
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Synth. Biol. , 18 March 2025
Sec. Ecosystems and Biodiversity Sustainability
Volume 3 - 2025 | https://doi.org/10.3389/fsybi.2025.1532846
This article is part of the Research Topic Harnessing Synthetic Biology for Ecosystem Sustainability and Biodiversity Conservation View all 3 articles
 Magdalena San Román†Andrea Arrabal†Belen Benitez-Dominguez†Isabel Quirós-Rodríguez†
Magdalena San Román†Andrea Arrabal†Belen Benitez-Dominguez†Isabel Quirós-Rodríguez† Juan Diaz-Colunga*†
Juan Diaz-Colunga*†Microbial communities are able to carry out myriad functions of biotechnological interest, ranging from the degradation of industrial waste to the synthesis of valuable chemical products. Over the past years, several strategies have emerged for the design of microbial communities and the optimization of their functions. Here we provide an accessible overview of these strategies. We highlight how principles of synthetic biology, originally devised for the engineering of individual organisms and sub-organismal units (e.g., enzymes), have influenced the development of the field of synthetic microbial ecology. With this, we aim to encourage readers to critically evaluate how insights from synthetic biology should guide our approach to community-level engineering.
Fulfilling the promise of a circular economy, where waste byproducts can be reutilized in closed-loop processes, relies on the development of sustainable alternatives to non-renewable resources. In recent years, it has become increasingly evident that addressing this challenge requires harboring the immense potential of microorganisms (Aswani et al., 2024). Microbes carry out a variety of functions that could be harnessed for biotechnological purposes. For example, some microbes are capable of degrading plastics (Taghavi et al., 2021), transforming complex compounds like lignocellulose (often present in municipal waste) into biofuels (Cragg et al., 2015; Ling et al., 2014; Prasad et al., 2019; Senne De Oliveira Lino et al., 2021), producing biomaterials (Laurent et al., 2024), or synthesizing high-value molecules including drugs (Zhang et al., 2022), plant natural products (Cravens et al., 2019; Walls et al., 2023), or vitamins (Fang et al., 2017). Microbes are now even emerging as a potential sustainable source of food (Graham and Ledesma-Amaro, 2023).
Unlocking the full biotechnological potential of microorganisms requires us to be able to optimize the relevant functions they provide. Humans have been harvesting microbial functions for millenia, with evidence that early agricultural societies already produced fermented beverages around 7,000 years ago (Wang et al., 2021). Yet, it was only in recent decades that the engineering of microbial functions began to be performed in a rational and methodical manner, driven by technological advances in microorganism identification, characterization, and manipulation. The development of genetic engineering tools (gene cloning, recombinant DNA technology, or more recently CRISPR-Cas9) fueled the establishment and expansion of the field of synthetic biology. Synthetic biology has aimed to systematize the engineering of microbial systems with a dual aim: understanding the principles by which living organisms process information, and constructing synthetic biosystems with enhanced (or even novel) traits for concrete biotechnological applications (Khalil and Collins, 2010; Leonard et al., 2008). While progress has been made over recent years, the latter objective has remained elusive (Hanson and Lorenzo, 2023; Zakeri and Carr, 2015).
Synthetic biologists have been mostly concerned with the engineering of individual strains or particular subcellular units, such as proteins or metabolic pathways. Some of the earliest examples include the genetic manipulation of yeast (Hansen and Kielland-Brandt, 1996; Romano et al., 1985) and bacteria (McKay and Baldwin, 1990) for food and beverage fermentation, the development of enzymes with high substrate specificity or catalytic activity through directed evolution (Schmidt-Dannert and Arnold, 1999; Yano et al., 1998; You and Arnold, 1996), or the optimization of intracellular biochemical reactions for increased enantioselectivity (Reetz et al., 1997). More recently, engineered microorganisms have been developed for modern applications such as carbon sequestration (Hu et al., 2019) or cancer immunotherapy (Chowdhury et al., 2019).
Engineered microorganisms, however, present various important limitations. The expression of synthetic genetic circuits is often inevitably noisy and subject to crosstalk with the native machinery of the host cells — a problem that is accentuated as these circuits become more complex (Kwok, 2010; Slusarczyk et al., 2012). In addition, heterologous expression often comes at a fitness cost, making engineered microbial functions sensitive to purging by evolution or to exclusion from better-adapted competitor species. Even when fitness costs are negligible, mere genetic drift may disrupt the expression of synthetic functions if they do not confer a benefit to the host. Synthetic biologists have proposed formal rules for the design of biosystems (Slusarczyk et al., 2012) which attempt to mitigate the effects of noise (Perrino et al., 2021), crosstalk (Müller et al., 2019), or evolution (Bull and Barrick, 2017); however challenges remain to this day.
The prospect of engineering microbial communities (as opposed to single organisms) has emerged as a promising alternative (Großkopf and Soyer, 2014; McCarty and Ledesma-Amaro, 2019; Shong et al., 2012). Communities present several advantages with respect to individual strains. They enable the compartmentalization of functional components (i.e., division of labor), alleviating fitness costs on individual strains (Beck et al., 2022; Wang M. et al., 2022; Roell et al., 2019). Functions that emerge at the community level may also be more robust (or at least change in predictable ways) in the face of evolution of constituent species (McEnany and Good, 2024; Venkataram and Kryazhimskiy, 2023). Communities may often be able to resist invasions from external species (Wagner, 2022; Mickalide and Kuehn, 2019) or groups of species (Diaz-Colunga et al., 2022; Lechón-Alonso et al., 2021). Furthermore, the composition and function of microbial communities may be modulated without the need to genetically engineer any member species, minimizing concerns on the environmental or health hazards that genetically modified organisms may pose (EFSA et al., 2020).
Here we review a variety of strategies which have been proposed for optimizing the functions of microbial communities, many of them inspired by synthetic biology at the level of organisms and subcellular units. These strategies range from bottom-up approaches, where defined sets of (typically few) species are combined into consortia with the aim of maximizing a function, to top-down approaches, where a community (which can be of high complexity and even of undefined composition) is manipulated through rational interventions (San León and Nogales, 2022). We also review methods based on mathematical modeling, including mechanistic models (most notably metabolic models) as well as more recent data-driven models. With this, we seek to provide an accessible overview of the rapidly-expanding field of synthetic ecology. We aim to prompt readers to carefully consider the extent to which the lessons learned from synthetic biology should guide our path towards community-level engineering.
Perhaps the most commonly used strategy for community-level engineering has relied on the rational, bottom-up assembly of synthetic consortia. In short, based on some known traits of a set of microbial species/strains, a consortium is constructed with the aim of maximizing a target function, as well as (potentially) its ecological and evolutionary stability (Krause et al., 2014; Lajoie and Kembel, 2019; Li et al., 2024).
This trait-based approach is reminiscent of how early efforts at protein design were based on searching for optimal amino acid sequences starting from the basic principles of amino acid biochemistry (Song et al., 2023; Bryson et al., 1995) — i.e., the biochemical “traits” of each amino acid. Using biochemical reasoning to predict protein function from sequence can be seen as solving a “puzzle” by carefully examining each of the pieces, with each piece being a specific amino acid (Figure 1A, top panel). In the case of synthetic organisms, the pieces could be genes within an artificial plasmid, while for communities they would represent the different member species (Figure 1A, bottom panel).
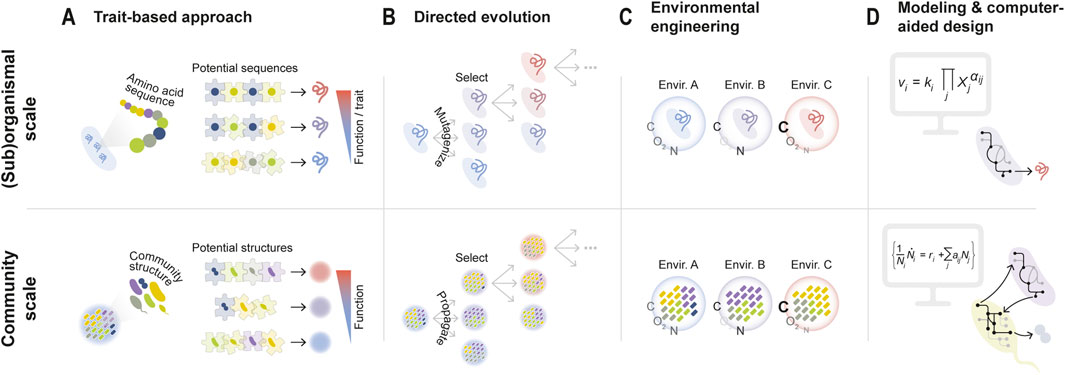
Figure 1. Approaches for microbial community-level design and optimization are often inspired by engineering strategies at the (sub)organismal scale. (A) In order to enhance the function of a protein, specific amino acid sequences can be rationally constructed based on fundamental knowledge of amino acid biochemistry. Analogously, specific microbes can be chosen to form a consortium based on their individual traits in order to optimize an ecological function. (B) Directed evolution, on the other hand, remains agnostic to the mechanisms of interaction between amino acids (or species). Instead, high-functioning sequences (or communities) are iteratively propagated and selected, re-introducing variation in each round. (C) Environmental variables (e.g., oxygen availability, carbon to nitrogen ratio, pH, temperature …) can strongly modulate organismal traits and community structures. Manipulating these variables can thus serve to optimize biological functions across scales, from molecules and organisms to communities. (D) Mathematical models can inform the design and construction of synthetic biological systems, from genetic, metabolic, and signaling pathways to entire communities.
The strategy of “solving the puzzle” has sometimes yielded good results. In some cases, consortia have been assembled by leveraging the natural capabilities of wild-type microbial species for performing specific tasks. For example, Park et al. used a two-species bacterial co-culture for the production of bioethanol by leveraging the natural ability of C. phytofermentans to hydrolyze cellulose and the potential of E. coli to ferment cellobiose catabolism byproducts into ethanol, respectively (Park et al., 2020). Alternatively, researchers have relied on the genetic manipulation of different member species/strains within a consortium. Examples include the construction of two-species/strain systems for the generation of photovoltaic energy (Zhu et al., 2019) or the production of resveratrol, a plant natural product (Camacho-Zaragoza et al., 2016). In this last example, two E. coli strains were engineered to express a complementary part of the resveratrol biosynthesis pathway. Genetic engineering has sometimes also served to enhance the stability of the community and facilitate the coexistence of its members, for instance through the imposition of obligate mutualisms (Pignon et al., 2024; Sgobba et al., 2018).
Beyond these and other biotechnological applications, it is worth noting that synthetic communities have also been used to address more fundamental questions in microbial ecology, such as how microbial interactions determine the structure and dynamics of communities (Hu et al., 2022; Van Vliet et al., 2022; Cordero and Datta, 2016). Researchers have used synthetic consortia as laboratory model systems to mimic natural communities in the soil (Coker et al., 2022) or associated with animal and human hosts (Bonillo-Lopez et al., 2024; Clark et al., 2021), among other contexts. This is again reminiscent of how synthetic organisms have also served purposes beyond biotechnology, such as providing insights on cellular information processing and signal transduction (Gao et al., 2023; Hanson and Lorenzo, 2023).
Yet, for biotechnology, the trait-based construction of synthetic communities presents important challenges. First, the traits expressed by an organism often depend on its ecological context, that is, which other species (and potentially at which abundances) may be present (Diaz-Colunga et al., 2024a; Yang, 2021). In addition, even if the contribution of each individual microbial cell to a community function was constant, species’ population sizes often vary differently across ecological contexts (Baichman-Kass et al., 2023; Sanchez et al., 2023), so the total functional contribution of a species may change depending on the presence/absence or abundance of other community members. Interactions between species can often present higher-order components, e.g., a third species may affect how two members of the community interact (Morin et al., 2022; Sanchez-Gorostiaga et al., 2019; Mickalide and Kuehn, 2019; Guo and Boedicker, 2016). In the “puzzle” analogy, it would be as if the shape of each piece changed every time we included a new one. Perhaps for this reason, the construction of synthetic consortia for biotechnology through trait-based approaches has been mostly limited to low-complexity communities, typically of two or three species/strains (Park et al., 2024; Park et al., 2020; Zhu et al., 2019). This also owes to the fact that the rational assembly of microbial consortia in high-throughput remains an experimentally tedious process. The development of new experimental methodologies (Diaz-Colunga et al., 2024a), including based on microfluidic devices (Kehe et al., 2019; Kehe et al., 2021), promises to facilitate this process and potentially expand the bottom-up approach to more complex communities.
