
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Synth. Biol. , 02 August 2024
Sec. Ecosystems and Biodiversity Sustainability
Volume 2 - 2024 | https://doi.org/10.3389/fsybi.2024.1419557
 Michael C. H. Wong1†
Michael C. H. Wong1† Thomas E. Grant2,3†
Thomas E. Grant2,3† Hamid Reza Karbalaei-Heidari1
Hamid Reza Karbalaei-Heidari1 Anna C. Robotham4Matthew E. Loewen5
Anna C. Robotham4Matthew E. Loewen5 Antony D. St-Jacques2
Antony D. St-Jacques2 Nediljko Budisa1
Nediljko Budisa1 Michele C. Loewen2,3*
Michele C. Loewen2,3*Introduction: Previous studies have shown how replacing canonical residues with isosteric amino acid analogs in enzymes can predictably alter biocatalytic activities by introducing variations in the first and second shell residues relative to the binding pocket. In this study, we explore the global integration of amino acid analogs into 1,2-catechol dioxygenase from Rhodococcus opacus (Rho 1,2-CTD), an industrial enzyme containing non-heme iron that is vital for phenol ring degradation.
Methods: We replaced tyrosine residues in the first shell of the binding pocket, phenylalanine residues in the second shell, and methionine residues near the binding pocket with m-fluorotyrosine (m-FY), m-fluorophenylalanine (m-FF), and Nle, respectively, using auxotrophic Escherichia coli strains.
Results: The expression, purification, and incorporation of m-FY and m-FF into the Rho 1,2-CTD was successful, but the Nle-insertion did not work. The structural characterization of the resulting m-FF- and m-FY-containing variants provided a mechanistic framework and plausible explanation for the results of the kinetic analyses of the native enzyme and fluorinated variants.
Discussion: Our findings demonstrate the impact of fluorination on the activity of 1,2-catechol dioxygenase, revealing its influence on residues near the substrate (first shell) as well as those distant from the binding pocket (second shell). This provides a robust foundation for future engineering activity.
Biocatalysis, a field of applied science, has undergone centuries of evolution, initially rooted in early practices such as fermentation in baking and brewing (Vasic-Racki, 2006). Over time, its applications expanded, spurred by the Industrial Revolution and enhanced by advancing insights into the molecular mechanisms driven by enzymes. The quest to enhance natural reactions led to the development of protein engineering techniques, including recombinant protein expression, purification, rational site-directed mutagenesis, and eventually, directed evolution. These methodologies have successfully facilitated enhancements in protein and enzyme properties such as activity, stability, binding affinity, and substrate specificity (Bornscheuer et al., 2012). Classical site-directed mutagenesis enables precise modifications of gene-encoded protein sequences by manipulating codons at the DNA level. While this method is effective for replacing structurally similar canonical amino acids like Ser/Ala/Cys, Thr/Val, Glu/Gln, Asp/Asn, and Tyr/Phe, it has limitations for amino acids such as Trp, Met, His, or Pro, which are crucial for protein structure and catalytic activity. To address this, non-disruptive isosteric modifications, achieved through the incorporation of non-canonical amino acids (ncAAs), allow for subtle changes at the atomic level—“atomic mutations” (Budisa et al., 1998). This molecular approach extends the scope of protein synthesis beyond classical DNA mutagenesis, enabling the study and engineering of protein structure, stability, and activity. Proteins generated through site-directed mutagenesis are commonly termed “mutants,” while those with substituted canonical amino acids are referred to as “variants”, “alloproteins”, or “protein congeners” (Thi To et al., 2022). Introducing ncAAs with reactive functional groups not only allows for the precise analysis of atomic-level changes but also broadens the chemical diversity of proteins. This expansion enables the development of novel protein functionalities, including enhanced redox activities, bioconjugation through click chemistry, specific and controllable enzyme immobilization, and the introduction of photoreactive groups for creating switches and cages (Budisa and Schneider, 2019; Hartman, 2022; Bednar et al., 2023).
The feasibility of incorporating various non-canonical amino acids (ncAAs; also referred to in the literature as “non-proteinogenic”, “non-natural”, or more commonly “unnatural amino acids” or “amino acid analogues”) into proteins has been demonstrated (Manandhar et al., 2021; Shandell et al., 2021; Goettig et al., 2023). These efforts have included both site-specific incorporation (by orthogonal translation with readthrough of in-frame stop codons) and global or residue-specific incorporation by selective pressure—known as selective pressure incorporation (SPI) (Hoesl and Budisa, 2012). Nevertheless, much of the research conducted thus far has been academic in nature, serving as proof-of-principle. The pivotal question persists: can the integration of ncAAs into enzymes yield tangible enhancements in industrial biocatalytic processes (Agostini et al., 2017; Birch-Price et al., 2023; Lugtenburg et al., 2023)?
In the context of understanding the potential utility of the global incorporation of ncAAs into industrially relevant biocatalysts, simpler ncAAs with single atom substitutions or small additions remain the focus. In particular, halogenated analogues as well as analogues that impart increased hydrophobicity such as norleucine (Nle) are of interest. Nle is a non-oxidizable analogue of methionine, with the sulfur atom replaced by a -CH2- group (Anderhuber et al., 2016). Fluorine is a preferred halogen for incorporation (Berger et al., 2017). It is not much larger than hydrogen, highly electronegative, and although highly hydrophobic, does not partition into hydrocarbons (Neil and Marsh, 2000).
We are inspired by the SPI method’s capacity to induce subtle modifications in protein residues, enhancing catalytic properties. For instance, replacing phenylalanine (Phe) residues with fluorinated variants, para- and meta-fluorophenylalanine (m-FF), in a lipase enzyme led to consistent pre-activation and a 25% boost in catalytic activity. Similarly, substituting Met residues with the isosteric analog norleucine (Nle) improved substrate specificity (Hoesl et al., 2011; Acevedo-Rocha et al., 2013). Another instance involves the global incorporation of norleucine (Nle), resulting in a twofold enhancement of the catalytic activity of a cytochrome P450 enzyme (Cirino et al., 2003). Furthermore, substituting Trp residues with 5-fluorotryptophan globally led to a fourfold increase in turnover and a twofold increase in catalytic efficiency in a glutathione transferase (Parsons et al., 1998), along with significant structural alterations in human recombinant annexin V (Minks et al., 1999). Similarly, the global substitution of Tyr-residues by meta-fluorotyrosine (m-FY) into an ω-transaminase resulted in a twofold enhancement in activity, particularly noticeable in the presence of organic solvents (Deepankumar et al., 2014). Furthermore, additional studies centered on site-specific integration have reinforced the potential advantages of incorporating such analogs for enzyme catalysis (reviewed by Agostini et al., 2017 and Birch-Price et al., 2023).
In this study, we explore the global integration of analogs into 1,2-catechol dioxygenase from Rhodococcus opacus (Rho 1,2-CTD; Matera et al., 2010), a non-heme iron-containing enzyme potentially significant for industrial applications in phenolic ring breakdown (Tian et al., 2017; Salvachúa et al., 2018; Kruyer et al., 2020). Specifically, tyrosine, phenylalanine, and methionine residues were globally substituted with m-FY, m-FF, and Nle, respectively (Figure 1; Supplementary Figure S1). After recombinant production and purification, mass spectrometry analyses confirmed the incorporation of m-FY and m-FF into Rho 1,2-CTD, while Met to Nle substitutions occurred at minimal levels, if any. Structural analyses affirmed that the overall fold remained largely unchanged, while modeling experiments established a reliable mechanistic framework for interpreting the outcomes of activity studies. Notably, our kinetic analyses of the resulting m-FF variants demonstrated behavior comparable to the wild-type enzyme, whereas the m-FY enzyme displayed modified activity in substrate specificity and catalytic turnover. We have examined these findings in the context of their importance for understanding structure–function relationships and their implications for industrial enzyme engineering.
