


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Synaptic Neurosci. , 26 January 2011
volume 3 - 2011 | https://doi.org/10.3389/fnsyn.2011.00001
Interactions between presynaptic and postsynaptic cellular adhesion molecules (CAMs) drive synapse maturation during development. These trans-synaptic interactions are regulated by alternative splicing of CAM RNAs, which ultimately determines neurotransmitter phenotype. The diverse assortment of RNAs produced by alternative splicing generates countless protein isoforms necessary for guiding specialized cell-to-cell connectivity. Failure to generate the appropriate synaptic adhesion proteins is associated with disrupted glutamatergic and gamma-aminobutyric acid signaling, resulting in loss of activity-dependent neuronal plasticity, and risk for developmental disorders, including autism. While the majority of genetic mutations currently linked to autism are rare variants that change the protein-coding sequence of synaptic candidate genes, regulatory polymorphisms affecting constitutive and alternative splicing have emerged as risk factors in numerous other diseases, accounting for an estimated 40–60% of general disease risk. Here, we review the relationship between aberrant RNA splicing of synapse-related genes and autism spectrum disorders.
Genome-wide association studies, analyses of copy number variants, DNA sequencing approaches, and familial linkage studies have revealed a number of autism risk genes encoding cellular adhesion molecules (CAMs) and synaptic scaffold proteins (for review, see Betancur et al., 2009). These include the neurexin, neuroligin, contactin, and cadherin gene families, members of the general immunoglobulin CAM superfamily, and SH3 and multiple ankyrin repeat domain proteins (SHANK). Subsequent attempts to find single nucleotide polymorphisms (SNPs) conferring risk within CAM and scaffold genes have revealed few disease-causing coding variants shared among affected individuals (Vincent et al., 2004; Gauthier et al., 2005, 2009; Yan et al., 2005, 2008; Ylisaukko-oja et al., 2005; Blasi et al., 2006; Feng et al., 2006; Durand et al., 2007; Berkel et al., 2010). These sequencing studies focused exclusively on protein-coding regions and adjacent constitutive splice sites, driven by the assumption that the pathogenic mutations are deleterious amino acid substitutions in these synaptic proteins. More recently, however, increased understanding of RNA biology continues to reveal mechanisms by which RNA processing defects cause or contribute to disease phenotypes in humans, especially in neurodegenerative and neuromuscular disorders, where dysfunctional RNA splicing is prominent (Cooper et al., 2009).
Under normal conditions, constitutive and alternative splicing is directed by a nuclear protein aggregate of more than 100 core proteins and secondary modulators, collectively known as the spliceosome, which interacts with heterogeneous nuclear RNA [hnRNA or pre-messenger RNA (pre-mRNA)] in a process highly conserved in eukaryotes (Zhou et al., 2002). This process produces tissue-specific RNA and protein isoforms that can vary throughout development or in response to synaptic activity (Li et al., 2007). Spliceosomal proteins recognize consensus cis-regulatory motifs, located in introns or exons, and subsequently govern the inclusion or exclusion of exons in the mature mRNA end-product. Pathogenic mutations often disrupt regulatory cis-binding splice motifs within known risk genes, resulting in aberrant splicing, and manifestation of the disease phenotype. Genetic, epigenetic, or environmental factors can also affect the capacity of trans-acting splice factors and produce aberrant splicing patterns contributing to disease. Figure 1 portrays an example of the relationship between trans- and cis-acting factors during alternative splicing. Global estimates based on mathematical models suggest up to 60% of all disease-causing mutations disrupt splicing in cis or trans (Lopez-Bigas et al., 2005; Wang and Cooper, 2007). When individual genes are assayed for disease-causing mutations, up to 50% of pathogenic mutations affect splicing, largely through cis-acting mechanisms. (Mayer et al., 1999, 2000; Teraoka et al., 1999; Ars et al., 2000; Messiaen et al., 2000; Cartegni et al., 2002; Pagenstecher et al., 2006).
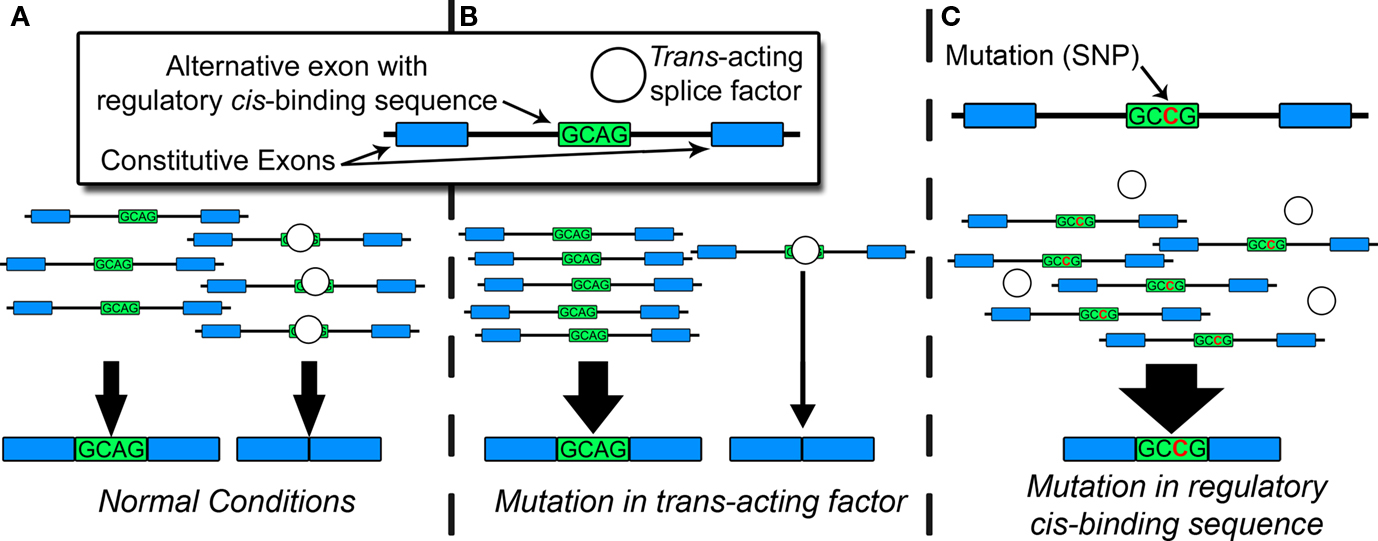
Figure 1. Factors in alternative splicing. (A) Under normal conditions, trans-acting splice factors bind cis-regulatory sequences (in introns or exons), resulting in typical patterns of spliceoform expression. (B) Genetic or epigenetic disruption of trans-acting splice factor alters the overall amount, activity, or affinity for the cis-regulatory sequence, resulting in changes in spliceoform quantities for multiple target RNAs. (C) Mutation in regulatory cis-binding sequence disrupts consensus binding site for trans-acting splice factor. This can result in quantitative or qualitative changes in spliceoform expression of a single target RNA.
