


- 1 Department of Pharmacology, Oxford University, Oxford, UK
- 2 Department of Clinical and Experimental Epilepsy, Institute of Neurology, University College London, London, UK
- 3 Department of Cell and Systems Biology, University of Toronto, Canada
Inhibitory circuits in the brain rely on GABA-releasing interneurons. For long, inhibitory circuits were considered weakly plastic in the face of patterns of neuronal activity that trigger long-term changes in the synapses between excitatory principal cells. Recent studies however have shown that GABAergic circuits undergo various forms of long-term plasticity. For the purpose of this review, we identify three major long-term plasticity expression sites. The first locus is the glutamatergic synapses that excite GABAergic inhibitory cells and drive their activity. Such synapses, on many but not all inhibitory interneurons, exhibit long-term potentiation (LTP) and depression (LTD). Second, GABAergic synapses themselves can undergo changes in GABA release probability or postsynaptic GABA receptors. The third site of plasticity is in the postsynaptic anion gradient of GABAergic synapses; coincident firing of GABAergic axons and postsynaptic neurons can cause a long-lasting change in the reversal potential of GABAA receptors mediating fast inhibitory postsynaptic potentials. We review the recent literature on these forms of plasticity by asking how they may be triggered by specific patterns of pre- and postsynaptic action potentials, although very few studies have directly examined spike-timing dependent plasticity (STDP) protocols in inhibitory circuits. Plasticity of interneuron recruitment and of GABAergic signaling provides for a rich flexibility in inhibition that may be central to many aspects of brain function. We do not consider plasticity at glutamatergic synapses on Purkinje cells and other GABAergic principal cells.
Plasticity in Excitatory Afferents of Inhibitory Circuits
Long-term potentiation (LTP) and depression (LTD) of glutamatergic synapses onto GABAergic interneurons has been discovered in several areas of the brain suggesting that plasticity in this locus is common in the CNS. Although these studies have concentrated heavily on the circuits of the hippocampal formation, there is evidence that similar plasticity takes place in the neocortex (Sarihi et al., 2008 ; Chen et al., 2009 ), as well as in striatum (Fino et al., 2008 , 2009 ) and amygdala (Mahanty and Sah, 1998 ). In addition there are reports on analogous long-term plasticity in the spinal cord (Santos et al., 2009 ) and in sensory pathways (Tzounopoulos et al., 2004 ).
Plasticity is Specific to GABAergic Interneuron Type
One of the striking features of plasticity in this locus is that it can be highly specific to an interneuron type (Kullmann and Lamsa, 2007 ; Sarihi et al., 2008 ; Oren et al., 2009 ; Nissen et al., 2010 ). Subpopulations of unidentified interneurons may show LTP, LTD or no plasticity at all, for stimulation of the same afferent glutamatergic pathway (Buzsaki and Eidelberg, 1982 ; Lei and McBain, 2004 ; Lamsa et al., 2007a ). Recent studies on identified interneurons in the hippocampus have revealed that these subpopulations are composed of anatomically distinct interneuron types. Plasticity has been shown to be consistent in individual anatomically identified GABAergic cell types (Alle et al., 2001 ; Oren et al., 2009 ; Nissen et al., 2010 ). More importantly, plastic properties can vary remarkably between two interneurons located in close proximity in the same hippocampal area, but showing different neurochemical marker expression or different axon distribution (Lamsa et al., 2007b ; Nissen et al., 2010 ) (Figure 1 ). A specific pattern of pre- and postsynaptic activity that induces plasticity in one interneuron type can fail or elicit a different plastic change in another interneuron type located in the same layer (Lamsa et al., 2005 , 2007b ; Nissen et al., 2010 ).
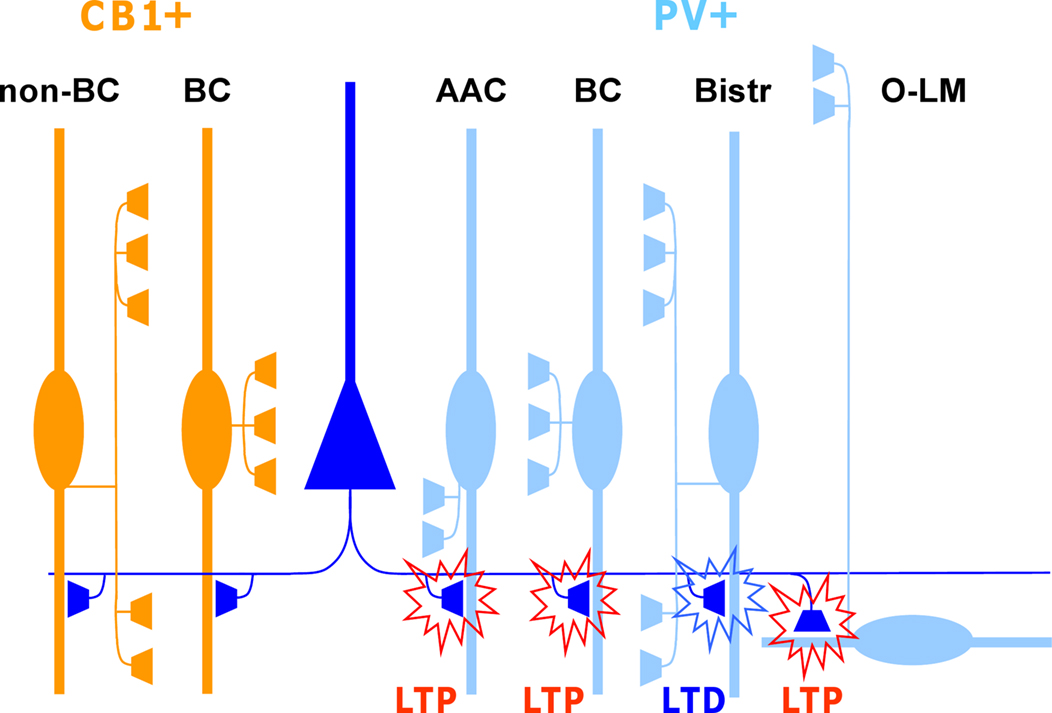
Figure 1. Glutamatergic fiber plasticity on hippocampal CA1 interneurons is specific to a GABAergic cell type. Schematic illustrates cell type-specific plasticity in six anatomically identified interneuron types. Repetitive high-frequency (100 Hz tetanic or theta burst) stimulation of pyramidal cell axons induces target cell-specific plasticity in parvalbumin-expressing (PV+) interneuron types, whereas interneurons expressing cannabinoid receptor type 1 (CB1R+) but not PV, show no plasticity with these protocols. PV and CB1R are mutually exclusive neurochemical markers and cell types belonging to these two groups form very similar inhibitory circuits in the CA1. Detailed anatomical analyses reveal that glutamatergic synapses onto PV+ axo-axonic (AAC), basket cells (BC) and oriens-lacunosum moleculare (O-LM) interneurons show consistently LTP, whereas bistratified cells (Bistr) show predominantly LTD. In contrast, synapses onto BCs expressing CB1R or dendrite-targeting CB1R+ non-basket cells (non-BC) show no long-term plasticity at all (Lamsa et al., 2007b ; Oren et al., 2009 ; Nissen et al., 2010 ).
