 Ying Yang
Ying Yang Nicole Calakos
Nicole Calakos
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Synaptic Neurosci. , 17 October 2013
Volume 5 - 2013 | https://doi.org/10.3389/fnsyn.2013.00008
Long-term synaptic plasticity is a major cellular substrate for learning, memory, and behavioral adaptation. Although early examples of long-term synaptic plasticity described a mechanism by which postsynaptic signal transduction was potentiated, it is now apparent that there is a vast array of mechanisms for long-term synaptic plasticity that involve modifications to either or both the presynaptic terminal and postsynaptic site. In this article, we discuss current and evolving approaches to identify presynaptic mechanisms as well as discuss their limitations. We next provide examples of the diverse circuits in which presynaptic forms of long-term synaptic plasticity have been described and discuss the potential contribution this form of plasticity might add to circuit function. Finally, we examine the present evidence for the molecular pathways and cellular events underlying presynaptic long-term synaptic plasticity.
Long-term synaptic plasticity is a fundamental property of the nervous system and is widely considered a primary mechanism for learning and memory (Kandel, 2001; Fusi et al., 2005). Long-term synaptic plasticity is defined by a long-lasting, activity-dependent change in synaptic efficacy. Long-term plasticity can bidirectionally modify synaptic strength—either enhancing (LTP, long-term potentiation) or depressing (LTD, long-term depression). Although early research focused on postsynaptic mechanisms, it is now clear that modification of synaptic strength may occur at either side of the synapse. Postsynaptic plasticity generally involves changes in postsynaptic receptor numbers or properties, while presynaptic plasticity involves an increase or decrease of neurotransmitter release.
Initial evidence for a presynaptic locus for long-term synaptic plasticity came from studies at hippocampal mossy fiber synapses and cerebellar parallel fiber synapses (Staubli et al., 1990; Zalutsky and Nicoll, 1990, 1992; Salin et al., 1996). In recent years, the list of brain regions found to express presynaptic LTP has markedly grown illustrating the prevalence of this form of synaptic plasticity. Moreover, the number of pathways implicated in the induction and expression has greatly expanded. Here we review the methodologies used to identify a presynaptic expression locus, describe the synapses where such plasticity exists, and discuss the molecular targets and putative cellular mechanisms responsible for presynaptic plasticity.
In the last decade of the twentieth century, scientists debated passionately a seemingly simple question—whether the change in synaptic strength during long-term plasticity is due primarily to presynaptic alterations in neurotransmitter release or to postsynaptic modifications in receptor numbers and/or biophysical properties (Malinow and Tsien, 1990; Malinow, 1991; Stevens, 1993; Bear and Malenka, 1994; Isaac et al., 1996; Manabe, 1997). Strong arguments were made on both sides, with disagreements arising from reproducibility of experimental results (Kullmann and Siegelbaum, 1995; Nicoll and Malenka, 1999a) to interpretations of the same observations (Korn and Faber, 1998; Malenka and Nicoll, 1999). These disagreements in part reflect the fact that the same synapse may undergo various forms of plasticity, depending on induction protocols and receptor compositions at the time. They also illustrate the importance of recognizing assumptions commonly held with particular methods used to obtain the experimental observations. In many cases, electrophysiological measurement of postsynaptic responses was the readout from which it was necessary to infer the nature of the synaptic change. This inference included assumptions of synaptic properties that broke down as exceptions were realized. In this section, we will review the general methodologies that have been employed to identify a presynaptic locus. We will discuss the basic assumptions and limitations of these methods and introduce more recent technical advances for monitoring neurotransmitter release and its alteration during long-term plasticity.
Initial attempts to dissect pre- or post-synaptic functions during long-term plasticity utilized conventional electrophysiological techniques. The approaches used generally relied upon the postsynaptic response as a reporter of synaptic activity, and typically included the aggregate response of a few to a few hundred synapses.
The idea of using quantal analysis to identify presynaptic mechanisms was proposed by Sir Bernard Katz (1971), following the discoveries of spontaneously occurring miniature synaptic potentials, or quanta, and the observation that evoked postsynaptic responses correspond to integral multiples of the quantal unit (Fatt and Katz, 1952; Kuno, 1964). Since its introduction, quantal analysis and its derivatives, such as coefficient of variation (CV) and minimal stimulation, have been used widely to investigate whether a change in neurotransmitter release accompanies long-term changes in synaptic strength. These methods were used in the first studies that proposed a presynaptic expression locus of LTP (at hippocampal mossy fiber-CA3 synapses) (Malinow and Tsien, 1990; Hirata et al., 1991), and have been subsequently employed to support a presynaptic expression locus during long-term plasticity in diverse brain regions which include hippocampal mossy fiber-interneuron synapses (Pelkey et al., 2005), hippocampal GABAergic synapses (Laezza et al., 1999), the amygdala (Tsvetkov et al., 2002; Fourcaudot et al., 2008), the neocortex (Torii et al., 1997; Sjöström et al., 2003, 2004; Huang et al., 2008a; Sarihi et al., 2012), striatum (Choi and Lovinger, 1997a), ventral tegmental area (VTA) (Pan et al., 2008) and cerebellum (Maejima et al., 2001).
The quantal analysis hypothesis proposes that the average amplitudes and fluctuations of postsynaptic responses follow simple or compound binomial statistics, which is supported by experimental evidence (Redman, 1990; Korn and Faber, 1991; Stevens, 1993). Under the simple binomial model, the average release probability, Pr, is assumed to be the same at all synapses. The CV is determined by the number of release sites, n, which is approximated by the number of synapses for most synapses in the CNS (Korn and Faber, 1991), and the average release probability, Pr, but is independent of quantal size, q, or the size of the postsynaptic response to any quanta of neurotransmitters1. Specifically,
Assuming a constant number of synapses, CV2 is negatively correlated with Pr—when Pr increases, CV2 will decrease and vice versa. In this way, a change in CV can be monitored as an indicator for changes in presynaptic release probability.
In minimal stimulation experiments, stimulation intensity is adjusted to a minimal level such that one or only a few presynaptic fibers are activated, and presumably only one or a few presynaptic release sites receive action potentials. Due to the stochastic nature of vesicle release (Katz, 1971), postsynaptic responses are not elicited with every stimulation. Failure rate, the average rate of not observing a response over multiple trials, is determined by the number of activated synapses, n, and the average release probability of these synapses, Pr,
Assuming a constant number of synapses, the failure rate is negatively correlated with average release probability, Pr. Therefore, failure rate in minimal stimulation experiments serve as an index of average release probability, with a higher failure rate corresponding to a lower release probability (Malinow and Tsien, 1990; Bekkers and Stevens, 1991).
As stated above, one of the assumptions for using a change in CV or failure rate to indicate a change in release probability is that the number of release sites remains constant. However, this assumption was called into question by the discovery of “silent synapses” (Malinow, 1991; Isaac et al., 1995; Liao et al., 1995). These synapses lack functional AMPA receptors but contain functional NMDA receptors postsynaptically. They were referred to as “silent” because at typical resting membrane potentials, NMDA receptors are blocked by magnesium and would not participate in synaptic responses under typical recording conditions. Changing the number of silent synapses is thus a postsynaptic mode of functionally changing, n, the number of release sites. In CV and minimal stimulation experiments, these synapses would not contribute to the initial number of release sites, n. However, upon LTP induction, functional AMPA receptors are recruited and inserted into the membrane of silent synapses and now contribute to the postsynaptic response. This postsynaptic phenomenon results in a decrease in CV2 and a decrease in failure rate. Without consideration of this possibility, the interpretation of these data would be (and was) that an increase in Pr had occurred, supporting the idea of presynaptic mechanisms for long-term plasticity (Malinow and Tsien, 1990; Bekkers and Stevens, 1991). In the early nineties, the expression locus for NMDA receptor-dependent LTP at hippocampal Schaffer collateral-CA1 synapses was debated fiercely. The accompanying decrease in CV2 and failure rate was viewed as support for a presynaptic locus (Malinow and Tsien, 1990; Bekkers and Stevens, 1991; Bolshakov and Siegelbaum, 1995). However, doubts started to emerge when it was noted that the relative contributions of AMPA and NMDA receptors to the postsynaptic response were also changing (Malinow and Tsien, 1990; Isaac et al., 1995; Kullmann and Siegelbaum, 1995). The subsequent discovery of postsynaptically silent synapses provided the necessary mechanistic insight to resolve these apparent discrepancies (Isaac et al., 1995; Liao et al., 1995). Postsynaptic unsilencing after LTP clearly reveals the postsynaptic expression locus of NMDA receptor-dependent LTP at hippocampal Schaffer collateral-CA1 synapses, with the exception of certain induction protocols (Zakharenko et al., 2002; Bayazitov et al., 2007) or in developing hippocampus (Bolshakov and Siegelbaum, 1995; Palmer et al., 2004).
To summarize, while quantal analysis and its derivatives (CV2 and failure rate) provide evidence for presynaptic long-term plasticity, they cannot stand alone as the sole evidence. The examples discussed above emphasize the importance of understanding the inherent assumptions underlying each approach to accurately guide interpretation and selection of complementary experiments.
A second experimental approach based on quantal analysis is examination of quantal synaptic events, namely, miniature postsynaptic currents and strontium evoked asynchronous postsynaptic currents. Miniature postsynaptic currents are useful when the majority of synapses assayed undergo plasticity, and asynchronous postsynaptic currents allows for analysis from the subset of synapses being stimulated (Oliet et al., 1996; Choi and Lovinger, 1997b). These methods have been employed to support a presynaptic expression locus in metabotropic glutamate receptor-dependent LTD at hippocampal Schaffer collateral-CA1 synapses (Oliet et al., 1997), and in the striatum (Choi and Lovinger, 1997b), as well as endocannabinoid-dependent LTD in the amygdala (Robbe et al., 2002b; Azad et al., 2004) and prefrontal cortex (Lafourcade et al., 2007). Early studies found that changing external calcium concentration, a manipulation known to alter presynaptic neurotransmitter release probability, changes the frequency of quantal events (del Castillo and Katz, 1954; Dodge and Rahamimoff, 1967). Therefore, a primary change in the frequency of quantal events is considered to represent a change in presynaptic neurotransmitter release probability (Choi and Lovinger, 1997b). However, this interpretation is also challenged by the existence of silent synapses. As reasoned above, postsynaptic unsilencing/silencing may increase/reduce the number of functional synapses as measured by postsynaptic currents, altering the frequency of quantal responses (Nicoll, 2003). Therefore, quantal analysis, too, cannot be used as the sole evidence for a presynaptic alteration. Though, consideration of the priniciples and assumptions underlying each assay can help in the selection of complementary assays with non-overlapping caveats.
MK801 is an irreversible, open channel blocker of the NMDA receptor (NMDAR) (Hessler et al., 1993; Rosenmund et al., 1993). Repeated activation of synapses in the presence of MK801 results in progressive decline of the NMDAR–mediated current, as each stimulus releases glutamate and opens NMDAR channels, the receptor becomes irreversibly blocked upon MK801 binding and thus are unavailable for subsequent responses. The rate of decline of the evoked NMDAR-mediated EPSC depends on the average release probability, Pr, because a higher Pr results in more frequent presynaptic release and consequently more rapid blockade of NMDARs. The rate of MK801 blockade of NMDAR-mediated current has been used to show that an increase in release probability is associated with LTP at mossy fiber-CA3 pyramidal synapses (Weisskopf and Nicoll, 1995), and that a decrease in release probability occurs with metabotropic glutamate receptor (mGluR)-mediated LTD at CA3-CA1 synapses of neonatal rats (Nosyreva and Huber, 2005). While this approach is not confounded by silent synapses, other limitations exist. First, this method relies upon the assumption that the activity of postsynaptic NMDARs is not altered as a result of the induction protocol. Should NMDAR plasticity exist and be unrecognized, the interpretation would erroneously attribute the changes to presynaptic release probability. Second, because MK801 blockade is irreversible, the same pool of synapses cannot be tested repeatedly, for example, before and after induction of LTP. Due to this limitation, evidence for a change in release probability is surmised by comparing the rate of blockade at a second set of stimulated synapses that have not undergone plasticity induction. In this condition, the assumption that the second set of stimulated afferents activates synapses with similar release probabilities must hold, and may not always be the case (Rosenmund et al., 1993; Dobrunz and Stevens, 1997). Third, if plasticity involved the addition of newly active presynaptic release sites but with the same release probability, the rate of MK801 blockade would not detect such a change (Malinow, 1994). Finally, this approach is only suitable for glutamatergic synapses.
When two stimuli are delivered to a presynaptic axon in rapid succession (fractions of a second), the second postsynaptic response often differs characteristically from the first. For example, being repeatedly larger or smaller by a certain degree. The ratio of the amplitude of the second EPSC response to that of the first is called the paired-pulse ratio (PPR). In the mammalian central nervous system, PPR inversely correlates with release probability (Zucker, 1989; Debanne et al., 1996; Dobrunz and Stevens, 1997; Zucker and Regehr, 2002) and is regularly used as an indicator of release probability. The mechanism for this change has been attributed to effects of presynaptic calcium dynamics and depletion of “release-ready” vesicles (Zucker, 1989; Debanne et al., 1996; Inchauspe et al., 2004; Catterall and Few, 2008). A decrease in PPR accompanies LTP at mossy fiber-CA3 pyramidal synapses (Zalutsky and Nicoll, 1990), corticothalamic synapses (Castro-Alamancos and Calcagnotto, 1999), cortical and thalamic synapses with lateral amygdala (Tsvetkov et al., 2002; Samson and Paré, 2005) and in other brain regions (Li et al., 2000; Lauri et al., 2007; Sarihi et al., 2012). Conversely, an increase in PPR accompanies LTD from electric stimulation or drug applications in various brain regions (Huang et al., 2001; Gerdeman et al., 2002; Chevaleyre and Castillo, 2003; Nosyreva and Huber, 2005). One caveat to the use of PPR as support for a presynaptic mechanism of plasticity is that changes in PPR are not exclusively mediated by changes in presynaptic release probability. For example, PPR can be influenced by postsynaptic receptor desensitization and lateral diffusion (Trussell et al., 1993; Heine et al., 2008; Frischknecht et al., 2009; Opazo et al., 2010). Additionally, modifications of short-term plasticity may occur that are not associated with a change in release probability but do affect PPR (Geppert et al., 1997; Basu et al., 2007; Shin et al., 2010b). These limitations again place PPR in the toolbox of reagents to suggest a presynaptic basis, but render this approach insufficient on its own to prove such a mechanism.
AMPA receptors and NMDA receptors are separately regulated at postsynaptic terminals. Forms of postsynaptic long-term plasticity, for example LTP involving delivery of AMPAR but not NMDAR to postsynaptic sites, commonly alter the ratio of activity between these two receptor types. Conversely, proportional changes in AMPA receptor-mediated and NMDA receptor-mediated responses have been used to support a presynaptic basis for long-term plasticity. Observations of a change in synaptic strength with a stable AMPA:NMDA ratio have been reported at hippocampal CA3-CA1 synapses and mossy fiber-interneuron synapses as supplemental evidence for a presynaptic locus of expression after long-term plasticity (Bayazitov et al., 2002; Lei and McBain, 2004). Conversely, if the population of presynaptic terminals being monitored does not have uniform AMPA:NMDA ratios, enhancing a fraction of synapses with distinct properties could change this ratio even though the mechanism was presynaptic.
The aforementioned methods are all based on electrophysiological analysis, which reports simultaneous activity of an unknown and potentially large number of synapses. As stated before, synapses from the same postsynaptic cell do not always share the same release probability presynaptically (Rosenmund et al., 1993; Dobrunz and Stevens, 1997). This heterogeneity in release probability may create artifacts in some scenarios while reduce the ability to detect a presynaptic alteration in others. To minimize interpretational difficulties, optical methods have been employed to provide single synapse resolution.
At an individual dendritic spine of an excitatory synapse, a calcium transient may be detected in response to a single afferent stimulus (Emptage et al., 1999; Oertner et al., 2002; Emptage et al., 2003; Reid et al., 2004; Enoki et al., 2009). Such events have been termed “EPSCaTs” for excitatory postsynaptic calcium transients. The probability of observing an EPSCaT at a synapse correlates with release probability at hippocampal CA3-CA1 and mossy fiber synapses (Emptage et al., 1999; Oertner et al., 2002; Emptage et al., 2003; Reid et al., 2004; Enoki et al., 2009), and the amplitude of EPSCaTs positively correlates with the amplitudes of excitatory postsynaptic potential (EPSP) at mossy fiber-CA3 pyramidal synapses (Reid et al., 2004). Monitoring EPSCaTs in rat organotypic hippocampal cultures, Emptage et al. (2003) showed that LTP at CA3-CA1 synapses is associated with a decrease in failure rate at single synapses, supporting the involvement of a presynaptic locus of expression. This approach overcomes limitations imposed by the presence of synapse unsilencing because the same active synapses are monitored before and after plasticity induction. Using this method, Reid et al. (2004) observed an increase in EPSCaT size in addition to a decrease in failure rate upon LTP induction at mossy fiber-CA3 pyramidal synapses, suggesting that LTP expression at single synapse may involve multi-vesicular release in addition to an increase in release probability.
EPSCaTs significantly advance the study of synaptic plasticity expression mechanisms by allowing interrogation of an individual synapse over time. Yet this approach has some limitations. For those spines showing an EPSCaT signal only after LTP induction (Reid et al., 2004), EPSCaT cannot distinguish whether the unsilencing reflects insertion of receptors postsynaptically or activation of new functional release sites presynaptically. Further, since the calcium transients include calcium influx through voltage-gated calcium channels (Yuste et al., 1999; Reid et al., 2001), the size of an EPSCaT could be altered by a modulation of postsynaptic calcium channel activity and/or back propagating action potentials (Yuste et al., 1999). If such mechanisms occurred, interpretations assigning amplitude changes to changes in the quanta of neurotransmitter released presynaptically would be erroneous. While the EPSCaT approach constitutes a major advance by allowing visualization of activity at single synapses, the main drawback of this approach is that the signal again arises from postsynaptic activity making it difficult to exclude with certainty contributions due to postsynaptic alterations.