Messenger RNA transcribed from CAM genes is extensively spliced to produce unique protein end-products necessary for specialized cell-to-cell interactions (Shapiro et al., 2007). In the central nervous system, alternative splicing of CAMs is associated with synapse maturation and the neurotransmitter to be used for synaptic signaling. For example, trans-synaptic binding of α-neurexin to neuroligin is determined by the expression of a neuroligin spliceoform lacking a critical N-glycosylation site that blocks α-neurexin binding (Boucard et al., 2005). In turn, interactions between these trans-synaptic binding partners determines phenotypic fate of maturing neuronal synapses, either toward excitatory glutamatergic or inhibitory gamma-aminobutyric acid (GABA)ergic signaling (Chih et al., 2006). Gene expression differences between ASD patients and controls – measured with microarrays – emphasize gene splicing and cellular adhesion as biological processes contributing to ASD (Abrahams and Geschwind, 2008).
At present, few studies have systematically examined CAM splicing in ASD, especially in tissues germane to the disorder, but atypical splicing patterns have been observed in autistic patients for at least five genes with significant synaptic functions (CADPS2, NLGN3, NLGN4X, NRXN1, and SHANK3). In addition, mutations affecting splicing have been frequently identified as pathogenic in genes responsible for some forms of syndromic autism (TSC1, TSC2, and NF1). Still other genes associated with syndromic autisms encode multifunctional proteins that have some capacity to perform alternative splicing in a trans-acting fashion (FMR1, MECP2, and SNRPN). With evidence for aberrant splicing accumulating in a multitude of other neurological disorder, it is prudent to critically review early splicing findings in ASD to determine whether similar dysfunctional processes are underlying ASD etiology or modifying autistic-like behaviors.
In humans, genetic variants that change or delete amino acid coding regions in NLGN3 or NLGN4 are sufficient to produce autistic phenotypes (Jamain et al., 2003; Laumonnier et al., 2004; Lawson-Yuen et al., 2008; Pampanos et al., 2009). One likely functional consequence of these mutations at the synaptic level, based on animal and in vitro human studies, is an imbalance in homeostatic glutamatergic, and GABAergic signaling (Chih et al., 2005; Chubykin et al., 2007; Tabuchi et al., 2007; Hines et al., 2008; Huang and Scheiffele, 2008; Blundell et al., 2009; Gibson et al., 2009). Neuroligins are crucial for synapse maturation, but not central to the actual formation of synapses. Neuroligin knockout mice, even triple knockouts for Nlgn1, Nlgn2, and Nlgn3, show no significant change in the total number of synapses formed, but the signaling intensity at formed glutamatergic and GABAergic synapses is reduced considerably, relative to wild-type animals, leading to respiratory failure, and early postnatal death (Varoqueaux et al., 2006). Alternative splicing of the acetylcholinesterase (AchE)-like domain in neuroligin mRNA transcripts mediates the affinity of the translated neuroligin protein to α-neurexins, which subsequently determines the neurotransmitter phenotype at the synapse (Boucard et al., 2005; Chih et al., 2006). The AchE-like domain is also crucial for neuroligin dimerization (Comoletti et al., 2003; Fabrichny et al., 2007). One study to date has examined neuroligin mRNA spliceoform expression in lymphoblastoid cell lines derived from autistic patients, revealing atypical splicing for neuroligin 3 (NLGN3) and X-linked neuroligin 4 (NLGN4X; Talebizadeh et al., 2006). One of 10 autistic samples lacked a truncated isoform of NGLN3, in which exon 7 is spliced out, that was present in all other samples and in female control brain tissue. A second patient from the autism cohort expressed a novel NLGN4X spliceoform lacking exon 4. The alternatively spliced exon for each of these spliceoforms encodes a portion of the AchE-like domain, potentially impacting dimerization and neurexin binding. However, the function of the constitutively expressed NLGN3 truncated isoform is unknown and no functional analyses were preformed for NLGN4X.
A second study, examining calcium-dependent secretion activator 2 (CADPS2) mRNA expression in autistic patient blood samples, noted expression of a novel splice variant lacking exon 3 in 4 of 16 autistic patients, but was not in 24 control samples (Sadakata et al., 2007a). However, a second independent laboratory found no difference between expression patterns of ASD and control patients, with both cohorts displaying a subset of samples expressing both isoforms (Eran et al., 2009). A significant difference between relative quantities of the two spliceoforms across the two studies is noted as a possible contributor to the discrepant results (Eran et al., 2009, authors reply). CADPS2 plays a crucial role in neuronal development through its modulation of brain-derived neurotrophic factor (BDNF) release and signaling properties at the synapse. Significant reductions in axonal CADPS2 transport for the novel spliceoform results in decreased CADPS2-mediated BDNF release (Sadakata et al., 2007a). Similarly, Cadps2 knockout mice have decreased BDNF release and significant morphological changes of cerebellar Purkinje cells (Sadakata et al., 2007b).
Important caveats need to be considered when drawing conclusions from the two aforementioned studies. First, no mutation was identified in any of the surveyed genes to explain the splicing differences, despite exon sequencing of NLGN3 and NLGN4X in the affected patients. Therefore, it is not clear whether these spliceoforms are the result of cis-acting genetic variants in unsequenced intronic regions or mutations that affect trans-acting splice factors. Second, the mRNA examined in each of these studies originated from peripheral blood tissues; either actively growing lymphoblastoid cell lines (Talebizadeh et al., 2006) or whole blood homogenates (Sadakata et al., 2007a). Global patterns of gene expression can substantially differ in these peripheral tissues, necessarily driving tissue differentiation and associated specialized tissue functions in the central nervous system. Even the media in which cells are cultured can contribute to isoforms-specific expression of splicing factors, which then have pleiotropic effects on downstream RNA targets (Lee et al., 2009). An unexpected spliceoform seen in blood tissues could be constitutively expressed in other tissues; such is the case with the CADPS2 truncated variant in the cerebellum versus blood (Eran et al., 2009). Furthermore, although reduced BDNF release was observed for the truncated version of CADPS2, no causative roles for the aberrant spliceoforms have been established in ASD. Therefore, caution must be exercised when extrapolating expression patterns observed in cultured tissues or whole blood to other tissues.