These findings, that the interneuron plasticity is diverse and cell type-specific, have important impacts on the dynamics of the cortical networks. First, specific patterns of pre- and postsynaptic action potentials are capable of triggering plasticity in only a subpopulation of inhibitory cells (Lamsa et al., 2005 , 2007b ). Second, plasticity is likely to be induced in different brain states in distinct interneuron types. It has been shown that the activity of interneurons and their glutamatergic afferents is highly specific to a cell type during neuronal oscillations related to different behavioral states (Klausberger and Somogyi, 2008 ). It is therefore tempting to speculate that the distinct forms of LTP and LTD discovered in interneurons in vitro might underlie the use-dependent dynamics reported in selective parts of the inhibitory networks in vivo (Buzsaki and Eidelberg, 1982 ; Csicsvari et al., 1998 ; Yazaki-Sugiyama et al., 2009 ). Several important questions remain to be answered on plasticity in interneurons and one of the most intriguing is related to the physiological activity patterns that might generate cell type-specific plasticities in vivo. To answer this question it will be important to reveal spike-timing dependent plasticity (STDP) properties of common cortical interneuron types and their identified glutamatergic afferents (Klausberger and Somogyi, 2008 ).
Given that GABAergic interneuron diversity is rich in many areas of the brain and full identification of cells is often challenging, it is understandable that STDP has not yet been extensively studied among GABAergic interneuron types. Instead, in most of these studies relatively robust extracellular stimulation protocols have been used. However, the major purpose of these pioneering studies has been to investigate a capacity for synaptic plasticity in inhibitory interneurons in distinct areas (Buzsaki and Eidelberg, 1982 ; Perez et al., 2001 ; Lei and McBain, 2004 ; Lamsa et al., 2005 , 2007b ; Galvan et al., 2008 ). Unfortunately, interneurons in many of these studies have been pooled together on the basis of their electrophysiological properties. However, these parameters correlate quite weakly with anatomical, neurochemical and gene expression patterns that are commonly used to identify the hippocampal and neocortical interneuron types (Ascoli et al., 2008 ). Yet, some STDP studies have been made in GABAergic interneurons and these results also suggest that ‘rules’ underlying long-term plasticity may differ between distinct interneuron types. Significant differences in STDP were found between fast- and regularly-spiking interneuron populations in the neocortex (Lu et al., 2007 ). However, as mentioned above firing patterns do not reveal inhibitory circuit wiring patterns or molecular profiles of the cells, and therefore cell type-specific STDP properties still remain to be elucidated in the hippocampus and neocortex. Noteworthy, interneuron subpopulation-specific plasticity has also been demonstrated in striatal GABAergic interneurons. Glutamatergic afferents from somatosensory cortex to striatal nitric oxide-synthase (NOS)-expressing GABAergic interneurons show relatively consistent STDP properties (Fino et al., 2008 , 2009 ). However, in many subcortical areas such as striatum the interneuron diversity is smaller than in the cortex (Freund and Buzsaki, 1996 ), which might explain homogeneity of striatal NOS-expressing GABAergic cells (Klausberger and Somogyi, 2008 ).
Plasticity Mechanisms
The induction and expression mechanisms of LTP and LTD of glutamatergic excitation of interneurons have attracted considerable attention. The emerging evidence points to substantial heterogeneity, most likely reflecting the diversity of interneuron types mentioned above. LTP at glutamatergic synapses on many interneurons in the hippocampal formation and neocortex exhibits a ‘Hebbian’ induction rule; that is, it can be evoked by the conjunction of presynaptic action potentials and postsynaptic depolarization (Alle et al., 2001 ; Perez et al., 2001 ; Lamsa et al., 2005 ; Galvan et al., 2008 ; Sarihi et al., 2008 ). Hebbian plasticity may even occur in the human motor cortex (Russmann et al., 2009 ). However, different studies have applied distinct patterns of presynaptic stimulation (high-frequency tetanization, ‘theta-burst’ or low-frequency stimulation), and have either allowed the postsynaptic neurons to be depolarized by the activated synapses, or have imposed action potentials via the recording pipette. It is therefore difficult to compare among studies. Nevertheless, some striking differences are emerging, both between LTP in interneurons and in principal cells, and among different interneurons.
NMDA receptor-dependent LTP
‘Hebbian’ LTP in pyramidal neurons is generally explained by the involvement of NMDA receptors, which require both presynaptic glutamate release and postsynaptic depolarization for their activation, and which trigger postsynaptic Ca2+ influx and down-stream activation of calcium/calmodulin-dependent kinase IIα (CaMKIIα). NMDA receptor-dependent LTP with very similar properties can be elicited at synapses made by Schaffer collaterals on a subset of interneurons in stratum radiatum of the rodent hippocampus (Lamsa et al., 2005 ). This phenomenon can be elicited by delivering postsynaptic depolarizing steps synchronously with low-frequency presynaptic stimulation, or by delivering a continuous postsynaptic depolarization to allow the presynaptic stimuli to evoke postsynaptic spiking. However, a pre- before post-protocol has not been explicitly tested. Although this shares many features with LTP in pyramidal neurons, including sensitivity to pharmacological blockers of CaMKIIα (Lamsa et al., 2007a ), it exhibits one striking difference: LTP is intact in mice harboring a mutation that prevents autophosphorylation of CaMKIIα, which has a profound deficit in NMDA receptor-dependent LTP in pyramidal neurons (Giese et al., 1998 ). This is perhaps unsurprising, because CaMKIIα has not been detected in interneurons, so the result of the pharmacological intervention suggests the involvement of another member of the calcium/calmodulin-dependent kinase family.
Non-NMDA receptor-dependent LTP
At many other synapses on interneurons, pharmacological blockade of NMDA receptors fails to prevent LTP induction. Postsynaptic chelation of Ca2+ is however effective (Alle et al., 2001 ; Perez et al., 2001 ), implying a critical role for Ca2+, which must enter the neuron from another source or be released from intracellular stores. Several studies have demonstrated an essential role for Ca2+-permeable AMPA receptors (Mahanty and Sah, 1998 ; Lamsa et al., 2007b ; Oren et al., 2009 ). A striking feature of such receptors, which are devoid of edited GluA2 subunits, is that they exhibit inward rectification, preferentially allowing Ca2+ influx at relatively negative potentials. Remarkably, LTP at synapses exhibiting strong inward rectification can be induced by pairing presynaptic action potentials with postsynaptic hyperpolarization. This phenomenon has been termed ‘anti-Hebbian LTP’ (Lamsa et al., 2007b ; Oren et al., 2009 ; Nissen et al., 2010 ). It is most readily observed at synapses formed by axon collaterals of pyramidal neurons on interneurons in the feedback circuit in stratum oriens of the hippocampus, in particular (although not exclusively) in oriens lacunosum-moleculare (O-LM) cells, which have dendrites extending parallel to stratum pyramidale and an axon extending to stratum lacunosum-moleculare (McBain et al., 1994 ). Although STDP protocols have not been explored in detail, this form of LTP can be elicited by delivering presynaptic stimuli at the trough, but not at the peak, of a 5-Hz sinusoidal membrane potential oscillation delivered via the postsynaptic pipette.