Given the limitations of inferring presynaptic changes through a readout involving the postsynaptic apparatus, newer methodologies have been developed to allow direct measurement of presynaptic neurotransmitter release. Early approaches used fluorescent lipophilic dyes (FM dyes) to image presynaptic vesicles (Stanton et al., 2001; Zakharenko et al., 2001). PHluorins, pH-sensitive GFP molecules fused to synaptic vesicle proteins (Miesenböck et al., 1998), were later employed to address the same question (Bayazitov et al., 2007).
FM dyes were first developed by Fei Mao (hence, FM) to image synaptic vesicle recycling (Betz and Bewick, 1992). These dyes are not fluorescent in aqueous solutions but become fluorescent when associated with lipids. Synaptic vesicles are loaded with dye by a protocol that stimulates high volumes of vesicle fusion and recycling typically using 10–12 K stimuli at 10 Hz (Harata et al., 2001; Zakharenko et al., 2001; Blundon and Zakharenko, 2008; Park et al., 2012). In this way, the inner leaflet of the synaptic vesicle lipid bilayer becomes loaded with dye. Because the dye cannot traverse the lipid bilayer, once a synaptic vesicle is loaded, it can only lose its fluorescence by exocytosis which allows the dye to diffuse away. This technique has been used to measure release probability at single synapses by monitoring the destaining rate of FM dyes in response to repeated axonal stimulation (Zakharenko et al., 2001). Using this methodology to determine whether presynaptic release probability was altered as a result of long-term plasticity, differences in FM dye destaining rates were observed before and after both LTP and LTD induction at CA3-CA1 synapses in hippocampal slices (Stanton et al., 2001; Zakharenko et al., 2001, 2002, 2003).
While the application of the FM assay is a great step forward for directly monitoring the presynaptic component, several practical limitations prevent its widespread utility for studying mechanisms of long-term synaptic plasticity. First, the populations of synaptic vesicles that are loaded must be reproducible across conditions being compared. There are multiple intracellular pools of synaptic vesicles (e.g., reserve pool, readily releasable pool). Changes in the distribution of newly endocytosed (and thus dye-loaded) vesicles among these pools could alter observed destaining rates independent of an effect on release probability (Maeno-Hikichi et al., 2011). Second, FM dyes require a few seconds to diffuse away from synaptic vesicles, as such, their destaining rate fails to reflect vesicle exocytosis under conditions of rapid vesicle recycling or kiss and run events (Maeno-Hikichi et al., 2011). Third, FM dyes cannot be used for continuous monitoring of presynaptic release because the dye is lost to the extracellular medium upon exocytosis. Thus, for experiments studying long-term plasticity, the loading procedure has to be repeated. This necessity raises another potential caveat because the loading protocol [typically 10 Hz trains for several minutes (Harata et al., 2001; Zakharenko et al., 2001; Blundon and Zakharenko, 2008; Park et al., 2012)] may induce its own plasticity, possibly occluding further plasticity. Mossy fiber-CA3 synapses are one example of synapses that are modified by 10 Hz activity (Nicoll and Schmitz, 2005).
The pHluorins are pH-sensitive GFP molecules that lose fluorescence as pH decreases. Exploiting the fact that synaptic vesicles maintain their lumen at a low pH, Miesenböck et al. (1998) linked pHluorins to a vesicle membrane protein to report transmission at individual presynaptic terminals. SynaptopHluorin (spH), a fusion protein of VAMP2/synaptobrevin and pHluorin, exhibits little fluorescence inside acidic synaptic vesicles, but increases its fluorescence when the synaptic vesicle lumen is exposed to the extracellular space during exocytosis. One advantage of this approach is that the dye can be used to monitor vesicle over multiple rounds of exo-/endo-cytosis because it is an integral membrane component of the vesicle. The pHluorin-based indicators are genetically encoded, and transgenic mice expressing these indicators have been developed (Araki et al., 2005; Li et al., 2005; Tabares et al., 2007). Using a transgenic mouse that expresses spH in CA3 pyramidal cells, Bayazitov et al. (2007) monitored presynaptic vesicle exocytosis for several hours while simultaneously measuring postsynaptic potentials. Because fluorescence from single vesicle fusion events could not be reliably monitored, presynaptic release probability was inferred from changes in the peak fluorescence in response to a test stimulus train (10 Hz for 5 s). Using this approach, Bayazitov et al. concluded that CA3-CA1 LTP induced by 200 Hz tetanization or theta-burst stimulation consisted of slow presynaptic and fast postsynaptic components. Enhancement of postsynaptic potentials (without changes in peak evoked spH fluorescence) was rapid (around 1 min); whereas changes in peak spH fluorescence in response to the test train increased gradually and stabilized approximately 90 min after induction. These experiments provide evidence that both presynaptic and postsynaptic modifications can be involved in the expression of LTP, albeit at different time scales.
An obvious limitation of this approach is the inability to monitor a single release event. Instead, conclusions rely on an inference of the average release probability derived from the behavior of a population of vesicles stimulated during a moderate frequency “test” train. Again, because the test train itself may induce short-term plasticity, it is difficult to distinguish whether observed differences in fluorescence are due to long-term changes in release probability or short-term plasticity effects. Newer generation pHluorins appear to be overcoming this obstacle. The pHluorins have been fused to two other synaptic vesicle proteins, vGluT1 and synaptophysin (Granseth et al., 2006; Voglmaier et al., 2006). These proteins both demonstrate significantly lower surface expression and enhanced signal-to-noise ratio relative to spH (Balaji and Ryan, 2007; Zhu et al., 2009). Both proteins have been proven useful for detecting single release events in cultured neurons (Balaji and Ryan, 2007; Matz et al., 2010). In principle these tools could be used in brain slice preparations to study mechanisms of long-term synaptic plasticity. More recent advances, such as a molecular glutamate sensor which has been used in tissue preparations and has excellent signal-to-noise properties (Marvin et al., 2013) or biotinylated vesicular neurotransmitter reporters (Xu et al., 2013), also show great promise for directly monitoring presynaptic function. Finally, optogenetic techniques (Boyden et al., 2005) can also be employed to achieve activation of single presynaptic boutons to facilitate the study of presynaptic plasticity mechanisms.
In summary, there is a wide array of conventional electrophysiological and newer imaging technologies that is valuable for examining presynaptic mechanisms of synaptic plasticity. Because no assay is sufficient to stand on its own to definitively implicate the presynaptic site, these are best used in combinations with the least overlapping caveats to support presynaptic plasticity mechanisms. In Table 1, we provide a summary of the assays that have been used to support a presynaptic locus for long-term synaptic plasticity in various brain regions.
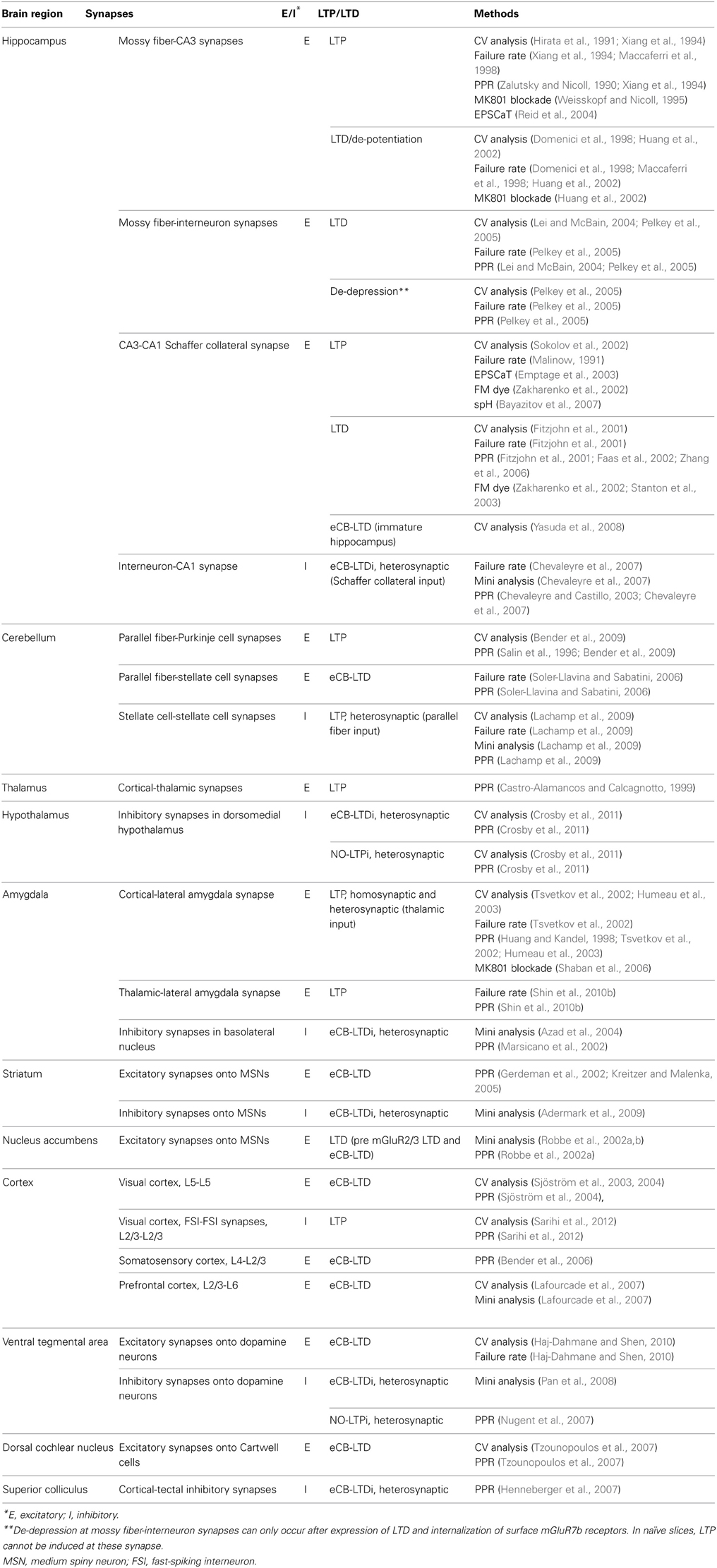
Table 1. Presynaptic long-term plasticity is a widespread phenomenon in the brain.
In this section, we will introduce the particular synapses at which presynaptic forms of long-term plasticity have been documented. We will review the evidence that supports a presynaptic expression locus and discuss the potential functional significance of presynaptic long-term plasticity at these synapses. We will also review briefly the induction mechanism (Figure 1); however, for a detailed discussion of induction requirements, please see two outstanding recent reviews by Castillo et al. (2011) and Castillo (2012).
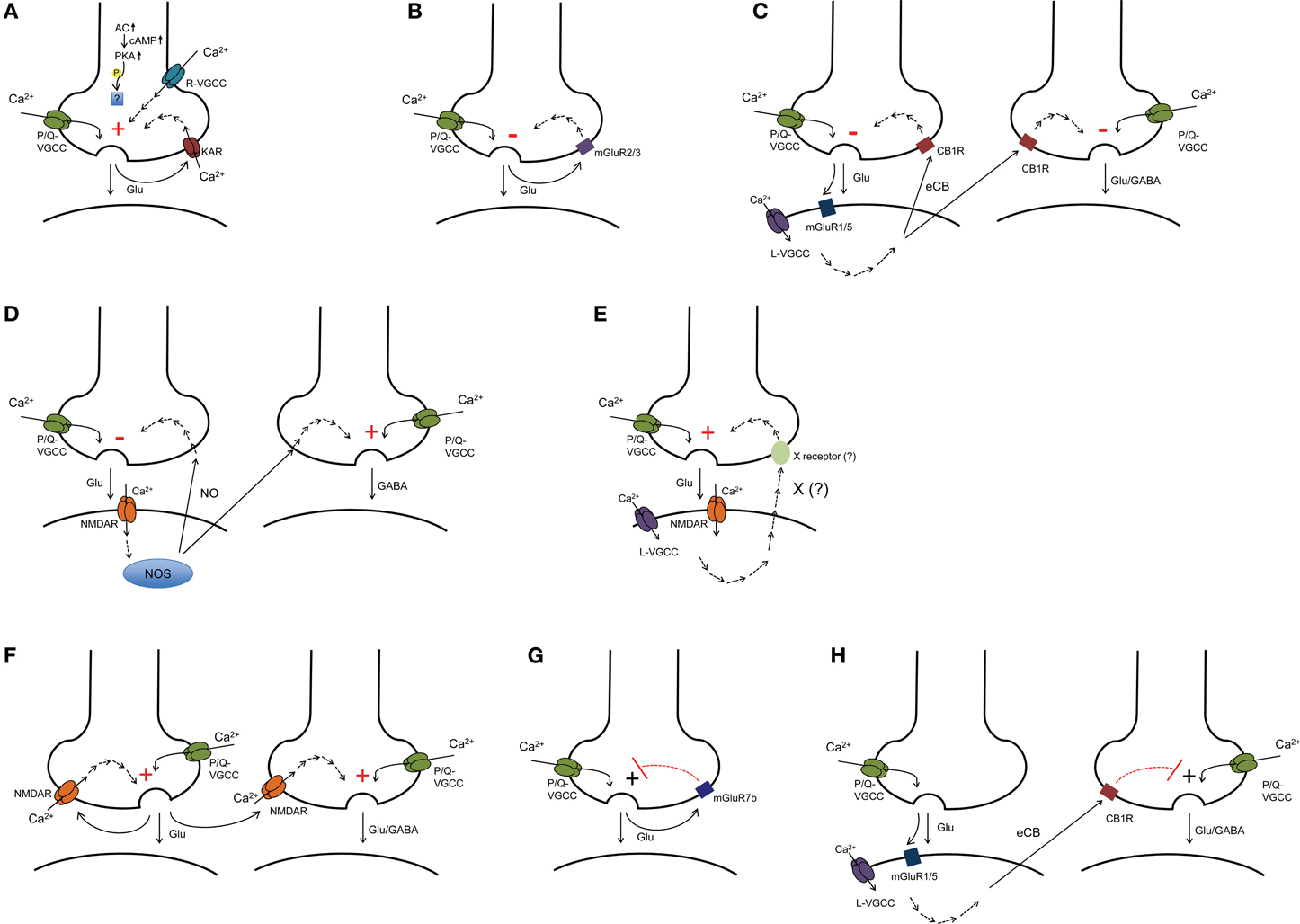
Figure 1. Induction mechanisms identified for presynaptic LTP and LTD. (A,B) presynaptic plasticity induced presynaptically (e.g., mossy fiber LTP and LTD). (C) eCB-dependent homosynaptic and heterosynaptic LTD. (D) NO-dependent LTD at excitatory synapses and LTP at inhibitory synapses. (E) postsynaptically induced LTP with as yet unidentified retrograde signaling mechanism (e.g., CA3-CA1 LTP). (F) presynaptic NMDAR-dependent homosynaptic and heterosynaptic LTP (e.g., LTP at cortico-LA synapses). (G) presynaptic LTP gated by mGluR7b at mossy fiber-SLIN synapses. (H) presynaptic LTP gated by CB1R (e.g., LTP at thalamic-LA synapses). AC, adenylate cyclase; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; VGCC, voltage-gated calcium channel; KAR, kainate receptor; mGluR, metabotropic glutamate receptor; eCB, endocannabinoid; CB1R, cannabinoid receptor type 1; NO, nitric oxide; NOS, nitric oxide synthase; CP-AMPAR SLIN, calcium permeable-AMPA receptor containing stratum lucidum interneurons.
Hebbian forms of synaptic plasticity are well described and have an intuitive appeal as to their role in learning. However, not all forms of long-term synaptic plasticity have properties consistent with Hebbian learning (which postulates a coordinated response of the postsynaptic site based upon the activity of a presynaptic afferent). For example, when long-term synaptic plasticity is both induced and expressed at the presynaptic terminal, Hebbian rules are violated. In these forms of plasticity, activation of the presynaptic neuron will alter the synaptic weight at all of its postsynaptic contacts, independent of postsynaptic coordinated activity. In this type of plasticity, the signaling efficacy of one presynaptic neuron may be increased relative to other presynaptic neurons. These forms of plasticity may be important for efficient circuit re-organization.
Mossy fiber synapses, the synapses between the axons of dentate granule cells and the proximal apical dendrites of CA3 pyramidal neurons, were the first synapses at which presynaptic long-term plasticity was described (Zalutsky and Nicoll, 1990; Kobayashi et al., 1996). Both LTP and LTD of mossy fiber-CA3 synapses are expressed by a change in release probability (Pr), as evidenced by quantal analysis, MK801 blockade of NMDA receptor-mediated current and PPR data (Zalutsky and Nicoll, 1990; Hirata et al., 1991; Xiang et al., 1994; Weisskopf and Nicoll, 1995; Domenici et al., 1998; Maccaferri et al., 1998). In addition to an increase in synaptic release probability, data from EPSCaTs imaged in slice cultures show that presynaptic unsilencing and an increase in quantal content may also occur (Reid et al., 2004). A presynaptic de-potentiation following LTP has also been described at these synapses and appears to follow a process similar to that involved in LTD (Tzounopoulos et al., 1998; Huang et al., 2002).
Induction of presynaptic long-term plasticity at these synapses requires action potential firing and calcium influx at the granule cell soma (Barnes et al., 2010), and calcium entry through calcium channels at the presynaptic terminal (Castillo et al., 1994; Domenici et al., 1998; Tzounopoulos et al., 1998; Kobayashi et al., 1999). Although no specific type of calcium channel is indispensable for plasticity induction (Castillo et al., 1994; Breustedt et al., 2003), the threshold for LTP induction is lowered by calcium influx through R-type calcium channels (Breustedt et al., 2003; Dietrich et al., 2003) or presynaptic kainate receptors and subsequent calcium release from intracellular calcium stores (Bortolotto et al., 1999; Lauri et al., 2003; Pinheiro et al., 2007). LTP induction has also been shown to require vesicular zinc and TrkB activation (Lu et al., 2000; Li et al., 2001; Huang et al., 2008b; Pan et al., 2011; but see Vogt et al., 2000; Matias et al., 2006). LTD induction requires activation of type II metabotropic glutamate receptors, mGluR 2/3, at presynaptic terminals (Yokoi et al., 1996; Tzounopoulos et al., 1998; Huang et al., 1999; Nicholls et al., 2006). Finally, two features that are commonly involved in postsynaptic forms of long-term plasticity, postsynaptic calcium influx and ionotropic glutamate receptor activity (NMDA- and AMPA-type receptors) are not required (Harris and Cotman, 1986; Zalutsky and Nicoll, 1990; Castillo et al., 1994; Langdon et al., 1995; Kobayashi et al., 1996; Tzounopoulos et al., 1998; Kobayashi et al., 1999; Mellor and Nicoll, 2001).