Mutations in cis-regulatory splice sites can disrupt the splicing process (Figure 1C), especially when present in consensus splice-donor, acceptor, or polypyrimidine branch sites recognized by the core components of the spliceosome. Exhaustive sequencing of two genes intimately linked to synaptogenesis, SHANK3 and NRXN1, uncovered rare splice-site mutations in ASD patients. For SHANK3, the protein-coding regions and flanking splice junctions from 427 ASD subjects and 190 controls were sequenced, revealing five novel non-synonymous variants and one splice-site mutation (Gauthier et al., 2009). Of these novel variants, only two were unique to autism, including the splice-site mutation characterized by the deletion of a guanine nucleotide crucial for definition of the splice-donor site preceding exon 19. Analysis of parental DNA suggests this mutation appeared de novo. This splice-site mutation produced a measurable change in gene expression, resulting in a longer transcript through the use of a cryptic downstream splice-donor site, confirmed by examining lymphoblast mRNA from this patient. Microdeletion or translocation of the chromosomal domain harboring SHANK3 (22q13.3) results in a consistent phenotype (Bonaglia et al., 2001, 2006), in which affected individuals often present with autistic-like behaviors (Cusmano-Ozog et al., 2007). During synaptogenesis, SHANK3 protein recruits essential glutamatergic components to the synapse, resulting in a greater number of functional synapses and increased excitatory signaling (Roussignol et al., 2005). More recently, exon sequencing also revealed protein-coding mutations in SHANK2 related to ASD and mental retardation (Berkel et al., 2010).
Yan et al. (2008) characterized NRXN1 mutations in 116 autistic patients and 192 controls by sequencing coding regions and adjacent splice junctions. This study yielded five autism-specific mutations within NRXN1, one of which disrupted the five’ splice-donor site following exon 4. Although the effect on mRNA expression is not confirmed in this study, the splice-donor site mutation could produce an effect similar to that observed for SHANK3, in which a frame shift compels use of a cryptic downstream splice site resulting in nonsense-mediated mRNA decay or a dysfunctional NRXN protein. A frame shift at exon 4 is predicted to disrupt the majority of the laminin neurexin sex hormone-binding protein (LNS) domains and epidermal growth factor (EGF)-like calcium binding domain. During synapse maturation, the sixth LNS domain is alternatively spliced to mediate GABAergic (Boucard et al., 2005; Kang et al., 2008) or glutamatergic postsynaptic development (Ko et al., 2009).
Sequencing efforts, like those put forth for SHANK3 and NRXN1, are crucial for identifying candidate mutations, although the focus on protein-coding mutations has yielded very few candidates unique to autism. Newer high-throughput sequencing technologies allow researchers to expand their search beyond the exome and detect intronic or intragenic mutations that also have the potential to contribute to ASD etiology. Still, estimating the contribution of any variant to ASD phenotype remains an important consideration.
Similar to the SHANK3 and NRXN1 mutations described above, gene-linked mutations that disrupt splicing are core components of tuberous sclerosis (TSC) and neurofibromatosis type 1 (NF-1), which contribute to syndromic ASD diagnoses. Mutations in one of two genes, TSC1 encoding hamartin or TSC2 encoding tuberin, cause TSC (Narayanan, 2003), while NF-1 results from a mutation in the neurofibromin 1 gene (NF1), usually producing a dysfunctional truncated protein (Heim et al., 1994). Loss of any one of these three genes is sufficient to alter neuron morphology (Li et al., 2001; Tavazoie et al., 2005; Hsueh, 2007) and perturb glutamatergic and GABAergic signaling (Husi et al., 2000; Costa et al., 2002; von der Brelie et al., 2006). Two separate studies examining mutations in TSC1 and TSC2 revealed a high proportion of pathogenic splice-site mutations that introduce premature stop codons and truncate the predicted protein sequence, disrupting normal function (Mayer et al., 1999, 2000). Similarly, cDNA screens of NF-1 patients reveal that splice-site mutations constitute the most common type of mutation causing NF-1, occurring in 28–50% of patients (Ars et al., 2000; Messiaen et al., 2000). Despite the high incidence of splice-site mutations in these three genes, the relationship between misspliced mRNA transcripts and ASD diagnosis remains to be investigated.
To this point, we have focused on cis-acting splice-site mutations that are potentially contributing to ASD. Here we focus on four multifunctional trans-acting proteins that contribute to syndromic ASDs that also demonstrate some capacity to perform alternative splicing. For these proteins, we note that alternative splicing is but one of their demonstrated functions and not necessarily driving the autistic phenotype.
Fragile X syndrome is responsible for approximately 2% of all ASD cases (Zafeiriou et al., 2007). The affected protein in fragile X (FMRP) contains a KH-type RNA binding domain that associates with structural “kissing complex” motifs present in the target mRNAs, which prevents polyribosome binding, thereby gating activity-dependent protein translation at the synapse (Darnell et al., 2005). Mutations within this domain can result in a mental retardation phenotype (De Boulle et al., 1993). FMRP can also bind RNAs containing structural motifs known as guanine(G)-quartets (Darnell et al., 2001) through which it acts as a splicing modulator in vitro (Didiot et al., 2008). The possibility that FMRP contributes to RNA splicing, in addition to its well-validated role in protein translation, is an attractive hypothesis that would add an additional layer of regulatory function to the repertoire of FMRP. The FMRP isoform responsible for translation regulation is located in the cytoplasm (Khandjian et al., 2004), but alternative splicing of FMR1 can result in nuclear localization of the protein (Sittler et al., 1996). In mouse, FMRP interacts with mRNA transcribed from neuroligin genes NLGN1 and NLGN2 in vivo, and FMRP null mice have reduced NLGN1 but not NLGN2 protein levels, suggesting a role of FMRP in NLGN1 translation (Dahlhaus and El-Husseini, 2010). However, the significance of FMRP on neuroligin transcript splicing remains to be investigated, especially considering that the capacity for FMRP to perform splicing has only been demonstrated in vitro on RNA transcribed from the fragile X gene (FMR1; Didiot et al., 2008).
