 Geoffrey P. Dobson
Geoffrey P. Dobson- Heart, Trauma and Sepsis Research Laboratory, Australian Institute of Tropical Health and Medicine, College of Medicine and Dentistry, James Cook University, Townsville, QLD, Australia
Despite a technically perfect procedure, surgical stress can determine the success or failure of an operation. Surgical trauma is often referred to as the “neglected step-child” of global health in terms of patient numbers, mortality, morbidity, and costs. A staggering 234 million major surgeries are performed every year, and depending upon country and institution, up to 4% of patients will die before leaving hospital, up to 15% will have serious post-operative morbidity, and 5–15% will be readmitted within 30 days. These percentages equate to around 1000 deaths and 4000 major complications every hour, and it has been estimated that 50% may be preventable. New frontline drugs are urgently required to make major surgery safer for the patient and more predictable for the surgeon. We review the basic physiology of the stress response from neuroendocrine to genomic systems, and discuss the paucity of clinical data supporting the use of statins, beta-adrenergic blockers and calcium-channel blockers. Since cardiac-related complications are the most common, particularly in the elderly, a key strategy would be to improve ventricular-arterial coupling to safeguard the endothelium and maintain tissue oxygenation. Reduced O2 supply is associated with glycocalyx shedding, decreased endothelial barrier function, fluid leakage, inflammation, and coagulopathy. A healthy endothelium may prevent these “secondary hit” complications, including possibly immunosuppression. Thus, the four pillars of whole body resynchronization during surgical trauma, and targets for new therapies, are: (1) the CNS, (2) the heart, (3) arterial supply and venous return functions, and (4) the endothelium. This is termed the Central-Cardio-Vascular-Endothelium (CCVE) coupling hypothesis. Since similar sterile injury cascades exist in critical illness, accidental trauma, hemorrhage, cardiac arrest, infection and burns, new drugs that improve CCVE coupling may find wide utility in civilian and military medicine.
As the patient goes to the operating room and anesthesia is induced, trauma is suffered and convalescence begins.
Moore (1) p. 291
Introduction
Major Surgery, Trauma and the Stress Response
Major surgery is defined “as any intervention in a hospital operating theater that requires incision, excision, manipulation or suturing of tissue occurring and requiring regional or general anesthesia or profound sedation to control pain” (2). Surgical trauma is defined as any injury produced by or related to major surgery. As recognized by surgeon Francis Moore over 60 years ago, trauma begins early in the operating room before general anesthesia and before the first surgical incision (1). The extent of injury will depend on the type and duration of surgery and anesthesia, the presence of cardiopulmonary bypass (CPB), a patient’s age, gender, pre-existing health status, medication profile, fluid loading and post-operative pain (3–6). Age is a particular concern because many older patients present with multiple comorbidities and have decreased reserves to cope with the stress of surgery (7–9) while the very young are often more sensitive to general anesthetics (10).
The stress response can be broadly defined as the body’s response to a “stressor,” which may be an injury, hemorrhage, infection or a burn. It is a physiological reaction of profound importance in nature and medicine. From an evolutionary perspective, the stress response would have been triggered by the central nervous system (CNS) to protect against macroscopic “external” threats (e.g., fight-or-flight response) followed by an underlying “internal” response to promote survival, which included activation of the acute inflammatory or immune systems to initiate wound repair and protect against pathogens (11). Rapid protective mechansisms would have conferred a profound “survival fitness” from events such as escaping predators, hunting accidents, or defending a territory. Although highly protective by design, there are chinks in the stress response’s “internal” evolutionary armor. Under certain conditions, such as major surgery, instead of inflammation being self-limiting and restorative, the response can exceed the body’s internal tolerances and become “overexpressed” and lead to further injury. The working hypothesis underpinning this review is that if surgical stress can be halted or diminished, patient outcomes will be improved since most cells in the body are genetically programed in that direction of repair. It is about “helping the body help itself.”
Objective and Scope of the Problem
Surgery has recently been referred to as the “neglected stepchild of global health”
Farmer and Kim (12) and Rose et al. (13)
Surgical stress is a global problem. In a recent modeling study, Weiser and colleagues estimated there are around 234 million non-cardiac surgical procedures performed annually around the world (2), 40 million in the USA (14) and 19 million in Europe (15). Of these patients, around 30% have an increased risk of cardiovascular complications, or 70 million patients per year. In the USA, Leape and colleagues recently analyzed nearly 500,000 surgeries and reported a 30-day readmission rate of 5.7%, with over 30% being surgical site infections (16). In another study, Dimick and Ghaferi analyzed nearly 60,000 patients from 112 Department of Veterans Affairs hospitals, and found that the overall 30-day readmission rate was two-times higher (11.9%) than Leape’s study, mostly from higher surgical site infections (56%) (17). These data, and others, led Bartels to write that: “The magnitude of all-cause perioperative mortality would make it the number three cause of death in the USA” (18). Moreover, in 28 European nations, Pearse and colleagues conducted a 7-day cohort study among 46,539 non-cardiac surgery patients from 498 hospitals, and found that 4% of total patients died before hospital discharge, and 8% were admitted to critical care with a median length stay of 1–2 days (19). The group concluded that new strategies are urgently required to improve perioperative outcomes because 73% of these surgical deaths occurred before admittance to critical care (19).
Combining the available data, surgery-related deaths range from 0.4 to 4% and post-operative morbidity from 5 to 15% (2, 19). Thus, up to 9 million patients die each year during or immediately after major surgery, and up to twice this number have post-operative complications. Since major surgery addresses around 11% of the global burden of disease (20), it can be deduced from the available statistics that: (1) surgical trauma already is in global crisis, (2) the large differences in mortality and morbidity within and across countries and institutions are unacceptable, and (3) around half of the deaths and complications may be preventable. New drug therapies could save up to 500 lives every hour.
Homeostasis, Trauma and the “Steady-State”
The coordinated physiological processes which maintain most of the steady states in the organism are so complex and so peculiar to living beings - involving, as they may, the brain and nerves, the heart, lungs, kidneys and spleen, all working cooperatively - that I have suggested a special designation for these states, homeostasis.
Cannon (21) p. 20–24
Todays’ understanding of the stress response has its roots firmly embedded in Walter B. Cannon’s concept of homeostasis. Cannon formally introduced the concept in 1926, which he cited in his 1929 review: Organization for Physiological Homeostasis (22, 23). Cannon’s homeostasis was built on Pfluger’s concept of “steady-state” (1877), Claude Bernard’s concept of “milieu intérieur” (1878), and Richet’s “stability of the organism” (1900) (22, 24). Cannon argued that a living organism was a system in dynamic state of constancy, with its constituent parts and processes being actively maintained in constant balance despite external fluctuations. This “balance” required a continuous exchange of matter and energy between the organism and the environment, and internal regulatory mechanisms to keep it in-check. Thus, Cannon’s homeostasis was not an equilibrium state, but a steady-state. This distinction is important because all living organisms are open systems that rely on a net flow of matter and energy with time whereas a strict equilibrium state, by definition, has no net flux (25). A stress, injury, or sickness was now seen in new light, and viewed as a challenge to the body’s dynamic steady-state. Thus a major goal of any drug therapy or treatment was to restore that balance.
Early History of the Surgical Stress Response
George Crile and “Stress-Free” Surgery
In the early 1900s, the idea of lowering surgical stress was spearheaded by neurosurgeon George W. Crile (1864–1943) at the Cleveland Clinic (26). Crile’s operative technique to improve recovery was revolutionary and included lightly anesthetizing the patient with mask inhalation of nitrous oxide and oxygen, and infiltrating all tissues with a dilute local anesthetic procaine before the first incision (27). General anesthesia was insufficient to reduce the patient’s stress response, and so he proposed the word anoci-association (anoci; noxious or harmful and associations; stimuli) to describe the potentially harmful “stressors” during surgery. For a “shockless” operation, Crile suggested a combination of sedation, local and regional anesthesia to reduce pain and improve recovery (28). Crile’s called his concept “stress-free anesthesia” (29), which formed the basis of modern “preemptive anesthesia” (see Optimizing Anesthesia and Analgesia to Reduce Pain and the Stress Response). Neurosurgeon Harvey Cushing (1869–1939) extended Crile’s revolutionary ideas and promoted the use of regional blocks before removing ether anesthesia from the patient to guarantee an optimal postoperative recovery (30). Cushing also confirmed Crile’s observation that surgical shock could be prevented by the careful monitoring of blood pressure and avoiding the “stressors” associated with surgery (30, 31). These century-old ideas and practices from two giants in medicine form the basis of modern-day anesthesiology.
Cuthbertson’s “Ebb and Flow” Injury Hypothesis and the HPA Axis
In the early 1930s, Scottish Chemist David Cuthbertson was among the first to characterize the “stress response to injury” (32). Cannon had already suggested a role for an activated sympathetic nervous system with adrenal secretions to increase cardiac output (CO) and mobilize energy stores in the “fight-or-flight” response (22, 33). However, Cuthbertson found in his patients with long bone fractures a dramatic rise in the loss of nitrogen (as urea), potassium, phosphorus, sulfur, creatine and creatinine compared with volunteers with no injury (32). This was groundbreaking because it suggested that trauma itself induced a “stress response.” Cuthbertson divided the body’s response into two quantifiable events: (1) An early “ebb” phase, which began 2 h post-injury and lasted 2–3 days; this was associated with a decrease in CO, reduced tissue perfusion, a lower metabolic rate and glucose intolerance, and: (2) a second “flow” phase lasting days and weeks, that was characterized by an increase in metabolic rate, a hyperdynamic circulation (higher CO, respiratory rate), hyperglycemia, a negative nitrogen balance, and muscle wasting (32, 34). The extent and duration of both “ebb” and “flow” phases depended upon the severity of the injury. Today, whole body energy consumption following major surgery (e.g., abdominal) can increase up to 1.5 times (up to 5 ml O2/kg/min) (35).
