 Seyed-Mostafa Razavi
Seyed-Mostafa Razavi Karen E. Lee
Karen E. Lee Benjamin E. Jin
Benjamin E. Jin Parvir S. Aujla
Parvir S. Aujla Sharareh Gholamin
Sharareh Gholamin Gordon Li
Gordon Li- 1Department of Neurosurgery, Stanford University School of Medicine, Stanford, CA, USA
- 2Institute of Stem Cell Biology and Regenerative Medicine, Stanford University School of Medicine, Stanford, CA, USA
Glioblastoma (GBM) is the most devastating brain tumor, with associated poor prognosis. Despite advances in surgery and chemoradiation, the survival of afflicted patients has not improved significantly in the past three decades. Immunotherapy has been heralded as a promising approach in treatment of various cancers; however, the immune privileged environment of the brain usually curbs the optimal expected response in central nervous system malignancies. In addition, GBM cells create an immunosuppressive microenvironment and employ various methods to escape immune surveillance. The purpose of this review is to highlight the strategies by which GBM cells evade the host immune system. Further understanding of these strategies and the biology of this tumor will pave the way for developing novel immunotherapeutic approaches for treatment of GBM.
Introduction
One of the challenges scientists face in the treatment of glioblastoma (GBM) is suboptimal responses to immunotherapy (1, 2). GBM is the most common adult brain tumor and patients usually succumb to the disease in <2 years. Despite significant improvement in chemo- and radiotherapy approaches for treatment of GBM, the median survival of one and a half years has not seen a significant change in the past few years (3, 4). Stagnation in the treatment of GBM is attributable to different challenges in therapy and our poor understating of both tumor biology and interactions with its microenvironment. Due to infiltrative growth, local microscopic metastases, and sometimes presence of multiple lesions at the time of diagnosis (5), complete surgical excision of the tumor is practically impossible and there is a strong need for new and effective therapies. With the introduction of immunotherapy as a novel and promising approach to cancer treatment, new hopes are raised for the management of brain tumors. However, as far as GBM is concerned, immunotherapeutic strategies so far have not been able to prompt a great change in survival. This article aims to review the mechanisms employed by GBM cells to suppress and evade the body’s immune responses. The collection of different molecules and mechanisms discussed in this review are summarized in Table 1 and a schematic representation of the GBM tumor cell interaction with the surrounding immune environment can be found in Figure 1.
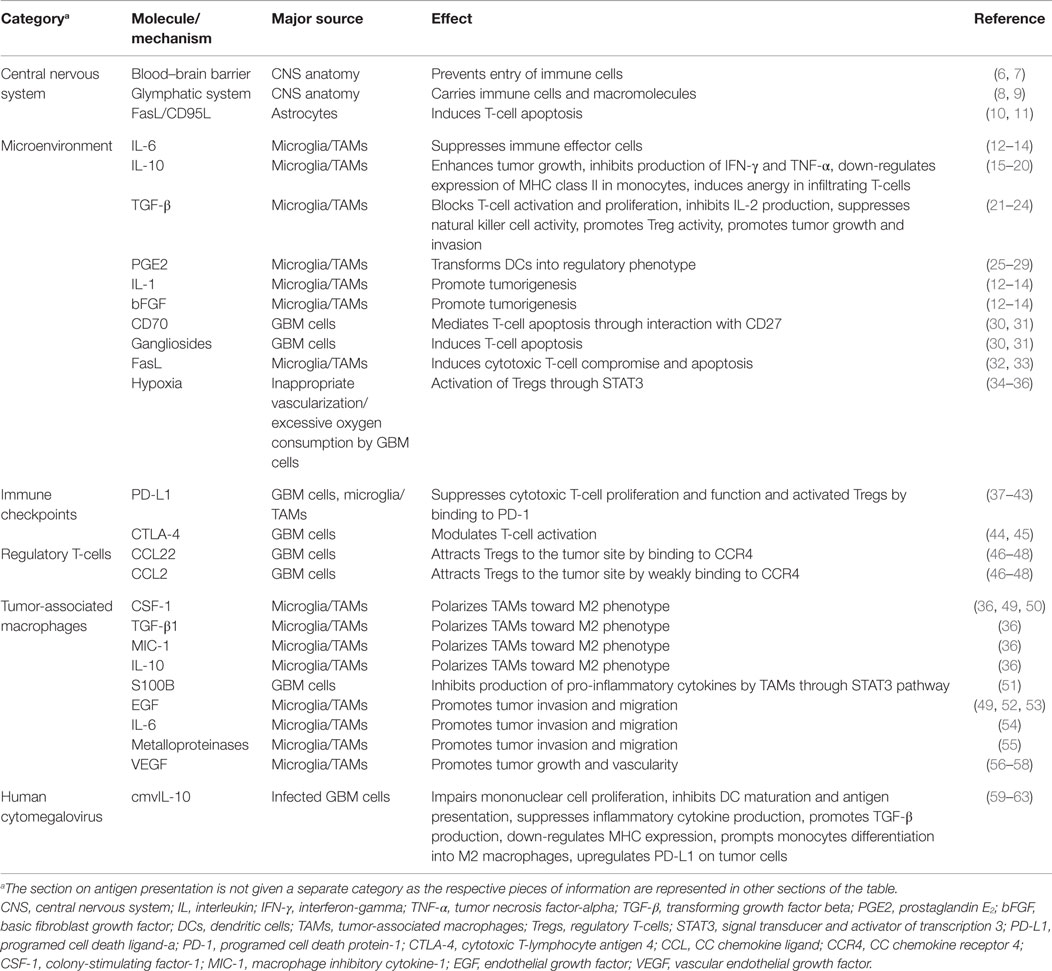
Table 1. Summary of mechanisms employed by GBM to evade the immune system.
