane complex and apical annuli proteins of the malaria parasite Plasmodium falciparum. Cell. Microbiol. 23 (9), e13341. doi:10.1111/cmi.13341
Wu, S. Z., Wei, H. X., Jiang, D., Li, S. M., Zou, W. H., and Peng, H. J. (2019). Genome-wide CRISPR screen identifies host factors required by Toxoplasma gondii infection. Front. Cell. Infect. Microbiol. 9, 460. doi:10.3389/fcimb.2019.00460
Wu, Y., Kirkman, L. A., and Wellems, T. E. (1996). Transformation of Plasmodium falciparum malaria parasites by homologous integration of plasmids that confer resistance to pyrimethamine. Proc. Natl. Acad. Sci. U. S. A. 93 (3), 1130–1134. doi:10.1073/pnas.93.3.1130
Xiao, T., and Zhou, W. (2020). The third-generation sequencing: The advanced approach to genetic diseases. Transl. Pediatr. 9 (2), 163–173. doi:10.21037/tp.2020.03.06
Yokota, H., van den EnGh, G., Hearst, J. E., Sachs, R. K., and Trask, B. J. (1995). Evidence for the organization of chromatin in megabase pair-sized loops arranged along a random walk path in the human G0/G1 interphase nucleus. J. Cell Biol. 130 (6), 1239–1249. doi:10.1083/jcb.130.6.1239
Zhang, C., Oguz, C., Huse, S., Xia, L., Wu, J., Peng, Y. C., et al. (2021). Genome sequence, transcriptome, and annotation of rodent malaria parasite Plasmodium yoelii nigeriensis N67. BMC Genomics 22 (1), 303. doi:10.1186/s12864-021-07555-9
Zhang, M., Wang, C., Otto, T. D., Oberstaller, J., Liao, X., Adapa, S. R., et al. (2018). Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360 (6388), eaap7847. doi:10.1126/science.aap7847
Zhao, Z., Tavoosidana, G., Sjolinder, M., Gondor, A., Mariano, P., Wang, S., et al. (2006). Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat. Genet. 38 (11), 1341–1347. doi:10.1038/ng1891
Zhou, Y., Ramachandran, V., Kumar, K. A., Westenberger, S., Refour, P., Zhou, B., et al. (2008). Evidence-based annotation of the malaria parasite's genome using comparative expression profiling. PLoS One 3 (2), e1570. doi:10.1371/journal.pone.0001570
Zhou, Z., Poe, A. C., Limor, J., Grady, K. K., Goldman, I., McCollum, A. M., et al. (2006). Pyrosequencing, a high-throughput method for detecting single nucleotide polymorphisms in the dihydrofolate reductase and dihydropteroate synthetase genes of Plasmodium falciparum. J. Clin. Microbiol. 44 (11), 3900–3910. doi:10.1128/JCM.01209-06
Keywords: systems biology, Plasmodium, Multiomic analysis, malaria, Omics
Citation: Chahine Z and Le Roch KG (2022) Decrypting the complexity of the human malaria parasite biology through systems biology approaches. Front. Syst. Biol. 2:940321. doi: 10.3389/fsysb.2022.940321
Received: 10 May 2022; Accepted: 22 August 2022;
Published: 16 September 2022.
Edited by:
Naveena Yanamala, The State University of New Jersey, United StatesReviewed by:
Catherine J. Merrick, University of Cambridge, United KingdomCopyright © 2022 Chahine and Le Roch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karine G. Le Roch, a2FyaW5lLmxlcm9jaEB1Y3IuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.