 Gengyin Guo
Gengyin Guo Jianfeng Zhuang2
Jianfeng Zhuang2 Zhen Zhang
Zhen Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Surg. , 16 May 2022
Sec. Neurosurgery
Volume 9 - 2022 | https://doi.org/10.3389/fsurg.2022.864518
Atypical teratoid/rhabdoid tumor (AT/RT) of the central nervous system is a highly malignant tumor that mainly occurs in children under the age of 3 and has only been rarely described in adults. The fact that AT/RT patients have such a terrible prognosis is even more regrettable. Herein, we reported two special cases of AT/RT, both of which were under 3 years. Symptoms at presentation included increased intracranial pressure and cerebellar symptoms such as headache, altered gait, and ataxia. As for the tumor location, one was infratentorial in the posterior fossa, and the other was the right lateral ventricle. Preoperative magnetic resonance imaging scans showed calcification and heterogeneous contrast enhancement in the lesions. The mass was excised surgically for the progression of symptoms. Postoperative pathologies of the tumors, combined with immunohistochemistry, revealed AT/RT. AT/RTs are often misdiagnosed as other types of brain tumors due to the lack of specific radiological features and other key characteristics. To improve awareness of AT/RT on the differential diagnosis of intracranial lesions among clinicians, we present this report and briefly summarize previous cases.
Atypical teratoid/rhabdoid tumor (AT/RT) is a highly malignant central nervous system (CNS) neoplasm predominantly found in children under the age of 3, and is extremely rare in adults (1, 2). In the year 1987, it was described for the first time (3). AT/RT usually occurs in posterior fossa for pediatric patients, most commonly in the cerebellum. By contrast, the most common locations in adults are the cerebral hemisphere and the sellar region (2, 4, 5). The pathological features of AT/RT are peculiar, consisting primarily of rhabdoid cells and heterogeneous portions containing mesenchymal, epithelial, and neuroectodermal cells (6).
According to the 2021 WHO Classification of CNS tumors, medulloblastoma and AT/RT were classified as embryonal tumors, and all embryonal tumors (except for cribriform neuroepithelial tumor and CNS tumor with BCL6 corepressor internal tandem duplication) were classified as Grade 4 tumor (7). Using DNA methylation and gene expression profiling, the relevant reports showed that AT/RTs are comprised of three epigenetic subgroups with distinct enhancer landscapes: ATRT-TYR, ATRT-SHH, and ATRT-MYC (8). AT/RTs are often misdiagnosed as other types of brain tumors due to the lack of specific radiological features and the heterogeneous nature of the tumor cells (6). Differential diagnoses should be noted for embryonal tumor, medulloblastoma, choroids plexus carcinoma, and high-grade glioma (9). Therefore, a diagnosis of AT/RTs requires the confirmation of specific genetic aberrations such as a loss of integrase interactor 1 (INI1) tumor suppressor gene on chromosome 22 or BRG1 gene (10, 11).
In addition, the treatment criteria (including stratification criteria) for patients with AT/RT are a controversial topic (10). The prognosis is grim, even if the treatment generally consists of surgical resection in combination with chemotherapy and radiation therapy (RT) (6). In order to better diagnose and treat AT/RT, more cases should be included in the study. In this report, we describe two patients who presented with intracranial masses in the left cerebellum and right ventricle, respectively, and show the clinical, imaging, pathological, and immunohistochemical aspects of AT/RTs.