An essential role for group I metabotropic glutamate receptors (mGluRs) has also been demonstrated in LTP (and LTD) induction in interneurons (Perez et al., 2001 ; Lapointe et al., 2004 ; Galvan et al., 2008 ; Gibson et al., 2008 ). Although many studies are difficult to compare because they have examined different brain regions, group I mGluR-dependent LTP has also been most extensively examined in O-LM cells. Indeed, these interneurons have abundant mGLuR1α, but also exhibit marked rectification of their synaptic AMPA receptors (Ferraguti et al., 2004 ; Oren et al., 2009 ). LTP at synapses made on O-LM cells by axon collaterals of local pyramidal neurons can be induced either with Hebbian or with anti-Hebbian protocols, and is sensitive to pharmacological blockade of either Ca2+-permeable AMPA receptors or group I mGluRs (Perez et al., 2001 ; Oren et al., 2009 ). Although both group I mGluR subtypes (mGluR1 and mGluR5) are linked to Ca2+, albeit via different cascades (Topolnik et al., 2005 ), how they interact with Ca2+ signaling triggered by AMPA receptor activation remains to be determined. LTP in stratum oriens interneurons can, furthermore, be facilitated by co-activation of nicotinic acetylcholine receptors (Jia et al., 2010 ).
An additional role for L-type Ca2+ channels in induction of NMDA receptor-independent LTP has been reported in interneurons in stratum lacunosum-moleculare, at synapses made by mossy fibers (Galvan et al., 2008 ). This phenomenon was elicited by high-frequency stimulation of presynaptic axons while allowing the postsynaptic neurons to fire. Here too, pharmacological dissection has revealed an essential role for mGluR1, blockade of which converted LTP into LTD. LTP however occurred at synapses with Ca2+-impermeable AMPA receptors, and was blocked by postsynaptic Ca2+ chelation or interference with intracellular Ca2+ stores.
In subtle contrast to LTP at mossy fiber synapses on interneurons in stratum lacunosum-moleculare of the hippocampus, Sarihi et al. (2008) found that LTP in fast-spiking in layer II/III interneurons of the visual cortex, elicited by theta-burst stimulation, depended on mGluR5, but not mGluR1, and that L-type Ca2+ channels were not involved. Nevertheless, this form of LTP was again independent of NMDA receptors, but was prevented by chelating postsynaptic Ca2+.
Unraveling how the different glutamate receptors and other sources of Ca2+ interact in LTP induction will require further work. Among possible factors influencing the outcome of different induction protocols is the method used to record from neurons: some studies resorted to perforated patch recordings to minimize disruption of the cytoplasm, but this approach renders voltage-clamping difficult, and prevents the routine introduction of markers for histological characterization (Kullmann and Lamsa, 2007 ). Furthermore, grouping together interneurons on the basis of their location or firing characteristics probably hides considerable diversity of subtypes (Klausberger and Somogyi, 2008 ).
Relatively little attention has thus far been given to the LTP expression mechanisms that maintain potentiation of synaptic transmission. NMDA receptor-dependent LTP in some interneurons appears not to be accompanied by changes in short-term plasticity (paired-pulse ratio), providing no evidence for an increase in presynaptic glutamate release probability (Lamsa et al., 2005 ). This phenomenon is therefore most simply explained by a postsynaptic insertion of AMPA receptors, much as has been reported for NMDA receptor-dependent LTP in pyramidal neurons. LTP dependent on group I mGluRs and Ca2+-permeable AMPA receptors, on the other hand, has been shown at several synapses to be accompanied by changes in paired-pulse ratio, failure rates, trial-to-trial fluctuations, or sensitivity to use-dependent blockers of Ca2+-permeable receptors (Perez et al., 2001 ; Oren et al., 2009 ). All of these observations point instead to an increase in presynaptic glutamate release (see also Alle et al., 2001 ; Galvan et al., 2008 ), implying the existence of a retrograde factor, the identity of which remains to be determined.
Another form of tetanic LTP at synapses on somatostatin-positive interneurons in the neocortex appears to be exclusively presynaptic, in that it does not depend on postsynaptic Ca2+ signaling (Chen et al., 2009 ). Instead, it depends on protein kinase A and therefore shares mechanisms in common with tetanus-induced mossy fiber LTP.
LTD
At several other synapses, LTD has been reported more robustly than LTP (Maccaferri et al., 1998 ; Laezza et al., 1999 ; Laezza and Dingledine, 2004 ). However, at least two forms of LTD have emerged. At synapses made by hippocampal mossy fibers equipped with calcium-impermeable AMPARs and NMDA receptors, high-frequency presynaptic stimulation leads to depression, which appears to share mechanisms with NMDA receptor-dependent LTD at glutamatergic synapses on pyramidal neurons (Lei and McBain, 2004 ). At other synapses equipped with rectifying AMPA receptors, similar stimuli trigger a form of LTD that is sensitive to postsynaptic Ca2+ chelation or blockade of group III mGluRs, and which appears to be expressed presynaptically. Synapses made by hippocampal mossy fibers can express both forms of plasticity although it remains to be determined to what extent the identity of the postsynaptic neurons determine the outcome of the pairing protocol. Importantly, the somata of the mossy fiber-LTD expressing interneurons are located in a different layer (mainly strata lucidum and radiatum) than the mossy fiber-LTP interneurons reported by Galvan et al. (2008) (stratum lacunosum-moleculare) and described earlier in this review. Interestingly, synapses where mGluR-dependent LTD has been elicited can be rendered capable of subsequent potentiation with repeated rounds of high-frequency stimulation (Pelkey et al., 2005 ). This phenomenon has been related to internalization of mGluR7, which plays a key role in triggering the initial depression.
High-frequency stimulation of Schaffer collaterals has also been shown to induce a form of NMDA receptor-independent LTD at interneurons in stratum radiatum (McMahon and Kauer, 1997 ). This too appears to be expressed presynaptically, and depends on mGluR1 receptors (Gibson et al., 2008 ). Interestingly, the phenomenon is also sensitive to manipulation of TRPV1 receptors, suggesting that these channels may act as presynaptic receptors for a retrograde messenger.
