

- 1 Department of Neuroscience, Physiology and Pharmacology, University College London, London, UK
- 2 Medical Research Council Centre for Synaptic Plasticity, Department of Anatomy, University of Bristol, Bristol, UK
Synaptic plasticity has historically been investigated most intensely in the hippocampus and therefore it is somewhat surprising that the majority of studies on spike timing-dependent plasticity (STDP) have focused not in the hippocampus but on synapses in the cortex. One of the major reasons for this bias is the relative ease in obtaining paired electrophysiological recordings from synaptically coupled neurons in cortical slices, in comparison to hippocampal slices. Another less obvious reason has been the difficulty in achieving reliable STDP in the hippocampal slice preparation and confusion surrounding the conditions required. The original descriptions of STDP in the hippocampus was performed on paired recordings from neurons in dissociated or slice cultures utilizing single pairs of presynaptic and postsynaptic spikes and were subsequently replicated in acute hippocampal slices. Further work in several laboratories using conditions that more closely replicate the situation in vivo revealed a requirement for multiple postsynaptic spikes that necessarily complicate the absolute timing rules for STDP. Here we review the hippocampal STDP literature focusing on data from acute hippocampal slice preparations and highlighting apparently contradictory results and the variations in experimental conditions that might account for the discrepancies. We conclude by relating the majority of the available experimental data to a model for STDP induction in the hippocampus based on a critical role for postsynaptic Ca2+ dynamics.
The classic asymmetrical spike timing curve between pairs of synaptically connected hippocampal neurons in dissociated culture described by Bi and Poo (1998) or in organotypic slice cultures by Debanne et al. (1998) have become synonymous with the field of spike timing-dependent plasticity (STDP). These data have been reproduced almost ubiquitously to demonstrate the elegance of spike timing plasticity induction. From this seemingly straightforward description the story of STDP in the hippocampus has followed a rather more complicated and tortuous route. Many groups have taken advantage of the stalwart of plasticity research, the Schaffer collateral to CA1 pyramidal cell synapse in the acute hippocampal slice preparation, to further characterize the requirements for STDP induction. At first glance the research that has emerged from this field is confusing with different groups showing different, seemingly contradictory results regarding the exact requirements for STDP induction. Now, as STDP in the hippocampus enters its second decade a clearer picture is starting to form and many of the previous controversies are helping to produce a more unified picture of STDP induction at this classic synapse and in the hippocampus as a whole.
STDP in the Hippocampus: the Data
Since the time of Hebb’s postulate many groups have investigated the timing requirements for plasticity induction in the hippocampus (Levy and Steward, 1979 ; Gustafsson et al., 1987 ; Stanton and Sejnowski, 1989 ; Debanne et al., 1994 ). Using pairs of single presynaptic and postsynaptic action potentials Bi and Poo (1998) and Debanne et al. (1998) were the first to fully characterize the timing window for the induction of what we now term STDP, in the hippocampus. They took advantage of cultured hippocampal preparations which allow pairs of connected cells to be recorded from with relative ease. This allows one to precisely control the spiking of both the presynaptic and the postsynaptic neuron. In a similar fashion, recordings between pairs of mono-synaptically connected pyramidal neurons in cortical slices also demonstrated reliable STDP (Markram et al., 1997 ; Sjostrom et al., 2001 ) which raised the possibility that STDP is a general phenomenon for all synapses exhibiting NMDA receptor-dependent synaptic plasticity. Bi and Poo and Debanne et al. demonstrated that the direction and magnitude of synaptic plasticity could be dictated by the precise millisecond timing of single presynaptic and postsynaptic spikes. If presynaptic spikes precede postsynaptic spikes by up to 30 ms (positive spike timing interval) then LTP was induced, whereas if the presynaptic spike occurred after the postsynaptic spike (negative spike timing interval) LTD ensued. The magnitude of LTP and LTD was greatest when the spikes were closest together leading to a switch from maximal LTD to maximal LTP over a narrow time window of only a few milliseconds. This spike timing window offered an extremely elegant model for plasticity induction in vivo and has proved popular with groups modeling information storage in the brain (e.g., Song et al., 2000 ; Drew and Abbott, 2006 ).