Case 1: The patient was a 19-month-old boy with a 1-month history of claudication before admission. Neurological examination verified that the left ankle clonus and the left Babinski sign were positive. Cerebral magnetic resonance imaging (MRI) revealed a mass in the right lateral ventricle with intraventricular extension. The size of the lump was about 6 × 5 × 2 cm, which was primarily isointense on both T1- and T2-weighted MRI. Additionally, the mass appeared hyperintense on enhanced MRI, with areas of calcification and heterogeneous contrast enhancement (Figures 1A–E). The patient underwent a right frontal approach for gross total resection in supine position. To observe the yellow and soft mass, we gently slit the epidermis, subcutaneous tissue, and other tissues to expose the skull, dissociated the skull with a milling cutter, and liberated the blood vessels. Grossly, the tumor had a tender fish flesh-like appearance with necrosis. However, the tumor was tightly connected to the lateral ventricle wall and the septum pellucidum and protruded into the third ventricle through the interventricular foramen. Then, piecemeal resection was performed on the tumor. Postoperative computed tomography (CT) showed that in the bilateral lateral ventricle walls, there were several irregular nodular, strip-like high-density foci with indistinct boundaries. The tumor was completely removed (Figures 1F–H). Postoperative pathological diagnosis showed embryonic tumors with multidirectional differentiation of immature small cells, epithelial, and neuron cells and revealed abundant eosinophilic cytoplasm and eccentric nucleus with prominent nucleoli. The tumors were negative for INI1 and positive for Synaptophysin (Syn) (Figures 3A–D). The Ki-67 labeling index was 50%. A case of AT/RT was ultimately diagnosed by histopathological examination of the resected specimens. Two months after the operation, the patient received two cycles of adjuvant chemotherapy including Vincristine, Methotrexate, Leucovorin, Cytoxan, Etoposide, and Cisplatin. Unfortunately, the patient died of tumor recurrence after 15 months.
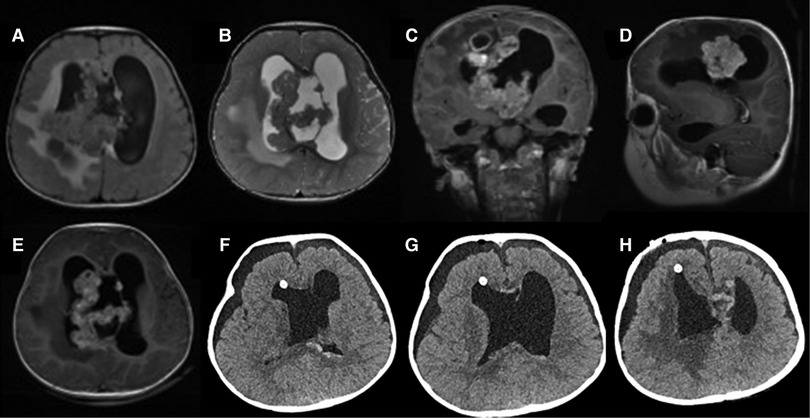
Figure 1. Preoperative MRI: (A) axial T1 image; (B) axial T2 image; (C) coronal T1-weighted image with gadolinium contrast; (D) sagittal T1-weighted image with gadolinium contrast; (E) axial T1-weighted image with gadolinium contrast; Postoperative CT: (F–H) axial scan.
Case 2: The patient was a 27-month-old boy, whose symptoms were unstable gait for a month, accompanied by nausea and vomiting for half a month. Physical examination did not reveal positive signs. An MRI revealed the presence of a mass measured 2.3 × 1.8 × 1.7 cm in the left cerebellum. MRI showed the isointense on T1-weighted images, heterogeneous iso/hyperintense on T2-weighted images and a pronounced enhancement after using an MR contrast agent (Figures 2A–E). The patient underwent a median suboccipital approach to the posterior fossa for tumor resection in the right lateral position. The epidermis and other tissues were slit progressively to reveal the tumor, just as we did in case 1. Then, we made a bone window along the squamous portion of the occipital bone. Unlike case 1, however, the tumor had a distinct capsule that separated it from the normal parenchyma. The tumor was red and soft, with abundant vascularity. A subtotal resection of the tumor was performed. Postoperative CT revealed that the left cerebellum was a low-density area, with an unclear boundary and sporadic pneumocephalus. The tumor was removed completely (Figures 2F–H). Tumor cells have eccentric nucleoli and abundant eosinophil cytoplasm. Brisk mitotic activity and apoptotic bodies are common. The tumors were partly positive for vimentin (Figures 3E–H) and the tumors were negative for INI1, Smooth Muscle Actin (SMA), and Glial Fibrillary Acidic Protein (GFAP). The Ki-67 labeling index was 40%. Postoperatively, the results of histopathological examinations demonstrated an AT/RT. After 2 months, the patient received four cycles of chemotherapy (cisplatin, cyclophosphamide, and vincristine). Despite these efforts, patient survival was less than 9 months. Notably, the two patients had no history of radiation or family history of hereditary diseases. Moreover, cerebrospinal fluid examinations for tumor markers were negative. Fortunately, no metastases occurred in the two patients. The relevant information on the patients is listed in Table 1.
