




94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Synaptic Neurosci., 22 December 2010
Volume 2 - 2010 | https://doi.org/10.3389/fnsyn.2010.00152
This article is part of the Research TopicOrganization of synaptic vesicle poolsView all 8 articles
Repeated release of transmitter from presynaptic elements depends on stimulus-induced Ca2+ influx together with recruitment and priming of synaptic vesicles from different vesicle pools. We have compared three different manipulations of synaptic strength, all of which are known to increase short-term synaptic efficacy through presynaptic mechanisms, in the glutamatergic CA3-to-CA1 stratum radiatum synapse in the mouse hippocampal slice preparation. Synaptic responses elicited from the readily releasable vesicle pool during low-frequency synaptic activation (0.1 Hz) were significantly enhanced by both the adenylate cyclase activator forskolin, the priming activator β-phorbol-12,13-dibutyrate (PDBu) and 4 mM [Ca2+]o, whereas during 20 Hz stimulation, the same manipulations reduced the time needed to reach the peak and increased the magnitude of the resulting frequency facilitation. In contrast, paired-pulse facilitations were unchanged in the presence of forskolin, decreased by 4 mM [Ca2+]o and essentially abolished by PDBu. The subsequent delayed response enhancement (DRE) responses, elicited during continuous 20 Hz stimulations and mediated by recruited vesicles, were enhanced by forskolin, essentially unchanged by PDBu and slightly decreased by 4 mM [Ca2+]o. Similar experiments done on slices devoid of the vesicle-associated synapsin I and II proteins indicated that synapsin I/II-induced enhancements of vesicle recruitment were restricted to Ca2+-induced frequency facilitations and forskolin-induced enhancements of the early DRE phase, whereas the proteins had minor effects during PDBu-treatment and represented constraints on late Ca2+-induced responses. The data indicate that in these glutamatergic synapses, the comparable enhancements of single synaptic responses induced by these biochemical mechanisms can be transformed during prolonged synaptic stimulation into highly distinct short-term plasticity patterns, which are partly dependent on synapsins I/II.
Presynaptic plasticity, which predominantly consists of rapid changes in transmitter release induced during synaptic activity and results in enhancements or depressions of synaptic responses, is partly caused by changing the release efficacy of synaptic vesicles originating in distinct vesicle pools (Attwood and Karunanithi, 2002; Zucker and Regehr, 2002; Rizzoli and Betz, 2005; Neher, 2006). Brief bursts of afferent stimulation often give rise to an early frequency facilitation deriving from the readily releasable vesicle pool (RRP; e.g., Zucker and Regehr, 2002; Neher and Sakaba, 2008). In contrast, contributions of vesicles from the reserve or resting pools are less well defined (Wesseling and Lo, 2002; Li et al., 2005; Fernandez-Alfonso and Ryan, 2008). In the well-characterized excitatory and facilitating CA3-to-CA1 hippocampal glutamatergic synapse, where the presynaptic elements contain RRPs of 5–10 vesicles and a total population of 80–100 vesicles (Harris and Sultan, 1995; Schikorski and Stevens, 1997), we have recently described a functionally defined phase designated the delayed response enhancement (DRE), which appears to represent a vesicle pool which begins to release transmitter when stimulation has lasted approximately 3.5 s. Around 75 stimuli contributed to this enhancement phase, which was followed by response decays (Jensen et al., 2007; see, however, Garcia-Perez et al., 2008). The putative vesicle pool involved in this response, which appeared to be dependent on the vesicle-associated synapsin I/II proteins, was found to be sensitive to temperature and F-actin cycling, and to be developmentally regulated (Jensen et al., 2007; Bogen et al., 2009). The exact identity and localization of these vesicles, as well as their physiological importance, remain, however, unclear.
In order to define possible physiological relationships between the vesicles present in the RRP and those responsible for the DRE phase, we have here examined these response phases during activation of three well-known intraterminal signaling pathways, including those regulated by cyclic AMP (cAMP), diacylglycerols (DAG) and increased levels of Ca2+, all of which may change vesicle recruitment, docking, priming, and/or exocytotic fusion of releasable synaptic vesicles (e.g., Hilfiker et al., 1999; Leenders and Sheng, 2005; Neher, 2006). For this purpose, we have used hippocampal slice preparations from wild-type (WT) and synapsin I/II double knock-out (DKO) mice, the latter showing a significant decrease in synaptic vesicles but no change in glutamate receptor levels (Owe et al., 2005; Bogen et al., 2006). These preparations were either equilibrated in the absence or presence of the adenylate cyclase activator forskolin or the DAG analog β-phorbol-12,13-dibutyrate, or were examined at different levels of [Ca2+]o (from 1 to 4 mM). Following equilibration, we examined responses obtained from the RRP vesicles by analyzing either fEPSPs obtained at 0.1 Hz stimulus frequency, paired-pulse facilitation (PPF) ratios obtained at 50 ms interstimulus intervals, or frequency facilitation magnitudes obtained during the initial part of prolonged 20 Hz stimulation trains. In contrast, responses from recruited vesicles giving rise to the delayed phases were examined by analyzing the DRE and subsequent responses during the final part of the 20 Hz stimulus trains. We show that stimulation-induced enhancements of synaptic responses induced by activation of the signaling pathways all gave comparable increases of fEPSPs during low-frequency stimulation, but that they gave different patterns of presynaptic modulation both during paired pulses, frequency facilitation and prolonged stimulation trains. Our data therefore indicate that in these prototypical glutamatergic excitatory synapses, different molecular stimuli which modulate temporally identical short-term plasticity phases may regulate functionally distinct vesicle pools.
Experiments were performed on hippocampal slices (Hvalby et al., 2006), prepared either from adult (3–6 months old) WT mice or from synapsin I and II DKO mice of the same age (Ferreira et al., 1998). The animals were killed with the general anesthetic Suprane (Baxter), the brains were removed and transverse slices (400 μm) were cut from the middle portion of each hippocampus with a vibroslicer in artificial cerebrospinal fluid (ACSF, 4°C, bubbled with 95% O2–5% CO2, pH 7.4) containing (in mM): 124 NaCl, 2 KCl, 1.25 KH2PO4, 2 MgSO4, 1 or 2 CaCl2, 26 NaHCO3, and 12 glucose. Slices were placed in a humidified interface chamber where the temperature was kept constant at 29°C. To avoid N-methyl-D-aspartic acid (NMDA) receptor-mediated synaptic plasticity, 50 μM DL-2-amino-5-phosphopentanoic acid (AP5; Sigma-Aldrich) was present throughout the experiments.
The effects of forskolin (50 μm; Sigma-Aldrich) and β-phorbol-12,13-dibutyrate (PDBu, 10 μM, Sigma-Aldrich) were analyzed following incubation of the slices with the drugs for at least 120 min, a time span which allowed the drugs to induce functional effects. During this period, the magnitudes of the field fEPSPs gradually increased in the presence of forskolin or PDBu until stable values were reached. The final concentrations of DMSO, which was present as a solvent, did not exceed 0.3%, which by itself had no effect on physiological responses (not shown). Similar incubations were performed on slice preparations in the absence of drugs.
Animal experiments were conducted according to the Norwegian Animal Welfare Act and the European Union’s Directive 86/609/EEC.
Orthodromic synaptic stimuli (50 μs, <300 μA, 0.1 Hz) were delivered alternately through two tungsten electrodes, one situated in the stratum radiatum and another in the stratum oriens (control pathway, used to continuously examine the general physiological stability of the preparation) of the CA1 region. Extracellular synaptic responses were monitored by two glass electrodes (filled with ACSF) placed in the corresponding synaptic layers. Following the presence of stable synaptic responses in both pathways (0.1 Hz stimulation) for at least 10–15 min, selective stimulation of the radiatum pathway was performed at 20 Hz for 60 s.