Over the next few decades, the body’s stress response to injury was identified to be under “neural control via the hypothalamus and the hypophyseal portal vessels of the pituitary stalk” (36). This grouping of the “responses” within the CNS and adrenal glands was termed the hypothalamic–pituitary-adrenal (HPA) axis (37). Cuthbertson lean muscle wasting was now viewed as a CNS-linked-catecholamine response, which could be blunted by beta-adrenergic, but not alpha-adrenergic blockers (3). Today, the HPA axis and catecholamines have many diverse functions from controlling CO and metabolism to selectively regulating the compliance, capacitance and blood volume of the systemic, splanchnic and venous vasculature (38). Within seconds of catecholamine release, nearly two-thirds of the splanchnic blood volume (~800 ml) can be autotransfused into the systemic circulation during times of stress (38). Thus catecholamine surges and changes in blood volume and shifts during surgery may be a potential target to improve patient outcomes following surgery (see Injury, Inflammation and Multiple Organ Failure, The First Incision, Effects of Anesthesia on the Surgical Stress Response, and Effects of Major Surgery on Other Organs) (7–9).
Injury, Inflammation and Multiple Organ Failure
Another major milestone in unraveling the stress response was the discovery of the relationship between injury, inflammation, infection and organ dysfunction (39). The history of inflammation dates back to the ancient Egyptians and Greeks, and the Roman medical writer Celsus in the first century AD who characterized injury by rubor (redness), tumor (swelling), calor (heat), dolor (pain) and functio lesa (loss of function) (40). Along with advances in immunology and molecular biology in the 1970s, a mechanistic link between injury and inflammation was serendipitously found by surgeon Arthur Baue who noticed a progressive, systemic organ failure in his intensive care patients (41). Two years later, Eiserman and colleagues termed this phenomenon multiple organ failure (MOF) and described it as a “fatal expression of uncontrolled infection” (42). Infection, however, was only part of the story.
In the 1980s, a German team led by Faist (43) and a Dutch group led by Goris (44) found that MOF developed in trauma patients, but curiously these patients did not have an infection. The new “stressor” was an “autodestructive inflammatory response,” and carried a mortality of over 50% (45). This devastating affliction was termed the systemic inflammatory response syndrome (SIRS) and was accompanied with delayed immunosuppression (46). Mild immunosuppression, like mild stress, is generally not harmful but if it progresses, secondary infections may occur followed by late MOF and death (47). Late MOF became so prevalent that Deitch wrote: “MOF has reached epidemic proportions in most intensive care units and is fast becoming the most common cause of death in the surgical intensive care unit” (39). Today, MOF remains the greatest contributor to late-trauma mortality and morbidity than any other cause (47).
Currently, animal studies to investigate MOF and SIRS are based on “one-hit” and “two-hit” models proposed by Ernest and Fred Moore (45, 48). In the “one hit” model, the initial stressor (trauma, hemorrhage, sepsis, or burn) is so massive that the subject is overwhelmed with SIRS and succumbs to MOF (49). In the “two-hit” model, less severe injury slowly develops into MOF from reactivation of an earlier “minor” inflammatory response (“secondary-hit”), which may cascade into a life-threatening situation (50). While significant overlap exists, the two models, and other variants, attribute MOF to an unchecked inflammation, cardiac depression, HPA axis activation, sympathetic discharge, coagulopathy, and impairment of mitochondrial function (47, 51). After severe to catastrophic trauma, loss of whole body homeostasis can develop into a lethal triad, which involves hypothermia, acidosis, and coagulopathy, and death is imminent (50, 52).
The First Incision
Besides cardiopulmonary bypass, the cutting, burning, fracturing and stretching of tissues induce an additional repair process, mediated by inflammatory responses and cell regeneration, which consumes energy and reserves. … The burden on some of them is substantial: a single sternotomy might reach the equivalence of a long bone fracture.
Prêtre (53)
Local Tissue Trauma
The first surgical incision induces localized injury to tissues, afferent nerves, pain receptors and blood vessels (Figure 1). Nerve damage leads to afferent signals from the injury site to the brain and stimulation of the HPA axis (Figures 1 and 2). Local coagulation necrosis, microparticle release, endothelial damage, immune cell activation, localized cell ischemia, edema and metabolic dysfunction all contribute to a succession of rapidly cascading events from a local to a systemic phenomenon (54–60). Hypoxia also contributes to injury, in part, through the activation of hypoxia-inducible factor (HIF-1) and potentiation of NF-kappaB, a master regulator of genes involved in innate immunity, inflammation, and apoptosis (59, 61). The degree of trauma will be affected by the invasive nature of surgical procedure (56). Laparoscopic procedures, for example, have lower levels of trauma compared with open procedures, and lower surgical stress (62). Open surgeries include a median sternotomy, thoracotomy, laparotomy, abdominal hysterectomy and orthopedic surgery (54, 63). Cardiac surgeon René Prêtre equated the severity of a median sternotomy in cardiothoracic surgery with a long bone fracture (quote above).
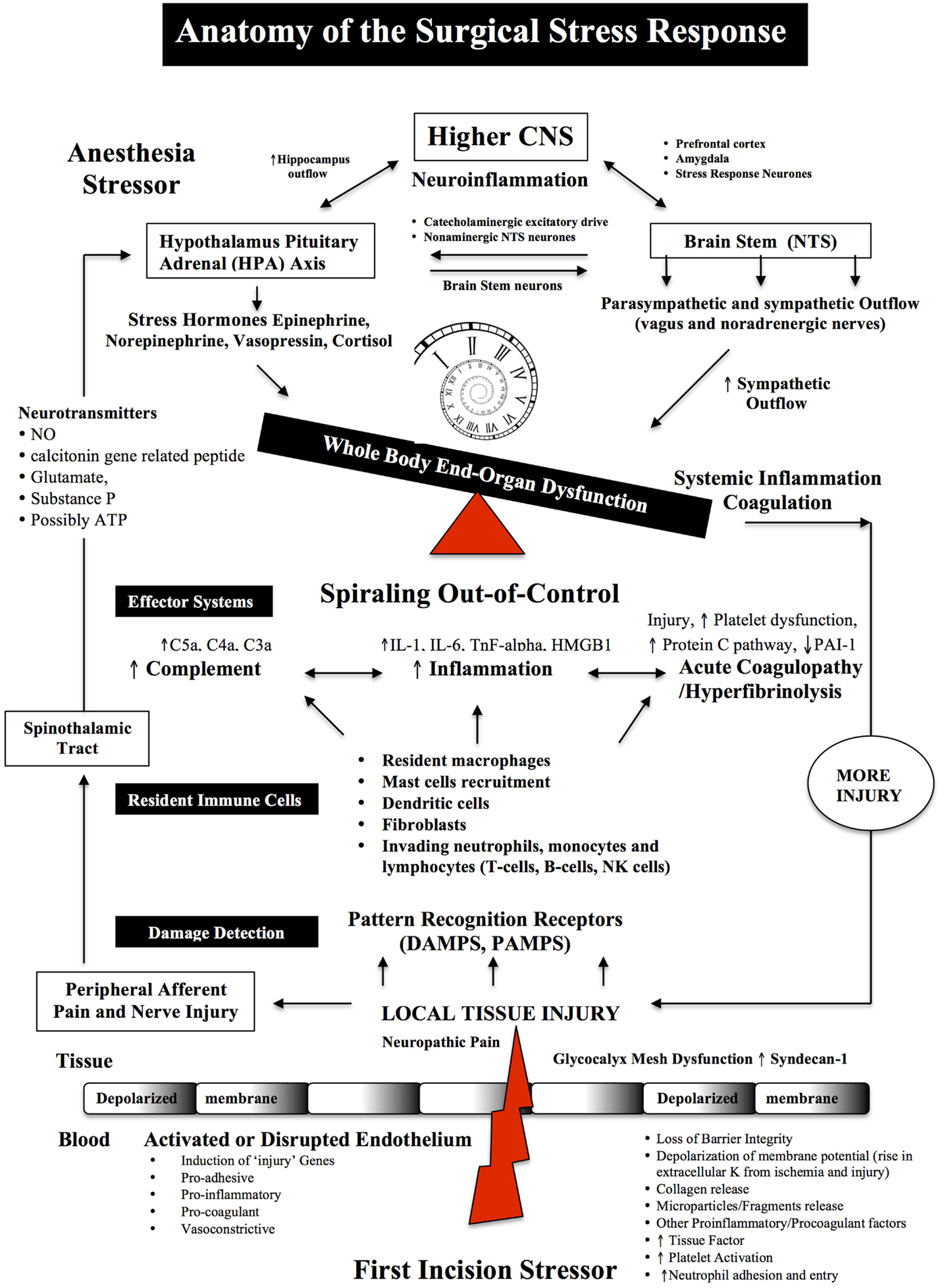
Figure 1. Anatomy of the Surgical Stress Response. Surgical stress triggers a wide and varied response at multiple levels depending on the type and duration of surgery, anesthesia and the patient’s age, gender and prior health status. The early drivers of the stress response are sterile local injury, afferent nerve cell firing, activation of the Hypothalamic-Pituitary-Adrenal (HPA) axis, Nucleus Tractus Solitarus (NTS), endothelial dysfunction and inflammation. Damage signals (also termed danger-associated molecular patterns or DAMPs and alarmins, e.g., heat shock proteins, adenosine, HMGB-1) are generated from tissue injury and detected by resident and non-resident immune cells. The key pro-inflammatory cytokines are IL-1, IL-6, and TnF-alpha and a complex interactions with complement. The primary goal of the acute immune response is wound healing and to prevent pathogen invasion. It is a restorative process that involves four phases: coagulation, inflammation, proliferation, and remodeling. Each phase of repair is predominately mediated by immune cells, cytokines, chemokines, transcription, and post-translational pathways (Tables 1 and 2). However, during major trauma, the early repair process can be overexpressed and lead to further injury, if not held in check. Peripheral nerve injury and pain induce afferent mediators and neurotransmitters to the spinal cord and central nervous system (CNS) and produce stress hormones, which exacerbate the stress response during major surgery.