Much like in the case of synthetic organisms, communities engineered through this trait-based approach may be disrupted by evolution, environmental fluctuations, or the influx of invader species (Amor et al., 2020; Shibasaki and Mitri, 2020). Several strategies have been proposed for enhancing stability. For example, it has been shown that synthetic communities engaging in division of labor exhibit increased stability when not only the fitness costs, but also the benefits of expressing a function are allocated evenly across member species (Wang M. et al., 2022). Stability may also be achieved by rationally modulating intercellular interactions (Deter and Lu, 2022; Karkaria et al., 2021; Kong et al., 2018; Wu et al., 2024), e.g., interspecies metabolic cross-feeding (Li et al., 2022; Park et al., 2024; Peng et al., 2024; Ziesack et al., 2019), or by imposing a defined spatial structure to physically separate different subpopulations (Wang L. et al., 2022).
It is notable how these challenges (eco-evolutionary stability, scalability beyond low-complexity constructs, etc.) are similarly faced when engineering single organisms. This is a direct consequence of the trait-based design of microbial communities being very explicitly based on engineering principles from synthetic biology (Johns et al., 2016; San León and Nogales, 2022) — which becomes particularly evident in the case of consortia made up by genetically engineered strains (Park et al., 2024; Sgobba et al., 2018; Camacho-Zaragoza et al., 2016). In this case, the reasoning is straightforward: the target function to express is encoded in a synthetic genetic circuit, but the burden of its expression is distributed across different strains (i.e., multiple “chassis”) instead of assigned to a single one. While the basic idea is appealing, we must consider its practical limitations if the end goal is to develop microbial communities for biotechnological purposes which can be deployed on a large scale. Over the past decades, synthetic biologists have devoted much effort to addressing the challenges of engineering single organisms (e.g., Perrino et al., 2021; Müller et al., 2019; Slusarczyk et al., 2012), with only limited practical success (Hanson and Lorenzo, 2023; Zakeri and Carr, 2015). As the field of synthetic ecology develops, we must carefully assess the risk of falling into a similar stage of stagnation.
Artificial selection, most notably directed evolution, has been used for decades to improve the traits of individual organisms or the properties of subcellular units such as enzymes (e.g., Schmidt-Dannert and Arnold, 1999; You and Arnold, 1996) (Figure 1B, top panel). Directed evolution mitigates many of the limitations of rational design approaches, as it bypasses the need for a detailed understanding of the underlying mechanisms that may determine the function/trait of a molecule/organism. The basic strategy can be conceptualized as an iterative exploration of a genotype space in search of optimal phenotypes, e.g., an exploration of a sequence-function landscape in the context of protein engineering.
Artificial selection at the microbiome level can be similarly framed as a guided exploration of a community structure space (defined as the set of all possible community compositions in terms of species presence/absence or abundance) in search of states that optimize a target community-level function. Such mappings between community compositions and functions have received the name of ecological landscapes or community-function landscapes (George and Korolev, 2023; Sanchez et al., 2023; Skwara et al., 2023), by analogy with the concept of genetic fitness landscapes.
In its most general form, an exploration of a community-function landscape through directed evolution would involve (i) ranking a set of microbial communities based on a target community-level function, (ii) selecting the best-performing ones, and (iii) propagating them into a new set of “offspring” communities, which can be ranked and propagated again in subsequent rounds of selection (Figure 1B, bottom panel). Despite its conceptual simplicity, this premise was not directly tested until the early 2000s, when Swenson et al. attempted to select for microbial ecosystems which efficiently performed functions such as the degradation of 3-chloroaniline (Swenson et al., 2000a) or the promotion of plant growth (Swenson et al., 2000b). More recent efforts have aimed to select microbiomes with the ability to induce early or late flowering in plant hosts (Panke-Buisse et al., 2015), or to degrade environmental pollutants (Arias-Sánchez et al., 2024). Community-level artificial selection has been typically implemented as a top-down strategy, where communities are selected and propagated without necessarily dissecting their species-level composition (Chang et al., 2021; Chang et al., 2020; Swenson et al., 2000a; Swenson et al., 2000b; Xie et al., 2019; Panke-Buisse et al., 2015). More recently, bottom-up variations have also been proposed, where the species-level composition of a community is well defined and experimentally manipulated in each round of selection (Arias-Sánchez et al., 2024; George and Korolev, 2023). While there exist promising studies of in silico community-level artificial selection (Chang et al., 2021; Lalejini et al., 2022; Xie et al., 2019), experiments have generally yielded only modest functional improvements, in some cases barely exceeding typical day-to-day fluctuations.
Selection (whether artificial or natural, at the level of organisms or groups) requires that there exists variation in the target trait/function, and that this variation can be passed from parents to offspring — i.e., that the trait is heritable to some extent (Lewontin, 1970). When organisms reproduce, they pass their genetic information to the next-generation, such that those phenotypes that are (at least partially) determined by the organism’s genotype can exhibit some degree of heritability. The very same process can also reintroduce trait variation through mutation or recombination, upon which selection can further act.
Directed evolution of communities, however, entails important nuances with respect to that of single organisms (Arias-Sánchez et al., 2019; Blouin et al., 2015; Sánchez et al., 2021; Xie et al., 2019). Communities, unlike organisms, cannot naturally self-replicate, and thus generating an “offspring” community from a “parental” one requires the intervention of the experimenter. The first attempts at microbiome breeding addressed the issue of community-level reproduction by taking inspiration from earlier experiments of group-level selection in small animal populations [e.g., of chickens (Muir, 1996) or beetles (Wade, 1976; Wade, 1977)]. In short, offspring populations were initialized from a random sample of individuals from the highest-functioning parental ones.
Yet, this approach was less successful when applied to microbial communities (Swenson et al., 2000a; Swenson et al., 2000b). This may be explained, at least partially, by an effect of population size (Sánchez et al., 2021): If the number of individuals sampled from a parent population is very large, all offspring populations may end up being compositionally very similar to one another, as the effect of stochastic sampling becomes negligible (Blouin et al., 2015). This issue is typically more prominent in microbial communities than in animal populations, as the former often have very large population sizes (e.g., a single colony can contain millions of microbial cells). The loss of structural (and therefore functional) variation across communities can naturally obstruct further selection (Blouin et al., 2015; Chang et al., 2020). Other mechanisms could also lead to the exhaustion of variation or even to functional collapse: for instance, evolution of member species within a community could have such effects under certain conditions (Shibasaki and Mitri, 2020; Venkataram and Kryazhimskiy, 2023; Xie et al., 2019).
Several strategies have been proposed to tackle this issue. Structural variation can be externally re-introduced into the offspring communities, for instance through the co-inoculation of invader species or groups of species, through the application of harsh population bottlenecks (Chang et al., 2021; Sánchez et al., 2021), or through the propagation of not only the highest-function community but also of sub-optimal ones (Xie et al., 2019). It has also been suggested that selection schemes inspired by evolutionary computing could aid in maintaining functional variation across communities, leading to better selection outcomes (Arias-Sánchez et al., 2024; Lalejini et al., 2022).
Community-level artificial selection, in principle, could substantially boost our ability to engineer microbiomes, the same way it revolutionized our ability to engineer enzymes (Schmidt-Dannert and Arnold, 1999). There is extensive theoretical and empirical evidence showing that artificial selection can act above the organismal level (Doulcier et al., 2020; Goodnight, 1990a; Goodnight, 1990b; Lewontin, 1970; Wade, 1976; Wade, 1977), and thus it is not immediately obvious why experiments of microbiome-level breeding have found only modest success in general. It is possible that current strategies for community-level selection may be limited by methodological aspects, such as the choice of community-level reproduction method or the maintenance of functional variation across communities. It is also possible that there exist more fundamental factors which may intrinsically limit community-level artificial selection. These could include stochastic fluctuations in species abundances, or the rapid emergence of “cheater” strains, especially when the expression of the target function is costly (Blouin et al., 2015; Shibasaki and Mitri, 2020; Smith and Schuster, 2019).
It is important to note that targets of selection, artificial or natural, do often have fundamental limits at the organismal scale. These limits are evidenced, for instance, by the common observation of diminishing returns at the level of organismal fitness — i.e., beneficial mutations having smaller positive fitness effects in genetic backgrounds which are already well-adapted (Chou et al., 2011; Schoustra et al., 2016; Wünsche et al., 2017). Diminishing returns have also been recently observed at the level of community function (Diaz-Colunga et al., 2024b), suggesting that when a community function is high, most interventions will tend to disrupt it rather than improve it. This could pose an obstacle towards community-level artificial selection. Such limitations may become more pronounced when the individual members of a community benefit from the expression of the target ecological function — for instance, when the function is the clearance of a toxin (Arias-Sánchez et al., 2024). When such organismal-level pressures exist, the function may strongly dictate the composition of the community in a very deterministic manner, leading to reduced across-community functional variation. This would then constrain further optimization through selection.
As promising as community-level directed evolution may be, further work is necessary before it can become a viable option for biotechnological applications. Are there intrinsic limits to selection at the level of microbial communities? If not, why has directed evolution not yet been successfully applied to microbiomes? If the reasons are purely methodological, there are perhaps reasons for optimism, as in silico simulations have proven useful to inform the design of selection protocols (Chang et al., 2021; Lalejini et al., 2022; Xie and Shou, 2021). These methods, however, remain to be tested empirically.
Most efforts for the optimization of microbial community functions have relied on manipulating its species/strain-level composition and/or the genetic architecture of community members. However, there is extensive evidence that abiotic environmental variables can strongly modulate the traits of individual microbes (Hu et al., 2021; Wasner et al., 2024; Yang, 2021), the interactions between species (Crocker et al., 2024; Ratzke and Gore, 2018), and therefore the dynamics, composition, and function of complex communities (Dal Bello et al., 2021; Estrela et al., 2021; Goldford et al., 2018; Hu et al., 2022; Silverstein et al., 2024; Sun et al., 2024). Thus, an alternative optimization strategy is to rationally engineer the environment that microorganisms inhabit (Sánchez et al., 2024; Silverstein et al., 2023; Silverstein et al., 2024) (Figure 1C).
One of the most paradigmatic examples of environmental engineering for microbial biotechnology is found in open fermentation systems (Li et al., 2014). In these, the premise is to manipulate a set of environmental conditions (e.g., pH, substrate availability, etc.) that will naturally select for microbial taxa which are able to carry out a desired function. The system is left unsterilized, open to the influx of environmental microbes, rather than inoculated with a specific set of strains previously chosen by the experimenter. This approach has been traditionally used in food and beverage fermentation, and has more recently been employed for applications such as the production of butanol from butyrate (Pinto et al., 2022) or the synthesis of enzymes (Qureshi et al., 2017) or lactic acid (Wang et al., 2016) from food waste.
The challenge of manipulating microbial environments lies in managing their highly multidimensional nature. Microbial growth and functional profiles may depend on a plethora of abiotic factors, including nutrient availability (Okano et al., 2019; Skonieczny and Yargeau, 2009; Zhu and Dai, 2024), temperature (Fu et al., 2022; Sun et al., 2024), pH (Pinto et al., 2022; Ratzke and Gore, 2018), the presence of antimicrobial compounds (Athamneh et al., 2014), or the spatial structure (or lack thereof) in their habitat (Pignon et al., 2024; Van Vliet et al., 2022). As an additional complication, the effect of these factors can often be highly non-additive. A paradigmatic example of this non-linearity is the observation that different antibiotics can act synergistically in combination (Cacace et al., 2023; Lázár et al., 2022; Yeh et al., 2006), or modify microbial responses to other environmental variables such as temperature (Cruz-Loya et al., 2021).
Ultimately, these challenges are similar to those faced by synthetic biologists when engineering single organisms (or communities) from the bottom-up. Interactions between environmental components are, at least conceptually, reminiscent of the interactions that exist between genes within an organism (or between member species within a community). These interaction networks can be very complex and of high dimensionality in all of these cases. Owing to this analogy, strategies for environmental design have drawn inspiration from the bioengineering of organisms or subcellular components (Sánchez et al., 2024). For example, genetic algorithms have been used to select optimal environments. In these cases, a set of environmental variables (pH, salinity, concentration of vitamins and minerals, etc.) were manipulated in each round of selection, and the best environments were propagated into subsequent generations. Vandecasteele et al. followed this principle to identify environments where a microbial community maximized the degradation of a synthetic dye (Vandecasteele et al., 2008). In another example, Kucharzyk et al. followed a similar approach to optimize the degradation of perchlorate, both when the function was performed by a single strain of Dechlorosoma sp. or by a complex microbial community (Kucharzyk et al., 2012).
