



- 1 Department of Cell and Molecular Physiology, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
- 2 Department of Otolaryngology/Head and Neck Surgery, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
- 3 Curriculum in Neurobiology, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
- 4 UNC Neuroscience Center, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
- 5 Neurodevelopmental Disorders Research Center, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Spike timing-dependent plasticity (STDP) has been proposed as a mechanism for optimizing the tuning of neurons to sensory inputs, a process that underlies the formation of receptive field properties and associative memories. The properties of STDP must adjust during development to enable neurons to optimally tune their selectivity for environmental stimuli, but these changes are poorly understood. Here we review the properties of STDP and how these may change during development in primary sensory cortical layers 2/3 and 4, initial sites for intracortical processing. We provide a primer discussing postnatal developmental changes in synaptic proteins and neuromodulators that are thought to influence STDP induction and expression. We propose that STDP is shaped by, but also modifies, synapses to produce refinements in neuronal responses to sensory inputs.
Introduction
A fundamental property of the brain is its ability to change in response to sensory stimuli. These adaptations to changes in either the sensory environment or sensory receptor function provide a substrate for the memory of sensory experiences and perceptual learning. A long-term goal of neuroscience research has been to determine the molecular mechanisms that underlie the formation of cortical responses to environmental stimuli. Changes in synaptic strength have been modeled in vitro using low- or high-frequency stimulation to produce long-term depression (LTD) and long-term potentiation (LTP), respectively (Bliss and Lomo, 1973 ; Dudek and Bear, 1992 ). While frequency-dependent plasticity has provided a wonderful tool to study the mechanism for the strengthening and weakening of cortical synapses during early stages of development, frequency-dependent plasticity is not sufficient to explain many modifications in synaptic strength that result from changes in sensory experience. Manipulations that produce synaptic plasticity in vivo are not always associated with significant changes in firing rates, and changes in firing rates that induce plasticity in vitro do not always produce plasticity when occurring naturally in vivo (Carandini and Ferster, 2000 ; Celikel et al., 2004 ; Fox and Wong, 2005 ). The discovery that the precise temporal precision of spiking between pre- and postsynaptic neurons in the hippocampus can dictate whether a synapse is strengthened or weakened raised great excitement, as this timing-dependent plasticity mechanism could readily account for changes observed in vivo that were not readily explained by frequency-dependent forms of plasticity (Levy and Steward, 1983 ; Magee and Johnston, 1997 ).
Since the initial discoveries of spike timing-dependent plasticity (STDP) (Levy and Steward, 1983 ; Bell et al., 1997 ; Magee and Johnston, 1997 ; Markram et al., 1997 ; Bi and Poo, 1998 ; Debanne et al., 1998 ), it has been proposed as a mechanism by which receptive field maps and sensory selectivity can be formed and modified in vivo (Song and Abbott, 2001 ; Clopath et al., 2010 ). STDP has been observed in sensory cortices just following birth, and is also thought to provide a mechanism for modifying synaptic strength in adulthood (Fu et al., 2002 ; Banerjee et al., 2009 ; Pellicciari et al., 2009 ). Although synaptic plasticity can occur throughout life, the induction and expression mechanisms of both frequency-dependent plasticity and STDP are believed to change over development. For example, adult plasticity in response to sensory deprivation is believed to result primarily from the potentiation of spared (sensory-driven) inputs and not by depression of the lost (sensory-independent) inputs (Glazewski and Fox, 1996 ). Similarly, both frequency- and spike timing-dependent LTD (tLTD) are difficult to induce following postnatal day 30 (P30) in rodents (Dudek and Bear, 1993 ; Fox, 2002 ; Corlew et al., 2007 ; Banerjee et al., 2009 ). This suggests that while the ability to strengthen and weaken sensory synapses remains throughout life, changes in synaptic proteins that occur throughout development may influence how plasticity is induced or expressed. Herein, we describe mechanisms by which STDP can be shaped through development via the expression of synaptic proteins in the cortices of the somatosensory, visual, and auditory systems (Table 1 ). Although STDP has been observed in many neocortical layers (Egger et al., 1999 ; Sjostrom et al., 2003 ; Kampa and Stuart, 2006 ; Letzkus et al., 2006 ), for simplicity we focus on the synaptic connection between cortical layer (L) 4 and L2/3 neurons and between L2/3 neurons. These synapses represent the major site of intracortical processing for inputs arriving from the thalamic relays. In addition, STDP in L2/3 synapses has been observed throughout development, is relatively well characterized in vitro, and occurs in response to sensory deprivation (Diamond et al., 1994 ; Drew and Feldman, 2009 ). We also consider the contribution of neuromodulation to the expression and development of cortical STDP. Although we emphasize changes in synaptic proteins between excitatory cortical connections that may influence STDP expression, considerable evidence demonstrates that STDP exists at inhibitory connections (Holmgren and Zilberter, 2001 ; Haas et al., 2006 ) and that there are considerable changes in inhibitory circuitry during development (Yazaki-Sugiyama et al., 2009 ) that are likely to be shaped by STDP.
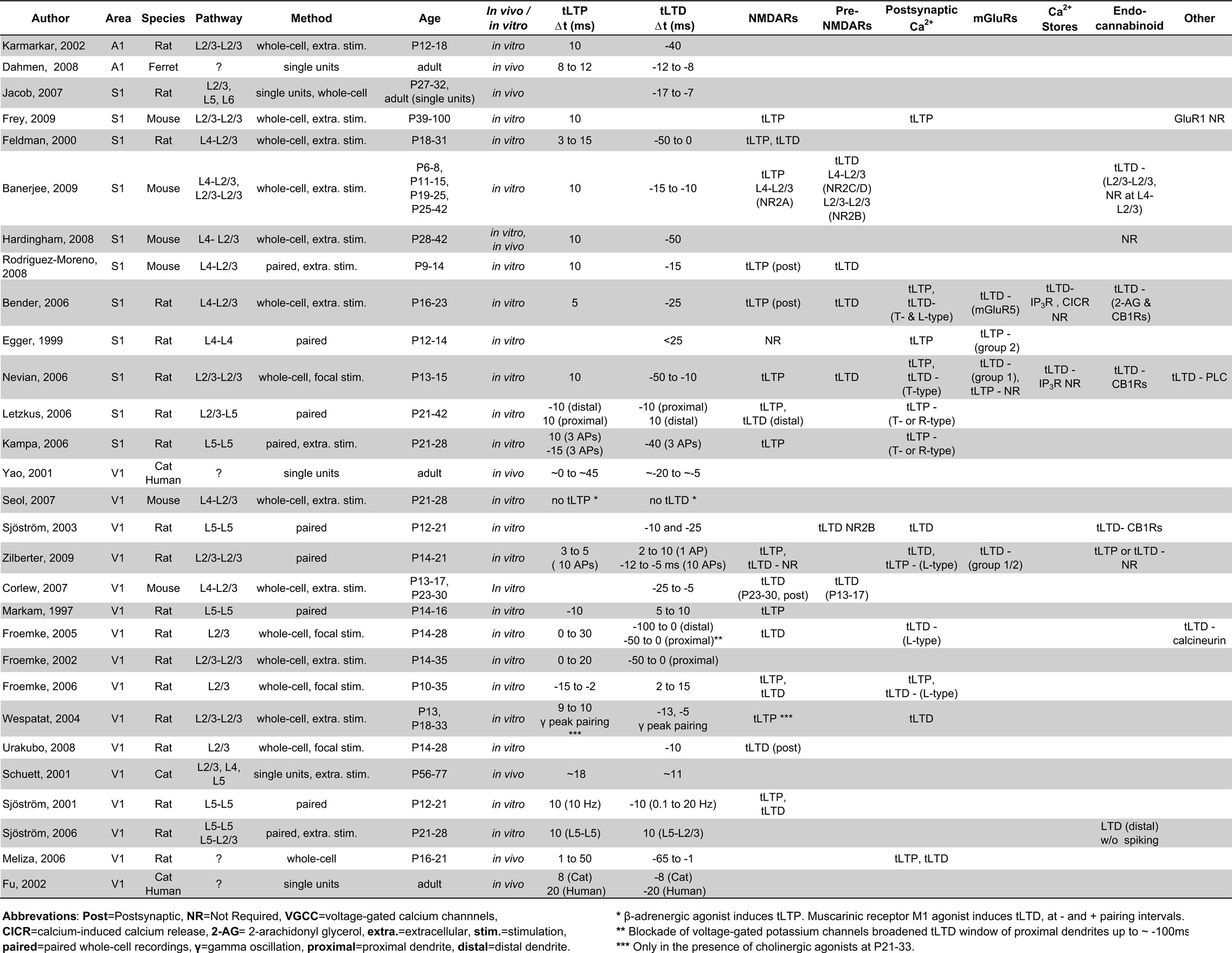
Table 1. Excitatory STDP in sensory neocortices.