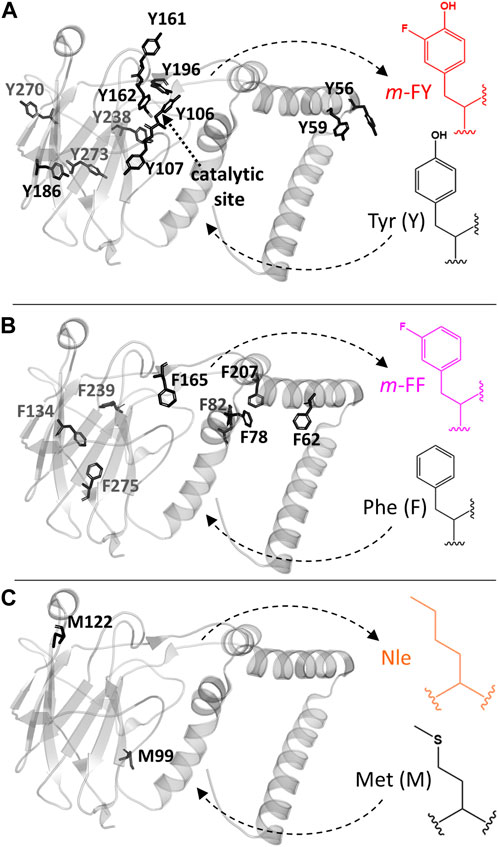
Figure 1. Rho 1,2-CTD engineering with noncanonical amino acids. The 3D crystal structure PDBID# 3HHY of the 1,2-catechol dioxygenase from Rhodococcus opacus is shown. (A) Tyrosine (Y), (B) phenyalanine (F), and (C) methionine (M) were globally replaced by related analogs as follows: Tyr by m-FY (meta-fluorotyrosine), Phe by m-FF (meta-fluorophenylalanine), and Met by Nle (norleucine). Tyr 162 and Tyr 196 contribute to Fe(III) coordination in the catalytic center. Phe 20 was not modeled in the 3HHY crystal structure.
All chemicals were obtained from MilliporeSigma® unless stated otherwise. Non-canonical amino acids meta-fluorotyrosine (m-FY), meta-fluoro-phenylalanine (m-FF), and norleucine (Nle) were obtained from ChemImpex. Auxotrophic strains, Escherichia coli B, BL21 (DE3)-ΔMetA::Tn10, BL21 (DE3)-ΔtyrA::Tn10, and E. coli BL21 (DE3)-ΔpheA::Tn10 were originally obtained from CGSC (The Coli Genetic Stock Center).
A gene encoding the R. opacus 1,2-catechol dioxygenase (Rho 1,2-CTD; also known as “pyrogallol dioxygenase”; GenBank ID: CAA67941) was synthesized (BioBasics), including an N-terminal His-tag, and cloned into the pQE80L vector (T5 promoter and ampicillin resistant) inserted between the BamHI and HindIII restriction sites. The resulting pQE80L-1,2-CTD expression construct was transformed into E. coli BL21 (DE3) cells. Recombinant wild-type Rho 1,2-CTD (WT-1,2-CTD) enzyme was produced using an Eppendorf New BrunswickTM Bioflo/Celligen 5L Bioreactor, with Terrific Broth (per L: 24 g yeast extract, 20 g tryptone, 4 mL glycerol, 100 mL phosphate buffer 0.17 M KH2PO4, 0.72 M K2HPO4), 100 mg/L of ampicillin, and 10 μL/L culture of antifoam. The temperature was set to 37 °C, agitation level to 150 rpm, and the dissolved oxygen minimum to 30%. Following inoculation with an overnight culture (10 mL/L culture) of E. coli BL21 (DE3) pQE80L-1,2-CTD, growth was monitored until an OD600 of 0.4 was achieved, at which time the temperature was decreased to 20 °C and growth further monitored until an OD600 of 0.6; isopropyl β-D-1-thiogalactopyranoside (IPTG) was the added to a final concentration of 0.7 mM. Following 18 h of additional fermentation, the cells were harvested by centrifugation at 3,250 g for 30 min at 4 °C and obtained cell pellets were frozen at −20 °C.
The E. coli [BL21 (DE3)] ΔtyrA::Tn10 (BU80), and E. coli [BL21 (DE3)] ΔpheA::Tn10 (BU24) strains harboring pQE80L-1,2-CTD vector were cultured in LB medium supplemented with 100 μg/mL ampicillin overnight for incorporation of 3-Fluoro-L-tyrosine (m-FY) and 3-Fluoro-L-phenylalanine (m-FF) analogs, respectively. The overnight auxotrophic E. coli cultures were washed twice with PBS buffer, and 1% inoculation was conducted in 0.5 L New Minimal Medium (NMM-19) (minimal media lacking the essential canonical amino acid (cAA); 7.5 mM (NH4)2SO4, 8.5 mM NaCl, 55 mM KH2PO4, 100 mM K2HPO4, 1 mM MgSO4, 1 mg/mL CaCl2, 1 mg/mL FeCl2, 1 ug/L CuCl2, 1 μg/L MnCl2, 1 μg/L ZnCl2, 1 μg/L Na2MoO4, 0.05 mg/mL of 19 amino acids, 10 mg/L biotin, 10 mg/L thiamine, 20 mM glucose), with 100 μg/mL ampicillin and containing a limiting concentration of either Tyr (35 μM) or Phe (20 μM), respectively. The cultures were incubated at 37 oC and 250 rpm for 10 h. After the corresponding canonical amino acids were completely exhausted, 1 mM m-FY or m-FF were added to the cultures while changing the temperature to 30 °C and 250 rpm. After 15 min incubation, 0.5 mM IPTG was added to the media and incubated under the same conditions overnight. Cells were then harvested by centrifugation at 3,250 g for 30 min at 4 °C and frozen at −20 °C. Total cell extracts were visualized by 12% SDS-PAGE. The same process was applied to the incorporation of Nle except that pQE80L-1,2-CTDwas transformed into E. coli B834 (DE3)-MetA (BU14).
Frozen cell pellets were resuspended at a ratio of 1 mL lysis buffer (50 mM Tris, pH 8.0, 300 mM NaCl, 10 mM imidazole, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 3 U/mL benzonase, and 1 mg/mL lysozyme) per 0.1 g of cell pellet (wet weight), and lysed by sonication 60× (for large scale wild-type preparation) or 12× (for small global incorporation preparations) cycles for 25 s each at 30% duty and 40 output power, with 30 s cooling periods (Vibra-Cell). The obtained lysates were clarified by centrifugation at 17,000 × g for 30 min at 4 °C. His-tagged 1,2-CTD (wild-type or globally incorporated) was purified from the clarified lysate using immobilized metal affinity chromatography with Ni-nitrilotriacetic acid (Ni-NTA) resin (Qiagen). Following lysate loading through the Ni-NTA resin, the column was washed with five column volumes of wash buffer (50 mM Tris, pH 8.0, 300 mM NaCl, 40 mM imidazole) and the 1,2-CTD eluted with three column volumes of the elution buffer (50 mM Tris pH 8.0, 300 mM NaCl, 300 mM imidazole). The obtained elution fractions were concentrated using an Amicon Ultra-15 centrifugal filter with a 10 kDa cutoff (MilliporeSigma). Selected samples were further subjected to fast performance liquid chromatography (FPLC; Amersham Pharmacia Biotech) using size-exclusion chromatography (SEC) with a HiLoad 16/600 Superdex 200 (Cytvia) column. Mobile phase buffer (10 mM Tris-HCl, 150 mM NaCl, pH 7.4) was degassed and the FPLC flowrate was set to 1 mL/min. Elution fractions were collected in increments of 1.4 mL. Fractions containing Rho 1,2-CTD were combined and concentrated using an Amicon Ultra-15 centrifugal filter with a 10 kDa cutoff (MilliporeSigma). The final concentrations of Rho 1,2-CTD (wild-type and globally incorporated variants) were determined using a Quick Start Bradford protein assay (Bio-Rad) according to the manufacturer’s protocol or based on the UV absorption of the sole tryptophan residue, based on ε280 = 5579 M−1 cm–1 (Budisa et al., 2004). Proteins samples were visualized by 12% SDS-PAGE.
To determine the degree of incorporation of non-canonical amino acids, the mass and apparent relative abundance of the sample components was assessed by electrospray ionization time of flight mass spectrometry (ESI-TOF-MS). The samples (∼5 µg) were injected onto a 2.1 × 30 mm Poros R2 reverse phase column (Applied Biosystems) and desalted using a fast (3 min, 3 mL/min) 0.1% formic acid aq/acetonitrile linear gradient (10%–75% acetonitrile). The column and solvents were heated to ∼80 °C to improve protein peak shape. The HPLC eluent was split to 100 μL/min just before the electrospray source. The LC-MS system performance was evaluated prior to sample analysis using intact myoglobin as a standard. The proteins were ionized in positive ion mode applying a cone voltage of 40 kV while scanning from 300 to 3,000 m/z. Raw data were analyzed employing the maximum entropy deconvolution algorithm with Bruker Data Analysis.