STDP at Excitatory Synapses on Interneurons
STDP protocols have not been tested systematically at all the synapses where LTP or LTD can be elicited. However, the patterns that are beginning to emerge from published studies of STDP suggest that multiple mechanisms interact to determine the outcome of the pairing, and that the relative importance of these mechanisms differs extensively among synapses. In a study of layer 2/3 interneurons of the somatosensory cortex, low-frequency pairing delivered with various pre-post intervals exclusively yielded LTD in fast-spiking cells, which could be prevented by postsynaptic Ca2+ chelation or broad-spectrum blockade of mGluRs (Lu et al., 2007 ). However, in the dorsal cochlear nucleus, presynaptic spikes delivered 8 ms before postsynaptic depolarization led to NMDA receptor-dependent LTP in low-threshold-spiking interneurons (Tzounopoulos et al., 2004 ). This work has shown that the outcome of pairing presynaptic action potentials with postsynaptic depolarization depends both on the repetition rate and on the spike-EPSP interval. A protocol that generally yields LTP in principal cells (pre before post) appears to engage both presynaptic cannabinoid-dependent LTD and postsynaptic CaMKII-dependent LTP in ‘cartwheel’ interneurons. STDP in striatal NOS- positive interneurons is equally puzzling, with pairing leading to LTD for most intervals between the presynaptic spike and the postsynaptic depolarization, and LTP only elicited when the postsynaptic depolarization is delivered approximately 40 ms after the spike (Fino et al., 2009 ).
Glutamatergic Afferent Plasticity in Network Function
A key feature of the plasticity in this locus is that it allows afferent-specific modulation of the inhibitory circuit. This means that a potentiated excitatory pathway has an increased contribution to firing of the inhibitory cell and consequently to the disynaptic inhibition of the GABAergic neurons’s target cells (Nissen et al., 2010 ). This is important because during cortical network oscillations, which provide spatiotemporal framework for information processing, synchronized cell assembly formation must be dynamic. Accordingly, the synaptic strengths between pyramidal cells and inhibitory interneurons must change in behaviorally relevant time scales (Csicsvari et al., 1998 ). Plasticity at glutamatergic afferents onto interneurons is a good candidate mechanism for maintaining the necessary flexibility in these connections during the formation or dissolution of cell assemblies.
In a recent study Yazaki-Sugiyame et al. (2009) demonstrated that inhibitory circuits of fast-spiking interneurons express robust use-dependent plasticity in the visual cortex by visual experience. By modeling a cortical circuit the authors conclude that the plasticity either locates in excitatory afferents onto PV+ inhibitory cells or in the GABAergic synapses made by these cells onto pyramidal cells. Interestingly, PV+ fast-spiking interneurons have recently been demonstrated to express robust LTP in the visual cortex and in the hippocampus (Lamsa et al., 2007b ; Sarihi et al., 2008 ; Nissen et al., 2010 ).
Lasting Plasticity of GABAergic Synapses is Robust at Early Developmental Stages
Long-lasting potentiation or depression of inhibitory synapses has also been reported in many areas of the brain. However, unlike plasticity at glutamatergic synapses reviewed above, LTP in hippocampal GABAergic synapses is almost exclusively restricted to early developmental stages (Gaiarsa et al., 2002 ). Yet, GABAergic LTP can be induced also in mature hippocampal circuits by certain extracellular stimulation patterns (such as theta-burst high-frequency stimulation) but even in these cases LTP is smaller than in neonatal synapses (Patenaude et al., 2003 , 2005 ). In neonatal hippocampus and in some areas of juvenile neocortex both LTP and LTD of GABAergic synapses are triggered by processes that depend on postsynaptic Ca2+ and involve either activation of NMDARs or voltage-gated calcium channels (VGCCs) (Holmgren and Zilberter, 2001 ; Haas et al., 2006 ). Interestingly, GABAergic LTP and LTD in neonatal hippocampus are expressed presynaptically (Caillard et al., 1999a ,b ; Gubellini et al., 2005 ) whereas LTD (and LTP) in mature hippocampus is postsynaptic (Stelzer et al., 1994 ; Lu et al., 2000 ; Patenaude et al., 2003 ; Maffei et al., 2006 ). In the visual cortex expression of LTP and LTD is also developmentally regulated (Maffei et al., 2006 ) and similar to hippocampus GABAergic LTP is less likely in mature animals (Komatsu, 1994 ). However, the expression site for the plasticity in the visual cortex is always postsynaptic, which suggests that the LTP is probably different from that described in the neonatal hippocampus. Induction of plasticity in GABAergic synapses with STDP protocols has been tested in a small number of studies only. In GABAergic synapses of juvenile rat entorhinal cortex, a pre-post-spiking sequence triggers LTP and a post-pre sequence elicits LTD. STDP plasticity of GABAergic synapses has also been demonstrated in GABAergic synapses in the Xenopus retino-tectal system during early development, where coincident pre- and post-activity leads to LTD (Lien et al., 2006 ). Interestingly in mature hippocampus STDP protocol induces mainly ionic shift plasticity, and changes in GABA release probability or postsynaptic receptor activity are negligible (Woodin et al., 2003 ).
In some GABAergic synapses a short or long-term suppression of GABA release can be triggered by postsynaptic activity without a specific requirement for presynaptic firing. Because many of these plasticity forms such as cannabinoid receptor-mediated LTP characteristically affect transmission in a large number of local GABAergic synapses without recognizing presynaptic terminal activity history, they are not reviewed any further here. This type of GABAergic synapse modulation has been reviewed recently elsewhere (Nugent and Kauer, 2008 ; Heifets and Castillo, 2009 ; McBain and Kauer, 2009 ).
Altogether, plasticity at the site of GABAergic synapses provides a powerful modulation mechanism of neuronal networks in particular during the development of cortical circuits. However, more knowledge on the plasticity in this locus especially concerning STDP and GABAergic cell type-specificity would be needed to understand its role widely in the hippocampal and neocortical networks.
‘Ionic Shift Plasticity’ Provides Modulation of GABAergic Inhibition
GABAergic synapses are susceptible to a form of plasticity dependent upon alterations in ion gradients (Fiumelli and Woodin, 2007 ). Low-frequency STDP induction protocols cause long-term changes in the postsynaptic chloride (Cl−) gradient. The Cl− gradient predominantly determines the reversal potential for GABAA receptor-mediated transmission (EGABA-A), which in turn is largely responsible for determining the strength of inhibition. At early developmental stages STDP induction at GABAergic synapses hyperpolarizes the reversal potential, which effectively strengthens inhibition (Balena and Woodin, 2008 ; Xu et al., 2008 ). In contrast, the same STDP induction protocol weakens inhibition in the mature CNS by depolarizing the reversal potential (Woodin et al., 2003 ; Fiumelli and Woodin, 2007 ; Ormond and Woodin, 2009 ). These bi-directional forms of GABAergic STDP are often referred to as ‘ionic shift plasticity’ (Figure 2 ).
