
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Synaptic Neurosci. , 17 May 2011
volume 3 - 2011 | https://doi.org/10.3389/fnsyn.2011.00003
 Herman Moreno1,2
Herman Moreno1,2 Soonwook Choi2,3 Eunah Yu2,3 Janaina Brusco2,4
Soonwook Choi2,3 Eunah Yu2,3 Janaina Brusco2,4 Jesus Avila5
Jesus Avila5 Jorge E. Moreira2,4 Mutsuyuki Sugimori2,3
Jorge E. Moreira2,4 Mutsuyuki Sugimori2,3 Rodolfo R. Llinás2,3*
Rodolfo R. Llinás2,3*Filamentous tau inclusions are hallmarks of Alzheimer’s disease and related neurodegenerative tauopathies, but the molecular mechanisms involved in tau-mediated changes in neuronal function and their possible effects on synaptic transmission are unknown. We have evaluated the effects of human tau protein injected directly into the presynaptic terminal axon of the squid giant synapse, which affords functional, structural, and biochemical analysis of its action on the synaptic release process. Indeed, we have found that at physiological concentration recombinant human tau (h-tau42) becomes phosphorylated, produces a rapid synaptic transmission block, and induces the formation of clusters of aggregated synaptic vesicles in the vicinity of the active zone. Presynaptic voltage clamp recordings demonstrate that h-tau42 does not modify the presynaptic calcium current amplitude or kinetics. Analysis of synaptic noise at the post-synaptic axon following presynaptic h-tau42 microinjection revealed an initial phase of increase spontaneous transmitter release followed by a marked reduction in noise. Finally, systemic administration of T-817MA, a proposed neuro-protective agent, rescued tau-induced synaptic abnormalities. Our results show novel mechanisms of h-tau42 mediated synaptic transmission failure and identify a potential therapeutic agent to treat tau-related neurotoxicity.
Classical neuropathological studies characterized the intracellular accumulation and aggregation of abnormal filaments composed primarily of the microtubule associated protein tau as a hallmark for a variety of neurodegenerative disorders known as tauopathies, as exemplified by progressive supranuclear palsy, Pick’s disease, corticobasal degeneration, and frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17; Lee et al., 2001). The most common tauopathy, Alzheimer’s disease (AD), is also characterized by additional filamentous structures paired helical filaments (PHFs) and straight filaments (SFs). These filaments eventually form large aggregations, known as neurofibrillary tangles (NFTs). In addition, diffuse and mature senile plaques, which are predominantly composed of amyloid beta (Aβ) peptides, are present in AD brains too (Selkoe, 2001).
A number of studies have investigated the role of different forms of Aβ peptides and aggregates in synaptic function (Arancio et al., 2004; Moreno et al., 2009), but so far the role of tau or tau aggregates in neurotransmission has not been elucidated.
The association between tau filaments, neuron loss, and brain dysfunction in vertebrates and invertebrates originally led to the hypothesis that NFTs invariably cause brain dysfunction and neurodegeneration. However, mouse tauopathy studies indicate that severe abnormalities in synaptic function can precede neuronal loss and even NFTs formation (LaFerla et al., 2007; Yoshiyama et al., 2007). While the molecular mechanisms responsible for this early malfunction (as well as those responsible for tau polymerization dependent pathogenesis) remain unknown (Marx, 2007). Neurofibrillary degeneration is accompanied by lysosomal hypertrophy (Nixon et al., 1992), beading and degeneration of distal dendrites (Braak et al., 1994; Marx, 2007) and axonal damage (Kowall and Kosik, 1987).
In the present study physiological concentrations of recombinant human tau isoform (full length h-tau42; Perez et al., 2001) was directly injected into the presynaptic terminal of the squid giant synapse, to examine possible acute effects of h-tau on the synaptic release mechanism. The results showed that heavy exogenous h-tau42 accumulation induces a rapid and short-lasting increase in spontaneous transmitter release followed by a drastic decrease and failure of synaptic transmission. This synaptic block does not affect presynaptic calcium current flow or spike generation at the presynaptic terminal. Immunohistochemistry, performed in h-tau42 injected synapses, demonstrated that h-tau42 becomes phosphorylated rapidly in good temporal agreement with the time course of the transmitter failure. Electron microscopy and electrophysiological experiments unambiguously indicate that h-tau42-mediated synaptic transmission block is due to exocytosis failure. Finally, systemic administration of the novel compound T-817MA prevented the structural, biochemical, and functional deleterious effects h-tau42 microinjection.
The present set of studies identifies several mechanisms of tau-mediated toxicity at the presynaptic terminal, and introduces a potential disease modifier for AD and other tauopathies for which there is no specific treatment presently.
Recombinant human tau, h-tau42 (isoform with four tubulin binding motifs and two extra exons in the N-terminal domain) was isolated as previously described (Perez et al., 2001).
A variation of the array tomography method by Micheva and Smith (2007) was followed. The ganglia were fixed by immersion in 4% paraformaldehyde (EM grade EM Sciences) plus 7.0% sucrose in calcium-free sea water for 3 h; rinsed with 7 % sucrose and 50 mM glycine in 0.1 M PBS; dehydrated with graded ethanol dilutions (50%, 70%, 90%, and 3× 100%), embedded in LR White resin (medium grade, SPI), and polymerized in gelatin capsules at 49°C for 48 h. Semithin sections (500 nm) were mounted on subbed slides and encircled on the slides with a PAP pen (EM Sciences, USA). The immunocytochemistry was done as follows: (a) blocked in 50 mM glycine in tris buffer pH 7.6, 5 min; (b) primary antibody incubation, anti-tau PHF (AT8, Thermoscientific, USA) diluted 1:50 in 1% BSA in tris (tris-BSA), 4 h; (c) rinses in tris-BSA 2× 5 min: (d) secondary antibody incubation, goat anti-mouse Alexa Fluor 594 diluted 1:150 in tris-BSA, 30 min; (e) tris and distilled water rinses, 4× 5 min each; (f) mounting of slides with coverslips and anti-fading mounting media; (g) image under fluorescent microscopy (Zeiss Axioimager, Germany). Controls were performed with the same protocol omitting the primary antibody. Primary antibody: Anti-tau PHF (AT8; Thermoscientific, USA), secondary antibody: Alexa Fluor 594 goat anti-mouse (Invitrogen).