Recordings in dissociated hippocampal cultures have their drawbacks, in particular the divergence of culture conditions from an intact hippocampal network. After the initial description of STDP in dissociated hippocampal cultures many groups investigated STDP timing curves in hippocampal slices. Paired Recording of connected CA3–CA1 pyramidal cells in acute hippocampal slices is extremely difficult owing to very low connectivity rates so this has been restricted to organotypic cultured slices. These experiments revealed that synchronous pairing of single presynaptic action potentials with postsynaptic bursts of action potentials lead to the induction of LTP whereas LTD could be induced if this stimulation was given asynchronously (Debanne et al., 1996 , 1999 ). The use of organotypic slice culture also has its drawbacks and it is unclear what developmental stage this cultured network represents. As a result many groups have resorted to investigating STDP in the acute hippocampal slice preparation through pairing extracellular Schaffer collateral stimulation with action potential initiation in patched CA1 pyramidal cells.
Nishiyama et al. witnessed the same STDP curve observed by Bi and Poo, when pairing Schaffer collateral stimulation with single post synaptic spikes in hippocampal slices from young adult rats. They also observed an additional LTD window at positive spike timing intervals between 15 and 20 ms. They argue that this additional LTD window may be due to the presence of inhibitory inputs that are lacking in cultured preparations (Nishiyama et al., 2000 ). Subsequent experiments have shown that the use of Cs+ ions in the internal electrode solution by Nishiyama et al. fundamentally alters the induction of synaptic plasticity by STDP protocols (Wittenberg and Wang, 2006 ; Isaac et al., 2009 ). Indeed, other groups have been unable to induce STDP with single postsynaptic spikes when using K+ ion based internal electrode solutions. Instead, the pairing of Schaffer collateral stimulation with a burst of postsynaptic action potentials is required for the induction of LTP in acute hippocampal slices (Pike et al., 1999 ; Watanabe et al., 2002 ; Meredith et al., 2003 ; Wittenberg and Wang, 2006 ; Buchanan and Mellor, 2007 ; Carlisle et al., 2008 ). The importance of postsynaptic bursting is also supported by the effectiveness of a variety of burst firing stimulation protocols to induce LTP at this synapse (Debanne et al., 1994 , 1998 ; Frick et al., 2004 ; Buchanan and Mellor, 2007 ). The requirement for postsynaptic burst firing appears to have a developmental profile and is critical for plasticity induction in slices from adult animals (Meredith et al., 2003 ; Buchanan and Mellor, 2007 ). Meredith et al. were able to induce LTP through pairing EPSPs with single postsynaptic spikes in adult slices if fast GABAergic inhibition was blocked. This suggests the maturation of inhibition may underlie the requirement for postsynaptic bursts, although this result was not replicated in other studies (Pike et al., 1999 ; Buchanan and Mellor, 2007 ). None of the aforementioned studies have systematically investigated the timing dependence of presynaptic spikes with postsynaptic burst firing. For this information we must turn to experiments on hippocampal slices taken from immature animals (<P21).