Modeling tools can also be used to inform environmental design strategies. For example, Pacheco and Segrè developed a computational method combining metabolic modeling and genetic algorithms to find optimal environmental compositions for target community functions (Pacheco and Segrè, 2021). Mathematical models of community dynamics that are extensively used by microbial ecologists may explicitly incorporate environmental variables, e.g., the secretion of metabolic byproducts to the environment is specifically included in microbial consumer-resource models (Marsland et al., 2020b; Marsland et al., 2020a) or in some dynamic community-level metabolic models (Dukovski et al., 2021). These could be useful to inform the construction of environments which optimize microbial community functions such as the efficiency of substrate utilization or the production of specific secondary metabolites.
Data-driven models have also been developed which aim to infer optimal environmental compositions from partial observations (Chen et al., 2009; Jiménez et al., 2014; Kikot et al., 2010; Skonieczny and Yargeau, 2009; Zhou et al., 2023). These methods have the advantage of not requiring information on the specific biological mechanisms which may drive microbial responses to their environment. Yet, the typically vast number of potential environmental factors makes it so these models often must be trained with very limited empirical data. They have thus generally taken very simple forms, e.g., linear regressions with the variables being the effects of each environmental factor, at most incorporating pairwise interactions between environmental components (Jiménez et al., 2014; Kikot et al., 2010). In practice, this has limited their application to relatively simple settings including few environmental components. As is the case with the bottom-up assembly of communities, recent methodological advances could facilitate the construction of environments in high throughput (Diaz-Colunga et al., 2024a; Sánchez et al., 2024) and the expansion of the scope of these models.
Mathematical and computational models have been widely used by microbial ecologists to address questions such as inferring species interactions from co-ocurrence networks (Faust and Raes, 2012), assessing microbial coexistence and community stability (Akjouj et al., 2024), explaining the emergence of community-level properties from complex interactions between species (Van Den Berg et al., 2022), or reproducing the relationships that exist between biodiversity and function within microbial communities (Marsland et al., 2020b). In the context of biotechnology, modeling has served to guide the construction of microbial consortia that efficiently deliver target functions (Figure 1D), such as the production of relevant metabolites (e.g., Jones et al., 2016; Clark et al., 2021).
Models can be cataloged according to different criteria (Van Den Berg et al., 2022). Here, for the sake of simplicity, we broadly classify them into two categories: (1) models based on species’ traits and interaction mechanisms, and (2) models which leverage statistical features of microbial communities to reproduce and predict community-level properties while remaining mechanism-agnostic (Figure 2).
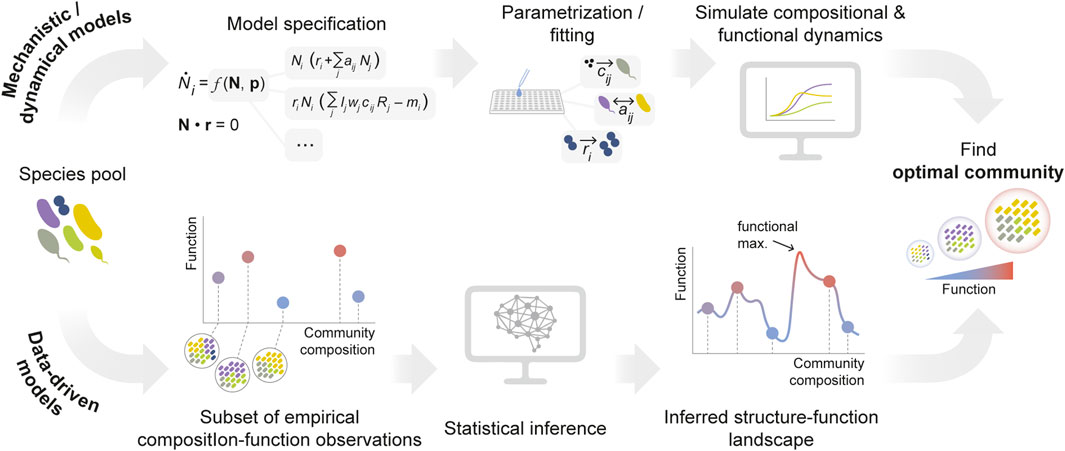
Figure 2. Modeling approaches for the optimization of microbial community functions. Mathematical/computational models can be broadly classified into two groups. Top: models of microbial communities can be based on the knowledge of the traits of a set of species and (potentially) their interactions. These models typically need to be parametrized from extensive empirical data (e.g., species growth rates, substrate preferences, interaction mechanisms …) or from genomic information. In order to inform the construction of optimal communities, they often (though not always) rely on simulating species’ dynamics. Bottom: alternatively, the second class of models are purely statistical, agnostic to the specific biological processes that underpin species’ traits and/or interactions. They rely on inference tools to learn the topography of the relationship between community composition and function (i.e., the community structure-function landscape).
Mechanistic, trait-based models often (though not always) represent the dynamics of the community, and can be expressed in the generic form
Where
With
Alternatively, the dynamics of the function to optimize can be explicitly incorporated into the model. This can be the case in microbial consumer-resource models (mCRM) (Marsland et al., 2020b; Marsland et al., 2020a), which explicitly model the dynamics of metabolite exchange between community members. For this reason, mCRMs can inform community design when the function of interest is the production of specific secondary metabolites or the rate of utilization of a substrate (Gowda et al., 2022; Van Den Berg et al., 2022). Still, using this type of models for biotechnology generally requires exhaustive characterization and quantification of species traits [although in some cases these may be inferred from genomic information (Gowda et al., 2022)] and/or their interactions.
An alternative modeling approach based on species-specific traits is metabolic modeling. Unlike gLV and mCRM models, metabolic modeling is not typically used to predict temporal changes in species abundances (although it can be adapted for this purpose, as discussed below). Instead, it leverages stoichiometric data inferred from species genomes and applies optimization criteria to predict metabolic fluxes. Given its extensive use in biotechnology, we describe this method in more detail in the following section.
Within the field of microbial biotechnology, arguably the most prominent class of mechanistic models are metabolic models. Metabolic modeling has become a cornerstone of systems biology, enabling researchers to simulate cellular metabolism, predict phenotypes, and guide metabolic engineering. Initially, metabolic modeling was utilized to investigate clonal populations (Varma and Palsson, 1994). For this, species-specific genome-scale metabolic models (GEMs) (Gu et al., 2019; Mendoza et al., 2019) together with Flux Balance Analysis (FBA) (Orth et al., 2010b) and related graph- and constraint-based techniques were used. The use of metabolic modeling to study clonal populations resulted in successful predictions of growth, secretion profiles (Jouhten et al., 2022; Neal et al., 2024; O’Brien et al., 2013), and the impact of genetic modifications (Edwards and Palsson, 2000; Segrè et al., 2002; Shlomi et al., 2005), making it invaluable for applications in bioproduction and strain engineering (Blazeck and Alper, 2010; Jiang et al., 2022) that considered isolated species.
In recent years, the use of metabolic modeling has expanded from individual species to microbial communities, whether they are small synthetic consortia or large natural microbiomes (Giordano et al., 2024; Machado et al., 2021; Zelezniak et al., 2015). This expansion has been made possible due to the development of tools that support the rapid reconstruction of GEMs (Heinken et al., 2023) and the adaptation of existing analytical methods as well as the development of new methods for studying communities instead of clonal populations (Chan et al., 2017; Diener et al., 2020; Heinken and Thiele, 2022; Khandelwal et al., 2013; Stolyar et al., 2007; Zomorrodi and Maranas, 2012). Additionally, the increasing development of easy-to-use software packages that implement complex metabolic modeling methods (Belcour et al., 2020; Dukovski et al., 2021; Ebrahim et al., 2013; Frioux et al., 2018; García-Jiménez et al., 2018; Zelezniak et al., 2015) has expanded the use of this approach, allowing non-experts to leverage these techniques for their own interests — ranging from studying the ecology of microbes to bioremediation, bioenergy production or personalized medicine.
Metabolic modeling relies on species-specific GEMs. These represent all known metabolites, metabolic genes, and reactions within a given organism. The process of reconstructing GEMs begins with draft model reconstruction. Using annotated genomes, metabolic genes and reactions are predicted to generate an initial draft. This draft model is then refined to improve accuracy in reproducing experimental data (Orth et al., 2010a; Mendoza et al., 2019). With recent software tools (Aite et al., 2018; Arkin et al., 2018; Karlsen et al., 2018; Machado et al., 2018; Olivier, 2018; Wang et al., 2018), high-quality draft models can now be created in minutes, expanding the application of metabolic modeling beyond a limited set of well-characterized, culturable species to include uncultured or lesser-known organisms as well.
Besides GEMs, metabolic modeling leverages methods for their analysis. Typically, these methods are categorized as graph-based and constraint-based. Adapting graph-based methods to investigate communities instead of clonal population is straightforward. On top of this, graph-based simulations are computationally efficient, enabling scalability and the analysis of large, complex microbial communities (Belcour et al., 2020). However, the main limitation of this approach is the loss of stoichiometric information which results in reduced output information and prediction accuracy.
As an alternative, constraint-based methods such as FBA, rely on species’ stoichiometric information. These methods rely on an optimization to find the flux through every reaction in the metabolic model which satisfies the stoichiometric and thermodynamic constraints imposed. This sets the main limitation of using metabolic modeling to investigate communities. While the optimization criteria when simulating clonal populations is straightforward - to maximize growth rate, it is far less evident when simulating communities where positive and negative interactions between community members determine total community biomass. To overcome this limitation, different methods have been developed (see for example Diener et al., 2020; Zomorrodi and Maranas, 2012) which account for potential trade-offs between species and community growth rate, improving predictions at community level.
In addition to graph and constraint-based methods, dynamic FBA (dFBA) was developed (Mahadevan et al., 2002) and soon adapted to investigate communities (Chiu et al., 2014; Hanly et al., 2012; Hanly and Henson, 2011; Salimi et al., 2010; Tzamali et al., 2011; Zhuang et al., 2011). dFBA combines FBA with ordinary differential equations to capture both environmental and growth dynamics. In essence, dFBA involves simulating a series of consecutive FBA calculations, updating species abundance and nutrient availability between simulations. This method has been further developed to model not only temporal changes but also spatial distributions of microbial populations (Bauer et al., 2017; Harcombe et al., 2014).
The application of metabolic modeling to microbial communities has opened new avenues for community design and optimization, with significant implications for industrial and environmental applications. For example, graph-based methods can be used to identify combinations of species that achieve a specific metabolic goal, such as the production of a valuable compound, by mapping metabolic pathways across community members (Belcour et al., 2020; Eng and Borenstein, 2016; Frioux et al., 2018; Julien-Laferrière et al., 2016). Constraint-based approaches, adapted for community-level analysis, enable researchers to optimize community composition and environmental conditions to enhance desired outputs (Benito-Vaquerizo et al., 2020; Jouhten et al., 2022), such as biofuel and bioplastic production, nutrient cycling, or pollutant degradation. Dynamic Flux Balance Analysis (dFBA) further extends these capabilities by incorporating temporal changes in nutrient availability and species abundance, allowing researchers to model how community function evolves over time.
Metabolic modeling is thus transforming the field of microbial community design, providing a framework for systematically engineering communities with tailored functions. Through in silico simulations, researchers can test multiple community configurations, explore various environmental conditions, and fine-tune community composition to achieve optimal performance.
Ultimately, the task of identifying optimal communities relies on our ability to accurately predict ecological function from composition — that is, to learn the topography of the community structure-function landscape (George and Korolev, 2023; Sanchez et al., 2023; Skwara et al., 2023). In the most general form, such a structure-function landscape can be expressed as a transformation (which we denote as
With
There exist many general methodologies designed for this task, with applications beyond microbial biotechnology. Perhaps some of the simplest examples are linear regression algorithms (Maulud and Abdulazeez, 2020), where the function
And the coefficients in Equation 4 (
Other data-driven methods, such as random forests, Bayesian inference approaches, or neural networks, have been often used to analyze the taxonomic structure, interactions, and dynamics of microbial communities (Baig et al., 2023; Bauer et al., 2017; DiMucci et al., 2018; Shafiei et al., 2014; Shafiei et al., 2015; Statnikov et al., 2013; Walsh et al., 2024). Yet, their use for predicting and optimizing biotechnologically relevant functions has remained more limited. A few notable exceptions include the use of random forests to predict the ability of soil microbiomes for decomposing plant litter (Thompson et al., 2019), the design of gut microbial consortia for the production of butyrate through a combination of linear regression, Bayesian inference, and gLV modeling (Clark et al., 2021), or the use of Bayesian optimization together with recurrent neural networks to predict and optimize species abundances and metabolite concentrations (Thompson et al., 2023).