STDP in Somatosensory Cortex
Spike timing-dependent plasticity in rodent primary somatosensory cortex (S1) has been proposed to underlie refinement of receptive fields in response to changes in whisker stimulation (Fox and Wong, 2005 ; Feldman, 2009 ). In support of this idea, whisker trimming during early life alters the firing sequence of L4–L2/3 synaptic connections in vivo to produce timing patterns known to weaken synapses in vitro, and this change in the temporal precision of spiking precedes the degradation of L2/3 receptive field maps (Feldman, 2000 ; Celikel et al., 2004 ). Response depression can also be produced in vivo by pairing natural spike trains with coincident whisker deflection to mimic the timing requirements for inducing tLTD in vitro (Jacob et al., 2007 ). Such findings suggest that STDP is likely to occur naturally during receptive field refinements through development and even into adulthood (Clark et al., 1988 ). Below, we discuss the molecular mechanisms underlying STDP and how they may be regulated to produce and tune STDP in developing S1.
tLTP in S1
In general, the induction of timing-dependent LTP (tLTP) in cortical areas requires glutamate binding of NMDA receptors (NMDARs) coincident with arrival of a backpropagating action potential (BAP) into the postsynaptic dendrite (Magee and Johnston, 1997 ; Froemke et al., 2005 ; Letzkus et al., 2006 ). The pairing of glutamate binding with the BAP causes the removal of Mg2+ from NMDARs and produces a supralinear summation of calcium entering through NMDARs and voltage-gated calcium channels (VGCCs) (Koester and Sakmann, 1998 ; Kampa and Stuart, 2006 ; Nevian and Sakmann, 2006 ). Despite the importance of both VGCCs and NMDAR activation for tLTP induction, postsynaptic NMDARs are believed to act as the sole coincidence detector for tLTP within the neocortex (Froemke et al., 2005 , 2006 ; Rodriguez-Moreno and Paulsen, 2008 ).
S1 pyramidal cells maintain the ability to express tLTP into adulthood, and many of the induction parameters appear to be similar throughout life. For example, the timing requirements for tLTP induction are largely unchanged across development, as pre–post pairings with positive intervals of ∼10 ms readily induce tLTP from P6–P100 (Feldman, 2000 ; Bender et al., 2006 ; Rodriguez-Moreno and Paulsen, 2008 ; Banerjee et al., 2009 ). The requirement for postsynaptic NMDAR activation is also maintained across development, because intracortical tLTP is blocked by the NMDAR antagonist APV in S1 in both younger (<P20) and older (>P35) rodents (Rodriguez-Moreno and Paulsen, 2008 ; Banerjee et al., 2009 ; Frey et al., 2009 ). The induction of tLTP between S1 L4–L2/3 synapses requires postsynaptic NMDARs, because selectively loading the postsynaptic recording pipette with the NMDAR antagonist MK-801 is sufficient to abolish tLTP (Bender et al., 2006 ; Rodriguez-Moreno and Paulsen, 2008 ). In addition to having many similar induction requirements across development, the magnitude of tLTP expression also does not correlate with age in rats across the P18–P32 developmental period (Feldman, 2000 ).
While many aspects of tLTP induction are similar throughout life, there are also likely to be important developmental differences. Because postsynaptic NMDARs are thought to be the sole coincidence detector for tLTP, developmental changes in NMDAR functions may by one important modulator of the properties of tLTP induction. In the neocortex, postsynaptic NMDARs undergo a developmental switch from primarily NR2B-containing to NR2A-containing receptors. In rodent S1, this switch to predominately NR2A-containing receptors occurs ∼P9 in L2/3 pyramidal cells (Flint et al., 1997 ; Liu et al., 2004 ). As would be predicted based on this expression pattern in S1, NR2A-, but not NR2B-containing, receptors are required for tLTP induction at L4–L2/3 synapses in P11–P15 mice (Banerjee et al., 2009 ). The contribution of NR2B to tLTP induction has not been studied at young ages (<P6), thus it is not yet clear how the developmental switch from NR2B to NR2A influences tLTP induction. There are currently two ideas as to how an increased NR2A/NR2B ratio would affect tLTP, with one prediction suggesting that a higher ratio would compress the tLTP timing window (Shouval et al., 2002 ) and the other suggesting that it will make tLTP less likely to be induced (Gerkin et al., 2007 ). Both predictions suggest that a shift in the NR2A/NR2B ratio would adjust the balance between tLTD and tLTP. Thus, further studies are warranted to determine how changes in S1 postsynaptic NMDAR composition and downstream signaling cascades at different ages influence the expression, magnitude, and timing requirements of tLTP.
tLTD
While postsynaptic NMDARs act as a coincidence detector for tLTP, they have not been shown to act as the coincidence detector for tLTD between L4 and L2/3 synapses. Instead, the near-simultaneous activation of postsynaptic mGluRs coincident with both postsynaptic depolarization and activation of VGCCs is thought to constitute a separate coincidence detector for tLTD (Karmarkar and Buonomano, 2002 ; Bender et al., 2006 ). In this model, tLTD is induced when postsynaptic group 1 mGluRs (likely mGluR5) are activated with T- or L-type VGCCs to increase PLC activity (Bender et al., 2006 ; Nevian and Sakmann, 2006 ). Activation of PLC leads to generation of inositol 1,4,5-triphosphate (IP3) and intracellular release of calcium from IP3-mediated internal stores (Bender et al., 2006 ; Nevian and Sakmann, 2006 ). This calcium, along with the calcium released from VGCCs, combines to trigger release of the endocannabinoid 2-arachidonyl glycerol (2-AG) from the postsynaptic neuron (Bender et al., 2006 ). Activation of presynaptic CB1 receptors and presynaptic NMDARs results in lasting reductions in release probability from the presynaptic neuron, although the time course and pathways by which this occurs remains to be determined. This type of LTD can become manifest with post-before-pre action potential pairings occurring with intervals up to 50 ms (Feldman, 2000 ; Bender et al., 2006 ; Nevian and Sakmann, 2006 ), which is much longer than the 10–20 ms pre-before-post timing window required for tLTP induction. It should be noted that tLTD in the visual cortex can also be induced in a manner thought to rely on postsynaptic NMDARs as the coincidence detector (Froemke et al., 2005 ; Urakubo et al., 2008 ), and a similar mechanism is likely to occur in S1. Exactly how these two forms of tLTD cooperate or are segregated is not clear, and it is possible that development influences tLTD in a location or spike-dependent manner.
While tLTP is thought to be inducible throughout life, a dramatic reduction in the ability to induce tLTD in vitro between L4 and L2/3 synapses in rodent S1 occurs by P25 (Banerjee et al., 2009 ). This decrease in tLTD magnitude is reminiscent of the developmental loss of frequency-dependent LTD in CA1 of the hippocampus (Dudek and Bear, 1993 ) and to the loss of LTP at S1 thalamocortical synapses (Crair and Malenka, 1995 ). A developmental reduction in tLTD magnitude has also been observed in L4–L2/3 synapses in primary visual cortex, and this loss is curiously dependent on inhibition (Corlew et al., 2007 ). This suggests that a developmental increase in inhibition might limit tLTD induction, perhaps through shunting inhibition, but this hypothesis has yet to be rigorously tested. Since standard experimental protocols do not reliably induce tLTD in mature neocortex, it is possible that the requirements for tLTD induction are different, and will require increasing the number or adjusting the timing of the pairings. In support of this idea, a very narrow window for inducing tLTD has been observed in adult rats in vivo (Jacob et al., 2007 ).