Squid received an oral dose of T-817MA (0.25 ml of 3.68 mmol l−1 T-817AM) every 12 h using a methodology recently developed by our group (Berk et al., 2009). At 24 h after the first dose a second dose was given, and the electrophysiological experiments were performed 1 h after the second feeding. The optimal dose, absorption, and concentration reached in the optic lobes of T-817MA in squid had been obtained previously (Berk et al., 2009). Indeed, it was determine that after 24 h of the double administration, a T817-MA concentration of 26.7 μmol 1−1 was measured in the CNS of the gastrically intubated squid (n = 10 squid). Synapses from T-817MA-treated squid were microinjected at the presynaptic axon with h-tau42 (an approximate 80 nM final concentration h-tau42 (n = 19), considering a 100× dilution factor. Pre- and post-synaptic potentials were recorded for 90 min.
The squid (Loligo paelli) stellate ganglia isolation from the mantle and the electrophysiological techniques used have been described previously (Llinas et al., 1985). Two glass micropipette electrodes impaled the largest (most distal) presynaptic terminal digit at the synaptic junction site while the post-synaptic axon was impaled by one microelectrode at the junctional site. One of the pre-electrodes was used for pressure microinjection of h-Tau 42 and also supported voltage clamp current feedback, while the second monitored membrane potential. The total volume injected fluctuated between 0.1 and 1 pl. (Llinas et al., 1985). The exact location of injection and the diffusion and steady-state distribution of the protein/fluorescent dye mix (0.001% dextran fluorescein) were monitored using a fluorescence microscope attached to a Hamamatsu camera system (Middlesex, NJ). In all experiments a good correlation was observed between the localization of the fluorescence and the electrophysiological findings.
Immediately following the electrophysiological study the ganglia were removed from the recording chamber, fixed by immersion in glutaraldehyde, post-fixed in osmium tetroxide, stained in block with uranium acetate, dehydrated and embedded in resin (Embed 812, EM Sciences). Ultrathin sections were collected on Pioloform (Ted Pella, Redding, CA) and carbon-coated single sloth grids, and contrasted with uranyl acetate and lead citrate. Morphometry and quantitative analysis of the synaptic vesicles were performed with the in house program developed with LabVIEW (National Instruments, Ostin, TX, USA). Electron micrographs were taken at an initial magnification of ×16,000 and ×31,500 and photographically enlarged to a magnification of ×40,000 and ×79,000 for synaptic vesicles and clathrin-coated vesicle (CCV) counting, respectively. Vesicle density at the synaptic active zones was determined as the number of vesicles per μm2, on an average area of 0.8 μm2 per active zone. CCV density was determined within the limits of the presynaptic terminal on an average terminal area of 3.3 μm2.
T-817MA (1-{3-[2-(1-benzothiophen-5-yl) ethoxy] propyl}azetidin-3-ol maleate), was synthesized at Toyama Chemical Co. Ltd., Japan.
Following presynaptic and post-synaptic axon impalements and the determination of normal synaptic transmission (Llinas et al., 1985, 1994; Lin et al., 1990), the effect of human tau on synaptic release was evaluated by presynaptic microinjections administered under direct visualization using a fluorescent dye/protein mix, reaching a final concentration after diffusion of approximately 80 nM (see Materials and Methods). Presynaptic and post-synaptic potentials were recorded simultaneously under current-clamp configuration. Presynaptic spikes were activated every 5 min (low-frequency protocol). With this paradigm, it was determined that 5–10 min after an injection of h-tau42, a reduction of transmitter release could be observed. With further time, a total block of transmission resulted, within 30–40 min depending on the length of the release zone in the preterminal axon (Figures 1A and 2 blue dots, n = 8). No modification of presynaptic spike amplitude or duration ensued (Figure 1A). By contrast, following T-817MA administration to the squid (see bellow) h-tau42-dependent transmitter block was prevented (Figures 1B and 2 red dots, n = 7).