Besides the presynaptically induced and expressed form of plasticity described above, the mossy fiber-CA3 synapses have also been reported to undergo spike-timing-dependent LTP and LTP selectively expressed by NMDA receptors; both forms of LTP require postsynaptic calcium rise during induction (Jaffe and Johnston, 1990; Urban and Barrionuevo, 1996; Kapur et al., 1998; Sokolov et al., 2003; Kwon and Castillo, 2008; Rebola et al., 2008). After potentiation of NMDA receptors, these synapse become capable of expressing NMDAR-dependent LTP of AMPA receptors (Rebola et al., 2011). Recently, it has been shown that a postsynaptic form of LTP can also be unmasked when synaptic zinc is chelated (Pan et al., 2011). The detailed mechanisms for these LTP are beyond the scope of the current review, which focuses on presynaptic forms of long-term plasticity. However, these observations demonstrate that even within a given synapse type, experimental conditions and induction protocols may elicit mechanistically distinct forms of long-term plasticity—such findings have been recapitulated in diverse brain circuits.
Cerebellar parallel fibers are the axon tracts from cerebellar granule cells that synapse onto cerebellar Purkinje cells. The strength of these synapses is dynamically regulated, and multiple forms of plasticity are required for motor learning (Boyden et al., 2006). The parallel fiber-Purkinje synapses exhibit a presynaptic LTP similar to that of the hippocampal mossy fiber-CA3 synapses (Salin et al., 1996; Castillo et al., 2002). This presynaptic LTP is expressed by an increase in neurotransmitter release, as evidenced by a reduction in synaptic failure rates and a decrease in PPR (Salin et al., 1996; Bender et al., 2009). The increase in neurotransmitter release is associated with an increase in quantal content, potentially indicating a shift from single to multi-vesicular release (Bender et al., 2009). Parallel fiber LTP is induced presynaptically; it is resistant to glutamate receptor antagonists and calcium chelation at postsynaptic Purkinje cells, but requires presynaptic calcium influx (Salin et al., 1996).
The thalamus and neocortex are two highly organized and complex brain structures that work in concert with each other. A majority of afferents to the neocortex originates in the thalamus, and in return, the neocortex has massive connectivity to the thalamus through cortico-thalamic synapses. This loop is critical for motor function and is also considered as an important means for cortico-cortical communication (Guillery and Sherman, 2002). At the glutamatergic synapses between cortico-thalamic fibers and the ventrobasal thalamus, LTP can be induced and expressed presynaptically. This presynaptic LTP has been shown to be associated with a decrease in PPR and is resistant to glutamate receptor antagonists (Castro-Alamancos and Calcagnotto, 1999).
Fast-spiking interneurons (FSI) are implicated in synchronizing activity among projection neurons and establishing gamma oscillations (Bartos et al., 2007; and Fries et al., 2007; Humphries et al., 2009). FSI dysfunction is implicated in diseases such as schizophrenia and Dravet Syndrome (Yamakawa, 2011; Nakazawa et al., 2012). A presynaptic form of LTP has recently been described in FSIs at layer II/III of the mouse visual cortex (Sarihi et al., 2012). Tetanic stimulation of the presynaptic FSI in a paired whole-cell recording configuration demonstrated potentiation of unitary IPSCs in FSI → FSI pairs, but not non-FSI → FSI pairs. A similar potentiation of FSI IPSPs was observed by tetanic stimulation of nearby afferents under conditions blocking glutamatergic transmission. This IPSP potentiation was associated with concomitant changes in PPR and CV2 to support a presynaptic mechanism. In addition, this form of FSI-FSI LTP was insensitive to manipulations that blocked rises in postsynaptic calcium or inhibited activity of mGluRs or L- or T-type voltage-gated calcium channels, suggesting that induction may also depend solely on presynaptic activity (Sarihi et al., 2012). Because LTP of an inhibitory synapse onto an FSI would result in long-lasting suppression of FS interneurons, this form of presynaptic LTP between FS interneurons may function to desynchronize the cortical network.
As mentioned earlier, activation of presynaptic mGluR2/3 is involved in LTD at the hippocampal mossy fiber-CA3 synapses (Tzounopoulos et al., 1998). This involvement is also observed in LTD at nucleus accumbens synapses (Robbe et al., 2002a). Bath application of an mGluR2/3 specific agonist, L-CCG-1 or LY354740, depresses synaptic transmission and occludes further tetanus-induced LTD at both synapses, suggesting that activation of type II mGluRs at the presynaptic terminals is sufficient for the induction of LTD (Tzounopoulos et al., 1998; Robbe et al., 2002b). Evidence for presynaptic expression for this form of LTD includes observations of an increased PPR and a decreased miniature EPSC frequency at both synapses (Tzounopoulos et al., 1998; Robbe et al., 2002a).
Retrograde signaling can be achieved by the diffusion of small molecules released from postsynaptic spines. One such example is through endocannabinoids (eCBs). Retrograde signaling by eCBs has become a heavily studied topic over the past decade and multiple excellent reviews have covered various aspects of this signaling pathway (Alger, 2002; Freund et al., 2003; Gerdeman and Lovinger, 2003; Chevaleyre et al., 2006; Lovinger, 2008; Heifets and Castillo, 2009; Kano et al., 2009). Endocannabinoid-mediated presynaptic LTD has been reported in many brain regions, such as the striatum (Gerdeman et al., 2002; Kreitzer and Malenka, 2005; Shen et al., 2008), the nucleus accumbens (Robbe et al., 2002b; Mato et al., 2008), amygdala (Marsicano et al., 2002; Huang et al., 2003; Azad et al., 2004), hippocampus (Chevaleyre and Castillo, 2003; Chevaleyre et al., 2007; Yasuda et al., 2008), cerebellum (Soler-Llavina and Sabatini, 2006), visual cortex (Sjöström et al., 2003, 2004; Crozier et al., 2007; Huang et al., 2008a), somatosensory cortex (Bender et al., 2006; Li et al., 2009), prefrontal cortex (Lafourcade et al., 2007), VTA (Pan et al., 2008; Haj-Dahmane and Shen, 2010), hypothalamus (Kuzmiski et al., 2009; Crosby et al., 2011), brain stem (Tzounopoulos et al., 2007) and superior colliculus (Henneberger et al., 2007).
In general, eCB-LTD requires the production of eCB from the postsynaptic neuron. However, the exact induction mechanisms differ widely across examples of eCB-LTD. These differences include different requirements of postsynaptic receptors or kinases to stimulate eCB production, as well as differences in the stimulation pattern required to induce LTD. For a detailed description of the induction mechanism in various synapses, please refer to two excellent reviews on the topic (Gerdeman and Lovinger, 2003; Heifets and Castillo, 2009).
Upon activation of the CB1R at presynaptic terminals, eCB-LTD is expressed as an overall decrease in neurotransmitter release, indicated by decreased frequency of quantal synaptic events (Robbe et al., 2002b; Huang et al., 2003; Sjöström et al., 2003; Bender et al., 2006; Lafourcade et al., 2007; Pan et al., 2008), increased PPR (Gerdeman et al., 2002; Chevaleyre and Castillo, 2003; Huang et al., 2003; Soler-Llavina and Sabatini, 2006; Tzounopoulos et al., 2007; Pan et al., 2008; Kuzmiski et al., 2009; Haj-Dahmane and Shen, 2010), increased CV2 (Sjöström et al., 2003; Tzounopoulos et al., 2007; Pan et al., 2008; Yasuda et al., 2008; Kuzmiski et al., 2009), and increased failure rate upon minimal stimulation (Soler-Llavina and Sabatini, 2006; Kuzmiski et al., 2009; Haj-Dahmane and Shen, 2010).
Not all forms of eCB-LTD are homosynaptic. Endocannabinoid-mediated heterosynaptic LTD is observed at GABAergic synapses in hippocampus, striatum, frontal cortex, VTA and superior colliculus, and is thought to arise from eCB spillover from nearby glutamatergic synapses expressing homosynaptic LTD (Chevaleyre and Castillo, 2003; Henneberger et al., 2007; Pan et al., 2008; Adermark et al., 2009; Chiu et al., 2010). Heterosynaptic eCB-LTD is also observed at glutamatergic synapses between layer 4 and layer 2/3 neurons in visual cortex of young mice (Huang et al., 2008a). These heterosynaptic forms of eCB-LTD do not require the activation of presynaptic neurons during induction, thus, not following the Hebbian rule. Both homo- and heterosynaptic eCB-LTD are important for normal brain functions and behaviors, and are implicated in disease models (Marsicano et al., 2002; Crozier et al., 2007; Huang et al., 2008a; Kuzmiski et al., 2009; Li et al., 2009; Lerner et al., 2010; Zhang and Alger, 2010).
A second example of retrograde signaling is nitric oxide. Nitric oxide is a small diffusible molecule with a short lifetime, and is generated by nitric oxide synthase (NOS) (Alderton et al., 2001). In neurons, NOS is coupled to NMDA receptors, and activated by Ca2+/calmodulin upon the opening of NMDA receptors (Marin et al., 1992). Due to its high mobility and ability to cross membranes, NO can act locally at the post-synaptic neuron, as an anterograde messeger, a retrograde messeger, or a volume messenger to nearby cells without synaptic activity or NOS expression (Arancio et al., 1996; Lev-Ram et al., 1997; Park et al., 1998; Ko and Kelly, 1999; Ledo et al., 2005; Garthwaite, 2008; Steinert et al., 2010). As a retrograde messeger, NO has been shown to reduce glutamate release at excitatory synapses (Gage et al., 1997; Stanton et al., 2003) and to increase GABA release at inhibitory synapses (Bains and Ferguson, 1997; Stern and Ludwig, 2001).
NO has been shown to play a role in presynaptic long-term plasticity at various synapses. It is required for a form of NMDA receptor-dependent presynaptic LTD at hippocampal CA3-CA1 synapses, and thought to serve as a Hebbian coincidence detector for the activation of both presynaptic and postsynaptic neurons (Stanton et al., 2003; Zhang et al., 2006). It also acts heterosynaptically to support presynaptic LTP at GABAergic synapses onto dopamine neurons in VTA (Nugent et al., 2007). Further, it is reported that NO is required heterosynaptically for presynaptic LTP and eCB-mediated presynaptic LTD at GABAergic synapses in the dorsomedial hypothalamus, suggesting a state-dependent action of NO and crosstalk between various signaling pathways [Crosby et al., 2011; will be discussed in State-Dependent LTP at Inhibitory Synapses in the Dorsomedial Nucleus of the Hypothalamus (DMH)].
Interestingly, although the heterosynaptic forms of NO-LTP at inhibitory synapses require activation of NMDA-R of the postsynaptic neurons (Nugent et al., 2007; Crosby et al., 2011), they do not require activity of the presynaptic neuron during induction (Nugent et al., 2007). These non-Hebbian forms of LTP may serve to maintain excitatory/inhibitory balance and circuit homeostasis, up-regulating the local inhibitory tone in response to elevated excitatory inputs.
The hippocampal CA3-CA1 Schaffer collateral-commissural synapse displays multiple forms of long-term plasticity. At this synapse, LTP can be readily induced in juvenile and adult animals with a variety of induction protocols, such as 50, 100, and 200 Hz train stimulation. LTP in adult animals induced with a single 50 or 100 Hz tetanus requires activation of NMDA receptors and postsynaptic calcium influx, and is expressed primarily by an addition of surface AMPA receptors to the postsynaptic spines (Nicoll and Malenka, 1999b; Malinow et al., 2000). However, other groups have found that LTP induced with a 200 Hz (or multiple trains of 100 Hz) tetanus or specific presynaptic and postsynaptic pairing protocols may involve an additional presynaptic component besides postsynaptic LTP of AMPA receptor insertion (Zakharenko et al., 2002; Emptage et al., 2003; Bayazitov et al., 2007; Enoki et al., 2009). This presynaptic component is suggested by a decrease in CV, PPR and minimal stimulation failure rate (Malinow, 1991; Sokolov et al., 2002; Emptage et al., 2003; but see Nicoll and Malenka, 1999b), and supported by evidence at the single synapse level of an increase in the number of post-synapses showing EPSCaT signal (Emptage et al., 2003; Enoki et al., 2009), an accelerated rate of FM dye destaining (Zakharenko et al., 2002, 2003), and an enhanced synapto-pHluorin signal (Bayazitov et al., 2007) after LTP. The induction of this presynaptic component requires postsynaptic depolarization and subsequent calcium influx through L-type calcium channels (Zakharenko et al., 2003; Bayazitov et al., 2007), yet the exact retrograde signal for this presynaptic component remains to be investigated. A presynaptically expressed CA3-CA1 LTP has also been described in neonatal animals (P6 rats) with presynaptic and postsynaptic pairing protocols; this LTP is expressed in presynaptic terminals with low initial release probability and is associated with a decrease in failure rate and PPR (Palmer et al., 2004).
Besides LTP, CA3-CA1 synapse can also undergo LTD with both presynaptic and postsynaptic expression, for example, in cases of mGluR-LTD. Postsynaptic activation of group 1 mGluRs, either by its selective agonist DHPG or by appropriate synaptic stimulation, leads to postsynaptic and presynaptic changes that reduce the efficacy of the CA3-CA1 synapses—a rapid removal of surface AMPARs postsynaptically (Snyder et al., 2001; Xiao et al., 2001), and a reduction in presynaptic vesicle release suggested by a slower rate of FM dye destaining, an increase in failure rate and an increase in both CV2 and PPR (Fitzjohn et al., 2001; Faas et al., 2002; Zakharenko et al., 2002). A slower rate of FM dye destaining and an increase in PPR also accompany a form of LTD induced by prolonged activation of NMDA receptors (Stanton et al., 2003; Zhang et al., 2006). It has been shown that bath application of selective NOS inhibitor L-nitroarginine partially reduced the degree of depression but completely reversed the change of FM dye destaining, suggesting that this LTD has postsynaptic and presynaptic components, with the presynaptic expression dependent on nitric oxide signaling (Stanton et al., 2003). It suggested that NO works as a retrograde messenger from activation of postsynaptic NMDA receptors because the presynaptic depression can be partially blocked by extracellular NO scavenger (Stanton et al., 2003). A third example of presynaptic LTD at CA3-CA1 synapses is induced by a specific presynaptic and postsynaptic pairing protocol and associated with a decrease in the number of post-synapses showing EPSCaT signal (Enoki et al., 2009).
These Hebbian forms of compound long-term plasticity ensure effective modulation of synaptic strength - the degree of modulation is magnified when both pre- and post-synaptic potentiation (or depression) occur together.
Once again, not all forms of synaptic plasticity are homo-synaptic. Heterosynaptic long-term plasticity, the induction of which requires activity of other synapses, may result from diffusible molecules binding to receptors at the presynaptic terminal. These diffusible molecules can be released from postsynaptic neurons, as with eCBs; but they can also arise from nearby presynaptic terminals, as in presynaptic NMDAR dependent LTP induced by glutamate. Heterosynaptic NMDAR-dependent LTP at excitatory neurons can serve as a coincidence detector for two different inputs, altering their relative strength and how the postsynaptic neurons integrate information from different sources. Heterosynaptic NMDAR-dependent LTP at inhibitory neurons may be involved in regulation of excitability and shifting the circuit balance for excitatory and inhibitory inputs.
Lateral amygdala (LA) receives input from both the cortex and thalamus, and is involved in mediating emotional responses, especially fear-associated learning and memory (LeDoux, 2000). Cortical afferents to the LA can undergo both homosynaptic and heterosynaptic LTP. Homosynaptic LTP can be induced with various protocols; both a Hebbian form and a non-Hebbian form have been described. The Hebbian form requires calcium influx through L-type calcium channel at the postsynaptic terminals and activation of NMDA receptors (Huang and Kandel, 1998; Tsvetkov et al., 2002), while the non-Hebbian form requires no ionotropic glutamatergic transmission but calcium influx into the presynaptic terminals and blockage of presynaptic GABAB receptors (Shaban et al., 2006). Heterosynaptic LTP requires activation of presynaptic NMDA receptors and presynaptic calcium influx in the cortical afferents at the same time, which is achieved only with simultaneous activation of both cortical and thalamic afferents, or upon a puff-application of NMDA coincident with train stimulation of cortical afferents (Humeau et al., 2003). Though induced differently, homosynaptic and heterosynaptic forms of LTP occlude each other, and are expressed presynaptically as an increase in neurotransmitter release, as evidenced by a decrease in PPR, CV2, a reduction in failure rate under minimal stimulation, and a faster MK801 blockade of NMDAR-mediating currents (Huang and Kandel, 1998; Tsvetkov et al., 2002; Humeau et al., 2003; Shaban et al., 2006; Fourcaudot et al., 2008). Both forms of LTP and corresponding presynaptic changes are occluded after fear conditioning, suggesting that presynaptically expressed LTP at the cortico-LA synapses may underlie fear memory (Tsvetkov et al., 2002; Shaban et al., 2006).
In the cerebellum, stellate cells form inhibitory synapses onto Purkinje cells, regulating synaptic integration properties as well as the timing and firing frequencies of Purkinje cells (Häusser and Clark, 1997). Stellate cells are also connected to each other via inhibitory synapses and form an inhibitory network (Mann-Metzer and Yarom, 1999). A heterosynaptic form of LTP has recently been reported at the stellate cell-stellate cell synapses (Lachamp et al., 2009). At these inhibitory synapses, NMDA receptors are present at presynaptic terminals and can be activated by glutamate released from parallel fibers (Glitsch and Marty, 1999; Liu and Lachamp, 2006). Activation of presynaptic NMDA receptors by tetanus of the parallel fibers or by brief application of NMDA greatly potentiates transmission at these synapses. This potentiation is due to an increase in GABA release, as evidenced by a reduction in PPR, CV2, failure rate under minimal stimulation, and an increased frequency of miniature IPSCs (Lachamp et al., 2009). Importantly, this form of LTP can be induced without firing of the presynaptic stellate cell (Lachamp et al., 2009). This type of presynaptic LTP might provide a mechanism for the inhibitory network to sense and balance excessive excitation in the circuit.