Before P25 in rodents, it is remarkable that the magnitude of tLTD is similar at all ages tested (P6–P32; Feldman, 2000 ; Banerjee et al., 2009 , despite large changes in many of the proteins involved in tLTD induction. Among these proteins, mGluRs and their downstream effectors are developmentally upregulated before P15. The requirement for group 1 mGluRs for tLTD at L2/3 synapses has been shown at P13–P23, when mGluR expression begins to plateau (Bender et al., 2006 ; Nevian and Sakmann, 2006 ). In S1, mGluR5 expression is uniform in all layers by P16 and remains constant at these levels through adulthood (Blue et al., 1997 ). Similarly, the expression of group 1 mGluR’s downstream effector, PLC, reaches stable expression in S1 by P14 (Hannan et al., 1998 ). The early developmental upregulation of mGluR5 and PLC expression do not seem to influence the magnitude or induction of tLTD, because mice aged P6–P8 show tLTD with a similar magnitude to mice at P11–P25 (Banerjee et al., 2009 ). This suggests that mGluRs do not developmentally gate tLTD induction, but may influence tLTD in other ways. It is clear that mGluRs and their downstream effectors play an important role in S1 development because the genetic deletion of mGluR5 or PLC causes barrels to form improperly (Hannan et al., 2001 ), yet whether this is a direct consequence of altered tLTD remains unknown.
Synaptic proteins involved in tLTD induction have also been suggested to be segregated based on synapse. For example, the requirement both for endocannabinoid signaling and specific preNMDAR subunits differs by synaptic pathway. In mice, CB1Rs are not required for tLTD between L4 and L2/3 synapses at either P11–P15 or P28–P42 but are required between L2/3 and L2/3 synapses at P11–P15 (Hardingham et al., 2008 ; Banerjee et al., 2009 ). In contrast, CB1Rs and postsynaptic endocannabinoid synthesis are required for tLTD induction between L4 and L2/3 rat neurons at P16–P23 (Bender et al., 2006 ). These differences may reflect laminar and species-specific differences in the activation of CB1Rs or their downstream signaling. The expression of CB1Rs reaches stable adult levels by P16 in rats and CB1R function is required during this period for barrel receptive field formation (Bodor et al., 2005 ; Deshmukh et al., 2007 ). Chronically blocking CB1Rs with the in vivo administration of the antagonist AM251 between P13 and P16 disrupts whisker tuning and results in the loss of experience-dependent plasticity in L2/3 rat S1 (Li et al., 2009 ). This demonstrates the importance of CB1R signaling in rats at a time when tLTD is readily inducible both in vivo and in vitro by CB1R activation, suggesting that there may be a causal relationship between tLTD induction and receptive field tuning in S1.
In a similar fashion to the segregation of endocannabinoid signaling across cortical layers, there are differing layer-dependent requirements for presynaptic NMDAR subunits. Moderately selective NR2C/D antagonists, but not NR2B or NR2A antagonists, block the induction of tLTD between L4 and L2/3 S1 synapses (Banerjee et al., 2009 ). In contrast, L2/3–L2/3 synapses show a requirement for NR2B-containing receptors, but not NR2C/D (Banerjee et al., 2009 ). The segregation of presynaptic NMDAR subunits may permit differential modulation of tLTD depending on the synaptic pathway, which is consistent with previous findings that the induction requirements and timing windows of STDP depend on dendritic location (Froemke et al., 2005 ; Letzkus et al., 2006 ). The mechanisms by which STDP are induced appear diverse and synapse-specific. Due to the wide variety of synaptic mechanisms for induction, tLTD may be developmentally regulated in a unique way at each synapse. Studies that compare the pathway-specific tLTD mechanisms could determine the exact requirements for tLTD induction at each S1 synapse. The existing evidence suggests that the molecular mechanisms of tLTD are not universal across synapses within sensory cortices.
STDP in Visual Cortex
The importance of coordinated activity in the developing visual cortex was first demonstrated in groundbreaking experiments by Hubel and Wiesel (1965) where binocular receptive fields were converted to monocular receptive fields by changing the synchrony of visual inputs in kittens with artificial strabismus. STDP within the visual cortex likely follows constraints unique to the environmental stimuli it receives, allowing this form of plasticity to modulate synaptic connectivity in a manner that is different from S1. Like plasticity in S1, STDP in V1 is a relevant mechanism for synaptic strengthening and weakening. Indeed, pairing action potentials with precisely timed visual stimuli induces STDP in vivo (Meliza and Dan, 2006 ). In further support of the idea that STDP can shape visual processing, manipulating the temporal order of spiking in V1 neurons is sufficient to change orientation preferences and receptive fields in vivo, and these modifications can occur in a bidirectional manner similar to STDP timing rules observed in vitro (Schuett et al., 2001 ). For example, when visual stimuli of a particular orientation are paired with electrical stimulation of a neuron, the orientation preference of that neuron shifts toward that of the given stimuli (Schuett et al., 2001 ). Reversing the pairing order (so that the neuron fires before the visual stimuli) weakens the orientation preference away from the given orientation in a tLTD-like manner. Additionally, the pairing of visual stimuli at two orientations shifts the orientation preference of V1 neurons depending on the temporal order of the pairings and can be predicted based on the temporal windows of STDP induced in vitro (Yao and Dan, 2001 ). The ability to modify visual responses via STDP learning rules exists through adulthood, as the pairing of visual stimuli can rapidly modify receptive fields and orientation preferences in adult cats (Yao and Dan, 2001 ; Fu et al., 2002 ). Lastly, STDP learning rules have been shown to be sufficient to segregate sensory inputs onto specific dendritic branches, underscoring how STDP may be essential for shaping cortical connectivity (Froemke et al., 2005 ). Overall these observations suggest that STDP provides a powerful mechanism by which visual cortical circuitry can be modeled and by which neurons can rapidly adapt to an ever-changing visual environment throughout life. Many synaptic proteins implicated in STDP induction or expression are developmentally regulated between P10 and P35 in rodents, overlapping with periods of receptive field development and the visual cortical critical period (Hensch, 2005 ; Smith and Trachtenberg, 2007 ). The regulation of these synaptic proteins may therefore favor the development and stability of visual circuits through adulthood by modulating STDP.
A surprising observation, which we will discuss below, is that the mechanisms of STDP appear largely similar between S1 and V1. The most pronounced differences in STDP between these regions are due to a developmental delay in V1 development compared to S1 development, and this delay is likely due to a delay in sensory-driven activity in V1.
tLTP
Similar to tLTP observed in the somatosensory cortex, tLTP in V1 is believed to rely on the interaction of BAPs with calcium influx through postsynaptic NMDARs and L-type VGCCs (Froemke et al., 2005 , 2006 ). NMDARs are required for tLTP induction between P12 and P35 at both L5 and L2/3 V1 synapses (Markram et al., 1997 ; Froemke et al., 2006 ; Zilberter et al., 2009 ). Unlike S1, the exact postsynaptic NMDAR subunits required for tLTP have not been investigated. Postsynaptic NMDARs in V1 show a developmental shift from NR2B to NR2A at a period later in development (∼P25) as compared to other cortical areas (de Marchena et al., 2008 ). This suggests that a greater proportion of NR2B-containing receptors may participate in tLTP induction before P25 in the visual cortex compared to somatosensory cortex, although it has been reported that the NR2B antagonist ifenprodil does not have a major impact on the NMDA:AMPA ratio in L5 neurons of P14–P15 rats (Sjostrom et al., 2003 ). How the switch in NMDAR subunits during the visual critical period influences STDP induction and expression is not known, but it may involve temporal changes in NMDAR glutamate binding (Laurie and Seeburg, 1994 ), magnesium sensitivity (Clarke and Johnson, 2006 ), or allosteric interactions (Urakubo et al., 2008 ) that could alter dendritic calcium and shape the temporal window for inducing STDP (Shouval et al., 2002 ).