Behavioral overlap exists between autism and Rett syndrome (Mount et al., 2003), but the presence of a causative genetic mutation precludes ASD diagnosis for this pervasive developmental disorder. The affected gene, MECP2, is also implicated as an ASD risk gene through haplotype analysis in families with autistic probands (Loat et al., 2008). MECP2 is most frequently studied for its role in epigenetic transcriptional repression, but it also binds RNA with high affinity via its RNA binding domains (Bienvenu and Chelly, 2006), through which MECP2 can act as an alternative splicing regulator in vitro (Young et al., 2005). In MeCP2 null mice, aberrant splicing patterns are observed for brain-expressed genes (Young et al., 2005), although it is not clear whether this is the direct results of MECP2 on RNA splicing or an indirect result of MECP2 acting as a transcriptional regulator of other undetermined splice factors.
Prader–Willi syndrome is a genetically complex disorder typically resulting from de novo deletion of paternally inherited genes on chromosomal region 15q13-11. Most notably, affected genes include the SNURF–SNRPN complex and small nucleolar RNAs (snoRNAs) also encoded within SNRPN. The snoRNAs encoded within SNRPN regulate the expression of the anti-sense encoded UBE3A (Runte et al., 2004), which is particularly significant given the gene-dose dependent effect of this region on autistic phenotypes; i.e., maternal uniparental disomy results in greater incidence of ASD (Veltman et al., 2004). The protein encoded by UBE3A localizes to synapses in cerebellar Purkinje cells and pyramidal neurons of the hippocampus and cortex where it contributes to dendritic spine development (Dindot et al., 2008). At least one snoRNA encoded in SNRPN, HBII-52, directs alternative splicing of numerous target genes (Kishore et al., 2010), including the serotonin 2C receptor (Kishore and Stamm, 2006). In addition to snoRNAs, the protein encoded by SNRPN is a member of the small nuclear ribonucleoprotein complex, which is formed on pre-mRNA and is responsible for constitutive RNA splicing (Horn et al., 1992). SNRPN and some specific snoRNAs are primarily expressed in neurons (Schmauss et al., 1992), suggesting a tissue-specific role in modulating gene expression and splicing, but no specific relationship to CAM splicing has been demonstrated.
Finally, the gene encoding RNA binding protein, FOX-1 homolog 1 (RBFOX1; also known as A2BP1 and FOX1) was found to be partially translocated and deleted in an autistic female patient (Martin et al., 2007; Sebat et al., 2007). In vertebrates, FOX-1 acts as a tissue-specific splicing regulator (Jin et al., 2003; Underwood et al., 2005) by binding pre-mRNA and preventing the formation of the spliceosomal early “E” complex (Fukumura et al., 2007; Zhou and Lou, 2008). Depending on the proximity of the FOX-1 protein to an alternative exon, it promotes either inclusion or exclusion of the target exon (Underwood et al., 2005). Analogous function in humans is inferred from the conserved binding properties of the human protein to specific cis-regulatory elements in RNA (Ponthier et al., 2006) that are spatially conserved in the human genome surrounding brain-specific alternative exons (Minovitsky et al., 2005), and the ability of the murine protein to direct the alternative splicing of human genes in vitro (Fukumura et al., 2007; Zhou and Lou, 2008). Apart from autism, de novo translocation/deletions of this gene have been associated with epilepsy and mental retardation (Bhalla et al., 2004). The extensive network of candidate genes targeted by FOX-1 includes the autism-associated genes NLGN3, NLGN4X, NRCAM, NRXN1, and PCDH9 (Zhang et al., 2008), supporting observations of diverse disease phenotypes when mutated.
At present, a role for trans-acting factors contributing to ASD through their alternative splicing capabilities is unsubstantiated. Two major questions need resolved before the role of these trans-acting factors in autism can be properly evaluated. First, do these proteins act upon ASD risk genes as splice factors in vivo? Second, even if they are shown to interact with RNA transcribed from risk genes to direct alternative splicing, is this relationship relevant to the manifestation of the disorder? Methods such as UV-based protein-RNA crosslinking, coupled with ultra high-throughput sequencing (Licatalosi et al., 2008) are available to begin answering these questions.
Identifying novel ASD risk genes remains an ongoing endeavor. As more susceptibility genes are verified, accumulating evidence suggests a critical role for disrupted synaptic signaling due to genetic mutations in CAMs. Observations of frequent and necessary alternative splicing of CAMs in synaptic signaling, coupled with a greater appreciation for the role RNA processing defects in disease, demands a critical review of early findings in autism. Studies where mutations were uncovered in cis-regulatory splice sites (SHANK3, NRXN1, TSC1, TSC2, and NF1) and characterizations of aberrant mRNA expression from autistic patients (NLGN3, NLGN4X, and CADPS2) support the possibility that splicing defects can contribute to autism, while more evidence is required to determine the splicing capacity of trans-acting ASD risk proteins (FMRP, MECP2, SNRPN, and FOX-1) and subsequent effects related to ASD behaviors or etiology. Genetic variants that cause aberrant splicing can fill part of the missing heritability in ASD, but this question remains to be addressed in a sufficient manner. First, however, we need to address whether disrupted alternative CAM splicing – while critical to synaptic function – can be responsible for behaviors inherent to autism.
A recent study found that several genes encoding CAMs expressed in cortical brain regions crucial to language development are subject to rapid evolutionary pressure specific to humans (Johnson et al., 2009). This finding is consistent with previous observations of accelerated evolution in non-coding sequences near neuronal CAM genes (Prabhakar et al., 2006). Alternatively spliced exons, in general, undergo accelerated evolution of the protein subsequences they encode in an inverse relationship to the frequency with which they are included in mature mRNA (Xing and Lee, 2005), i.e., alternative exons with low mRNA inclusion have a high rate of protein-altering mutations. If one considers that alternative splicing is largely directed through intronic cis-acting regulatory sequences, perhaps subject to accelerated evolution, it is conceivable that higher mutation rates in these regulatory sequences result in greater variability in alternative splicing, and when alternative exons are included, they are more likely to contain a mutation that changes the encoded protein. Because CAM gene expression is enriched in brain regions crucial for language processing, sociability, and emotion processing (Johnson et al., 2009), selection pressures related to the rapid evolution of these genes could preferentially impact these human functions. In fact, CAMs were frequently identified as differentially expressed and alternatively spliced in human versus mouse brains, as shown with CNTNAP2 in the prefrontal cortex (Johnson et al., 2009), which is directly implicated in familial autism (Arking et al., 2008). From the studies presented in this review, one can speculate that the alternative splicing dysfunction must have a measurable effect on signaling at glutamatergic and/or GABAergic synapses in brain areas related to the affected behaviors, thereby producing an autistic phenotype. At present, quantitative and qualitative measures of spliceoform expression across CAM genes are lacking; nevertheless, the high prevalence of aberrant splicing in synaptic genes and their role in behaviors related to ASD demands further attention.