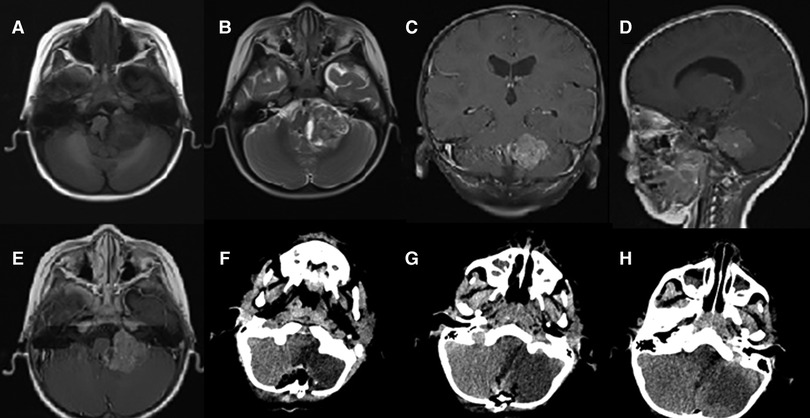
Figure 2. Preoperative MRI: (A) axial T1 image; (B), axial T2 image; (C) coronal T1-weighted image with gadolinium contrast; (D) sagittal T1-weighted image with gadolinium contrast; (E) axial T1-weighted image with gadolinium contrast; Postoperative CT: (F–H) axial scan.
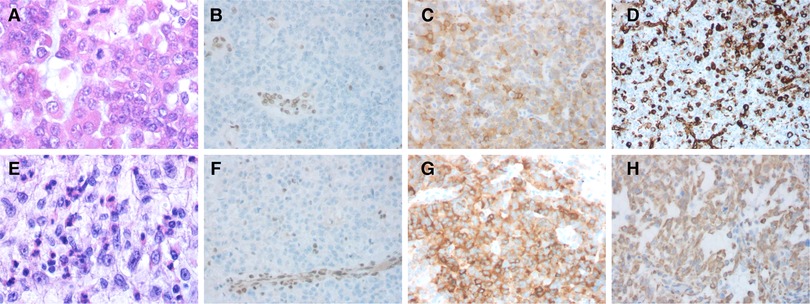
Figure 3. Hematoxylin and eosin stain (H&E) and immunohistochemical features of the cases. Case 1: (A) The majority of the tumor cells displayed eccentric round nuclei and prominent nucleolus; (B) IHC showing the tumor is negative for integrase interactor 1 (INI1), with vascular endothelial cell internal control being positive; (C) positive for Synaptophysin; and (D) partially positive for vimentin. Case 2: (E) Brisk mitotic activity is common; (F) Immunostaining showed a lack of INI1 protein expression in the tumor cells and apoptotic bodies; (G) positive for Synaptophysin; and (H) partially positive for vimentin.

Table 1. Clinical features, imaging findings, and therapy in two cases of AT/RTs.
As in prior cases, the pathology of tumors showed that cells had a prominent nucleus with eccentric nucleoli and abundant cytoplasm of a somewhat “rhabdoid” morphology (6). However, pathological features cannot diagnose AT/RT. The final diagnosis of the disease still depends on the loss of nuclear INI1 expression. However, INI1 expression may be retained in a small percentage of AT/RTs, and INI1 deletion may also occur in other tumors. In our case, postoperative immunohistochemistry of all patients showed that the tumor cells were negative for INI1, OLIG2, and GFAP. The SMA was positive in the first patient. Meanwhile, immunostaining of the two patients’ tumor tissue showed that the tumor cells were positive for epithelial membrane antigen (EMA). The Ki-67 labeling indexes were 40% and 50%. The results of immunohistochemical staining are listed in Table 2.