It is well known that synapses are capable of expressing multiple forms of long-term plasticity. Why might there be multiple forms, and how might the synapse regulate which are used? Behaviorally, depending on past experience and the internal state of an animal, we know that the same stimulus can evoke different responses. Recent studies have found that this is also true at the synaptic level. Here we review three examples of how changes in presynaptic surface receptors can gate the ability of a synapse to express certain forms of long-term plasticity, such that the same induction protocol elicits different plasticity. This state-dependent plasticity may underlie hierarchical information processing and is important for proper behavioral responses.
Besides CA3 pyramidal neurons, mossy fibers also synapse onto CA3 stratum lucidum interneurons (SLIN). Unlike mossy fiber-CA3 synapses that are large (~3–8 μm in diameter) with multiple release sites (Rollenhagen and Lübke, 2010), mossy fiber-SLIN synapses are small terminations (~1 μm) of numerous fine filopodia that radiate from mossy fiber boutons (Acsády et al., 1998). After HFS of mossy fiber inputs, which produces presynaptic LTP at the CA3 pyramidal synapses, those SLINs which contain calcium-permeable AMPA receptors will exhibit LTD presynaptically (Maccaferri et al., 1998; Lei and McBain, 2004; Pelkey et al., 2005). This presynaptically expressed LTD follows a Hebbian learning rule, as it requires calcium influx through postsynaptic AMPA receptors and activation of presynaptic mGluR7b receptors (Laezza et al., 1999; Pelkey et al., 2005). Upon activation, presynaptic mGluR7bs are internalized. Without surface mGluR7b, the same synapses will now exhibit de-depression with a second train of HFS. Although not a mechanistic reversal of LTD, de-depression is induced presynaptically and expressed as an increase in neurotransmitter release (Pelkey et al., 2005; Pelkey and McBain, 2008). The ability of the mossy fiber-SLIN filopodia synapses to undergo presynaptic LTD and meta-plasticity may allow for profound and rapid state-dependent control of feed-forward inhibition in the hippocampal network, and is implicated in spatial and temporal information processing (Kullmann and Lamsa, 2008; McBain and Kauer, 2009).
A second class of presynaptic receptors gating the polarity of presynaptic long-term plasticity is CB1 receptors. Activation of CB1 receptors by eCBs has been shown to prevent the expression of presynaptic LTP at multiple synapses with behavioral significance, and is discussed in the following 2 subsections.
LTP of thalamic synapses onto LA has generally been found to be expressed postsynaptically through changes in AMPA receptor trafficking (Humeau et al., 2005; Rumpel et al., 2005). One recent study, however, provides evidence for conditions under which thalamic-LA synapses may also undergo presynaptic LTP, which is gated by CB1 receptor activation (Shin et al., 2010b). When postsynaptic LA neurons were held at a hyperpolarized potential (−70 mV), repetitive stimulation of thalamic afferents resulted in presynaptic LTP, as evidenced by a reduction in PPR as well as failure rate under minimal stimulation. This LTP was induced presynaptically; it required activation of presynaptic kainate receptors (KAR) and calcium influx into the presynaptic terminals, but not NMDAR activation nor calcium influx into the postsynaptic LA neurons. Interestingly, when postsynaptic LA neurons were held at depolarized potential (+30 mV) during induction, the same presynaptic stimulation for KAR-dependent presynaptic LTP would instead lead to postsynaptic LTP and suppression of presynaptic LTP. The suppression of presynaptic LTP appears to be mediated by activation of presynaptic CB1Rs. When CB1Rs are blocked, the presynaptic LTP is unmasked and can be expressed simultaneously with postsynaptic LTP at these synapses (Shin et al., 2010b).
These findings suggest that the eCB system is employed to report activation of postsynaptic LA neurons and prevent over potentiation of the thalamic afferents. When thalamic input fails to depolarize postsynaptic LA neurons, presynaptic LTP is employed to ensure efficient synaptic transmission. However, because glutamate released from thalamic afferents can activate presynaptic NMDA receptor to promote heterosynaptic LTP at cortico-LA synapses (Humeau et al., 2003), unchecked potentiation of the thalamic afferents may lead to inappropriate potentiation of cortico-LA synapses during fear conditioning, potentially resulting in increased fear experience and fear generalization. Gating of presynaptic LTP at thalamic-LA synapses by eCB and CB1R may thus serve as an activity-dependent constraint to prevent the generalization of conditioned fear. Indeed, behavioral studies have shown that CB1R antagonism enhanced baseline freezing, which is usually attributed to fear generalization (Reich et al., 2008).
Studies of the DMH provide some of the first direct evidence that the internal state of an animal can alter presynaptic surface receptors and gate presynaptic plasticity. The DMH has roles in signaling satiety and regulating food intake (Bellinger and Bernardis, 2002). Recent work provides evidence that the feeding state of an animal reduces CB1R signaling and determines the polarity of long-term plasticity in inhibitory synapses in DMH (Crosby et al., 2011). When mice are given ad libitum access to food, repeated stimulus trains (100 Hz, 4 s) induces presynaptic LTD at inhibitory synaspes in DMH, as supported by an increase in PPR and CV. This LTD requires activation of presynaptic CB1R by eCBs and is dependent on NO production. Both eCBs and NO are thought to be produced heterosynaptically by excitatory synapses. When mice were food deprived, however, the same tetanus produces presynaptic LTP at these synapses, which is dependent on the production of NO (Crosby et al., 2011). It was further shown that food deprivation dampens CB1R signaling at inhibitory synapses in DMH and blockade of CB1R in naïve mice shifts presynaptic LTD to LTP similar to that in food deprivation, suggesting that food availability is encoded at these synapses by presynaptic CB1Rs which gates their ability of to undergo LTP. Because DMH neurons are thought to send excitatory projections to the paraventral nucleus of the hypothalamus (PVN) that suppresses food intake, an increase in inhibitory drive in DMH would suppress neuronal activities in PVN, ultimately leading to an elevated drive to eat when food is limited (Crosby et al., 2011).
CB1 receptors are widely expressed in the brain and highly regulated by neuronal activities and behavioral states of an animal. It is possible that presynaptic CB1R may affect the expression of different forms of long-term plasticity in other brain regions besides mediating eCB-LTD. Other receptors at the presynaptic terminals, such as NMDA receptors, kainate receptors, mGluRs and GABAB receptors, can also be regulated by activity and influence the plasticity decision of a synapse. As such, gating of synaptic plasticity by presynaptic surface receptors may turn out to be a more widespread phenomenon.
The molecular mechanisms underlying presynaptically expressed long-term plasticity and their maintenance have been a focus of the field for decades. It was recognized early on that the cyclic adenosine monophosphate (cAMP) and cAMP-dependent protein kinase A (PKA) pathway is required for mossy fiber LTP in guinea pigs (Weisskopf et al., 1994), rats (Huang et al., 1994) and mice (Huang et al., 1995). Here, we review evidence for the involvement of the cAMP/PKA pathway for various forms of presynaptically expressed LTP and the notion that a shift in the balance of kinase and phosphatase activity underlies presynaptically expressed LTD. We then review what processes downstream of, or in parallel with, these signaling pathways can account for the long-term alteration of neurotransmitter release. Changes in neurotransmitter release efficacy can theoretically result from diverse mechanisms, such as modifications of the release machinery, alterations in calcium influx through voltage-gated calcium channels, or changes in intrinsic membrane excitability of the presynaptic terminal.
The first evidence implicating cAMP/PKA signaling in presynaptic LTP was observed at hippocampal mossy fiber-CA3 synapses. In guinea pigs, the Nicoll group (Weisskopf et al., 1994) discovered that brief application of the adenylyl cyclase activator, forskolin, increased basal transmission at mossy fiber synapses in an enduring manner, and occluded electrically stimulated presynaptic LTP. Sp-cAMPs, analogs of cAMP that activate PKA more selectively, also enhanced basal transmission long-lastingly following brief application, and this effect was occluded by prior application of forskolin. These findings indicated that brief elevation of PKA activity might be sufficient for mossy fiber LTP. Weisskopf and colleagues further established the necessity of the cAMP/PKA cascade by using three different PKA inhibitors (Weisskopf et al., 1994). Blockers of the catalytic subunit of PKA, KT5720 and H-89, as well as an inhibitor of the regulatory subunit, Rp-cAMPs, all antagonized mossy fiber LTP, without any effect on basal transmission. These effects were localized to the presynaptic terminal by demonstrating that a reduction of PPR accompanied forskolin-enhanced transmission and that postsynaptic responses to iontophoretic pulse of glutamate remained constant. Together, these results suggested that a brief elevation of cAMP causes persistent increase in neurotransmitter release and that PKA activation is an essential step in mossy fiber LTP (Weisskopf et al., 1994). Similar results were observed at hippocampal mossy fiber-CA3 synapses in rats and mice (Huang et al., 1994; Spillane et al., 1995; Castillo et al., 1997).
The cAMP/PKA pathway has subsequently been shown to be involved in presynaptically expressed LTP in many brain regions. Forskolin enhances basal neurotransmitter release and occludes LTP at cerebellar parallel fiber-Purkinje cell synapses (Salin et al., 1996), cortico-thalamic synapses (Domenici et al., 1998), the cortical afferents to LA (Huang and Kandel, 1998), as well as inhibitory synapses between cerebellar stellate cells (Lachamp et al., 2009). Similarly, LTP at these synapses is blocked by the PKA inhibitor, Rp-cAMPs (Salin et al., 1996; Domenici et al., 1998; Huang and Kandel, 1998; Lachamp et al., 2009). These results support that cAMP/PKA activation underlies presynaptically expressed LTP. Interestingly, the cAMP/PKA pathway also functions at the hippocampal mossy fiber-interneuron synapses, but only under certain conditions. As reviewed earlier, these synapses undergo presynaptic LTD when presynaptic mGluR7b receptors are at the surface, but exhibit de-depression once those receptors are internalized (Pelkey et al., 2008). Besides gating the direction of presynaptic plasticity, surface mGluR7b also gates the sensitivity to cAMP and PKA. When mGluR7b are at the surface, forskolin has no effect on basal transmission. Only after mGluR7b are internalized, does forskolin enhance basal transmission (Pelkey et al., 2008). While the adenylyl cyclase inhibitor, DDOA, or the PKA inhibitor, H-89, did not affect mGluR7b mediated presynaptic depression, both drugs abolished subsequent de-depression after mGluR7b internalization. In all, these results showed that, following internalization of presynaptic surface mGluR7b receptors, the cAMP/PKA pathway is involved in presynaptic de-depression at the hippocampal mossy fiber-interneuron synapses (Pelkey et al., 2008).
One note about the forskolin effect of enhancing transmission is warranted. Although this phenomenon completely occluded subsequent tetanus-induced LTP, tetanus-induced LTP only partially reduced the enhancement by brief application of forskolin (Weisskopf et al., 1994). This observation can be readily explained if there are multiple degrees of potentiation and/or heterogeneous induction thresholds for different afferents, such that not all electrically tetanized synapses are potentiated to their maximum. Alternatively, forskolin may activate multiple mechanisms to enhance neurotransmitter release (Lonart et al., 1998), some of which may not normally be engaged by tetanus, the cAMP/PKA pathway, or under physiological conditions. Therefore, one has to be cautious when comparing pharmacological- vs. activity-induced long-term plasticity results.
A requirement for the cAMP/PKA pathway in presynaptic LTP has also been shown genetically in studies using mice lacking the genes encoding adenylyl cyclase or PKA. In the type-1 adenylyl cyclase knockout mouse, hippocampal mossy fiber LTP was selectively impaired, leaving the prefrontal pathway LTP and the Schaffer collateral LTP intact (Villacres et al., 1998). In this mouse, high concentrations of forskolin were still capable of enhancing transmission, suggesting that downstream signaling of cAMP was intact. In the PKA knockout mouse, both electrically stimulated mossy fiber LTP and forskolin enhanced synaptic transmission were abolished, confirming the essential role of cAMP/PKA signaling in presynaptically expressed LTP (Huang et al., 1995).
Cyclic AMP/PKA activity is thought to be down-regulated in presynaptically expressed LTD at hippocampal mossy fiber-CA3 synapses (Tzounopoulos et al., 1998), hippocampal interneuron-pyramidal cell synapses (Chevaleyre et al., 2007), nucleus accumbens synapses (Robbe et al., 2002a; Mato et al., 2008), and excitatory synapses onto dopamine neurons in the VTA (Haj-Dahmane and Shen, 2010). PKA inhibitors, KT5720, H-89 or PKI have been shown to occlude tetanus-induced or pharmacologically-induced LTD by suppressing basal transmission at the nucleus accumbens synapses and the hippocampal interneuron-pyramidal cell synapses (Robbe et al., 2002a; Chevaleyre et al., 2007; Mato et al., 2008). The down-regulation of PKA is localized to the presynaptic neuron by the findings that neither basal transmission nor LTD was altered when a membrane impermeable form of PKI was delivered to the postsynaptic cell via the recording pipette (Chevaleyre et al., 2007).
Interestingly, presynaptic LTD at these three synapses requires activation of G-protein coupled receptors, mGluR2/3s or CB1Rs. At the nucleus accumbens synapses, where both receptors are present at the presynaptic terminals, mGluR2/3-LTD and eCB-LTD mutually occlude, suggesting that both forms of presynaptic LTD share a common mechanism (Mato et al., 2005). Indeed, both receptors are coupled to Gαi. The αi effector limb shared by these two receptor types would inhibit adenylyl cyclase activity and thereby the cAMP/PKA pathway (Pin and Duvoisin, 1995; Childers and Deadwyler, 1996). The involvement of Gαi-coupled receptors and their known inhibition of adenylyl cyclase further suggest that presynaptic LTD may be expressed through a reduction in cAMP/PKA kinase activity. Importantly, brief activation of CB1R alone is not sufficient to induce presynaptic LTD, suggesting that other signaling pathways might be involved besides a reduction in cAMP/PKA kinase activity (Gerdeman et al., 2002; Heifets et al., 2008; Heifets and Castillo, 2009; Lovinger, 2010). At hippocampal interneuron-pyramidal cell synapses, Heifets et al. (2008) found that, calcineurin, a calcium-activated phosphatase, is required for eCB-LTD. This finding supports a model whereby the balance of kinase and phosphatase activity is shifted in presynaptically expressed long-term plasticity to favor dephosphorylation in LTD.
The involvement of the cAMP/PKA pathway in presynaptically expressed long-term plasticity has been firmly established in the last decades. However, the downstream effector has not yet been fully identified. Two presynaptic proteins, synapsin and rabphilin, that are both phosphorylated by PKA are not required for presynaptic long-term plasticity (Spillane et al., 1995; Lonart and Südhof, 1998; Hosaka et al., 1999; Schlüter et al., 1999). Another PKA substrate at the presynaptic terminal, RIM1α, is required for many forms of presynaptic long-term plasticity (reviewed in the next section). However, in the RIM1α knockout mouse, despite the lack of mossy fiber LTP, basal transmission can still be enhanced by forskolin, suggesting that PKA may function in parallel with RIM1α (Castillo et al., 2002). Furthermore, the significance of a PKA phosphorylation site on RIM1α, Serine 413, although shown to be necessary for presynaptic LTP in a cerebellar culture model (Lonart et al., 2003; Simsek-Duran et al., 2004), was not supported when presynaptic LTP was studied in more in vivo brain preparations. Using acute brain slices from a genetic knockin model (S413A) and in an acute in vivo rescue approach, mutation of the S413 site did not affect expression of presynaptic LTP (Kaeser et al., 2008b; Yang and Calakos, 2010). Therefore, the PKA targets underlying presynaptic LTP remain to be elucidated.
Vesicle exocytosis is tightly regulated at the presynaptic terminal and a major determinant of synaptic release probability. In this section, we will discuss molecular targets for presynaptic long-term plasticity that affect synaptic vesicle release efficacy (Figure 2).
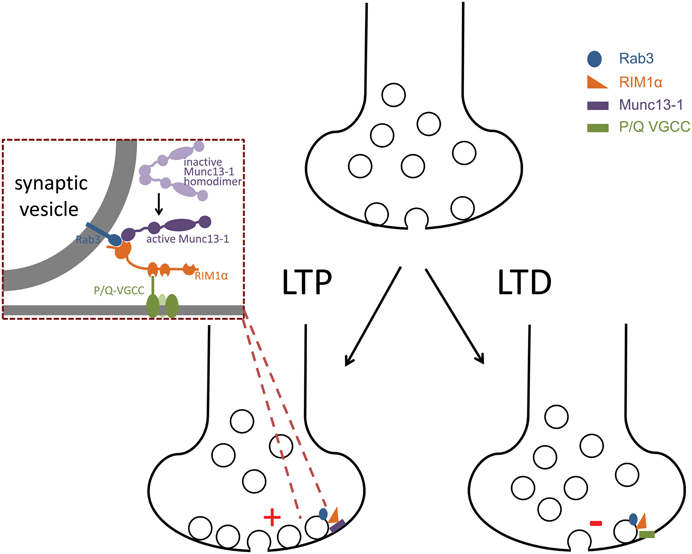
Figure 2. Working models of expression mechanisms for presynaptic LTP and LTD. In this schematic, presynaptic molecules implicated in the expression of changes in presynaptic release probability associated with LTP and LTD are shown. In LTP at mossy fiber synapses, the specific interaction of RIM1a with Munc13-1 that promotes activation of Munc13 for vesicle priming is required, indicating that a likely cellular mechanism for increasing release probability may be through changes in the number of primed vesicles (indicated by vesicles adjacent to the terminal membrane). In LTD at hippocampal mossy fiber—stratum lucidum interneuron synapses and the nucleus accumbens, inhibition of P/Q type voltage-gated calcium channel (VGCC) activity is associated with expression of LTD. Whether this modulation of channel activity requires interaction with RIM1a has not been tested. At other synapses, a requirement for Rab3 and RIM1a for LTD has been described. Therefore, in the LTD working model, we also hypothesize that LTD mechanisms opposite to LTP may occur and thus show fewer primed vesicles to lead to a decrease in release probability. However, a requirement for Munc13 in LTD has not yet been tested. In addition, while Rab3a and RIM1a are both implicated in LTP and LTD, whether their interaction is required has also not been tested. Lastly, whether multiple expression mechanisms co-exist within a synapse or occur at distinct subsets of synapses remains to be determined.