In slices from juvenile animals spike timing induction protocols have produced a variety of results. Pairs of single presynaptic and postsynaptic spikes given at positive spike timing intervals have been found to induce either; no plasticity (Buchanan and Mellor, 2007 ), LTD (Wittenberg and Wang, 2006 ; Campanac and Debanne, 2008 ) or LTP (Meredith et al., 2003 ; Buchanan and Mellor, 2007 ; Campanac and Debanne, 2008 ) dependent on specific experimental conditions. Several groups have described a frequency dependency to the induction of STDP where LTP is only induced when positive spike timing pairs are repeated at 10 Hz or greater and no plasticity or a small amount of LTD is observed when positive spike pairs are repeated at lower frequencies (5 Hz). In these cases the spike pair repetition rate becomes the dominating factor and the resultant plasticity is timing independent (Wittenberg and Wang, 2006 ; Buchanan and Mellor, 2007 ). Timing dependence can be reintroduced when single EPSPs are paired with a postsynaptic burst (Wittenberg and Wang, 2006 ). In contrast, two other studies were able to induce LTP with positive spike timing pairs repeated at lower frequencies (Meredith et al., 2003 ; Campanac and Debanne, 2008 ). The reasons for the discrepancies between results from different groups is not immediately apparent although it is of note that only two of the studies (Meredith et al., 2003 ; Buchanan and Mellor, 2007 ) made use of a control input pathway to determine the induction of synaptic plasticity. In addition, it has been shown that postsynaptic spiking is relatively less important than EPSP amplitude for the induction of STDP in the immature hippocampus compared to the mature network (Buchanan and Mellor, 2007 ). This suggests that differences in the EPSP amplitude used during STDP induction could explain observed discrepancies.
In all cases, although the significance of precise spike timing in the induction of synaptic plasticity in the hippocampus is questionable, the plasticity observed is still dependent on the coincidence of presynaptic and postsynaptic activity as EPSPs or postsynaptic action potentials given on their own fail to induce plasticity. Also, a timing window is still observed as spike timing intervals of ±100 ms fail to induce any change in synaptic strength (Meredith et al., 2003 ; Wittenberg and Wang, 2006 ; Buchanan and Mellor, 2007 ; Campanac and Debanne, 2008 ).
So, the pursuit of the elegant hippocampal spike timing curve described by Bi and Poo seems to have lost its way. In its place we have a variety of spike timing window shapes described by different groups under different experimental conditions (Figure 1 ). But out of this seemingly contradictory mess there seems to be a common underlying theme that is starting to unveil a clearer picture of STDP rules in the hippocampus.
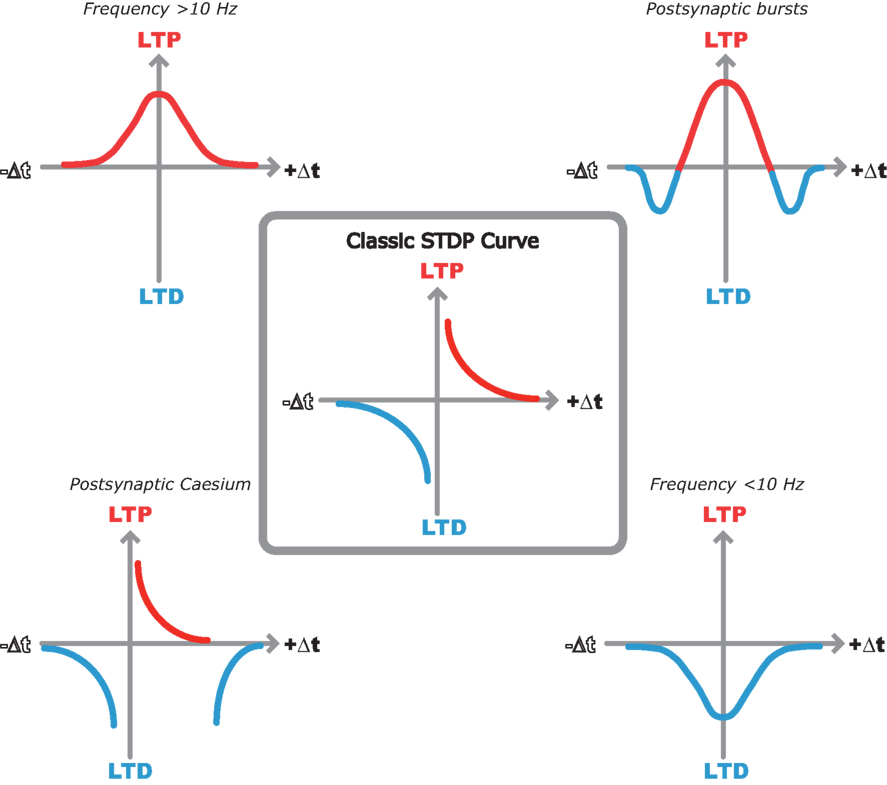
Figure 1. Multiple STDP timing windows in the hippocampus. Center, classic STDP curve originally described by Bi and Poo (1998) in dispersed hippocampal cultures. Top left, LTP only window when spike timing pairs are repeated at 10 Hz or greater in hippocampal slices (Buchanan and Mellor, 2007 ). Top right, bidirectional STDP window observed when single EPSPs are paired with postsynaptic bursts (Wittenberg and Wang, 2006 ). Bottom right, LTD only window when spike timing pairs are repeated at less than 10 Hz (Wittenberg and Wang, 2006 ). Bottom left, classic STDP window with additional LTD window observed with postsynaptic cesium in hippocampal slices (Nishiyama et al., 2000 ).