If we once again turn our attention to the organismal scale and below, we find a much broader variety of data-driven methods which have been used to infer the relationship between structure and function, including in the context of protein sequence-structure-function landscapes (e.g., Barrio-Hernandez et al., 2023; Otwinowski, 2018; Romero et al., 2013) and organismal genotype-phenotype maps (e.g., Sailer et al., 2020; Tareen et al., 2022; Tonner et al., 2022). Perhaps the most notable recent example is the development of AlphaFold, a deep learning-based method for predicting protein structure from sequence (Jumper et al., 2021). These methods bypass the need for a priori knowledge on the mechanisms of interaction between genetic components or amino acids, allowing for the optimization of biological function much further beyond what rational design strategies can achieve.
Can we apply similar principles at the ecological scale? Microbial community functions emerge from interactions between species, similar to how protein function emerges from biochemical interactions between amino acids. The mechanistic basis underpinning interactions at these two scales is in principle very different. However, recent research has shown that the interactions between species within a community often follow similar statistical patterns than those between genetic components (Diaz-Colunga et al., 2024b; Eble et al., 2023; Gould et al., 2018; Morris et al., 2020; Sanchez-Gorostiaga et al., 2019). Furthermore, microbial communities may in many cases be well represented by low-dimensional statistical models with few parameters (Arya et al., 2023; Arya et al., 2024; Skwara et al., 2023; Zhao et al., 2024) — that is, despite the underlying complexity of species interactions, there often appears to be an emergent simplicity at the level of community function (Bergelson et al., 2021; Goldford et al., 2018). This strongly suggests that models akin to those used to predict function from structure at the scale of genes and proteins could also fare well for predicting microbial community function from composition, even when trained on very sparse data. Still, the application of such methodologies for the optimization of microbial community functions remains to be tested in practice.
Throughout this review, we have discussed the parallels that exist between the engineering of biological systems at the ecological scale and below. It is clear that synthetic biology tools have had an enormous influence on the development of strategies for the design and optimization of community-level microbial functions. In particular, a very common strategy has been to assemble synthetic consortia from the bottom-up, building on the available phenotypic information of the constituent parts (i.e., the species or strains). This approach has sometimes been informed by mechanistic models of ecological interactions, and/or relied on the genetic engineering of community members. While this is naturally a reasonable starting point when attempting to engineer microbiomes, it is also important to acknowledge its limitations.
Communities, like all biological entities, are complex systems, and thus their properties and functions are often difficult to explain simply from those of their parts. Applying engineering principles to their design is therefore nuanced, as in doing so there is an underlying assumption of modularity and scalability that may often not hold. This, of course, also applies at the organismal scale and below: a single cell is a very complex system in itself, and thus combining different organism types into a community means building complexity on top of complexity. Synthetic biology has arguably not yet fulfilled its promise of developing practical solutions for biotechnology on a large scale. It is thus imperative that we ask whether applying similar principles at the level of communities will yield better results, or if, on the other hand, there are fundamental limitations to this approach when it comes to biological systems.
Top-down strategies, as well as data-driven modeling, could in principle be more suitable for the task of prediction and optimization. Their main advantage lies in the fact that these methods do not rely on mechanistic information. For example, community-level directed evolution could be implemented without even characterizing the composition of the community nor the interactions between its member species. In practice, however, this strategy has only yielded modest results, and further work is necessary to identify the factors which may have limited its success. Data-driven models have been used extensively for predicting biological function below the ecological scale, but more rarely for optimizing microbial community functions. Their main limitation is the difficulty in extracting relevant biological insights from them, as these models often tend to operate as “black boxes”. In any case, this may be a lesser consideration if our primary focus is to optimize biotechnological processes.
The variety of available strategies for community-level engineering underscore the importance of choosing the appropriate approach if we wish to develop practical, viable, and sustainable solutions for open challenges in biotechnology. Taking inspiration in other areas of biology beyond microbial ecology can be fruitful, but we must carefully consider the limitations we may face. Synthetic ecology emerged as an extension to synthetic biology that promised to alleviate the limitations of the latter, in particular with respect to scalability and robustness. Yet, the rational design of microbial consortia has faced similar obstacles, perhaps because it has been approached using similar bottom-up thinking. As the field of synthetic ecology develops, it will be important to devise new optimization strategies that embrace and deal with the underlying complexity of microbial communities — and of biological systems at all scales.
MS: Investigation, Methodology, Writing–original draft, Writing–review and editing. AA: Investigation, Methodology, Writing–original draft, Writing–review and editing. BB-D: Investigation, Methodology, Writing–original draft, Writing–review and editing. IQ-R: Investigation, Methodology, Writing–original draft, Writing–review and editing. JD-C: Funding acquisition, Investigation, Methodology, Supervision, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. MS acknowledges support from a “Juan de la Cierva” Fellowship (ref. FJC2021-046960-I). JD-C acknowledges support from the “Ramón y Cajal” program (ref. RYC2023-045580-I) funded by MICIU/AEI/10.13039/501100011033 and by FSE+, and from a RyC-MaX Excellence Grant (ref. 20252MAX002) funded by the Spanish National Research Council (CSIC).
We thank D. Bajic for helpful feedback on the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aite, M., Chevallier, M., Frioux, C., Trottier, C., Got, J., Cortés, M. P., et al. (2018). Traceability, reproducibility and wiki-exploration for “à-la-carte” reconstructions of genome-scale metabolic models. PLOS Comput. Biol. 14 (5), e1006146. doi:10.1371/journal.pcbi.1006146
Akjouj, I., Barbier, M., Clenet, M., Hachem, W., Maïda, M., Massol, F., et al. (2024). Complex systems in ecology: a guided tour with large Lotka–Volterra models and random matrices. Proc. R. Soc. A Math. Phys. Eng. Sci. 480 (2285), 20230284. doi:10.1098/rspa.2023.0284
Amor, D. R., Ratzke, C., and Gore, J. (2020). Transient invaders can induce shifts between alternative stable states of microbial communities. Sci. Adv. 6 (8), eaay8676. doi:10.1126/sciadv.aay8676
Arias-Sánchez, F. I., Vessman, B., Haym, A., Alberti, G., and Mitri, S. (2024). Artificial selection improves pollutant degradation by bacterial communities. Nat. Commun. 15 (1), 7836. doi:10.1038/s41467-024-52190-z
Arias-Sánchez, F. I., Vessman, B., and Mitri, S. (2019). Artificially selecting microbial communities: if we can breed dogs, why not microbiomes? PLOS Biol. 17 (8), e3000356. doi:10.1371/journal.pbio.3000356
Arkin, A. P., Cottingham, R. W., Henry, C. S., Harris, N. L., Stevens, R. L., Maslov, S., et al. (2018). KBase: the United States department of energy systems biology knowledgebase. Nat. Biotechnol. 36 (7), 566–569. doi:10.1038/nbt.4163
Arya, S., George, A., and O’Dwyer, J. (2024). The architecture of theory and data in microbiome design: towards an S-matrix for microbiomes. Ecol. Evol. Biol. doi:10.32942/X2SD0W
Arya, S., George, A. B., and O’Dwyer, J. P. (2023). Sparsity of higher-order landscape interactions enables learning and prediction for microbiomes. Proc. Natl. Acad. Sci. 120 (48), e2307313120. doi:10.1073/pnas.2307313120
Aswani, R., Soni, K. B., and Radhakrishnan, E. K. (2024). “Introduction to circular economy—a unique approach,” in The potential of microbes for a circular economy (Elsevier), 1–24. doi:10.1016/B978-0-443-15924-4.00011-4
Athamneh, A. I. M., Alajlouni, R. A., Wallace, R. S., Seleem, M. N., and Senger, R. S. (2014). Phenotypic profiling of antibiotic response signatures in Escherichia coli using Raman spectroscopy. Antimicrob. Agents Chemother. 58 (3), 1302–1314. doi:10.1128/AAC.02098-13
Baichman-Kass, A., Song, T., and Friedman, J. (2023). Competitive interactions between culturable bacteria are highly non-additive. eLife 12, e83398. doi:10.7554/eLife.83398
Baig, Y., Ma, H. R., Xu, H., and You, L. (2023). Autoencoder neural networks enable low dimensional structure analyses of microbial growth dynamics. Nat. Commun. 14 (1), 7937. doi:10.1038/s41467-023-43455-0
Barrio-Hernandez, I., Yeo, J., Jänes, J., Mirdita, M., Gilchrist, C. L. M., Wein, T., et al. (2023). Clustering predicted structures at the scale of the known protein universe. Nature 622 (7983), 637–645. doi:10.1038/s41586-023-06510-w
Bauer, E., Zimmermann, J., Baldini, F., Thiele, I., and Kaleta, C. (2017). BacArena: individual-based metabolic modeling of heterogeneous microbes in complex communities. PLOS Comput. Biol. 13 (5), e1005544. doi:10.1371/journal.pcbi.1005544
Beck, A. E., Pintar, K., Schepens, D., Schrammeck, A., Johnson, T., Bleem, A., et al. (2022). Environment constrains fitness advantages of division of labor in microbial consortia engineered for metabolite push or pull interactions. mSystems 7 (4), e0005122–22. doi:10.1128/msystems.00051-22
Belcour, A., Frioux, C., Aite, M., Bretaudeau, A., Hildebrand, F., and Siegel, A. (2020). Metage2Metabo, microbiota-scale metabolic complementarity for the identification of key species. eLife 9, e61968. doi:10.7554/eLife.61968
Benito-Vaquerizo, S., Diender, M., Parera Olm, I., Martins Dos Santos, V. A. P., Schaap, P. J., Sousa, D. Z., et al. (2020). Modeling a co-culture of Clostridium autoethanogenum and Clostridium kluyveri to increase syngas conversion to medium-chain fatty-acids. Comput. Struct. Biotechnol. J. 18, 3255–3266. doi:10.1016/j.csbj.2020.10.003