Surprisingly little is known about how the properties of tLTP adjust over development in the visual cortex, but some assumptions can be made based on known tLTP mechanisms. In addition to the aforementioned changes in NMDAR subunit expression, there are other developmental changes in tLTP-related proteins that can affect tLTP induction across the length of the dendrite. The magnitude of tLTP in L2/3 pyramidal neurons varies with location of the stimulated inputs, such that stimulation of synapses on the proximal dendrite produce a larger magnitude of tLTP than stimulation of synapses on more distal dendrites (Froemke et al., 2005 ). This effect probably depends on the attenuation of the BAP along the extent of the dendrite (Magee and Johnston, 1997 ; Froemke et al., 2005 ; Sjostrom and Hausser, 2006 ), which would be predicted to affect the supralinear potentiation of calcium that has been observed with tLTP induction (Nevian and Sakmann, 2006 ). Such an interpretation is consistent with studies in the somatosensory cortex showing that voltage-gated sodium channel dependent action potentials, in turn activate VGCCs (Kampa and Stuart, 2006 ; Komai et al., 2006 ). Consequently, any developmental changes in the magnitude or localization of dendritic sodium or calcium channels would be expected to alter the timing requirements and magnitude of tLTP, perhaps by changing the resulting calcium transient. Developmental changes in other dendritic proteins that can affect the shape or size of the BAP, such as A-type potassium channels (Hoffman et al., 1997 ; Froemke et al., 2005 ), would likewise be expected to alter tLTP induction and expression.
tLTD
Like tLTP, NMDAR activation is required for the induction of tLTD. Unlike tLTP, there appears to be both a presynaptic and a postsynaptic contribution of NMDARs. The relative contribution of pre- and postsynaptic NMDARs may vary by age and pathway. Initial studies using bath-applied APV to globally block NMDARs led to the assumption that the NMDARs relevant to tLTD were exclusively postsynaptic (Markram et al., 1997 ; Feldman, 2000 ). Later studies in ∼P14–P18 rodents found that tLTD could still be induced when postsynaptic (but not presynaptic) NMDARs were blocked (Sjostrom et al., 2003 ; Corlew et al., 2007 ). This form of tLTD appeared to have a dual requirement for presynaptic NMDAR and CB1R activation, similar to what has been described for S1 (Sjostrom et al., 2003 ; Bender et al., 2006 ; Rodriguez-Moreno and Paulsen, 2008 ), although whether preNMDARs are acting on a rapid or slow time scale has been debated (Sjostrom et al., 2003 ; Bender et al., 2006 ).
While some studies have shown that tLTD at L5–L5 and L4–L2/3 synapses requires presynaptic NMDAR activation (Sjostrom et al., 2003 ; Corlew et al., 2007 ), others have shown that tLTD is fully blocked by postsynaptic inhibition of NMDARs in L2/3 V1 neurons (Froemke et al., 2005 ; Urakubo et al., 2008 ). This apparent discrepancy might be explained by age-related modifications in the mechanisms of tLTD. Presynaptic NMDARs, which are required for tLTD during early life, are sharply downregulated between P16 and P27 (Corlew et al., 2007 ). Remarkably, this anatomical reduction in presynaptic NMDARs coincides with the loss of presynaptically expressed tLTD between L4 and L2/3 synapses, which occurs around 3 weeks of age, suggesting that there may be a causal relationship between the two events. In support of this idea, studies showing a requirement for presynaptic NMDARs in tLTD have been performed in P14–P18 rodents, while those that support a postsynaptic requirement for NMDARs have been performed in rodents including ages >P21. The form of tLTD involving postsynaptic NMDARs requires activation of the phosphatase calcineurin (Froemke et al., 2005 ; Urakubo et al., 2008 ), but it is not known if this is a requirement in younger rodents (Figure 1 ).
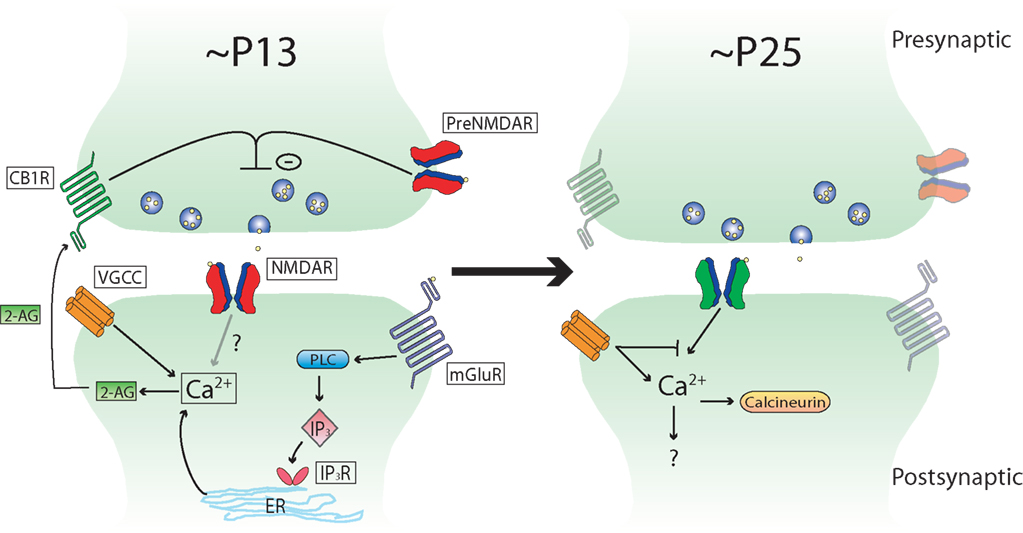
Figure 1. Schematic depicting developmental changes in known tLTD induction mechanisms at L4–L2/3 synapses in rodent sensory neocortex. Note that the mechanisms are very similar between the different sensory areas, and that this scheme could apply to primary visual cortex as well as primary somatosensory cortex (see text for details).
In addition to developmental changes in the contribution of presynaptic NMDARs to tLTD, there are also likely to be differences in the role that inhibition plays in tLTD. A developmental loss of tLTD at L4–L2/3 synapses is evident in V1 by ∼P23 (Corlew et al., 2007 ), similar to that observed in S1 by ∼P25 (Banerjee et al., 2009 ). However, the induction of tLTD can be restored in older mice by performing the post-before-pre pairing protocol in the presence of GABAA receptor antagonists (Corlew et al., 2007 ). When inhibition is blocked at these older ages, tLTD requires postsynaptic NMDARs instead of presynaptic NMDARs. This suggests development may shape the mechanism by which tLTD is induced from one that is predominately presynaptic to one that is predominately postsynaptic. It is interesting that tLTD that relies on postsynaptic NMDAR activation in older animals is smaller in magnitude than that induced at younger ages, suggesting development may also subtly affect tLTD magnitude in V1 (Corlew et al., 2007 ). As the loss of presynaptically expressed tLTD coincides with a period of rapid inhibitory development (Hensch, 2005 ), it suggests that inhibition may influence the mechanisms underlying tLTD. An unresolved issue is whether tLTD requires one or two coincidence detectors. While inhibition is one factor that influences tLTD induction mechanisms, others such as dendritic location (Froemke et al., 2005 ) and dendritic calcium buffering (Kampa and Stuart, 2006 ; Nevian and Sakmann, 2006 ) may also influence how tLTD is induced.
STDP in Auditory Cortex
Sounds in the acoustic environment have complex temporal structures that overlap in time, space, and frequency content. Cortical lesion studies demonstrate the importance of the auditory cortex in the perception of time-varying sounds across a large range of time scales (Whitfield, 1980 ). As in visual cortex, coordinated activity may play a role in plasticity in auditory cortex. Raising rats in a noisy environment devoid of structured spectral and temporal cues delays the refinement of the tonotopic map in primary auditory cortex (A1), and this can be reversed by experience in an acoustic environment with tonal structure (Chang and Merzenich, 2003 ). Neurons in A1 can fire with millisecond precision to the fine temporal structure of acoustic stimuli (for example, Eggermont, 2007 ), and it was recently shown that millisecond differences in neural activity in A1 can be exploited to guide decisions (Yang et al., 2008 ). Given the robust plasticity, importance of temporal features in sound identification and discrimination, and the precision of spiking timing in A1 (Recanzone et al., 1993 ; Kudoh and Shibuki, 1994 ; Bao et al., 2004 ), it is natural to wonder whether A1 has unique timing rules for STDP. Although surprisingly few studies of STDP have been performed in A1, the studies to date suggest that the properties of STDP in A1 are fundamentally similar to those observed in other sensory cortices.
Spike timing-dependent plasticity-like rules have been observed in a variety of species (Gerstner et al., 1996 ) throughout the auditory pathway, including brainstem (Tzounopoulos et al., 2004 ) and cortical areas (Schnupp et al., 2006 ). STDP in the dorsal cochlear nucleus appears to follow Hebbian and anti-Hebbian patterns in a cell-specific manner (Tzounopoulos et al., 2004 ). In contrast, STDP in the auditory cortex, at least at some synapses onto pyramidal cells, appears to follow a traditional Hebbian rule. In P12–P18 rat auditory cortical slices, repetitive pairing of pre-before-post spiking activity at 10 ms intervals produces tLTP and post-before-pre intervals at 40 ms produces tLTD at L2/3–L2/3 synapses (Karmarkar and Buonomano, 2002 ). Although the entire STDP window in A1 was not investigated in this study, the results are consistent with findings at similar synapses in P10–P35 rat V1 (Froemke et al., 2006 ) and in P13–P15 rat S1 (Nevian and Sakmann, 2006 ). These data suggest that, at least in vitro, STDP rules between L2/3 neurons appear roughly similar in all sensory cortices.