RIM1α is a presynaptic scaffolding protein of the active zone; it binds to many molecules involved in neurotransmitter release, such as Munc13 (Betz et al., 2001; Schoch et al., 2002; Deng et al., 2011), Synaptotagmin1 (Coppola et al., 2001; Schoch et al., 2002), Rab3 (Wang et al., 1997; Schoch et al., 2002) and voltage-gated calcium channles (VGCCs) (Coppola et al., 2001; Wang et al., 2001; Kiyonaka et al., 2007; Kaeser et al., 2011; Han et al., 2011). RIM1α is required for presynaptic LTP at hippocampal mossy fiber-CA3 synapses, Schaffer collateral-CA1 synapses, cerebellar parallel fiber synapses, cortical LA synapses, as well as presynaptic LTD at inhibitory synapses in the hippocampus, in basolateral amygdala, and at cerebellar stellate cell synapses (Castillo et al., 2002; Huang et al., 2005; Chevaleyre et al., 2007; Fourcaudot et al., 2008; Lachamp et al., 2009). Interestingly, while deleting RIM1α abolishes presynaptic LTD at inhibitory synapses, RIM1α deletion enhances presynaptic LTD at hippocampal mossy fiber-CA3 synapses (Castillo et al., 2002). These observations indicate that RIM1α may have different functions at excitatory and inhibitory synapses. Nevertheless, changes in the release machinery via RIM1α may be a general mechanism underlying presynaptic long-term plasticity. RIM1α has been shown to enhance neurotransmission by promoting vesicle priming, a process by which vesicles at the active zone become fusion competent (Calakos et al., 2004). The ability of RIM1α to promote vesicle priming is mediated through its interaction with the priming protein, Munc13 (Betz et al., 2001; Deng et al., 2011). Of the myriad potential protein interactions and synaptic modifications that RIM1a may influence, we recently showed that the interaction of RIM1α and Munc13 required to modulate vesicle priming is also required for presynaptic LTP at hippocampal mossy fiber synapses (Yang and Calakos, 2011). These data indicate that regulation of vesicle priming via a RIM-Munc13 interaction is a likely cellular substrate for enhancement of presynaptic strength in presynaptic LTP.
Rab3 proteins (Rab3A/B/C/D) are a family of small GTPases located on synaptic vesicles. They cycle between the GDP-bound inactive form and GTP-bound active form and are associated with synaptic vesicle membranes when GTP is bound (Fischer von Mollard et al., 1991; Sudhof, 2004). Removal of all four Rab3 proteins leads to perinatal lethality and abnormal short-term plasticity and vesicle dynamics in neuronal cultures (Schlüter et al., 2006). In Drosophila, Rab3 proteins are implicated in active zone dynamics. At the Drosophila neuromuscular junction, Rab3 deletion resulted in presynaptic terminals devoid of active zone proteins and calcium channels, which could be reversed within 24 h after re-introduction of Rab3 to mutant Drosophila larvae (Graf et al., 2009). Rab3 proteins interact with RIM1α in their GTP-bound form (Wang et al., 1997), and two Rab3 isoforms have been shown to be required for presynaptic long-term plasticity at various synapses. Rab3A deletion has been shown to abolish both presynaptic LTP and LTD at hippocampal mossy fiber-CA3 synapses (Castillo et al., 1997), as well as presynaptic LTP at cortical–lateral amygdala synapses (Tzounopoulos et al., 1998) and late phase LTP at CA3-CA1 synapses (Huang et al., 2005). Rab3B deletion has been shown to eliminate presynaptic LTD at inhibitory synapses in the hippocampus (Tsetsenis et al., 2011). Due to its interaction with RIM1α and their dual requirement for presynaptic LTP at excitatory synapses, Rab3 proteins are believed to function together with RIM1α during presynaptic long-term plasticity (Sudhof, 2004; García-Junco-Clemente et al., 2005; Kaeser et al., 2008a; Tsetsenis et al., 2011). However, Rab3A knockout mice and RIM1α knockout mice display different presynaptic LTD phenotypes at hippocampal mossy fiber-CA3 synapses (Tzounopoulos et al., 1998; Castillo et al., 2002), suggesting that Rab3A and RIM1α may play different roles in different forms of presynaptic long-term plasticity. How Rab3 proteins regulate presynaptic strength during plasticity and whether its interaction with RIM1α is required remain to be determined.
Munc13 proteins (Munc13-1, Munc13-2, Munc13-3, Munc13-4) are active zone priming proteins required for neurotransmission. In the absence of full-length Munc13 proteins (Munc13-1 and Munc13-2 double KO mice), no vesicle priming was detected in hippocampal excitatory synapses of autaptic cultured pyramidal neurons, resulting in a complete loss of spontaneous and evoked synaptic transmission (Augustin et al., 1999; Rosenmund et al., 2002; Varoqueaux et al., 2002). Munc13 proteins have multiple functional domains. Current evidence supports an essential role for the MUN domain in vesicle priming (Basu et al., 2005; Stevens et al., 2005), a regulatory role for the C2A domain in vesicle priming (Betz et al., 2001; Deng et al., 2011), and roles for the C2 domains, C1 domain, and calmodulin (CaM)-binding sequence in short-term synaptic plasticity (Junge et al., 2004; Basu et al., 2007; Shin et al., 2010a). The C2A domain of Munc13 interacts with RIM1α. Disruption of this interaction resulted in autoinhibitory homo-dimerization of Munc13 which reduced the size of the readily releasable pool and evoked EPSC in cultured neurons (Betz et al., 2001; Deng et al., 2011). Munc13-1, but not Munc13-2, is required for presynaptic LTP at hippocampal mossy fiber synapses (Breustedt et al., 2010), suggesting different roles of the two full-length Munc13 proteins in long-term plasticity. Further, it has been shown that the requirement for Munc13-1 in hippocampal mfLTP is through its interaction with RIM1α, supporting regulation of vesicle priming as the cellular basis for long-term presynaptic plasticity (Yang and Calakos, 2011).
Calcium influx through VGCCs is an essential step in triggering fast synchronous neurotransmitter release. The magnitude and duration of calcium transients as well as the basal intracellular calcium concentration greatly shape synaptic release probability (Sudhof, 2004; Neher and Sakaba, 2008). Therefore, factors affecting intracellular calcium concentration are well suited to mediate a change in neurotransmitter release. Such factors include the open probability and open duration of VGCCs, the intracellular calcium stores, and the calcium buffers that shape the calcium microdomains. To date, only the calcium transients and the functions of VGCCs have been studied in the expression of presynaptic long-term plasticity. At nucleus accumbens synapses, where mGluR2/3- and eCB-LTD are present and may both function through suppression of the cAMP/PKA pathway, P/Q-type VGCCs are selectively inhibited to reduce neurotransmission in presynaptic LTD. At the plateau phase of agonist-induced mGluR2/3-LTD, the relative contributions of P/Q-type VGCC to field excitatory post-synaptic potentials were greatly reduced (Robbe et al., 2002a). Further, blockade of P/Q-type throughout the experiment, but not L- or N-type VGCCs, occluded agonist-induced mGluR2/3-LTD and tetanus-induced eCB-LTD (Robbe et al., 2002a; Mato et al., 2008). These findings indicate that presynaptic LTD in the nucleus accumbens is expressed by inhibition of P/Q-type VGCCs. Selective inhibition of P/Q-type VGCC has also been reported in presynaptic LTD at the mossy fiber-stratum lucidum interneuron synapses. At these synapses, calcium transients are reduced after HFS-induced LTD; this reduction is due to a reduction of calcium influx through the P/Q-type VGCCs, as shown by pharmacological experiments (Pelkey et al., 2006).
Presynaptic LTP, however, is not always associated with an enhancement of VGCCs. Multiple groups have shown with different calcium indicators that calcium transients are unaltered at the mossy fiber bouton after LTP expression (Regehr and Tank, 1991; Kamiya et al., 2002; Pelkey et al., 2006). At the mossy fiber-stratum lucidum interneuron synapses, where LTD results from a reduction in calcium influx, de-depression or LTP of the same synapse does not involve a reversal (i.e., increase) of the calcium transient, suggesting that LTP and LTD are not mere reciprocals of each other mechanistically (Pelkey et al., 2008). This observation also excludes the slight possibility that none of the calcium indicators were sensitive enough to detect a change in calcium transients before and after long-term plasticity, confirming that regulation of calcium transients does not appear to be involved in these forms of presynaptic LTP.
Long-term forms of presynaptic plasticity are abundant and contribute unique computational features to circuit function. In this review, we have discussed methods typically used for identifying presynaptic mechanisms in long-term plasticity, those synapses at which this process has been observed, and our current understanding of the molecular mechanisms underlying presynaptic long-term plasticity. Historically, the necessity of using postsynaptic neurons as “reporters” of presynaptic activity has imposed an additional obstacle specific to studying presynaptic forms of long-term plasticity. Going forward, however, technical advances in the ability to image single synapses, vesicles and molecules are removing this obstacle. These technical advances hold great promise to accelerate our understanding of the detailed cellular and molecular mechanisms of presynaptic long-term plasticity. Major questions to resolve in the near future include identifying the specific synaptic processes and molecular events that produce an increase in release probability and discretely manipulating presynaptic long-term plasticity in vivo to demonstrate its behavioral significance in various circuits.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to acknowledge Dr. Enhui Pan and members of the Calakos laboratory for helpful discussions and critical readings of the manuscript. The authors would like to acknowledge funding for work on presynaptic plasticity from the following sources: NINDS (NS053862, NS054840, NS041344) and The Ruth K. Broad Foundation.
1^Note that for CV to be independent of quantal size q, it is required that q remains constant for the repeated measurements needed to calculate CV. Rapid changes in q may come from postsynaptic receptor desensitization or trafficking. Moreover, it has been recently shown that CV is altered by crosslinking AMPA receptors to inhibit lateral diffusion (Heine et al., 2008). The possibility of such postsynaptic processes must be kept in mind in interpreting CV changes.
Acsády, L., Kamondi, A., Sík, A., Freund, T., and Buzsáki, G. (1998). GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J. Neurosci. 18, 3386–3403.
Adermark, Louise, Talani, G., and Lovinger, D. M. (2009). Endocannabinoid-dependent plasticity at GABAergic and glutamatergic synapses in the striatum is regulated by synaptic activity. Eur. J. Neurosci. 29, 32–41. doi: 10.1111/j.1460-9568.2008.06551.x
Alderton, W. K., Cooper, C. E., and Knowles, R. G. (2001). Nitric oxide synthases: structure, function and inhibition. Biochem. J. 357, 593–615. doi: 10.1042/0264-6021:3570593
Alger, B. E. (2002). Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog. Neurobiol. 68, 247–286. doi: 10.1016/S0301-0082(02)00080-1
Araki, R., Sakagami, H., Yanagawa, Y., Hikima, T., Ishizuka, T., and Yawo, H. (2005). Transgenic mouse lines expressing synaptophluorin in hippocampus and cerebellar cortex. Genesis 42, 53–60. doi: 10.1002/gene.20125
Arancio, O., Kiebler, M., Lee, C. J., Lev-Ram, V., Tsien, R. Y., Kandel, E. R., et al. (1996). Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell 87, 1025–1035. doi: 10.1016/S0092-8674(00)81797-3
Augustin, I., Rosenmund, C., Südhof, T. C., and Brose, N. (1999). Munc13-11 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400, 457–461. doi: 10.1038/22768
Azad, S. C., Monory, K., Marsicano, G., Cravatt, B. F., Lutz, B., Zieglgänsberger, W., et al. (2004). Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J. Neurosci. 24, 9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004
Bains, J. S., and Ferguson, A. V. (1997). Nitric oxide regulates NMDA-driven GABAergic inputs to type I neurones of the rat paraventricular nucleus. J. Physiol. 499, 733–746.
Balaji, J., and Ryan, T. A. (2007). Single-vesicle imaging reveals that synaptic vesicle exocytosis and endocytosis are coupled by a single stochastic mode. Proc. Natl. Acad. Sci. U.S.A. 104, 20576–20581. doi: 10.1073/pnas.0707574105
Barnes, S. J., Opitz, T., Merkens, M., Kelly, T., von der Brelie, C., Krueppel, R., et al. (2010). Stable mossy fiber long-term potentiation requires calcium influx at the granule cell soma, protein synthesis, and microtubule-dependent axonal transport. J. Neurosci. 30, 12996–13004. doi: 10.1523/JNEUROSCI.1847-10.2010
Bartos, M., Vida, I., and Jonas, P. (2007). Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 8, 45–56. doi: 10.1038/nrn2044
Basu, J., Betz, A., Brose, N., and Rosenmund, C. (2007). Munc13-11 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J. Neurosci. 27, 1200–1210. doi: 10.1523/JNEUROSCI.4908-06.2007
Basu, J., Shen, N., Dulubova, I., Lu, J., Guan, R., et al. (2005). A minimal domain responsible for Munc13 activity. Nat. Struct. Mol. Biol. 12, 1017–1018. doi: 10.1038/nsmb1001
Bayazitov, I. T., Richardson, R. J., Fricke, R. G., and Zakharenko, S. S. (2007). Slow presynaptic and fast postsynaptic components of compound long-term potentiation. J. Neurosci. 27, 11510–11521. doi: 10.1523/JNEUROSCI.3077-07.2007
Bayazitov, I. T., Voronin, L. L., Kas'yanov, A. M., Kleshchevnikov, A. M., Kul'hitskii, S. V., and Sametskii, E. A. (2002). Long-term potentiation of the AMPA and NMDA components of minimal postsynaptic currents in rat hippocampal field CA1. Neurosci. Behav. Physiol. 32, 533–540. doi: 10.1023/A:1019867808419
Bear, M. F., and Malenka, R. C. (1994). Synaptic plasticity: LTP and LTD. Curr. Opin. Neurobiol. 4, 389–399. doi: 10.1016/0959-4388(94)90101-5
Bekkers, J. M., and Stevens, C. F. (1991). Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc. Natl. Acad. Sci. U.S.A. 88, 7834–7838. doi: 10.1073/pnas.88.17.7834
Bellinger, L. L., and Bernardis, L. L. (2002). The dorsomedial hypothalamic nucleus and its role in ingestive behavior and body weight regulation: lessons learned from lesioning studies. Physiol. Behav. 76, 431–442. doi: 10.1016/S0031-9384(02)00756-4
Bender, K. J., Allen, C. B., Bender, V. A., and Feldman, D. E. (2006). Synaptic basis for whisker deprivation-induced synaptic depression in rat somatosensory cortex. J. Neurosci. 26, 4155–4165. doi: 10.1523/JNEUROSCI.0175-06.2006
Bender, V. A., Pugh, J. R., and Jahr, C. E. (2009). Presynaptically expressed long-term potentiation increases multivesicular release at parallel fiber synapses. J. Neurosci. 29, 10974–10978. doi: 10.1523/JNEUROSCI.2123-09.2009
Betz, A., Thakur, P., Junge, H. J., Ashery, U., Rhee, J. S., Scheuss, V., et al. (2001). Functional interaction of the active zone proteins Munc13-11 and RIM1 in synaptic vesicle priming. Neuron 30, 183–196. doi: 10.1016/S0896-6273(01)00272-0
Betz, W. J., and Bewick, G. S. (1992). Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science 255, 200–203. doi: 10.1126/science.1553547
Blundon, J. A., and Zakharenko, S. S. (2008). Dissecting the components of long-term potentiation. Neuroscientist 14, 598–608. doi: 10.1177/1073858408320643
Bolshakov, V. Y., and Siegelbaum, S. A. (1995). Regulation of hippocampal transmitter release during development and long-term potentiation. Science 269, 1730–1734. doi: 10.1126/science.7569903
Bortolotto, Z. A., Clarke, V. R., Delany, C. M., Parry, M. C., Smolders, I., Vignes, M., et al. (1999). Kainate receptors are involved in synaptic plasticity. Nature 1999, 297–301.
Boyden, E. S., Katoh, A., Pyle, J. L., Chatila, T. A., Tsien, R. W., and Raymond, J. L. (2006). Selective engagement of plasticity mechanisms for motor memory storage. Neuron 51, 823–834. doi: 10.1016/j.neuron.2006.08.026
Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G., and Deisseroth, K. (2005). Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268. doi: 10.1038/nn1525
Breustedt, J., Gundlfinger, A., Varoqueaux, F., Reim, K., Brose, N., and Schmitz, D. (2010). Munc13-12 differentially affects hippocampal synaptic transmission and plasticity. Cereb. Cortex 20, 1109–1120. doi: 10.1093/cercor/bhp170
Breustedt, J., Vogt, K. E., Miller, R. J., Nicoll, R. A., and Schmitz, D. (2003). Alpha1E-containing Ca(2+) channels are involved in synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 100, 12450–12455. doi: 10.1073/pnas.2035117100
Calakos, N., Schoch, S., Sudhof, T., and Malenka, R. (2004). Multiple roles for the active zone protein RIM1alpha in late stages of neurotransmitter release. Neuron 42, 889–896. doi: 10.1016/j.neuron.2004.05.014
Castillo, P. E. (2012). Presynaptic LTP and LTD of excitatory and inhibitory synapses. Cold Spring Harb. Perspect. Biol. 4, pii: a005728. doi: 10.1101/cshperspect.a005728
Castillo, P. E., Chiu, C. Q., and Carroll, R. C. (2011). Long-term plasticity at inhibitory synapses. Curr. Opin. Neurobiol. 21, 328–338. doi: 10.1016/j.conb.2011.01.006
Castillo, P. E., Janz, R., Sudhof, T. C., Tzounopoulos, T., Malenka, R., and Nicoll, R. (1997). Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature 388, 590–593. doi: 10.1038/41574
Castillo, P. E., Schoch, S., Schmitz, F., Sudhof, T., and Malenka, R. (2002). RIM1alpha is required for presynaptic long-term potentiation. Nature 415, 327–330. doi: 10.1038/415327a
Castillo, P. E., Weisskopf, M. G., and Nicoll, R. A. (1994). The role of Ca(2+) channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron 12, 261–269. doi: 10.1016/0896-6273(94)90269-0
Castro-Alamancos, M. A., and Calcagnotto, M. E. (1999). Presynaptic long-term potentiation in corticothalamic synapses. J. Neurosci. 19, 9090–9097.