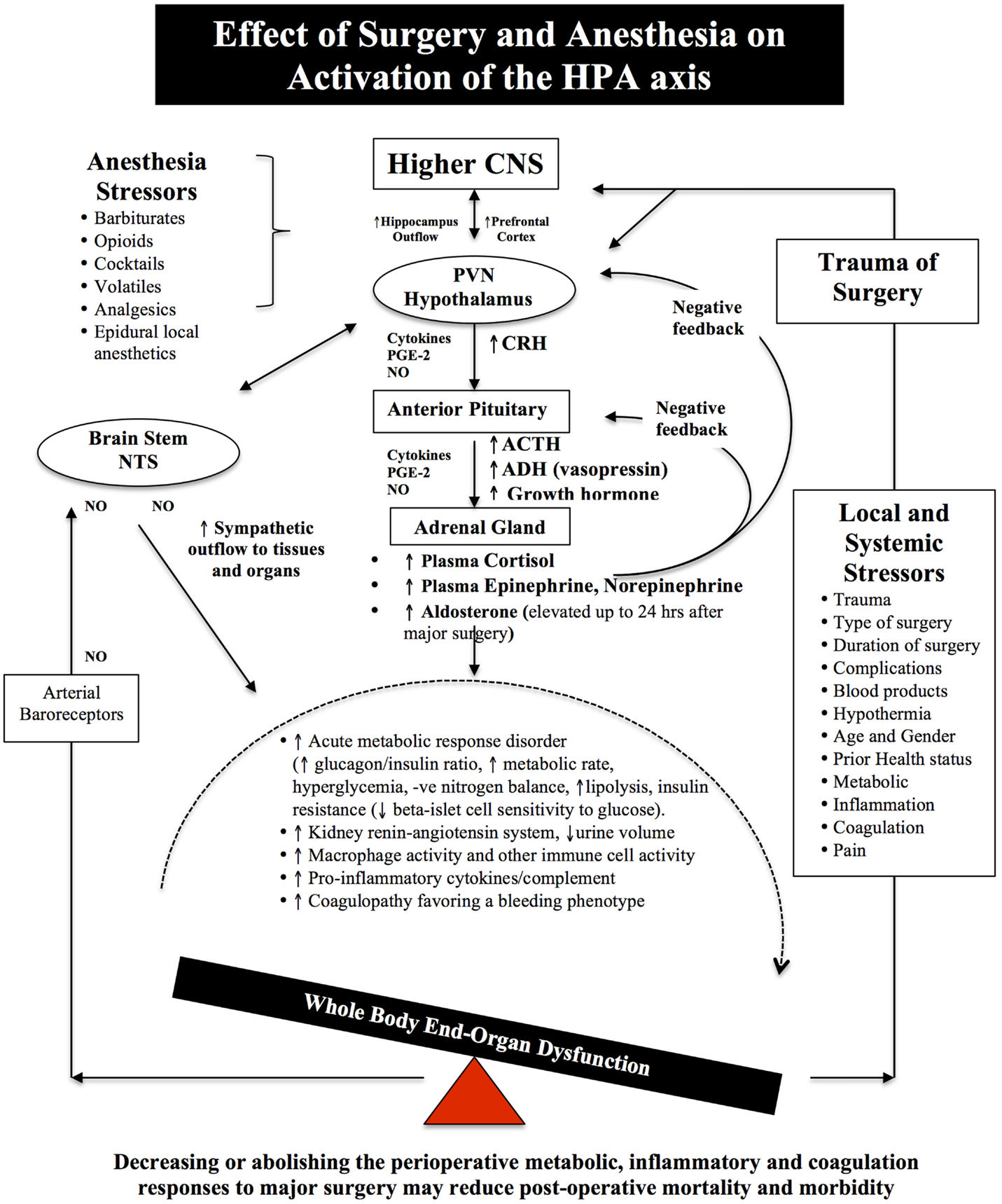
Figure 2. Schematic of the HPA axis and the Stress Response to Surgery. Different anesthetics have different effects on the HPA axis and immune system (see Effects of Anesthesia on the Surgical Stress Response). During surgical stress, activation of the HPA axis is controlled by a relatively small number of neurons located in the paraventricular nucleus (PVN) of the hypothalamus. These neurons release neural factors, such as corticotrophin-releasing hormone (CRH) and arginine vasopressin (AVP) into the hypophyseal portal circulation, which stimulates the anterior pituitary gland to release ACTH into the blood and activates the adrenal gland to release the stress hormones, catecholamines, and cortisol. Under normal exposure to stress, the HPA axis is held in check via multiple negative feedback mechanisms. However, during major surgery, trauma, infection or burns, imbalances occur and the action of stress hormones are potentiated by cytokines IL-1beta, IL-6 and TNF-alpha, prostaglandin-2 (PGE-2), and nitric oxide (NO), which predispose the body to further injury from ischemia, inflammation, and coagulopathy. Older surgical patients appear to be more vulnerable to surgical stress because their hypothalamus and pituitary are less sensitive to negative feedback from both cortisol and ACTH (7–9). The medullary Nucleus Tractus Solitarus (NTS) is also influenced by the stress response as it receives sensory neural inputs from the arterial baroreceptors, integrates this information with the hypothalamus, and other parts of the brain, and regulates the sympathetic and parasysmpathetic outflows to the body (7, 64–72).
Activation of Resident Immune Cells
At the incision site, resident and non-resident immune cells are the “first responders” to danger signals (Table 1) (60, 73–77). Resident immune cells are the macrophages, mast cells, dendritic cells, fibroblasts and lymphocytes (via lymph), and the non-resident blood-borne cells such as neutrophils and lymphocytes, which all produce a variety of cytokines, chemokines, proteases, leukotrienes, and nitric oxide (NO) in response to local injury (Tables 1 and 2) (75, 78–80). Mast cells are normally found in close association with blood vessels, lymphatic vessels, pain receptors and nerves, and on activation they degranulate with the release of inflammatory cytokines, histamine, and prostaglandins that initiate a neuropathic and nociceptive pain response and local changes in blood flow (60, 81–83). Circulating platelets also contribute to immune surveillance, damage control and amplification response to local injury (84–86). At the injury site, tissue factor (TF) activates platelets and coagulation pathways to reduce further blood loss. Normally, an effective inflammatory response isolates the area of injury, clears the injured cells/debris or kills invading pathogens and restores tissue function. However, the stress response can quickly become over-expressed and promotes further injury.
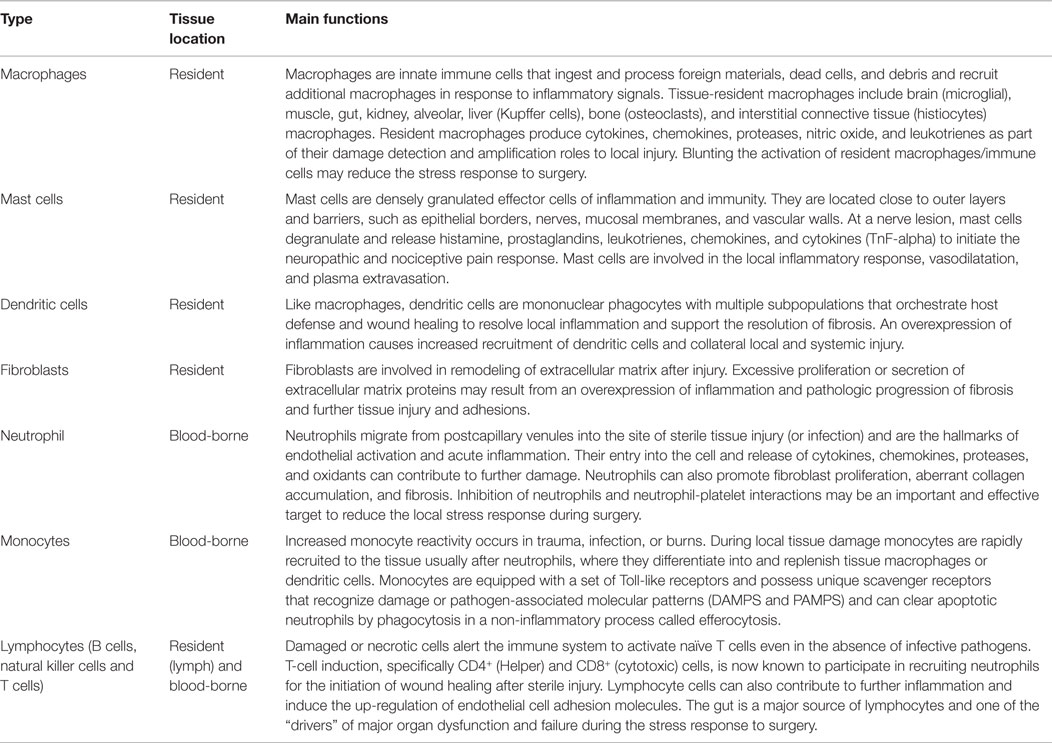
Table 1. Immune cells involved in the detection, integration and healing following localized sterile injury.

Table 2. Major cytokines, chemokines and danger signals involved in the acute stress response to sterile injury.