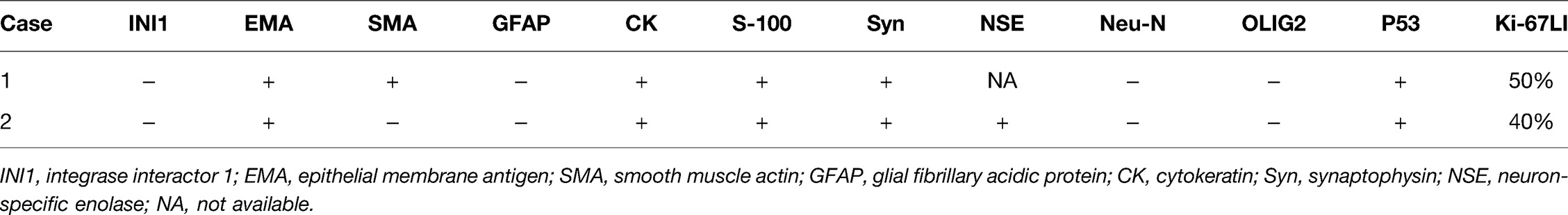
Table 2. Immunohistochemical staining in two cases of AT/RTs.
AT/RT is an extremely rare and highly aggressive CNS tumor, especially in children under 3 years of age (median age 2 years) (1, 2, 12). The prevalence of AT/RT is estimated to be approximately 1%–2% among all pediatric CNS tumors (13). The incidence of AT/RT is not high, but large sample studies are currently lacking. The prognosis of this tumor is extremely poor, with the historic median overall survival (OS) ranging from 6 to 18 months (14). The different clinical manifestations, such as vomiting, lethargy, or cranial nerve palsies, relies predominantly on the location of the tumor (15). The distribution of AT/RT is as follows: 52% in the posterior fossa (the cerebellum being the predominant site); 39% supratentorial; 5% in the pineal region; 2% in the spine; and 2% are multifocal (16). Males have a higher incidence rate than females, with the reported ratio being 3:2 to 2:1 (12). The incidence is increasing as a result of improved diagnostic techniques, especially the introduction of the biological marker INI1 (17).
Histopathologically, AT/RT is characterized by the presence of rhabdoid cells, with or without fields resembling a typical embryonal tumor, epithelial tissue, and neoplastic mesenchyme (16–18). The rhabdoid cell has an eccentric round nucleus in an abundant eosinophilic cytoplasm and a prominent nucleolus (19). Necrosis and brisk mitotic activity are common in the rhabdoid cell (3, 19). In general, immunohistochemistry contains staining of vimentin, EMA, and other markers such as INI1, MyoD1, S-100, SMA, and GFAP (6).
The cellular origin of AT/RT is unknown, but inactivating mutations of the hSNF5/INI1 gene in chromosomal region 22q11.2 are regarded as a critical step in its molecular pathogenesis (20). AT/RT is characterized by a loss of the long arm of the chromosome 22, which results in a loss of the hSNF5/INI1 gene, causing the INI1 protein expression to be negative (21). The hSNF5/INI1 gene is considered as a tumor suppressor gene in peripheral AT/RTs of the CNS (22, 23). Previous studies have suggested that INI1 suppresses tumor formation by regulating cell proliferation via the Rb cell cycle checkpoint (9). But the loss of INI1 expression is not a certainty. Others have previously reported that an AT/RT retained INI1 expression staining was detected by immunohistochemistry and genetic testing. This reminds us that we cannot diagnose AT/RT merely on the basis of a loss of nuclear INI1 expression. Furthermore, they estimated that tumors that retain INI1 staining account for roughly 2% of AT/RTs. Even though the loss of nuclear INI1 expression is the most frequent surrogate marker for AT/RT diagnosis, a minor subset may have retained INI1 expression and should be evaluated for the loss of nuclear BGR1 (SMARCA4) expression. Conversely, we also bear in mind that INI1 loss may occur in other neoplasms, including poorly differentiated chordomas, and schwannomatosis-associated schwannomas (in a mosaic pattern). Therefore, correlations with morphological and clinicopathological features should be noted as well (24). In addition, the essential diagnostic criteria for AT/RT include: (1) a CNS embryonal tumor with a polyimmunophenotype AND (2) Loss of nuclear SMARCB1 (INI1) or SMARCA4 (BRG1) expression in tumor cells OR A DNA methylation profile aligned with AT/RT.