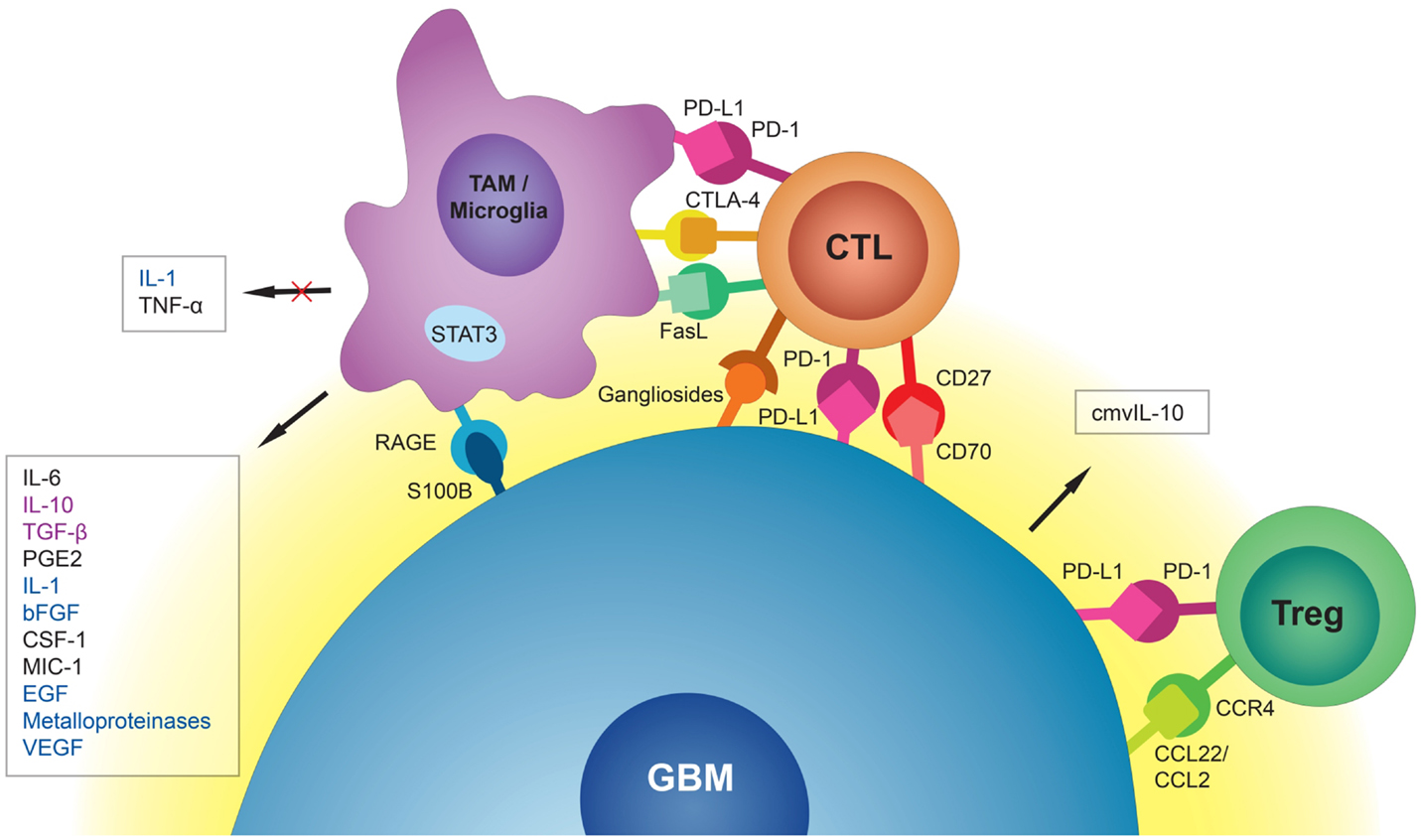
Figure 1. Schematic representation of the GBM tumor cell interaction with surrounding immune environment. Tumor-associated macrophages (TAMs) and microglia release immunosuppressive and pro-tumorigenic cytokines into the GBM microenvironment. They also induce cytotoxic T-cell (CTL) apoptosis via PD-L1, CTLA-4, and FasL. GBM cells, through interaction of S100B protein with receptor for advanced glycation end products (RAGE), inhibit production of immunostimulatory cytokines by TAMs and microglia. CMV-infected GBM cells secrete cmvIL-10 into their microenvironment with a range of immunosuppressive properties. Through interaction of CC chemokine ligand 22 (CCL22) and the weaker CC chemokine ligand 2 (CCL2) with CC chemokine receptor 4 (CCR4), GBM cells attract regulatory T-cells (Tregs) to the tumor site. Interaction of PD-L1 on GBM cells with PD-1 on Tregs promotes immunoregulatory functions of these cells. Immunosuppressive signals (black) could be distinguished from tumorigenic signals (blue) and signals that are both immunosuppressive and tumorigenic (purple).
Central Nervous System and the Immune System
The central nervous system (CNS), and more specifically the brain, has been historically presumed as the “immune privileged” organ of the body due to an intact blood–brain barrier (BBB). Absence of a usual lymphatic system and paucity of antigen-presenting cells (APCs) in brain tissue have also fueled this notion (6, 7). This assumption has been questioned in light of recent discoveries. The CNS possesses a functional “glymphatic system” located within the walls of dural sinuses and connected to the deep cervical lymph nodes capable of carrying immune cells and macromolecules (6, 8, 9). Immune cells can migrate into the brain parenchyma by chemotaxis, in which interferon-gamma (IFN-γ) and integrins play a major role (64, 65). Antigens can pass through walls of cerebral arteries and enter cervical lymph nodes through the Virchow–Robin perivascular spaces (66). By attaching to FcRn, a receptor found on a variety of body tissues, immunoglobulins are also able to cross the BBB via carrier-mediated transport (67, 68). APCs are present in many areas of the brain, including leptomeninges, ventricles, and perivascular spaces (69, 70). Via the rostral migratory stream, dendritic cells (DCs) can travel outside the brain and present antigens to T-cells located in the cervical lymph nodes (71). Peripheral immune cells can migrate to the CNS perivascular spaces but not into the brain parenchyma, thanks to the BBB. Tight junctions between foot processes of astrocytes form the physical BBB between perivascular spaces and parenchyma, while FasL/CD95L, expressed on these processes, induces apoptosis of T-cells that express the Fas receptor (10, 11). In disease states however, the integrity of the barrier is compromised, enabling immune cells to migrate past the BBB (72). During clinical trials for DC vaccines in patients with brain tumors, tumor-infiltrating lymphocytes have been observed in GBM samples (73, 74).
Microenvironment
Functional immunosuppression in the GBM microenvironment is characterized by production of immunosuppressive cytokines, inhibition of T-cell proliferation and effector responses, activation of FoxP3+ regulatory T-cells (Tregs), and tissue hypoxia. Immunosuppressive cytokines, including interleukin (IL)-6, IL-10, transforming growth factor-beta (TGF-β), and prostaglandin E2 (PGE2), as well as tumor-promoting cytokines, IL-1, and basic fibroblast growth factor (bFGF), are present in the GBM microenvironment and dampen the antitumor immune response (12–14). TGF-β promotes immunosuppression in GBM by blocking T-cell activation and proliferation, inhibiting IL-2 production, suppressing natural killer cell activity, and promoting Tregs (21). In addition, TGF-β has been shown to promote tumor growth and invasion by supporting GBM stem cells and enhancing angiogenesis (22–24).