Catterall, W. A., and Few, A. P. (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901. doi: 10.1016/j.neuron.2008.09.005
Chevaleyre, V., and Castillo, P. E. (2003). Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38, 461–472. doi: 10.1016/S0896-6273(03)00235-6
Chevaleyre, V., Heifets, B. D., Kaeser, P. S., Südhof, T. C., Purpura, D. P., and Castillo, P. E. (2007). Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron 54, 801–812. doi: 10.1016/j.neuron.2007.05.020
Chevaleyre, V., Takahashi, K. A., and Castillo, P. E. (2006). Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 29, 37–46. doi: 10.1146/annurev.neuro.29.051605.112834
Childers, S. R., and Deadwyler, S. A. (1996). Role of cyclic AMP in the actions of cannabinoid receptors. Biochem. Pharmacol. 52, 819–827. doi: 10.1016/0006-2952(96)00419-4
Chiu, C. Q., Puente, N., Grandes, P., and Castillo, P. E. (2010). Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. J. Neurosci. 30, 7236–7248. doi: 10.1523/JNEUROSCI.0736-10.2010
Choi, S., and Lovinger, D. M. (1997a). Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc. Natl. Acad. Sci. U.S.A. 94, 2665–2670. doi: 10.1073/pnas.94.6.2665
Choi, S., and Lovinger, D. M. (1997b). Decreased frequency but not amplitude of quantal synaptic responses associated with expression of corticostriatal long-term depression. J. Neurosci. 17, 8613–8620.
Coppola, T., Magnin-Luthi, S., Perret-Menoud, V., Gattesco, S., Schiavo, G., and Regazzi, R. (2001). Direct interaction of the Rab3 effector RIM with Ca(2+) channels, SNAP-25, and synaptotagmin. J. Biol. Chem. 276, 32756–32762. doi: 10.1074/jbc.M100929200
Crosby, K. M., Inoue, W., Pittman, Q. J., and Bains, J. S. (2011). Endocannabinoids gate state-dependent plasticity of synaptic inhibition in feeding circuits. Neuron 71, 529–541. doi: 10.1016/j.neuron.2011.06.006
Crozier, R. A., Wang, Y., Liu, C., and Bear, M. F. (2007). Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proc. Natl. Acad. Sci. U.S.A. 104, 1383–1388. doi: 10.1073/pnas.0609596104
Debanne, D., Guérineau, N. C., Gähwiler, B. H., and Thompson, S. M. (1996). Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J. Physiol. 491, 163–176.
del Castillo, J., and Katz, B. (1954). Quantal components of the end-plate potential. J. Physiol. 124, 560–573.
Deng, L., Kaeser, P. S., Xu, W., and Südhof, T. C. (2011). RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 69, 317–331. doi: 10.1016/j.neuron.2011.01.005
Dietrich, D., Kirschstein, T., Kukley, M., Pereverzev, A., von der Brelie, C., Schneider, T., et al. (2003). Functional specialization of presynaptic Cav2.3 Ca(2+) channels. Neuron 39, 483–496. doi: 10.1016/S0896-6273(03)00430-6
Dobrunz, L. E., and Stevens, C. F. (1997). Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 18, 995–1008. doi: 10.1016/S0896-6273(00)80338-4
Dodge, F. A., and Rahamimoff, R. (1967). Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J. Physiol. 193, 419–432.
Domenici, M. R., Berretta, N., and Cherubini, E. (1998). Two Distinct Forms of Long-Term Depression Coexist at the Mossy Fiber-CA3 Synapse in the Hippocampus During Development. Proc. Natl. Acad. Sci. U.S.A. 95, 8310–8315. doi: 10.1073/pnas.95.14.8310
Emptage, N. J., Bliss, T. V., and Fine, A. (1999). Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22, 115–124. doi: 10.1016/S0896-6273(00)80683-2
Emptage, N. J., Reid, C. A., Fine, A., and Bliss, T. (2003). Optical quantal analysis reveals a presynaptic component of LTP at hippocampal schaffer-associational synapses. Neuron 38, 797–804. doi: 10.1016/S0896-6273(03)00325-8
Enoki, R., Hu, Y.-L., Hamilton, D., and Fine, A. (2009). Expression of long-term plasticity at individual synapses in hippocampus is graded, bidirectional, and mainly presynaptic: optical quantal analysis. Neuron 62, 242–253. doi: 10.1016/j.neuron.2009.02.026
Faas, G. C., Adwanikar, H., Gereau, R. W., and Saggau, P. (2002). Modulation of presynaptic calcium transients by metabotropic glutamate receptor activation: a differential role in acute depression of synaptic transmission and long-term depression. J. Neurosci. 22, 6885–6890.
Fatt, P., and Katz, B. (1952). Spontaneous subthreshold activity at motor nerve endings. J. Physiol. 117, 109–128.
Fischer von Mollard, G., Südhof, T. C., and Jahn, R. (1991). A small GTP-binding protein dissociates from synaptic vesicles during exocytosis. Nature 349, 79–81. doi: 10.1038/349079a0
Fitzjohn, S. M., Palmer, M. J., May, J. E., Neeson, A., Morris, S. A., and Collingridge, G. L. (2001). A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J. Physiol. 537, 421–430. doi: 10.1111/j.1469-7793.2001.00421.x
Fourcaudot, E., Gambino, F., Humeau, Y., Casassus, G., Shaban, H., Poulain, B., et al. (2008). cAMP/PKA signaling and RIM1alpha mediate presynaptic LTP in the lateral amygdala. Proc. Natl. Acad. Sci. U.S.A. 105, 15130–15135. doi: 10.1073/pnas.0806938105
Freund, T. F., Katona, I., and Piomelli, D. (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 83, 1017–1066.
Fries, P., Nikolić, D., and Singer, W. (2007). The gamma cycle. Trends Neurosci. 30, 309–316. doi: 10.1016/j.tins.2007.05.005
Frischknecht, R., Heine, M., Perrais, D., Seidenbecher, C. I., Choquet, D., and Gundelfinger, E. D. (2009). Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat. Neurosci. 12, 897–904. doi: 10.1038/nn.2338
Fusi, S., Drew, P. J., and Abbott, L. F. (2005). Cascade models of synaptically stored memories. Neuron 45, 599–611. doi: 10.1016/j.neuron.2005.02.001
Gage, A. T., Reyes, M., and Stanton, P. K. (1997). Nitric-oxide-guanylyl-cyclase-dependent and -independent components of multiple forms of long-term synaptic depression. Hippocampus 7, 286–295. doi: 10.1002/(SICI)1098-1063(1997)7:3<286::AID-HIPO4>3.0.CO;2-J
García-Junco-Clemente, P., Linares-Clemente, P., and Fernández-Chacón, R. (2005). Active zones for presynaptic plasticity in the brain. Mol. Psychiatry 10, 185–200. doi: 10.1038/sj.mp.4001628
Garthwaite, J. (2008). Concepts of neural nitric oxide-mediated transmission. Eur. J. Neurosci. 27, 2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x
Geppert, M., Goda, Y., Stevens, C., and Sudhof, T. C. (1997). The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature 387, 810–814. doi: 10.1038/42954
Gerdeman, G. L., and Lovinger, D. M. (2003). Emerging roles for endocannabinoids in long-term synaptic plasticity. Br. J. Pharmacol. 140, 781–789. doi: 10.1038/sj.bjp.0705466
Gerdeman, G. L., Ronesi, J., and Lovinger, D. M. (2002). Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat. Neurosci. 5, 446–451. doi: 10.1038/nn832
Glitsch, M., and Marty, A. (1999). Presynaptic effects of NMDA in cerebellar purkinje cells and interneurons. J. Neurosci. 19, 511–519.
Graf, E. R., Daniels, R. W., Burgess, R. W., Schwarz, T. L., and DiAntonio, A. (2009). Rab3 dynamically controls protein composition at active zones. Neuron 64, 663–677. doi: 10.1016/j.neuron.2009.11.002
Granseth, B., Odermatt, B., Royle, S. J., and Lagnado, L. (2006). Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron 51, 773–786. doi: 10.1016/j.neuron.2006.08.029
Guillery, R. W., and Sherman, S. W. (2002). Thalamic relay functions and their role in corticocortical communication: generalizations from the visual system. Neuron 33, 163–175. doi: 10.1016/S0896-6273(01)00582-7
Haj-Dahmane, S., and Shen, R.-Y. (2010). Regulation of plasticity of glutamate synapses by endocannabinoids and the cyclic-AMP/protein kinase a pathway in midbrain dopamine neurons. J. Physiol. 588, 2589–2604. doi: 10.1113/jphysiol.2010.190066
Han, Y., Kaeser, P. S., Südhof, T. C., and Schneggenburger, R. (2011). RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron 69, 304–316. doi: 10.1016/j.neuron.2010.12.014
Harata, N., Ryan, T. A., Smith, S. J., Buchanan, J., and Tsien, R. W. (2001). Visualizing recycling synaptic vesicles in hippocampal neurons by FM 1-43 photoconversion. Proc. Natl. Acad. Sci. U.S.A. 98, 12748–12753. doi: 10.1073/pnas.171442798
Harris, E. W., and Cotman, C. W. (1986). Long-Term Potentiation of Guinea Pig Mossy Fiber Responses Is Not Blocked by N-Methyl D-Aspartate Antagonists. Neurosci. Lett. 70, 132–137. doi: 10.1016/0304-3940(86)90451-9
Häusser, M., and Clark, B. A. (1997). Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron 19, 665–678. doi: 10.1016/S0896-6273(00)80379-7
Heifets, B. D., and Castillo, P. E. (2009). Endocannabinoid Signaling and Long-Term Synaptic Plasticity. Annu. Rev. Physiol. 71, 283–306. doi: 10.1146/annurev.physiol.010908.163149
Heifets, B. D., Chevaleyre, V., and Castillo, P. E. (2008). Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proc. Natl. Acad. Sci. U.S.A. 105, 10250–10255. doi: 10.1073/pnas.0711880105
Heine, M., Groc, L., Frischknecht, R., Béïque, J., Lounis, B., Rumbaugh, G. et al., and Choquet, D. (2008). Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science 320, 201–205. doi: 10.1126/science.1152089
Henneberger, C., Redman, S. J., and Grantyn, R. (2007). Cortical efferent control of subcortical sensory neurons by synaptic disinhibition. Cereb. Cortex 17, 2039–2049. doi: 10.1093/cercor/bhl112
Hessler, N. A., Shirke, A. M., and Malinow, R. (1993). The probability of transmitter release at a mammalian central synapse. Nature 366, 569–572. doi: 10.1038/366569a0
Hirata, K., Sawada, S., and Yamamoto, C. (1991). Enhancement of transmitter release accompanying with long-term potentiation in synapses between mossy fibers and CA3 neurons in hippocampus. Neurosci. Lett. 123, 73–76. doi: 10.1016/0304-3940(91)90161-L
Hosaka, M., Hammer, R. E., and Südhof, T. C. (1999). A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron 24, 377–387. doi: 10.1016/S0896-6273(00)80851-X
Huang, C. C., Chen, Y. L., Liang, Y. C., and Hsu, K. S. (2002). Role for cAMP and protein phosphatase in the presynaptic expression of mouse hippocampal mossy fibre depotentiation. J. Physiol. 543, 767–778. doi: 10.1113/jphysiol.2002.025668
Huang, C. C., Lo, S. W., and Hsu, K. S. (2001). Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J. Physiol. 532, 731–748. doi: 10.1111/j.1469-7793.2001.0731e.x
Huang, L., Killbride, J., Rowan, M. J., and Anwyl, R. (1999). Activation of mGluRII induces LTD via activation of protein kinase a and protein Kinase C in the Dentate gyrus of the hippocampus in vitro. Neuropharmacology 38, 73–83. doi: 10.1016/S0028-3908(98)00168-3
Huang, Y., Yasuda, H., Sarihi, A., and Tsumoto, T. (2008a). Roles of endocannabinoids in heterosynaptic long-term depression of excitatory synaptic transmission in visual cortex of young mice. J. Neurosci. 28, 7074–7083. doi: 10.1523/JNEUROSCI.0899-08.2008
Huang, Y., Zakharenko, S., Schoch, S., Kaeser, P. S., Janz, R., Südhof, T. C., et al. (2005). Genetic evidence for a protein-kinase-a-mediated presynaptic component in NMDA-receptor-dependent forms of long-term synaptic potentiation. Proc. Natl. Acad. Sci. U.S.A. 102, 9365–9370. doi: 10.1073/pnas.0503777102
Huang, Y.-C., Wang, S.-J., Chiou, L.-C., and Gean, P.-W. (2003). Mediation of amphetamine-induced long-term depression of synaptic transmission by CB1 cannabinoid receptors in the rat amygdala. J. Neurosci. 23, 10311–10320.
Huang, Y.-Y., and Kandel, E. R. (1998). Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron 21, 169–178. doi: 10.1016/S0896-6273(00)80524-3
Huang, Y.-Y., Kandel, E. R., Varshavsky, L., Brandon, E., Qi, M., Idzerda, R., et al. (1995). A Genetic test of the effects of mutations in PKA on mossy fiber LTP and its relation to spatial and contextual learning. Cell 83, 1211–1222. doi: 10.1016/0092-8674(95)90146-9
Huang, Y.-Y., Li, X., and Kandel, E. R. (1994). cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell 79, 69–79. doi: 10.1016/0092-8674(94)90401-4
Huang, Y. Z., Pan, E., Xiong, Z.-Q., and McNamara, J. O. (2008b). Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy Fiber-CA3 pyramid synapse. Neuron 57, 546–558. doi: 10.1016/j.neuron.2007.11.026
Humeau, Y., Herry, C., Kemp, N., Shaban, H., Fourcaudot, E., Bissière, S., et al. (2005). Dendritic spine heterogeneity determines afferent-specific hebbian plasticity in the amygdala. Neuron 45, 119–131. doi: 10.1016/j.neuron.2004.12.019
Humeau, Y., Shaban, H., Bissière, S., and Lüthi, A. (2003). Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature 426, 841–845. doi: 10.1038/nature02194
Humphries, M. D., Wood, R., and Gurney, K. (2009). Dopamine-modulated dynamic cell assemblies generated by the GABAergic striatal microcircuit. Neural Netw. 22, 1174–1188. doi: 10.1016/j.neunet.2009.07.018
Inchauspe, C. G., Martini, F. J., Forsythe, I. D., and Uchitel, O. D. (2004). Functional compensation of P/Q by N-type channels blocks Short-term plasticity at the calyx of held presynaptic terminal. J. Neurosci. 24, 10379–10383. doi: 10.1523/JNEUROSCI.2104-04.2004
Isaac, J. T., Nicoll, R. A., and Malenka, R. C. (1995). Evidence for silent synapses: implications for the expression of LTP. Neuron 15, 427–434. doi: 10.1016/0896-6273(95)90046-2
Isaac, J. T., Oliet, S. H., Hjelmstad, G. O., Nicoll, R. A., and Malenka, R. C. (1996). Expression mechanisms of long-term potentiation in the hippocampus. J. Physiol. Paris 90, 299–303. doi: 10.1016/S0928-4257(97)87901-6
Jaffe, D., and Johnston, D. (1990). Induction of long-term potentiation at hippocampal mossy-fiber synapses follows a Hebbian rule. J. Neurophysiol. 64, 948–960.
Junge, H., Rhee, J., Jahn, O., Varoqueaux, F., Spiess, J., Waxham, M., et al. (2004). Calmodulin and Munc13 form a Ca(2+) sensor/effector complex that controls short-term synaptic plasticity. Cell 118, 389–401. doi: 10.1016/j.cell.2004.06.029
Kaeser, P. S., Deng, L., Wang, Y., Dulubova, I., Liu, X., Rizo, J., et al. (2011). RIM proteins tether Ca(2+) channels to presynaptic active zones via a direct PDZ-domain interaction. Cell, 282–295. doi: 10.1016/j.cell.2010.12.029
Kaeser, P. S., Kwon, H.-B., Blundell, J., Chevaleyre, V., Morishita, W., Malenka, R. C., et al. (2008b). RIM1alpha phosphorylation at Serine-413 by protein Kinase a is not required for presynaptic long-term plasticity or learning. Proc. Natl. Acad. Sci. U.S.A. 105, 14680–14685. doi: 10.1073/pnas.0806679105
Kaeser, P. S., Kwon, H.-B., Chiu, C. Q., Deng, L., Castillo, P. E., and Südhof, T. C. (2008a). RIM1alpha and RIM1beta are synthesized from distinct promoters of the RIM1 gene to mediate differential but overlapping synaptic functions. J. Neurosci. 28, 13435–13447. doi: 10.1523/JNEUROSCI.3235-08.2008
Kamiya, H., Umeda, K., Ozawa, S., and Manabe, T. (2002). Presynaptic Ca(2+) entry is unchanged during hippocampal mossy fiber long-term potentiation. J. Neurosci. 22, 10524–10528.
Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. doi: 10.1126/science.1067020
Kano, M., Ohno-Shosaku, T., Hashimotodani, Y., Uchigashima, M., and Watanabe, M. (2009). Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 89, 309–380. doi: 10.1152/physrev.00019.2008
Kapur, A., Yeckel, M. F., Gray, R., and Johnston, D. (1998). L-type calcium channels are required for one form of hippocampal mossy fiber LTP. J. Neurophysiol. 79, 2181–2190.
Katz, B. (1971). Quantal mechanism of neural transmitter release. Science 173, 123–126. doi: 10.1126/science.173.3992.123
Kiyonaka, S., Wakamori, M., Miki, T., Uriu, Y., Nonaka, M., Bito, H., et al. (2007). RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca(2+) channels. Nat. Neurosci. 10, 691–791. doi: 10.1038/nn1904
Ko, G. Y., and Kelly, P. T. (1999). Nitric oxide acts as a postsynaptic signaling molecule in calcium/calmodulin-induced synaptic potentiation in hippocampal CA1 pyramidal neurons. J. Neurosci. 19, 6784–6794.