Enzyme assay reactions were performed using 96-well UV star microplates (Griener Bio) with a total reaction volume of 400 µL including 0.75 μg/mL of enzyme, reaction buffer (50 mM Tris-HCl, pH 7.2), and a range of concentrations of the 3-methylcatechol substrate (0.54–70 µM). The absorbance at 260 nm was monitored using a SpectraMax M5e spectrophotometer (Molecular Devices) for 30 min. The absorption coefficient used for 3-methylcatechol was 18,000 L/(mol*cm) or M−1 cm−1. Initial reaction rates at varying substrate concentrations were fit to the Michaelis–Menten equation using nonlinear regression as implemented by GraphPad Prism v9.1.2 to determine kcat and KM. The reaction rate at each substrate concentration was evaluated in triplicate.
Far-UV CD spectra were recorded from 260 to 190 nm using a Jasco J-810 spectropolarimeter with an instrumental time constant of 1 s, and a scan speed of 20 nm/min. Reported spectra were recorded at 25 °C using a 1.0 mm cuvette. A 10 mM phosphate buffer was used during analysis.
The effect of the m-FF and m-FY residues’ incorporation in the Rho 1,2-CTD variants’ tertiary structure was assessed by aromatic (exc. at 280 nm) or only tryptophan (exc. at 295 nm) fluorescence measurement after 15-min incubation of the protein samples at 25 °C before measurement. The fluorescence emission spectra (300–500 nm) were recorded after excitation at 280 or 295 nm and band slit of 5 nm at 25 °C.
The substrate 3-methylcatechol was generated using Avogadro, an open-source molecular builder and visualization tool, version 1.2.0 (Hanwell et al., 2012), with energy minimization conducted using the universal force field (Rappé et al., 1992). Water molecules and ligands were removed from the 3D crystal structure PDBID# 3HHY using PyMOL (Molecular Graphics System, Version 2.5.2). Coordinate B for Asp246, Thr253, and Thr263 were eliminated using Swiss-PdbViewer 4.1.0 (Guex and Peitsch, 1997), with hydrogens added subsequently. The fluorine atom was manually introduced at the meta position of all Tyr or Phe residues using the PyMOL build function to generate m-FY-1,2-CTD or m-FF-1,2-CTD models, respectively. We docked 3-methylcatechol into the models using Vinawizard in PyRx software (Dallakyan and Olson, 2015) with all Tyr and Phe residues within 8 Å of the Iron III ion set to be flexible. The confirmation with the lowest binding affinity of 27 docked models was selected as the optimal structure. Figures were generated using PyMOL and LigPlot + v.2.2.8 (Dallakyan and Olson, 2015).
The production of WT-1,2-CTD enzyme was scaled up to a 5 L fermentative format using the pQE80L-1,2-CTD vector transformed into E. coli BL21. Following overnight induction with IPTG, cells were harvested by centrifugation and the obtained cell pellet re-suspended in lysis buffer for several rounds of sonication to extract proteins. The obtained clarified supernatant was collected and WT-1,2-CTD was enriched by Ni-NTA affinity chromatography. Thick bands at the expected MW of approximately 33 kDa are visible upon SDS-PAGE analysis in the three elution fractions (Supplementary Figure S2A). Significant amounts of protein were also detected at 33 kDa in the washes and flow-through, suggesting some loss during enrichment. Moreover, a small amount of WT-1,2-CTD was detected in the lysis pellet, indicating that cell lysis may not have proceeded to completion or that some amount of the enzyme ended up in inclusion bodies. The protein obtained from Ni-NTA enrichment was subjected to further purification by size exclusion chromatography. The chromatographic separation curve for WT-1,2-CTD (Supplementary Figure S2B) showed a clear and steep peak at 57 mL with a minor shoulder at 48 mL. Fractions spanning the 57 mL peak (omitting the shoulder) were collected and pooled for a final yield of 94 mg of WT-1,2-CTD/L of cell culture, despite significant losses during the Ni-NTA affinity chromatography washing steps (Supplementary Figure S2A).
To enable global incorporation of amino acid analogues into Rho 1,2-CTD, the pQE80L-1,2-CTD expression construct was transformed into the auxotrophic E. coli strains for the canonical amino acid to be replaced. Thus, for incorporation of m-FF and m-FY, the corresponding E. coli BL21 (DE3) strains (BU80 and BU24, auxotrophic for Phe and Tyr) were used. For Nle incorporation, an E. coli strain auxotrophic for Met (BU14) was used. In all cases, global incorporation was achieved by initially growing the transformed auxotrophic cells in minimal media supplemented with the essential amino acid (either Phe and Tyr or Met) until growth arrest was achieved upon depletion of the essential amino acid. The culture supernatant was removed, and the cell pellet was resuspended in fresh minimal medium supplemented with the desired amino acid analog. The production of the recombinant Rho 1,2-CTD variants was induced overnight with IPTG. After harvesting and lysis of the cells, the presence of the globally incorporated 1,2-CTD was examined.
SDS-PAGE analysis of soluble fractions revealed bands of similar intensity at ∼ 35 kDa for recombinant Rho 1,2-CTD protein production in the presence of either cAAs (WT-1,2-CTD), m-FF (m-FF-1,2-CTD), or m-FY (m-FY-1,2-CTD) (Figure 2A). In comparing the intensity of overexpression, the m-FY-1,2-CTD soluble fraction strikingly showed the highest expression, followed by WT-1,2-CTD and the m-FF-1,2-CTD prior to enrichment. Notably, expression was lost upon omission of either the ncAA or the IPTG (Supplementary Figure S3), giving confidence that incorporation was successful. To confirm analogue incorporation, the expressed enzymes were purified by Ni-NTA affinity chromatography. Overall yields after purification were 51 mg and 31 mg/L of culture respectively for m-FY-1,2-CTD and m-FY-1,2-CTD (Figure 2A) in the shake flask format.
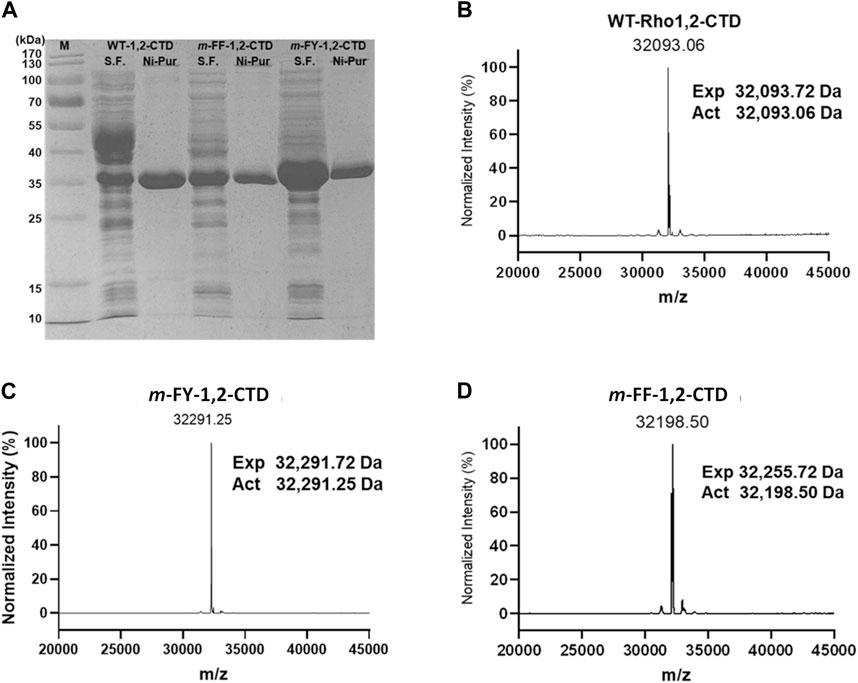
Figure 2. SDS-PAGE and ESI-MS of WT- and its non-canonical amino acid incorporated variants. (A) 12% SDS-PAGE analysis of soluble fraction (S.F.) and Ni-NTA purified form (Ni-Pur) of the wild-type, m-FY- and m-FF- 1,2-CTD. (B–D) ESI-MS analysis pattern of N-purified protein samples.