Generally known as an immunosuppressive cytokine, IL-10 is found at high levels in a variety of neoplasms (15, 16). This cytokine is secreted by various immune cells (mainly macrophages, but also helper and cytotoxic T-cells, DCs, B-cells, monocytes, and mast cells) as well as GBM cells (16, 17). IL-10 associated with GBM is shown to enhance tumor growth (18), inhibit production of IFN-γ and tumor necrosis factor-alpha (TNF-α) by the immune system, downregulate expression of MHC class II in monocytes, and, via the co-stimulatory CD28-CD80/86 pathway, induce anergy in infiltrating T-cells (19, 20).
PGE2 is known to promote regulatory immune response in cancers and stimulate tumor cell growth (25). Together with TGF-β, it transforms DCs into a regulatory phenotype that suppresses T-cell proliferation (26, 27). In the GBM microenvironment, however, the concentration of PGE2 is not found to be high enough to suppress T-cell functions on its own (28, 29).
The GBM microenvironment also mediates immunosuppression via mechanisms that increase T-cell propensity to apoptosis through a cooperative interaction between CD70 and gangliosides (30, 31). CD70, through interaction with CD27, a member of TNF receptor family proteins, mediates apoptosis in T-cells. Inhibition of gangliosides, components of the plasma membrane that modulate signal transduction events, causes GBM cells to be significantly less efficient at inducing T-cell apoptosis. It has been shown that blocking both CD70 and ganglioside function produces an additive effect on provoking T-cell apoptosis (31). Programed cell death protein-1 ligand (PD-L1, B7-H1, or CD274), a potent immunosuppressive molecule, is expressed on microglia. The expression of PD-L1 on microglial cells is increased when in proximity to GBM cells that can induce T-cell apoptosis (37–39). The role of PD-L1 as an immune checkpoint is discussed further in the respective section. Another immunoinhibitory molecule expressed on tumor-associated microglia is FasL, which can induce cytotoxic T-cell compromise and apoptosis. Inhibition of FasL has resulted in an increased number of immune cells within the tumor (32, 33).
Lack of oxygen in the GBM microenvironment is the result of morphologically inappropriate neovascularization, irregular blood flow, and excessive consumption of oxygen from rapidly proliferating tumor cells. Hypoxia is a strong stimulus for expression of genes involved in tumor cell growth and angiogenesis (34). Specifically, the hypoxic GBM microenvironment activates signal transducer and activator of transcription 3 (STAT3), an immunosuppressive pathway and potent regulator of anti-inflammatory responses, which triggers the synthesis of hypoxia-inducible factor-1α (HIF-1α) that subsequently induces activation of Tregs and production of vascular endothelial growth factor (VEGF) (34). Tregs are modulators of the immune response, and VEGF is known for its immunosuppressive effects. Additionally, the hypoxic microenvironment triggers CNS macrophages to transform into tumor-associated macrophages (TAMs), which then adopt immunosuppressive and tumor-supportive phenotypes (M2). This transformation, via the STAT3 pathway, induces TAMs to promote angiogenesis and tumor cell invasion (35). Additionally, it has been shown that TAMs are modulated by GBM cancer stem cells (gCSCs) through induction of an immunosuppressive phenotype via the STAT3 pathway (36). Furthermore, since HIF-1α promotes gCSCs, hypoxia likely causes a feed-forward mechanism in tumor-mediated immunosuppression.
Antigen Presentation
Despite tremendous research, the mechanisms involved in developing tumor-sensitized immune effector cells are not well understood. Antigens from dead tumor cells are collected and processed by APCs and “cross-presented” on MHC class I to cytotoxic T-cells (75). Whether this antigen presentation for GBM occurs mainly in the brain or in the periphery is a subject of ongoing research (76). Microglia are the major myeloid immunocompetent cells of the brain, and scientists have elaborated their ability to present antigens to cytotoxic T-cells within the CNS (77, 78). However, the immunosuppressive microenvironment of GBM down-regulates MHC expression and compromises the antigen-presenting ability of microglia (79–83). GBM cells also stimulate secretion of IL-10 and inhibit production of TNF-α by microglia, further promoting suppression of the immune response (84). In fact, studies suggest that tumor-infiltrating DCs have a bigger part in GBM antigen presentation. In a 2008 study, Beauvillain et al. discovered that tumor-infiltrating DCs were more efficient than neonatal microglia in priming cytotoxic T-cells with exogenous antigens and could trigger higher levels of IL-2 and IFN-γ secretion by these cells (85). Presence of tumor-infiltrating DCs in the brain alongside microglia would prompt a better immune response in the CNS (77). Both glioma-associated antigen-pulsed and tumor-lysate-pulsed DCs have been successful in eliciting T-cell response in GBM patients (73, 74, 86). Wilms’ tumor 1 (WT1)-pulsed DC vaccine could improve neurological findings and shrink the tumor in a recent study (87). Nonetheless, tumor microenvironments would also blunt the action of tumor-infiltrating DCs and further investigation is needed to optimize this therapeutic technique (14, 20).
Macrophages are the major population of immune cells infiltrating solid tumors and GBM (88, 89). These cells are involved in antigen presentation, immune induction, cytotoxicity, removal of debris, regulation of inflammatory response, and thrombosis. Macrophages derived from monocyte precursors polarize into two distinct categories based on signals from the environment: M1, with a pro-inflammatory cytokine profile, and M2, with overall anti-inflammatory properties. Exposure to IFN-γ or bacterial lipopolysaccharide polarizes monocytes toward M1 macrophages. An alternate activation process happens by exposure to IL-4, resulting in the M2 category (90, 91). TAMs are believed to be of the latter population as they share many functions and surface proteins with M2 macrophages. While TAMs are known to be capable of cross-presenting tumor antigens to T-cells and prime antitumor immune response (92) due to limitations in histologic differentiation of TAMs from microglia, there is no definite answer to their importance in tumor antigen presentation in the brain (93, 94).