Inflammatory Cytokines: IL-1, IL-6, and TnF-Alpha
The main pro-inflammatory cytokines are IL-1, IL-6, and TnF-alpha (Table 2) (7, 76, 80, 87–91). IL-1 beta can directly activate nociceptive fibers within 60 s, and indirectly elicit the production of prostaglandins in response to injury (60, 78, 81, 88, 92). In addition to local effects, plasma IL-1, IL-6, and TnF-alpha stimulate the HPA axis leading to the release of cortisol and catecholamines and systematization of the stress response (68, 93). Interestingly, plasma IL-6 levels appear to correlate with the severity of surgical injury and post-operative complications (94). In 2006, Ishibashi and colleagues showed that IL-6 levels correlated with the length of a surgical incision (1.0 cm vs. 3.0 cm) with the larger incision leading to doubling of IL-6 values at 3 and 6 h (95). During colorectal surgery, plasma IL-6 and P-selectin were also shown to increase during surgery and peaked at day one after surgery (96). IL-6 also appears to play a dual role by providing a later “immunological brake” to restore homeostatic balance by up-regulating anti-inflammatory mediators and cytokine inhibitors, such as prostaglandin PGI2, IL-1 receptor antagonist (IL-1ra), IL-10, and transforming growth factor (TGF-b) (40, 77, 97). Thus, resolution of the local and systemic inflammatory responses is an emerging area of increasing complexity and involves immune cells, cytokines, the endothelium, and a multitude of negative feedback new mediators such as the resolvins, lipoxins, maresins, and NF-kappa beta pathways (40).
TNF-alpha is another major cytokine and participates in a diverse number of cellular and molecular events such as neutrophil-endothelial adhesive interactions, vasodilatation, microvascular leakage, edema, and oxidative stress (Table 2) (98–100). TnF-alpha, and other cytokines, activate the hepatic acute phase protein response with the release of C-reactive protein (CRP), pro-calcitonin, and C-3 complement factor (70, 92). Complement activation is an integral part of the stress response to surgical injury and intersects and amplifies the coagulation activation pathways (see Complement Injury Cascade) (84). Like IL-6, TNF-alpha serves a dual role in resolution of the inflammatory response via signaling through the TNF-R2 (p75) receptor (101). The timing of these pro- and anti-inflammatory roles during surgical stress and recovery are not well understood.
Local Coagulation and Systemic Coagulopathy
Local clotting, as mentioned, is initiated by exposure of platelets to the subendothelial components and TF activation of plasma factors VII/VIIa via the extrinsic pathway, which in turn leads to the generation of thrombin, fibrin deposition, and clot formation (102–104). Other proteins, such as von Willebrand factor, facilitate the binding of platelets to the injured vessel wall. TF is expressed by the injured blood vessel wall (adventitial fibroblasts, smooth muscle cells and pericytes), platelets, activated endothelium, macrophages, neutrophils, circulating microparticles, and epithelial cells (105, 106). Importantly, TF is essential for hemostasis, but uncontrolled expression of TF can lead to coagulopathy (107). A second clotting pathway, known as the Intrinsic Pathway, appears to be more important in the growth, amplification and elongation of clot formation, rather than the TF-activated initiation (108). The intrinsic pathway involves clotting factors within the blood itself combined with cell fragments, microparticles or damage-associated molecular pattern molecules (109). The relationships between the extrinsic and intrinsic pathways, and their different timings and systemic manifestation during major surgery are not well understood.
In major surgery, a common complication is hyperfibrinolysis (110). According to the STS database, up to 50% of cardiac surgery patients require blood product transfusions (111), and 5–7% will lose in excess of 2 l in the first day postoperatively (112, 113). About 50% of blood loss is due to identifiable surgical bleeding, and the other 50% is due to a hypocoagulopathy associated with surgical trauma, heparinization, and CPB (112, 113). Surgical hypothermia further exacerbates hemorrhagic, cardiovascular, and infectious complications compared to normothermia (114). In non-cardiac surgery, coagulopathic perioperative bleeding is also a major problem. In a recent prospective, multicenter observational cohort study of 1134 consecutive patients with coronary stents, 9.5% experienced postoperative hemorrhagic complications after non-cardiac surgery, and 12% of these died (115). In general, trauma-induced postoperative coagulopathy is associated with extended hospital stays and three times higher healthcare costs (116). The impact of the first incision on triggering the local inflammatory and coagulation responses appears to be overlooked, as there is surprisingly little information on the nature and timing of these events, and when they become systemically expressed. New pharmacological therapies for prevention or early correction of inflammation and coagulopathy are urgently sought.
Complement Injury Cascade
Complement activation is another integral part of the stress response to surgical injury (Figure 1). The blood complement system comprises a group of at least 30 soluble plasma and membrane bound proteins and can be activated in two main ways; (1) as a component of the innate immune cascade in response to aseptic tissue injury (i.e., antibodies or T cell receptors not involved), and (2) from the adaptive immune response when antibodies (IgG or IgM) binds to antigen at the surface of a cell (117–121). Along with the local inflammatory and coagulation responses to injury, the complement system is part of an ancient innate immune response to trauma, ischemia, hemorrhage, burns, infection, and autoimmunity (120, 121).
Complement proteins are synthesized in the liver and by extra-hepatic tissue macrophages, blood monocytes, and epithelial cells. Local production and activation of complement makes a significant contribution to local tissue inflammatory injury (necrosis), coagulopathy, and augmentation of the adaptive immune response (117, 122). The two main mechanisms of complement cell damage include membrane attack complex (MAC) and cell bound ligands C4b and C3b that activate innate immune cells such as neutrophils bearing complement receptor (117), and second, from small activation fragments known as anaphylatoxins (C3 and C5 convertases) that further promote the influx and activation of neutrophils, cytokines, chemokines, adhesion molecules, and cascade mechanisms (117). Tissue ischemia and reperfusion can also activate complement by exposing cell phospholipids and mitochondrial proteins that are recognized by natural and locally occurring antibodies and autoantibodies (118, 120, 122, 123). CRP, an acute phase pro-inflammatory mediator and activator of the classical complement pathway, has been shown to increase up to 1000-fold in human plasma following tissue injury (120, 124).
In cardiac surgery, the timing of complement system activation was thought to occur only when the patient’s blood was in contact with pro-inflammatory plastic tubing of the CPB circuit (125, 126). However, Gu and colleagues found that plasma complement and inflammation occurred soon after the first chest incision (127). This was further supported by their observation that complement was activated in those patients who did not receive CPB but related to the first incision (127). In addition, a smaller anterolateral thoracotomy was associated with reduced complement activation, and lower IL-6, compared to the median sternotomy. Interestingly, IL-6 was not elevated in plasma until the end of the operation, and Gu’s group suggested that the “incision” trigger was a tissue type plasminogen activator, which is known to stimulate complement (127). Unfortunately, the study did not measure plasma IL-1, TnF-alpha, or other markers of inflammation and coagulation at baseline and before or after CBP. As with other components of the innate immune response, excessive activation of complement pathways damages healthy tissues from “friendly fire,” and exacerbates the surgical insult (121). Further research is required to better understand the mechanisms of hyper-expression, the timing of systemic organ damage and failure, and to develop novel therapeutic strategies, such as complement inhibitors, to possibly improve surgical outcomes (126).
Role of Histamine, Nerve Growth Factors and Local Opioids
Histamine is locally released from mast cell granules, and nerve growth factor is released from damaged nerves, which activate peripheral nerves that either terminate in the brain or spinal cord dorsal horn resulting in pain facilitation (128). Histamine release also appears to be directly related to changes in the cardiovascular system that are often seen during anesthesia (129). Simultaneously, endogenous analgesic mechanisms are activated including anti-inflammatory cytokines, endocannabinoids, and opioid peptides. Opioid peptides such as endorphins, enkephalins, and dynorphins are produced by immune cells such as leukocytes and can be released locally in the inflamed tissue on stimulation with IL-1 or from corticotropin releasing factor that drives the body’s response to stress (130). Following release, opioid peptides bind to receptors on peripheral sensory neurons and produce analgesia in animal models and humans (131).
Effects of Anesthesia on the Surgical Stress Response
It is critical to recognize that certain stressors may act on the brain even in the unconscious state - this is true of the anesthetic itself and the hormonal, metabolic and inflammatory mediators of the surgical stress response.
Borsook et al. (132) p. 607
Blunting the Stress Hormones
Notwithstanding the difficulty in separating the stress effects of anesthesia from the surgery itself, there is a general consensus that most anesthetics reduce the neuroendocrine response. The degree of reduction is difficult to assess and most likely depends upon the anesthetic’s mode of action, dose and duration of use (Figure 2). As mentioned in Section “Cuthbertson’s “Ebb and Flow” Injury Hypothesis and the HPA axis,” the central integrative “hub” controlling the stress response is the HPA axis that controls catecholamine and cortisol production, and the Nucleus Tractus Solitarus (NTS) that control sympathetic-parasympathetic outflows (7, 64–72). In addition, prostaglandin and NO production in higher centers, such as the cortex, hippocampus, and amygdala and periphery, are all involved in the regulation of HPA axis and NTS under basal and stress conditions (71) (Figures 1 and 2).
Blunting the stress response includes a reduction in the production of the following major hormones or autocoids as possible clinical endpoints:
• pituitary hormones: adrenocorticotropic hormone (ACTH), growth hormone (GH) and antidiuretic vasopressin, beta-endorphins;
• adrenal catabolic hormones: cortisol and catecholamines;
• pancreatic hormone: glucagon;
• prostaglandins (e.g., PGE-2).
The most common general anesthetics include the barbiturates (thiopental), opioids (fentanyl, remifentanil, sufentanil), benzodiazepines (midazolam), dissociative anesthetic agents (ketamine) and intravenous propfofol, etomidate, and clonidine (7, 68). No sedation or drug cocktail, however, offers complete “stress-free anesthesia” (133). Opioids appear to be the most powerful suppressors of HPA, and particularly short-acting etomidate, which suppresses corticosteroid production, cortisol release, catecholamines, and aldosterone release, which may last between 8 and 22 h after surgery (68, 134). Propofol-remifentanil cocktail also blunts the stress response but to a lesser degree (135). Despite their individual effects, many of the current anesthetic combinations, particularly used in cardiac surgery, still lead to persistent elevations of 2– 6 fold in cortisol, GH, and norepinephrine lasting around 2 days (136). The volatile anesthetics halothane, isoflurane, sevoflurane, and nitrous oxide have been reported to be less effective in blunting the stress response (137, 138). However, sevoflurane appears to be more effective at blunting the inflammatory response than isoflurane (139). Other anesthetics, such as thiopental, ketamine, and opioids, possess some anti-inflammatory and anti-oxidant properties (140), however, prospective, randomized clinical trials are required to examine if these differences are clinically significant (138, 141).