The purified m-FY-1,2-CTD sample was analyzed by intact mass ESI-MS and compared to purified WT-1,2-CTD (Figures 2B, C). WT-1,2-CTD yielded a signal at the expected calculated mass based on protein sequence (observed mass = 32,093.72 Da, calculated mass = 32,093.06 Da; method accuracy ± 1 Da). However, some protein heterogeneity was observed, likely due to partial oxidation or other minor post-translational modifications. For m-FY-1,2-CTD, the most abundant mass was shifted upwards +198 Da to 32,291.25 Da, which is equal to the difference if 11 Tyr were substituted with 11 m-FY [expected mass difference: 11 x (181–163 Da) = 198 Da], consistent with the primary amino acid sequence of Rho 1,2 CTD including 11 Tyr. Essentially, no signal was detected at 32,094 Da (the mass of wild-type 1,2-CTD) in the m-FY-1,2-CTD sample. Thus, 32,291 Da likely represents fully m-FY-incorporated Rho 1,2-CTD. That said, given the heterogeneity of WT-1,2-CTD, the possibility of other post-translational modifications (e.g., oxidations, dehydrations) of partially or fully m-FY-incorporated Rho 1,2-CTD contributing to some signals cannot be excluded. Nonetheless, these results are strong evidence that most of the isolated m-FY-1,2-CTD has 11 m-FY incorporated, making the fully incorporated version the most abundant.
The purified m-FF-1,2-CTD was also submitted for ESI-MS analysis. As for the mFY-1,2-CTD, the observed mass signal arising from the m-FF-1,2-CTD sample was also shifted upwards (Figure 2D). However, the incorporation does not appear to be as complete as it was for m-FY-1,2-CTD. The most abundant mass for m-FF-1,2-CTD is 32,198.50 Da, which is +105 Da greater than the mass of wild-type Rho1,2-CTD and could represent replacement of 6 Phe with m-FF (expected mass difference: 6 x (165–147 Da) = 108 Da). However, the primary amino acid sequence of Rho 1,2-CTD includes 9 Phe. The less abundant mass at 32,255.72 Da is shifted 162 Da from the expected WT-1,2-CTD signal, consistent with all 9 Phe replaced with m-FF (9 × (165–157 Da) = 162 Da). Minor signals are observed in the m-FF-1,2-CTD sample, including at 32,092 Da, the mass of WT-1,2-CTD with no m-FF incorporation. This suggests that there was some residual Phe in the expression system. Additional observed species, separated by ∼18 Da, at 32,216 Da, 32,234 Da, and 32,252 Da, are likely Rho 1,2-CTD with a mixture of m-FF and Phe (i.e. 7, 8, and 9 m-FF, respectively).
The putative Nle-incorporated Nle-1,2-CTD was purified by Ni-NTA affinity chromatography (Supplementary Figure S4A) yielding 61 mg of enzyme/L of culture and submitted for ESI-MS analysis. In contrast to m-FY-1,2-CTD and m-FF-1,2-CTD, no obvious single peaks was observed in the Nle-1,2-CTD MS profile (Supplementary Figure S4B). The most abundant mass, 32,093 Da, matches the calculated mass of WT-1,2-CTD. The primary amino acid sequence of 1,2 CTD includes four methionines. If all methionines were replaced with Nle, the expected mass should be 32,022 Da (expected mass difference: 4 x (113–131 Da) = −72 Da). Some signal is observed at approximately 32,022 Da and other masses between 32,022 Da and 32,093 Da (e.g., 32,074 Da =–1 putative incorporation, 32,052 Da =–2 putative incorporations, 32,033 Da =–3 putative incorporations); however, these species cannot be distinguished from the heterogeneity observed in the WT-1,2-CTD control. Given the ambiguous data, it is not possible to say whether any Nle was successfully incorporated into Rho 1,2-CTD. Thus, we conclude that under the conditions that enabled a high degree of incorporation of other analogues, global incorporation of Nle into Rho 1,2-CTD was poor.
To assess the impact of m-FF and m-FY incorporation on the protein structure of the variant compared to WT-1,2-CTD, we conducted UV-Vis absorption, UV-circular dichroism (CD) spectra, and fluorescence analyses (Figure 3). Our examination of the secondary structure profiles of the mutant proteins via CD and potential tertiary structural changes through fluorescence confirmed the protein variants’ structural integrity in solution. The observed spectral shifts remained largely marginal, indicating that the structures are not strongly affected by the global fluorination of all Tyr and most Phe residues. Far-UV CD analysis within the 190–260 nm range (Figure 3A) corroborated that the global substitution of all Phe and Tyr residues by m-FY and m-FF in Rho 1,2-CTD had minimal impact on the enzyme’s secondary structure. However, the UV-Vis spectra of the fluorinated variants exhibited a slight blue-shift towards 230 nm compared to WT-1,2-CTD (Figure 3B). Conversely, a subtle red shift (5–10 nm) in the maximum fluorescence profiles (Figures 3C,D) suggested alterations in the microenvironment of the Try and Phe residues, likely attributed to fluorination.
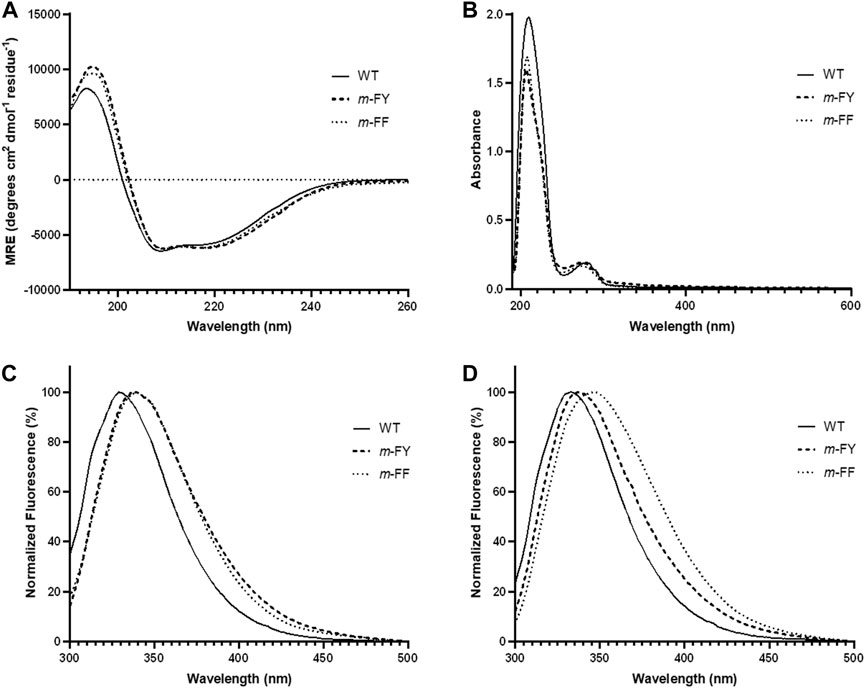
Figure 3. Absorption, circular dichroic, and fluorescence profiles of native and substituted proteins. (A) Far-UV CD profiles of the WT and fluorinated variants of Rho 1,2-CTD protein forms at room temperature. (B) UV spectra of WT and fluorinated variants of Rho 1,2-CTD measured in the range 195–700 nm. Fluorescence emission spectra of WT and fluorinated variants excited at (C) 280 nm and (D) 295 nm. Spectroscopic conditions are described in “Materials and Methods”. In Rho 1,2 CTD, 6–9 Phe-residues and 11 Tyr-residues were substituted by m-FF and m-FY, respectively.
The SEC purified WT-1,2 CTD was used to conduct a kinetic analysis of the enzyme’s activity against varying concentrations of the known substrate, 3-methylcatechol, yielding a saturable curve (Figure 4A). A Michaelis–Menten analysis of the obtained data yielded a KM of 3.6 ± 0.3 µM, a turnover number (kcat) of 1.02 ± 0.02 s−1, and a catalytic efficiency (kcat/KM) of 0.28 ± 0.02 μM−1 s−1. Kinetic analysis of the SEC purified m-FY-1,2-CTD variant showed a slight decrease in overall catalytic efficiency compared to recombinant WT-1,2-CTD (Figure 4B; Table 1). This is largely attributable to the ten-fold reduction in kcat (turnover number) alongside six-fold higher productive substrate binding. Kinetic analysis of the Ni-NTA affinity purified m-FF-1,2-CTD variant showed a five-fold improvement in overall catalytic efficiency (Figure 4C; Table 1). With the kcat virtually the same as wild-type, this improved efficiency must be largely attributable to the four-fold increase in productive substrate binding.