While mainly involved in humoral immune response, B-cells can also act as APCs and directly present antigens to T-cells via both MHC class I and II (95–97). Interaction of GBM cells with tumor-infiltrating B-cells has not been thoroughly investigated. Candolfi et al. studied the role of B-cells in a GBM murine model. After treatment of mice with intratumoral adenovector and immunostimulatory cytokines, B-cells were found to have remnants of tumor antigens in their cytoplasm and the ability to stimulate T-cell proliferation in vitro (98).
Tumor antigen presentation can also occur in peripheral lymph nodes. Activated T-cells have been found in the cervical lymph nodes of murine GBM models (99). Evidence exists that CNS antigens can move out of the CNS through perivascular spaces and be collected by resident DCs in cervical lymph nodes (100). Immunosuppressive cytokines secreted by GBM cells do not have a high enough systemic concentration to justify impairment of peripheral immune cell functions (101, 102). Engineered CTLs targeting IL-13 receptor 2 have shown promise in GBM in vivo models (103). Regardless of the underlying cause, vitiated cell-mediated immunity in GBM patients can compromise antigen presentation and T-cell activation even in the peripheral lymphatic tissue, adding to the challenges of immunotherapeutic efforts.
Immune Checkpoints
Immune checkpoint molecules, a group of co-stimulatory and co-inhibitory pathways that limit the function of immune system, have recently been targets for extensive research. By inhibition of immune checkpoints, researchers were able to reverse immunoresistance of cancer cells and activate the immune cells against tumors (104).
A major immune checkpoint molecule implicated in GBM immune evasion is PD-L1. Modulated by the PI(3)K–Akt–mTOR pathway (38), PD-L1 suppresses proliferation and function of cytotoxic T-cells and promotes Tregs activity by binding to programed cell death-1 (PD-1) (40). Expression of PD-L1 on tumor cells and T-cells is correlated with tumor grade (41) and poor survival of GBM patients (42). Microglia and TAMs are also known to express PD-L1 on their surface and at the same time promote PD-L1 expression on GBM cells (37, 43, 105). Collectively, these findings have made this immune checkpoint a prime target for GBM immunotherapy. Pre-clinical studies have been promising (106, 107) with plans for clinical trials on GBM patients currently under way.
Another immune checkpoint molecule, cytotoxic T-lymphocyte antigen 4 (CTLA-4) expressed on activated T-cells and Tregs could play a role in GBM immune evasion. Targeting CTLA-4 in GBM models might be able to enhance antitumor activity by T-cells (44, 45). Immune checkpoint inhibitors as targeted cancer therapeutics have shown promise in recent years with researchers trying to find new checkpoints as immunotherapeutic targets.
Regulatory T-Cells
Tregs, a small population of CD4+ T-cells that specifically express FoxP3 transcription factor, are a group of circulating lymphocytes with suppressive effects on various immune cells (108, 109). Other markers that help distinguish Treg subpopulations are CD25 (high-affinity IL-2 receptor), CTLA-4, and glucocorticoid-induced tumor necrosis factor receptor (110). Tregs can be divided into two major subpopulations based on their origin. Thymus-derived Tregs, developed from naïve CD4+ cells after antigen presentation in the thymus, express high levels of FoxP3. By contrast, under IL-10 and TGF-β signaling in the periphery, conventional CD4+ T-cells differentiate into peripherally induced Tregs with negligible FoxP3 expression (109). Tregs are commonly known to regulate immune response against tumor cells and to shift the tumor cytokine milieu toward immunosuppression. The presence of Tregs in GBM patients was described years ago (111), but their intricate function and interaction with other cells is a matter of ongoing investigation. A higher population of Tregs is demonstrated in GBM patients, reported to comprise up to 25% of tumor-infiltrating lymphocytes, and their abundance is associated with poor prognosis (112–114). Studies have revealed that glioma-associated Tregs are mostly of thymic origin rather than tumor-derived (115), suggesting that the abundance of Tregs in GBM is a result of chemotactic attraction of the thymus-derived subpopulation rather than local differentiation in the tumor (116). The CC chemokine ligand 22 (CCL22) and the weaker CC chemokine ligand 2 (CCL2) are among the first molecules revealed to attract Tregs to the tumor site by binding to CC chemokine receptor 4 (CCR4) (46, 47). Further studies revealed that blocking this receptor cannot completely abrogate Treg infiltration into GBM tumor mass, suggesting involvement of other secretory molecules in Treg chemoattraction (48). Peripherally derived Tregs are not believed to be the major population of Tregs in GBM, but presence of IL-10 and TGF-β at high levels in the GBM microenvironment suggests the possibly noticeable role of these cells in immune evasion of the tumor (14, 109). Further studies are needed to reveal the holistic picture of Tregs recruitment mechanisms into GBM.
Tumor-Associated Macrophages
Involvement of macrophages in GBM progression is a question to be further investigated. Recent studies provide significant evidence in contextual response of macrophages in tumor progression, highly modulated by the tumor microenvironment and tumor response to conventional treatments. Distinguishing TAMs from microglia in the brain is still a challenge for researchers. While TAMs are found to have a high expression of CD11b and CD45 compared to microglia, which have high expression of CD11b but low expression of CD45, there is still disagreement over a universally accepted histological marker that distinguishes the two cell types (117, 118).
Tumor-associated macrophages are usually linked to accelerated disease progression and poor outcome in cancer patients (119–121). Recently, several approaches have been investigated to abrogate tumor progression through ablating TAMs. Modulating the routes involved in macrophage polarization has provided insight into the regulatory effect of these cells in the GBM microenvironment (122).
Innate immunosuppressive properties of gliomas are derived from the regulatory cross-talk between M2 phenotype macrophages and tumor cells (93). Macrophages and microglia as dominant populations of tumor-infiltrating immune cells are, to a great extent, regulated by glioma initiating cells. Upon chemoattraction into the tumor environment (47, 49, 123, 124) with a high concentration of colony-stimulating factor-1 (CSF-1), TGF-β1, macrophage inhibitory cytokine-1 (MIC-1), and IL-10, TAMs are polarized toward the M2 phenotype, subsequently inhibiting their phagocytic ability and enhancing their capacity to inhibit cytotoxic T-cell proliferation and increase the effect of Tregs (36). Inhibiting the CSF-1 receptor can shift the polarization of TAMs away from M2, hinder their tumor-promoting functions, and increase survival of the GBM-bearing mice (50). Another protein recently found on GBM cells to induce innate immune suppression is S100B. Through interaction of S100B with receptor for advanced glycation end products (RAGE) on macrophages, GBM cells induce the STAT3 pathway in TAMs and inhibit the production of IL-1β, TNF-α, and other pro-inflammatory cytokines by these cells (51).