It is increasingly evident that genetic variation outside of coding regions contributes to phenotypic diversity and disease. Now that candidate genes are identified as having pathogenic mutations in coding regions, the next steps require analysis of intronic and intergenic regions that regulate the transcription and processing of the risk gene RNAs. Understanding the genetic mechanisms contributing to a phenotype (i.e., increased versus decreased expression of any given spliceoform) provides a strong basis for forming hypotheses about the microenvironment where the expressed gene has influence, and eventually even hypotheses about entire biological systems or neural circuits. Furthermore, in highly heritable disorders, the most immediate phenotype resulting from a genetic mutation is observable at the RNA level. Examining more distant phenotypes to uncover genetic contributions to ASD, such as complex behaviors, introduces more opportunity for environmental influence, consequently diluting the ability to detect subtle genetic factors.
Sequence variants that affect splicing quantitatively or those producing an unexpected novel spliceoform expressed at low levels are difficult to detect, especially when expressed in discrete cell types. Even when the unique spliceoforms are known, measuring their expression requires tissues relevant to the disorder from affected individuals. To that end, the Autism Tissue Program was established by Autism Speaks (http://www.autismtissueprogram.org). Measuring allelic differences in mRNA splicing in brain tissues is a powerful approach for uncovering splicing polymorphisms in ASD risk genes. For example, using this approach in human brain tissues, we have identified two frequent intronic SNPs in the dopamine D2 receptor (DRD2) that alter the splicing ratios between the short (D2S) and long (D2L) isoforms of the receptor, which correlated with working memory and activation of the striatum (Zhang et al., 2007). Our laboratory is now coupling this allele-specific approach with high-throughput sequencing to assay the entire transcriptome for the presence of functional genetic variants that affect RNA expression and alternative splicing of genes expressed in human cortex.
Newfound genetic candidates require validation of their function at the level of cellular signaling. A more cohesive analysis of genetics and synaptic function can be accomplished through the use of inducible pluripotent stem cells derived from affected individuals, which allows an ex vivo analysis of specialized differentiated cells, such as neurons, from living subjects (for review, see Pfannkuche et al., 2010). Advances in understanding alternative gene splicing have clarified the consequences of aberrant splicing in many neurological diseases (Cooper et al., 2009). Autism appears to be another disorder in which disrupted mRNA splicing possibly contributes to etiology, although it remains to be seen whether aberrant splicing is recapitulated in tissues germane to the disorder, such as the brain.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355.
Arking, D. E., Cutler, D. J., Brune, C. W., Teslovich, T. M., West, K., Ikeda, M., Rea, A., Guy, M., Lin, S., Cook, E. H., and Chakravarti, A. (2008). A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 82, 160–164.
Ars, E., Serra, E., Garcia, J., Kruyer, H., Gaona, A., Lazaro, C., and Estivill, X. (2000). Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum. Mol. Genet. 9, 237–247.
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., Volker, E., Roberts, W., Szatmari, P., Pinto, D., Bonin, M., Riess, A., Engels, H., Sprengel, R., Scherer, S. W., and Rappold, G. A. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491.
Betancur, C., Sakurai, T., and Buxbaum, J. D. (2009). The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 32, 402–412.
Bhalla, K., Phillips, H. A., Crawford, J., McKenzie, O. L., Mulley, J. C., Eyre, H., Gardner, A. E., Kremmidiotis, G., and Callen, D. F. (2004). The de novo chromosome 16 translocations of two patients with abnormal phenotypes (mental retardation and epilepsy) disrupt the A2BP1 gene. J. Hum. Genet. 49, 308–311.
Bienvenu, T., and Chelly, J. (2006). Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 7, 415–426.
Blasi, F., Bacchelli, E., Pesaresi, G., Carone, S., Bailey, A. J., and Maestrini, E. (2006). Absence of coding mutations in the X-linked genes neuroligin 3 and neuroligin 4 in individuals with autism from the IMGSAC collection. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141B, 220–221.
Blundell, J., Tabuchi, K., Bolliger, M. F., Blaiss, C. A., Brose, N., Liu, X., Sudhof, T. C., and Powell, C. M. (2009). Increased anxiety-like behavior in mice lacking the inhibitory synapse cell adhesion molecule neuroligin 2. Genes Brain Behav. 8, 114–126.
Bonaglia, M. C., Giorda, R., Borgatti, R., Felisari, G., Gagliardi, C., Selicorni, A., and Zuffardi, O. (2001). Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am. J. Hum. Genet. 69, 261–268.
Bonaglia, M. C., Giorda, R., Mani, E., Aceti, G., Anderlid, B. M., Baroncini, A., Pramparo, T., and Zuffardi, O. (2006). Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J. Med. Genet. 43, 822–828.
Boucard, A. A., Chubykin, A. A., Comoletti, D., Taylor, P., and Sudhof, T. C. (2005). A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 48, 229–236.
Cartegni, L., Chew, S. L., and Krainer, A. R. (2002). Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 3, 285–298.
Chih, B., Engelman, H., and Scheiffele, P. (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307, 1324–1328.
Chih, B., Gollan, L., and Scheiffele, P. (2006). Alternative splicing controls selective trans-synaptic interactions of the neuroligin–neurexin complex. Neuron 51, 171–178.
Chubykin, A. A., Atasoy, D., Etherton, M. R., Brose, N., Kavalali, E. T., Gibson, J. R., and Sudhof, T. C. (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931.
Comoletti, D., Flynn, R., Jennings, L. L., Chubykin, A., Matsumura, T., Hasegawa, H., Sudhof, T. C., and Taylor, P. (2003). Characterization of the interaction of a recombinant soluble neuroligin-1 with neurexin-1beta. J. Biol. Chem. 278, 50497–50505.
Costa, R. M., Federov, N. B., Kogan, J. H., Murphy, G. G., Stern, J., Ohno, M., Kucherlapati, R., Jacks, T., and Silva, A. J. (2002). Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 415, 526–530.