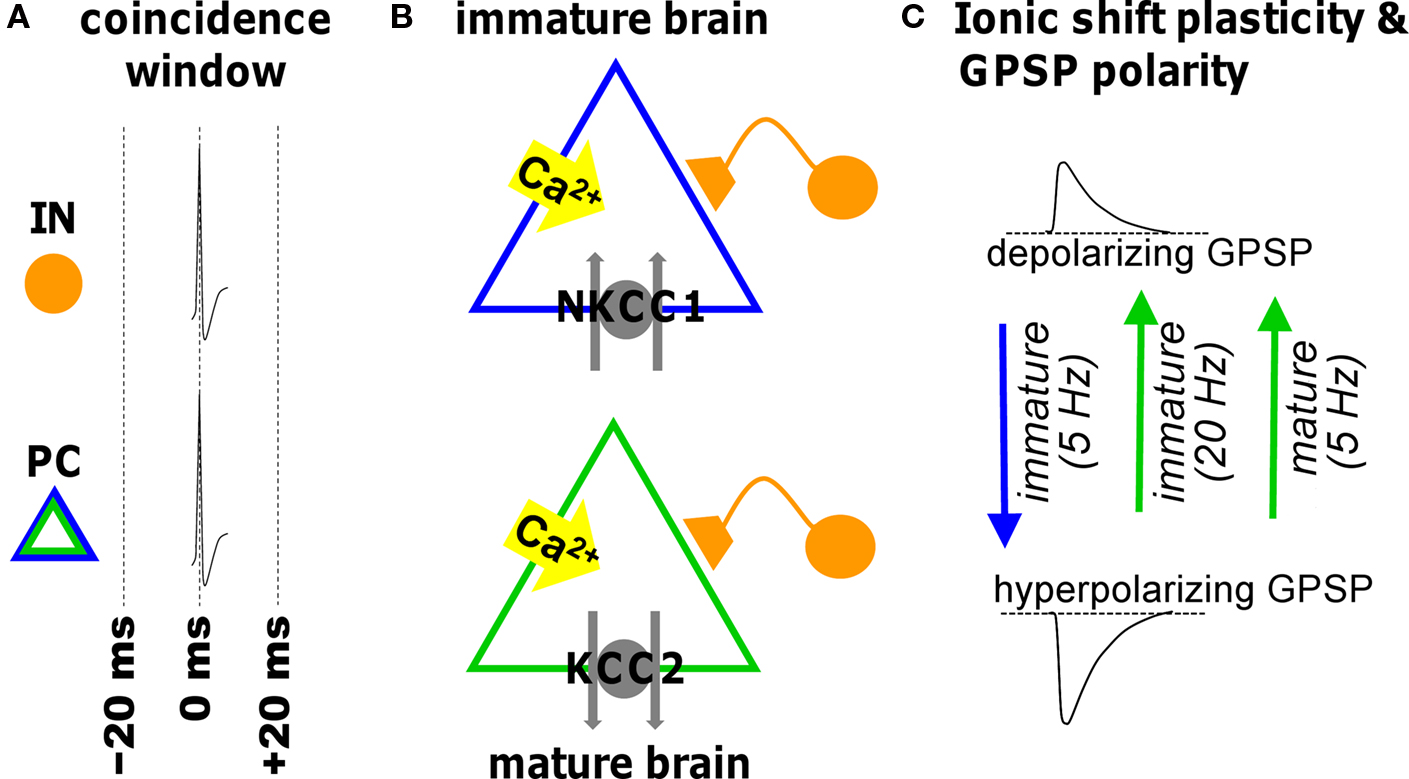
Figure 2. Illustration of ionic shift plasticity in synaptically connected interneuron (IN) -principal cell (PC) pair in immature and mature brain. (A) Repetitive concurrent firing of the two cells inside the coincidence window activates a cascade that shifts the reversal potential of GABAergic PSPs (GPSP). (B) Spiking-associated calcium influx regulates cation-chloride cotransporter function altering neuronal Cl− transport. (C) The consequent shift in transmembrane chloride gradient shifts the reversal potential in either the positive or negative direction (indicated by arrows) depending on the developmental stage and the frequency. This can convert a depolarizing GPSP to a hyperpolarizing GPSP (and vice versa).
Mechanisms Regulating the GABAA Reversal Potential
Fast inhibitory GABAergic transmission in the mature CNS is largely mediated by GABAARs. These are ionotropic receptors permeable to Cl− and HCO3−, but due to differences in the permeabilities and reversal potentials of these two ions it is Cl− that plays the most important role in determining the strength of GABAergic transmission under normal physiological conditions (Farrant and Kaila, 2007 ). The regulation of this gradient depends on the balance and function of the prominent neuronal Cl− transporters: KCC2 and NKCC1. They are both members of the cation-chloride cotransporter gene family SLC12a1-9 (Payne et al., 2003 ; Mercado et al., 2004 ; Gamba, 2005 ). They are also both secondary active transporters that depend on ionic gradients established by the primary active transporter Na+-K+-ATPase; KCC2 derives energy from the K+ gradient to transport Cl− out of the cell, while NKCC1 derives energy for the Na+ gradient to transport Cl− inward.
During embryonic development and early postnatal life, NKCC1 is the dominantly expressed Cl−-cotransporter (Xu et al., 1994 ; Plotkin et al., 1997 ; Russell, 2000 ; Dzhala et al., 2005 ), and as a result neuronal Cl− is high and GABAergic transmission causes postsynaptic depolarization that is sometimes excitatory (although EGABA-A may also be affected by recording conditions, see Rheims et al., 2009 ). However during early postnatal development there is a significant increase in KCC2 expression that results in a hyperpolarizing shift in EGABA-A below the resting membrane potential that produces synaptic inhibition (Rivera et al., 1999 ; Blaesse et al., 2009 ). This dynamic regulation of the sign of GABAergic transmission (excitatory vs. inhibitory) is also observed during neuronal injury, epilepsy, and neuropathic pain (van den Pol et al., 1996 ; Cohen et al., 2002 ; Rivera et al., 2002 ; Coull et al., 2005 ; Blaesse et al., 2009 ). In these pathophysiological cases KCC2 expression dramatically decreases, rending GABAergic transmission excitatory – in what has been proposed to be a recapitulation of developmental programs (Payne et al., 2003 ; Blaesse et al., 2009 ).
Developmentally Regulated Ionic Shift Plasticity
It is not just developmental and pathophysiological events that alter the electrochemical gradient for Cl−. Physiologically normal patterns of neuronal activity such as those used during the induction of STDP can also shift the Cl− gradient, resulting in changes in the strength of GABAergic transmission. Coincident pre- and postsynaptic activity within ±20 ms (at 5 Hz for 30 s) depolarizes EGABA-A (with no corresponding change in conductance) (Woodin et al., 2003 ). Beyond ±50 ms GABAergic inhibition is weakened by a decrease in conductance (but no change in EGABA-A). The result is a symmetrical spike timing window which leads to two initial observations: (1) GABAergic synapses are also sensitive to spike timing; and (2) the shape of the spike timing window differs from the asymmetrical window for glutamatergic synapses. This last point also implies that the pre-post order is not essential during GABAergic STDP induction, as it is during glutamatergic STDP. Rather during GABAergic STDP the essential requirement is correlated timing.