Optimizing Anesthesia and Analgesia to Reduce Pain and the Stress Response
Nearly 90% of surgical patients claim to experience moderate-to-severe postoperative pain after major surgery (142). Neuropathic and nociceptive pain contributes to and amplifies the stress response by increasing inflammation, coagulation disorders, organ hypoperfusion, decreasing wound healing, and possibly cognitive dysfunction (77, 143, 144). To address this problem, the concept of pre-emptive analgesia has been introduced in recent years (142, 145, 146), which combines general anesthesia with epidural and/or intravenous (IV) infusion agents (e.g., opioids, local anesthetics). This analgesic strategy also reduces the need for steroids, which have been associated with increased infections (147, 148), and opioid use, which reduces GI post-operative complications (149). The recent emphasis on pre-emptive analgesia for procedure-specific pain management via CNS and peripheral desensitization appears to be improving post-operative care (150), and similar to George Crile’s anoci-association and “stress-free surgery” proposed over 100 years ago (see George Crile and “Stress-Free” Surgery).
Stress-Induced Diabetes
Stress hormones from the HPA axis and increased sympathetic outflows from the NTS also lead to hyperglycemia and insulin resistance (Figure 2), which may persist for several days from higher levels of cortisol, catecholamines, GH, and pro-inflammatory cytokines (66, 72, 151). This condition has been termed the “diabetes of injury” (5, 6) or “critical illness diabetes,” and is common after severe surgical trauma, multiple injury trauma, burns, or infection (5). Surgical stress leads to net glucose production, a decrease in uptake and tissue utilization and/or a decrease in pancreatic β-cell responsiveness to insulin signaling (68, 152, 153). It is also associated with endothelial activation, coagulopathy, cardiac dysfunction, arrhythmias, immunosuppression, and slower wound healing times (72, 154). In addition, those patients who are already diabetic and undergo cardiac surgery, have a 24% higher risk of readmission for cardiac-related issues, deep sternal wound infections and post-operative strokes, and a 44% higher risk for rehospitalization for any cause (155).
Cognitive Dysfunction
The pathogenesis of postoperative cognitive dysfunction (POCD) is multifactorial and future studies should focus on evaluating the role of postoperative sleep disturbances, inflammatory stress responses, pain and environmental factors.
Krenk et al. (156), p. 951
A Persistent Complication of Major Surgery
Cognitive dysfunction remains a continuing complication in the aged and very young. In patients 60 years or older undergoing cardiac surgery, cognitive dysfunction occurs in 30–52% of cases (157), and may last up to 5 years (158, 159). In non-cardiac surgery patients, Price and colleagues reported cognitive dysfunction in 56% of cases, and 25% in older patients after 3 months (160). A number of systematic reviews generally support these findings but cautioned that differences may also reflect differences in the neuropsychological tests to assess cognitive dysfunction (161). In pediatric patients after cardiac surgery, 5–10% acquire some form of cerebral dysfunction (162), although it usually resolves faster than in older patients (156). In addition, major surgery in very low-birth-weight infants is independently associated with a greater than 50% increased risk of death or neurodevelopmental impairment and anesthetics are believed to be involved (163).
The factors responsible for cognitive dysfunction are complex and include transient hypoperfusion, hypoxia, ischemia-reperfusion injury, low preoperative hemoglobin levels, fluid overload, blood transfusions, microemboli, perioperative pain, hyperglycemia, and large swings in CNS temperature (77, 157, 164–167). However, the underlying factor believed to be responsible for cognitive dysfunction underpinning all these appears to be inflammation (Figure 3).
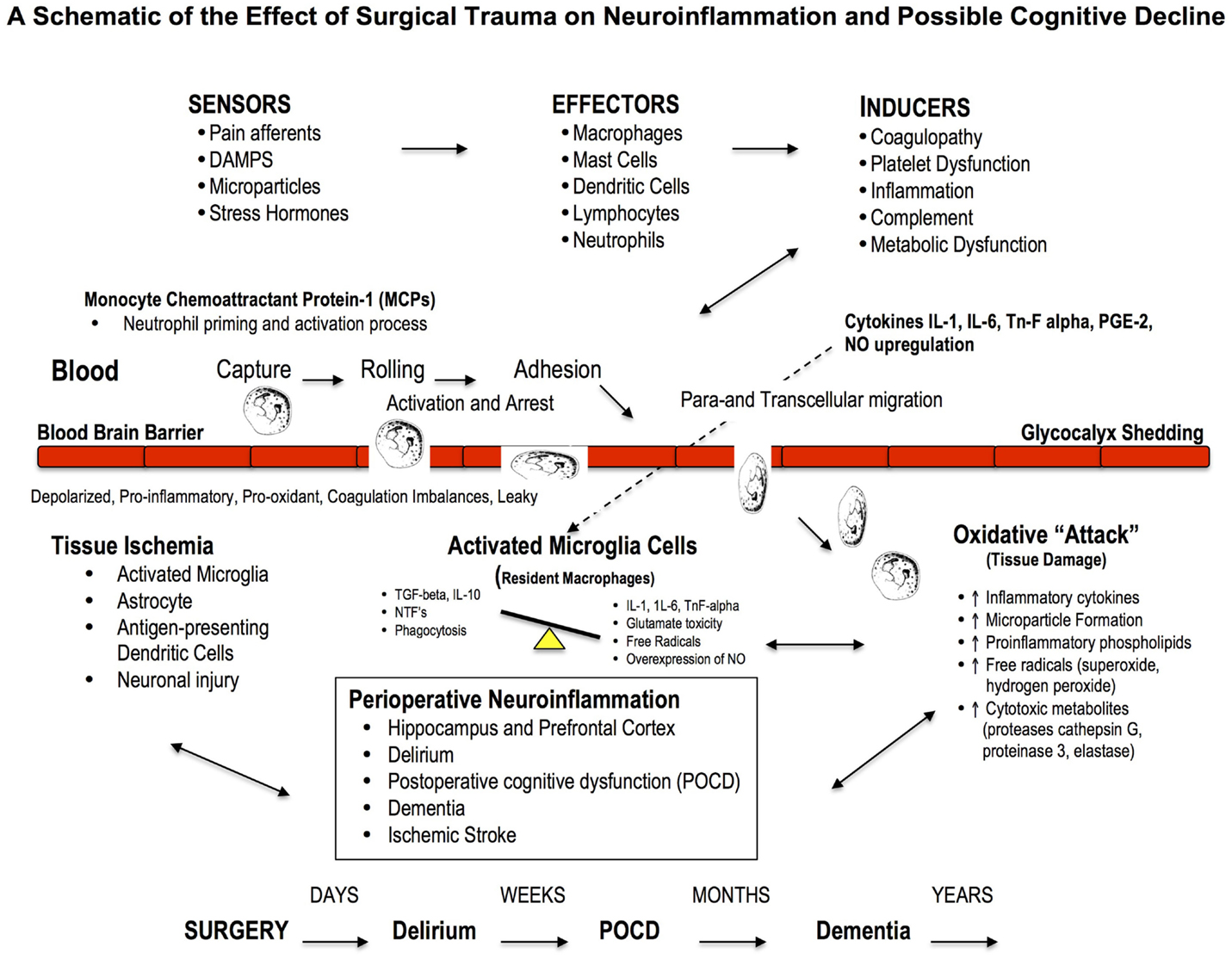
Figure 3. Effect of Major Surgery on the Blood Brain Barrier and Neuroinflammation. The blood brain barrier (BBB) is the body’s natural “firewall” to protect against unwanted incoming agents entering the CNS from the general circulation. During major stress and trauma, the BBB is particularly vulnerable to attack from inflammatory cells and cytokines (156). A breach can lead to neuroinflammation and activation of microglial cells, the brain’s resident macrophages, which may lead to further injury and cognitive dysfunction. The three most common types of cognitive dysfunction are delirium, postoperative cognitive dysfunction (POCD) and dementia (168). Delirium is normally defined as a transient loss of mental attention and orientation in the hours after surgery; dementia is a series of syndromes associated with global deterioration of cognitive ability lasting months to years; and POCD is the deterioration in performance (156, 169). POCD is a more subtle condition and longer lasting than delirium and can include post-traumatic stress disorder after an ICU stay (156). Stroke is another leading cause of severe, long-term cognitive disability after major surgery and occurs in around 2% of patients (167, 170), and 20% of these occur within the first two postoperative days (171).
Neuroinflammation: A Driver of Cognitive Loss
Neuroinflammation is associated with local or widespread activation of microglia, which are the resident macrophages in the brain and spinal cord, and early responders to injury (77, 86, 172, 173). A large part of early cognitive decline appears to occur within the hippocampal and prefrontal cortex areas (174, 175), and may involve: (1) the acute activation of the phagocyte NADPH oxidase (PHOX) found in microglia, (2) expression of the inducible nitric oxide synthase (iNOS) in glia, and (3) microglial phagocytosis of neurons (176). In addition, an acute overexpression and secretion of brain-derived neurotrophic factor (BDNF) by microglial cells, and their intraneuronal pathways, have been implicated as potential mediators of inflammation and hippocampal neuronal dysfunction (e.g., decreased neurogenesis, synaptic plasticity and long-term potentiation of stable memory formation) (174, 175, 177). Recently, Hovens further showed microglia activation in the hippocampus and prefrontal cortex persisted for one week after major surgery (175).