Tumor-associated macrophages and microglia can also play a role in GBM growth, invasion, and angiogenesis. Endothelial growth factor (EGF), CSF-1, TGF-β1, IL-6, and metalloproteinases originating from TAMs and microglia are instrumental for glioma invasion and migration (49, 52, 54, 55, 125). Inhibition of the EGF receptor (EGFR) on GBM cells has been associated with antiangiogenic and proapoptotic effects on the tumor (53). Inhibition of VEGF signaling in TAMs and microglia leads to decreased GBM growth and vascularity (56), but addition of anti-VEGF-A antibody to standard treatment has not improved patient survival (57, 58). Other populations of cells from myeloid lineage have been found in gliomas, including tumor-associated neutrophils, angiogenic monocytes, and immunosuppressive myelomonocytic cells, the importance of which is yet to be elucidated (126).
Human Cytomegalovirus Infection
Human cytomegalovirus (HCMV) is a β-herpesvirus implicated in GBM pathogenesis. Different studies have found HCMV genome in most tested GBM samples with no trace of the virus in surrounding brain tissue (59, 60). The role of HCMV in GBM development and pathogenesis is not yet clarified. What is clear though is that HCMV infection could play a role in immunosuppression in the context of GBM microenvironment.
Human cytomegalovirus genome encodes an IL-10 homolog (cmvIL-10) – a product of UL111A gene – that could impair mononuclear cell proliferation, inhibit DC maturation and antigen presentation, suppress inflammatory cytokine production, and down-regulate MHC expression (61, 62). Moreover, it has been demonstrated that cmvIL-10 prompts monocytes to differentiate into M2 macrophages and up-regulates the immunoinhibitory PD-L1 protein on GBM cells. Additionally, monocytes treated with cmvIL-10 produce TGF-β, augmenting the immunosuppressive microenvironment (63).
Summary and Future Prospects
The interaction of GBM with the immune system is intricate at every level. Any of the various mechanisms employed by this tumor to evade and suppress the immune response could be targeted with immunotherapy. To date, trials of immunotherapeutic modalities for GBM have not been as successful as promised. As different mechanisms of GBM immune resistance are revealed, scientists could have a better understanding of the pitfalls in GBM immunotherapy. GBM strategies for immune evasion are diverse and the key to successful immunotherapeutic treatment seems to be in targeting several pathways at the same time.
Author Contributions
SMR organized and wrote major parts of the article. KL helped with writing the article. BJ helped with writing the article. PA helped with designing the figure. SG helped with writing the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Xu LW, Chow KKH, Lim M, Li G. Current vaccine trials in glioblastoma: a review. J Immunol Res (2014) 2014:796856. doi: 10.1155/2014/796856
2. Ampie L, Woolf EC, Dardis C. Immunotherapeutic advancements for glioblastoma. Front Oncol (2015) 5:12. doi:10.3389/fonc.2015.00012
3. Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol (2012) 107:359–64. doi:10.1007/s11060-011-0749-4
4. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10:459–66. doi:10.1016/S1470-2045(09)70025-7
5. Thomas RP, Xu LW, Lober RM, Li G, Nagpal S. The incidence and significance of multiple lesions in glioblastoma. J Neurooncol (2013) 112:91–7. doi:10.1007/s11060-012-1030-1
6. Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol (2015) 36:569–77. doi:10.1016/j.it.2015.08.006
7. Forrester JV, Xu H, Lambe T, Cornall R. Immune privilege or privileged immunity? Mucosal Immunol (2008) 1:372–81. doi:10.1038/mi.2008.27
8. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523:337–41. doi:10.1038/nature14432
9. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med (2015) 212:991–9. doi:10.1084/jem.20142290
10. Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol (2008) 67:1113–21. doi:10.1097/NEN.0b013e31818f9ca8
11. Chow BW, Gu C. The molecular constituents of the blood-brain barrier. Trends Neurosci (2015) 38:598–608. doi:10.1016/j.tins.2015.08.003
12. Hao C, Parney IF, Roa WH, Turner J, Petruk KC, Ramsay DA. Cytokine and cytokine receptor mRNA expression in human glioblastomas: evidence of Th1, Th2 and Th3 cytokine dysregulation. Acta Neuropathol (2002) 103:171–8. doi:10.1007/s004010100448
13. Rodrigues JC, Gonzalez GC, Zhang L, Ibrahim G, Kelly JJ, Gustafson MP, et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol (2010) 12:351–65. doi:10.1093/neuonc/nop023
14. Jackson C, Ruzevick J, Phallen J, Belcaid Z, Lim M. Challenges in immunotherapy presented by the glioblastoma multiforme microenvironment. Clin Dev Immunol (2011) 2011:732413–20. doi:10.1155/2011/732413
15. Mittal SK, Roche PA. Suppression of antigen presentation by IL-10. Curr Opin Immunol (2015) 34:22–7. doi:10.1016/j.coi.2014.12.009
16. Trifunović J, Miller L, Debeljak Ž, Horvat V. Pathologic patterns of interleukin 10 expression – a review. Biochem Med (Zagreb) (2015) 25:36–48. doi:10.11613/BM.2015.004
17. Huettner C, Paulus W, Roggendorf W. Messenger RNA expression of the immunosuppressive cytokine IL-10 in human gliomas. Am J Pathol (1995) 146:317–22.
18. Huettner C, Czub S, Kerkau S, Roggendorf W, Tonn JC. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res (1997) 17:3217–24.
19. Van Meir EG. Cytokines and tumors of the central nervous system. Glia (1995) 15:264–88. doi:10.1002/glia.440150308
20. Strle K, Zhou JH, Shen WH, Broussard SR, Johnson RW, Freund GG, et al. Interleukin-10 in the brain. Crit Rev Immunol (2001) 21:427–49. doi:10.1615/CritRevImmunol.v21.i5.20
21. Fontana A, Bodmer S, Frei K, Malipiero U, Siepl C. Expression of TGF-beta 2 in human glioblastoma: a role in resistance to immune rejection? Ciba Found Symp (1991) 157:232–8.