In vivo studies also support a role for STDP in A1. In anesthetized and awake adult ferrets, repetitive and asynchronous pairings of pure tones of different frequencies produce shifts in the frequency selectivity of neurons recorded extracellularly (Dahmen et al., 2008 ), and the temporal specificity of these shifts is similar to that observed in vitro. In this study, a non-preferred tone frequency was paired with a preferred tone frequency with an 8- to 12-ms time delay between the two tones. When the non-preferred tone was presented before the preferred tone, there was a shift in the neuronal best frequency toward the non-preferred tone frequency. Conversely, when the non-preferred tone frequency was played after the preferred tone frequency, then the neuronal best frequency shifted away from the non-preferred tone. The duration of STDP in A1 observed in vivo is similar to that reported in visual receptive fields of V1 in anesthetized cats (Yao and Dan, 2001 ) and for STDP in whisker-evoked responses of barrel cortex in rats (Jacob et al., 2007 ). Interestingly, in A1, the shifts in cortical frequency tuning are restricted to cortical L2/3 and L4 (Dahmen et al., 2008 ). These observations highlight that the temporal relationships among the components of acoustic stimuli on a millisecond scale to influence auditory processing and suggest that STDP is a relevant mechanism for plasticity in the auditory cortex.
To date, little is known about the mechanistic pathway or developmental modifications of STDP in A1. As STDP displays components of frequency-dependent LTP and LTD, it is rational to speculate that it may use the same mechanisms known to underlie associative LTP and LTD (Malenka and Bear, 2004 ). Indeed, many of the mechanisms for frequency-dependent plasticity in S1 and V1 are similar to those demonstrated for STDP, it is reasonable to assume that the same may be true for A1. In A1, frequency-dependent LTP and LTD have been demonstrated at thalamocortical synapses and at excitatory intracortical synapses (Kudoh and Shibuki, 1994 , 1996 , 1997 ; Bandrowski et al., 2001 ). The induction of frequency-dependent LTP is regulated by age and experience (Speechley et al., 2007 ), suggesting that the same may be true for tLTP. Frequency-dependent LTP of thalamocortical synapses requires activation of NMDARs (Kudoh and Shibuki, 1994 , 1996 ), while LTD at the same synapse requires activation of mGluRs receptors and protein kinase C (Bandrowski et al., 2001 ). It might be expected in A1 that tLTD requires mGluR activation and activation of a PKC pathway, while tLTP may involve the classic postsynaptic NMDAR pathway. While such a finding would be consistent with tLTP and tLTD mechanisms in V1 and S1, there is not yet experimental evidence that this is true.
Neuromodulation of STDP in Sensory Cortices
Neuromodulators alter receptive field plasticity in sensory cortices by expanding the cellular representation of sensory stimuli (Weinberger, 2003 ). Examples thought to engage neuromodulators include the observations that (1) classical conditioning using whisker stimuli expands the representation of trained whiskers in S1 (Siucinska and Kossut, 2004 ), (2) perceptual training on visual stimulus orientation discrimination tasks alters V1 tuning for the trained feature (Fu et al., 2002 ), and (3) activation of cholinergic or dopaminergic inputs during tonal stimuli increases A1 responses to the tone frequency (Bakin and Weinberger, 1996 ; Bao et al., 2001 ; Weinberger et al., 2006 ). At the cellular level, neuromodulators have both facilitating and depressing effects on cortical activity that depend on the type of neuromodulators and the pattern of neuromodulator receptors expressed in sensory cortices (Spehlmann, 1971 ).
The effects of neuromodulators on receptive field plasticity appear to depend on the engagement of STDP-like mechanisms. The properties of STDP can be powerfully adjusted by neuromodulators, which can control the polarity, magnitude, or even the ability to induce STDP through development. In the absence of neuromodulators, tLTD and tLTP can be induced in L2/3–L2/3 synapses in developing V1 by temporally pairing EPSPs to β or γ oscillations produced by injected sinusoidal currents, such that EPSPs synchronous with hyperpolarizing and depolarizing membrane potentials produced tLTD and tLTP, respectively (Wespatat et al., 2004 ). This form of tLTP is impaired in V1 slices from older rats (>P21), but it can be rescued when pairings are made while muscarinic receptors are activated (Wespatat et al., 2004 ). Similarly, both tLTP and tLTD are impaired at L4–L2/3 synapses in older rat V1, but both tLTD and tLTP can be recovered when AP–EPSP pairings are made in the presence of M1 muscarinic or β-adrenergic receptor activation, respectively (Seol et al., 2007 ). These results demonstrate that neuromodulators gate STDP in the adult brain. In addition to their role as permissive gatekeepers for STDP induction, neuromodulators are also likely to control the polarity and temporal requirements for inducing STDP plasticity in sensory cortices, as such roles for neuromodulators have been observed in other areas of the brain. For example, in L2/3–L5 synapses in prefrontal cortex, nicotine application converts tLTP to tLTD (Couey et al., 2007 ). In hippocampal CA1, a β-adrenergic receptor agonist broadens the tLTP window from 3–10 to 15 ms without affecting tLTP magnitude (Lin et al., 2003 ). Also at hippocampal synapses, dopamine agonists not only extends the tLTP window from 20 ms to at least 45 ms but, also converts tLTD to tLTP (Zhang et al., 2009 ). Thus, neuromodulators can adjust multiple aspects of STDP induction, and the precise effects of neuromodulators on STDP induction likely depend on the neuromodulator, receptor types, synaptic pathway, and age.
How might neuromodulators alter the properties of STDP? Although there are many targets of neuromodulators, the common denominator for most of these mechanisms is that they ultimately influence local calcium levels associated with AP–EPSP pairings. There are several mechanisms by which neuromodulators bring about their effects on calcium levels. First, neuromodulators can activate kinases and phosphatases that regulate the kinetics and availability of dendritic ion channels, such as transient (IA) and Ca2+-activated K+ channels (Watanabe et al., 2002 ). Such modulation brings about profound changes in the width and amplitude of BAPs, ultimately influencing dendritic calcium (Magee and Johnston, 1997 ; Froemke et al., 2006 ). For example, β-adrenergic and muscarinic receptor agonists enhance spike backpropagating efficacy by phosphorylating protein kinase A and protein kinase C that result in reduction of IA channel availability (Tsubokawa and Ross, 1997 ; Hoffman and Johnston, 1998 , 1999 ). Such changes in IA might contribute to the observations that M1 muscarinic receptors promote tLTD induction through a PLC-dependent pathway, while β-adrenergic receptor activation promotes tLTP through the adenylate cyclase cascade (Seol et al., 2007 ). Second, neuromodulators can target IP3 receptors and activate calcium-induced-calcium-release from intracellular stores, thereby influencing polarity and input-specificity of STDP (Nishiyama et al., 2000 ). Third, neuromodulators can facilitate NMDAR currents (Brocher et al., 1992 ; Kirkwood et al., 1999 ) and presumably directly regulate STDP induction. Although it has not yet been investigated, developmental changes in neuromodulator influences are also likely to affect the timing rules for inducing STDP in sensory cortices and could play a role in defining critical periods. In support of this possibility, the expression of certain neuromodulator receptor families, such as alpha 7 nicotinic receptors and 5HT receptors, exhibit dramatic regulation around the critical period for receptive field plasticity in sensory cortices (Broide et al., 1995 , 1996 ; Aramakis and Metherate, 1998 ; Basura et al., 2008 ).
Conclusion
The studies discussed here support the argument that STDP is a key mechanism used in sensory processing in somatosensory, visual, and auditory cortices, both for the establishment of circuits during development, and for the storage and processing of sensory information later in life. At a cellular level, STDP is shaped by, but also modifies, specific synapses to produce refinements in neuronal responses to sensory stimuli. While we have emphasized the role of synaptic proteins in shaping STDP, very little is known about how these changes influence the exact characteristics of induction, expression, and timing of STDP. As STDP depends not just on timing, but on spike patterning (Sjostrom et al., 2001 ; Froemke and Dan, 2002 ; Nelson et al., 2002 ; Froemke et al., 2006 ), dendritic location (Froemke et al., 2005 ; Letzkus et al., 2006 ; Sjostrom and Hausser, 2006 ), and previous neuronal activity (Zilberter et al., 2009 ), the roles of specific synaptic proteins in regulating STDP are likely both state- and context-dependent. These changes likely coincide with developmental changes in inhibition and neuromodulation that also shape how STDP learning rules are applied to sensory information (Kirkwood et al., 1999 ; Meredith et al., 2003 ). Therefore, STDP refines sensory inputs in a manner that is dependent on the developmental context while providing feedback that further changes cortical structure and function.