Bergelson, J., Kreitman, M., Petrov, D. A., Sanchez, A., and Tikhonov, M. (2021). Functional biology in its natural context: a search for emergent simplicity. eLife 10, e67646. doi:10.7554/eLife.67646
Blazeck, J., and Alper, H. (2010). Systems metabolic engineering: genome-scale models and beyond. Biotechnol. J. 5 (7), 647–659. doi:10.1002/biot.200900247
Blouin, M., Karimi, B., Mathieu, J., and Lerch, T. Z. (2015). Levels and limits in artificial selection of communities. Ecol. Lett. 18 (10), 1040–1048. doi:10.1111/ele.12486
Bonillo-Lopez, L., Rouam-el Khatab, O., Obregon-Gutierrez, P., Florez-Sarasa, I., Correa-Fiz, F., Sibila, M., et al. (2024). In vitro metabolic interaction network of a rationally designed nasal microbiota community. bioRxiv. doi:10.1101/2024.10.23.619785
Bryson, J. W., Betz, S. F., Lu, H. S., Suich, D. J., Zhou, H. X., O’Neil, K. T., et al. (1995). Protein design: a hierarchic approach. Science 270 (5238), 935–941. doi:10.1126/science.270.5238.935
Bull, J. J., and Barrick, J. E. (2017). Arresting evolution. Trends Genet. 33 (12), 910–920. doi:10.1016/j.tig.2017.09.008
Cacace, E., Kim, V., Varik, V., Knopp, M., Tietgen, M., Brauer-Nikonow, A., et al. (2023). Systematic analysis of drug combinations against Gram-positive bacteria. Nat. Microbiol. 8 (11), 2196–2212. doi:10.1038/s41564-023-01486-9
Camacho-Zaragoza, J. M., Hernández-Chávez, G., Moreno-Avitia, F., Ramírez-Iñiguez, R., Martínez, A., Bolívar, F., et al. (2016). Engineering of a microbial coculture of Escherichia coli strains for the biosynthesis of resveratrol. Microb. Cell Factories 15 (1), 163. doi:10.1186/s12934-016-0562-z
Chan, S. H. J., Simons, M. N., and Maranas, C. D. (2017). SteadyCom: predicting microbial abundances while ensuring community stability. PLOS Comput. Biol. 13 (5), e1005539. doi:10.1371/journal.pcbi.1005539
Chang, C., Osborne, M. L., Bajic, D., and Sanchez, A. (2020). Artificially selecting bacterial communities using propagule strategies. Evolution 74 (10), 2392–2403. doi:10.1111/evo.14092
Chang, C.-Y., Vila, J. C. C., Bender, M., Li, R., Mankowski, M. C., Bassette, M., et al. (2021). Engineering complex communities by directed evolution. Nat. Ecol. and Evol. 5 (7), 1011–1023. doi:10.1038/s41559-021-01457-5
Chen, Y., Lin, C.-J., Jones, G., Fu, S., and Zhan, H. (2009). Enhancing biodegradation of wastewater by microbial consortia with fractional factorial design. J. Hazard. Mater. 171 (1–3), 948–953. doi:10.1016/j.jhazmat.2009.06.100
Chiu, H.-C., Levy, R., and Borenstein, E. (2014). Emergent biosynthetic capacity in simple microbial communities. PLoS Comput. Biol. 10 (7), e1003695. doi:10.1371/journal.pcbi.1003695
Chou, H.-H., Chiu, H.-C., Delaney, N. F., Segrè, D., and Marx, C. J. (2011). Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 332 (6034), 1190–1192. doi:10.1126/science.1203799
Chowdhury, S., Castro, S., Coker, C., Hinchliffe, T. E., Arpaia, N., and Danino, T. (2019). Programmable bacteria induce durable tumor regression and systemic antitumor immunity. Nat. Med. 25 (7), 1057–1063. doi:10.1038/s41591-019-0498-z
Clark, R. L., Connors, B. M., Stevenson, D. M., Hromada, S. E., Hamilton, J. J., Amador-Noguez, D., et al. (2021). Design of synthetic human gut microbiome assembly and butyrate production. Nat. Commun. 12 (1), 3254. doi:10.1038/s41467-021-22938-y
Coker, J., Zhalnina, K., Marotz, C., Thiruppathy, D., Tjuanta, M., D’Elia, G., et al. (2022). A reproducible and tunable synthetic soil microbial community provides new insights into microbial ecology. mSystems 7 (6), e0095122–22. doi:10.1128/msystems.00951-22
Connors, B. M., Thompson, J., Ertmer, S., Clark, R. L., Pfleger, B. F., and Venturelli, O. S. (2023). Control points for design of taxonomic composition in synthetic human gut communities. Cell Syst. 14 (12), 1044–1058.e13. doi:10.1016/j.cels.2023.11.007
Cordero, O. X., and Datta, M. S. (2016). Microbial interactions and community assembly at microscales. Curr. Opin. Microbiol. 31, 227–234. doi:10.1016/j.mib.2016.03.015
Cragg, S. M., Beckham, G. T., Bruce, N. C., Bugg, T. D., Distel, D. L., Dupree, P., et al. (2015). Lignocellulose degradation mechanisms across the tree of life. Curr. Opin. Chem. Biol. 29, 108–119. doi:10.1016/j.cbpa.2015.10.018
Cravens, A., Payne, J., and Smolke, C. D. (2019). Synthetic biology strategies for microbial biosynthesis of plant natural products. Nat. Commun. 10 (1), 2142. doi:10.1038/s41467-019-09848-w
Crocker, K., Lee, K. K., Chakraverti-Wuerthwein, M., Li, Z., Tikhonov, M., Mani, M., et al. (2024). Environmentally dependent interactions shape patterns in gene content across natural microbiomes. Nat. Microbiol. 9 (8), 2022–2037. doi:10.1038/s41564-024-01752-4
Cruz-Loya, M., Tekin, E., Kang, T. M., Cardona, N., Lozano-Huntelman, N., Rodriguez-Verdugo, A., et al. (2021). Antibiotics shift the temperature response curve of Escherichia coli growth. mSystems, 6(4), e0022821. doi:10.1128/msystems.00228-21
Dal Bello, M., Lee, H., Goyal, A., and Gore, J. (2021). Resource–diversity relationships in bacterial communities reflect the network structure of microbial metabolism. Nat. Ecol. and Evol. 5 (10), 1424–1434. doi:10.1038/s41559-021-01535-8
Deter, H. S., and Lu, T. (2022). Engineering microbial consortia with rationally designed cellular interactions. Curr. Opin. Biotechnol. 76, 102730. doi:10.1016/j.copbio.2022.102730
Diaz-Colunga, J., Catalan, P., San Roman, M., Arrabal, A., and Sanchez, A. (2024a). Full factorial construction of synthetic microbial communities. eLife. 13, RP101906. doi:10.7554/eLife.101906.1
Diaz-Colunga, J., Lu, N., Sanchez-Gorostiaga, A., Chang, C.-Y., Cai, H. S., Goldford, J. E., et al. (2022). Top-down and bottom-up cohesiveness in microbial community coalescence. Proc. Natl. Acad. Sci. 119 (6), e2111261119. doi:10.1073/pnas.2111261119
Diaz-Colunga, J., Skwara, A., Vila, J. C. C., Bajic, D., and Sanchez, A. (2024b). Global epistasis and the emergence of function in microbial consortia. Cell 187 (12), 3108–3119.e30. doi:10.1016/j.cell.2024.04.016
Diener, C., Gibbons, S. M., and Resendis-Antonio, O. (2020). MICOM: metagenome-scale modeling to infer metabolic interactions in the gut microbiota. mSystems 5 (1), e00606-19. doi:10.1128/mSystems.00606-19
DiMucci, D., Kon, M., and Segrè, D. (2018). Machine learning reveals missing edges and putative interaction mechanisms in microbial ecosystem networks. mSystems 3 (5), 001811-18. doi:10.1128/msystems.00181-18
Doulcier, G., Lambert, A., De Monte, S., and Rainey, P. B. (2020). Eco-evolutionary dynamics of nested Darwinian populations and the emergence of community-level heredity. eLife 9, e53433. doi:10.7554/eLife.53433
Dukovski, I., Bajić, D., Chacón, J. M., Quintin, M., Vila, J. C. C., Sulheim, S., et al. (2021). A metabolic modeling platform for the computation of microbial ecosystems in time and space (COMETS). Nat. Protoc. 16 (11), 5030–5082. doi:10.1038/s41596-021-00593-3
Eble, H., Joswig, M., Lamberti, L., and Ludington, W. B. (2023). Master regulators of biological systems in higher dimensions. Proc. Natl. Acad. Sci. 120 (51), e2300634120. doi:10.1073/pnas.2300634120
Ebrahim, A., Lerman, J. A., Palsson, B. O., and Hyduke, D. R. (2013). COBRApy: COnstraints-based reconstruction and analysis for Python. BMC Syst. Biol. 7 (1), 74. doi:10.1186/1752-0509-7-74
Edwards, J. S., and Palsson, B. O. (2000). Metabolic flux balance analysis and the in silico analysis of Escherichia coli K-12 gene deletions. BMC Bioinforma. 1 (1), 1. doi:10.1186/1471-2105-1-1
Efsa, S. C., More, S., Bampidis, V., Benford, D., Bragard, C., Halldorsson, T., et al. (2020). Evaluation of existing guidelines for their adequacy for the microbial characterisation and environmental risk assessment of microorganisms obtained through synthetic biology. EFSA J. 18 (10), e06263. doi:10.2903/j.efsa.2020.6263
Eng, A., and Borenstein, E. (2016). An algorithm for designing minimal microbial communities with desired metabolic capacities. Bioinformatics 32 (13), 2008–2016. doi:10.1093/bioinformatics/btw107
Estrela, S., Sanchez-Gorostiaga, A., Vila, J. C., and Sanchez, A. (2021). Nutrient dominance governs the assembly of microbial communities in mixed nutrient environments. eLife 10, e65948. doi:10.7554/eLife.65948
Fang, H., Kang, J., and Zhang, D. (2017). Microbial production of vitamin B12: a review and future perspectives. Microb. Cell Factories 16 (1), 15. doi:10.1186/s12934-017-0631-y
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10 (8), 538–550. doi:10.1038/nrmicro2832
Frioux, C., Fremy, E., Trottier, C., and Siegel, A. (2018). Scalable and exhaustive screening of metabolic functions carried out by microbial consortia. Bioinformatics 34 (17), i934–i943. doi:10.1093/bioinformatics/bty588
Fu, F., Tschitschko, B., Hutchins, D. A., Larsson, M. E., Baker, K. G., McInnes, A., et al. (2022). Temperature variability interacts with mean temperature to influence the predictability of microbial phenotypes. Glob. Change Biol. 28 (19), 5741–5754. doi:10.1111/gcb.16330
Gao, Y., Wang, L., and Wang, B. (2023). Customizing cellular signal processing by synthetic multi-level regulatory circuits. Nat. Commun. 14 (1), 8415. doi:10.1038/s41467-023-44256-1
García-Jiménez, B., García, J. L., and Nogales, J. (2018). FLYCOP: metabolic modeling-based analysis and engineering microbial communities. Bioinformatics 34 (17), i954–i963. doi:10.1093/bioinformatics/bty561
George, A. B., and Korolev, K. S. (2023). Ecological landscapes guide the assembly of optimal microbial communities. PLOS Comput. Biol. 19 (1), e1010570. doi:10.1371/journal.pcbi.1010570
Giménez-Palomares, F., Fernández De Córdoba, P., Mejuto, J. C., Bendaña-Jácome, R. J., and Pérez-Guerra, N. (2022). Evaluation and mathematical analysis of a four-dimensional lotka–volterra-like equation designed to describe the batch nisin production system. Mathematics 10 (5), 677. doi:10.3390/math10050677
Giordano, N., Gaudin, M., Trottier, C., Delage, E., Nef, C., Bowler, C., et al. (2024). Genome-scale community modelling reveals conserved metabolic cross-feedings in epipelagic bacterioplankton communities. Nat. Commun. 15 (1), 2721. doi:10.1038/s41467-024-46374-w
Goldford, J. E., Lu, N., Bajić, D., Estrela, S., Tikhonov, M., Sanchez-Gorostiaga, A., et al. (2018). Emergent simplicity in microbial community assembly. Science 361 (6401), 469–474. doi:10.1126/science.aat1168
Goodnight, C. J. (1990a). Experimental studies of community evolution i: the response to selection at the community level. Evolution 44 (6), 1614–1624. doi:10.1111/j.1558-5646.1990.tb03850.x
Goodnight, C. J. (1990b). Experimental studies of community evolution ii: the ecological basis of the response to community selection. Evolution 44 (6), 1625–1636. doi:10.1111/j.1558-5646.1990.tb03851.x
Gould, A. L., Zhang, V., Lamberti, L., Jones, E. W., Obadia, B., Korasidis, N., et al. (2018). Microbiome interactions shape host fitness. Proc. Natl. Acad. Sci. 115 (51), E11951–E11960. doi:10.1073/pnas.1809349115