Cusmano-Ozog, K., Manning, M. A., and Hoyme, H. E. (2007). 22q13.3 deletion syndrome: a recognizable malformation syndrome associated with marked speech and language delay. Am. J. Med. Genet. C Semin. Med. Genet. 145C, 393–398.
Dahlhaus, R., and El-Husseini, A. (2010). Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav. Brain Res. 208, 96–105.
Darnell, J. C., Fraser, C. E., Mostovetsky, O., Stefani, G., Jones, T. A., Eddy, S. R., and Darnell, R. B. (2005). Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 19, 903–918.
Darnell, J. C., Jensen, K. B., Jin, P., Brown, V., Warren, S. T., and Darnell, R. B. (2001). Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107, 489–499.
De Boulle, K., Verkerk, A. J., Reyniers, E., Vits, L., Hendrickx, J., Van Roy, B., Van den Bos, F., de Graaff, E., Oostra, B. A., and Willems, P. J. (1993). A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet. 3, 31–35.
Didiot, M. C., Tian, Z., Schaeffer, C., Subramanian, M., Mandel, J. L., and Moine, H. (2008). The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res. 36, 4902–4912.
Dindot, S. V., Antalffy, B. A., Bhattacharjee, M. B., and Beaudet, A. L. (2008). The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 17, 111–118.
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., Nygren, G., Rastam, M., Gillberg, I. C., Anckarsater, H., Sponheim, E., Goubran-Botros, H., Delorme, R., Chabane, N., Mouren-Simeoni, M. C., de Mas, P., Bieth, E., Roge, B., Heron, D., Burglen, L., Gillberg, C., Leboyer, M., and Bourgeron, T. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27.
Eran, A., Graham, K. R., Vatalaro, K., McCarthy, J., Collins, C., Peters, H., Brewster, S. J., Hanson, E., Hundley, R., Rappaport, L., Holm, I. A., Kohane, I. S., and Kunkel, L. M. (2009). Comment on “Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients.” J. Clin. Invest. 119, 679–680; author reply 680–671.
Fabrichny, I. P., Leone, P., Sulzenbacher, G., Comoletti, D., Miller, M. T., Taylor, P., Bourne, Y., and Marchot, P. (2007). Structural analysis of the synaptic protein neuroligin and its beta-neurexin complex: determinants for folding and cell adhesion. Neuron 56, 979–991.
Feng, J., Schroer, R., Yan, J., Song, W., Yang, C., Bockholt, A., Cook, E. H. Jr., Skinner, C., Schwartz, C. E., and Sommer, S. S. (2006). High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett. 409, 10–13.
Fukumura, K., Kato, A., Jin, Y., Ideue, T., Hirose, T., Kataoka, N., Fujiwara, T., Sakamoto, H., and Inoue, K. (2007). Tissue-specific splicing regulator Fox-1 induces exon skipping by interfering E complex formation on the downstream intron of human F1gamma gene. Nucleic Acids Res. 35, 5303–5311.
Gauthier, J., Bonnel, A., St-Onge, J., Karemera, L., Laurent, S., Mottron, L., Fombonne, E., Joober, R., and Rouleau, G. A. (2005). NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 132B, 74–75.
Gauthier, J., Spiegelman, D., Piton, A., Lafreniere, R. G., Laurent, S., St-Onge, J., Lapointe, L., Hamdan, F. F., Cossette, P., Mottron, L., Fombonne, E., Joober, R., Marineau, C., Drapeau, P., and Rouleau, G. A. (2009). Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 421–424.
Gibson, J. R., Huber, K. M., and Sudhof, T. C. (2009). Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons. J. Neurosci. 29, 13883–13897.
Heim, R. A., Silverman, L. M., Farber, R. A., Kam-Morgan, L. N., and Luce, M. C. (1994). Screening for truncated NF1 proteins. Nat. Genet. 8, 218–219.
Hines, R. M., Wu, L., Hines, D. J., Steenland, H., Mansour, S., Dahlhaus, R., Singaraja, R. R., Cao, X., Sammler, E., Hormuzdi, S. G., Zhuo, M., and El-Husseini, A. (2008). Synaptic imbalance, stereotypies, and impaired social interactions in mice with altered neuroligin 2 expression. J. Neurosci. 28, 6055–6067.
Horn, D. A., Suburo, A., Terenghi, G., Hudson, L. D., Polak, J. M., and Latchman, D. S. (1992). Expression of the tissue specific splicing protein SmN in neuronal cell lines and in regions of the brain with different splicing capacities. Brain Res. Mol. Brain Res. 16, 13–19.
Huang, Z. J., and Scheiffele, P. (2008). GABA and neuroligin signaling: linking synaptic activity and adhesion in inhibitory synapse development. Curr. Opin. Neurobiol. 18, 77–83.
Husi, H., Ward, M. A., Choudhary, J. S., Blackstock, W. P., and Grant, S. G. (2000). Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 3, 661–669.
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., Soderstrom, H., Giros, B., Leboyer, M., Gillberg, C., and Bourgeron, T. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29.
Jin, Y., Suzuki, H., Maegawa, S., Endo, H., Sugano, S., Hashimoto, K., Yasuda, K., and Inoue, K. (2003). A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 22, 905–912.
Johnson, M. B., Kawasawa, Y. I., Mason, C. E., Krsnik, Z., Coppola, G., Bogdanovic, D., Geschwind, D. H., Mane, S. M., State, M. W., and Sestan, N. (2009). Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509.
Kang, Y., Zhang, X., Dobie, F., Wu, H., and Craig, A. M. (2008). Induction of GABAergic postsynaptic differentiation by alpha-neurexins. J. Biol. Chem. 283, 2323–2334.
Khandjian, E. W., Huot, M. E., Tremblay, S., Davidovic, L., Mazroui, R., and Bardoni, B. (2004). Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proc. Natl. Acad. Sci. U.S.A. 101, 13357–13362.
Kishore, S., Khanna, A., Zhang, Z., Hui, J., Balwierz, P. J., Stefan, M., Beach, C., Nicholls, R. D., Zavolan, M., and Stamm, S. (2010). The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing. Hum. Mol. Genet. 19, 1153–1164.
Kishore, S., and Stamm, S. (2006). The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 311, 230–232.
Ko, J., Fuccillo, M. V., Malenka, R. C., and Sudhof, T. C. (2009). LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron 64, 791–798.