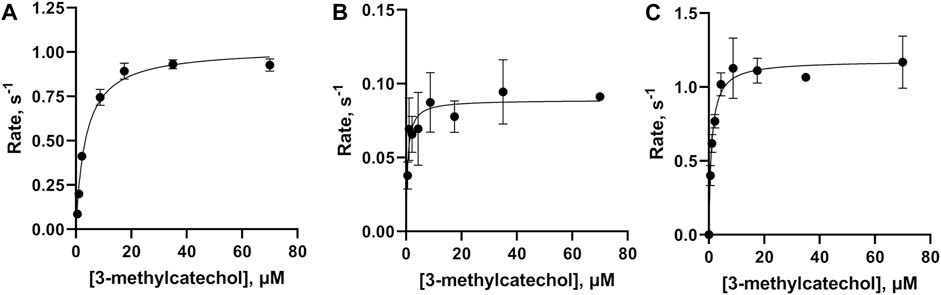
Figure 4. Kinetic analysis of the activities of wild-type, m-FY, and m-FF globally incorporated Rho 1,2-CTD variants. Michaelis–Menten kinetic plots, obtained using 3-methylcatechol as the substrate for (A) WT-1,2-CTD, (B) m-FY-1,2-CTD, and (C) m-FF-1,2-CTD. Points represent the average of three replicates plotted with standard deviation.
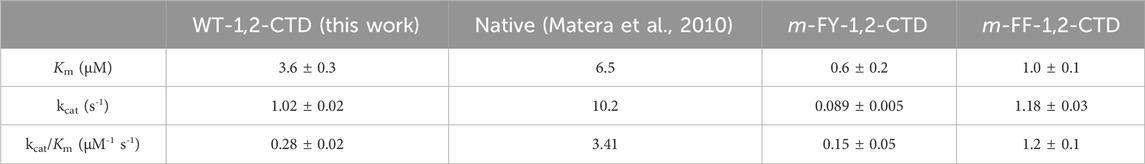
Table 1. Kinetic values obtained in a Michaelis–Menten analysis for wild-type 1,2 CTD compared to mFY-1,2-CTD and mFF-1,2-CTD variants. Kinetic values obtained from a similar analysis of wild-type 1,2-CTD in an earlier report are provided for comparison. 3-methylcathecol was used as the substrate in all cases.
The binding energy determined in the docking experiment for the substrate 3-methylcatechol to WT-1,2-CTD, m-FF-1,2-CTD, m-FY-1,2-CTD, and Nle-1,2-CTD was calculated as −7.1 kcal/mol, −7.4 kcal/mol, −6.7 kcal/mol, and −7.0 kcal/mol, respectively. Similar results were obtained for catechol docking, but the effect was not as pronounced as with methylated substrate (unpublished material). Our modeling of the binding pocket, in the case of 3-methylcatechol (the substrate used in the kinetic analysis herein) with m-FY106 and m-FY196, shows that fluorination has “pushed” the substrate so far as to create a new hydrophobic contact with Asp86 (cf. Figures 5A,B). At the same time, modeling also shows that the fluorination of tyrosine residues disrupts the coordination geometry of the 3-methylcatechol with the iron cofactor (cf. Figures 5C,D). Interestingly, the number of hydrophobic contacts with the substrate is also increased in the binding pocket of m-FY-1,2-CTD and includes residues Asp80 and His222 (Supplementary Figure S5). It is important to note that the Phe residues in the second shell are placed relative to the binding pocket of Rho 1,2-CTD and that our modeling approach did not capture the potential impact of the fluorination of these residues on the overall activity (see “Discussion”).
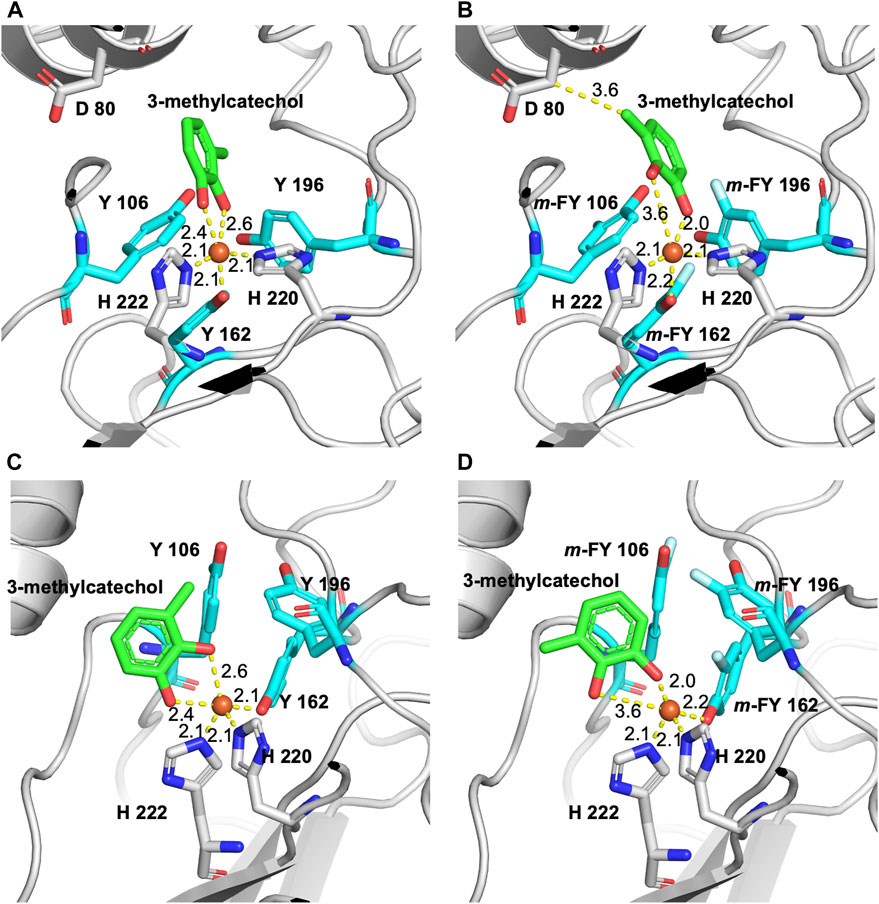
Figure 5. Model of fluorine m-FY and m-FF incorporated 3D-structure of Rho 1,2-CTD in the active site pocket using the RCSB PBD entry 3HHY as a template. (A) The binding pocket of the wild-type enzyme with Y106 and Y196 and the 3-methylcatechol substrate in coordination with the iron cofactor. (B) Binding pocket with m-FY106 and m-FY196 shows that fluorination has “pushed” the 3-methylcatechol substrate to create a hydrophobic contact with Asp86. (C) Alternate view of the binding pocket of the wild-type enzyme with Y106 and Y196 and the 3-methylcatechol substrate coordinated with the iron cofactor. (D) Alternate view of the binding pocket with m-FY106 and m-FY196 shows that fluorination distorts the coordination geometry of 3-methylcatechol to the iron cofactor.
Selective pressure incorporation (SPI) facilitates the in vivo incorporation of various non-canonical amino acids into recombinant proteins, closely resembling their canonical counterparts in shape, size, and chemical properties, with yields comparable to wild-type forms. Based on the chemical properties of the incorporated analogues and the positions of these residues within the protein (Figure 1), Tyr, Phe, and Met residues were individually targeted for replacement with m-FY, m-FF and Nle, respectively, in Rho 1,2 CTD. The enzyme contains 9 Phe and 11 Tyr residues distributed throughout, but only four Met residues, with two playing structural or catalytic roles and the others involved in the initiation of translation, rendering them irrelevant for this study.
With respect to the replacement of all Phe and Tyr residues with their fluorinated analogs, fluorine—the most electronegative element in a C–X bond context—significantly alters the electronic properties of the residue compared to a C–H bond, resulting in a reverse polarity at the replacement site (Bae et al., 2004). The isosteric H→F substitutions (“atomic mutations”; Minks et al., 1999) lead to limited changes in bond lengths and Van der Waals radii (the C–H bond is 0.25 Å shorter than the C–F bond, with a difference of approximately 0.15 Å in Van der Waals radii). Moreover, fluorine’s presence as a hydrogen bond acceptor may promote the formation of additional hydrogen bonds in the protein structure, influencing local conformations and overall stability to some extent (Minks et al., 2000). However, in most cases such minute changes do not significantly affect the overall structure, stability, and activity of the protein, as functionally and catalytically important residues are typically unaffected. The impact of the global replacement of all Tyr and Phe side chains in Rho 1,2-CTD is evident, for example, in the UV and fluorescence profiles of the fluorinated variants (Figure 3). Both variants exhibit a blue shift (3–6 nm) in absorbance, while the fluorescence emission maxima are slightly red shifted under neutral pH conditions.