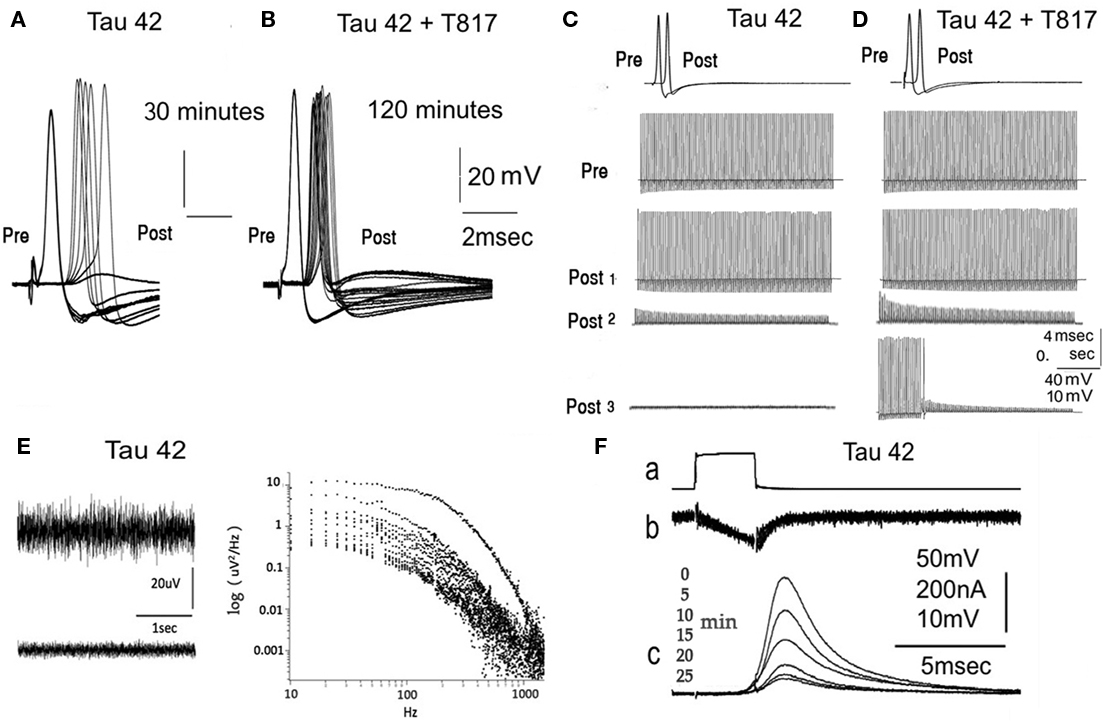
Figure 1. Electrophysiological Findings following presynaptic h-tau42 injection. (A) Pre- and post-synaptic potential following a direct electrical stimulation of the presynaptic axon. Synaptic transmission fails in 30 min following h-tau42 preinjection (six subsequent stimuli at 5-min intervals). (B) Similar experiment as in (A) (24 subsequent stimulus are shown at 5-min interval) following h-tau42 injection in a T-817MA-treated squid. Transmission block does not occur within the 2-h period. (C) Repetitive stimulation at 100 Hz following h-tau42 injection results in a step like decrease in post-synaptic response amplitude that slowly progresses (post 1,2,3) but shows small variation in amplitude during a given stimulus train. (D) Similar train stimulus paradigm produced reduction of transmitter release, which recuperated to the 80% amplitude in 15 min (post 1, 2, and 3). (E) Power spectra of spontaneous post-synaptic noise. Noise recording at the post-synaptic terminal (upper and lower panels on the left) were taken at 1-min intervals following h-tau42preinjection. Spontaneous release as determined by synaptic noise power spectrum (right panel) in (E), showed rapid reduction within a 12-min interval (reading taken at a 1/minrate). (F) h-tau42 preinjection produces block of presynatic release (c) without affecting presynaptic inward calcium current (b). Preterminal voltage step were repeated every minutes. For further detail see text and Figure 2.
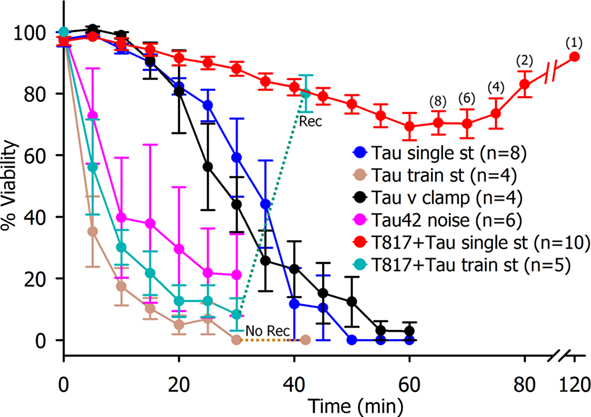
Figure 2. Time course of synaptic transmission reduction following presynaptic Tau injection. Single spike transmission, (blue circles n = 8), voltage clamp release (black circles, n = 4), post-synaptic spectral noise (purple circles n = 3) and repetitive presynaptic stimulation (brown circles n = 3) resulted in a rapid reduction of transmitter release without recovery after 15-min rest period. Parenteral T-817MA administration results in the block of h-tau42 effect on transmitter release reduction (red circles n = 7) and in the recovery of synaptic transmission from train stimulation after 15-min rest period.
To determine whether the transmission block described above was produced by a reduction of transmitter availability (as would be expected by inhibition of any step in the synaptic vesicle recycling pathway, e.g., endocytosis, refilling of vesicles with transmitter, or docking.) or by a defect in synaptic vesicle fusion, the effect of trains of presynaptic high frequency stimuli (100 Hz) was tested. This form of activation rapidly depleted the transmitter, as evidence by the rapid decrease in post-synaptic potential amplitude during a stimulus train (Figure 1D Post 2). The rapid time course of this decay has been shown to give an estimate of transmitter availability (Llinás et al., 1994), and is a reflection of a decrease in either synaptic vesicle mobilization or docking. On the other hand, synaptic block accompanied by a slow, progressive reduction of post-synaptic amplitude, without amplitude reduction during the tetanic stimulus, is a direct indication that the block is due to a defect in transmitter release (Llinás et al., 1994), i.e., vesicular fusion.
The test paradigm implemented to address this query consisted of synaptic high frequency (100 Hz) spike activation of the presynaptic axon of synapses preinjected with h-tau42 (Figure 1C, 1 Pre Figure 2 light brown dots, n = 4) which initially generated post-synaptic repetitive activation (Figure 1C, Post 1). This train stimulation was repeated once a minute until post-synaptic spike failure occurred from the first stimulus in the train. Note that the amplitude of the subthreshold synaptic potentials showed little reduction during the stimulus train itself but steadily reduced in amplitude as h-tau42 mobilizes into the preterminal. The fact that the amplitude of the evoked post-synaptic potentials remained unchanged during the duration of a given repetitive stimulation barrage indicates that the limiting factor was the vesicular release process (Figure 1C Post 2). This synaptic failure did not recuperate after 15 min of rest (as normally occur in this preparation; Llinas et al., 1985, 1994; Lin et al., 1990), but rather diminished to just noticeable amplitude (Post 3). In synapses from squid pretreated with T-817MA, microinjection with h-tau42 (Figures 1D and 2, green dots n = 6), show the normal reduction of the EPSP amplitude during the repetitive stimulus barrage, however, the treated animals showed normal recovery after a 15-min rest period. These findings indicate that T-817MA administration results in the normal recovery of synaptic transmission after high frequency stimulation, suggesting that the h-tau 42 effects could relate to vesicular availability and not to the actual vesicular release process (Figure 1D Post 3 and Figure 2, green dot, n = 5).