It is clear that there are large gaps in our knowledge regarding STDP in sensory cortices as well. For example, STDP timing windows, as measured experimentally, are quite noisy, which may reflect either the basal state of synapses prior to the experimental measurement, or the specific selection of synaptic pathways during recording. This variability makes it difficult to discern whether the timing windows are truly different between sensory areas or within different local circuit pathways, as might be predicted from the different temporal dynamics of the incoming sensory information versus the temporal dynamics of local and long-distance intracortical pathways. This biological variability is confounded by inevitable discrepancies in experimental approaches. Second, the reversibility of STDP in sensory cortex has not been investigated. In one of the first investigations of STDP (Bell et al., 1997 ) in the electrosensory lobe of mormyrid fish, STDP could be induced rapidly and was readily reversed within minutes with appropriate pairing patterns. Reversibility of timing-dependent plasticity has also been observed in the xenopus retinotectal system (Zhou et al., 2003 ). Is STDP in sensory cortex similarly reversible? Such rapid and reversible plasticity would seem to have clear utility in sensory processing, independent of a role in establishing longer sensory memory or shaping response maps. Third, the role of STDP in the inhibitory network is far from understood. Developmental changes in inhibition help drive the establishment of cortical circuits, and may involve STDP (Kanold and Shatz, 2006 ). Within established cortical networks, both inhibition and excitation exhibit plasticity (Froemke et al., 2007 ; Lu et al., 2007 ; Galindo-Leon et al., 2009 ), but it is not known whether this in vivo plasticity is spike-timing based or not. Given the critical role of inhibition in shaping response maps and spike timing, the role of STDP at inhibitory synapses merits greater investigation. Fourth, the role of STDP in the development and establishment of sensory response maps, while an attractive hypothesis supported by computational studies (Song and Abbott, 2001 ) and consistent with the available data, has not yet been unequivocally tested. To clarify the role of STDP, rate-based versus timing-based plasticity mechanisms must be disentagled to identify their respective roles in receptive field plasticity in vivo. Experimental approaches that allow manipulation of spike trains with millisecond precision in vivo are just emerging (Boyden et al., 2005 ; Chow et al., 2010 ; Gunaydin et al., 2010 ). These technologies can be leveraged in cleverly-designed experiments and carefully-posed questions to answer the major issues outstanding in the field of STDP.
Conflict of Interest Statement
The authors declare the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Thorfinn Riday for assistance with the making of the figure and table, and Dr. Serena Dudek and Dr. Rob Froemke for helpful discussions. Our work was funded by the National Science Foundation grant # 0822969 and the National Eye Institute R01EY018323 (BDP), and NIDCD grant R01DC000425 (PBM).
References
Aramakis, V. B., and Metherate, R. (1998). Nicotine selectively enhances NMDA receptor-mediated synaptic transmission during postnatal development in sensory neocortex. J. Neurosci. 18, 8485–8495.
Bakin, J. S., and Weinberger, N. M. (1996). Induction of a physiological memory in the cerebral cortex by stimulation of the nucleus basalis. Proc. Natl. Acad. Sci. U.S.A. 93, 11219–11224.
Bandrowski, A. E., Ashe, J. H., and Crawford, C. A. (2001). Tetanic stimulation and metabotropic glutamate receptor agonists modify synaptic responses and protein kinase activity in rat auditory cortex. Brain Res. 894, 218–232.
Banerjee, A., Meredith, R. M., Rodriguez-Moreno, A., Mierau, S. B., Auberson, Y. P., and Paulsen, O. (2009). Double dissociation of spike timing-dependent potentiation and depression by subunit-preferring NMDA receptor antagonists in mouse barrel cortex. Cereb. Cortex 19, 2959–2969.
Bao, S., Chan, V. T., and Merzenich, M. M. (2001). Cortical remodelling induced by activity of ventral tegmental dopamine neurons. Nature 412, 79–83.
Bao, S., Chang, E. F., Woods, J., and Merzenich, M. M. (2004). Temporal plasticity in the primary auditory cortex induced by operant perceptual learning. Nat. Neurosci. 7, 974–981.
Basura, G. J., Abbas, A. I., O’Donohue, H., Lauder, J. M., Roth, B. L., Walker, P. D., and Manis, P. B. (2008). Ontogeny of serotonin and serotonin2A receptors in rat auditory cortex. Hear. Res. 244, 45–50.
Bell, C. C., Han, V. Z., Sugawara, Y., and Grant, K. (1997). Synaptic plasticity in a cerebellum-like structure depends on temporal order. Nature 387, 278–281.
Bender, V. A., Bender, K. J., Brasier, D. J., and Feldman, D. E. (2006). Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J. Neurosci. 26, 4166–4177.
Bi, G. Q., and Poo, M. M. (1998). Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 18, 10464–10472.
Bliss, T. V., and Lomo, T. (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. (Lond.) 232, 331–356.
Blue, M. E., Martin, L. J., Brennan, E. M., and Johnston, M. V. (1997). Ontogeny of non-NMDA glutamate receptors in rat barrel field cortex: I. Metabotropic receptors. J. Comp. Neurol. 386, 16–28.
Bodor, A. L., Katona, I., Nyiri, G., Mackie, K., Ledent, C., Hajos, N., and Freund, T. F. (2005). Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J. Neurosci. 25, 6845–6856.
Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G., and Deisseroth, K. (2005). Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268.
Brocher, S., Artola, A., and Singer, W. (1992). Agonists of cholinergic and noradrenergic receptors facilitate synergistically the induction of long-term potentiation in slices of rat visual cortex. Brain Res. 573, 27–36.
Broide, R. S., O’Connor, L. T., Smith, M. A., Smith, J. A., and Leslie, F. M. (1995). Developmental expression of alpha 7 neuronal nicotinic receptor messenger RNA in rat sensory cortex and thalamus. Neuroscience 67, 83–94.
Broide, R. S., Robertson, R. T., and Leslie, F. M. (1996). Regulation of alpha7 nicotinic acetylcholine receptors in the developing rat somatosensory cortex by thalamocortical afferents. J. Neurosci. 16, 2956–2971.
Carandini, M., and Ferster, D. (2000). Membrane potential and firing rate in cat primary visual cortex. J. Neurosci. 20, 470–484.
Celikel, T., Szostak, V. A., and Feldman, D. E. (2004). Modulation of spike timing by sensory deprivation during induction of cortical map plasticity. Nat. Neurosci. 7, 534–541.
Chang, E. F., and Merzenich, M. M. (2003).Environmental noise retards auditory cortical development. Science 18, 498–502.
Chow, B. Y., Han, X., Dobry, A. S., Qian, X., Chuong, A. S., Li, M., Henninger, M. A., Belfort, G. M., Lin, Y., Monahan, P. E., and Boyden, E. S. (2010). High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 463, 98–102.
Clark, S. A., Allard, T., Jenkins, W. M., and Merzenich, M. M. (1988). Receptive fields in the body-surface map in adult cortex defined by temporally correlated inputs. Nature 332, 444–445.
Clarke, R. J., and Johnson, J. W. (2006). NMDA receptor NR2 subunit dependence of the slow component of magnesium unblock. J. Neurosci. 26, 5825–5834.
Clopath, C., Busing, L., Vasilaki, E., and Gerstner, W. (2010). Connectivity reflects coding: a model of voltage-based STDP with homeostasis. Nat. Neurosci. 13, 344–352.
Corlew, R., Wang, Y., Ghermazien, H., Erisir, A., and Philpot, B. D. (2007). Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J. Neurosci. 27, 9835–9845.
Couey, J. J., Meredith, R. M., Spijker, S., Poorthuis, R. B., Smit, A. B., Brussaard, A. B., and Mansvelder, H. D. (2007). Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron 54, 73–87.
Crair, M. C., and Malenka, R. C. (1995). A critical period for long-term potentiation at thalamocortical synapses. Nature 375, 325–328.
Dahmen, J. C., Hartley, D. E., and King, A. J. (2008). Stimulus-timing-dependent plasticity of cortical frequency representation. J. Neurosci. 28, 13629–13639.
de Marchena, J., Roberts, A. C., Middlebrooks, P. G., Valakh, V., Yashiro, K., Wilfley, L. R., and Philpot, B. D. (2008). NMDA receptor antagonists reveal age-dependent differences in the properties of visual cortical plasticity. J. Neurophysiol. 100, 1936–1948.