Kobayashi, K., Manabe, T., and Takahashi, T. (1996). Presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. Science 273, 648–650. doi: 10.1126/science.273.5275.648
Kobayashi, K., Manabe, T., and Takahashi, T. (1999). Calcium-dependent mechanisms involved in presynaptic long-term depression at the hippocampal mossy fibre-CA3 synapse. Eur. J. Neurosci. 11, 1633–1638. doi: 10.1046/j.1460-9568.1999.00578.x
Korn, H., and Faber, D. S. (1991). Quantal analysis and synaptic efficacy in the CNS. Trends Neurosci. 14, 439–445. doi: 10.1016/0166-2236(91)90042-S
Korn, H., and Faber, D. S. (1998). Quantal analysis and long-term potentiation. Comptes Rendus De l'Académie Des Sciences Série III, Sciences De La Vie 321, 125–130. doi: 10.1016/S0764-4469(97)89811-3
Kreitzer, A. C., and Malenka, R. C. (2005). Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J. Neurosci. 25, 10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005
Kullmann, D. M., and Lamsa, K. (2008). Roles of distinct glutamate receptors in induction of anti-hebbian long-term potentiation. J. Physiol. 586, 1481–1486. doi: 10.1113/jphysiol.2007.148064
Kullmann, D. M., and Siegelbaum, S. A. (1995). The site of expression of NMDA receptor-dependent LTP: new fuel for an old fire. Neuron 15, 997–1002. doi: 10.1016/0896-6273(95)90089-6
Kuno, M. (1964). Quantal components of excitatory synaptic potentials in spinal motoneurones. J. Physiol. 175, 81–99.
Kuzmiski, B. J., Pittman, Q. J., and Bains, J. S. (2009). Metaplasticity of hypothalamic synapses following in vivo challenge. Neuron 62, 839–849. doi: 10.1016/j.neuron.2009.05.027
Kwon, H.-B., and Castillo, P. E. (2008). Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron 57, 108–120. doi: 10.1016/j.neuron.2007.11.024
Lachamp, P. M., Liu, Y., and Liu, S. J. (2009). Glutamatergic modulation of cerebellar interneuron activity is mediated by an enhancement of GABA release and requires protein Kinase a/RIM1alpha signaling. J. Neurosci. 29, 381–392. doi: 10.1523/JNEUROSCI.2354-08.2009
Laezza, F., Doherty, J. J., and Dingledine, R. (1999). Long-term depression in hippocampal interneurons: joint requirement for pre- and postsynaptic events. Science 285, 1411–1414. doi: 10.1126/science.285.5432.1411
Lafourcade, M., Elezgarai, I., Mato, S., Bakiri, Y., Grandes, P., and Manzoni, O. J. (2007). Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS ONE 2: e709. doi: 10.1371/journal.pone.0000709
Langdon, R., Johnson, J., and Barrionuevo, G. (1995). Posttetanic potentiation and presynaptically induced long-term potentiation at the mossy fiber synapse in rat hippocampus. J. Neurobiol. 26, 370–385. doi: 10.1002/neu.480260309
Lauri, S. E., Bortolotto, Z. A., Nistico, R., Bleakman, D., Ornstein, P. L., Lodge, D., et al. (2003). A role for Ca2+ stores in kainate receptor-dependent synaptic facilitation and LTP at mossy fiber synaspes in the hippocampus. Neuron 39, 327–341. doi: 10.1016/S0896-6273(03)00369-6
Lauri, S. E., Palmer, M., Segerstrale, M., Vesikansa, A., Taira, T., and Collingridge, G. L. (2007). Presynaptic mechanisms involved in the expression of STP and LTP at CA1 synapses in the hippocampus. Neuropharmacology 52, 1–11. doi: 10.1016/j.neuropharm.2006.06.017
Ledo, A., Barbosa, R. M., Gerhardt, G. A., Cadenas, E., and Laranjinha, J. (2005). Concentration dynamics of nitric oxide in rat hippocampal subregions evoked by stimulation of the NMDA glutamate receptor. Proc. Natl Acad. Sci. U.S.A. 102, 17483–17488. doi: 10.1073/pnas.0503624102
LeDoux, J. E. (2000). Emotion circuits in the brain. Annu. Rev. Neurosci. 23, 155–184. doi: 10.1146/annurev.neuro.23.1.155
Lei, S., and McBain, C. J. (2004). Two loci of expression for long-term depression at hippocampal mossy fiber-interneuron synapses. J. Neurosci. 24, 2112–2121. doi: 10.1523/JNEUROSCI.4645-03.2004
Lerner, T. N., Horne, E. A., Stella, N., and Kreitzer, A. C. (2010). Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. J. Neurosci. 30, 2160–2164. doi: 10.1523/JNEUROSCI.5844-09.2010
Lev-Ram, V., Jiang, T., Wood, J., Lawrence, D. S., and Tsien, R. Y. (1997). Synergies and coincidence requirements between NO, cGMP, and Ca2+ in the induction of cerebellar long-term depression. Neuron 18, 1025–1038. doi: 10.1016/S0896-6273(00)80340-2
Li, L., Bender, K. J., Drew, P. J., Jadhav, S. P., Sylwestrak, E., and Feldman, D. E. (2009). Endocannabinoid signaling is required for development and critical period plasticity of the Whisker Map in somatosensory cortex. Neuron 64, 537–549. doi: 10.1016/j.neuron.2009.10.005
Li, S., Anwyl, R., and Rowan, M. J. (2000). A persistent reduction in short-term facilitation accompanies long-term potentiation in the CA1 area in the intact hippocampus. Neuroscience 100, 213–220. doi: 10.1016/S0306-4522(00)00267-0
Li, Y., Hough, C. J., Frederickson, C. J., and Sarvey, J. M. (2001). Induction of mossy fiber –> Ca3 long-term potentiation requires translocation of synaptically released Zn(2+). J. Neurosci. 21, 8015–8025.
Li, Z., Burrone, J., Tyler, W. J., Hartman, K. N., Albeanu, D. F., and Murthy, V. N. (2005). Synaptic vesicle recycling studied in transgenic mice expressing synaptopHluorin. Proc. Natl. Acad. Sci. U.S.A. 102, 6131–6136. doi: 10.1073/pnas.0501145102
Liao, D., Hessler, N. A., and Malinow, R. (1995). Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 375, 400–404. doi: 10.1038/375400a0
Liu, S. J., and Lachamp, P. M. (2006). The activation of excitatory glutamate receptors evokes a long-lasting increase in the release of GABA from cerebellar stellate cells. J. Neurosci. 26, 9332–9339. doi: 10.1523/JNEUROSCI.2929-06.2006
Lonart, G., Janz, R., Johnson, K., and Südhof, T. C. (1998). Mechanism of action of rab3A in mossy fiber LTP. Neuron 21, 1141–1150. doi: 10.1016/S0896-6273(00)80631-5
Lonart, G., Schoch, S., Kaeser, P. S., Larkin, C., Südhof, T. C., and Linden, D. (2003). Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115, 49–60. doi: 10.1016/S0092-8674(03)00727-X
Lonart, G., and Südhof, T. C. (1998). Region-specific phosphorylation of rabphilin in mossy fiber nerve terminals of the hippocampus. J. Neurosci. 18, 634–640.
Lovinger, D. M. (2008). Presynaptic modulation by endocannabinoids. Handb. Exp. Pharmacol. 184, 435–477. doi: 10.1007/978-3-540-74805-2_14
Lovinger, D. M. (2010). Neurotransmitter roles in synaptic modulation, plasticity and learning in the Dorsal striatum. Neuropharmacology 58, 951–961. doi: 10.1016/j.neuropharm.2010.01.008
Lu, Y. M., Taverna, F. A., Tu, R., Ackerley, C. A., Wang, Y. T., and Roder, J. (2000). Endogenous Zn(2+) is required for the induction of long-term potentiation at rat hippocampal mossy fiber-CA3 synapses. Synapse 38, 187–197. doi: 10.1002/1098-2396(200011)38:2<187::AID-SYN10>3.0.CO;2-R
Maccaferri, G., Tóth, K., and McBain, C. J. (1998). Target-specific expression of presynaptic mossy fiber plasticity. Science 279, 1368–1370. doi: 10.1126/science.279.5355.1368
Maejima, T., Hashimoto, K., Yoshida, T., Aiba, A., and Kano, M. (2001). Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31, 463–475. doi: 10.1016/S0896-6273(01)00375-0
Maeno-Hikichi, Y., Polo-Parada, L., Kastanenka, K. V., and Landmesser, L. T. (2011). Frequency-dependent modes of synaptic vesicle endocytosis and exocytosis at adult mouse neuromuscular junctions. J. Neurosci. 31, 1093–1105. doi: 10.1523/JNEUROSCI.2800-10.2011
Malenka, R. C., and Nicoll, R. A. (1999). Long-term potentiation–a decade of progress? Science 285, 1870–1874. doi: 10.1126/science.285.5435.1870
Malinow, R. (1991). Transmission between pairs of hippocampal slice neurons: quantal levels, oscillations, and LTP. Science 252, 722–724. doi: 10.1126/science.1850871
Malinow, R. (1994). LTP: desperately seeking resolution. Science 266, 1195–1196. doi: 10.1126/science.7973700
Malinow, R., Mainen, Z. F., and Hayashi, Y. (2000). LTP mechanisms: from silence to four-lane traffic. Curr. Opin. Neurobiol. 10, 352–357. doi: 10.1016/S0959-4388(00)00099-4
Malinow, R., and Tsien, R. W. (1990). Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature 346, 177–180. doi: 10.1038/346177a0
Manabe, T. (1997). Two forms of hippocampal long-term depression, the counterpart of long-term potentiation. Rev. Neurosci. 8, 179–193. doi: 10.1515/REVNEURO.1997.8.3-4.179
Mann-Metzer, P., and Yarom, Y. (1999). Electrotonic coupling interacts with intrinsic properties to generate synchronized activity in cerebellar networks of inhibitory interneurons. J. Neurosci. 19, 3298–3306.
Marin, P., Lafon-Cazal, M., and Bockaert, J. (1992). A nitric oxide synthase activity selectively stimulated by NMDA receptors depends on protein Kinase C activation in mouse striatal neurons. Eur. J. Neurosci. 4, 425–432. doi: 10.1111/j.1460-9568.1992.tb00892.x
Marsicano, G., Wotjak, C. T., Azad, S. C., Bisogno, T., Rammes, G., Cascio, M. G., et al. (2002). The endogenous cannabinoid system controls extinction of aversive memories. Nature 418, 530–534. doi: 10.1038/nature00839
Marvin, J. S., Borqhuis, B. G., Tian, L., Cichon, J., Harnett, M. T., Akerboom, J., et al. (2013). An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 10, 162–170. doi: 10.1038/nmeth.2333
Matias, C. M., Matos, N. C., Arif, M., Dionisio, J. C., and Quinta-Ferreira, E. M. (2006). Effect of the zinc chelator N,N,N',N'-Tetrakis (2-Pyridylmethyl)Ethylenediamine (TPEN) on hippocampal mossy fiber calcium signals and on synaptic transmission. Biol. Res. 39, 521–530. doi: 10.4067/S0716-97602006000300013
Mato, S., Lafourcade, M., Robbe, D., Bakiri, Y., and Manzoni, O. J. (2008). Role of the cyclic-AMP/PKA cascade and of P/Q-Type Ca(2+) channels in endocannabinoid-mediated long-term depression in the nucleus accumbens. Neuropharmacology 54, 87–94. doi: 10.1016/j.neuropharm.2007.04.014
Mato, S., Robbe, D., Puente, N., Grandes, P., and Manzoni, O. J. (2005). Presynaptic Homeostatic plasticity rescues long-term depression after chronic delta 9-Tetrahydrocannabinol exposure. J. Neurosci. 25, 11619–11627. doi: 10.1523/JNEUROSCI.2294-05.2005
Matz, J., Gilyan, A., Kolar, A., McCarvill, T., and Krueger, S. R. (2010). Rapid structural alterations of the active zone lead to sustained changes in neurotransmitter release. Proc. Natl. Acad. Sci. U.S.A. 107, 8836–8841. doi: 10.1073/pnas.0906087107
McBain, C. J., and Kauer, J. A. (2009). Presynaptic plasticity: targeted control of inhibitory networks. Curr. Opin. Neurobiol. 19, 254–262. doi: 10.1016/j.conb.2009.05.008
Mellor, J., and Nicoll, R. A. (2001). Hippocampal mossy fiber LTP is independent of postsynaptic calcium. Nat. Neurosci. 4, 125–126. doi: 10.1038/83941
Miesenböck, G., De Angelis, D. A., and Rothman, J. E. (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195. doi: 10.1038/28190
Nakazawa, K., Zsiros, V., Jiang, Z., Nakao, K., Kolata, S., Zhang, S., et al. (2012). GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 62, 1574–1583. doi: 10.1016/j.neuropharm.2011.01.022
Neher, E., and Sakaba, T. (2008). Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59, 861–872. doi: 10.1016/j.neuron.2008.08.019
Nicholls, R., Zhang, X., Bailey, C., Conklin, B., Kandel, E., and Stanton, P. (2006). mGluR2 acts through inhibitory galpha subunits to regulate transmission and long-term plasticity at hippocampal mossy fiber-CA3 synapses. Proc. Natl. Acad. Sci. U.S.A. 103, 6380–6385. doi: 10.1073/pnas.0601267103
Nicoll, R. A. (2003). Expression mechanisms underlying long-term potentiation: a postsynaptic view. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 721–726. doi: 10.1098/rstb.2002.1228
Nicoll, R. A., and Malenka, R. C. (1999a). Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann. N.Y. Acad. Sci. 868, 515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x
Nicoll, R. A., and Malenka, R. C. (1999b). Leaky synapses. Neuron 23, 197–198. doi: 10.1016/S0896-6273(00)80769-2
Nicoll, R. A., and Schmitz, D. (2005). Synaptic plasticity at hippocampal mossy fibre synapses. Nat. Rev. Neurosci. 6, 863–876. doi: 10.1038/nrn1786
Nosyreva, E. D., and Huber, K. M. (2005). Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 25, 2992–3001. doi: 10.1523/JNEUROSCI.3652-04.2005
Nugent, F. S., Penick, E. C., and Kauer, J. A. (2007). Opioids block long-term potentiation of inhibitory synapses. Nature 446, 1086–1090. doi: 10.1038/nature05726
Oertner, T. G., Sabatini, B. L., Nimchinsky, E. A., and Svoboda, K. (2002). Facilitation at single synapses probed with optical quantal analysis. Nat. Neurosci. 5, 657–664. doi: 10.1038/nn867
Oliet, S. H., Malenka, R. C., and Nicoll, R. A. (1996). Bidirectional control of quantal size by synaptic activity in the hippocampus. Science 271, 1294–1297. doi: 10.1126/science.271.5253.1294
Oliet, S. H., Malenka, R. C., and Nicoll, R. A. (1997). Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18, 969–982. doi: 10.1016/S0896-6273(00)80336-0
Opazo, P., Labrecque, S., Tigaret, C. M., Frouin, A., Wiseman, P. W., Koninck, P. D., et al. (2010). CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 67, 239–252. doi: 10.1016/j.neuron.2010.06.007
Palmer, M. J., Isaac, J. T., and Collingridge, G. L. (2004). Multiple, developmentally regulated expression mechanisms of long-term potentiation at CA1 synapses. J. Neurosci. 24, 4903–4911. doi: 10.1523/JNEUROSCI.0170-04.2004
Pan, B., Hillard, C. J., and Liu, Q. (2008). Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J. Neurosci. 28, 1385–1397. doi: 10.1523/JNEUROSCI.4033-07.2008
Pan, E., Zhang, X., Huang, Z., Krezel, A., Zhao, M., Tinberg, C. E., et al. (2011). Vesicular zinc promotes presynaptic and inhibits postsynaptic long-term potentiation of mossy fiber-CA3 synapse. Neuron 71, 1116–1126. doi: 10.1016/j.neuron.2011.07.019
Park, H., Li, Y., and Tsien, R. W. (2012). Influence of synaptic vesicle position on release probability and exocytotic fusion mode. Science 335, 1362–1366. doi: 10.1126/science.1216937
Park, J. H., Straub, V. A., and O'Shea, M. (1998). Anterograde signaling by nitric oxide: characterization and in vitro reconstitution of an identified nitrergic synapse. J. Neurosci. 18, 5463–5476.
Pelkey, K. A., Lavezzari, G., Racca, C., Roche, K. W., and McBain, C. J. (2005). mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron 46, 89–102. doi: 10.1016/j.neuron.2005.02.011
Pelkey, K. A., and McBain, C. J. (2008). Target-cell-dependent plasticity within the mossy fibre-CA3 circuit reveals compartmentalized regulation of presynaptic function at divergent release sites. J. Physiol. 586, 1495–1502. doi: 10.1113/jphysiol.2007.148635
Pelkey, K. A., Topolnik, L., Lacaille, J.-C., and McBain, C. J. (2006). Compartmentalized Ca(2+) channel regulation at divergent mossy-fiber release sites underlies target cell-dependent plasticity. Neuron 52, 497–510. doi: 10.1016/j.neuron.2006.08.032
Pelkey, K. A., Topolnik, L., Yuan, X.-Q., Lacaille, J.-C., and McBain, C. J. (2008). State-dependent cAMP sensitivity of presynaptic function underlies metaplasticity in a hippocampal feedforward inhibitory circuit. Neuron 60, 980–987. doi: 10.1016/j.neuron.2008.11.018
Pin, J. P., and Duvoisin, R. (1995). The Metabotropic Glutamate Receptors: Structure and Functions. Neuropharmacology 34, 1–26. doi: 10.1016/0028-3908(94)00129-G
Pinheiro, P. S., Perrais, D., Coussen, F., Barhanin, J., Bettler, B., Mann, J. R., et al. (2007). GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc. Natl. Acad. Sci. U.S.A. 104, 12181–12186. doi: 10.1073/pnas.0608891104
Rebola, N., Carta, M., Lanore, F., Blanchet, C., and Mulle, C. (2011). NMDA receptor-dependent metaplasticity at hippocampal mossy fiber synapses. Nat. Neurosci. 14, 691–693. doi: 10.1038/nn.2809
Rebola, N., Lujan, R., Cunha, R. A., and Mulle, C. (2008). Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 57, 121–134. doi: 10.1016/j.neuron.2007.11.023
Redman, S. (1990). Quantal analysis of synaptic potentials in neurons of the central nervous system. Physiol. Rev. 170, 165–198.