Beyond spike-initiated release, the possibility that h-tau42 may affect spontaneous transmitter release was directly evaluated using post-synaptic noise analysis. A detailed description of the technique has been published by our team (Lin et al., 1990). In all preparations tested (n = 6) the membrane noise recorded from the post-synaptic axon increased during the initial 5 ± 1 min after presynaptic microinjection of h-tau42, this was followed by rapid noise level reduction in parallel with decreased amplitude of the evoked transmitted release. An example of the time course of noise level changes is illustrated in Figure 1E (left panel and Figure 2 purple dots n = 3). Note the biphasic changes in noise levels. A frequency spectrum of the membrane noise at different times after the h-tau42 injection is shown in Figure 1E (right panel). This pattern indicates that block by h-tau42 interferes with both spontaneous and evoked transmitter release.
Following the initial finding that transmission is rapidly blocked by h-tau42; the possibility that this block was associated with changes in presynaptic calcium currents (ICa2+) was tested. The amplitude and time course of ICa2+ (Figure 1F) were directly determined by presynaptic voltage clamp steps, after blocking voltage dependent K+ and Na+ currents (n = 6), as previously described (Llinas et al., 1985). ICa2+ amplitude and time course were determined at 5-min intervals over a period of 25 min following presynaptic injection of h-tau42, 80 nM.
Voltage clamp experiments were implemented with presynaptic voltage steps that generated presynaptic inward calcium current and post-synaptic EPSPs. Following h-tau42 injection presynaptic voltage steps were repeated at 5 min intervals (low-frequency stimuli), which resulted in a progressive reduction of post-synaptic response amplitude, to total failure, without a change in the amplitude or time course of the presynaptic ICa2+ (Figure 1F). Under normal conditions this paradigm results in transmitter release that last, unaltered, for up to 2 h, the maximum period utilized (Llinas et al., 1985). The results show, therefore, that neither the time course nor the amplitudes of the presynaptic calcium currents were altered concomitantly with the transmitter release block induced by h-tau42 (Figures 1F and 2). The time course and number of electrophysiological experiments are summarized in Figure 2.
Since the aggregation of typical tau filaments is accompanied by the development of tau hyperphosphorylation, we investigated whether h-tau42 residues serine 202, threonine 205 and/or 231were phosphorylated in the squid synapse. We used AT8 antibodies, as commonly used in neuropathological studies (Goedert et al., 1995). Here, we used immunohistochemistry in a variance of the array tomography technique (Micheva and Smith, 2007); we found that single sections (500 nm) allowed a clear view of the pre- and post-synaptic compartments (Figure 3A). Anti-phospho-tau immunohistochemistry was detected as dot-like profiles in the presynaptic compartment in h-tau42 injected synapses but drastically reduced in T-817 treated squid (Figure 3B lower panel). These were absent in synapses injected with vehicle (Figure 3D lower panel). This finding demonstrates that h-tau42 becomes phosphorylated in the squid axon.
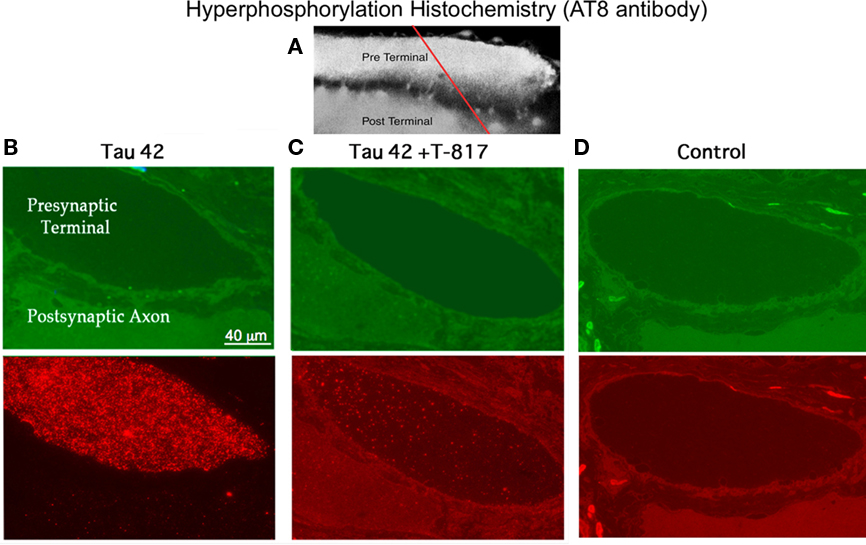
Figure 3. Presynaptic hyperphosphorylation immunohistochemistry. (A) Confocal image of the giant synapse showing the presynaptic terminal (Pre Terminal) and the post-synaptic axon (Post Terminal). The red line indicates plane of section for the images in (B), (C), and (D). (B) Light microscopical image of the two cross-sected giant axons showing the pre- and post-synaptic axoplasmic cytosol. Upper left panel (green): section of the synapse showing the presynaptic terminal (dark). Lower left panel: Fluorescence immunohistochemistry using AT8 antibodies, demonstrating h-tau42 phosphorylation level (red). (C) Upper middle panel: Transected synapse in a T-817MA-treated squid (green). Lower middle panel: Immunohistochemistry demonstrating a highly reduced level of h-tau42 phosphorylation (red). (D) Upper right panel: Transected synapse, injected with vehicle, to serve as a control, using the same protocol as in (C). Lower panel immunohistochemical absence of h-tau42 phosphorylation in the h-tau42 uninjected synapse.