Debanne, D., Gahwiler, B. H., and Thompson, S. M. (1998). Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J. Physiol. (Lond.) 507 (Pt 1), 237–247.
Deshmukh, S., Onozuka, K., Bender, K. J., Bender, V. A., Lutz, B., Mackie, K., and Feldman, D. E. (2007). Postnatal development of cannabinoid receptor type 1 expression in rodent somatosensory cortex. Neuroscience 145, 279–287.
Diamond, M. E., Huang, W., and Ebner, F. F. (1994). Laminar comparison of somatosensory cortical plasticity. Science 265, 1885–1888.
Drew, P. J., and Feldman, D. E. (2009). Intrinsic signal imaging of deprivation-induced contraction of whisker representations in rat somatosensory cortex. Cereb. Cortex 19, 331–348.
Dudek, S. M., and Bear, M. F. (1992). Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc. Natl. Acad. Sci. U.S.A. 89, 4363–4367.
Dudek, S. M., and Bear, M. F. (1993). Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J. Neurosci. 13, 2910–2918.
Egger, V., Feldmeyer, D., and Sakmann, B. (1999). Coincidence detection and changes of synaptic efficacy in spiny stellate neurons in rat barrel cortex. Nat. Neurosci. 2, 1098–1105.
Eggermont, J. J. (2007). Correlated neural activity as the driving force for functional changes in auditory cortex. Hear Res. 229, 69–80.
Feldman, D. E. (2000). Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron 27, 45–56.
Feldman, D. E. (2009). Synaptic mechanisms for plasticity in neocortex. Annu. Rev. Neurosci. 32, 33–55.
Flint, A. C., Maisch, U. S., Weishaupt, J. H., Kriegstein, A. R., and Monyer, H. (1997). NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J. Neurosci. 17, 2469–2476.
Fox, K. (2002). Anatomical pathways and molecular mechanisms for plasticity in the barrel cortex. Neuroscience 111, 799–814.
Fox, K., and Wong, R. O. (2005). A comparison of experience-dependent plasticity in the visual and somatosensory systems. Neuron 48, 465–477.
Frey, M. C., Sprengel, R., and Nevian, T. (2009). Activity pattern-dependent long-term potentiation in neocortex and hippocampus of GluA1 (GluR-A) subunit-deficient mice. J. Neurosci. 29, 5587–5596.
Froemke, R. C., and Dan, Y. (2002). Spike-timing-dependent synaptic modification induced by natural spike trains. Nature 416, 433–438.
Froemke, R. C., Merzenich, M. M., and Schreiner, C. E. (2007). A synaptic memory trace for cortical receptive field plasticity. Nature 450, 425–429.
Froemke, R. C., Poo, M. M., and Dan, Y. (2005). Spike-timing-dependent synaptic plasticity depends on dendritic location. Nature 434, 221–225.
Froemke, R. C., Tsay, I. A., Raad, M., Long, J. D., and Dan, Y. (2006). Contribution of individual spikes in burst-induced long-term synaptic modification. J. Neurophysiol. 95, 1620–1629.
Fu, Y. X., Djupsund, K., Gao, H., Hayden, B., Shen, K., and Dan, Y. (2002). Temporal specificity in the cortical plasticity of visual space representation. Science 296, 1999–2003.
Galindo-Leon, E. E., Lin, F. G., and Liu, R. C. (2009). Inhibitory plasticity in a lateral band improves cortical detection of natural vocalizations. Neuron 62, 705–716.
Gerkin, R. C., Lau, P. M., Nauen, D. W., Wang, Y. T., and Bi, G. Q. (2007). Modular competition driven by NMDA receptor subtypes in spike-timing-dependent plasticity. J. Neurophysiol. 97, 2851–2862.
Gerstner, W., Kempter, R., van Hemmen, J. L., and Wagner, H. (1996). A neuronal learning rule for sub-millisecond temporal coding. Nature 383, 76–81.
Glazewski, S., and Fox, K. (1996). Time course of experience-dependent synaptic potentiation and depression in barrel cortex of adolescent rats. J. Neurophysiol. 75, 1714–1729.
Gunaydin, L. A., Yizhar, O., Berndt, A., Sohal, V. S., Deisseroth, K., and Hegemann, P. (2010). Ultrafast optogenetic control. Nat. Neurosci. 13, 387–392.
Haas, J. S., Nowotny, T., and Abarbanel, H. D. (2006). Spike-timing-dependent plasticity of inhibitory synapses in the entorhinal cortex. J. Neurophysiol. 96, 3305–3313.
Hannan, A. J., Blakemore, C., Katsnelson, A., Vitalis, T., Huber, K. M., Bear, M., Roder, J., Kim, D., Shin, H. S., and Kind, P. C. (2001). PLC-beta1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat. Neurosci. 4, 282–288.
Hannan, A. J., Kind, P. C., and Blakemore, C. (1998). Phospholipase C-beta1 expression correlates with neuronal differentiation and synaptic plasticity in rat somatosensory cortex. Neuropharmacology 37, 593–605.
Hardingham, N., Wright, N., Dachtler, J., and Fox, K. (2008). Sensory deprivation unmasks a PKA-dependent synaptic plasticity mechanism that operates in parallel with CaMKII. Neuron 60, 861–874.
Hensch, T. K. (2005). Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 6, 877–888.
Hoffman, D. A., and Johnston, D. (1998). Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J. Neurosci. 18, 3521–3528.
Hoffman, D. A., and Johnston, D. (1999). Neuromodulation of dendritic action potentials. J. Neurophysiol. 81, 408–411.
Hoffman, D. A., Magee, J. C., Colbert, C. M., and Johnston, D. (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387, 869–875.
Holmgren, C. D., and Zilberter, Y. (2001). Coincident spiking activity induces long-term changes in inhibition of neocortical pyramidal cells. J. Neurosci. 21, 8270–8277.
Hubel, D. H., and Wiesel, T. N. (1965). Binocular interaction in striate cortex of kittens reared with artificial squint. J. Neurophysiol. 28, 1041–1059.
Jacob, V., Brasier, D. J., Erchova, I., Feldman, D., and Shulz, D. E. (2007). Spike timing-dependent synaptic depression in the in vivo barrel cortex of the rat. J. Neurosci. 27, 1271–1284.
Kampa, B. M., and Stuart, G. J. (2006). Calcium spikes in basal dendrites of layer 5 pyramidal neurons during action potential bursts. J. Neurosci. 26, 7424–7432.
Kanold, P. O., and Shatz, C. J. (2006). Subplate neurons regulate maturation of cortical inhibition and outcome of ocular dominance plasticity. Neuron 51, 627–638.
Karmarkar, U. R., and Buonomano, D. V. (2002). A model of spike-timing dependent plasticity: one or two coincidence detectors? J. Neurophysiol. 88, 507–513.
Kirkwood, A., Rozas, C., Kirkwood, J., Perez, F., and Bear, M. F. (1999). Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. J. Neurosci. 19, 1599–1609.
Koester, H. J., and Sakmann, B. (1998). Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc. Natl. Acad. Sci. U.S.A. 95, 9596–9601.
Komai, S., Licznerski, P., Cetin, A., Waters, J., Denk, W., Brecht, M., and Osten, P. (2006). Postsynaptic excitability is necessary for strengthening of cortical sensory responses during experience-dependent development. Nat. Neurosci. 9, 1125–1133.
Kudoh, M., and Shibuki, K. (1994). Long-term potentiation in the auditory cortex of adult rats. Neurosci. Lett. 171, 21–23.
Kudoh, M., and Shibuki, K. (1996). Long-term potentiation of supragranular pyramidal outputs in the rat auditory cortex. Exp. Brain Res. 110, 21–27.
Kudoh, M., and Shibuki, K. (1997). Importance of polysynaptic inputs and horizontal connectivity in the generation of tetanus-induced long-term potentiation in the rat auditory cortex. J. Neurosci. 17, 9458–9465.
Laurie, D. J., and Seeburg, P. H. (1994). Ligand affinities at recombinant N-methyl-d-aspartate receptors depend on subunit composition. Eur. J. Pharmacol. 268, 335–345.
Letzkus, J. J., Kampa, B. M., and Stuart, G. J. (2006). Learning rules for spike timing-dependent plasticity depend on dendritic synapse location. J. Neurosci. 26, 10420–10429.
Levy, W. B., and Steward, O. (1983). Temporal contiguity requirements for long-term associative potentiation/depression in the hippocampus. Neuroscience 8, 791–797.