The synaptic strength was assessed by measuring the maximal slope of the rising phase (V/s) of the fEPSPs and normalizing the value of each response to the first response in the stimulation train. For every experiment, the PPF ratio (50 ms interstimulus interval) was calculated as fEPSP2 slope/fEPSP1 slope, i.e., the value of the second response in the stimulation train was divided by the value of the first response (Jensen et al., 2007). Three distinct time points were also used to characterize the time courses of the processes studied, i.e., (a) the time needed to reach the maximal magnitude of the initial frequency facilitation, (b) the transition point where the minimal value of the response during the subsequent decay period was observed, and (c) the time point of the maximal value reached during the DRE phase. Time points were determined in each of the similarly treated experiments and data were pooled. Values are presented as mean ± S.E.M., and statistical significance of differences was evaluated using a two-tailed, unpaired t-test.
In preparations from both WT and synapsin I/II DKO mice, low-frequency synaptic activation (0.1 Hz) of the afferent fibers in the hippocampal CA1 region (stratum radiatum) led to stable synaptic responses (Figure 1A). Presynaptic short-term forms of synaptic plasticity can be induced by repetitive Ca2+-influx occurring during presynaptic stimulation trains (Zucker and Regehr, 2002; Schneggenburger and Neher, 2005; Klyachko and Stevens, 2006). In agreement, application of a 20-Hz stimulation train (1 min duration) to the afferent fibers under standard conditions (2 mM CaCl2, 29°C, 50 μM AP5) induced a transient frequency facilitation, which reached a peak after a seconds, followed by a steep decay which reached an apparent minimum value after b seconds. Continuous stimulation of WT slices then gave a DRE phase which reached a maximum value at c seconds (Figure 2). In the DKO, the latter phase was virtually absent (Figure 1B). In earlier studies, the presence of DRE was found to be independent of stimulus frequency variation between 5 and 20 Hz, inhibitory GABAergic synaptic activity, or stimulus-induced desensitization of the α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA)-receptors (Jensen et al., 2007), but to be sensitive to actin filament turnover, developmental stage, and incubation temperature between 24 to 37°C (Jensen et al., 2007; Bogen et al., 2009). Other analyses of this synapse done in adult animals have also failed to reveal differences in either PPF or frequency facilitation when these genotypes were compared (Rosahl et al., 1995; Jensen et al., 2007; Bogen et al., 2009). Hence, present knowledge indicates that the major effects of synapsins I/II on presynaptic plasticity may predominantly be restricted to response enhancements during the DRE phase and the subsequent late response period (Figure 1B).
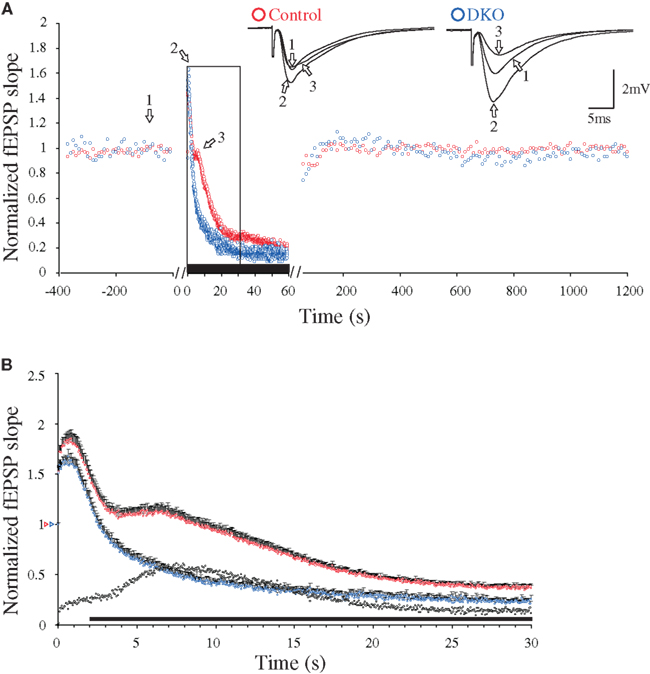
Figure 1. Changes in 20 Hz stimulation-induced delayed response enhancement (DRE) in hippocampal excitatory CA3-to-CA1 synapses, caused by inactivation of the synapsin I/II genes. (A) Normalized fEPSP slope measurements during afferent stimulation in stratum radiatum at 0.1 Hz, followed by 20 Hz for 60 s (indicated by black bar, note difference in time scales), and reversal to 0.1 Hz, obtained in two experiments performed at 29°C during NMDA receptor blockade (see Materials and Methods). Symbols in blue are from a synapsin I/II double knock-out (DKO) mouse and symbols in red are from a WT mouse, respectively. The insets show superimposed synaptic responses at the stimulation times indicated by arrows and numbers (left traces from the control mouse, right traces from the synapsin I/II DKO mouse). (B) Normalized and pooled fEPSP slope measurements during the initial 30 s of 20 Hz stimulation [boxed area in (A)] from WT (n = 22, red symbols) and DKO mice (n = 18, blue symbols). Vertical bars indicate S.E.M. Subtractions of the values obtained in synapsin I/II DKO experiments from those obtained in WT experiments are represented by the black, open symbols. The black horizontal bar along the abscissa indicates p < 0.05 when comparing the two genotypes. Colored open triangles on the ordinate point to the starting values for the different conditions.
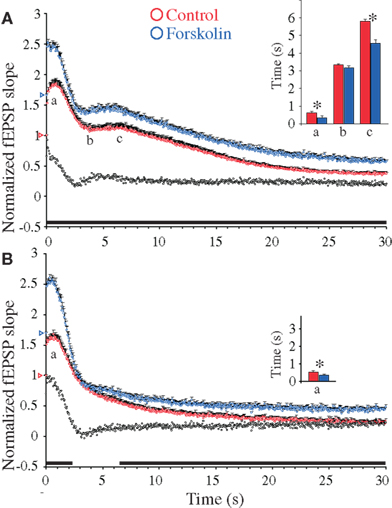
Figure 2. Forskolin-induced synaptic enhancements during 20 Hz stimulation in hippocampal excitatory CA3-to-CA1 synapses. (A) Normalized fEPSP slope measurements from CA3-to-CA1 synapses during 20 Hz stimulation in slices (n = 17) from WT mice treated with forskolin (50 μM, blue symbols) and in experiments without forskolin (n = 22, red symbols). Vertical bars indicate S.E.M. Subtractions of the values without forskolin from those obtained with forskolin are represented by the black, open symbols. The black horizontal bar along the abscissa indicates p < 0.05 when comparing the two situations. Colored open triangles on the ordinate point to the starting values for the different conditions. a Indicates time point of the maximum magnitude of the initial frequency facilitation; b time to the transition point between the initial frequency facilitation and the DRE; c time needed to reach the peak of the DRE. The average measurements from individual experiments are shown by inset histogram with corresponding colored vertical bars. Vertical bars indicate S.E.M. *p < 0.05 when compared to the control situation. (B) As in (A) but the slice experiments are from DKO mice treated with forskolin (50 μM, blue symbols) (n = 18) compared to experiments without the compound (n = 18, red symbols).