22. Tritschler I, Gramatzki D, Capper D, Mittelbronn M, Meyermann R, Saharinen J, et al. Modulation of TGF-beta activity by latent TGF-beta-binding protein 1 in human malignant glioma cells. Int J Cancer (2009) 125:530–40. doi:10.1002/ijc.24443
23. Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci (2003) 116:217–24. doi:10.1242/jcs.00229
24. Kirkbride KC, Blobe GC. Inhibiting the TGF-beta signalling pathway as a means of cancer immunotherapy. Expert Opin Biol Ther (2003) 3:251–61. doi:10.1517/14712598.3.2.251
25. Sheng KC, Wright MD, Apostolopoulos V. Inflammatory mediators hold the key to dendritic cell suppression and tumor progression. Curr Med Chem (2011) 18:5507–18. doi:10.2174/092986711798347207
26. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med (2005) 202:919–29. doi:10.1084/jem.20050463
27. Whiteside TL, Mandapathil M, Schuler P. The role of the adenosinergic pathway in immunosuppression mediated by human regulatory T cells (Treg). Curr Med Chem (2011) 18:5217–23. doi:10.1007/s00262-010-0924-z
28. Couldwell WT, Dore-Duffy P, Apuzzo ML, Antel JP. Malignant glioma modulation of immune function: relative contribution of different soluble factors. J Neuroimmunol (1991) 33:89–96. doi:10.1016/0165-5728(91)90052-9
29. Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol (1999) 100:216–32. doi:10.1016/S0165-5728(99)00203-9
30. Wischhusen J, Jung G, Radovanovic I, Beier C, Steinbach JP, Rimner A, et al. Identification of CD70-mediated apoptosis of immune effector cells as a novel immune escape pathway of human glioblastoma. Cancer Res (2002) 62:2592–9.
31. Chahlavi A, Rayman P, Richmond AL, Biswas K, Zhang R, Vogelbaum M, et al. Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Cancer Res (2005) 65:5428–38. doi:10.1158/0008-5472.CAN-04-4395
32. Badie B, Schartner J, Prabakaran S, Paul J, Vorpahl J. Expression of Fas ligand by microglia: possible role in glioma immune evasion. J Neuroimmunol (2001) 120:19–24. doi:10.1016/S0165-5728(01)00361-7
33. Walker DG, Chuah T, Rist MJ, Pender MP. T-cell apoptosis in human glioblastoma multiforme: implications for immunotherapy. J Neuroimmunol (2006) 175:59–68. doi:10.1016/j.jneuroim.2006.03.006
34. Wei J, Wu A, Kong L-Y, Wang Y, Fuller G, Fokt I, et al. Hypoxia potentiates glioma-mediated immunosuppression. PLoS One (2011) 6:e16195. doi:10.1371/journal.pone.0016195
35. Lin EY, Li J-F, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66:11238–46. doi:10.1158/0008-5472.CAN-06-1278
36. Wu A, Wei J, Kong L-Y, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol (2010) 12:1113–25. doi:10.1093/neuonc/noq082
37. Magnus T, Schreiner B, Korn T, Jack C, Guo H, Antel J, et al. Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. J Neurosci (2005) 25:2537–46. doi:10.1523/JNEUROSCI.4794-04.2005
38. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med (2007) 13:84–8. doi:10.1038/nm1517
39. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi:10.1038/nm730
40. Lipson EJ, Forde PM, Hammers H-J, Emens LA, Taube JM, Topalian SL. Antagonists of PD-1 and PD-L1 in cancer treatment. Semin Oncol (2015) 42:587–600. doi:10.1053/j.seminoncol.2015.05.013
41. Wilmotte R, Burkhardt K, Kindler V, Belkouch M-C, Dussex G, Tribolet Nd, et al. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport (2005) 16:1081–5. doi:10.1097/00001756-200507130-00010
42. Wei B, Wang L, Zhao X, Du C, Guo Y, Sun Z. The upregulation of programmed death 1 on peripheral blood T cells of glioma is correlated with disease progression. Tumour Biol (2014) 35:2923–9. doi:10.1007/s13277-013-1376-9
43. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res (2013) 19:3165–75. doi:10.1158/1078-0432.CCR-12-3314
44. Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res (2007) 13:2158–67. doi:10.1158/1078-0432.CCR-06-2070
45. Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLoS One (2014) 9:e101764. doi:10.1371/journal.pone.0101764
46. Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother (2008) 57:123–31. doi:10.1007/s00262-007-0336-x
47. Desbaillets I, Tada M, de Tribolet N, Diserens AC, Hamou MF, Van Meir EG. Human astrocytomas and glioblastomas express monocyte chemoattractant protein-1 (MCP-1) in vivo and in vitro. Int J Cancer (1994) 58:240–7. doi:10.1002/ijc.2910580216
48. Crane CA, Ahn BJ, Han SJ, Parsa AT. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol (2012) 14:584–95. doi:10.1093/neuonc/nos014
49. Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med (2012) 18:519–27. doi:10.2119/molmed.2011.00217
50. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) 19:1264–72. doi:10.1038/nm.3337
51. Zhang L, Liu W, Alizadeh D, Zhao D, Farrukh O, Lin J, et al. S100B attenuates microglia activation in gliomas: possible role of STAT3 pathway. Glia (2011) 59:486–98. doi:10.1002/glia.21118
52. Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci USA (2009) 106:12530–5. doi:10.1073/pnas.0804273106
53. Camara-Quintana JQ, Nitta RT, Li G. Pathology: commonly monitored glioblastoma markers: EFGR, EGFRvIII, PTEN, and MGMT. Neurosurg Clin N Am (2012) 23:237–46. doi:10.1016/j.nec.2012.01.011
54. Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, Yong VW. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis (2012) 33:312–9. doi:10.1093/carcin/bgr289
55. Lin H-C, Song T-Y, Hu M-L. S-adenosylhomocysteine promotes the invasion of C6 glioma cells via increased secretion of matrix metalloproteinase-2 in murine microglial BV2 cells. Toxicol Sci (2009) 112:322–30. doi:10.1093/toxsci/kfp218