Neuroinflammation and cognitive loss may also occur from damage to the blood brain barrier (BBB), which forms the nexus between the central and peripheral nervous and circulatory systems. A breach to the BBB may facilitate entry of blood-borne immune cells (e.g., neutrophils, lymphocytes) and cytokines (e.g., IL-1, IL-6, or TnF-alpha) and prostaglandins (PGE-2), which all exacerbate inflammation (166, 178) (Figure 3). Based on cerebral MRI, BBB disruption and leakiness have been reported in around 50% of cardiac surgery patients with CPB (179, 180). Excessive leakage of fluid and proteins is also a common occurrence following traumatic and ischemic brain injury, and is more pronounced in the elderly (181). The presence of cerebrospecific protein S100β in serum is an important indicator of cerebral damage (182). Platelets also accumulate in the CNS parenchyma and release pro-inflammatory factors, and play a role in the pathogenesis of cognitive decline (86). In 2012, He and colleagues found in aged rats after a splenectomy that the BBB was damaged, and the hippocampus had high levels of upregulated HMGB1 and the receptor for advanced glycation end products (RAGE) compared to controls (183). Since control rats received the same general anesthesia, the cognitive dysfunction was attributed to the surgery. Serum HMGB1 and IL-6 levels increase significantly after major gastrointestinal (GI) surgery in elderly patients and have been associated with cognitive decline after surgery (184). Unfortunately serum S100β was not measured. Currently there is no adequate protective therapy for cognitive decline.
Effects of Major Surgery on the Other Organs
Myocardial Injury
Among adults undergoing non-cardiac surgery, myocardial injury after non-cardiac surgery is common and associated with substantial mortality.
Botto and Alonso-Coello (185), p. 564
Globally each day over 700,000 adult patients undergo non-cardiac surgery, and around 30% will have some form of pre-existing coronary artery disease (14). In this pre-existing coronary artery disease group, 3.9% or over 8000 patients per day will carry the risk of suffering a major perioperative cardiac event (186). The most common event is a myocardial infarction (MI), which is associated with an in-hospital mortality of 15–25% (186, 187). In a recent international prospective cohort study of over 15,000 non-cardiac surgery patients, 8% of patients over 45 years suffered myocardial injury, based on elevated blood troponin T levels, and 10% of these (120 patients) died from a cardiac event within 30 days (185).
Myocardial ischemia can also result from excessive catecholamine production, alterations in baroreceptor set-point receptivity, and reduced heart rate variability, especially in older patients (188). Plasma catecholamines are associated with higher cardiac troponin levels, and some perioperative thrombotic states can lead to coronary plaque disruption and MI (187). Recent trials using low-dose presynaptic alpha2-adrenergic agonist, clonidine, designed to blunt norepinephrine production, failed to reduce the incidence of perioperative MI (189). However, pretreatment with cyclosporine-A in patients undergoing elective CABG surgery did reduce the perioperative myocardial injury during longer CPB operations (190). Further drug discovery to reduce perioperative “adrenergic stress” and protect the heart from myocardial injury is urgently needed (185).
Another adverse event of major surgery is low CO syndrome, particularly following pediatric and adult cardiac surgery (191). Cardiac depression occurs in many other acute critical states, such as hemorrhagic shock, infection, sepsis, or burns (192–194). While the mechanisms are not fully understood, a cardiac inflammatory response and altered myocardial Ca2+ handling are thought to be involved (191). Interestingly, in animal models, infusion of pro-inflammatory cytokine TNF-alpha has been shown to induce myocardial depression and ischemic-reperfusion injury (195, 196). Similarly, during major surgery it is proposed that the gut may be a possible source of high levels of TnF-alpha because in acute injury models, the gut and mesenteric lymphatics are major contributors to systemic inflammation and multiple organ dysfunction (39, 192, 197, 198). In addition, rat studies show that ligating the mesenteric duct has improved CO during acute burn trauma, further suggesting a “gut-derived factor” may be responsible for low CO syndrome (192, 193). The heart itself may contribute to depressed function because TnF-alpha can also be produced by resident macrophages (199). Other cardiovascular complications during major surgery include arrhythmias, unstable angina, cardiac arrest, heart failure, hypertension, and stroke (200).
Renal Dysfunction
Postoperative acute deterioration in renal function, producing oliguria and/or increase in serum creatinine, is one of the most serious complication in surgical patients.
Brienza et al. (201), p. 2079
Cardiac dysfunction can negatively impact on every organ and tissue of the body. The kidneys normally receive 20% of the CO and insufficient flow can lead to an abrupt reduction in glomerular filtration rate and acute kidney injury (AKI) (202). AKI occurs in 1% of non-cardiac surgical patients, and up to 30% in cardiac surgery patients and around 3% of these patients may require dialysis (203, 204). Patients with AKI requiring renal replacement therapies have mortality rates in excess of 40–50% (202). This condition is strongly associated with ischemia, systemic inflammation, emboli, GI bleeding, respiratory infections, and sepsis (205). To date, no single perioperative strategy has demonstrated a therapeutic benefit to improve CO and prevent renal injury after CPB surgery (202, 203).
Perioperative Pulmonary Injury
Perioperative pulmonary complications may equal or outnumber cardiac events.
Johnson and Kaplan (206)
An unappreciated fact is that pulmonary perioperative complications may equal or outnumber cardiac complications (206–208). Postoperative lung dysfunction occurs in 3–10% of patients after elective abdominal surgery (209), 2–7% after thoracic surgery (210), and 30–50% in patients after cardiac surgery (211). Anesthesia induction itself can lead to ventilation/perfusion mismatch, atelectasis and impaired oxygenation (208, 209). Atelectasis occurs in around 90% of all major surgeries and can persist for several days and predispose the patient to pulmonary infection (212). Abdominal or thoracic surgery is often associated with 20% or more loss of functional residual capacity from diaphragmatic dysfunction, decreased chest wall compliance, and pain-limited inspiration, which may not resolve itself for a week postoperatively (212).
A devastating complication of cardiac surgery is acute respiratory distress syndrome (ARDS), which occurs in 2–3% of low to medium risk patients, and up to 20% in high-risk patients (213, 214). Post-operative ARDS carries a mortality of 40–80% (214). A milder form of ARDS is acute lung injury (ALI) and it generally appears within 2 days of surgery (137, 215). Both ARDS and ALI are part of a systemic disorder associated with microvascular endothelial permeability dysfunction, inflammation, and widespread organ involvement including the heart and cytokines from the GI tract (216, 217). A recent study involving 1817 patients found that transfusion-related ALI, termed TRALI, occurred in 1.4% of patients undergoing major surgery with higher incidences reported (2–3%) after vascular and transplant surgery (218). In addition, the same group found that transfusion-associated circulatory overload (TACO) was a leading cause of transfusion-related fatalities with an overall incidence of 5.5% and highest in vascular (12.1%), transplant (8.8%), and thoracic surgeries (7.2%) (219).
Pulmonary complications after CPB have been known since the mid-1950s because the lungs are almost entirely excluded from the systemic circulation, and alveolar blood is nearly “static” other than receiving residual blood flow from the bronchial arteries (137). During long cross-clamp times, the lungs are under enormous ischemic and inflammatory stress, which is further exacerbated when the cross-clamp is released and the heart and lungs are “reperfused” with oxygenated blood. Re-oxygenation, mitochondrial free oxygen radicals and increased “gut” cytokines further exacerbate the inflammatory response. Neutrophils and macrophage infiltration damage Type I cells leading to alveolar flooding with protein-rich fluid (edema) and Type II cells leading to reduced surfactant production, both of which predispose the lung to ALI and ARDS (214). Recent clinical trials with β2 agonists to increase alveolar fluid clearance and “immunonutrition” with omega-3 fatty acids have been disappointing (215). Novel therapies are required to protect the lung from injury during major surgery, and in particular CPB surgery, and the possibility of using mesenchymal stem cells to form new Type 1 and 2 cells are being investigated.
Gastrointestinal Injury and the “Cytokine Storm”
We believe that there is currently no unifying hypothesis that encompasses the diverse ways in which the gut influences outcome in critical illness.
Clark and Coopersmith (198), p. 385
During major surgery, excessive sympathetic activation, and inflammatory and coagulation imbalances can cause gut dysfunction (220). The GI tract is a powerful immunologically active organ, and plays a key role in maintaining the health of the brain, heart, and lung under normal conditions (38). GI dysfunction ranges from mild complications such as ileus to less common but severe hypoperfusion and ischemic complications, which all carry a high mortality of 60–80% (221). Like the kidneys, the GI tract receives about 20–25% of the resting CO (38). However, during surgical stress sympathetic alpha-adrenergic stimulation can constrict the mesenteric artery and intestinal and intrahepatic portal veins leading to hypoperfusion of the gut and associated organs (liver, pancreas, and spleen), and this response may be potentiated by the posterior pituitary hormone vasopressin (38, 222). During CPB, the initial rise in circulating catecholamines can decrease hepatic perfusion by 20–45% and splanchnic blood flow by approximately 20% (38, 222). These falls in blood perfusion can lead to ischemic complications especially during longer more complex operations. GI injury is an important component of the SIRS and can lead to sepsis, MOF, and death.
The gut is so critically important to the health of the surgical patient that Meakins and Marshall described it as the “motor” of MOF (198) p384. This “motor” can magnify the systemic inflammatory response from: (1) a breach in intestinal epithelium permeablity, (2) activation of the cytokine-mediated GI immune system, and (3) bacterial and endotoxin translocation from the lumen into the peritoneum via the portal circulation (198, 223). A breach can have such a profound effect because in a normal healthy intestine there are more than a hundred trillion (~1015) bacteria, viruses, and fungi, which outnumbers the cells in the human body by tenfold (224, 225). In addition, there are more lymphocytes associated with the intestine than there are in the rest of the human body. Commensal bacteria at the intestinal epithelial interface are believed to regulate the level of NF-κB activity and thereby affect the GI mucosal immune balance (226). During major surgery, and other forms of trauma, if this balance is perturbed complications can arise from a “cytokine storm.” As the “storm” develops more circulating neutrophils are attracted to the interstitial compartments and damaging O2 free radicals, proteolytic enzymes and more cytokines are produced, tight junctions are breached, mucosal cells proceed to necrosis and apoptosis and endotoxemia and sepsis can develop.