The structural changes that follow h-tau42 injection were addressed by rapidly fixing stellate ganglia (see Materials and Methods) after high or low-frequency stimulation protocols. The material consisted of injected synapses (62 synaptic active zones from 10 different squid) and vehicle-injected synapses (control, 27 active zones in five synapses). The synapses were fixed ∼75–90 min after h-tau42 injection and processed for ultrastructural microscopy (see Materials and Methods). There was a statistically significant reduction in the number of “docked vesicles” in h-tau42-injected synapses compared to axons injected with vehicle (Table 1). This reduction was not seen in T817MA-treated squid following h-tau42 injection (Table 1).
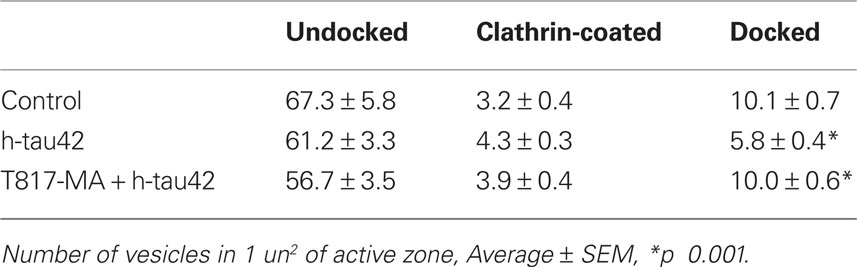
Table 1. Quantification of preterminal docked vesicles in: control, h-tau42 injected and h-tau42 injected in T-817MA pretreated squid.
As shown in representative control synapses (Figure 4A), vesicles are normally present at the active zone, some in contact with the presynaptic terminal membrane (docked). By contrast, in h-tau42-injected synapses (Figure 4B) vesicles were often closely aggregated with electron dense material serving as a bonding matrix (red dot). Similar electron dense material was also observed around vesicles in contact with the active zone (red arrows). At a lower magnification (Figure 4C) a large number of aggregated vesicular profiles are evident in the vicinity of the active zone (red dots). In T-817MA administered squid, the synaptic morphology was quite similar to the vehicle-injected synapses (compare Figures 4A,D). Quantification of number of undocked, docked, and clathrin-coated vesicles (CCV) among the three groups (control, h-tau42 injected, and h-tau42 injected + T-817MA) is tabulated in Table 1.
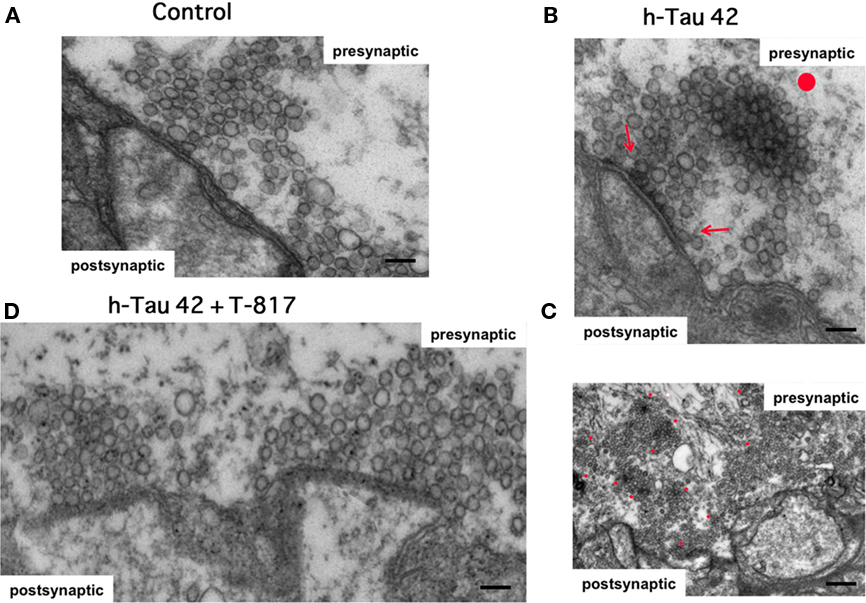
Figure 4. Ultrastructure of the synaptic junction demonstrating differences in normal and h-tau42 injected synapses in untreated and T-817MA-treated squid. (A) Control synapse showing the typical presynaptic ultrastructure with synaptic vesicles grouped at the synaptic active zone (scale bar: 100 nm). (B) Ultrastructure of ah-tau42 injected synapse showing a synaptic vesicle cluster congealed around electron dense material (red dot, scale bar: 100 nm). (C) low magnification image of a h-tau42 injected synapse showing a large number of vesicular clusters as in the upper panel also marked with red dots (scale bar: 500 nm). (D): Ultrastructural image of a T-817MA-treated squid demonstrating normal synaptic morphology and properly docked vesicles at the presynaptic active zone (scale bar: 100 nm).