The adenylate cyclase activator forskolin enhances glutamatergic transmission in the CA1 stratum radiatum synapse in the hippocampal slice (Chavez-Noriega and Stevens, 1992), with a substantial contribution made by presynaptic mechanisms (Chavez-Noriega and Stevens, 1994). Early work suggested that activation of cAMP-dependent protein kinase (PKA) also enhanced postsynaptic AMPA-receptor-mediated responses (Greengard et al., 1991), thereby making it difficult to distinguish pre- from postsynaptic effects of cAMP. However, present knowledge suggests that the postsynaptic effects of PKA on glutamatergic transmission predominantly occur through NMDA-receptor stimulated trafficking of AMPA receptors to the synaptic membrane (Gao et al., 2006; Oh et al., 2006; Chen and Roche, 2007). Therefore, inclusion of AP5, a specific and competitive NMDA-receptor blocker, in our standard incubation medium, together with the use of DKO mice devoid of the presynaptically localized synapsin proteins, should minimize incorrect interpretation concerning the locus of forskolin effects.
In our experiments, addition of forskolin (50 μM) led to fEPSP responses being significantly enhanced to similar levels in both the WT (1.66 ± 0.11, n = 18) and DKO (1.70 ± 0.07, n = 16) preparations. Hence, forskolin induced an enhancement of basal, low-frequency-stimulated synaptic transmission irrespective of the synapsins I/II. In contrast, during a similar period when slices from both genotypes were equilibrated in the absence of drugs, we observed no significant changes in baseline responses at 120 min, either in the WT (1.07 ± 0.02, n = 19) or the DKO (1.03 ± 0.02, n = 20) preparations. Further analysis of synaptic release probability was done by giving a pair of subsequent stimuli, identical in strength and with interstimulus intervals of 50 ms, to the afferent fibers in stratum radiatum. In this situation, the second fEPSP was increased when compared to the first (Table 1). This PPF, believed to reside presynaptically, is thought to inversely reflect the release probability (Katz and Miledi, 1968; Zucker and Regehr, 2002). In the present experiments, the PPF ratios in forskolin-treated slices showed only statistically insignificant decreases when compared to untreated preparations in both genotypes (Table 1). Therefore, the probability of release of RRP vesicles appeared to be unchanged by forskolin, irrespective of the absence or presence of the synapsins I/II proteins.
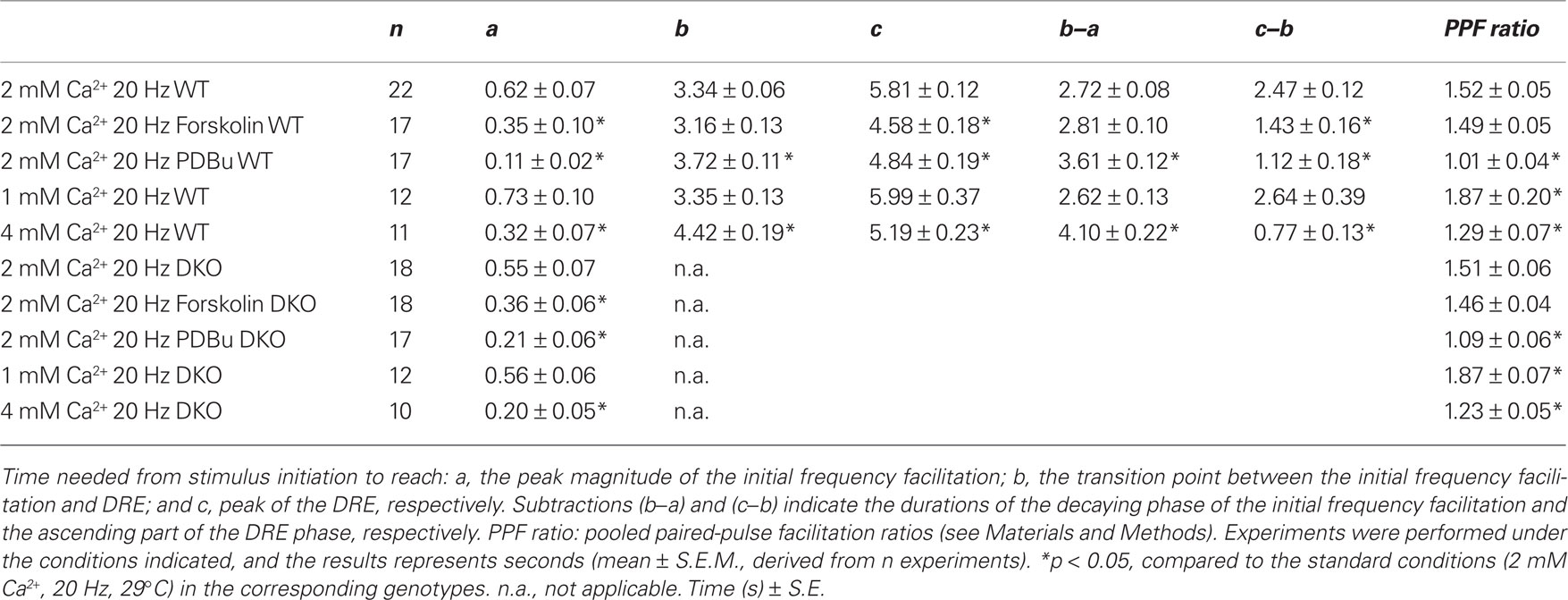
Table 1. Characterization of response transformations during stimulation of CA1 synapses in mouse hippocampus.
In contrast, analysis of the frequency facilitation which occurred during application of 20 Hz stimulation trains in the presence of forskolin showed strong response enhancements during the frequency facilitation in both WT and DKO mice, which reached peak magnitudes of 2.57 ± 0.10 and 2.60 ± 0.10, respectively (Figures 2A,B). Moreover, these forskolin-induced peaks occurred significantly earlier than those in untreated preparations, again through mechanisms that were independent of synapsins I/II (see Table 1 and inset histograms in Figures 2A,B). These results indicated that forskolin continuously induced enhanced release from RRP pools, without changing the probability of release from individual RRP vesicles.
In contrast, protracted 20 Hz stimulation gave continuous forskolin-induced response enhancements of both the DRE and the late responses in the WT preparation. This did not occur in the DKO preparation, thereby indicating that the forskolin- induced response enhancements occurring during the rising phase of the DRE depended entirely on the presence of synapsin I/II (compare Figures 2A,B). Thereafter, a forskolin effect reappeared in the DKO preparation after 6.5 s stimulation, when responses again became significantly increased in both genotypes. Hence, synapsins I/II appear pivotal for a forskolin-induced increase in transmitter release during a period restricted to the rising phase of the DRE, whereas forskolin-induced response enhancements occurring either prior to or subsequent to the enhancing part of the DRE phase all appeared to be mediated through synapsin I/II-independent mechanisms.
Tumor-promoting phorbol esters, stable analogs of the DAGs which are generated upon activation of phospholipase C, are known to activate both the calmodulin- and DAG-binding munc-13 proteins (Rhee et al., 2002; Brose et al., 2004, Junge et al., 2004) and the multifunctional protein kinase C family, all of which have been implicated in regulation of transmitter release (e.g., Billiard et al., 1997; Walaas, 1999; Searl and Silinsky, 2003; Wierda et al., 2007). In the present study, incubation of the slices with PDBu (10 μM) led to major and unexpected response patterns.
First, addition of PDBu during 0.1 Hz stimulation substantially enhanced the fEPSPs to comparable levels in the two genotypes (WT: 2.26 ± 0.35, n = 17; DKO: 1.95 ± 0.26, n = 16; p = 0.46). These PDBu-induced fEPSP enhancements were accompanied by almost abolished PPF ratios in both genotypes (Table 1), in accordance with earlier reports (Malenka et al., 1986; Gustafsson et al., 1988). Furthermore, during 20 Hz stimulation trains, the presence of PDBu led to small frequency facilitation responses which reached peak values more rapidly in the presence than in the absence of PDBu in both genotypes (Table 1). They also showed only modest magnitude enhancements, in the WT preparations reaching 2.34 ± 0.11 (n = 17) and in the DKO preparations reaching 2.21 ± 0.12 (n = 17; Figures 3A,B). Finally, after approximately 0.5–1 s, the effects of PDBu on response magnitudes disappeared in both genotypes, and during the subsequent decaying phase, the responses rapidly merged with the responses obtained in the untreated control preparations, without any difference occurring between the WT and DKO preparations.