The cytokine “storm” from the gut may also be responsible for perioperative cardiac, lung, brain, and other organ dysfunction. As mentioned, cardiac depression, ALI and ARDS are related to an overexpression of the acute immune response. Following major GI surgery, Takahata and colleagues correlated the duration of SIRS and pulmonary dysfunction with the appearance of serum cytokine HMGB-1 levels (227). Other studies implicate intestinal phospholipase A2 generated arachidonic acid and its subsequent 5-lipoxygenase products to pulmonary injury (216), while others suggest an alteration of the T-helper 1/T-helper 2 cytokine lymphocyte balance (228). It is also becoming apparent that CNS imbalances leading to low heart rate variability (229, 230) are associated with increases in systemic inflammation, and possibly involve TNF-alpha production by spleen macrophages (231). Interestingly, increasing parasympathethic outflow via vagal cholinergic stimulation appears to reduce inflammation in ileus following intestinal manipulation in animal models (229, 230). Other factors that modulate the gut may come into play and include vasoactive intestinal peptide, glutamate, and NO, all of which have been shown to modulate immune cells (230). Maintaining a healthy GI tract is imperative to bolstering a patient’s defense against the stress of major surgery.
Liver Injury during Major Surgery
The clinical task to minimize perioperative hepatic cellular injury is challenging for anaesthetists and intensivists alike.
Beck et al. (232), p. 1070
Mild liver dysfunction in patients without liver disease is common following major surgery (232, 233). Acute “ischemic hepatitis,” as it is termed, is a diffuse injury that arises secondary to hypoperfusion (hemodynamic instability) and hypoxia, and is exacerbated by systemic inflammation. Typically, this condition resolves within a few days. More serious complications can arise in patients with preexisting liver disease and cardiac dysfunction. The liver is particularly vulnerable to low flow because of its high O2 requirement and complex portal vein and hepatic artery network (38, 232, 234). The portal system from the stomach, spleen, pancreas, intestines, and omentum supplies 70–80% of blood to the liver at very low pressures (5–10 mmHg) (234), and patients with chronic hepatic congestion or cirrhosis are vulnerable to hypoxic liver injury (232). Other risk factors include the use of CPB, total time on bypass, non-pulsatile flows, fluid overload, and the type of anesthetic and perioperative vasopressor support (233). As mentioned above (see Gastrointestinal Injury and the “Cytokine Storm”), CPB can lead to decreases in splanchnic blood flow by ~20% and hepatic arterial blood flow by up to 45% (222), which may lower venous return and therefore CO resulting in systemic ischemia (38, 235). Anesthetic agents can also reduce hepatic artery blood flow by 50–70% (232, 233). Agents such as isoflurane, desflurane, sevoflurane, and propofol are preferred in patients with liver disease because they have less impact to reduce blood flow compared to other inhaled anesthetic agents (232, 233).
Systemic inflammation is perhaps the most common underlying factor leading to acute and chronic liver dysfunction (236). The patient with liver disease is already in a pro-inflammatory state of “rebalanced hemostasis” that can lead either to excessive bleeding or thrombotic complications (237). Like the GI tract, the liver plays a critical role in immune defense against surgical stress (238, 239), and is involved in maintaining adequate venous return and CO (38, 235). New therapies are urgently required to protect the liver and maintain adequate blood flow through the splanchnic system to support cardiac function during major surgery.
Immunosuppression and Susceptibility to Infection
General anesthesia accompanied by surgical stress is considered to suppress immunity, presumably by directly affecting the immune system or activating the hypothalamic-pituitary-adrenal axis, and the sympathetic nervous system.
Kurosawa and Kato (240)
Immunosuppression is common following major surgery secondary to systemic inflammation and coagulopathy (87, 97). A dramatic depression of cell-mediated immunity predisposes the patient to slow healing, multiple organ injury, infection, and sepsis (58, 241, 242). In cancer patients, emboli dispersal from surgery along with post-operative immunosuppression can lead to further tumor metastases (243). Recent studies implicate impaired natural killer (NK) cell response, lymphocyte depression, and monocyte deactivation as playing major roles in mediating immunosuppression after major surgery (57, 244). Trauma appears to increase the expression of T-helper 2 (Th2)-stimulated lymphocytes and their cytokines resulting in a lower plasma Th1/Th2 cytokine ratio, which is believed to be associated with immune paralysis (228, 239).
A lower plasma Th1/Th2 cytokine ratio is also mediated by the stress hormones glucocorticoids and norepinephrine via activation of the HPA axis and mast cell-histamine reactions (239, 245, 246), and possibly from increased NTS sympathetic outflows. Immunosuppression may be exacerbated by persistent perioperative splanchnic and liver hypoperfusion and gut cytokine production (247–249). Suppressed cellular immunity can continue for 3–10 days post-operatively in patients who have undergone major surgery, but not minor surgery (57). As noted in the Section “Inflammatory Cytokines IL-1, IL-6, and TnF alpha” and Table 2, post-operative immune-competence can be routinely evaluated by measuring plasma levels of interleukins (1β, 2, 6, 8, 10, 12), TNF-α, stress hormones, CRP, and the T-lymphocyte profile (62).
Current Perioperative Therapies: The Good, Bad and the Ugly
Cardiologists frequently advise on perioperative care for non-cardiac surgery and require guidance based on randomized controlled trials that are not discredited by misconduct or misreporting.
Nowbar et al. (250), p. 138
The goal of perioperative therapies is to reduce or prevent surgical “stressors” from developing and to accelerate recovery (251–253). Three major therapies that have attracted a lot of clinical interest to improve perioperative protection are: (1) statins, (2) beta-adrenergic blockers, and (3) calcium-channel blockers.
Statins
Statins are hydroxymethylglutaryl (HMG)-CoA reductase inhibitors and powerful cholesterol-lowering agents (253). Perioperative interest comes from their pleiotropic ability to potentially decrease oxidative stress, inflammation and thrombosis via inhibition of G proteins and induction of transcription factors (251, 254). The first line of evidence supporting statin therapy originated from a landmark, non-surgical Heart Protection Study involving over 20,000 high-risk patients with coronary artery disease or diabetes. In those patients who received 40 mg simvastatin daily there was a significant reduction in all-cause mortality (12.9 vs. 14.7%) from MI, stroke, and the need for coronary and non-coronary revascularization (255, 256). However, these data were challenged in a meta-regression analysis of Robinson and colleagues who compared non-statin and statin trials between 1966 to October 2004 and concluded statins do not appear to contribute a cardiovascular benefit beyond their well proven lipid lowering abilities (257). More recently, in high-risk patients undergoing non-cardiac surgery, de Waal and colleagues concluded there is insufficient data to support final recommendations on perioperative statin therapy (258).
Despite the controversy, some groups argue that statins are underutilized during major surgery (253). For example, Paraskevas and colleagues concluded from Medline searches that statins reduce the incidence of postoperative and postprocedural renal insufficiency and they assist in the earlier recovery of kidney function in vascular patients (259). In another Medline search comparing any statin treatment before cardiac surgery, Liakopoulos and colleagues supported Paraskevas’ findings and further discovered that preoperative statin therapy reduced the risk of post-operative AF and shortened ICU and hospital stay (260). Sanders and colleagues examined Cochrane Central Register of Controlled Trials and reported that short-term statin therapy, commenced before or on the day of non-cardiac vascular surgery and continuing for at least 48 h afterward, improved patient outcomes but had no influence on the risk of MI, stroke, renal disease, pain, or length of hospital stay (261). In a prospective randomized trial of 418 consecutive patients undergoing CABG surgery, Ouattara and colleagues concluded that statin therapy was associated with a significant and dose-dependent reduction in adverse cardiovascular events such as heart failure, malignant arrhythmia, and cardiac death after surgery (262). However, they recommended more trials are required including an evaluation of patient tolerance to the therapy (262). Kulik and Ruel in a review of the Medline data (1987 to January 2009) concluded that the benefits of statin use seem to outweigh the risks in CABG surgery, both in the preoperative and postoperative period. In the absence of contraindications, they argued nearly all CABG patients are candidates for life-long statin therapy, which ideally should be started before surgery (263). Chopra and colleagues’ also undertook a meta-analysis and concluded perioperative statin treatment in statin-naive patients reduced atrial fibrillation, MI, and duration of hospital stay (264).
In summary, while statins appear to be well tolerated during surgery, their use has largely come from retrospective and subgroup analysis of large studies from Medline searches. Statins themselves are diverse in their actions and some clinical trials have demonstrated potential benefits while others have not. For example, pravastatin appears to promote risk reduction in the occurrence of new onset diabetes, whereas atorvastatin, rosuvastatin, and simvastatin increase the risk (265). Other questions on whether patients who are already on statin therapy should remain on statin therapy during surgery or those statin-naïve patients should continue after surgery are clinically important to answer (252, 266). Whether statins reduce mortality and morbidity after major surgery or not can only be answered by clear questions and performing properly designed, prospective, randomized, multi-center clinical trials.
Beta-Adrenergic Blockers
β-Blockers have a long history of potential beneficial effects in patients with a cardiac risk profile. Some of the benefits include: (1) reducing sympathetic nervous system activity, (2) improving myocardial O2 supply/demand ratio from decreased heart rate, systolic blood pressure and myocardial contractility, and (3) having antiarrhythmic properties (267–269). There is also some evidence that beta-blockers may blunt the inflammatory response after injury by reducing the expression of cytokines IL-1, IL-6, and TnF-alpha and CRP (270). Beta-blockers have also been reported in animal and human studies to reduce myocardial ischemia, infarction, and death (269).