Using DNA methylation and gene expression profiling, AT/RTs are comprised of three subgroups ATRT-TYR, ATRT-SHH, and ATRT-MYC (8). Furthermore, the ATRT-SHH subgroup exhibited further heterogeneity, segregating further into two subtypes: ATRT-SHH-1 and ATRT-SHH-2 (25). ATRT-TYR is characterized by tyrosinase overexpression (25, 26). The whole or partial deletion of one copy of chromosome 22, accompanied by an inactivating mutation in INI1 on the other allele, is the prototypic type of biallelic SMARCB1 inactivation in the ATRT-TYR group. ATRT-TYR patients are the youngest patient group clinically, with a median age of 12 months at diagnosis (25). Protein expression of achaete-scute homolog 1 (ASCL1) has been suggested as an immunohistochemical marker for the ATRT-SHH subgroup. But, ASCL1 may not be expressed in all ATRT-SHH patients. ASCL1-positive ATRT-SHH patients had greater OS than ASCL1-negative ATRTs (27). The ATRT-SHH subgroup was reported to be less likely to display no enhancement (28). In comparison with ATRT-TYR and ATRT-MYC, homo- or heterozygous SMARCB1 deletions are less frequently found in ATRT-SHH. ATRT-SHH is a more intermediate grouping in terms of age (median age 20 months) (25). Withholding radiotherapy in ATRT-SHH tumors appears to have no detrimental influence on OS (27). Another report found a robust trend toward longer event-free survival (EFS) and OS in ATRT-SHH tumors, with a 6-month EFS of 100% (29). MYC oncogene expression is elevated in ATRT-MYC. Point mutations are uncommon in ATRT-MYC tumors in contrast to ATRT-TYR or ATRT-SHH tumors (25). MRI studies have demonstrated that ATRT-MYC tumors are characterized by the presence of intense peritumoral edema (28). The median age of ATRT-MYC patients is significantly higher than that of the other subgroups (27 months) (25). Both ATRT-TYR and ATRT-SHH tumors tend to have focal aberrations in SMARCB1, whereas ATRT-MYC tumors tend to have broad deletions affecting Chr22q11.2. Unlike ATRT-SHH cancers, ATRT-MYC and ATRT-TYR tumors have a strong reliance on receptor tyrosine kinase pathways (27).
At present, it is indistinct to formulate a vintage treatment for AT/RT. Therapeutic strategies of AT/RT are largely dependent on the age of the patient, the location of the tumor in the CNS, and the stage of the disease at the time of diagnosis (25). In general, surgical resection of AT/RT is the principal treatment. Surgical treatment generally guarantees most of the removal of the tumor according to the relationship between the location of the tumor and the blood vessels in the surrounding tissues. Taking into account the blood vessels around the tumor, especially the tumor in difficult location, angiography and preoperative embolization may yield some effects on surgery (30). Adjuvant therapy includes RT, high-dose chemotherapy, and autologous stem cell transplantation (12, 31). RT is an effective treatment but can be avoided in patients under 3 years of age due to long-term neurocognitive sequelae. Patients receiving RT earlier in the treatment plan may not experience early relapse or progression during induction chemotherapy and may show improvement in outcomes (12, 32). In addition, it has been demonstrated that antisense-mediated downregulation of insulin-like growth factor I receptor (IGF-IR) results in sensitization to doxorubicin and cisplatin (18). The use of high-dose chemotherapy and autologous stem cell transplantation (HDCT/auto-SCT) has shown clinical benefits in children with brain tumors (33). Moreover, existing literature has shown that a tumor vaccination strategy using lysate-loaded autologous dendritic cells is feasible and safe even in small children with AT/RT (34). When the prognosis is poor, people may try to regain control through the use of alternative therapies, including dietary changes. Complementary and alternative medicine (CAM) may not be limited to prayer, spiritual healing, vitamins, herbs, and dietary changes (15).