Li, L., Bender, K. J., Drew, P. J., Jadhav, S. P., Sylwestrak, E., and Feldman, D. E. (2009). Endocannabinoid signaling is required for development and critical period plasticity of the whisker map in somatosensory cortex. Neuron 64, 537–549.
Lin, Y. W., Min, M. Y., Chiu, T. H., and Yang, H. W. (2003). Enhancement of associative long-term potentiation by activation of beta-adrenergic receptors at CA1 synapses in rat hippocampal slices. J. Neurosci. 23, 4173–4181.
Liu, X. B., Murray, K. D., and Jones, E. G. (2004). Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J. Neurosci. 24, 8885–8895.
Lu, J. T., Li, C. Y., Zhao, J. P., Poo, M. M., and Zhang, X. H. (2007). Spike-timing-dependent plasticity of neocortical excitatory synapses on inhibitory interneurons depends on target cell type. J. Neurosci. 27, 9711–9720.
Magee, J. C., and Johnston, D. (1997). A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275, 209–213.
Markram, H., Lubke, J., Frotscher, M., and Sakmann, B. (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215.
Meliza, C. D., and Dan, Y. (2006). Receptive-field modification in rat visual cortex induced by paired visual stimulation and single-cell spiking. Neuron 49, 183–189.
Meredith, R. M., Floyer-Lea, A. M., and Paulsen, O. (2003). Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J. Neurosci. 23, 11142–11146.
Nelson, S. B., Sjostrom, P. J., and Turrigiano, G. G. (2002). Rate and timing in cortical synaptic plasticity. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 357, 1851–1857.
Nevian, T., and Sakmann, B. (2006). Spine Ca2+ signaling in spike-timing-dependent plasticity. J. Neurosci. 26, 11001–11013.
Nishiyama, M., Hong, K., Mikoshiba, K., Poo, M. M., and Kato, K. (2000). Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408, 584–588.
Pellicciari, M. C., Miniussi, C., Rossini, P. M., and De Gennaro, L. (2009). Increased cortical plasticity in the elderly: changes in the somatosensory cortex after paired associative stimulation. Neuroscience 163, 266–276.
Recanzone, G. H., Schreiner, C. E., and Merzenich, M. M. (1993). Plasticity in the frequency representation of primary auditory cortex following discrimination training in adult owl monkeys. J. Neurosci. 13, 87–103.
Rodriguez-Moreno, A., and Paulsen, O. (2008). Spike timing-dependent long-term depression requires presynaptic NMDA receptors. Nat. Neurosci. 11, 744–745.
Schnupp, J. W., Hall, T. M., Kokelaar, R. F., and Ahmed, B. (2006). Plasticity of temporal pattern codes for vocalization stimuli in primary auditory cortex. J. Neurosci. 26, 4785–4795.
Schuett, S., Bonhoeffer, T., and Hubener, M. (2001). Pairing-induced changes of orientation maps in cat visual cortex. Neuron 32, 325–337.
Seol, G. H., Ziburkus, J., Huang, S., Song, L., Kim, I. T., Takamiya, K., Huganir, R. L., Lee, H. K., and Kirkwood, A. (2007). Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron 55, 919–929.
Shouval, H. Z., Bear, M. F., and Cooper, L. N. (2002). A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 99, 10831–10836.
Siucinska, E., and Kossut, M. (2004). Experience-dependent changes in cortical whisker representation in the adult mouse: a 2-deoxyglucose study. Neuroscience 127, 961–971.
Sjostrom, P. J., and Hausser, M. (2006). A cooperative switch determines the sign of synaptic plasticity in distal dendrites of neocortical pyramidal neurons. Neuron 51, 227–238.
Sjostrom, P. J., Turrigiano, G. G., and Nelson, S. B. (2001). Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron 32, 1149–1164.
Sjostrom, P. J., Turrigiano, G. G., and Nelson, S. B. (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39, 641–654.
Smith, S. L., and Trachtenberg, J. T. (2007). Experience-dependent binocular competition in the visual cortex begins at eye opening. Nat. Neurosci. 10, 370–375.
Song, S., and Abbott, L. F. (2001). Cortical development and remapping through spike timing-dependent plasticity. Neuron 32, 339–350.
Speechley, W. J., Hogsden, J. L., and Dringenberg, H. C. (2007). Continuous white noise exposure during and after auditory critical period differentially alters bidirectional thalamocortical plasticity in rat auditory cortex in vivo. Eur. J. Neurosci. 26, 2576–2584.
Spehlmann, R. (1971). Acetylcholine and the synaptic transmission of non-specific impulses to the visual cortex. Brain 94, 139–150.
Tsubokawa, H., and Ross, W. N. (1997). Muscarinic modulation of spike backpropagation in the apical dendrites of hippocampal CA1 pyramidal neurons. J. Neurosci. 17, 5782–5791.
Tzounopoulos, T., Kim, Y., Oertel, D., and Trussell, L. O. (2004). Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat. Neurosci. 7, 719–725.
Urakubo, H., Honda, M., Froemke, R. C., and Kuroda, S. (2008). Requirement of an allosteric kinetics of NMDA receptors for spike timing-dependent plasticity. J. Neurosci. 28, 3310–3323.
Watanabe, S., Hoffman, D. A., Migliore, M., and Johnston, D. (2002). Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 99, 8366–8371.
Weinberger, N. M. (2003). The nucleus basalis and memory codes: auditory cortical plasticity and the induction of specific, associative behavioral memory. Neurobiol. Learn. Mem. 80, 268–284.
Weinberger, N. M., Miasnikov, A. A., and Chen, J. C. (2006). The level of cholinergic nucleus basalis activation controls the specificity of auditory associative memory. Neurobiol. Learn. Mem. 86, 270–285.
Wespatat, V., Tennigkeit, F., and Singer, W. (2004). Phase sensitivity of synaptic modifications in oscillating cells of rat visual cortex. J. Neurosci. 24, 9067–9075.
Whitfield, I. C. (1980). Auditory cortex and the pitch of complex tones. J. Acoust. Soc. Am. 67, 644–647.
Yang, Y., DeWeese, M. R., Otazu, G. H., and Zador, A. M. (2008). Millisecond-scale differences in neural activity in auditory cortex can drive decisions. Nat. Neurosci. 11, 1262–1263.
Yao, H., and Dan, Y. (2001). Stimulus timing-dependent plasticity in cortical processing of orientation. Neuron 32, 315–323.
Yazaki-Sugiyama, Y., Kang, S., Cateau, H., Fukai, T., and Hensch, T. K. (2009). Bidirectional plasticity in fast-spiking GABA circuits by visual experience. Nature 462, 218–221.
Zhang, J. C., Lau, P. M., and Bi, G. Q. (2009). Gain in sensitivity and loss in temporal contrast of STDP by dopaminergic modulation at hippocampal synapses. Proc. Natl. Acad. Sci. U.S.A. 106, 13028–13033.
Zhou, Q., Tao, H. W., and Poo, M. M. (2003). Reversal and stabilization of synaptic modifications in a developing visual system. Science 300, 1953–1957.
Zilberter, M., Holmgren, C., Shemer, I., Silberberg, G., Grillner, S., Harkany, T., and Zilberter, Y. (2009). Input specificity and dependence of spike timing-dependent plasticity on preceding postsynaptic activity at unitary connections between neocortical layer 2/3 pyramidal cells. Cereb. Cortex 19, 2308–2320.
Keywords: spike timing-dependent plasticity, presynaptic NMDA receptor, endocannabinoid, visual cortex, auditory cortex, somatosensory cortex, neuromodulation
Citation: Larsen RS, Rao D, Manis PB and Philpot BD (2010) STDP in the developing sensory neocortex. Front. Syn. Neurosci. 2:9. doi: 10.3389/fnsyn.2010.00009
Received: 12 February 2010;
Paper pending published: 01 March 2010;
Accepted: 17 May 2010;
Published online: 09 June 2010
Edited by:
Per Jesper Sjöström, University College London, UKReviewed by:
Ole Paulsen, University of Oxford, UKVincent Jacob, Cardiff University, UK
Copyright: © 2010 Larsen, Rao, Manis and Philpot. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence:
Benjamin D. Philpot, University of North Carolina, Campus Box 7545, 115 Mason Farm Road, Chapel Hill, NC 27599-7545, USA. e-mail:YnBoaWxwb3RAbWVkLnVuYy5lZHU=;
Paul B. Manis, University of North Carolina, G127 Physician’s Office Building, Campus Box 7070, 170 Manning Drive, Chapel Hill, NC 27599-7070, USA. e-mail:cG1hbmlzQG1lZC51bmMuZWR1