During the induction of GABAergic STDP postsynaptic Ca2+ influx through VGCCs leads to a decrease in the function of KCC2 (Woodin et al., 2003 ). This results in less outward Cl− transport and thus a depolarization of EGABA-A that weakens inhibition. This plasticity occurs in cultured hippocampal neurons (Woodin et al., 2003 ), and hippocampal slices prepared from juveniles and mature rats (Woodin et al., 2003 ; Ormond and Woodin, 2009 ). The central requirement for the induction of ionic shift plasticity appears to be an appropriate level of Ca2+ influx, and not the Ca2+ source. In hippocampal cultures and slices from juveniles, Ca2+ influx via VGCCs is sufficient (Woodin et al., 2003 ). However in slices from adults, Ca2+ influx through both VGCCs and NMDARs is required (Ormond and Woodin, 2009 ). In these experiments STDP was induced by paired stimulation of the Schaeffer collaterals and pyramidal neurons with both glutamatergic and GABAergic transmission intact. Thus in the adult there is cooperation between glutamatergic and GABAergic transmission; glutamate facilitates the opening of NMDARs, which is required for GABAergic STDP. Regardless of the source of Ca2+ influx, the mechanism appears to be a posttranslational modification of KCC2 (Acton et al., 2009 ; Ormond and Woodin, 2009 ). Ionic shift plasticity can also be induced by repetitive prolonged stimulation of the postsynaptic neuron (Fiumelli et al., 2005 ; Brumback and Staley, 2008 ). However there is discrepancy regarding the mechanism of this plasticity. In one scenario, the required Ca2+ influx occurs via release from internal stores, and results in a PKC-dependent regulation of KCC2 (Fiumelli et al., 2005 ). In the other scenario, the spiking resets the thermodynamic equilibrium for NKCC1 transport which indirectly lead to changes in neuronal Cl− (Brumback and Staley, 2008 ).
During early postnatal life when NKCC1 is the dominantly expressed Cl−-transporter, the same STDP induction protocol that induces ionic shift plasticity in the mature CNS also modifies immature synapses (Balena and Woodin, 2008 ; Xu et al., 2008 ). The difference is that at immature synapses we see the direction of the plasticity reverse. Coincident pre- and postsynaptic activity at 5 Hz hyperpolarizes EGABA-A through a regulation of NKCC1 which strengths inhibition. In the developing CNS ionic shift plasticity appears to be frequency dependent. Increasing the stimulation frequency above 20 Hz (while maintaining the spike timing interval) produces a GABABR and CaMKII-dependent depolarization of EGABA-A, likely due to GABA spillover that occurs at higher frequencies (Xu et al., 2008 ).
Synapse-Specificity of Ionic Shift Plasticity
One of the most prominent differences between ionic shift plasticity and glutamatergic plasticity onto interneurons is that while the latter is mainly homosynaptic, ionic shift plasticity has the potential to be heterosynaptic. Ionic shift plasticity produces a change in the Cl− gradient at the activated synapse; because Cl− is a diffusible ion we can expect this gradient change to also affect neighboring synapses within the same neuronal compartment. Moreover, given that GABAergic synapses terminate onto pyramidal cell dendritic shafts rather than spines, any postsynaptic change in the Cl− concentration is unlikely to be restricted to the activated postsynaptic site. Because basket cell interneurons primarily target the principal cell perisomatic region, we would expect the shift in the Cl− gradient to extend throughout the somatic compartment. However we would not necessarily expect the change in the Cl− gradient to extend beyond the somatic compartment to dendrites or axon because it is known that pyramidal cells can maintain domain-specific intracellular Cl− gradients (Szabadics et al., 2006 ; Khirug et al., 2008 ).
Regulation of Inhibitory Strength in Neuronal Networks
What is the function of this ionic shift plasticity? Neuronal patterns of activity that induce glutamatergic STDP in the CA1 region of the hippocampus can also induce GABAergic STDP of interneurons onto pyramidal neurons (Ormond and Woodin, 2009 ). It has been demonstrated both computationally and experimentally that GABAergic STDP regulates pyramidal neuron spiking (Saraga et al., 2008 ) and produces a disinhibition-mediated LTP (Ormond and Woodin, 2009 ). This is perhaps not surprising given that GABAergic inhibition in the CA1 is known to regulate synaptic integration and spike timing (Pouille and Scanziani, 2001 ; Lamsa et al., 2005 ).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Original work of the authors cited in this review has been supported by the Wellcome Trust, the UK Medical Research Council, the European Research Council, the Natural Sciences Research Council of Canada, and the DAAD German Academic Exchange Service.
References
Acton, B. A., Mercado, A., Mount, D. B., and Woodin, M. A. (2009). Activity-Dependent Modification of the K+/Cl− Cotransporter KCC2 in the Hippocampus. Chicago, IL: Society for Neuroscience. Online.
Alle, H., Jonas, P., and Geiger, J. R. (2001). PTP and LTP at a hippocampal mossy fiber-interneuron synapse. Proc. Natl. Acad. Sci. U.S.A. 98, 14708–14713.
Ascoli, G. A., Alonso-Nanclares, L., Anderson, S. A., Barrionuevo, G., Benavides-Piccione, R., Burkhalter, A., Buzsaki, G., Cauli, B., Defelipe, J., Fairen, A., Feldmeyer, D., Fishell, G., Fregnac, Y., Freund, T. F., Gardner, D., Gardner, E. P., Goldberg, J. H., Helmstaedter, M., Hestrin, S., Karube, F., Kisvarday, Z. F., Lambolez, B., Lewis, D. A., Marin, O., Markram, H., Munoz, A., Packer, A., Petersen, C. C., Rockland, K. S., Rossier, J., Rudy, B., Somogyi, P., Staiger, J. F., Tamas, G., Thomson, A. M., Toledo-Rodriguez, M., Wang, Y., West, D. C., and Yuste, R. (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 9, 557–568.
Balena, T., and Woodin, M. A. (2008). Coincident pre- and postsynaptic activity downregulates NKCC1 to hyperpolarize E(Cl) during development. Eur. J. Neurosci. 27, 2402–2412.
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838.
Brumback, A. C., and Staley, K. J. (2008). Thermodynamic regulation of NKCC1-mediated Cl-cotransport underlies plasticity of GABA(A) signaling in neonatal neurons. J. Neurosci. 28, 1301–1312.
Buzsaki, G., and Eidelberg, E. (1982). Direct afferent excitation and long-term potentiation of hippocampal interneurons. J. Neurophysiol. 48, 597–607.
Caillard, O., Ben-Ari, Y., and Gaiarsa, J. L. (1999a). Mechanisms of induction and expression of long-term depression at GABAergic synapses in the neonatal rat hippocampus. J. Neurosci. 19, 7568–7577.
Caillard, O., Ben-Ari, Y., and Gaiarsa, J. L. (1999b). Long-term potentiation of GABAergic synaptic transmission in neonatal rat hippocampus. J. Physiol. (Lond.) 518(Pt 1), 109–119.
Chen, H. X., Jiang, M., Akakin, D., and Roper, S. N. (2009). Long-term potentiation of excitatory synapses on neocortical somatostatin-expressing interneurons. J. Neurophysiol. 102, 3251–3259.
Cohen, I., Navarro, V., Clemenceau, S., Baulac, M., and Miles, R. (2002). On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298, 1418–1421.
Coull, J. A., Beggs, S., Boudreau, D., Boivin, D., Tsuda, M., Inoue, K., Gravel, C., Salter, M. W., and De Koninck, Y. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021.