A Unifying Theory to Describe STDP in the Hippocampus
Due to a long history of plasticity research at the Schaffer collateral-CA1 pyramidal cell synapse much is known about the downstream mechanisms that determine the expression of synaptic plasticity. The critical trigger for plasticity is the influx of Ca2+ through NMDA receptors where local peak [Ca2+] is crucial in setting levels of CAMKII and PP1 activity (Lisman and Zhabotinsky, 2001 ) and therefore determining both the magnitude and direction of the resultant plasticity (Bienenstock et al., 1982 ; Lisman, 1989 ; Yang et al., 1999 ). This increase in postsynaptic [Ca2+] is dependent on the level of postsynaptic depolarization to relieve the Mg2+ block from NMDA receptors and many of the apparent controversies regarding STDP induction in the hippocampus may be explained in this context. In turn, postsynaptic depolarization will be influenced by a number of factors such as action potential back-propagation, modulation of dendritic membrane potential and excitability, EPSP amplitude, presence of inhibitory synaptic transmission and frequency of stimulation. We shall consider each of these factors in turn.
Bursts of action potentials produce a larger and more prolonged postsynaptic depolarization and therefore a much greater spine [Ca2+] than single spikes. This results in efficient induction of LTP (Pike et al., 1999 ; Buchanan and Mellor, 2007 ; Carlisle et al., 2008 ) or conversion of LTD to LTP (Wittenberg and Wang, 2006 ). At longer positive spike timing intervals the postsynaptic burst lags too far behind the EPSP to reach the threshold for LTP. Although calcium levels are still elevated beyond those observed for EPSPs or postsynaptic bursts alone leading to the observation of a second LTD window (Figure 1 ; Wittenberg and Wang, 2006 ). In the absence of bursts, other mechanisms for enhancing postsynaptic excitability are required to activate NMDARs and induce synaptic plasticity.
Depolarizing the membrane by perfusing Cs+ into the neuron from the patch pipette will broaden and increase the back- propagation of somatic action potentials (Wittenberg and Wang, 2006 ) as well as depolarizing the membrane allowing single postsynaptic spikes to induce STDP in adult hippocampal slices. An additional LTD window is observed at longer positive STIs due to the calcium levels dropping below the threshold for LTP but not LTD (Nishiyama et al., 2000 ; Wittenberg and Wang, 2006 ). Similarly, enhancing excitability by activation of neuromodulatory receptors, for example muscarinic acetylcholine receptors, reduces spike attenuation (Tsubokawa and Ross, 1997 ) and facilitates LTP induction (Isaac et al., 2009 ). This may be particularly critical in the slice preparation where external neuromodulatory inputs are removed. Modulation of other ionic conductances such as the sAHP can also regulate the induction of STDP again illustrating the critical role played by membrane excitability (Fuenzalida et al., 2007 ).