56. Kerber M, Reiss Y, Wickersheim A, Jugold M, Kiessling F, Heil M, et al. Flt-1 signaling in macrophages promotes glioma growth in vivo. Cancer Res (2008) 68:7342–51. doi:10.1158/0008-5472.CAN-07-6241
57. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med (2014) 370:709–22. doi:10.1056/NEJMoa1308345
58. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med (2014) 370:699–708. doi:10.1056/NEJMoa1308573
59. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol (2008) 10:10–8. doi:10.1215/15228517-2007-035
60. Scheurer ME, Bondy ML, Aldape KD, Albrecht T, El-Zein R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol (2008) 116:79–86. doi:10.1007/s00401-008-0359-1
61. Chang WLW, Baumgarth N, Yu D, Barry PA. Human cytomegalovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. J Virol (2004) 78:8720–31. doi:10.1128/JVI.78.16.8720-8731.2004
62. Spencer JV, Lockridge KM, Barry PA, Lin G, Tsang M, Penfold MET, et al. Potent immunosuppressive activities of cytomegalovirus-encoded interleukin-10. J Virol (2002) 76:1285–92. doi:10.1128/JVI.76.3.1285-1292.2002
63. Dziurzynski K, Wei J, Qiao W, Hatiboglu MA, Kong L-Y, Wu A, et al. Glioma-associated cytomegalovirus mediates subversion of the monocyte lineage to a tumor propagating phenotype. Clin Cancer Res (2011) 17:4642–9. doi:10.1158/1078-0432.CCR-11-0414
64. Gorbachev AV, Kobayashi H, Kudo D, Tannenbaum CS, Finke JH, Shu S, et al. CXC chemokine ligand 9/monokine induced by IFN-gamma production by tumor cells is critical for T cell-mediated suppression of cutaneous tumors. J Immunol (2007) 178:2278–86. doi:10.4049/jimmunol.178.4.2278
65. Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, Aurrand-Lions M, et al. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity (2005) 22:175–84. doi:10.1016/j.immuni.2004.12.008
66. Cserr HF, Harling-Berg CJ, Knopf PM. Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol (1992) 2:269–76. doi:10.1111/j.1750-3639.1992.tb00703.x
67. Ohtsuki S, Terasaki T. Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm Res (2007) 24:1745–58. doi:10.1007/s11095-007-9374-5
68. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol (2007) 7:715–25. doi:10.1038/nri2155
69. D’Agostino PM, Gottfried-Blackmore A, Anandasabapathy N, Bulloch K. Brain dendritic cells: biology and pathology. Acta Neuropathol (2012) 124:599–614. doi:10.1007/s00401-012-1018-0
70. Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol (2007) 28:5–11. doi:10.1016/j.it.2006.11.007
71. Mohammad MG, Tsai VWW, Ruitenberg MJ, Hassanpour M, Li H, Hart PH, et al. Immune cell trafficking from the brain maintains CNS immune tolerance. J Clin Invest (2014) 124:1228–41. doi:10.1172/JCI71544
72. Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol (2012) 33:579–89. doi:10.1016/j.it.2012.07.004
73. Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res (2011) 17:1603–15. doi:10.1158/1078-0432.CCR-10-2563
74. Prins RM, Wang X, Soto H, Young E, Lisiero DN, Fong B, et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J Immunother (2013) 36:152–7. doi:10.1097/CJI.0b013e3182811ae4
75. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol (2012) 12:557–69. doi:10.1038/nri3254
76. McDonnell AM, Robinson BWS, Currie AJ. Tumor antigen cross-presentation and the dendritic cell: where it all begins? Clin Dev Immunol (2010) 2010:539519. doi:10.1155/2010/539519
77. Jarry U, Jeannin P, Pineau L, Donnou S, Delneste Y, Couez D. Efficiently stimulated adult microglia cross-prime naive CD8+ T cells injected in the brain. Eur J Immunol (2013) 43:1173–84. doi:10.1002/eji.201243040
78. Flügel A, Labeur MS, Grasbon-Frodl EM, Kreutzberg GW, Graeber MB. Microglia only weakly present glioma antigen to cytotoxic T cells. Int J Dev Neurosci (1999) 17:547–56. doi:10.1016/S0736-5748(99)00020-9
79. Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia (2002) 40:252–9. doi:10.1002/glia.10147
80. Kjellman C, Olofsson SP, Hansson O, Schantz Von T, Lindvall M, Nilsson I, et al. Expression of TGF-beta isoforms, TGF-beta receptors, and SMAD molecules at different stages of human glioma. Int J Cancer (2000) 89:251–8. doi:10.1002/1097-0215(20000520)89:3<251:AID-IJC7>3.0.CO;2-5
81. Mieczkowski J, Kocyk M, Nauman P, Gabrusiewicz K, Sielska MG, Przanowski P, et al. Down-regulation of IKKβ expression in glioma-infiltrating microglia/macrophages is associated with defective inflammatory/immune gene responses in glioblastoma. Oncotarget (2015) 6(32):33077–90. doi:10.18632/oncotarget.5310
82. Schartner JM, Hagar AR, Van Handel M, Zhang L, Nadkarni N, Badie B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia (2005) 51:279–85. doi:10.1002/glia.20201
83. Badie B, Bartley B, Schartner J. Differential expression of MHC class II and B7 costimulatory molecules by microglia in rodent gliomas. J Neuroimmunol (2002) 133:39–45. doi:10.1016/S0165-5728(02)00350-8
84. Kostianovsky AM, Maier LM, Anderson RC, Bruce JN, Anderson DE. Astrocytic regulation of human monocytic/microglial activation. J Immunol (2008) 181:5425–32. doi:10.4049/jimmunol.181.8.5425
85. Beauvillain C, Donnou S, Jarry U, Scotet M, Gascan H, Delneste Y, et al. Neonatal and adult microglia cross-present exogenous antigens. Glia (2008) 56:69–77. doi:10.1002/glia.20565
86. Lasky JL, Panosyan EH, Plant A, Davidson T, Yong WH, Prins RM, et al. Autologous tumor lysate-pulsed dendritic cell immunotherapy for pediatric patients with newly diagnosed or recurrent high-grade gliomas. Anticancer Res (2013) 33:2047–56.