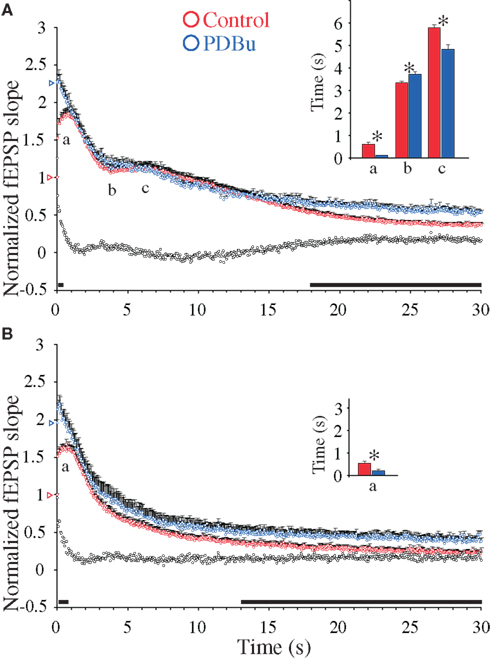
Figure 3. Phorbol ester-induced synaptic enhancements during 20 Hz stimulation in hippocampal excitatory CA3-to-CA1 synapses. (A) Normalized fEPSP slope measurements from CA3-to-CA1 synapses during 20 Hz stimulation in slices (n = 17) from WT mice treated with β-phorbol-12,13-dibutyrate (PDBu, 10 μM, blue symbols) and in experiments without PDBu (n = 22, red symbols). Vertical bars indicate S.E.M. Subtractions of the values without PDBu from those obtained with PDBu present are represented by the black, open symbols. The black horizontal bar along the abscissa indicates p < 0.05 when comparing the two situations. Colored open triangles on the ordinate point to the starting value for the different conditions. a Indicates time point of the maximum magnitude of the initial frequency facilitation; b time to the transition point between the initial frequency facilitation and the DRE; c time needed to reach the peak of the DRE. The average measurements from individual experiments are shown in inset histogram with corresponding colored vertical bars. Vertical bars indicate S.E.M. *p < 0.05 when compared to the control situation. (B) As in (A) but the slice experiments are from DKO mice treated with PDBu (10 μM, blue symbols; n = 17) compared to experiments without PDBu (n = 18, red symbols).
During subsequent stimulation, both the WT and DKO responses occurred at slightly different time points in the PDBu-treated as compared to the untreated preparations (Figures 3A,B). However, neither the WT (where a small DRE occurred) nor the DKO preparation (which is devoid of the DRE phase, see Figure 1B) showed significant changes in response magnitudes during PDBu treatment. In contrast, during the late response period a small, but significant PDBu-dependent response enhancement occurred, which lasted during the remaining part of the stimulation train. Since a similar late PDBu effect also occurred in the DKO, this effect was not dependent on synapsins I/II (Figures 3A,B).
Ca2+ influx through voltage-dependent channels followed by binding to synaptotagmin in the vesicle–SNARE complex are thought to be predominant factors in determining presynaptic strength (Lisman et al., 2007; Sun et al., 2007), and increasing [Ca2+]o has therefore been used to enhance synaptic responses (Zucker and Regehr, 2002; Schneggenburger and Neher, 2005). We compared the effects of varying [Ca2+]o between 1 and 4 mM, and standardized these experiments according to the normalized fEPSP values obtained in ACSF containing 2 mM CaCl2 in order to be able to compare the results to the ones obtained with forskolin and PDBu.
Equilibration of slices from WT and DKO mice in 1 mM CaCl2 significantly reduced the fEPSPs (0.53 ± 0.05, n = 12 and 0.50 ± 0.06, n = 10, respectively), whereas equilibration in 4 mM CaCl2 significantly increased the fEPSPs (1.25 ± 0.04, n = 19 and 1.33 ± 0.06, n = 16, respectively; Hvalby et al., 2006). Thus, these Ca2+-induced fEPSP changes were synapsin I/II-independent. Furthermore, both the increases in PPF ratios seen at 1 mM [Ca2+]o and the decreases in PPF ratios seen at 4 mM [Ca2+]o were synapsin I/II-independent (Table 1).
In contrast, previous analysis of responses obtained at 20 Hz frequency in this synapse has demonstrated the existence of a synapsin I/II-dependent frequency facilitation enhancement during the initial 40 stimuli which was present in 4 mM [Ca2+]o but absent in 1 mM [Ca2+]o (Hvalby et al., 2006). The present analysis demonstrates that after 4–6 stimulations in our preparations, a frequency facilitation was obtained which at 4 mM [Ca2+]o reached a peak magnitude of 1.86 ± 0.06 in WT, whereas a magnitude of 1.62 ± 0.04 was reached in the DKO (Figures 4A,B; Table 1). Moreover, the subsequent response decreases were also different in the two genotypes, being both more rapid and extensive in the DKO than in the WT (Figure 4). Taken together (Hvalby et al., 2006; and this study), these data indicate that in these synapses, the presence of synapsin I/II proteins induced an enhancement of the Ca2+-induced frequency facilitation. However, under the present experimental conditions this effect appeared to be restricted to supraphysiological levels of [Ca2+]o.
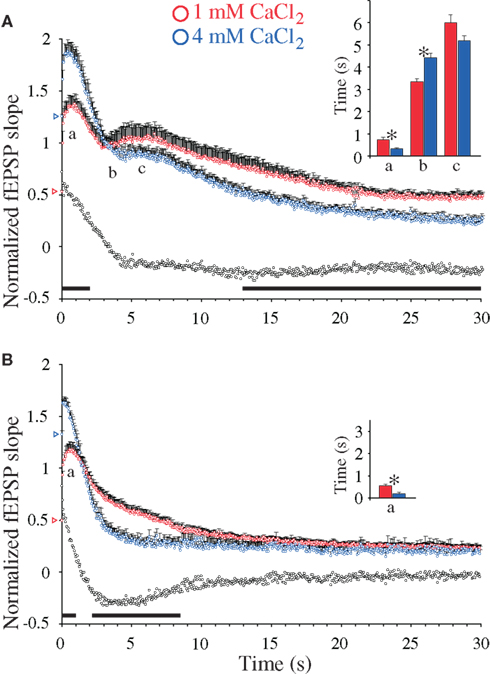
Figure 4. Calcium-induced synaptic enhancements during 20 Hz stimulation in hippocampal excitatory CA3-to-CA1 synapses. (A) Normalized fEPSP slope measurements from CA3-to-CA1 synapses during 20 Hz stimulation in slices (n = 12) from WT mice treated with 4 mM CaCl2 (blue symbols) and in experiments with 1 mM CaCl2 (n = 11, red symbols). Vertical bars indicate S.E.M. Subtractions of the values at 1 mM CaCl2 from those obtained at 4 mM CaCl2 are represented by the black, open symbols. The black horizontal bar along the abscissa indicates p < 0.05 when comparing the two situations. Colored open triangles on the ordinate point to the starting value for the different conditions. a Indicates time point of the maximum magnitude of the initial frequency facilitation; b time to the transition point between the initial frequency facilitation and the DRE; c time needed to reach the peak of the DRE. The average measurements from individual experiments are shown in inset histogram with corresponding colored vertical bars. Vertical bars indicate S.E.M. *p < 0.05 when compared to the control situation. (B) As in (A) but the slice experiments are from DKO mice treated with 4 mM CaCl2 (n = 10, blue symbols) compared to experiments with 1 mM CaCl2 (n = 12, red symbols).