The magnitude of the EPSP used to induce STDP will contribute to the depolarization seen within the spine and therefore to NMDA receptor activation during STDP induction. This could explain discrepancies between reports since synaptic response amplitude varies from EPSCs of ∼50 pA (Buchanan and Mellor, 2007 ) to ∼50–150 pA (Wittenberg and Wang, 2006 ) to EPSPs of ∼3–5 mV (Meredith et al., 2003 ; Campanac and Debanne, 2008 ). Larger EPSPs could produce sufficient depolarization during single pairs of presynaptic and postsynaptic stimulation to induce LTP (Meredith et al., 2003 ; Campanac and Debanne, 2008 ). Interestingly, repetitive stimulation of individual suprathreshold EPSPs (EPSPs that are large enough to induce an action potential) induces LTD (Wittenberg and Wang, 2006 ) which predicts that the same will occur for suprathreshold stimulation during extracellular recording even at low stimulation frequencies. Also the driving of multiple action potentials by bursts of EPSPs induces LTP (Buchanan and Mellor, 2007 ). Conversely, in dissociated culture conditions EPSC amplitude was found to be inversely correlated with LTP induction although the EPSC amplitude range is much greater (30–2000 pA) than that used in acute slices (Bi and Poo, 1998 ).
Blockade of GABAA receptors can also enhance excitability and therefore allow the induction of LTP by single pairs of spikes (Meredith et al., 2003 ). This could also explain some age dependent effects on STDP induction since the mature GABAergic network in adults may increase the threshold for action potential back-propagation whereas in younger animals a less mature GABAergic network allows single spikes to back-propagate fully. However, there is also evidence that somatically induced action potentials are unable to provide the postsynaptic depolarization required for the induction of LTP in slices from juvenile animals. When somatic action potentials are blocked by focal TTX application LTP can still be induced if the level of presynaptic stimulation is increased. This suggests that dendritically initiated spikes may play a critical role in the induction of LTP in slices from younger animals (Buchanan and Mellor, 2007 ).
The frequency dependence of STDP in juvenile hippocampal slices can also be explained through differences in the levels of postsynaptic depolarization. Above a frequency of 10 Hz individual action potentials do not repolarize back to the resting membrane potential before the next action potential in the train (Buchanan and Mellor, 2007 ). This results in a residual level of depolarization throughout the train of spike pairings which may increase postsynaptic depolarization and enhance NMDAR activation. In addition, [Ca2+] will summate at higher frequencies ensuring spike timing pairs reach the threshold for LTP. Below 10 Hz there is little or no residual depolarization and the coincidence of EPSPs and action potentials are only able to produce enough depolarization to reach the threshold for LTD (Sjostrom et al., 2001 ; Wittenberg and Wang, 2006 ; Buchanan and Mellor, 2007 ).
The timing independence of STDP observed by several groups can also be explained through differences in postsynaptic depolarization. At higher frequencies the level of residual depolarization may mean that any degree of near coincidence is capable of increasing the postsynaptic calcium above the threshold for LTP (Buchanan and Mellor, 2007 ). Also the majority of experiments in hippocampal slices are done in the presence of GABAA receptor blockers which could prolong the duration of the postsynaptic depolarization caused by both the EPSP and the back-propagating action potential. This could result in a much broader timing window for the induction of plasticity.