Replacing Phe and Tyr residues with m-FF and m-FY in Rho 1,2-CTD—a non-heme iron-containing enzyme—marked the initial use of amino acid analogs in non-heme iron-containing dioxygenases. This substitution not only served as a model for examining the impact of these residues on enzyme catalytic properties but also aimed to elucidate the extent to which these isosteric analogs with defined physicochemical properties could aid in understanding the enzyme’s catalytic mechanism.
The family of catechol dioxygenases is divided into subfamilies based on substrate specificity and bond cleavage selectivity (Borowski and Siegbahn, 2006; Burroughs et al., 2019; Fetzner, 2012). Substrates generally range from catechol to various methylated and halogenated analogues thereof, and either intradiol or extradiol bonds are differentially prioritized for cleavage. Rho 1,2-CTD from R. opacus maintains its highest catalytic efficiency against catechol, with some moderate activity detected against methylated catechols and pyrogallol and only limited activity detected against chlorinated catechols (Matera et al., 2010). Rho 1,2-CTD preferentially cleaves the intradiol bond between the hydroxylated ring carbons (Supplementary Figure S6). Analysis of an X-ray crystallographic-derived structure of this Rho 1,2-CTD in the presence of catechol highlights the coordination of the Fe(III) by two His residues (His220 and His222) and one Tyr (Tyr162) as well as the two ring hydroxyl groups of catechol (Figure 6A; PDBID# 3HHY; Matera et al., 2010). This orientation of the substrate dictates the intradiol cleavage selectivity. It is notable that, in the absence of catechol, Tyr196 is rotated such that it also contributes to metal coordination (PDBID #3HGI; Matera et al., 2010). More broadly Rho 1,2-CTD maintains an overall cupin-like fold (Fetzner, 2012).
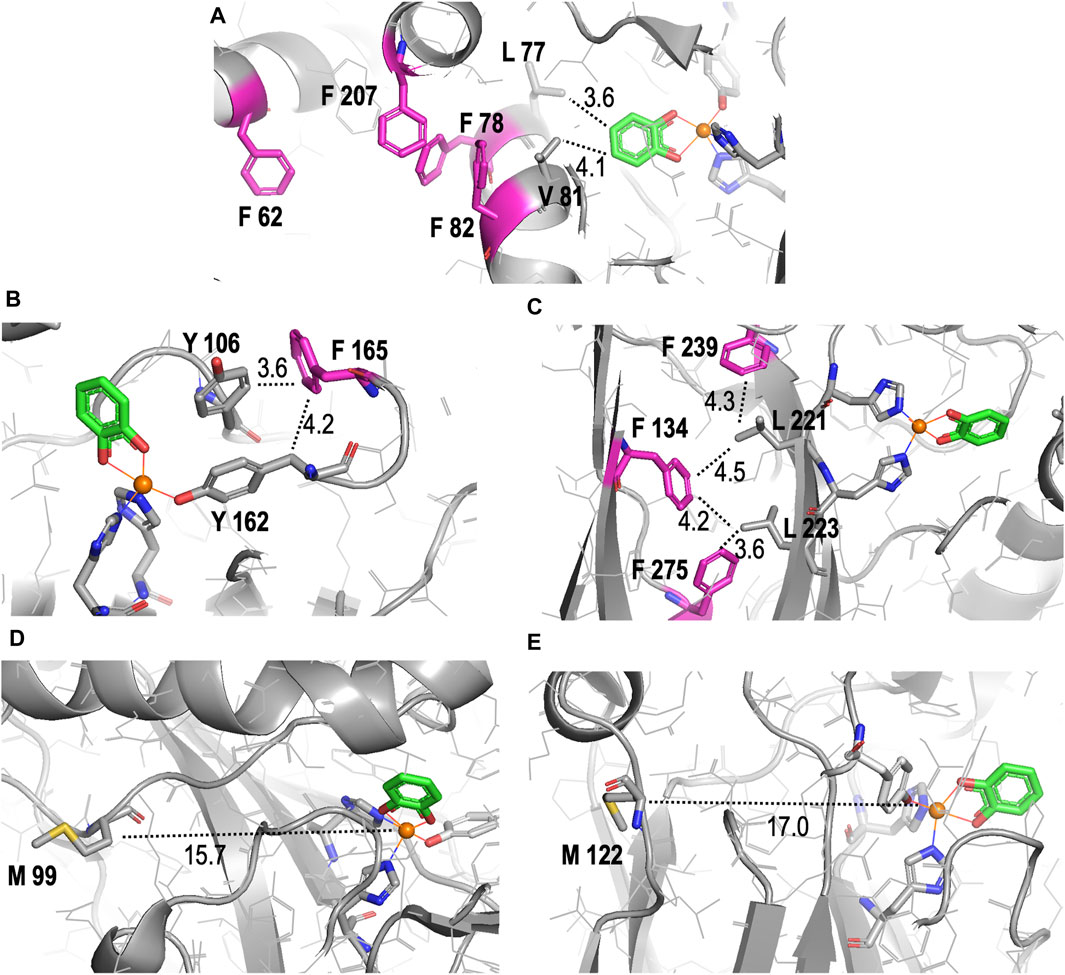
Figure 6. Detailed structural representations of the proximities of substituted amino acids in the crystal structure of Rho 1,2-CTD to the active site pocket. All structures shown are derived from the RCSB PBD entry 3HHY. (A–C) Proximities of different Phe (F) residues (pink) in the enzyme, highlighting their roles in hydrophobic networks stabilizing of secondary structural elements that host the active site. (D, E) Proximities of Met (M) residues (yellow). Nitrogens are shown in dark blue, carbon in grey, oxygen in red, and Fe(III) in orange in all images. Distances in angstroms are indicated with dotted lines. All images were generated and distances measured using PyMOL.
A comparison of the kinetic results obtained for Rho 1,2-CTD with a previous report highlights some differences (Table 1), most likely attributable to the previous kinetic analysis having been conducted with native Rho 1,2-CTD purified from R. opacus, and thus lacking the fused N-terminal His-tag. Speculatively, existing crystal structures place the N-terminus (residue Ala 25) in relative proximity to the entrance of the substrate binding pocket (Matera et al., 2010), potentially enabling the N-terminal His-tag to contribute to the overall decreased catalytic efficiency.
The ten-fold reduction in kcat for the m-FY-1,2-CTD variant (Table 1) can be most obviously be explained by the presence of five potentially substituted Tyr residues in the active site of the enzyme (Figures 1–5). Most notable are Tyr162, which comprises one of the primary iron coordinating residues, and Tyr196, which also contributes to iron coordination but only in the absence of substrate. Adding the fluorine groups could have altered the physical position and/or conformation of these m-FYs in the Fe(III) coordination sphere compared to the Tyr positions or the electronic nature of the coordination interaction between the amino acid and Fe(III). The latter effect altered the electronegativity of the phenolic oxygen of m-FY. In either case, modification of the electronic nature of the Fe itself and/or reduced Fe occupancy could be the cause of the reduced kcat. Interestingly, the Km for m-FY-1,2-CTD was also significantly reduced, indicating higher productive substrate binding. Most notably the catechol substrate ring is located only 3.6 Å from the meta-carbon of the Tyr106 ring and 3.7 Å from the meta-carbon of the Tyr196 ring (Figure 5). The orientations of these rings do not support any contribution of π-interactions here. We therefore hypothesize that this increased affinity is rather related to local changes in polarity caused by numerous H→F exchanges in the microenvironment of the active site. However, despite the enhanced productive substrate binding, it was insufficient to offset the decrease in turnover, resulting in an overall lower catalytic efficiency for this variant. In this case, with the energetic stability of the Michaelis complex compromised due to the presence of fluorinated Tyr and without a corresponding decrease in the energy of the transition state of the rate-determining step, an elevation in the overall energy barrier of the catalytic mechanism would be expected, consequently reducing catalytic turnover despite heightened substrate affinity. Additionally, the increased productive substrate binding might result in product release being the rate-determining step rather than a chemical step.