The magnitude of the subsequent DRE responses seen in the WT preparation showed a tendency of becoming reduced at high Ca2+, albeit not significantly, when results obtained at 4 mM [Ca2+]o during 4–15 s stimulations were compared to those seen at 1 mM [Ca2+]o during the same period. Surprisingly, during the same period the DKO preparations showed a pronounced and unexpected decrease in Ca2+-dependent response magnitudes, which during the period between ∼2 and ∼8 s stimulation transformed the Ca2+-induced responses in the DKO from response enhancements to decreases. During the remaining stimulation period, Ca2+-induced responses obtained at 4 mM Ca2+ in the WT preparation were indeed significantly smaller than those seen at 1 mM Ca2+, whereas no differences between the different Ca2+ levels were seen in the DKO preparations (Figures 4A,B). These synapses therefore appear to have highly complex relationships between Ca2+ levels, synapsin proteins and synaptic responses, with brief, early responses deriving from RRP vesicles and prolonged delayed responses deriving from other vesicle pools, both being under the control of the synapsin I/II proteins. None of these effects could be mimicked by addition of forskolin or PDBu.
Although the major synapsin I/II-specific effects appear restricted to the late frequency facilitation and DRE phases, more subtle effects may occur outside this time period (Hilfiker et al., 1998; Hvalby et al., 2006; Coleman and Bykovskaia, 2009b). We therefore summarized to what extent the individual effects induced by forskolin, PDBu, and 4 mM [Ca2+]o were dependent on synapsin-mediated mechanisms throughout the different response phases by subtracting the effects of the treatments during the 20 Hz stimulations in the DKO from those seen in the WT preparations. This analysis (Figure 5) showed that partial but distinct effects of synapsin I/II could be seen under all stimulatory periods. The synapsin I/II effects on forskolin-induced response modulations comprised a brief response decrease during the initial 2 s and a synapsin-dependent response enhancement during the next 8 s. No synapsin I/II-dependent effect was seen after 10 s stimulations. In the PDBu-treated samples, synapsin I/II effects comprised a small, initial decrease in response magnitude which lasted for 10 s, followed by a slow reversal to baseline, after which synapsins were without effects. In contrast, the changes induced by 4 mM Ca2+ included a rapid synapsin-dependent response enhancement during the first 2 s, i.e., during release of RRP vesicles, followed by a decrease to baseline during the next 6 s, and a steady, synapsin I/II-dependent negative effect on Ca2+-induced synaptic responses which lasted during the remaining stimulation period (Figure 5).
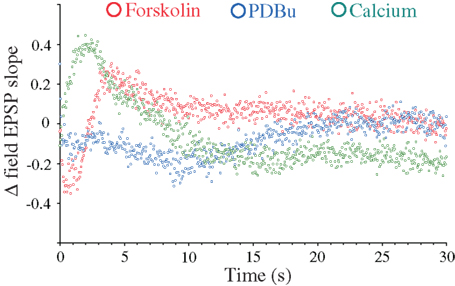
Figure 5. Synapsin-dependence of frequency facilitation and DRE as a function of treatment with forskolin, PDBu, and 4 mM CaCl2. Subtraction of differences between measurements obtained in the presence of forskolin (red symbols), PDBu (blue symbols), and 4 mM CaCl2 (green symbols) and measurements obtained in control solutions in the synapsin DKO mice, from the corresponding differences seen in WT mice.
Activations of the intracellular cAMP-, DAG-, and Ca2+-mediated second messenger systems in the glutamatergic CA3-to-CA1 synapse in mouse hippocampus induce similar increases in baseline responses. The major findings from this study show that during subsequent repetitive stimulations, where both vesicle docking and priming as well as recruitment from additional vesicle pools become more important, they induce different responses. These conclusions depend on experimentally obtained changes in postsynaptic responses caused by transmitter released from the RRP and/or other vesicle pools. Moreover, some but not all of the effects appear to depend on the synapsin I/II proteins, which may be involved both in response enhancements and depressions, depending on both the identity of the specific second messenger system activated and the vesicle pool involved.
Multiple effects of the 3 second messenger systems are known to occur throughout all synaptic compartments, and a variety of presynaptic and postsynaptic structures are well known as targets for the biochemical systems we examine (e.g., Walaas and Greengard, 1991). These include presynaptic and postsynaptic ion channels and postsynaptic receptors, the modification of which could change postsynaptic responses. However, given the exclusive presynaptic location of the synapsin I/II proteins, and the comparative analysis of the two genotypes, the most parsimonious explanation for the results obtained is that they represent putative modulations of endo- and exocytotic trafficking and recruitment of presynaptic vesicle pools. However, minor contributions from functional changes occurring elsewhere cannot be completely excluded, particularly those observed in the absence of synapsin I/II.
During stimulation at low frequency (0.1 Hz), where already primed vesicles are activated, both forskolin, PDBu and 4 mM Ca2+ induced increases in fEPSP magnitudes. Moreover, these changes in synaptic strength were essentially identical in WT and DKO preparations, demonstrating that despite the ability of, e.g., synapsin I to modulate exocytotic kinetics (Hilfiker et al., 1998; Coleman and Bykovskaia, 2009b), second messenger-induced modulations of single vesicle exocytosis were not targets for the synapsins I/II proteins. These data therefore tend to exclude that phosphorylation of domain A in the synapsin proteins, catalyzed by cAMP-or Ca2+-dependent protein kinases (Walaas and Greengard, 1991; Cesca et al., 2010), represents important regulators of the already primed vesicles that participate during Ca2+-induced membrane fusion and exocytosis (e.g., Lisman et al., 2007).
In contrast to the effect on single responses, the PPF patterns, which depend on the ability of the next RRP vesicles to be released upon the arrival of the second stimulus, were dependent on the distinct second messenger system, although to varying extents. Firstly, in the absence of any pharmacological treatment no effects were seen following removal of synapsin I/II, indicating that synapsin-dependent vesicle recruitment (Chi et al., 2001, 2003) is not a limiting factor. In contrast, the fEPSP enhancers had clear but different effects, with forskolin enhancing both the first and second fEPSP but leaving the PPF ratio unchanged, 4 mM [Ca2+]o significantly decreasing and 1 mM Ca2+ significantly increasing the PPF ratio, and PDBu essentially abolishing the PPF and rather retaining the second fEPSP at baseline levels. An inverse relationship has been proposed to exist between release probability and subsequent changes in synaptic efficacy during chemical neurotransmission (Zucker and Regehr, 2002). Therefore, our PPF data suggest that in the CA1 synapse forskolin probably enhances synaptic responses without changing release probability (and therefore may enhance synaptic strength by increasing the number of functional RRP vesicles). Previous analyses of forskolin effects on PPF ratio in the same synapses are in essential agreement with our data, with minor PPF decreases of 4–10% being reported (Dumas and Foster, 1998; Kameyama et al., 1998). In contrast, other CNS synapses show highly significant decreases in PPF following forskolin treatment, including cerebellar parallel fiber-to Purkinje cell synapses (Chen and Regehr, 1997) and hippocampal mossy fiber-to CA3 pyramidal cell synapses (Weisskopf et al., 1994), both of which therefore appear to respond to increases in cAMP by increasing synaptic release probability. The different forskolin effects between these synapses and the synapses examined in this study indicate important differences in molecular or structural properties in different presynaptic populations (Bogen et al., 2006; Bragina et al., 2007). In contrast, the very low PPF ratio induced by PDBu treatment suggests that the enhanced responses in these synapses predominantly were caused by inducing major increases in vesicle release probability. Although this interpretation is at variance with previous studies performed in a number of species, where phorbol esters appeared to induce significant increases in RRP size (e.g., Gillis et al., 1996; Waters and Smith, 2000; Searl and Silinsky, 2003; Junge et al., 2004), our data are in agreement with recent studies (Basu et al., 2007; Lou et al., 2008), which conclude that PDBu, working through munc-13, preferentially induces a specific increase in release probability.