A Ca2+ hypothesis for the induction of synaptic plasticity by pairs of presynaptic and postsynaptic spikes was first formalized in a model for STDP based on [Ca2+] (Shouval et al., 2002 ). Since then it has been revised to include the activation of calmodulin and CAMKII (Shouval et al., 2002 ; Rubin et al., 2005 ; Graupner and Brunel, 2007 ; Helias et al., 2008 ; Urakubo et al., 2008 ) and in this issue a model is presented specifically designed to model the Ca2+ dynamics within the spines of CA1 pyramidal cell dendrites during STDP at Schaffer collateral synapses in the hippocampus (Rackham et al., 2010 under review). This paper illustrates how most of the current data on synaptic plasticity induction can be replicated by such a model and can potentially unify current thinking on STDP in the hippocampus. It will be interesting to see if future experiments measuring spine calcium dynamics during STDP, similar to those performed at cortical synapses (Nevian and Sakmann, 2006 ), confirm such a Ca2+ based model for the induction of synaptic plasticity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bi, G. Q., and Poo, M. M. (1998). Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 18, 10464–10472.
Bienenstock, E. L., Cooper, L. N., and Munro, P. W. (1982). Theory for the development of neuron selectivity – orientation specificity and binocular interaction in visual-cortex. J. Neurosci. 2, 32–48.
Buchanan, K. A., and Mellor, J. R. (2007). The development of synaptic plasticity induction rules and the requirement for postsynaptic spikes in rat hippocampal CA1 pyramidal neurones. J. Neurosci. 585, 429–445.
Campanac, E., and Debanne, D. (2008). Spike timing-dependent plasticity: a learning rule for dendritic integration in rat CA1 pyramidal neurons. J. Physiol. (Lond.) 586, 779–793.
Carlisle, H. J., Fink, A. E., Grant, S. G., and O’Dell, T. J. (2008). Opposing effects of PSD-93 and PSD-95 on long-term potentiation and spike timing- dependent plasticity. J. Physiol. (Lond.) 586, 5885–5900.
Debanne, D., Gahwiler, B. H., and Thompson, S. M. (1994). Asynchronous pre- and postsynaptic activity induces associative long-term depression in area CA1 of the rat hippocampus in vitro. Proc. Natl. Acad. Sci. U.S.A. 91, 1148–1152.
Debanne, D., Gahwiler, B. H., and Thompson, S. M. (1996). Cooperative interactions in the induction of long-term potentiation and depression of synaptic excitation between hippocampal CA3–CA1 cell pairs in vitro. Proc. Natl. Acad. Sci. U.S.A. 93, 11225–11230.
Debanne, D., Gahwiler, B. H., and Thompson, S. M. (1998). Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J. Physiol. (Lond.) 507, 237–247.
Debanne, D., Gahwiler, B. H., and Thompson, S. M. (1999). Heterogeneity of synaptic plasticity at unitary CA3-CA1 and CA3-CA3 connections in rat hippocampal slice cultures. J. Neurosci. 19, 10664–10671.
Drew, P. J., and Abbott, L. F. (2006). Extending the effects of spike-timing-dependent plasticity to behavioral timescales. Proc. Natl. Acad. Sci. U.S.A. 103, 8876–8881.
Frick, A., Magee, J., and Johnston, D. (2004). LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat. Neurosci. 7, 126–135.
Fuenzalida, M., Fernandez de Sevilla, D., and Buno, W. (2007). Changes of the EPSP waveform regulate the temporal window for spike-timing-dependent plasticity. J. Neurosci. 27, 11940–11948.
Graupner, M., and Brunel, N. (2007). STDP in a bistable synapse model based on CaMKII and associated signaling pathways. PLoS Comput. Biol. 3, e221. doi: 10.1371/journal.pcbi.0030221.
Gustafsson, B., Wigstrom, H., Abraham, W. C., and Huang, Y. Y. (1987). Long-term potentiation in the hippocampus using depolarizing current pulses as the conditioning stimulus to single volley synaptic potentials. J. Neurosci. 7, 774–780.
Helias, M., Rotter, S., Gewaltig, M. O., and Diesmann, M. (2008). Structural plasticity controlled by calcium based correlation detection. Front. Comput. Neurosci. 2:7. doi:10.3389/neuro.10.007.2008.