With respect to the reduced KM value detected in the m-FF-1,2-CTD variant, our modeling shows that no Phe residues are localized to the first shell of the catalytic site (Supplementary Figure S5). However visual inspection of the crystal structure highlighted several Phe residues in the second shell. Of these, the most likely residues contributing to the observed increased productive substrate binding are residues 78 and 82 (Figure 6A). These are both located in α-helix 3, which is the side of the substrate binding pocket, contributing Van der Waals interactions with the catechol phenolic ring via Leu77 (3.6 Å) and Val81 (4.1 Å; Figure 6A; Supplementary Figure S5). While residues 78 and 82 are not predicted to make any direct interactions with the substrate due to their orientation on the other side of the helix, they likely contribute to stabilizing the position of α-helix 3, along with the overall fold of the enzyme, by forming a hydrophobic pocket with residues 207 located on α-helix 5 and 62 located on α-helix 2. Substitutions with m-FF for Phe in this hydrophobic pocket may contribute to shifting the position, or rotation, of α-helix 3, altering interactions with the substrate. Of course, this remains to be structurally validated. Phe residue 165 also cannot be ignored in its position at the entrance of the active site pocket, only 4.2 Å from the β-carbon of the Fe(III) coordinating Tyr162, and 3.6 Å from the Tyr106 (Figure 6B). Increased electronegativity at this site upon substitution with m-FF could also contribute to modulated binding. The remaining Phe residues are stabilizing the β-sheet stacking, notably located in proximity to the main chain of the β-strand harboring the Fe(III) coordinating residues His220 and His222 (Figure 6C). In particular, Phe275 is 3.6 Å from Leu223, Phe239 is 4.5 Å from Leu221, while Phe134 is 4.2 Å from Leu223 and 4.3 Å Leu221, together contributing Van der Waals interactions to stabilize the positing of the Fe(III) coordinating histidines. However, how increased electronegativity at these sites upon substitution of Phe with m-FF could contribute to modulation of substrate affinity is not directly evident.
The apparent failure of Nle incorporation is notable. Originally, we hypothesized that Nle, being a hydrophobic amino acid, could potentially enhance Rho 1,2-CTD functionality akin to the fluorinated analogs. The substitution of Met-resides with Nle in human prion protein resulted in “antiaggregating” variants as this analogue is strictly hydrophobic and non-oxidizable (Wolschner et al., 2009). Following this logic, all moderately hydrophobic Met-residues in the protein interior once replaced by strictly hydrophobic Nle should increase hydrophobic core interactions for improved functionality (Moroder and Budisa, 2010). Thus, having only two anticipated structural substitutions, the unsuccessful incorporation of Nle is surprising. The presence of considerable WT-1,2-CTD in the sample, as detected in the MS profile, suggests significant residual Met contamination during protein expression induction. However, SDS-PAGE analysis indicates less WT-1,2-CTD production than the positive control, implying that the culture likely depleted all Met before protein expression was complete, potentially allowing Nle-1,2-CTD synthesis. Alternatively, Nle might have been incompatible with the endogenous translation machinery in E. coli BL21 (DE3), causing an issue with translational mechanisms or co-translational folding (Walker et al., 2022). It is also relevant that the surface localization of Met99 and Met122 (Figures 6E,F), situated 15.7 Å and 17.0 Å away from Fe(III), might increase local hydrophobicity detrimental for forming the active and folded protein scaffold.
The observed enhancements in substrate affinities with both fluorinated analogs align with the anticipated potential offered by the expanded chemistries of amino acid analogs. However, this study also underscores the limitations inherent in incorporation technologies which persistently hinder widespread application. Encouragingly, global incorporation generally has a less detrimental impact on the recombinant yields of target proteins compared to site-specific incorporation. Notably, working in our own laboratory with recombinant Rho 1,2-CTD, site-specific incorporation of Nδ-methylhistidine resulted in up to a ∼10,000-fold decrease in enzyme yield (Lister, 2024). In contrast, yields observed here for m-FF and m-FY incorporation were comparable to WT-1,2-CTD yields. Nevertheless, the incorporation of Nle into Rho 1,2-CTD was unsuccessful, likely attributable to the specific positioning of Met residues on the surface of the enzyme in the specific microenvironment that do not tolerate a dramatic increase in the local hydrophobicity. Additionally, it is crucial to acknowledge the cost associated with synthetic analogs, whether for global or site-specific incorporation. While there are initiatives aimed at developing strains capable of biosynthesizing certain analogs for simultaneous production and incorporation (e.g., Anderhuber et al., 2016), addressing this remains a long-term challenge for the future.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
MW: Writing–review and editing, Formal Analysis, Investigation, Methodology, Validation. TG: Formal Analysis, Investigation, Methodology, Writing–original draft. HK: Methodology, Supervision, Writing–review and editing. AR: Formal Analysis, Methodology, Validation, Writing–review and editing. ML: Conceptualization, Funding acquisition, Project administration, Writing–review and editing. AS-J: Formal Analysis, Methodology, Supervision, Writing–review and editing. NB: Conceptualization, Formal Analysis, Methodology, Supervision, Writing–review and editing. ML: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Writing–review and editing.
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This project was funded by the Natural Sciences and Engineering Research Council–Alliance Program [Project # ALLRP 550057-20] for ML and ML; NB and HK thank the Canada Research Chairs Program (Grant Nr. 950–231971) and MW thanks the Natural Sciences and Engineering Research Council (NSERC) of Canada through the Discovery Grant (RGPIN-05669-2020) for support. We are grateful to the National Research Council of Canada for providing access to facilities and in-kind operational.
We are grateful to Luana Porto (University of Ottawa) and John Kelly (National Research Council of Canada) for critically reading the manuscript. This report represents National Research Council of Canada Communication # 58478.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsybi.2024.1419557/full#supplementary-material
Rho 1,2-CTD, Rhodococcus opacus 1,2-catechol dioxygenase; cAA, canonical amino acid; ncAA, non-canonical amino acid; m-FY, meta-fluorotyrosine; m-FF, meta-fluorophenylalanine; Nle, norleucine.