T-817MA (1-{3-[2-(1-benzothiophen-5-yl)ethoxy]propyl} azetidin-3-ol maleate) is a newly developed neuro-protective agent that prevents oxidative stress, nitric oxide-induced neurotoxicity, and acts as a neurotrophic factor (Hirata et al., 2005). Although it has been shown that T-817MA administration may ameliorate amyloid beta related neuronal dysfunction (Nguyen et al., 2007; Kimura et al., 2009), its effect on tau related pathology is unknown. To study the potential effects of T-817MA on tau neuropathology, squids received an oral dose of T-817MA as described in the Section “Materials and Methods”. Electrophysiologically, as shown in Figure 1B no significant changes in the amplitude or time course of the pre- or post-synaptic potentials were observed. Further, ultrastructural studies in synapses used for the electrophysiological experiments demonstrated the number of docked vesicles recovered to the normal range in h-tau42/T-817MA squid (31 active zones in six synapses; 10.0 ± 0.6) compared to control synapses (10.1 ± 0.7), with the presence of normal CCV profiles (3.9 ± 0.4). Also clear was a significant reduction, of electron dense vesicles clusters and electron dense active zones (see Table 1; Figure 4). It is thus concluded that T-817MA prevented the h-tau42 dependent synaptic vesicle clustering, indicating a close relation between such morphology and the synaptic transmitter release block observed electrophysiologically. Finally squid pretreated with T-817MA showed a significantly reduced signal of intra-axonal h-tau42 phosphorylation, as detected by AT8 immunohistochemistry (Figure 3).
While the underlying pathogenic mechanisms of tau related neuronal abnormalities remain obscure, the acute effects of preterminal injection of h-tau42 presented here suggest that synaptic dysfunction is an early mechanism in AD and other tauopathies. We used the squid giant synapse in this study because it provides unique advantages in addressing the cellular and molecular mechanisms involved in chemical synaptic transmission. In this set of experiments we determined that h-tau42 produces a rapid failure in exocytosis. Our results also indicate that h-tau42 has previously unknown physiological properties that may be relevant in tau related neurodegenerative process.
The data presented here indicate that an excess of h-tau42 protein produces synaptic transmission block by interfering with a mechanism of synaptic vesicle exocytosis. Our conclusion, that h-tau42 induces a failure in neurotransmitter availability due to reduced synaptic vesicle release, is derived from morphological (Figures 4B,C), high frequency stimulation (Figure 1C), and spontaneous neurotransmitter release data (Figure 1E). Moreover, all the h-tau-42 injected synapses demonstrated a drastic block of both spontaneous and evoked transmitted release, without affecting presynaptic spike generation (Figure 1A) or the associated calcium current (Figure 1F). We interpret these findings as reflecting the reduced vesicle count at the active zone, the vesicles being instead concentrated in groups away from the active zone. It was also noted that these electron dense vesicular congregations were characterized by profiles resembling vesicular adhesions to microfilaments as would be expected if synapsin 1 were to be dephosphorylated affording a strong adhesion to such microfilaments (Llinas et al., 1985). As a result h-tau42 would lead to the failure in exocytosis due to both a defect in the release mechanism and a reduction in vesicular availability (Llinas et al., 1991).
Previous experiments in drosophila have shown that misexpression of human – tau (h-tau), the same isoform as the one used in our experiments, produced significant neurodegeneration (Jackson et al., 2002; Avila et al., 2004; Steinhilb et al., 2007). In the drosophila model tau co-expressed with Shaggy, which generated a single fly homolog of GSK-3β, the phenotype was aggravated (Jackson et al., 2002). Dysfunctional phenotypes were also found in the central neurons of lamprey, where long-term expression (2–38 days) of several h-tau isoforms produced neurodegenerative changes as a result of accumulation of h-hyperphosphorylated tau which correlated with the appearance of structures that resemble AD characteristic – “straight like filaments” (Hall et al., 1997). In the latter experiments the isoform hyperphosphorylated moiety was, to a larger extent, the long form of h-tau (h-tau42; Hall et al., 1997; Lee et al., 2009). It has been proposed that the physiological function of tau is adversely affected by excess phosphorylation resulting in tau being displaced from microtubules and aggregating, which in turn leads to microtubule disassembly, disruption of axonal transport, and finally synaptic failure (Stamer et al., 2002).
Concerning cephalopods, it has been demonstrated that h-tau binds to the squid axonal microtubules, but monomeric h-tau did not affect fast axonal transport (FAT), while filamentous h-tau42 did block anterograde FAT (LaPointe et al., 2009). However, other studies have found that flies misexpressing tau show defects in neuronal traffic without evidence of tau aggregation (Jackson et al., 2002; Mudher et al., 2004). Finally, extracellular applied h-tau42 to cell cultures produced aberrant signaling through muscarinic receptor activation (Gomez-Ramos et al., 2008; Diaz-Hernandez et al., 2010), suggesting that even “normal” tau may be detrimental when its expression becomes elevated or when it accumulates extracellularly. From these observations, it appears that an optimal level of tau phosphorylation is required to achieve the balance in the level of “free” and “microtubule bound” tau that is essential in maintaining microtubule dynamics and subsequent axonal transport.
As in the experiments mentioned above, in our experiments h-tau42 also became phosphorylated in the isolated axon (separated from the cell body) as demonstrated by immunohistochemistry using AT8 antibodies (Figure 3B). AT8 recognizes epitopes phosphorylated by GSK3 and cdk5 kinases both of which are found in squid axoplasm (Takahashi et al., 1995; Morfini et al., 2002; Hanger et al., 2009), suggesting that either one or both kinases may be involved in the effects of h-tau42 in the presynaptic terminal. Whichever the specific kinase, the results demonstrate that isolated axons have the complete machinery to produce local post translational modifications and that these changes may explain, in part, the detrimental effects of excessive “normal tau” on the function of the presynaptic terminal.
Moreover, the vesicle clustering observed in h-tau42 injected synapses, resembled the effect of unphosphorylated synapsin 1 on synaptic vesicle (Jackson et al., 2002). The fact that h-tau42 is phosphorylated intra-axonally and that unphosphorylated synapsin 1 restrains the vesicle pool to the cytoskeleton – producing a decreased number of vesicles available for exocytosis was actually demonstrated some time ago (Llinas et al., 1985).