Gowda, K., Ping, D., Mani, M., and Kuehn, S. (2022). Genomic structure predicts metabolite dynamics in microbial communities. Cell 185 (3), 530–546.e25. doi:10.1016/j.cell.2021.12.036
Graham, A. E., and Ledesma-Amaro, R. (2023). The microbial food revolution. Nat. Commun. 14 (1), 2231. doi:10.1038/s41467-023-37891-1
Großkopf, T., and Soyer, O. S. (2014). Synthetic microbial communities. Curr. Opin. Microbiol. 18, 72–77. doi:10.1016/j.mib.2014.02.002
Gu, C., Kim, G. B., Kim, W. J., Kim, H. U., and Lee, S. Y. (2019). Current status and applications of genome-scale metabolic models. Genome Biol. 20 (1), 121. doi:10.1186/s13059-019-1730-3
Guo, X., and Boedicker, J. (2016). High-order interactions between species strongly influence the activity of microbial communities. Biophysical J. 110 (3), 143a. doi:10.1016/j.bpj.2015.11.811
Hanly, T. J., and Henson, M. A. (2011). Dynamic flux balance modeling of microbial co-cultures for efficient batch fermentation of glucose and xylose mixtures. Biotechnol. Bioeng. 108 (2), 376–385. doi:10.1002/bit.22954
Hanly, T. J., Urello, M., and Henson, M. A. (2012). Dynamic flux balance modeling of S. cerevisiae and E. coli co-cultures for efficient consumption of glucose/xylose mixtures. Appl. Microbiol. Biotechnol. 93 (6), 2529–2541. doi:10.1007/s00253-011-3628-1
Hansen, J., and Kielland-Brandt, M. C. (1996). Modification of biochemical pathways in industrial yeasts. J. Biotechnol. 49 (1–3), 1–12. doi:10.1016/0168-1656(96)01523-4
Hanson, A. D., and Lorenzo, V. D. (2023). Synthetic Biology─High time to deliver? ACS Synth. Biol. 12 (6), 1579–1582. doi:10.1021/acssynbio.3c00238
Harcombe, W. R., Riehl, W. J., Dukovski, I., Granger, B. R., Betts, A., Lang, A. H., et al. (2014). Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 7 (4), 1104–1115. doi:10.1016/j.celrep.2014.03.070
Heinken, A., Hertel, J., Acharya, G., Ravcheev, D. A., Nyga, M., Okpala, O. E., et al. (2023). Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nat. Biotechnol. 41 (9), 1320–1331. doi:10.1038/s41587-022-01628-0
Heinken, A., and Thiele, I. (2022). Microbiome Modelling Toolbox 2.0: efficient, tractable modelling of microbiome communities. Bioinformatics 38 (8), 2367–2368. doi:10.1093/bioinformatics/btac082
Hu, A., Ren, M., and Wang, J. (2021). Microbial species performance responses to environmental changes: genomic traits and nutrient availability. Ecology 102 (7), e03382. doi:10.1002/ecy.3382
Hu, G., Li, Y., Ye, C., Liu, L., and Chen, X. (2019). Engineering microorganisms for enhanced CO2 sequestration. Trends Biotechnol. 37 (5), 532–547. doi:10.1016/j.tibtech.2018.10.008
Hu, J., Amor, D. R., Barbier, M., Bunin, G., and Gore, J. (2022). Emergent phases of ecological diversity and dynamics mapped in microcosms. Science 378 (6615), 85–89. doi:10.1126/science.abm7841
Jiang, S., Otero-Muras, I., Banga, J. R., Wang, Y., Kaiser, M., and Krasnogor, N. (2022). OptDesign: identifying optimum design strategies in strain engineering for biochemical production. ACS Synth. Biol. 11 (4), 1531–1541. doi:10.1021/acssynbio.1c00610
Jiménez, J., Guardia-Puebla, Y., Romero-Romero, O., Cisneros-Ortiz, M. E., Guerra, G., Morgan-Sagastume, J. M., et al. (2014). Methanogenic activity optimization using the response surface methodology, during the anaerobic co-digestion of agriculture and industrial wastes. Microbial community diversity. Biomass Bioenergy 71, 84–97. doi:10.1016/j.biombioe.2014.10.023
Johns, N. I., Blazejewski, T., Gomes, A. L., and Wang, H. H. (2016). Principles for designing synthetic microbial communities. Curr. Opin. Microbiol. 31, 146–153. doi:10.1016/j.mib.2016.03.010
Jones, J. A., Vernacchio, V. R., Sinkoe, A. L., Collins, S. M., Ibrahim, M. H. A., Lachance, D. M., et al. (2016). Experimental and computational optimization of an Escherichia coli co-culture for the efficient production of flavonoids. Metab. Eng. 35, 55–63. doi:10.1016/j.ymben.2016.01.006
Jouhten, P., Konstantinidis, D., Pereira, F., Andrejev, S., Grkovska, K., Castillo, S., et al. (2022). Predictive evolution of metabolic phenotypes using model-designed environments. Mol. Syst. Biol. 18 (10), e10980. doi:10.15252/msb.202210980
Julien-Laferrière, A., Bulteau, L., Parrot, D., Marchetti-Spaccamela, A., Stougie, L., Vinga, S., et al. (2016). A combinatorial algorithm for microbial consortia synthetic design. Sci. Rep. 6 (1), 29182. doi:10.1038/srep29182
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Karkaria, B. D., Fedorec, A. J. H., and Barnes, C. P. (2021). Automated design of synthetic microbial communities. Nat. Commun. 12 (1), 672. doi:10.1038/s41467-020-20756-2
Karlsen, E., Schulz, C., and Almaas, E. (2018). Automated generation of genome-scale metabolic draft reconstructions based on KEGG. BMC Bioinforma. 19 (1), 467. doi:10.1186/s12859-018-2472-z
Kehe, J., Kulesa, A., Ortiz, A., Ackerman, C. M., Thakku, S. G., Sellers, D., et al. (2019). Massively parallel screening of synthetic microbial communities. Proc. Natl. Acad. Sci. 116 (26), 12804–12809. doi:10.1073/pnas.1900102116
Kehe, J., Ortiz, A., Kulesa, A., Gore, J., Blainey, P. C., and Friedman, J. (2021). Positive interactions are common among culturable bacteria. Sci. Adv. 7 (45), eabi7159. doi:10.1126/sciadv.abi7159
Khalil, A. S., and Collins, J. J. (2010). Synthetic biology: applications come of age. Nat. Rev. Genet. 11 (5), 367–379. doi:10.1038/nrg2775
Khandelwal, R. A., Olivier, B. G., Röling, W. F. M., Teusink, B., and Bruggeman, F. J. (2013). Community flux balance analysis for microbial consortia at balanced growth. PLoS ONE 8 (5), e64567. doi:10.1371/journal.pone.0064567
Kikot, P., Viera, M., Mignone, C., and Donati, E. (2010). Study of the effect of pH and dissolved heavy metals on the growth of sulfate-reducing bacteria by a fractional factorial design. Hydrometallurgy 104 (3–4), 494–500. doi:10.1016/j.hydromet.2010.02.026
Kirwan, L., Connolly, J., Finn, J. A., Brophy, C., Lüscher, A., Nyfeler, D., et al. (2009). Diversity–interaction modeling: estimating contributions of species identities and interactions to ecosystem function. Ecology 90 (8), 2032–2038. doi:10.1890/08-1684.1
Kong, W., Meldgin, D. R., Collins, J. J., and Lu, T. (2018). Designing microbial consortia with defined social interactions. Nat. Chem. Biol. 14 (8), 821–829. doi:10.1038/s41589-018-0091-7
Krause, S., Le Roux, X., Niklaus, P. A., Van Bodegom, P. M., Lennon, J. T., Bertilsson, S., et al. (2014). Trait-based approaches for understanding microbial biodiversity and ecosystem functioning. Front. Microbiol. 5, 251. doi:10.3389/fmicb.2014.00251
Kucharzyk, K. H., Crawford, R. L., Paszczynski, A. J., Soule, T., and Hess, T. F. (2012). Maximizing microbial degradation of perchlorate using a genetic algorithm: media optimization. J. Biotechnol. 157 (1), 189–197. doi:10.1016/j.jbiotec.2011.10.011
Kwok, R. (2010). Five hard truths for synthetic biology. Nature 463 (7279), 288–290. doi:10.1038/463288a
Lajoie, G., and Kembel, S. W. (2019). Making the most of trait-based approaches for microbial ecology. Trends Microbiol. 27 (10), 814–823. doi:10.1016/j.tim.2019.06.003
Lalejini, A., Dolson, E., Vostinar, A. E., and Zaman, L. (2022). Artificial selection methods from evolutionary computing show promise for directed evolution of microbes. eLife 11, e79665. doi:10.7554/eLife.79665
Laurent, J. M., Jain, A., Kan, A., Steinacher, M., Enrriquez Casimiro, N., Stavrakis, S., et al. (2024). Directed evolution of material-producing microorganisms. Proc. Natl. Acad. Sci. 121 (31), e2403585121. doi:10.1073/pnas.2403585121
Lázár, V., Snitser, O., Barkan, D., and Kishony, R. (2022). Antibiotic combinations reduce Staphylococcus aureus clearance. Nature 610 (7932), 540–546. doi:10.1038/s41586-022-05260-5
Lechón-Alonso, P., Clegg, T., Cook, J., Smith, T. P., and Pawar, S. (2021). The role of competition versus cooperation in microbial community coalescence. PLOS Comput. Biol. 17 (11), e1009584. doi:10.1371/journal.pcbi.1009584
Leonard, E., Nielsen, D., Solomon, K., and Prather, K. J. (2008). Engineering microbes with synthetic biology frameworks. Trends Biotechnol. 26 (12), 674–681. doi:10.1016/j.tibtech.2008.08.003
Lewontin, R. C. (1970). The units of selection. Annu. Rev. Ecol. Syst. 1 (1), 1–18. doi:10.1146/annurev.es.01.110170.000245
Li, C., Han, Y., Zou, X., Zhang, X., Ran, Q., and Dong, C. (2024). A systematic discussion and comparison of the construction methods of synthetic microbial community. Synthetic Syst. Biotechnol. 9 (4), 775–783. doi:10.1016/j.synbio.2024.06.006
Li, T., Chen, X., Chen, J., Wu, Q., and Chen, G. (2014). Open and continuous fermentation: products, conditions and bioprocess economy. Biotechnol. J. 9 (12), 1503–1511. doi:10.1002/biot.201400084
Li, X., Zhou, Z., Li, W., Yan, Y., Shen, X., Wang, J., et al. (2022). Design of stable and self-regulated microbial consortia for chemical synthesis. Nat. Commun. 13 (1), 1554. doi:10.1038/s41467-022-29215-6
Ling, H., Teo, W., Chen, B., Leong, S. S. J., and Chang, M. W. (2014). Microbial tolerance engineering toward biochemical production: from lignocellulose to products. Curr. Opin. Biotechnol. 29, 99–106. doi:10.1016/j.copbio.2014.03.005
Machado, D., Andrejev, S., Tramontano, M., and Patil, K. R. (2018). Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 46 (15), 7542–7553. doi:10.1093/nar/gky537
Machado, D., Maistrenko, O. M., Andrejev, S., Kim, Y., Bork, P., Patil, K. R., et al. (2021). Polarization of microbial communities between competitive and cooperative metabolism. Nat. Ecol. and Evol. 5 (2), 195–203. doi:10.1038/s41559-020-01353-4
Mahadevan, R., Edwards, J. S., and Doyle, F. J. (2002). Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophysical J. 83 (3), 1331–1340. doi:10.1016/S0006-3495(02)73903-9
Marsland, R., Cui, W., Goldford, J., and Mehta, P. (2020a). The Community Simulator: a Python package for microbial ecology. PLOS ONE 15 (3), e0230430. doi:10.1371/journal.pone.0230430
Marsland, R., Cui, W., and Mehta, P. (2020b). A minimal model for microbial biodiversity can reproduce experimentally observed ecological patterns. Sci. Rep. 10 (1), 3308. doi:10.1038/s41598-020-60130-2
Maulud, D., and Abdulazeez, A. M. (2020). A review on linear regression comprehensive in machine learning. J. Appl. Sci. Technol. Trends 1 (2), 140–147. doi:10.38094/jastt1457
McCarty, N. S., and Ledesma-Amaro, R. (2019). Synthetic biology tools to engineer microbial communities for biotechnology. Trends Biotechnol. 37 (2), 181–197. doi:10.1016/j.tibtech.2018.11.002
McEnany, J., and Good, B. H. (2024). Predicting the first steps of evolution in randomly assembled communities. Nat. Commun. 15 (1), 8495. doi:10.1038/s41467-024-52467-3
McKay, L. L., and Baldwin, K. A. (1990). Applications for biotechnology: present and future improvements in lactic acid bacteria. FEMS Microbiol. Lett. 87 (1–2), 3–14. doi:10.1111/j.1574-6968.1990.tb04876.x