The PPF ratios induced by changes in [Ca2+]o are in agreement with the proposed relation between facilitation and synaptic release probability (Zucker and Regehr, 2002). These changes may partly be due to initial dose-dependent effects on the vesicular Ca2+ sensor synaptotagmin, which determines the baseline fEPSP (e.g., Sun et al., 2007). Since no differences were seen between the second fEPSPs in the WT and DKO preparations, our data exclude synapsin I/II from participating in these changes. However, Ca2+ may have other modulatory effects on presynaptic vesicle functions, mediated by, e.g., Ca2+-calmodulin modulating a variety of protein phosphorylation systems (Walaas and Greengard, 1991; Jovanovic et al., 2001), the munc-13 priming proteins (Brose et al., 2004; Junge et al., 2004), and the vesicle-associated rab3A protein (Geppert et al., 1997; Park et al., 1997; Schlüter et al., 2006; Coleman and Bykovskaia, 2009a). In the present context, however, the contribution and importance of these separate modulatory systems on synaptic facilitation remain uncertain.
Analysis of the frequency facilitation induced by continuous afferent 20 Hz stimulation showed fEPSP enhancements which under standard conditions peaked after ∼12 stimuli, i.e., after 0.6 s stimulation (Hvalby et al., 2006; Jensen et al., 2007), with similar response magnitudes seen in the WT and DKO preparations. In contrast to the PPF responses, all three experimental manipulations induced substantial increases and decreased the time needed to reach the peak of this frequency facilitation. Hence, prolonged recruitment of RRP vesicles appeared to be both accelerated and enhanced by all treatments. However, subtle differences were observed. Forskolin-induced frequency facilitation showed a considerable magnitude increase and accelerated enhancement, in a manner not dependent on synapsin I/II, suggesting that the vesicles involved are not dependent on synapsin I/II proteins, despite these proteins being excellent substrates for cAMP-dependent protein kinase (Walaas and Greengard, 1991). Rather, other active zone components may be targets for cAMP-induced regulation, e.g., SNAP-25 (Nagy et al., 2002), the phosphorylation of which may be involved in regulating vesicle priming (Neher, 2006).
In contrast to the forskolin-induced effects, PDBu induced only a very brief, although accelerated increase in frequency facilitation, which also was independent on the synapsins I/II. Therefore, independently of the synapsin I/II proteins, PDBu induced a limited number of vesicles from the RRP to become available for exocytosis. Phorbol esters may mediate effects on release through activation of the munc-13 priming protein family (Rhee et al., 2002; Wierda et al., 2007; Lou et al., 2008), most probably by increasing release probability (Basu et al., 2007). The PDBu-induced small size and short duration of the frequency facilitation observed in this synapse appear consistent with the interpretation that DAG activation is restricted to recruitment of only a limited number of already primed, synapsin I/II-independent RRP vesicles.
Furthermore, increasing [Ca2+]o from 1 to 4 mM in WT preparations also gave an increased frequency facilitation, which rapidly was transformed into a steep decay. However, in contrast to the frequency facilitations induced either under standard conditions, with forskolin or with PDBu, the 4 mM Ca2+-induced enhancements of the frequency facilitation were highly dependent on the synapsin I/II proteins. At this level of Ca2+, the frequency facilitation was enhanced by a factor of approx. 1.5 in the WT preparation, whereas without synapsins I/II, frequency facilitation in the DKO was increased by a factor of less than 1.3 (Figure 4). Hence, the presence of synapsins I/II led to almost a doubling of the frequency facilitation enhancement induced by the 20 Hz train. In contrast, at 1 mM Ca2+, the presence of synapsins I/II did not lead to significant differences in the frequency facilitation when compared to that seen in the DKO.
Given that frequency facilitations are thought to represent increased release of vesicles deriving from the RRP (Zucker and Regehr, 2002), these data indicate that a combination of high levels of Ca2+ and synapsins I/II proteins can modulate RRP vesicles. This unexpected conclusion is in contrast to the suggestion that synapsin-mediated functions would be restricted to reserve vesicles (Cesca et al., 2010), but appears to be partly supported by recent analysis of the postnatal development of these synapses (Bogen et al., 2009), where a transient synapsin I/II-induced enhancement of frequency facilitation induced at 2 mM Ca2+ occurred at developmental stages p18–27, following which it disappeared in adults. Apparently, in this synapse increasing the concentration of Ca2+ to 4 mM in adult mice appears to be sufficient to recreate this Ca2+- and-synapsin I/II-dependent response from RRP vesicles. Since neither forskolin (through cAMP) nor PDBu were able to mimic this effect, a unique relation between Ca2+-regulated biochemical mechanism(s) and synapsin I/II-dependent RRP vesicles clearly exists. Possible mechanisms may include Ca2+-induced activation of calcium-calmodulin-dependent protein kinase II (CaM kinase II) present in the active zone of excitatory synapses together with synapsin I/II (Tao-Cheng, 2006; Tao-Cheng et al., 2006), which, by phosphorylating synapsin I at sites 2,3, could induce vesicle–cytoskeletal dissociation and subsequent vesicle exocytosis (Llinas et al., 1991; Jovanovic et al., 2001; Chi et al., 2001, 2003; Hilfiker et al., 2005).
Finally, comparisons between the amplitudes of the frequency facilitations reached during the different pharmacological treatments indicate that forskolin, which led to the frequency facilitation reaching ∼2.6-fold increase of the baseline response, represents the most efficient modulator of the RRP vesicles in this synapse. The exact mechanisms for this cAMP-induced effect remain unclear, but could include both phosphorylation of multiple cAMP-dependent protein kinase substrates (Nagy et al., 2002, 2004; Leenders and Sheng, 2005), and activation of cAMP-induced release through mechanisms distinct from phosphorylation (e.g., Gekel and Neher, 2008).
The effects of second messenger modulations of other vesicle pools were somewhat unexpected, with both the DRE and the late response phases being potently regulated by some but not by other treatments. The DRE phase, which is restricted to WT preparations and may represent a pool of specific synapsin I/II-dependent (Jensen et al., 2007; Bogen et al., 2009) vesicles, responded to forskolin by being enhanced in a synapsin I/II-dependent manner, suggesting that phosphorylation of the synapsin domain A (Hilfiker et al., 2005) stimulated by cAMP (Walaas and Greengard, 1991) may be necessary for recruitment of vesicles involved in the DRE phase. In contrast, forskolin-induced enhancement of the previous or subsequent response phases occurred in a synapsin I/II-independent manner and would therefore rather be expected to be mediated through proteins distinct from the synapsins (Leenders and Sheng, 2005). In contrast to forskolin, PDBu showed essentially no effect on the DRE phase, but showed a small, synapsin-independent response enhancement during the later short-term plasticity phase. Indeed, following the major increase in single fEPSP and the small frequency facilitation induced by PDBu, the subsequent response patterns induced by PDBu mostly resembled the pattern induced in standard solution. Hence, treatment with PDBu may not activate major novel enhancement mechanisms, but rather activate and thereby occlude the mechanisms normally induced by 20 Hz stimulation. In contrast, the small but significant PDBu-induced final response enhancements which occurred in both genotypes were similar to the late response enhancement observed in forskolin-treated preparations (Figures 2A,B), suggesting the existence of distinct, late response enhancements, where both cAMP and DAGs may employ synapsin I/II-insensitive mechanisms. Finally, 4 mM [Ca2+]o showed a tendency for decreasing the fEPSP during the DRE phase, followed by a significant Ca2+- and synapsin I/II-dependent fEPSP decrease. The latter may be related to the inverse effects of Ca2+-dependent enzymes on transmitter release observed during different stimulatory conditions (Jovanovic et al., 2001; Chi et al., 2003). Interestingly, this phase also coincided with the period during which these synapses showed both PDBu- and forskolin-induced response enhancements, raising the question of whether recruitment of one single pool of reserve vesicles can be enhanced by forskolin- or PDBu-dependent, synapsin I/II-independent mechanisms, and at the same time be decreased by Ca2+-synapsin I/II-dependent mechanisms.