A possible explanation of our findings is that h-tau42 induces changes in the balance of kinases and phosphatases, perhaps influenced by the concentration of h-tau aggregates. We hypothesize that this could decrease the phosphorylated/dephosphorylated ratio in proteins involved in synaptic vesicle function, such as synapsin 1, which would result in a reduction in the available vesicles and ultimately synaptic transmission failure. Tau is phosphorylated by several protein kinases and this is balanced by protein phosphatases dephosphorylation. The potential kinases and phosphatases involved are so numerous that biochemical experiments dedicated to solve this issue are necessary. If this process were to involve also constitutive vesicular dynamics a secondary dying-back event (Moreno et al., 2009) would results in the synaptic disconnection encountered in AD pathomorphology.
The present results identify the protective effect of systemic administration of T-817MA on h-tau42 mediated axonal/synaptic dysfunction as shown in Figures 1B,D, 2, and 3C. This finding is highly significant, as it has been demonstrated that reduction of endogenous tau in an AD mouse model, ameliorates amyloid beta induced neurodegeneration at several levels (Roberson et al., 2007). Therefore, T-817MA has a potential to treat tau pathology. Addressing the mechanisms of T-817MA neuro-protection in relation to tau pathology remains to be elucidated. But the fact that both functional and biochemical h-tau42 induced abnormalities in the presynaptic axon are prevented/ameliorated by this compound is a fundamental initial step toward the identification of therapeutic candidates to treat tauopathies.
Our results indicate that h-tau42 affects synaptic release by modifying intracellular phosphorylation homeostasis as a resultant of the h-tau42 hyperphosphorylation. This dynamic change, we propose, leads to a marked reduction of synaptic vesicle availability, most probably due to the reduction of synapsin 1 phosphorylation, known to be a powerful modulator of synaptic release (Llinas et al., 1985). Beyond affecting synaptic release the reduction of such vesicular fusion on constitutive vesicular dynamics would also result in a disconnection event ultimately generating a “dying-back” phenomenon (Stamer et al., 2002; Pigino et al., 2007; Serulle et al., 2007). Systemic administration of T-817MA, a proposed neuro-protective agent, rescued tau-induced synaptic abnormalities, markedly reduced h-tau42 hyperphosphorylation and prevented synaptic vesicle clustering, as determine by ultrastructural analysis.
Herman Moreno, Soonwook Choi, Eunah Yu, Mutsuyuki Sugimori, and Rodolfo R. Llinás designed research; Herman Moreno, Soonwook Choi, Eunah Yu, Jorge E. Moreira, Mutsuyuki Sugimori, and Rodolfo R. Llinás performed research; Herman Moreno, Soonwook Choi, Eunah Yu, Janaina Brusco, Jorge E. Moreira, Mutsuyuki Sugimori, and Rodolfo R. Llinás analyzed data; Herman Moreno and Jesus Avila implemented molecular biology, and Herman Moreno, Soonwook Choi, G.P., Jorge E. Moreira, Jesus Avila, Mutsuyuki Sugimori, and Rodolfo R. Llinás wrote the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Grant sponsors: NIH AG027476 to Herman Moreno; NS13742/NS/NINDS/NIH HHS to RLl; FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo, Brasil) projects 2009/01571-6 and CinAPSe 05/56447-7 to Jorge E. Moreira.
Arancio, O., Zhang, H. P., Chen, X., Lin, C., Trinchese, F., Puzzo, D., Liu, S., Hegde, A., Yan, S. F., Stern, A., Luddy, J. S., Lue, L. F., Walker, D. G., Roher, A., Buttini, M., Mucke, L., Li, W., Schmidt, A. M., Kindy, M., Hyslop, P. A., Stern, D. M., and Yan, S. S. (2004). RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 23, 4096–4105.
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384.
Berk, W., Teperman, J., Walton, K. D., Hirata, K., Sugimori, M., and Llinas, R. R. (2009). Oral administration of pharmacologically active substances to squid: a methodological description. Biol. Bull. 216, 1–6.
Braak, E., Braak, H., and Mandelkow, E. M. (1994). A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 87, 554–567.
Diaz-Hernandez, M., Gómez-Ramos, A., Rubio, A., Gómez-Villafuertes, R., Naranjo, J. R., Miras-Portugal, M. T., and Avila, J. (2010). Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J. Biol. Chem. 285, 32539–32548.
Goedert, M., Jakes, R., and Vanmechelen, E. (1995). Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci. Lett. 189, 167–169.
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Miras-Portugal, M. T., and Avila, J. (2008). Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 37, 673–681.
Hall, G. F., Yao, J., and Lee, G. (1997). Human tau becomes phosphorylated and forms filamentous deposits when overexpressed in lamprey central neurons in situ. Proc. Natl. Acad. Sci. U.S.A. 94, 4733–4738.
Hanger, D. P., Seereeram, A., and Noble, W. (2009). Mediators of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Expert Rev. Neurother. 9, 1647–1666.
Hirata, K., Yamaguchi, H., Takamura, Y., Takagi, A., Fukushima, T., Iwakami, N., Saitoh, A., Nakagawa, M., and Yamada, T. (2005). A novel neurotrophic agent, T-817MA [1-{3-[2-(1-benzothiophen-5-yl) ethoxy] propyl}-3-azetidinol maleate], attenuates amyloid-beta-induced neurotoxicity and promotes neurite outgrowth in rat cultured central nervous system neurons. J. Pharmacol. Exp. Ther. 314, 252–259.
Jackson, G. R., Wiedau-Pazos, M., Sang, T. K., Wagle, N., Brown, C. A., Massachi, S., and Geschwind, D. H. (2002). Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 34, 509–519.
Kimura, T., Hong Nguyen, P. T., Ho, S. A., Tran, A. H., Ono, T., and Nishijo, H. (2009). T-817MA, a neurotrophic agent, ameliorates the deficits in adult neurogenesis and spatial memory in rats infused i.c.v. with amyloid-beta peptide. Br. J. Pharmacol. 157, 451–463.