However, there are some limitations in this report. First, the number of cases included in the report is relatively small. So far, there is still a lack of large sample studies, and no mature therapeutic plan has been formed. Furthermore, the figures of postoperative MRI images are missing. Finally, according to multicenter data, AT/RT is still a rare tumor with poor prognosis in children. In general, most of the patients died of local relapse, metastatic lesions through the subarachnoid space, or distant metastasis (35).
AT/RT is a rare disease condition, and a preoperative diagnosis is difficult without an adequate knowledge of the disease. To date, despite multiple advanced diagnoses and treatments being available, the prognosis of patients with AT/RT remains grim. More AT/RT case reports should be included, which will lead to greater diagnostic accuracy as well as improvement in treatment and prognosis. Due to its aggressiveness and poor prognosis, AT/RT cannot be ruled out in the differential diagnosis of intracranial lesions. The case reports replenish the existing literature with new knowledge and hopefully will draw the attention of doctors to the rare tumor.
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author/s.
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
GG drafted the manuscript. KZ and ZZho was involved in the acquisition of data. JZ, and YW performed analysis or interpretation of the data. ZZha performed the surgeries and devised the study concept and design. All authors contributed to the article and approved the submitted version.
This work was supported by the Projects of Medical and Health Technology Development Program in Shandong province (grant number: 202004041407), the Natural Science Foundation of Shandong Province (grant number: ZR2021MH228), and the Projects of Traditional Medical and Health Technology Development Program in Shandong province (grant number: 2021M185).
ZZha was employed by the company Jiangsu Simcere Pharmaceutical Co. Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Meel MH, Guillén Navarro M, de Gooijer MC, Metselaar DS, Waranecki P, Breur M, et al. MEK/MELK inhibition and blood-brain barrier deficiencies in atypical teratoid/rhabdoid tumors. Neuro-Oncology. (2020) 22(1):58–69. doi: 10.1093/neuonc/noz151
2. Ostrom QT, Chen Y, MdB P, Ondracek A, Farah P, Gittleman H, et al. The descriptive epidemiology of atypical teratoid/rhabdoid tumors in the United States, 2001–2010. Neuro-Oncology. (2014) 16(10):1392–9. doi: 10.1093/neuonc/nou090
3. Bhattacharjee M, Hicks J, Langford L, Dauser R, Strother D, Chintagumpala M, et al. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. Ultrastruct Pathol. (1997) 21(4):369–78. doi: 10.3109/01913129709021935
4. Chan V, Marro A, Findlay JM, Schmitt LM, Das S. A systematic review of atypical teratoid rhabdoid tumor in adults. Front Oncol. (2018) 8:567. doi: 10.3389/fonc.2018.00567
5. Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. (1996) 85(1):56–65. doi: 10.3171/jns.1996.85.1.0056
6. Mathkour M, Carsky K, Chabot AB, Werner C, Berry JF, Carr C, et al. Adult pineal region atypical teratoid rhabdoid tumor: a case for aggressive surgical and chemoradiation management with comprehensive literature review. World Neurosurg. (2020) 142:117–27. doi: 10.1016/j.wneu.2020.06.144
7. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncology. (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
8. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. (2016) 29(3):379–93. doi: 10.1016/j.ccell.2016.02.001
9. Coccé MC, Lubieniecki F, Kordes U, Alderete D, Gallego MS. A complex karyotype in an atypical teratoid/rhabdoid tumor: case report and review of the literature. J Neurooncol. (2011) 104(1):375–80. doi: 10.1007/s11060-010-0478-0
10. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
11. Taschner U, Diebold M, Shah MJ, Prinz M, Urbach H, Erny D, et al. Freiburg neuropathology case conference: a 6-year-old girl presenting with vomiting and right-sided facial paresis. Clin Neuroradiol. (2021) 31(3):885–92. doi: 10.1007/s00062-021-01069-3
12. Wang X, Liu X, Lin Z, Chen Y, Wang P, Zhang S. Atypical teratoid/rhabdoid tumor (AT/RT) arising from the acoustic nerve in a young adult: a case report and a review of literature. Medicine. (2015) 94(4):e439. doi: 10.1097/MD.0000000000000439
13. Woehrer A, Slavc I, Waldhoer T, Heinzl H, Zielonke N, Czech T, et al. Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer. (2010) 116(24):5725–32. doi: 10.1002/cncr.25540
14. Buscariollo DL, Park HS, Roberts KB, Yu JB. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a surveillance, epidemiology, and end results analysis. Cancer. (2012) 118(17):4212–9. doi: 10.1002/cncr.27373
15. Howes TL, Buatti JM, O’Dorisio MS, Kirby PA, Ryken TC. Atypical teratoid/rhabdoid tumor case report: treatment with surgical excision, radiation therapy, and alternative medicines. J Neurooncol. (2005) 72(1):85–8. doi: 10.1007/s11060-004-3120-1
16. Dang T, Vassilyadi M, Michaud J, Jimenez C, Ventureyra EC. Atypical teratoid/rhabdoid tumors. Childs Nerv Syst. (2003) 19(4):244–8. doi: 10.1007/s00381-003-0731-3
17. Yang M, Chen X, Wang N, Zhu K, Hu YZ, Zhao Y, et al. Primary atypical teratoid/rhabdoid tumor of central nervous system in children: a clinicopathological analysis and review of literature in China. Int J Clin Exp Pathol. (2014) 7(5):2411–20. https://pubmed.ncbi.nlm.nih.gov/24966951/ PMID: 24966951; PMCID: PMC4069879
18. D’Cunja J, Shalaby T, Rivera P, von Büren A, Patti R, Heppner FL, et al. Antisense treatment of IGF-IR induces apoptosis and enhances chemosensitivity in central nervous system atypical teratoid/rhabdoid tumours cells. Eur J Cancer. (2007) 43(10):1581–9. doi: 10.1016/j.ejca.2007.03.003
19. Strother D. Atypical teratoid rhabdoid tumors of childhood: diagnosis, treatment and challenges. Expert Rev Anticancer Ther. (2005) 5(5):907–15. doi: 10.1586/14737140.5.5.907
20. Jin S, Sun C, Yu S, Wang Q, An T, Wen Y. Atypical teratoid/rhabdoid tumor of the brain in an adult with 22q deletion but no absence of INI1 protein: a case report and review of the literature. Folia Neuropathol. (2015) 53(1):80–5. doi: 10.5114/fn.2015.49977
21. Ginn KF, Gajjar A. Atypical teratoid rhabdoid tumor: current therapy and future directions. Front Oncol. (2012) 2:114. doi: 10.3389/fonc.2012.00114
22. Haberler C, Laggner U, Slavc I, Czech T, Ambros IM, Ambros PF, et al. Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors: lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype. Am J Surg Pathol. (2006) 30(11):1462–8. doi: 10.1097/01.pas.0000213329.71745.ef
23. Biegel JA, Kalpana G, Knudsen ES, Packer RJ, Roberts CW, Thiele CJ, et al. The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res. (2002) 62(1):323–8. https://pubmed.ncbi.nlm.nih.gov/11782395/ PMID: 11782395
24. Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. (2011) 35(6):933–5. doi: 10.1097/PAS.0b013e3182196a39
25. Ho B, Johann PD, Grabovska Y, De Dieu Andrianteranagna MJ, Yao F, Frühwald M, et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors - a reinvestigation and current consensus. Neuro-Oncology. (2020) 22(5):613–24. doi: 10.1093/neuonc/noz235
26. Hasselblatt M, Thomas C, Nemes K, Monoranu CM, Riemenschneider MJ, Koch A, et al. Tyrosinase immunohistochemistry can be employed for the diagnosis of atypical teratoid/rhabdoid tumours of the tyrosinase subgroup (ATRT-TYR). Neuropathol Appl Neurobiol. (2020) 46(2):186–9. doi: 10.1111/nan.12560
27. Torchia J, Picard D, Lafay-Cousin L, Hawkins CE, Kim SK, Letourneau L, et al. Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol. (2015) 16(5):569–82. doi: 10.1016/S1470-2045(15)70114-2
28. Nowak J, Nemes K, Hohm A, Vandergrift LA, Hasselblatt M, Johann PD, et al. Magnetic resonance imaging surrogates of molecular subgroups in atypical teratoid/rhabdoid tumor. Neuro-Oncology. (2018) 20(12):1672–9. doi: 10.1093/neuonc/noy111
29. Reddy AT, Strother DR, Judkins AR, Burger PC, Pollack IF, Krailo MD, et al. Efficacy of high-dose chemotherapy and three-dimensional conformal radiation for atypical teratoid/rhabdoid tumor: a report from the Children's Oncology Group Trial ACNS0333. J Clin Oncol. (2020) 38(11):1175–85. doi: 10.1200/JCO.19.01776
30. Chan V, Marro A, Chainey J, Schmitt L, Das S. Intraventricular atypical teratoid rhabdoid tumour in an adult: a case report and literature review. Folia Neuropathol. (2019) 57(3):295–300. doi: 10.5114/fn.2019.88460
31. Lee J, Kim DS, Han JW, Suh CO. Atypical teratoid/rhabdoid tumors in children treated with multimodal therapies: the necessity of upfront radiotherapy after surgery. Pediatr Blood Cancer. (2017) 64(12). doi: 10.1002/pbc.26663
32. Park ES, Sung KW, Baek HJ, Park KD, Park HJ, Won SC, et al. Tandem high-dose chemotherapy and autologous stem cell transplantation in young children with atypical teratoid/rhabdoid tumor of the central nervous system. J Korean Med Sci. (2012) 27(2):135–40. doi: 10.3346/jkms.2012.27.2.135
33. Sung KW, Lim DH, Yi ES, Choi YB, Lee JW, Yoo KH, et al. Tandem high-dose chemotherapy and autologous stem cell transplantation for atypical teratoid/rhabdoid tumor. Cancer Res Treat. (2016) 48(4):1408–19. doi: 10.4143/crt.2015.347
34. van Gool SW, Holm S, Rachor J, Adamson L, Technau A, Maass E, et al. Immunotherapy in atypical teratoid-rhabdoid tumors: data from a survey of the HGG-immuno group. Cytotherapy. (2016) 18(9):1178–86. doi: 10.1016/j.jcyt.2016.06.004
Keywords: atypical teratoid/rhabdoid tumor, pediatric brain tumors, integrase interactor 1, adjuvant therapy, molecular subgrouping
Citation: Guo G, Zhuang J, Zhang K, Zhou Z, Wang Y and Zhang Z (2022) Atypical Teratoid/Rhabdoid Tumor of the Central Nervous System in Children: Case Reports and Literature Review. Front. Surg. 9:864518. doi: 10.3389/fsurg.2022.864518
Received: 28 January 2022; Accepted: 25 April 2022;
Published: 16 May 2022.
Edited by:
Roberto Colasanti, University Hospital of Padua, ItalyReviewed by:
M. Adelita Vizcaino, Mayo Clinic, United StatesCopyright © 2022 Guo, Zhuang, Zhang, Zhou, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhen Zhang emhhbmd6aGVuQHNkZm11LmVkdS5jbg==
Specialty section: This article was submitted to Neurosurgery, a section of the journal Frontiers in Surgery
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.