In conclusion, our data demonstrate that in these small, prototypic excitatory glutamatergic synapses, different pools of synaptic vesicles are sensitive to a number of distinct regulatory mechanisms during continuous evoked glutamate release. In particular, we show that the RRP vesicles which mediate both single and paired synaptic responses as well as frequency facilitations induced in the absence or presence of forskolin or PDBu, are all essentially synapsin I/II-independent. In contrast, both the vesicles responsible for Ca2+-induced frequency facilitations, as well as vesicles involved in forskolin-induced response enhancements during the DRE phase were dependent on the presence of synapsin I/II. Further characterization of these response patterns remains an interesting challenge.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank prof. Paul Greengard, Rockefeller University, NY, USA, for gift of the DKO mice. Financial support was given by the Jahre Foundation for Medical Research, Oslo, Norway.
Attwood, H. L., and Karunanithi, S. (2002). Diversification of synaptic strength: presynaptic elements. Nat. Rev. Neurosci. 3, 497–516.
Basu, J., Betz, A., Brose, N., and Rosenmund, C. (2007). Munc13-1 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J. Neurosci. 27, 1200–1210.
Billiard, J., Koh, D. S., Babcock, D. F., and Hille, B. (1997). Protein kinase C as a signal for exocytosis. Proc. Natl. Acad. Sci. U.S.A. 94, 12192–12197.
Bogen, I. L., Boulland, J.-L., Mariussen, E., Wright, M. S., Fonnum, F., and Walaas, S. I. (2006). Absence of synapsins I and II is accompanied by decreases in vesicular transport of specific neurotransmitters. J. Neurochem. 96, 1458–1466.
Bogen, I. L., Jensen, V., Hvalby, O., and Walaas, S. I. (2009). Synapsin-dependent development of glutamatergic synaptic vesicles and presynaptic plasticity in postnatal mouse brain. Neuroscience 158, 231–241.
Bragina, L., Candiracci, C., Barbaresi, P., Giovedi, S., Benfenati, F., and Conti, F. (2007). Heterogeneity of glutamatergic and GABAergic release machinery in cerebral cortex. Neuroscience 146, 1829–1840.
Brose, N., Betz, A., and Wegmeyer, H. (2004). Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Curr. Opin. Neurobiol. 14, 328–340.
Cesca, F., Baldelli, P., Valtorta, F., and Benfenati, F. (2010). The synapsins: key actors of synaptic function and plasticity. Prog. Neurobiol. 91, 313–348.
Chavez-Noriega, L. E., and Stevens, C. F. (1992). Modulation of synaptic efficacy in field CA1 of the rat hippocampus by forskolin. Brain Res. 574, 85–92.
Chavez-Noriega, L. E., and Stevens, C. F. (1994). Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J. Neurosci. 14, 310–317.
Chen, B.-S., and Roche, K. W. (2007). Regulation of NMDA receptors by phosphorylation. Neuropharmacol. 53, 362–368.
Chen, C., and Regehr, W. G. (1997). The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J. Neurosci. 17, 8687–8694.
Chi, P., Greengard, P., and Ryan, T. A. (2001). Synapsin dispersion and reclustering during synaptic activity. Nat. Neurosci. 4, 1187–1193.
Chi, P., Greengard, P., and Ryan, T. A. (2003). Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 38, 69–78.
Coleman, W. L., and Bykovskaia, M. (2009a). Rab3a-mediated vesicle recruitment regulates short-term plasticity at the mouse diaphragm synapse. Mol. Cell. Neurosci. 41, 286–296.
Coleman, W. L., and Bykovskaia, M. (2009b). Synapsin I accelerates the kinetics of neurotransmitter release in mouse motor terminals. Synapse 63, 531–533.
Dumas, T. C., and Foster, T. C. (1998). Late developmental changes in the ability of A1 adenosine receptors to regulate synaptic transmission in the hippocampus. Dev. Brain Res. 105, 137–139.
Fernandez-Alfonso, T., and Ryan, T. A. (2008). A heterogeneous “resting pool” of synaptic vesicles that is dynamically interchanged across boutons in mammalian CNS synapses. Brain Cell Biol. 36, 87–100.
Ferreira, A., Chin, L. S., Lanier, L. M., Kosik, K. S., and Greengard, P. (1998). Distinct roles of synapsin I and synapsin II during neuronal development. Mol. Med. 4, 22–28.
Gao, C., Sun, X., and Wolf, M. E. (2006). Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J. Neurochem. 98, 1664–1677.
Garcia-Perez, E., Lo, D. C., and Wesseling, J. F. (2008). Kinetic isolation of a slowly recovering component of short-term depression during exhaustive use at excitatory hippocampal synapses. J. Neurophysiol. 100, 781–795.
Gekel, I., and Neher, E. (2008). Application of an epac activator enhances neurotransmitter release at excitatory central synapses. J. Neurosci. 28, 7991–8002.
Geppert, M., Goda, Y., Stevens, C. F., and Südhof, T. C. (1997). The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature 387, 810–814.
Gillis, K. D., Mossner, R., and Neher, E. (1996). Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron 16, 1209–1220.
Greengard, P., Jen, J., Nairn, A. C., and Stevens, C. F. (1991). Enhancement of the glutamate response by cAMP-dependent protein kinase in hippocampal neurons (1991). Science 253, 1135–1138.
Gustafsson, B., Huang, Y. Y., and Wigström, H. (1988). Phorbol ester-induced synaptic potentiation differs from long-term potentiation in the guinea pig hippocampus in vitro. Neurosci. Lett. 85, 77–81.
Harris, K. M., and Sultan, P. (1995). Variation in the number, location and size of synaptic vesicles provides an anatomical basis for the nonuniform probability of release at hippocampal CA1 synapses. Neuropharmacol. 34, 1387–1395.
Hilfiker, S., Benfenati, F., Doussaut, F., Nairn, A. C., Czernik, A. J., Augustine, G. J., and Greengard, P. (2005). Structural domains involved in the regulation of transmitter release by synapsins. J. Neurosci. 25, 2658–2669.
Hilfiker, S., Pieribone, V. A., Czernik, A. J., Kao, H. T., Augustine, G. J., and Greengard, P. (1999). Synapsins as regulators of neurotransmitter release. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 28, 269–279.
Hilfiker, S., Schweizer, F. E., Kao, H. T., Czernik, A. J., Greengard, P., and Augustine, G. J. (1998). Two sites of action for synapsin domain E in regulating neurotransmitter release. Nature Neurosci. 1, 29–35.