Isaac, J. T., Buchanan, K. A., Muller, R. U., and Mellor, J. R. (2009). Hippocampal place cell firing patterns can induce long-term synaptic plasticity in vitro. J. Neurosci. 29, 6840–6850.
Levy, W. B., and Steward, O. (1979). Synapses as associative memory elements in the hippocampal formation. Brain Res. 175, 233–245.
Lisman, J. (1989). A Mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc. Natl. Acad. Sci. U.S.A. 86, 9574–9578.
Lisman, J. E., and Zhabotinsky, A. M. (2001). A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 31, 191–201.
Markram, H., Lubke, J., Frotscher, M., and Sakmann, B. (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215.
Meredith, R. M., Floyer-Lea, A. M., and Paulsen, O. (2003). Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J. Neurosci. 23, 11142–11146.
Nevian, T., and Sakmann, B. (2006). Spine Ca2+ signaling in spike-timing-dependent plasticity. J. Neurosci. 26, 11001–11013.
Nishiyama, M., Hong, K., Mikoshiba, K., Poo, M., and Kato, K. (2000). Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408, 584–588.
Pike, F. G., Meredith, R. M., Olding, A. W. A., and Paulsen, O. (1999). Postsynaptic bursting is essential for ‘Hebbian’ induction of associative long-term potentiation at excitatory synapses in rat hippocampus. J. Physiol. (Lond.) 518, 571–576.
Rubin, J. E., Gerkin, R. C., Bi, G. Q., and Chow, C. C. (2005). Calcium time course as a signal for spike-timing-dependent plasticity. J. Neurophysiol. 93, 2600–2613.
Shouval, H. Z., Bear, M. F., and Cooper, L. N. (2002). A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 99, 10831–10836.
Sjostrom, P. J., Turrigiano, G. G., and Nelson, S. B. (2001). Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron 32, 1149–1164.
Song, S., Miller, K. D., and Abbott, L. F. (2000). Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat. Neurosci. 3, 919–926.
Stanton, P. K., and Sejnowski, T. J. (1989). Associative long-term depression in the hippocampus induced by Hebbian covariance. Nature 339, 215–218.
Tsubokawa, H., and Ross, W. N. (1997). Muscarinic modulation of spike backpropagation in the apical dendrites of hippocampal CA1 pyramidal neurons. J. Neurosci. 17, 5782–5791.
Urakubo, H., Honda, M., Froemke, R. C., and Kuroda, S. (2008). Requirement of an allosteric kinetics of NMDA receptors for spike timing-dependent plasticity. J. Neurosci. 28, 3310–3323.
Watanabe, S., Hoffman, D. A., Migliore, M., and Johnston, D. (2002). Dendritic K+channels contribute to spike- timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 99, 8366–8371.
Wittenberg, G. M., and Wang, S. S. H. (2006). Malleability of spike-timing-dependent plasticity at the CA3–CA1 synapse. J. Neurosci. 26, 6610–6617.
Keywords: hippocampus, synaptic plasticity, STDP
Citation: Buchanan KA and Mellor JR (2010) The activity requirements for spike timing-dependent plasticity in the hippocampus. Front. Syn. Neurosci. 2:11. doi: 10.3389/fnsyn.2010.00011
Received: 22 January 2010;
Paper pending published: 15 February 2010;
Accepted: 17 May 2010;
Published online: 07 June 2010
Edited by:
Per Jesper Sjöström, University College London, UKReviewed by:
Wickliffe C. Abraham, University of Otago, New ZealandGuo-Qiang Bi, University of Pittsburgh, USA
Dominique Debanne, Université de la Méditerranée, France
Copyright: © 2010 Buchanan and Mellor. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Jack R. Mellor, Medical Research Council Centre for Synaptic Plasticity, Department of Anatomy, University of Bristol, Bristol BS8 1TD, UK. e-mail:amFjay5tZWxsb3JAYnJpc3RvbC5hYy51aw==