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M. P., Raynaud, M., Ronce, N., Lemonnier, E., Calvas, P., Laudier, B., Chelly, J., Fryns, J. P., Ropers, H. H., Hamel, B. C., Andres, C., Barthelemy, C., Moraine, C., and Briault, S. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557.
Lawson-Yuen, A., Saldivar, J. S., Sommer, S., and Picker, J. (2008). Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur. J. Hum. Genet. 16, 614–618.
Lee, J. A., Tang, Z. Z., and Black, D. L. (2009). An inducible change in Fox-1/A2BP1 splicing modulates the alternative splicing of downstream neuronal target exons. Genes Dev. 23, 2284–2293.
Li, C., Cheng, Y., Gutmann, D. A., and Mangoura, D. (2001). Differential localization of the neurofibromatosis 1 (NF1) gene product, neurofibromin, with the F-actin or microtubule cytoskeleton during differentiation of telencephalic neurons. Brain Res. Dev. Brain Res. 130, 231–248.
Li, Q., Lee, J. A., and Black, D. L. (2007). Neuronal regulation of alternative pre-mRNA splicing. Nat. Rev. Neurosci. 8, 819–831.
Licatalosi, D. D., Mele, A., Fak, J. J., Ule, J., Kayikci, M., Chi, S. W., Clark, T. A., Schweitzer, A. C., Blume, J. E., Wang, X., Darnell, J. C., and Darnell, R. B. (2008). HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–469.
Loat, C. S., Curran, S., Lewis, C. M., Duvall, J., Geschwind, D., Bolton, P., and Craig, I. W. (2008). Methyl-CpG-binding protein 2 polymorphisms and vulnerability to autism. Genes Brain Behav. 7, 754–760.
Lopez-Bigas, N., Audit, B., Ouzounis, C., Parra, G., and Guigo, R. (2005). Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 579, 1900–1903.
Martin, C. L., Duvall, J. A., Ilkin, Y., Simon, J. S., Arreaza, M. G., Wilkes, K., Alvarez-Retuerto, A., Whichello, A., Powell, C. M., Rao, K., Cook, E., and Geschwind, D. H. (2007). Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 869–876.
Mayer, K., Ballhausen, W., Leistner, W., and Rott, H. (2000). Three novel types of splicing aberrations in the tuberous sclerosis TSC2 gene caused by mutations apart from splice consensus sequences. Biochim. Biophys. Acta 1502, 495–507.
Mayer, K., Ballhausen, W., and Rott, H. D. (1999). Mutation screening of the entire coding regions of the TSC1 and the TSC2 gene with the protein truncation test (PTT) identifies frequent splicing defects. Hum. Mutat. 14, 401–411.
Messiaen, L. M., Callens, T., Mortier, G., Beysen, D., Vandenbroucke, I., Van Roy, N., Speleman, F., and Paepe, A. D. (2000). Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 15, 541–555.
Minovitsky, S., Gee, S. L., Schokrpur, S., Dubchak, I., and Conboy, J. G. (2005). The splicing regulatory element, UGCAUG, is phylogenetically and spatially conserved in introns that flank tissue-specific alternative exons. Nucleic Acids Res. 33, 714–724.
Mount, R. H., Charman, T., Hastings, R. P., Reilly, S., and Cass, H. (2003). Features of autism in Rett syndrome and severe mental retardation. J. Autism Dev. Disord. 33, 435–442.
Narayanan, V. (2003). Tuberous sclerosis complex: genetics to pathogenesis. Pediatr. Neurol. 29, 404–409.
Pagenstecher, C., Wehner, M., Friedl, W., Rahner, N., Aretz, S., Friedrichs, N., Sengteller, M., Henn, W., Buettner, R., Propping, P., and Mangold, E. (2006). Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum. Genet. 119, 9–22.
Pampanos, A., Volaki, K., Kanavakis, E., Papandreou, O., Youroukos, S., Thomaidis, L., Karkelis, S., Tzetis, M., and Kitsiou-Tzeli, S. (2009). A substitution involving the NLGN4 gene associated with autistic behavior in the Greek population. Genet. Test Mol. Biomarkers 13, 611–615.
Pfannkuche, K., Hannes, T., Khalil, M., Noghabi, M. S., Morshedi, A., Hescheler, J., and Droge, P. (2010). Induced pluripotent stem cells: a new approach for physiological research. Cell. Physiol. Biochem. 26, 105–124.
Ponthier, J. L., Schluepen, C., Chen, W., Lersch, R. A., Gee, S. L., Hou, V. C., Lo, A. J., Short, S. A., Chasis, J. A., Winkelmann, J. C., and Conboy, J. G. (2006). Fox-2 splicing factor binds to a conserved intron motif to promote inclusion of protein 4.1R alternative exon 16. J. Biol. Chem. 281, 12468–12474.
Prabhakar, S., Noonan, J. P., Paabo, S., and Rubin, E. M. (2006). Accelerated evolution of conserved noncoding sequences in humans. Science 314, 786.
Roussignol, G., Ango, F., Romorini, S., Tu, J. C., Sala, C., Worley, P. F., Bockaert, J., and Fagni, L. (2005). Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J. Neurosci. 25, 3560–3570.
Runte, M., Kroisel, P. M., Gillessen-Kaesbach, G., Varon, R., Horn, D., Cohen, M. Y., Wagstaff, J., Horsthemke, B., and Buiting, K. (2004). SNURF-SNRPN and UBE3A transcript levels in patients with Angelman syndrome. Hum. Genet. 114, 553–561.
Sadakata, T., Washida, M., Iwayama, Y., Shoji, S., Sato, Y., Ohkura, T., Katoh-Semba, R., Nakajima, M., Sekine, Y., Tanaka, M., Nakamura, K., Iwata, Y., Tsuchiya, K. J., Mori, N., Detera-Wadleigh, S. D., Ichikawa, H., Itohara, S., Yoshikawa, T., and Furuichi, T. (2007a). Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients. J. Clin. Invest. 117, 931–943.
Sadakata, T., Kakegawa, W., Mizoguchi, A., Washida, M., Katoh-Semba, R., Shutoh, F., Okamoto, T., Nakashima, H., Kimura, K., Tanaka, M., Sekine, Y., Itohara, S., Yuzaki, M., Nagao, S., and Furuichi, T. (2007b). Impaired cerebellar development and function in mice lacking CAPS2, a protein involved in neurotrophin release. J. Neurosci. 27, 2472–2482.