Kowall, N. W., and Kosik, K. S. (1987). Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann. Neurol. 22, 639–643.
LaFerla, F. M., Green, K. N., and Oddo, S. (2007). Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509.
LaPointe, N. E., Morfini, G., Pigino, G., Gaisina, I. N., Kozikowski, A. P., Binder, L. I., and Brady, S. T. (2009). The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J. Neurosci. Res. 87, 440–451.
Lee, S., Jung, C., Lee, G., and Hall, G. F. (2009). Exonic point mutations of human tau enhance its toxicity and cause characteristic changes in neuronal morphology, tau distribution and tau phosphorylation in the lamprey cellular model of tauopathy. J. Alzheimers Dis. 16, 99–111.
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159.
Lin, J. W., Sugimori, M., Llinas, R. R., McGuinness, T. L., and Greengard, P. (1990). Effects of synapsin I and calcium/calmodulin-dependent protein kinase II on spontaneous neurotransmitter release in the squid giant synapse. Proc. Natl. Acad. Sci. U.S.A. 87, 8257–8261.
Llinas, R., Gruner, J. A., Sugimori, M., McGuinness, T. L., and Greengard, P. (1991). Regulation by synapsin I and Ca(2+)-calmodulin-dependent protein kinase II of the transmitter release in squid giant synapse. J. Physiol. 436, 257–282.
Llinas, R., McGuinness, T. L., Leonard, C. S., Sugimori, M., and Greengard, P. (1985). Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc. Natl. Acad. Sci. U.S.A. 82, 3035–3039.
Llinás, R., Sugimori, M., Lang, E. J., Morita, M., Fukuda, M., Ninobe, M., and Mikoshiba, K. (1994). The inositol high-polyphosphate series blocks synaptic transmission by preventing vesiculat fusion: a squid giant synapse study. Proc. Natl. Acad. Sci. U.S.A. 91, 12990–12993.
Micheva, K. D., and Smith, S. J. (2007). Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron 55, 25–36.
Moreno, H., Yu, E., Pigino, G., Hernandez, A. I., Kim, N., Moreira, J. E., Sugimori, M., and Llinás, R. R. (2009). Synaptic transmission block by presynaptic injection of oligomeric amyloid beta. Proc. Natl. Acad. Sci. U.S.A. 106, 5901–5906.
Morfini, G., Szebenyi, G., Elluru, R., Ratner, N., and Brady, S. T. (2002). Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 21, 281–293.
Mudher, A., Shepherd, D., Newman, T. A., Mildren, P., Jukes, J. P., Squire, A., Mears, A., Drummond, J. A., Berg, S., MacKay, D., Asuni, A. A., Bhat, R., and Lovestone, S. (2004). GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry 9, 522–530. [In English].
Nguyen, P. T., Kimura, T., Ho, S. A., Tran, A. H., Ono, T., and Nishijo, H. (2007). Ameliorative effects of a neuroprotective agent, T-817MA, on place learning deficits induced by continuous infusion of amyloid-beta peptide (1-40) in rats. Hippocampus 17, 443–455.
Nixon, R. A., Cataldo, A. M., Paskevich, P. A., Hamilton, D. J., Wheelock, T. R., and Kanaley-Andrews, L. (1992). The lysosomal system in neurons. Involvement at multiple stages of Alzheimer’s disease pathogenesis. Ann. N. Y. Acad. Sci. 674, 65–88.
Perez, M., Arrasate, M., Montejo De Garcini, E., Munoz, V., and Avila, J. (2001). In vitro assembly of tau protein: mapping the regions involved in filament formation. Biochemistry 40, 5983–5991.
Pigino, G., Opalach, K., Serulle, Y., Moreira, J. E., Sugimori, M., Llinas, R. R., and Brady, S. T. (2007). 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 104, 2442–2447.
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., Gerstein, H., Yu, G. Q., and Mucke, L. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754.
Serulle, Y., Morfini, G., Pigino, G., Moreira, J. E., Sugimori, M., Brady, S. T., and Llinas, R. R. (2007). 1-Methyl-4-phenylpyridinium induces synaptic dysfunction through a pathway involving caspase and PKC∂ enzymatic activities. Proc. Natl. Acad. Sci. U.S.A. 104, 2437–2441.
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002). Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063.
Steinhilb, M. L., Dias-Santagata, D., Fulga, T. A., Felch, D. L., and Feany, M. B. (2007). Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell 18, 5060–5068.
Takahashi, M., Amin, N., Grant, P., and Pant, H. C. (1995). P13suc1 associates with a cdc2-like kinase in a multimeric cytoskeletal complex in squid axoplasm. J. Neurosci. 15, 6222–6229.
Keywords: tauopathies, synaptic transmission, hyperphosphorylation, Altzheimer’s disease, neurodegenerative, human tau, synaptic vesicles, neuro-protective agent
Citation: Moreno H, Choi S, Yu E, Brusco J, Avila J, Moreira JE, Sugimori M and Llinás RR (2011) Blocking effects of human tau on squid giant synapse transmission and its prevention by T-817 MA. Front. Syn. Neurosci. 3:3. doi: 10.3389/fnsyn.2011.00003
Received: 24 January 2011;
Paper pending published: 04 April 2011;
Accepted: 05 May 2011;
Published online: 17 May 2011.
Edited by:
Joël Bockaert, INSERM, FranceCopyright: © 2011 Moreno, Choi, Yu, Brusco, Avila, Moreira, Sugimori and Llinás. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with
*Correspondence: Rodolfo R. Llinás, Department of Physiology and Neuroscience, New York University School of Medicine, 550 First Avenue, New York, NY 10016, USA. e-mail:bGxpbmFyMDFAbWVkLm55dS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.