87. Sakai K, Shimodaira S, Maejima S, Udagawa N, Sano K, Higuchi Y, et al. Dendritic cell-based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent malignant glioma. J Neurosurg (2015) 123:989–97. doi:10.3171/2015.1.JNS141554
88. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol (2002) 196:254–65. doi:10.1002/path.1027
89. Morimura T, Neuchrist C, Kitz K, Budka H, Scheiner O, Kraft D, et al. Monocyte subpopulations in human gliomas: expression of Fc and complement receptors and correlation with tumor proliferation. Acta Neuropathol (1990) 80:287–94. doi:10.1007/BF00294647
90. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol (2006) 177:7303–11. doi:10.4049/jimmunol.177.10.7303
91. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol (2010) 11:889–96. doi:10.1038/ni.1937
92. Tseng D, Volkmer J-P, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci USA (2013) 110:11103–8. doi:10.1073/pnas.1305569110
93. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol (2006) 8:261–79. doi:10.1215/15228517-2006-008
94. Randolph GJ, Jakubzick C, Qu C. Antigen presentation by monocytes and monocyte-derived cells. Curr Opin Immunol (2008) 20:52–60. doi:10.1016/j.coi.2007.10.010
95. Carrasco YR, Batista FD. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity (2007) 27:160–71. doi:10.1016/j.immuni.2007.06.007
96. Hon H, Oran A, Brocker T, Jacob J. B lymphocytes participate in cross-presentation of antigen following gene gun vaccination. J Immunol (2005) 174:5233–42. doi:10.4049/jimmunol.174.9.5233
97. Clark MR, Massenburg D, Siemasko K, Hou P, Zhang M. B-cell antigen receptor signaling requirements for targeting antigen to the MHC class II presentation pathway. Curr Opin Immunol (2004) 16:382–7. doi:10.1016/j.coi.2004.03.007
98. Candolfi M, Curtin JF, Yagiz K, Assi H, Wibowo MK, Alzadeh GE, et al. B cells are critical to T-cell-mediated antitumor immunity induced by a combined immune-stimulatory/conditionally cytotoxic therapy for glioblastoma. Neoplasia (2011) 13:947–60. doi:10.1593/neo.11024
99. Okamoto Y, Yamashita J, Hasegawa M, Fujisawa H, Yamashima T, Hashimoto T, et al. Cervical lymph nodes play the role of regional lymph nodes in brain tumour immunity in rats. Neuropathol Appl Neurobiol (1999) 25:113–22. doi:10.1046/j.1365-2990.1999.00165.x
100. Laman JD, Weller RO. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J Neuroimmune Pharmacol (2013) 8:840–56. doi:10.1007/s11481-013-9470-8
101. Waziri A. Glioblastoma-derived mechanisms of systemic immunosuppression. Neurosurg Clin N Am (2010) 21:31–42. doi:10.1016/j.nec.2009.08.005
102. Gustafson MP, Lin Y, New KC, Bulur PA, O’Neill BP, Gastineau DA, et al. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol (2010) 12:631–44. doi:10.1093/neuonc/noq001
103. Kahlon KS, Brown C, Cooper LJN, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res (2004) 64:9160–6. doi:10.1158/0008-5472.CAN-04-0454
104. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27:450–61. doi:10.1016/j.ccell.2015.03.001
105. Nduom EK, Wei J, Yaghi NK, Huang N, Kong L-Y, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol (2016) 18(2):195–205. doi:10.1093/neuonc/nov172
106. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys (2013) 86:343–9. doi:10.1016/j.ijrobp.2012.12.025
107. Tang X, Li Q, Zhu Y, Zheng D, Dai J, Ni W, et al. The advantages of PD1 activating chimeric receptor (PD1-ACR) engineered lymphocytes for PDL1(+) cancer therapy. Am J Transl Res (2015) 7:460–73.
108. Whiteside TL. Induced regulatory T cells in inhibitory microenvironments created by cancer. Expert Opin Biol Ther (2014) 14:1411–25. doi:10.1517/14712598.2014.927432
109. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6:295–307. doi:10.1038/nri1806
110. Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev (2008) 223:371–90. doi:10.1111/j.1600-065X.2008.00637.x
111. Elliott LH, Brooks WH, Roszman TL. Activation of immunoregulatory lymphocytes obtained from patients with malignant gliomas. J Neurosurg (1987) 67:231–6. doi:10.3171/jns.1987.67.2.0231
112. Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res (2008) 14:5166–72. doi:10.1158/1078-0432.CCR-08-0320
113. Waziri A, Killory B, Ogden AT, Canoll P, Anderson RCE, Kent SC, et al. Preferential in situ CD4+CD56+ T cell activation and expansion within human glioblastoma. J Immunol (2008) 180:7673–80. doi:10.4049/jimmunol.180.11.7673
114. Ebert LM, Tan BS, Browning J, Svobodova S, Russell SE, Kirkpatrick N, et al. The regulatory T cell-associated transcription factor FoxP3 is expressed by tumor cells. Cancer Res (2008) 68:3001–9. doi:10.1158/0008-5472.CAN-07-5664
115. Wainwright DA, Nigam P, Thaci B, Dey M, Lesniak MS. Recent developments on immunotherapy for brain cancer. Expert Opin Emerg Drugs (2012) 17:181–202. doi:10.1517/14728214.2012.679929
116. Jacobs JFM, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJM, Wesseling P, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol (2010) 225:195–9. doi:10.1016/j.jneuroim.2010.05.020
117. Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol (1995) 154:4309–21.
118. Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery (2000) 46:957–61. doi:10.1097/00006123-200004000-00035
119. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer (2004) 4:71–8. doi:10.1038/nrc1256
120. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res (2006) 66:605–12. doi:10.1158/0008-5472.CAN-05-4005
121. Siveen KS, Kuttan G. Role of macrophages in tumour progression. Immunol Lett (2009) 123:97–102. doi:10.1016/j.imlet.2009.02.011
122. da Fonseca AC, Badie B. Microglia and macrophages in malignant gliomas: recent discoveries and implications for promising therapies. Clin Dev Immunol (2013) 2013:264124–5. doi:10.1155/2013/264124
123. Badie B, Schartner J, Klaver J, Vorpahl J. In vitro modulation of microglia motility by glioma cells is mediated by hepatocyte growth factor/scatter factor. Neurosurgery (1999) 44:1077–82. doi:10.1097/00006123-199905000-00075
124. Held-Feindt J, Hattermann K, Müerköster SS, Wedderkopp H, Knerlich-Lukoschus F, Ungefroren H, et al. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp Cell Res (2010) 316:1553–66. doi:10.1016/j.yexcr.2010.02.018
125. Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, et al. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion – an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene (2008) 27:918–30. doi:10.1038/sj.onc.1210683
Keywords: glioblastoma, immune system, immunosuppression, immune evasion, cancer immunotherapy
Citation: Razavi SM, Lee KE, Jin BE, Aujla PS, Gholamin S and Li G (2016) Immune Evasion Strategies of Glioblastoma. Front. Surg. 3:11. doi: 10.3389/fsurg.2016.00011
Received: 04 November 2015; Accepted: 10 February 2016;
Published: 02 March 2016
Edited by:
William Tupper Couldwell, University of Utah, USACopyright: © 2016 Razavi, Lee, Jin, Aujla, Gholamin and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gordon Li, Z29yZG9ubGlAc3RhbmZvcmQuZWR1