Mendoza, S. N., Olivier, B. G., Molenaar, D., and Teusink, B. (2019). A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 20 (1), 158. doi:10.1186/s13059-019-1769-1
Mickalide, H., and Kuehn, S. (2019). Higher-order interaction between species inhibits bacterial invasion of a phototroph-predator microbial community. Cell Syst. 9 (6), 521–533.e10. doi:10.1016/j.cels.2019.11.004
Morin, M. A., Morrison, A. J., Harms, M. J., and Dutton, R. J. (2022). Higher-order interactions shape microbial interactions as microbial community complexity increases. Sci. Rep. 12 (1), 22640. doi:10.1038/s41598-022-25303-1
Morris, A., Meyer, K., and Bohannan, B. (2020). Linking microbial communities to ecosystem functions: what we can learn from genotype–phenotype mapping in organisms. Philosophical Trans. R. Soc. B Biol. Sci. 375 (1798), 20190244. doi:10.1098/rstb.2019.0244
Muir, W. M. (1996). Group selection for adaptation to multiple-hen cages: selection program and direct responses. Poult. Sci. 75 (4), 447–458. doi:10.3382/ps.0750447
Müller, I. E., Rubens, J. R., Jun, T., Graham, D., Xavier, R., and Lu, T. K. (2019). Gene networks that compensate for crosstalk with crosstalk. Nat. Commun. 10 (1), 4028. doi:10.1038/s41467-019-12021-y
Neal, M., Brakewood, W., Betenbaugh, M., and Zengler, K. (2024). Pan-genome-scale metabolic modeling of Bacillus subtilis reveals functionally distinct groups. mSystems 9 (11), e0092324–24. doi:10.1128/msystems.00923-24
O’Brien, E. J., Lerman, J. A., Chang, R. L., Hyduke, D. R., and Palsson, B. Ø. (2013). Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 9 (1), 693. doi:10.1038/msb.2013.52
Okano, H., Hermsen, R., Kochanowski, K., and Hwa, T. (2019). Regulation underlying hierarchical and simultaneous utilization of carbon substrates by flux sensors in Escherichia coli. Nat. Microbiol. 5 (1), 206–215. doi:10.1038/s41564-019-0610-7
Olivier, B. (2018). SystemsBioinformatics/cbmpy-metadraft: metaDraft is now available (Version v0.9.0-rc.1). Zenodo. [Computer software]. doi:10.5281/ZENODO.2398336
Orth, J. D., Fleming, R. M. T., and Palsson, B. Ø. (2010a). Reconstruction and use of microbial metabolic networks: the core Escherichia coli metabolic model as an educational guide. EcoSal Plus. 4(1), doi:10.1128/ecosalplus.10.2.1
Orth, J. D., Thiele, I., and Palsson, B. Ø. (2010b). What is flux balance analysis? Nat. Biotechnol. 28 (3), 245–248. doi:10.1038/nbt.1614
Otwinowski, J. (2018). Biophysical inference of epistasis and the effects of mutations on protein stability and function. Mol. Biol. Evol. 35 (10), 2345–2354. doi:10.1093/molbev/msy141
Pacheco, A. R., and Segrè, D. (2021) An evolutionary algorithm for designing microbial communities via environmental modification. J. R. Soc. Interface. 18: 20210348. doi:10.1098/rsif.2021.0348
Panke-Buisse, K., Poole, A. C., Goodrich, J. K., Ley, R. E., and Kao-Kniffin, J. (2015). Selection on soil microbiomes reveals reproducible impacts on plant function. ISME J. 9 (4), 980–989. doi:10.1038/ismej.2014.196
Park, H., Patel, A., Hunt, K. A., Henson, M. A., and Carlson, R. P. (2020). Artificial consortium demonstrates emergent properties of enhanced cellulosic-sugar degradation and biofuel synthesis. Npj Biofilms Microbiomes 6 (1), 59. doi:10.1038/s41522-020-00170-8
Park, Y.-K., Peng, H., Hapeta, P., Sellés Vidal, L., and Ledesma-Amaro, R. (2024). Engineered cross-feeding creates inter- and intra-species synthetic yeast communities with enhanced bioproduction. Nat. Commun. 15 (1), 8924. doi:10.1038/s41467-024-53117-4
Peng, H., Darlington, A. P. S., South, E. J., Chen, H.-H., Jiang, W., and Ledesma-Amaro, R. (2024). A molecular toolkit of cross-feeding strains for engineering synthetic yeast communities. Nat. Microbiol. 9 (3), 848–863. doi:10.1038/s41564-023-01596-4
Perrino, G., Hadjimitsis, A., Ledesma-Amaro, R., and Stan, G.-B. (2021). Control engineering and synthetic biology: working in synergy for the analysis and control of microbial systems. Curr. Opin. Microbiol. 62, 68–75. doi:10.1016/j.mib.2021.05.004
Pignon, E., Holló, G., Steiner, T., Van Vliet, S., and Schaerli, Y. (2024). Engineering microbial consortia: uptake and leakage rate differentially shape community arrangement and composition. doi:10.1101/2024.07.19.604250
Pinto, T., Grimalt-Alemany, A., Flores-Alsina, X., Gavala, H. N., Gernaey, K. V., and Junicke, H. (2022). Shaping an open microbiome for butanol production through process control. Fermentation 8 (7), 333. doi:10.3390/fermentation8070333
Prasad, R. K., Chatterjee, S., Mazumder, P. B., Gupta, S. K., Sharma, S., Vairale, M. G., et al. (2019). Bioethanol production from waste lignocelluloses: a review on microbial degradation potential. Chemosphere 231, 588–606. doi:10.1016/j.chemosphere.2019.05.142
Qureshi, A. S., Khushk, I., Naqvi, S. R., Simiar, A. A., Ali, C. H., Naqvi, M., et al. (2017). Fruit waste to energy through open fermentation. Energy Procedia 142, 904–909. doi:10.1016/j.egypro.2017.12.145
Ratzke, C., and Gore, J. (2018). Modifying and reacting to the environmental pH can drive bacterial interactions. PLOS Biol. 16 (3), e2004248. doi:10.1371/journal.pbio.2004248
Reetz, M. T., Zonta, A., Schimossek, K., Jaeger, K., and Liebeton, K. (1997). Creation of enantioselective biocatalysts for organic chemistry by in vitro evolution. Angewandte Chemie Int. Ed. Engl. 36 (24), 2830–2832. doi:10.1002/anie.199728301
Roell, G. W., Zha, J., Carr, R. R., Koffas, M. A., Fong, S. S., and Tang, Y. J. (2019). Engineering microbial consortia by division of labor. Microb. Cell Factories 18 (1), 35. doi:10.1186/s12934-019-1083-3
Romano, P., Soli, M. G., Suzzi, G., Grazia, L., and Zambonelli, C. (1985). Improvement of a wine Saccharomyces cerevisiae strain by a breeding program. Appl. Environ. Microbiol. 50 (4), 1064–1067. doi:10.1128/aem.50.4.1064-1067.1985
Romero, P. A., Krause, A., and Arnold, F. H. (2013). Navigating the protein fitness landscape with Gaussian processes. Proc. Natl. Acad. Sci. 110 (3), E193–E201. doi:10.1073/pnas.1215251110
Sailer, Z. R., Shafik, S. H., Summers, R. L., Joule, A., Patterson-Robert, A., Martin, R. E., et al. (2020). Inferring a complete genotype-phenotype map from a small number of measured phenotypes. PLOS Comput. Biol. 16 (9), e1008243. doi:10.1371/journal.pcbi.1008243
Salimi, F., Zhuang, K., and Mahadevan, R. (2010). Genome-scale metabolic modeling of a clostridial co-culture for consolidated bioprocessing. Biotechnol. J. 5 (7), 726–738. doi:10.1002/biot.201000159
Sánchez, Á., Arrabal, A., San Román, M., and Díaz-Colunga, J. (2024). The optimization of microbial functions through rational environmental manipulations. Mol. Microbiol. 122 (3), 294–303. doi:10.1111/mmi.15236
Sanchez, A., Bajic, D., Diaz-Colunga, J., Skwara, A., Vila, J. C. C., and Kuehn, S. (2023). The community-function landscape of microbial consortia. Cell Syst. 14 (2), 122–134. doi:10.1016/j.cels.2022.12.011
Sánchez, Á., Vila, J. C. C., Chang, C.-Y., Diaz-Colunga, J., Estrela, S., and Rebolleda-Gomez, M. (2021). Directed evolution of microbial communities. Annu. Rev. Biophysics 50 (1), 323–341. doi:10.1146/annurev-biophys-101220-072829
Sanchez-Gorostiaga, A., Bajić, D., Osborne, M. L., Poyatos, J. F., and Sanchez, A. (2019). High-order interactions distort the functional landscape of microbial consortia. PLOS Biol. 17 (12), e3000550. doi:10.1371/journal.pbio.3000550
San León, D., and Nogales, J. (2022). Toward merging bottom–up and top–down model-based designing of synthetic microbial communities. Curr. Opin. Microbiol. 69, 102169. doi:10.1016/j.mib.2022.102169
Schmidt-Dannert, C., and Arnold, F. H. (1999). Directed evolution of industrial enzymes. Trends Biotechnol. 17 (4), 135–136. doi:10.1016/S0167-7799(98)01283-9
Schoustra, S., Hwang, S., Krug, J., and De Visser, J. A. G. M. (2016). Diminishing-returns epistasis among random beneficial mutations in a multicellular fungus. Proc. R. Soc. B Biol. Sci. 283 (1837), 20161376. doi:10.1098/rspb.2016.1376
Segrè, D., Vitkup, D., and Church, G. M. (2002). Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. 99 (23), 15112–15117. doi:10.1073/pnas.232349399
Senne De Oliveira Lino, F., Bajic, D., Vila, J. C. C., Sánchez, A., and Sommer, M. O. A. (2021). Complex yeast–bacteria interactions affect the yield of industrial ethanol fermentation. Nat. Commun. 12 (1), 1498. doi:10.1038/s41467-021-21844-7
Sgobba, E., Stumpf, A. K., Vortmann, M., Jagmann, N., Krehenbrink, M., Dirks-Hofmeister, M. E., et al. (2018). Synthetic Escherichia coli-Corynebacterium glutamicum consortia for l-lysine production from starch and sucrose. Bioresour. Technol. 260, 302–310. doi:10.1016/j.biortech.2018.03.113
Shafiei, M., Dunn, K. A., Boon, E., MacDonald, S. M., Walsh, D. A., Gu, H., et al. (2015). BioMiCo: a supervised Bayesian model for inference of microbial community structure. Microbiome 3 (1), 8. doi:10.1186/s40168-015-0073-x
Shafiei, M., Dunn, K. A., Chipman, H., Gu, H., and Bielawski, J. P. (2014). BiomeNet: a bayesian model for inference of metabolic divergence among microbial communities. PLoS Comput. Biol. 10 (11), e1003918. doi:10.1371/journal.pcbi.1003918
Shibasaki, S., and Mitri, S. (2020). Controlling evolutionary dynamics to optimize microbial bioremediation. Evol. Appl. 13 (9), 2460–2471. doi:10.1111/eva.13050
Shlomi, T., Berkman, O., and Ruppin, E. (2005). Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc. Natl. Acad. Sci. 102 (21), 7695–7700. doi:10.1073/pnas.0406346102
Shong, J., Jimenez Diaz, M. R., and Collins, C. H. (2012). Towards synthetic microbial consortia for bioprocessing. Curr. Opin. Biotechnol. 23 (5), 798–802. doi:10.1016/j.copbio.2012.02.001
Silverstein, M. R., Bhatnagar, J. M., and Segrè, D. (2024). Metabolic complexity drives divergence in microbial communities. Nat. Ecol. and Evol. 8 (8), 1493–1504. doi:10.1038/s41559-024-02440-6
Silverstein, M. R., Segrè, D., and Bhatnagar, J. M. (2023). Environmental microbiome engineering for the mitigation of climate change. Glob. Change Biol. 29 (8), 2050–2066. doi:10.1111/gcb.16609
Skonieczny, M. T., and Yargeau, V. (2009). Biohydrogen production from wastewater by Clostridium beijerinckii: effect of pH and substrate concentration. Int. J. Hydrogen Energy 34 (8), 3288–3294. doi:10.1016/j.ijhydene.2009.01.044
Skwara, A., Gowda, K., Yousef, M., Diaz-Colunga, J., Raman, A. S., Sanchez, A., et al. (2023). Statistically learning the functional landscape of microbial communities. Nat. Ecol. and Evol. 7 (11), 1823–1833. doi:10.1038/s41559-023-02197-4
Slusarczyk, A. L., Lin, A., and Weiss, R. (2012). Foundations for the design and implementation of synthetic genetic circuits. Nat. Rev. Genet. 13 (6), 406–420. doi:10.1038/nrg3227
Smith, P., and Schuster, M. (2019). Public goods and cheating in microbes. Curr. Biol. 29 (11), R442–R447. doi:10.1016/j.cub.2019.03.001