Acevedo-Rocha, C. G., Hoesl, M. G., Nehring, S., Royter, M., Wolschner, C., Wiltschi, B., et al. (2013). Non-canonical amino acids as a useful synthetic biological tool for lipase-catalysed reactions in hostile environments. Catal. Sci. Technol. 3 (5), 1198. doi:10.1039/c3cy20712a
Agostini, F., Voller, J. S., Koksch, B., Acevedo-Rocha, C. G., Kubyshkin, V., and Budisa, N. (2017). Biocatalysis with unnatural amino acids: enzymology meets xenobiology. Angew. Chem. Int. Ed. Engl. 56 (33), 9680–9703. doi:10.1002/anie.201610129
Anderhuber, N., Fladischer, P., Gruber-Khadjawi, M., Mairhofer, J., Striedner, G., and Wiltschi, B. (2016). High-level biosynthesis of norleucine in E. coli for the economic labeling of proteins. J. Biotechnol. 235, 100–111. doi:10.1016/j.jbiotec.2016.04.033
Bae, J. H., Paramita Pal, P., Moroder, L., Huber, R., and Budisa, N. (2004). Crystallographic evidence for isomeric chromophores in 3-fluorotyrosyl-green fluorescent protein. Chembiochem 5 (5), 720–722. doi:10.1002/cbic.200300818
Bednar, R. M., Karplus, P. A., and Mehl, R. A. (2023). Site-specific dual encoding and labeling of proteins via genetic code expansion. Cell Chem. Biol. 30 (4), 343–361. doi:10.1016/j.chembiol.2023.03.004
Berger, A. A., Voller, J. S., Budisa, N., and Koksch, B. (2017). Deciphering the fluorine code-the many hats fluorine wears in a protein environment. Acc. Chem. Res. 50 (9), 2093–2103. doi:10.1021/acs.accounts.7b00226
Birch-Price, Z., Taylor, C. J., Ortmayer, M., and Green, A. P. (2023). Engineering enzyme activity using an expanded amino acid alphabet. Protein Eng. Des. Sel. 36, gzac013. doi:10.1093/protein/gzac013
Bornscheuer, U. T., Huisman, G. W., Kazlauskas, R. J., Lutz, S., Moore, J. C., and Robins, K. (2012). Engineering the third wave of biocatalysis. Nature 485 (7397), 185–194. doi:10.1038/nature11117
Budisa, N., Huber, R., Golbik, R., Minks, C., Weyher, E., and Moroder, L. (1998). Atomic mutations in annexin V--thermodynamic studies of isomorphous protein variants. Eur. J. Biochem. 253 (1), 1–9. doi:10.1046/j.1432-1327.1998.2530001.x
Budisa, N., Pal, P. P., Alefelder, S., Birle, P., Krywcun, T., Rubini, M., et al. (2004). Probing the role of tryptophans in Aequorea victoria green fluorescent proteins with an expanded genetic code. Biol. Chem. 385 (2), 191–202. doi:10.1515/BC.2004.038
Budisa, N., and Schneider, T. (2019). Expanding the DOPA universe with genetically encoded, mussel-inspired bioadhesives for material sciences and medicine. Chembiochem 20 (17), 2163–2190. doi:10.1002/cbic.201900030
Cirino, P. C., Tang, Y., Takahashi, K., Tirrell, D. A., and Arnold, F. H. (2003). Global incorporation of norleucine in place of methionine in cytochrome P450 BM-3 heme domain increases peroxygenase activity. Biotechnol. Bioeng. 83 (6), 729–734. doi:10.1002/bit.10718
Dallakyan, S., and Olson, A. J. (2015). “Small-molecule library screening by docking with PyRx,” in Chemical biology: methods and protocols. Editors J. E. Hempel, C. H. Williams, and C. C. Hong (New York: Springer), 243–250. doi:10.1007/978-1-4939-2269-7_19
Deepankumar, K., Shon, M., Nadarajan, S. P., Shin, G., Mathew, S., Ayyadurai, N., et al. (2014). Enhancing thermostability and organic solvent tolerance of ω-transaminase through global incorporation of fluorotyrosine. Adv. Synthesis Catal., 356(5), 993–998. doi:10.1002/adsc.201300706
Goettig, P., Koch, N. G., and Budisa, N. (2023). Non-canonical amino acids in analyses of protease structure and function. Int. J. Mol. Sci. 24 (18), 14035. doi:10.3390/ijms241814035
Guex, N., and Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18 (15), 2714–2723. doi:10.1002/elps.1150181505
Hanwell, M. D., Curtis, D. E., Lonie, D. C., Vandermeersch, T., Zurek, E., and Hutchison, G. R. (2012). Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 4, 17. doi:10.1186/1758-2946-4-17
Hartman, M. C. T. (2022). Non-canonical amino acid substrates of E. coli aminoacyl-tRNA synthetases. Chembiochem 23 (1), e202100299. doi:10.1002/cbic.202100299
Hoesl, M. G., Acevedo-Rocha, C. G., Nehring, S., Royter, M., Wolschner, C., Wiltschi, B., et al. (2011). Lipase congeners designed by genetic code engineering. ChemCatChem 3 (1), 213–221. doi:10.1002/cctc.201000253
Hoesl, M. G., and Budisa, N. (2012). Recent advances in genetic code engineering in Escherichia coli. Curr. Opin. Biotechnol. 23 (5), 751–757. doi:10.1016/j.copbio.2011.12.027
Kruyer, N. S., Wauldron, N., Bommarius, A. S., and Peralta-Yahya, P. (2020). Fully biological production of adipic acid analogs from branched catechols. Sci. Rep. 10 (1), 13367. doi:10.1038/s41598-020-70158-z
Lister, J. (2024). Using genetic code expansion and rational disulfide bond design to engineer improved activity and (thermo)stability of Rhodococcus opacus cathecol 1. USA: dioxygenase University of Ottawa, 2.
Lugtenburg, T., Gran-Scheuch, A., and Drienovska, I. (2023). Non-canonical amino acids as a tool for the thermal stabilization of enzymes. Protein Eng. Des. Sel. 36, gzad003. doi:10.1093/protein/gzad003
Manandhar, M., Chun, E., and Romesberg, F. E. (2021). Genetic code expansion: inception, development, commercialization. J. Am. Chem. Soc. 143 (13), 4859–4878. doi:10.1021/jacs.0c11938
Matera, I., Ferraroni, M., Kolomytseva, M., Golovleva, L., Scozzafava, A., and Briganti, F. (2010). Catechol 1,2-dioxygenase from the Gram-positive Rhodococcus opacus 1CP: quantitative structure/activity relationship and the crystal structures of native enzyme and catechols adducts. J. Struct. Biol. 170 (3), 548–564. doi:10.1016/j.jsb.2009.12.023
Minks, C., Huber, R., Moroder, L., and Budisa, N. (1999). Atomic mutations at the single tryptophan residue of human RecombinantAnnexin V: effects on structure, stability, and activity. Biochemistry 38 (33), 10649–10659. doi:10.1021/bi990580g
Minks, C., Huber, R., Moroder, L., and Budisa, N. (2000). Noninvasive tracing of recombinant proteins with "fluorophenylalanine-fingers". Anal. Biochem. 284 (1), 29–34. doi:10.1006/abio.2000.4667
Moroder, L., and Budisa, N. (2010). Synthetic biology of protein folding. Chemphyschem 11 (6), 1181–1187. doi:10.1002/cphc.201000035
Neil, E., and Marsh, G. (2000). Towards the nonstick egg: designing fluorous proteins. Chem. Biol. 7, R153–R157. doi:10.1016/s1074-5521(00)00139-3
Parsons, J. F., Xiao, G., Gilliland, G. L., and Armstrong, R. N. (1998). Enzymes harboring unnatural amino acids: mechanistic and structural analysis of the enhanced catalytic activity of a glutathione transferase containing 5-fluorotryptophan. Biochemistry 37, 6286–6294. doi:10.1021/bi980219e
Rappé, A. K., Casewit, C. J., Colwell, K. S., Goddard III, W. A., and Skiff, W. M. (1992). UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024–10035. doi:10.1021/ja00051a040
Salvachúa, D., Johnson, C. W., Singer, C. A., Rohrer, H., Peterson, D. J., Black, B. A., et al. (2018). Bioprocess development for muconic acid production from aromatic compounds and lignin. Green Chem. 20 (21), 5007–5019. doi:10.1039/c8gc02519c
Shandell, M. A., Tan, Z., and Cornish, V. W. (2021). Genetic code expansion: a brief history and perspective. Biochemistry 60 (46), 3455–3469. doi:10.1021/acs.biochem.1c00286
Thi To, T. M., Kubyshkin, V., Schmitt, F.-J., Budisa, N., and Friedrich, T. (2022). Residue-specific exchange of proline by proline analogs in fluorescent proteins: how "molecular surgery" of the backbone affects folding and stability. J. Vis. Exp.(180), e63320. doi:10.3791/63320
Tian, M., Du, D., Zhou, W., Zeng, X., and Cheng, G. (2017). Phenol degradation and genotypic analysis of dioxygenase genes in bacteria isolated from sediments. Braz J. Microbiol. 48 (2), 305–313. doi:10.1016/j.bjm.2016.12.002
Vasic-Racki, D. (2006). History of industrial biotransformations – dreams and realities. In Ind. Biotransformations (pp. 1–36). doi:10.1002/9783527608188.ch1
Walker, E. J., Bettinger, J. Q., Welle, K. A., Hryhorenko, J. R., Molina Vargas, A. M., O'Connell, M. R., et al. (2022). Protein folding stabilities are a major determinant of oxidation rates for buried methionine residues. J. Biol. Chem. 298 (5), 101872. doi:10.1016/j.jbc.2022.101872
Keywords: global incorporation, non-canonical amino acid, meta-fluorotyrosine, norleucine, 1,2-catechol dioxygenase, substrate affinity, meta-fluorophenylalanine
Citation: Wong MCH, Grant TE, Karbalaei-Heidari HR, Robotham AC, Loewen ME, St-Jacques AD, Budisa N and Loewen MC (2024) Global incorporation of meta-fluorotyrosine or meta-fluorophenylalanine into 1,2-catechol dioxygenase modulates the binding affinities of substrates . Front. Synth. Biol. 2:1419557. doi: 10.3389/fsybi.2024.1419557
Received: 18 April 2024; Accepted: 21 June 2024;
Published: 02 August 2024.
Edited by:
Youran Li, Jiangnan University, ChinaReviewed by:
Amreesh Parvez, University of the Western Cape, South AfricaCopyright © 2024 His Majesty the King in Right of Canada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michele C. Loewen, bWljaGVsZS5sb2V3ZW5AbnJjLmNh
†These authors have contributed equally to this work and share first authorship.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.