Csicsvari, J., Hirase, H., Czurko, A., and Buzsaki, G. (1998). Reliability and state dependence of pyramidal cell-interneuron synapses in the hippocampus: an ensemble approach in the behaving rat. Neuron 21, 179–189.
Dzhala, V. I., Talos, D. M., Sdrulla, D. A., Brumback, A. C., Mathews, G. C., Benke, T. A., Delpire, E., Jensen, F. E., and Staley, K. J. (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 11, 1205–1213.
Farrant, M., and Kaila, K. (2007). The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog. Brain Res. 160, 59–87.
Ferraguti, F., Cobden, P., Pollard, M., Cope, D., Shigemoto, R., Watanabe, M., and Somogyi, P. (2004). Immunolocalization of metabotropic glutamate receptor 1alpha (mGluR1alpha) in distinct classes of interneuron in the CA1 region of the rat hippocampus. Hippocampus 14, 193–215.
Fino, E., Deniau, J. M., and Venance, L. (2008). Cell-specific spike-timing-dependent plasticity in GABAergic and cholinergic interneurons in corticostriatal rat brain slices. J. Physiol. (Lond.) 586, 265–282.
Fino, E., Paille, V., Deniau, J. M., and Venance, L. (2009). Asymmetric spike-timing dependent plasticity of striatal nitric oxide-synthase interneurons. Neuroscience 160, 744–754.
Fiumelli, H., Cancedda, L., and Poo, M. M. (2005). Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron 48, 773–786.
Fiumelli, H., and Woodin, M. A. (2007). Role of activity-dependent regulation of neuronal chloride homeostasis in development. Curr. Opin. Neurobiol. 17, 81–86.
Gaiarsa, J. L., Caillard, O., and Ben-Ari, Y. (2002). Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. Trends Neurosci. 25, 564–570.
Galvan, E. J., Calixto, E., and Barrionuevo, G. (2008). Bidirectional Hebbian plasticity at hippocampal mossy fiber synapses on CA3 interneurons. J. Neurosci. 28, 14042–14055.
Gamba, G. (2005). Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 85, 423–493.
Gibson, H. E., Edwards, J. G., Page, R. S., Van Hook, M. J., and Kauer, J. A. (2008). TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron 57, 746–759.
Giese, K. P., Fedorov, N. B., Filipkowski, R. K., and Silva, A. J. (1998). Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873.
Gubellini, P., Ben-Ari, Y., and Gaiarsa, J. L. (2005). Endogenous neurotrophins are required for the induction of GABAergic long-term potentiation in the neonatal rat hippocampus. J. Neurosci. 25, 5796–5802.
Haas, J. S., Nowotny, T., and Abarbanel, H. D. (2006). Spike-timing-dependent plasticity of inhibitory synapses in the entorhinal cortex. J. Neurophysiol. 96, 3305–3313.
Heifets, B. D., and Castillo, P. E. (2009). Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 71, 283–306.
Holmgren, C. D., and Zilberter, Y. (2001). Coincident spiking activity induces long-term changes in inhibition of neocortical pyramidal cells. J. Neurosci. 21, 8270–8277.
Jia, Y., Yamazaki, Y., Nakauchi, S., Ito, K., and Sumikawa, K. (2010). Nicotine facilitates long-term potentiation induction in oriens–lacunosum moleculare cells via Ca entry through non-alpha7 nicotinic acetylcholine receptors. Eur. J. Neurosci. 31, 463–476.
Khirug, S., Yamada, J., Afzalov, R., Voipio, J., Khiroug, L., and Kaila, K. (2008). GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J. Neurosci. 28, 4635–4639.
Klausberger, T., and Somogyi, P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57.
Komatsu, Y. (1994). Age-dependent long-term potentiation of inhibitory synaptic transmission in rat visual cortex. J. Neurosci. 14, 6488–6499.
Kullmann, D. M., and Lamsa, K. P. (2007). Long-term synaptic plasticity in hippocampal interneurons. Nat. Rev. Neurosci. 8, 687–699.
Laezza, F., and Dingledine, R. (2004). Voltage-controlled plasticity at GluR2-deficient synapses onto hippocampal interneurons. J. Neurophysiol. 92, 3575–3581.
Laezza, F., Doherty, J. J., and Dingledine, R. (1999). Long-term depression in hippocampal interneurons: joint requirement for pre- and postsynaptic events. Science 285, 1411–1414.
Lamsa, K., Heeroma, J. H., and Kullmann, D. M. (2005). Hebbian LTP in feed-forward inhibitory interneurons and the temporal fidelity of input discrimination. Nat. Neurosci. 8, 916–924.
Lamsa, K., Irvine, E. E., Giese, K. P., and Kullmann, D. M. (2007a). NMDA receptor-dependent long-term potentiation in mouse hippocampal interneurons shows a unique dependence on Ca(2+)/calmodulin-dependent kinases. J. Physiol. (Lond.) 584, 885–894.
Lamsa, K. P., Heeroma, J. H., Somogyi, P., Rusakov, D. A., and Kullmann, D. M. (2007b). Anti-Hebbian long-term potentiation in the hippocampal feedback inhibitory circuit. Science 315, 1262–1266.
Lapointe, V., Morin, F., Ratte, S., Croce, A., Conquet, F., and Lacaille, J. C. (2004). Synapse-specific mGluR1-dependent long-term potentiation in interneurones regulates mouse hippocampal inhibition. J. Physiol. (Lond.) 555, 125–135.
Lei, S., and McBain, C. J. (2004). Two loci of expression for long-term depression at hippocampal mossy fiber-interneuron synapses. J. Neurosci. 24, 2112–2121.
Lien, C. C., Mu, Y., Vargas-Caballero, M., and Poo, M. M. (2006). Visual stimuli-induced LTD of GABAergic synapses mediated by presynaptic NMDA receptors. Nat. Neurosci. 9, 372–380.
Lu, J. T., Li, C. Y., Zhao, J. P., Poo, M. M., and Zhang, X. H. (2007). Spike-timing-dependent plasticity of neocortical excitatory synapses on inhibitory interneurons depends on target cell type. J. Neurosci. 27, 9711–9720.
Lu, Y. M., Mansuy, I. M., Kandel, E. R., and Roder, J. (2000). Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron 26, 197–205.
Maccaferri, G., Toth, K., and McBain, C. J. (1998). Target-specific expression of presynaptic mossy fiber plasticity. Science 279, 1368–1370.
Maffei, A., Nataraj, K., Nelson, S. B., and Turrigiano, G. G. (2006). Potentiation of cortical inhibition by visual deprivation. Nature 443, 81–84.
Mahanty, N. K., and Sah, P. (1998). Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 394, 683–687.
McBain, C. J., DiChiara, T. J., and Kauer, J. A. (1994). Activation of metabotropic glutamate receptors differentially affects two classes of hippocampal interneurons and potentiates excitatory synaptic transmission. J. Neurosci. 14, 4433–4445.