Song, B., Shang, S., Cai, F. M., Liu, Z., Fang, J., Li, N., et al. (2023). Microbial resistance in rhizosphere hotspots under biodegradable and conventional microplastic amendment: community and functional sensitivity. Soil Biol. Biochem. 180, 108989. doi:10.1016/j.soilbio.2023.108989
Statnikov, A., Henaff, M., Narendra, V., Konganti, K., Li, Z., Yang, L., et al. (2013). A comprehensive evaluation of multicategory classification methods for microbiomic data. Microbiome 1 (1), 11. doi:10.1186/2049-2618-1-11
Stein, R. R., Tanoue, T., Szabady, R. L., Bhattarai, S. K., Olle, B., Norman, J. M., et al. (2018). Computer-guided design of optimal microbial consortia for immune system modulation. eLife 7, e30916. doi:10.7554/eLife.30916
Stolyar, S., Van Dien, S., Hillesland, K. L., Pinel, N., Lie, T. J., Leigh, J. A., et al. (2007). Metabolic modeling of a mutualistic microbial community. Mol. Syst. Biol. 3 (1), 92. doi:10.1038/msb4100131
Sun, X., Favier, A., Folmar, J., Pyenson, N. C., Sanchez, A., and Rebolleda-Gómez, M. (2024). Metabolic plasticity shapes microbial communities across a temperature gradient. Am. Nat. 204 (4), 381–399. doi:10.1086/731997
Swenson, W., Arendt, J., and Wilson, D. S. (2000a). Artificial selection of microbial ecosystems for 3-chloroaniline biodegradation. Environ. Microbiol. 2 (5), 564–571. doi:10.1046/j.1462-2920.2000.00140.x
Swenson, W., Wilson, D. S., and Elias, R. (2000b). Artificial ecosystem selection. Proc. Natl. Acad. Sci. 97 (16), 9110–9114. doi:10.1073/pnas.150237597
Taghavi, N., Singhal, N., Zhuang, W.-Q., and Baroutian, S. (2021). Degradation of plastic waste using stimulated and naturally occurring microbial strains. Chemosphere 263, 127975. doi:10.1016/j.chemosphere.2020.127975
Tareen, A., Kooshkbaghi, M., Posfai, A., Ireland, W. T., McCandlish, D. M., and Kinney, J. B. (2022). MAVE-NN: learning genotype-phenotype maps from multiplex assays of variant effect. Genome Biol. 23 (1), 98. doi:10.1186/s13059-022-02661-7
Thompson, J., Johansen, R., Dunbar, J., and Munsky, B. (2019). Machine learning to predict microbial community functions: an analysis of dissolved organic carbon from litter decomposition. PLOS ONE 14 (7), e0215502. doi:10.1371/journal.pone.0215502
Thompson, J. C., Zavala, V. M., and Venturelli, O. S. (2023). Integrating a tailored recurrent neural network with Bayesian experimental design to optimize microbial community functions. PLOS Comput. Biol. 19 (9), e1011436. doi:10.1371/journal.pcbi.1011436
Tonner, P. D., Pressman, A., and Ross, D. (2022). Interpretable modeling of genotype–phenotype landscapes with state-of-the-art predictive power. Proc. Natl. Acad. Sci. 119 (26), e2114021119. doi:10.1073/pnas.2114021119
Tzamali, E., Poirazi, P., Tollis, I. G., and Reczko, M. (2011). A computational exploration of bacterial metabolic diversity identifying metabolic interactions and growth-efficient strain communities. BMC Syst. Biol. 5 (1), 167. doi:10.1186/1752-0509-5-167
Vandecasteele, F. P. J., Crawford, R. L., and Hess, T. F. (2008). Using a genetic algorithm to drive a microbial ecosystem in a desirable direction. Environ. Microbiol. 10 (7), 1823–1830. doi:10.1111/j.1462-2920.2008.01603.x
Van Den Berg, N. I., Machado, D., Santos, S., Rocha, I., Chacón, J., Harcombe, W., et al. (2022). Ecological modelling approaches for predicting emergent properties in microbial communities. Nat. Ecol. and Evol. 6 (7), 855–865. doi:10.1038/s41559-022-01746-7
Van Vliet, S., Hauert, C., Fridberg, K., Ackermann, M., and Dal Co, A. (2022). Global dynamics of microbial communities emerge from local interaction rules. PLOS Comput. Biol. 18 (3), e1009877. doi:10.1371/journal.pcbi.1009877
Varma, A., and Palsson, B. O. (1994). Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl. Environ. Microbiol. 60 (10), 3724–3731. doi:10.1128/aem.60.10.3724-3731.1994
Venkataram, S., and Kryazhimskiy, S. (2023). Evolutionary repeatability of emergent properties of ecological communities. Philosophical Trans. R. Soc. B Biol. Sci. 378 (1877), 20220047. doi:10.1098/rstb.2022.0047
Wade, M. J. (1976). Group selections among laboratory populations of Tribolium. Proc. Natl. Acad. Sci. 73 (12), 4604–4607. doi:10.1073/pnas.73.12.4604
Wade, M. J. (1977). An experimental study of group selection. Evolution 31 (1), 134–153. doi:10.1111/j.1558-5646.1977.tb00991.x
Wagner, A. (2022). Competition for nutrients increases invasion resistance during assembly of microbial communities. Mol. Ecol. 31 (15), 4188–4203. doi:10.1111/mec.16565
Walls, L. E., Otoupal, P., Ledesma-Amaro, R., Velasquez-Orta, S. B., Gladden, J. M., and Rios-Solis, L. (2023). Bioconversion of cellulose into bisabolene using Ruminococcus flavefaciens and Rhodosporidium toruloides. Bioresour. Technol. 368, 128216. doi:10.1016/j.biortech.2022.128216
Walsh, C., Stallard-Olivera, E., and Fierer, N. (2024). Nine (not so simple) steps: a practical guide to using machine learning in microbial ecology. mBio 15 (2), e0205023–23. doi:10.1128/mbio.02050-23
Wang, H., Marcišauskas, S., Sánchez, B. J., Domenzain, I., Hermansson, D., Agren, R., et al. (2018). RAVEN 2.0: a versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLOS Comput. Biol. 14 (10), e1006541. doi:10.1371/journal.pcbi.1006541
Wang, J., Gao, M., Wang, Q., Zhang, W., and Shirai, Y. (2016). Pilot-scale open fermentation of food waste to produce lactic acid without inoculum addition. RSC Adv. 6 (106), 104354–104358. doi:10.1039/C6RA22760K
Wang, J., Jiang, L., and Sun, H. (2021). Early evidence for beer drinking in a 9000-year-old platform mound in southern China. PLOS ONE 16 (8), e0255833. doi:10.1371/journal.pone.0255833
Wang, L., Zhang, X., Tang, C., Li, P., Zhu, R., Sun, J., et al. (2022a). Engineering consortia by polymeric microbial swarmbots. Nat. Commun. 13 (1), 3879. doi:10.1038/s41467-022-31467-1
Wang, M., Chen, X., Liu, X., Fang, Y., Zheng, X., Huang, T., et al. (2022b). Even allocation of benefits stabilizes microbial community engaged in metabolic division of labor. Cell Rep. 40 (13), 111410. doi:10.1016/j.celrep.2022.111410
Wasner, D., Schnecker, J., Han, X., Sun, Y., Frossard, A., Zagal Venegas, E., et al. (2024). Environment and microbiome drive different microbial traits and functions in the macroscale soil organic carbon cycle. Glob. Change Biol. 30 (8), e17465. doi:10.1111/gcb.17465
Wu, S., Zhou, Y., Dai, L., Yang, A., and Qiao, J. (2024). Assembly of functional microbial ecosystems: from molecular circuits to communities. FEMS Microbiol. Rev. 48, fuae026. doi:10.1093/femsre/fuae026
Wünsche, A., Dinh, D. M., Satterwhite, R. S., Arenas, C. D., Stoebel, D. M., and Cooper, T. F. (2017). Diminishing-returns epistasis decreases adaptability along an evolutionary trajectory. Nat. Ecol. and Evol. 1 (4), 0061. doi:10.1038/s41559-016-0061
Xie, L., and Shou, W. (2021). Steering ecological-evolutionary dynamics to improve artificial selection of microbial communities. Nat. Commun. 12 (1), 6799. doi:10.1038/s41467-021-26647-4
Xie, L., Yuan, A. E., and Shou, W. (2019). Simulations reveal challenges to artificial community selection and possible strategies for success. PLOS Biol. 17 (6), e3000295. doi:10.1371/journal.pbio.3000295
Yang, Y. (2021). Emerging patterns of microbial functional traits. Trends Microbiol. 29 (10), 874–882. doi:10.1016/j.tim.2021.04.004
Yano, T., Oue, S., and Kagamiyama, H. (1998). Directed evolution of an aspartate aminotransferase with new substrate specificities. Proc. Natl. Acad. Sci. 95 (10), 5511–5515. doi:10.1073/pnas.95.10.5511
Yeh, P., Tschumi, A. I., and Kishony, R. (2006). Functional classification of drugs by properties of their pairwise interactions. Nat. Genet. 38 (4), 489–494. doi:10.1038/ng1755
You, L., and Arnold, F. H. (1996). Directed evolution of subtilisin E in Bacillus subtilis to enhance total activity in aqueous dimethylformamide. Protein Eng. Des. Sel. 9 (1), 77–83. doi:10.1093/protein/9.1.77
Zakeri, B., and Carr, P. A. (2015). The limits of synthetic biology. Trends Biotechnol. 33 (2), 57–58. doi:10.1016/j.tibtech.2014.10.008
Zelezniak, A., Andrejev, S., Ponomarova, O., Mende, D. R., Bork, P., and Patil, K. R. (2015). Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl. Acad. Sci. 112 (20), 6449–6454. doi:10.1073/pnas.1421834112
Zhang, J., Hansen, L. G., Gudich, O., Viehrig, K., Lassen, L. M. M., Schrübbers, L., et al. (2022). A microbial supply chain for production of the anti-cancer drug vinblastine. Nature 609 (7926), 341–347. doi:10.1038/s41586-022-05157-3
Zhao, Y., Cordero, O. X., and Tikhonov, M. (2024). Linear-regression-based algorithms can succeed at identifying microbial functional groups despite the nonlinearity of ecological function. PLOS Comput. Biol. 20 (11), e1012590. doi:10.1371/journal.pcbi.1012590
Zheng, X., and Li, D. (2016). Interaction of Acidithiobacillus ferrooxidans, Rhizobium phaseoli and Rhodotorula sp. in bioleaching process based on Lotka–Volterra model. Electron. J. Biotechnol. 22, 90–97. doi:10.1016/j.ejbt.2016.06.004
Zhou, H., Gao, X., Wang, S., Zhang, Y., Coulon, F., and Cai, C. (2023). Enhanced bioremediation of aged polycyclic aromatic hydrocarbons in soil using immobilized microbial consortia combined with strengthening remediation strategies. Int. J. Environ. Res. Public Health 20 (3), 1766. doi:10.3390/ijerph20031766
Zhu, H., Meng, H., Zhang, W., Gao, H., Zhou, J., Zhang, Y., et al. (2019). Development of a longevous two-species biophotovoltaics with constrained electron flow. Nat. Commun. 10 (1), 4282. doi:10.1038/s41467-019-12190-w
Zhu, M., and Dai, X. (2024). Shaping of microbial phenotypes by trade-offs. Nat. Commun. 15 (1), 4238. doi:10.1038/s41467-024-48591-9
Zhuang, K., Izallalen, M., Mouser, P., Richter, H., Risso, C., Mahadevan, R., et al. (2011). Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 5 (2), 305–316. doi:10.1038/ismej.2010.117
Ziesack, M., Gibson, T., Oliver, J. K. W., Shumaker, A. M., Hsu, B. B., Riglar, D. T., et al. (2019). Engineered interspecies amino acid cross-feeding increases population evenness in a synthetic bacterial consortium. mSystems 4. doi:10.1128/mSystems.00352-19
Keywords: microbial ecology, synthetic ecology, synthetic microbial communities, synthetic and systems biology, microbial communities
Citation: San Román M, Arrabal A, Benitez-Dominguez B, Quirós-Rodríguez I and Diaz-Colunga J (2025) Towards synthetic ecology: strategies for the optimization of microbial community functions. Front. Synth. Biol. 3:1532846. doi: 10.3389/fsybi.2025.1532846
Received: 22 November 2024; Accepted: 03 March 2025;
Published: 18 March 2025.
Edited by:
Alicia Sanchez-Gorostiaga, Instituto Madrileño de Investigación y Desarrollo Rural, Agrario y Alimentario, SpainReviewed by:
Finn Edward Stirling, University of Cambridge, United KingdomCopyright © 2025 San Román, Arrabal, Benitez-Dominguez, Quirós-Rodríguez and Diaz-Colunga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Diaz-Colunga, amRjQHVzYWwuZXM=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.