Hvalby, Ø., Jensen, V., Kao, H. T., and Walaas, S. I. (2006). Synapsin-regulated synaptic transmission from readily releasable synaptic vesicles in excitatory hippocampal synapses in mice. J. Physiol. 571, 75–82.
Jensen, V., Walaas, S. I., Hilfiker, S., Ruiz, A., and Hvalby, Ø. (2007). A delayed response enhancement during hippocampal presynaptic plasticity in mice. J. Physiol. 583, 129–143.
Jovanovic, J. N., Sihra, T. S., Nairn, A. C., Hemmings, H. C. Jr., Greengard, P., and Czernik, A. J. (2001). Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J. Neurosci. 21, 7944–7953.
Junge, H. J., Rhee, J. S., Jahn, O., Varoqeaux, F., Spiess, J., Waxham, M. N., Rosenmund, C., and Brose, N. (2004). Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell 118, 389–401.
Kameyama, K., Lee, H.-K., Bear, M. F., and Huganir, R. L. (1998). Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron 21, 1163–1175.
Katz, B., and Miledi, R. (1968). Role of calcium in neuromuscular facilitation. J. Physiol. 195, 481–492.
Klyachko, V. A., and Stevens, C. F. (2006).Temperature-dependent shift of balance among the components of short-term plasticity in hippocampal synapses. J. Neurosci. 26, 6945–6957.
Leenders, A. G. M., and Sheng, Z. H. (2005). Modulation of neurotransmitter release by the second messenger-activated protein kinases: implications for presynaptic plasticity. Pharmacol. Ther. 105, 69–84.
Li, Z., Burrone, J., Tyler, W. J., Hartman, K. N., Albeanu, D. F., and Murthy, V. (2005). Synaptic vesicle recycling studied in transgenic mice expressing synaptopHluorin. Proc. Natl. Acad. Sci. U.S.A. 102, 6131–6136.
Lisman, J. E., Raghavachari, S., and Tsien, R. W. (2007). The sequence of events that underlie quantal transmission at central glutamatergic synapses. Nat. Rev. Neurosci. 8, 597–609.
Llinas, R., Gruner, J. A., Sugimori, M., McGuinness, T. M., and Greengard, P. (1991). Regulation by synapsin I and Ca(2+)-calmodulin-dependent protein kinase II of the transmitter release in squid giant synapse. J. Physiol. 436, 257–282.
Lou, X., Korogod, N., Brose, N., and Schneggenburger, R. (2008). Phorbol esters modulate spontaneous and Ca2+-evoked transmitter release via acting on both Munc13 and protein kinase C. J. Neurosci. 28, 8257–8267.
Malenka, R. C., Madison, D. V., and Nicoll, R. A. (1986). Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature 321, 175–177.
Nagy, G., Matti, U., Nehring, R. B., Binz, T., Rettig, J., Neher, E., and Sorensen, J. B. (2002). Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J. Neurosci. 22, 9278–9286.
Nagy, G., Reim, K., Matti, U., Brose, N., Binz, T., Rettig, J., Neher, E., and Sorensen, J. B. (2004). Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron 41, 351–365.
Neher, E. (2006). A comparison between control mechanisms in adrenal chromaffin cells and a glutamatergic synapse. Pflugers Arch. Eur. J. Physiol. 453, 261–268.
Neher, E., and Sakaba, T. (2008). Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59, 861–871.
Oh, M. C., Derkach, V. A., Guire, E. S., and Soderling, T. R. (2006). Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem. 281, 752–758.
Owe, S. G., Bogen, I. L., Walaas, S. I., Storm-Mathisen, J., and Bergersen, L. H. (2005). Ultrastructural quantification of glutamate receptors at excitatory synapses in hippocampus of synapsin I+II double knock out mice. Neuroscience 136, 769–777.
Park, J. B., Farnsworth, C. C., and Glomseth, J. A. (1997). Ca2+/calmodulin causes rab3A to dissociate from synaptic membranes. J. Biol. Chem. 272, 20857–20865.
Rhee, J. S., Betz, A., Pyott, S., Reim, K., Varoqueaux, F., Augustin, I., Hesse, D., Südhof, T. C., Takahashi, M., Rosenmund, C., and Brose, N. (2002). Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 108, 121–133.
Rosahl, T. W., Spillane, D., Missler, M., Herz, J., Selig, D. K., Wolff, J. R., Hammer, R. E., Malenka, R. C., and Südhof, T. C. (1995). Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375, 488–493.
Schikorski, T., and Stevens, C. F. (1997). Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 17, 5858–5867.
Schlüter, O. M., Basu, J., Südhof, T. C., and Rosenmund, C. (2006). Rab3 superprimes synaptic vesicles for release: implications for short-term synaptic plasticity. J. Neurosci 26, 1239–1246.
Schneggenburger, R., and Neher, E. (2005). Presynaptic calcium and control of vesicle fusion. Curr. Opin. Neurobiol. 3, 266–274.
Searl, T. J., and Silinsky, E. M. (2003). Phorbol esters and adenosine affect the readily releasable neurotransmitter pool by different mechanisms at amphibian motor nerve endings. J. Physiol. 553, 445–456.
Sun, J., Pang, Z. P., Qin, D., Fahim, A. T., Adachi, R., and Südhof, T. C. (2007). A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450, 676–682.
Tao-Cheng, J. H. (2006). Active zone redistribution of presynaptic proteins at the active zone. Neuroscience 141, 1217–1224.
Tao-Cheng, J. H., Dosemeci, A., Winters, C. A., and Reese, T. S. (2006). Changes in the distribution of calcium calmodulin–dependent protein kinase II at the presynaptic bouton after depolarization. Brain Cell Biol. 35, 117–124.
Walaas, S. I. (1999). Regulation of calcium-dependent [3H]-noradrenaline release from rat cerebrocortical synaptosomes by protein kinase C and modulation of the actin cytoskeleton. Neurochem. Int. 34, 221–233.
Walaas, S. I., and Greengard, P. (1991). Protein phosphorylation and neuronal function. Pharmacol. Rev. 43, 299–349.
Waters, J., and Smith, S. J. (2000). Phorbol esters potentiate evoked and spontaneous release by different presynaptic mechanisms. J. Neurosci. 20, 7863–7870.
Weisskopf, M. G., Castillo, P. E., Zalutsky, R. A., and Nicoll, R. A. (1994). Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science 265, 1878–1882.
Wesseling, J. F., and Lo, D. C. (2002). Limit on the role of activity in controlling the release-ready supply of synaptic vesicles. J. Neurosci. 22, 9708–9720.
Wierda, K. D., Toonen, R. F., de Wit, H., Brussaard, A. B., and Verhage, M. (2007). Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron 54, 275–290.
Keywords: presynaptic plasticity, glutamate, calcium, forskolin, phorbol ester, synapsin
Citation: Hvalby Ø, Jensen V, Kao H-T and Walaas SI (2010) Synapsin-dependent vesicle recruitment modulated by forskolin, phorbol ester and Ca2+ in mouse excitatory hippocampal synapses. Front. Syn. Neurosci. 2:152. doi: 10.3389/fnsyn.2010.00152
Received: 07 June 2010;
Accepted: 09 December 2010;
Published online: 22 December 2010.
Edited by:
Robert Renden, UCB Pharma SA, BelgiumReviewed by:
Stephen James Mennerick, Washington University in St. Louis, USACopyright: © 2010 Hvalby, Jensen, Kao and Walaas. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Sven Ivar Walaas, Department of Biochemistry, Institute of Basic Medical Sciences, University of Oslo, PO Box 1112, Blindern, N-0316 Oslo, Norway. e-mail:cy5pLndhbGFhc0BtZWRpc2luLnVpby5ubw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.