Schmauss, C., Brines, M. L., and Lerner, M. R. (1992). The gene encoding the small nuclear ribonucleoprotein-associated protein N is expressed at high levels in neurons. J. Biol. Chem. 267, 8521–8529.
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T., Yamrom, B., Yoon, S., Krasnitz, A., Kendall, J., Leotta, A., Pai, D., Zhang, R., Lee, Y. H., Hicks, J., Spence, S. J., Lee, A. T., Puura, K., Lehtimaki, T., Ledbetter, D., Gregersen, P. K., Bregman, J., Sutcliffe, J. S., Jobanputra, V., Chung, W., Warburton, D., King, M. C., Skuse, D., Geschwind, D. H., Gilliam, T. C., Ye, K., and Wigler, M. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449.
Shapiro, L., Love, J., and Colman, D. R. (2007). Adhesion molecules in the nervous system: structural insights into function and diversity. Annu. Rev. Neurosci. 30, 451–474.
Sittler, A., Devys, D., Weber, C., and Mandel, J. L. (1996). Alternative splicing of exon 14 determines nuclear or cytoplasmic localisation of fmr1 protein isoforms. Hum. Mol. Genet. 5, 95–102.
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., and Sudhof, T. C. (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76.
Talebizadeh, Z., Lam, D. Y., Theodoro, M. F., Bittel, D. C., Lushington, G. H., and Butler, M. G. (2006). Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J. Med. Genet. 43, e21.
Tavazoie, S. F., Alvarez, V. A., Ridenour, D. A., Kwiatkowski, D. J., and Sabatini, B. L. (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734.
Teraoka, S. N., Telatar, M., Becker-Catania, S., Liang, T., Onengüt, S., Tolun, A., Chessa, L., Sanal, O., Bernatowska, E., Gatti, R. A., and Concannon, P. (1999). Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am. J. Hum. Genet. 64, 1617–1631.
Underwood, J. G., Boutz, P. L., Dougherty, J. D., Stoilov, P., and Black, D. L. (2005). Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol. Cell. Biol. 25, 10005–10016.
Varoqueaux, F., Aramuni, G., Rawson, R. L., Mohrmann, R., Missler, M., Gottmann, K., Zhang, W., Sudhof, T. C., and Brose, N. (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754.
Veltman, M. W., Thompson, R. J., Roberts, S. E., Thomas, N. S., Whittington, J., and Bolton, P. F. (2004). Prader-Willi syndrome – a study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur. Child. Adolesc. Psychiatry 13, 42–50.
Vincent, J. B., Kolozsvari, D., Roberts, W. S., Bolton, P. F., Gurling, H. M., and Scherer, S. W. (2004). Mutation screening of X-chromosomal neuroligin genes: no mutations in 196 autism probands. Am. J. Med. Genet. B Neuropsychiatr. Genet. 129B, 82–84.
von der Brelie, C., Waltereit, R., Zhang, L., Beck, H., and Kirschstein, T. (2006). Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur. J. Neurosci. 23, 686–692.
Wang, G. S., and Cooper, T. A. (2007). Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 8, 749–761.
Xing, Y., and Lee, C. (2005). Evidence of functional selection pressure for alternative splicing events that accelerate evolution of protein subsequences. Proc. Natl. Acad. Sci. U.S.A. 102, 13526–13531.
Yan, J., Noltner, K., Feng, J., Li, W., Schroer, R., Skinner, C., Zeng, W., Schwartz, C. E., and Sommer, S. S. (2008). Neurexin 1alpha structural variants associated with autism. Neurosci. Lett. 438, 368–370.
Yan, J., Oliveira, G., Coutinho, A., Yang, C., Feng, J., Katz, C., Sram, J., Bockholt, A., Jones, I. R., Craddock, N., Cook, E. H., Jr., Vicente, A., and Sommer, S. S. (2005). Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 10, 329–332.
Ylisaukko-oja, T., Rehnstrom, K., Auranen, M., Vanhala, R., Alen, R., Kempas, E., Ellonen, P., Turunen, J. A., Makkonen, I., Riikonen, R., Nieminen-von Wendt, T., von Wendt, L., Peltonen, L., and Jarvela, I. (2005). Analysis of four neuroligin genes as candidates for autism. Eur. J. Hum. Genet. 13, 1285–1292.
Young, J. I., Hong, E. P., Castle, J. C., Crespo-Barreto, J., Bowman, A. B., Rose, M. F., Kang, D., Richman, R., Johnson, J. M., Berget, S., and Zoghbi, H. Y. (2005). Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. U.S.A. 102, 17551–17558.
Zafeiriou, D. I., Ververi, A., and Vargiami, E. (2007). Childhood autism and associated comorbidities. Brain Dev. 29, 257–272.
Zhang, C., Zhang, Z., Castle, J., Sun, S., Johnson, J., Krainer, A. R., and Zhang, M. Q. (2008). Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 22, 2550–2563.
Zhang, Y., Bertolino, A., Fazio, L., Blasi, G., Rampino, A., Romano, R., Lee, M. L., Xiao, T., Papp, A., Wang, D., and Sadee, W. (2007). Polymorphisms in human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity during working memory. Proc. Natl. Acad. Sci. U.S.A. 104, 20552–20557.
Zhou, H. L., and Lou, H. (2008). Repression of prespliceosome complex formation at two distinct steps by Fox-1/Fox-2 proteins. Mol. Cell. Biol. 28, 5507–5516.
Keywords: autism spectrum disorder, synaptogenesis, alternative RNA splicing, cellular adhesion molecules, neurexin, neuroligin, gene expression, neural development
Citation: Smith RM and Sadee W (2011) Synaptic signaling and aberrant RNA splicing in autism spectrum disorders. Front. Syn. Neurosci. 3:1. doi: 10.3389/fnsyn.2011.00001
Received: 25 April 2010;
Accepted: 12 January 2011;
Published online: 26 January 2011.
Edited by:
Kelsey Martin, University of California Los Angeles, USAReviewed by:
Peter Scheiffele, University of Basel, SwitzerlandCopyright: © 2011 Smith and Sadee. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Wolfgang Sadee, Department of Pharmacology, The Ohio State University, 333 West 10th Avenue, 5072 Graves Hall, Columbus, OH 43210, USA. e-mail:d29sZmdhbmcuc2FkZWVAb3N1bWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.