McBain, C. J., and Kauer, J. A. (2009). Presynaptic plasticity: targeted control of inhibitory networks. Curr. Opin. Neurobiol. 19, 254–262.
McMahon, L. L., and Kauer, J. A. (1997). Hippocampal interneurons express a novel form of synaptic plasticity. Neuron 18, 295–305.
Mercado, A., Mount, D. B., and Gamba, G. (2004). Electroneutral cation-chloride cotransporters in the central nervous system. Neurochem. Res. 29, 17–25.
Nissen, W., Szabo, A., Somogyi, J., Somogyi, P., and Lamsa, K. P. (2010). Cell type-specific long-term plasticity at glutamatergic synapses onto hippocampal interneurons expressing either parvalbumin or CB1 cannabinoid receptor. J. Neurosci. 30, 1337–1347.
Nugent, F. S., and Kauer, J. A. (2008). LTP of GABAergic synapses in the ventral tegmental area and beyond. J. Physiol. (Lond.) 586, 1487–1493.
Oren, I., Nissen, W., Kullmann, D. M., Somogyi, P., and Lamsa, K. P. (2009). Role of ionotropic glutamate receptors in long-term potentiation in rat hippocampal CA1 oriens-lacunosum moleculare interneurons. J. Neurosci. 29, 939–950.
Ormond, J., and Woodin, M. A. (2009). Disinhibition mediates a form of hippocampal long-term potentiation in area CA1. PLoS ONE 4, e7224. doi: 10.1371/journal.pone.0007224.
Patenaude, C., Chapman, C. A., Bertrand, S., Congar, P., and Lacaille, J. C. (2003). GABAB receptor- and metabotropic glutamate receptor-dependent cooperative long-term potentiation of rat hippocampal GABAA synaptic transmission. J. Physiol. (Lond.) 553, 155–167.
Patenaude, C., Massicotte, G., and Lacaille, J. C. (2005). Cell-type specific GABA synaptic transmission and activity-dependent plasticity in rat hippocampal stratum radiatum interneurons. Eur. J. Neurosci. 22, 179–188.
Payne, J. A., Rivera, C., Voipio, J., and Kaila, K. (2003). Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 26, 199–206.
Pelkey, K. A., Lavezzari, G., Racca, C., Roche, K. W., and McBain, C. J. (2005). mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron 46, 89–102.
Perez, Y., Morin, F., and Lacaille, J. C. (2001). A hebbian form of long-term potentiation dependent on mGluR1a in hippocampal inhibitory interneurons. Proc. Natl. Acad. Sci. U.S.A. 98, 9401–9406.
Plotkin, M. D., Snyder, E. Y., Hebert, S. C., and Delpire, E. (1997). Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J. Neurobiol. 33, 781–795.
Pouille, F., and Scanziani, M. (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293, 1159–1163.
Rheims, S., Holmgren, C. D., Chazal, G., Mulder, J., Harkany, T., Zilberter, T., and Zilberter, Y. (2009). GABA action in immature neocortical neurons directly depends on the availability of ketone bodies. J. Neurochem. 110, 1330–1338.
Rivera, C., Li, H., Thomas-Crusells, J., Lahtinen, H., Viitanen, T., Nanobashvili, A., Kokaia, Z., Airaksinen, M. S., Voipio, J., Kaila, K., and Saarma, M. (2002). BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol. 159, 747–752.
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., Pirvola, U., Saarma, M., and Kaila, K. (1999). The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255.
Russmann, H., Lamy, J. C., Shamim, E. A., Meunier, S., and Hallett, M. (2009). Associative plasticity in intracortical inhibitory circuits in human motor cortex. Clin. Neurophysiol 120, 1204–1212.
Santos, S. F., Luz, L. L., Szucs, P., Lima, D., Derkach, V. A., and Safronov, B. V. (2009). Transmission efficacy and plasticity in glutamatergic synapses formed by excitatory interneurons of the substantia gelatinosa in the rat spinal cord. PLoS ONE 4, e8047. doi: 10.1371/journal.pone.0008047.
Saraga, F., Balena, T., Wolansky, T., Dickson, C. T., and Woodin, M. A. (2008). Inhibitory synaptic plasticity regulates pyramidal neuron spiking in the rodent hippocampus. Neuroscience 155, 64–75.
Sarihi, A., Jiang, B., Komaki, A., Sohya, K., Yanagawa, Y., and Tsumoto, T. (2008). Metabotropic glutamate receptor type 5-dependent long-term potentiation of excitatory synapses on fast-spiking GABAergic neurons in mouse visual cortex. J. Neurosci. 28, 1224–1235.
Stelzer, A., Simon, G., Kovacs, G., and Rai, R. (1994). Synaptic disinhibition during maintenance of long-term potentiation in the CA1 hippocampal subfield. Proc. Natl. Acad. Sci. U.S.A. 91, 3058–3062.
Szabadics, J., Varga, C., Molnar, G., Olah, S., Barzo, P., and Tamas, G. (2006). Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311, 233–235.
Topolnik, L., Congar, P., and Lacaille, J. C. (2005). Differential regulation of metabotropic glutamate receptor- and AMPA receptor-mediated dendritic Ca2+ signals by presynaptic and postsynaptic activity in hippocampal interneurons. J. Neurosci. 25, 990–1001.
Tzounopoulos, T., Kim, Y., Oertel, D., and Trussell, L. O. (2004). Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat. Neurosci. 7, 719–725.
van den Pol, A. N., Obrietan, K., and Chen, G. (1996). Excitatory actions of GABA after neuronal trauma. J. Neurosci. 16, 4283–4292.
Woodin, M. A., Ganguly, K., and Poo, M. M. (2003). Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl− transporter activity. Neuron 39, 807–820.
Xu, C., Zhao, M. X., Poo, M. M., and Zhang, X. H. (2008). GABA(B) receptor activation mediates frequency-dependent plasticity of developing GABAergic synapses. Nat. Neurosci. 11, 1410–1418.
Xu, J. C., Lytle, C., Zhu, T. T., Payne, J. A., Benz, E., Jr., and Forbush, B., 3rd. (1994). Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc. Natl. Acad. Sci. U.S.A. 91, 2201–2205.
Keywords: interneuron, GABA, fast-spiking, oscillation, chloride, NKCC1, KCC2
Citation: Lamsa KP, Kullmann DM and Woodin MA (2010) Spike-timing dependent plasticity in inhibitory circuits. Front. Syn. Neurosci. 2:8. doi: 10.3389/fnsyn.2010.00008
Received: 31 January 2010;
Paper pending published: 15 February 2010;
Accepted: 17 May 2010;
Published online: 21 June 2010
Edited by:
Per Jesper Sjöström, University College London, UKReviewed by:
Julie Kauer, Brown University, USAYuri Zilberter, Institut de Neurobiologie de la MEDiterranée, France
Copyright: © 2010 Lamsa, Kullmann and Woodin. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Karri P. Lamsa, Department of Pharmacology, Oxford University, Mansfield Road, Oxford OX1 3QT, UK. e-mail:a2FycmkubGFtc2FAcGhhcm0ub3guYWMudWs=