Regehr, W., and Tank, D. (1991). The maintenance of LTP at hippocampal mossy fiber synapses is independent of sustained presynaptic calcium. Neuron 7, 451–459. doi: 10.1016/0896-6273(91)90297-D
Reich, C. G., Mohammadi, M. H., and Alger, B. E. (2008). Endocannabinoid modulation of fear responses: learning and state-dependent performance effects. J. Psychopharmacol. 22, 769–777. doi: 10.1177/0269881107083999
Reid, C. A., Dixon, D., Takahashi, M., Bliss, T., and Fine, A. (2004). Optical quantal analysis indicates that long-term potentiation at single hippocampal mossy fiber synapses is expressed through increased release probability, recruitment of new release sites, and activation of silent synapses. J. Neurosci. 24, 3618–3626. doi: 10.1523/JNEUROSCI.3567-03.2004
Reid, C. A., Fabian-Fine, R., and Fine, A. (2001). Postsynaptic calcium transients evoked by activation of individual hippocampal mossy fiber synapses. J. Neurosci. 21, 2206–2214.
Robbe, D., Alonso, G., Chaumont, S., Bockaert, J., and Manzoni, O. J. (2002a). Role of P/Q-Ca(2+) channels in metabotropic glutamate receptor 2/3-dependent presynaptic long-term depression at nucleus accumbens synapses. J. Neurosci. 22, 4346–4356.
Robbe, D., Kopf, M., Remaury, A., Bockaert, J., and Manzoni, O. J. (2002b). Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc. Natl. Acad. Sci. U.S.A. 99, 8384–8388. doi: 10.1073/pnas.122149199
Rollenhagen, A., and Lübke, J. H. R. (2010). The mossy fiber bouton: the “Common” or the “Unique” synapse? Front. Synaptic Neurosci. 2:2. doi: 10.3389/fnsyn.2010.00002
Rosenmund, C., Clements, J. D., and Westbrook, G. L. (1993). Nonuniform probability of glutamate release at a hippocampal synapse. Science 262, 754–757. doi: 10.1126/science.7901909
Rosenmund, C., Sigler, A., Augustin, I., Reim, K., Brose, N., and Rhee, J. S. (2002). Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron 33, 411–424. doi: 10.1016/S0896-6273(02)00568-8
Rumpel, S., LeDoux, J., Zador, A., and Malinow, R. (2005). Postsynaptic receptor trafficking underlying a form of associative learning. Science 308, 83–88. doi: 10.1126/science.1103944
Salin, P., Malenka, R. C., and Nicoll, R. A. (1996). Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron 16, 797–803. doi: 10.1016/S0896-6273(00)80099-9
Samson, R. D., and Paré, D. (2005). Activity-dependent synaptic plasticity in the central nucleus of the amygdala. J. Neurosci. 25, 1847–1855. doi: 10.1523/JNEUROSCI.3713-04.2005
Sarihi, A., Mirnajafi-Zadeh, J., Jiang, B., Sohya, K., Safari, M.-S., Arami, M. K., et al. (2012). Cell type-specific, presynaptic LTP of inhibitory synapses on fast-spiking GABAergic neurons in the mouse visual cortex. J. Neurosci. 32, 13189–13199. doi: 10.1523/JNEUROSCI.1386-12.2012
Schlüter, O. M., Basu, J., Südhof, T. C., and Rosenmund, C. (2006). Rab3 superprimes synaptic vesicles for release: implications for short-term synaptic plasticity. J. Neurosci. 26, 1239–1246. doi: 10.1523/JNEUROSCI.3553-05.2006
Schlüter, O. M., Schnell, E., Verhage, M., Tzonopoulos, T., Nicoll, R. A., Janz, R., et al. (1999). Rabphilin knock-out mice reveal that rabphilin is not required for Rab3 function in regulating neurotransmitter release. J. Neurosci. 19, 5834–5846.
Schoch, S., Castillo, P. E., Jo, T., Mukherjee, K., Geppert, M., Wang, Y., et al. (2002). RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415, 321–326. doi: 10.1038/415321a
Shaban, H., Humeau, Y., Herry, C., Cassasus, G., Shigemoto, R., Ciocchi, S., et al. (2006). Generalization of amygdala LTP and conditioned fear in the absence of presynaptic inhibition. Nat. Neurosci. 9, 1028–1035. doi: 10.1038/nn1732
Shen, W., Flajolet, M., Greengard, P., and Surmeier, D. J. (2008). Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321, 848–851. doi: 10.1126/science.1160575
Shin, O.-H., Lu, J., Rhee, J.-S., Tomchick, D. R., Pang, Z. P., Wojcik, S. M., et al. (2010a). Munc13 C2B domain is an activity-dependent Ca(2+) regulator of synaptic exocytosis. Nat. Struct. Mol. Biol. 17, 280–288. doi: 10.1038/nsmb.1758
Shin, R.-M., Tully, K., Li, Y., Cho, J.-H., Higuchi, M., Suhara, T., et al. (2010b). Hierarchical order of coexisting pre- and postsynaptic forms of long-term potentiation at synapses in amygdala. Proc. Natl. Acad. Sci. U.S.A. 107, 19073–19078. doi: 10.1073/pnas.1009803107
Simsek-Duran, F., Linden, D., and Lonart, G. (2004). Adapter protein 14-13-3 is required for a presynaptic form of LTP in the cerebellum. Nat. Neurosci. 7, 1296–1298. doi: 10.1038/nn1348
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39, 641–654. doi: 10.1016/S0896-6273(03)00476-8
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2004). Endocannabinoid-dependent neocortical layer-5 LTD in the absence of postsynaptic spiking. J. Neurophysiol. 92, 3338–3343. doi: 10.1152/jn.00376.2004
Snyder, E. M., Philpot, B. D., Huber, K. M., Dong, X., Fallon, J. R., and Bear, M. F. (2001). Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat. Neurosci. 4, 1079–1085. doi: 10.1038/nn746
Sokolov, M. V., Rossokhin, A. V., Astrelin, A. V., Frey, J. U., and Voronin, L. L. (2002). Quantal analysis suggests strong involvement of presynaptic mechanisms during the initial 3 H maintenance of long-term potentiation in rat hippocampal CA1 area in vitro. Brain Res. 957, 61–75. doi: 10.1016/S0006-8993(02)03600-4
Sokolov, M. V., Rossokhin, A. V., Kasyanov, A. M., Gasparini, S., Berretta, N., Cherubini, E., et al. (2003). Associative mossy fibre LTP induced by pairing presynaptic stimulation with postsynaptic hyperpolarization of CA3 neurons in rat hippocampal slice. Eur. J. Neurosci. 17, 1425–1437. doi: 10.1046/j.1460-9568.2003.02563.x
Soler-Llavina, G. J., and Sabatini, B. L. (2006). Synapse-specific plasticity and compartmentalized signaling in cerebellar stellate cells. Nat. Neurosci. 9, 798–806. doi: 10.1038/nn1698
Spillane, D., Rosahl, T., Sudhof, T. C., and Malenka, R. C. (1995). Long-term potentiation in mice lacking synapsins. Neuropharmacology 34, 1573–1579. doi: 10.1016/0028-3908(95)00107-H
Stanton, P. K., Heinemann, U., and Muller, W. (2001). FM1-43 imaging reveals cGMP-dependent long-term depression of presynaptic transmitter release. J. Neurosci. 21, RC167.
Stanton, P. K., Winterer, J., Bailey, C. P., Kyrozis, A., Raginov, I., Laube, G., et al. (2003). Long-term depression of presynaptic release from the readily releasable vesicle pool induced by NMDA receptor-dependent retrograde nitric oxide. J. Neurosci. 23, 5936–5944.
Staubli, U., Larson, J., and Lynch, G. (1990). Mossy fiber potentiation and long-term potentiation involve different expression mechanisms. Synapse 5, 333–335. doi: 10.1002/syn.890050410
Steinert, J. R., Chernova, T., and Forsythe, I. D. (2010). Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist 16, 435–452. doi: 10.1177/1073858410366481
Stern, J. E., and Ludwig, M. (2001). NO inhibits supraoptic oxytocin and vasopressin neurons via activation of GABAergic synaptic inputs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R1815–R1822.
Stevens, C. F. (1993). Quantal release of neurotransmitter and long-term potentiation. Cell 72, 55–63. doi: 10.1016/S0092-8674(05)80028-5
Stevens, D. R., Wu, Z.-X., Matti, U., Junge, H. J., Schirra, C., Becherer, U., et al. (2005). Identification of the minimal protein domain required for priming activity of Munc13-11. Curr. Biol. 15, 2243–2248. doi: 10.1016/j.cub.2005.10.055
Sudhof, T. C. (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547. doi: 10.1146/annurev.neuro.26.041002.131412
Tabares, L., Ruiz, R., Linares-Clemente, P., Gaffield, M. A., de Toledo, G. A., Fernandez-Chacón, R., et al. (2007). Monitoring synaptic function at the neuromuscular junction of a mouse expressing synaptopHluorin. J. Neurosci. 27, 5422–5430. doi: 10.1523/JNEUROSCI.0670-07.2007
Torii, N., Tsumoto, T., Uno, L., Astrelin, A. V., and Voronin, L. L. (1997). Quantal analysis suggests presynaptic involvement in expression of neocortical short- and long-term depression. Neuroscience 79, 317–321.
Trussell, L. O., Zhang, S., and Raman, I. M. (1993). Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron 10, 1185–1196. doi: 10.1016/0896-6273(93)90066-Z
Tsetsenis, T., Younts, T. J., Chiu, C. Q., Kaeser, P. S., Castillo, P. E., and Südhof, T. C. (2011). Rab3B protein is required for long-term depression of hippocampal inhibitory synapses and for normal reversal learning. Proc. Natl. Acad. Sci. U.S.A. 108, 14300–14305. doi: 10.1073/pnas.1112237108
Tsvetkov, E., Carlezon, W. A., Benes, F. M., Kandel, E. R., and Bolshakov, V. Y. (2002). Fear Conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron 34, 289–300. doi: 10.1016/S0896-6273(02)00645-1
Tzounopoulos, T., Janz, R., Sudhof, T. C., Nicoll, R., and Malenka, R. C. (1998). A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron 21, 837–845. doi: 10.1016/S0896-6273(00)80599-1
Tzounopoulos, T., Rubio, M. E., Keen, J. E., and Trussell, L. O. (2007). Coactivation of pre- and postsynaptic signaling mechanisms determines cell-specific spike-timing-dependent plasticity. Neuron 54, 291–301. doi: 10.1016/j.neuron.2007.03.026
Urban, N. N., and Barrionuevo, G. (1996). Induction of hebbian and non-hebbian mossy fiber long-term potentiation by distinct patterns of high-frequency stimulation. J. Neurosci. 16, 4293–4299.
Varoqueaux, F., Sigler, A., Rhee, J.-S., Brose, N., Enk, C., Reim, K., et al. (2002). Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. U.S.A. 99, 9037–9042. doi: 10.1073/pnas.122623799
Villacres, E., Wong, S., Chavkin, C., and Storm, D. (1998). Type I adenylyl cyclase mutant mice have impaired mossy fiber long-term potentiation. J. Neurosci. 18, 3186–3194.
Voglmaier, S. M., Kam, K., Yang, H., Fortin, D. L., Hua, Z., Nicoll, R. A., et al. (2006). Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron 51, 71–84. doi: 10.1016/j.neuron.2006.05.027
Vogt, K., Mellor, J., Tong, G., and Nicoll, R. A. (2000). The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 26, 187–196. doi: 10.1016/S0896-6273(00)81149-6
Wang, X., Hu, B., Zimmermann, B., and Kilimann, M. (2001). Rim1 and Rabphilin-3 bind Rab3-GTP by composite determinants partially related through N-terminal alpha -Helix motifs. J. Biol. Chem. 276, 32480–32488. doi: 10.1074/jbc.M103337200
Wang, Y., Okamoto, M., Schmitz, F., Hofmann, K., and Sudhof, T. C. (1997). Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature 388, 593–598. doi: 10.1038/41580
Weisskopf, M. G., and Nicoll, R. A. (1995). Presynaptic changes during mossy fibre LTP revealed by NMDA receptor-mediated synaptic responses. Nature 376, 256–259. doi: 10.1038/376256a0
Weisskopf, M., Castillo, P. E., Zalutsky, R., and Nicoll, R. (1994). Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science 265, 1878–1882. doi: 10.1126/science.7916482
Xiang, Z., Greenwood, A., Kairiss, E., and Brown, T. (1994). Quantal mechanism of long-term potentiation in hippocampal mossy-fiber synapses. J. Neurophysiol. 71, 2552–2556.
Xiao, M. Y., Q Zhou, Q., and Nicoll, R. A. (2001). Metabotropic glutamate receptor activation causes a rapid redistribution of AMPA receptors. Neuropharmacology 41, 664–671. doi: 10.1016/S0028-3908(01)00134-4
Xu, W., Tse, Y. C., Dobie, F. A., Baudry, M., Craig, A. M., Wong, T. P., et al. (2013). Simultaneous monitoring of presynaptic transmitter release and postsynaptic receptor trafficking reveals an enhancement of presynaptic activity in metabotropic glutamate receptor-mediated long-term depression. J. Neurosci. 27, 5867–5877. doi: 10.1523/JNEUROSCI.1508-12.2013
Yamakawa, K. (2011). Molecular and cellular basis: insights from experimental models of Dravet syndrome. Epilepsia 52, 70–71. doi: 10.1111/j.1528-1167.2011.03006.x
Yang, Y., and Calakos, N. (2010). Acute in vivo genetic rescue demonstrates that phosphorylation of RIM1alpha Serine 413 is not required for mossy fiber long-term potentiation. J. Neurosci. 30, 2542–2546. doi: 10.1523/JNEUROSCI.4285-09.2010
Yang, Y., and Calakos, N. (2011). Munc13-11 is required for presynaptic long-term potentiation. J. Neurosci. 31, 12053–12057. doi: 10.1523/JNEUROSCI.2276-11.2011
Yasuda, H., Huang, Y., and Tsumoto, T. (2008). Regulation of excitability and plasticity by endocannabinoids and PKA in developing hippocampus. Proc. Natl. Acad. Sci. U.S.A. 105, 3106–3111. doi: 10.1073/pnas.0708349105
Yokoi, M., Kobayashi, K., Manabe, T., Takahashi, T., Sakaguchi, I., Katsuura, G., et al. (1996). Impairment of hippocampal mossy fiber LTD in mice lacking mGluR2. Science 273, 645–647. doi: 10.1126/science.273.5275.645
Yuste, R., Majewska, A., Cash, S. S., and Denk, W. (1999). Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J. Neurosci. 19, 1976–1987.
Zakharenko, S. S., Patterson, S. L., Dragatsis, I., Zeitlin, S. O., Siegelbaum, S. A., Kandel, E. R., et al. (2003). Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron 39, 975–990. doi: 10.1016/S0896-6273(03)00543-9
Zakharenko, S. S., Zablow, L., and Siegelbaum, S. A. (2001). Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat. Neurosci. 4, 711–717. doi: 10.1038/89498
Zakharenko, S. S., Zablow, L., and Siegelbaum, S. A. (2002). Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron 35, 1099–1110. doi: 10.1016/S0896-6273(02)00898-X
Zalutsky, R. A., and Nicoll, R. A. (1990). Comparison of two forms of long-term potentiation in single hippocampal neurons. Science 248, 1619–1624. doi: 10.1126/science.2114039
Zalutsky, R. A., and Nicoll, R. A. (1992). Mossy fiber long-term potentiation shows specificity but no apparent cooperativity. Neurosci. Lett. 138, 193–197. doi: 10.1016/0304-3940(92)90503-Y
Zhang, L., and Alger, B. E. (2010). Enhanced endocannabinoid signaling elevates neuronal excitability in Fragile X syndrome. J. Neurosci. 30, 5724–5729. doi: 10.1523/JNEUROSCI.0795-10.2010
Zhang, X. L., Zhou, Z. Y., Winterer, J., Muller, W., and Stanton, P. K. (2006). NMDA-dependent, but not group I metabotropic glutamate receptor-dependent, long-term depression at Schaffer collateral CA1 synapses is associated with long-term reduction of release from the rapidly recycling presynaptic vesicle pool. J. Neurosci. 26, 10270–10280. doi: 10.1523/JNEUROSCI.3091-06.2006
Zhu, Y., Xu, J., and Heinemann, S. F. (2009). Two pathways of synaptic vesicle retrieval revealed by single-vesicle imaging. Neuron 61, 397–411. doi: 10.1016/j.neuron.2008.12.024
Zucker, R. S. (1989). Short-term synaptic plasticity. Annu. Rev. Neurosci. 12, 13–31. doi: 10.1146/annurev.ne.12.030189.000305
Keywords: synaptic plasticity, neurotransmitter release, presynaptic plasticity, synaptic vesicle, long-term potentiation, long-term depression
Citation: Yang Y and Calakos N (2013) Presynaptic long-term plasticity. Front. Synaptic Neurosci. 5:8. doi: 10.3389/fnsyn.2013.00008
Received: 23 April 2013; Paper pending published: 14 May 2013;
Accepted: 09 September 2013; Published online: 17 October 2013.
Edited by:
Alanna Watt, McGill University, CanadaReviewed by:
Martin Heine, Leibniz Institute for Neurobiology, GermanyCopyright © 2013 Yang and Calakos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Yang, Department of Pediatrics, Stanford University School of Medicine, 1291 Welch Rd., CCSR 2100, Stanford, CA 94305-5164, USA e-mail:eWFuZ3lpbmdAc3RhbmZvcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.