Non-Cardiac Surgery
Since the late 1990s, multiple retrospective analyses have supported perioperative benefits of β-blockers following surgery (271). However, after a literature search of eleven large databases up to October 2005, Wiesbauer and colleagues concluded that β-blockers did not reduce the incidence of MI, length of hospitalization or mortality (272). They did report there was a trend toward reduced myocardial ischemia and perioperative arrhythmias. On the basis of retrospective analyses, and two small relevant clinical trials, the American College of Cardiology and American Heart Association (ACC/AHA) in 2007 published a set of guidelines recommending perioperative β-blockers for non-cardiac surgery (267, 273). In the following year, a number of groups argued against these guidelines claiming that they were premature and the ACC/AHA should “soften their advocacy” because past trials suffered from a high risk of bias (273).
Moreover, in 2008, the Perioperative Ischemic Evaluation (POISE) trial indicated that long-acting β-blocker metoprolol succinate increased mortality, ischemic stroke, hypotension, and bradycardia in patients at high risk of atherosclerotic disease (274). A possible weakness of the POISE trial was its fixed and relatively high-dose of metoprolol that was started shortly before surgery, and this strategy was not consistent with optimal current practice (267, 269). However, despite differences in dose and timing of delivery compared to current practice, the take home message was clear. From retrospective analysis of larger trials and the POISE trial, Devereaux (co-principal investigator of POISE) argued that urgent attention is required to assess the safety and efficacy of perioperative β-blockers (267). The ACC/AHA committee eventually yielded to the mounting pressure and softened their guidelines (269). More recent studies by Angeli and colleagues concluded that β-blockers reduced total mortality in patients who underwent high-risk non-cardiac surgery but not lower risk surgery (275). Indeed, the data suggests from low risk of bias trials, an increase in all-cause mortality and stroke with β-blocker use (276).
The controversy regarding perioperative β-blockers reached new heights after 2011 with the discovery of scientific misconduct and fabrication in the “Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography (DECREASE)” trials. This was tragic news because the DECREASE family of studies provided much of the original evidence for prophylactic β-blockade use in non-cardiac surgery, and shaped the European Society Cardiology (ESC) Guidelines (250, 269). At the center of the controversy was Don Polderman, chairperson of the ESC guidelines and taskforce on “Pre-operative cardiac risk assessment and perioperative cardiac management in non-cardiac surgery.” Polderman lost his position at the Erasmus Medical Center in Rotterdam for scientific misconduct and the institution released a note of his dismissal on November 16, 2011 stating that he was:
“careless in collecting the data for his research. In one study it was found that he used patient data without written permission, used fictitious data and that two reports were submitted to conferences which included knowingly unreliable data”.
http://www.erasmusmc.nl/corp_home/corp_news-center/2011/2011-11/ontslag.hoogleraar/?lang=en
In 2014, the European Society of Cardiology and European Society of Anesthesiology (ESC/ESA) released joint guidelines with new recommendations stating that β-blockers are not recommended in patients without clinical risk factors, given that the drugs do not decrease the risk of cardiac complications and “may even increase this risk” (276). Despite this warning, the guidelines continue to recommend β-blocker use as reasonable in patients with intermediate- or high-risk myocardial ischemia documented prior to surgery (class IIb, level of evidence C), and for those with three or more risk factors, such as diabetes, heart failure, or coronary artery disease. This “relaxing” of the guidelines appears to ignore the fact that the DECREASE family studies, on which many of the guidelines are based, were deemed “unreliable” and contained “fictitious data” (277). In 2013, Bouri, Francis, Cole, and colleagues argued that initiation of β-blockers in patients undergoing non-cardiac surgery increased the risk of mortality by 27%, potentially resulting in the deaths of as many as 10,000 patients per year in the UK alone (278).
Cardiac Surgery
In cardiac surgery, β-blockers are generally recommended to reduce postoperative atrial fibrillation (AF) and cardiovascular ischemic events and have been used for more than 40 years (269, 271, 279). Ogawa and colleagues showed in 136 patients undergoing off-pump CABG that administration of low-dose continuous infusion of ultra short-acting landiolol from the beginning of the operation until postoperative day 2, significantly reduced the incidence of postoperative atrial fibrillation by nearly 50% (19 vs. 37%) and also significantly suppressed systemic inflammation during CABG from a reduced postoperative peak in CRP compared to the non-landiolol group (268, 280). Recently, Blessberger and colleagues concluded after examining 89 randomized controlled trials with 19,211 participants that β-blockers in cardiac surgery can substantially reduce the high burden of supraventricular and ventricular arrhythmias following surgery (276, 281). However, they found that the influence of β-blockers on mortality, AMI, stroke, congestive heart failure, hypotension, and bradycardia in this setting remained unclear (276). In another meta-analysis with more than 100,000 study participants, Bangalore and colleagues warned against use of beta-blockers in post-MI patients because of a possible increase in the risk of heart failure and cardiogenic shock (282).
As with statin use, it appears that the potential benefits of beta-blockers in cardiac or non-cardiac surgery has largely been filtered from retrospective and subgroup analysis of large studies. Well-designed, prospective, randomized clinical trials, with the appropriate statistical power and relevant primary endpoints such as perioperative MI, ischemic stroke, cardiovascular death, and all-cause death are urgently required.
Calcium-Channel Blockers
Calcium blockers were originally introduced in surgery to reduce intracellular Ca2+ loading and protect against myocardial ischemia and angina pectoris. Prior to 2004, Wijeysundera and colleagues undertook a meta-analysis involving forty-one clinical trials using Ca2+ blockers (e.g., amlodipine, nifedipine, nicardipine) and beta-blockers, and concluded that the short-acting Ca2+ blockers dihydropyridines were associated with anti-ischemic effects and a trend toward reduced mortality after CABG surgery (283). Over the past decade, the safety and efficacy of Ca2+ channel blockers as a group has been controversial and it appears that they have little cardiac benefit in patients undergoing non-cardiac surgery or cardiac surgery (284). In 2008, Kertai and colleagues’ retrospective analysis of a large database showed that dihydropiridines were independently associated with increased 30-day mortality in patients undergoing aortic aneurysm surgery than non-users (285).
Today, there appears to be a growing consensus that Ca2+ blockers may be harmful in the perioperative setting in patients undergoing major non-cardiac surgery. Of particular concern is peripheral vasodilation causing a reflex adrenergic activation resulting in an increase in heart rate, which may be associated with myocardial ischemia (284). Thus, in patients with unstable angina, dihydropyridines are contraindicated in the absence of beta-blockade (284). Unfortunately, there are few prospective randomized, prospective, trials that specifically examine hard outcomes associated with Ca2+ channel blockers and perioperative hemodynamics, because patients with different hemodynamic profiles may respond with different outcomes (284).
Where Do We Stand Today?
In those patients already on chronic β-blockers, statins and Ca2+ blocker therapies, the general consensus among anesthesiologists and surgeons is to continue their use before and after major surgery (286). With respect to patients not on these drugs and who require major surgery, the data are much less clear. In 2014 Francis, Cole and colleagues argued that the Guideline bodies should retract their recommendations based on fictitious data (278). To this end, Nowbar and colleagues examined 14 such recommendations and dismissed 11 of them based on lack of data or past associations with bias or misconduct. They concluded that there is insufficient evidence to recommend statins, beta-blockers or Ca2+ blockers without properly designed trials (250). Bouri, Francis, Cole and colleagues also proposed that: “any remaining enthusiasts might best channel their energy into a further randomized trial which should be designed carefully and conducted honestly” (278). The controversy continues.
Search for New Therapies from a Systems-Based Approach
What we anticipate seldom occurs; what we least expect generally happens.
Benjamin Disraeli (1804-81) Henrietta Temple
Frontline Protection Begins before the First Incision
For a Kuhnian revolution to occur in surgical protection, it is proposed that new drugs and treatment strategies must embrace the CNS control of whole body function. A highly reductionist approach leveled at single drug targets ignores the complexity of biological systems. Reductionism is important in breaking a system into its constituent parts for study, however, it does not do away with the system (287). Thus current practice of identifying, documenting and treating a single perturbation during or following an operation, and then the next defect, and so on down the line, is not working and may result in what US surgeon William C. Shoemaker termed “an uncoordinated and sometimes contradictory therapeutic outcome” (287, 288).
Protection should begin early before the first incision to prevent the body from overshooting its normal homeostatic tolerance limits. Drug targets include the regions of tissue injury, the CNS response to that injury and their systemic manifestations (Table 3). No drug or drug management strategy currently exists to effectively blunt or prevent these stressors and responders to major surgery.

Table 3. Three potential perioperative targets for reducing the stress response to major surgery.
Toward Stress-Free Surgery in the 21st Century: A Working Hypothesis
It is proposed that targeting local tissue injury, the CNS response to that injury and systemic manifestations may improve patient outcomes by reducing surgical trauma and minimizing “secondary-hit” complications from neuroendocrine, inflammatory, coagulation, and metabolic imbalances. The key to maintaining or restoring cellular homeostasis is to provide material exchange between the blood and the tissues. As a working hypothesis, the four pillars of whole body resynchronization during surgical trauma are:
• CNS as central controller;
• Heart as pressure generator;
• Arterial supply venous capacitance as pressure/volume regulators;
• Vascular endothelium as the systemic integrator.
If imbalances or uncoupling occurs to any of these pillars beyond their normal design tolerances, perioperative complications may arise (Figure 4). This stress-induced mismatch is termed Central-CardioVascular-Endothelium (CCVE) uncoupling. If central and local control of CO and ventricular-arterial coupling are impaired, endothelial and micro-vascular function may be impaired and tissue O2 delivery compromised. A stress-induced sympathetic discharge results in loss of heart rate variability and changes to baroreceptor sensitivities, which profoundly impacts CO and hemodynamics and whole body function. If CO is reduced, and the ability of the arterial system to receive blood from the heart is impaired, splanchnic venous capacitance may be diminished and venous return (and CO) will drop further leading to tissue hypoperfusion, endothelium damage, systemic inflammation, and coagulopathy (Figure 4). Maintenance of cardiac preload thus depends on the ability of the CNS to control venous compliance and hence to redistribute blood volume between peripheral organs and the cardiopulmonary compartment.
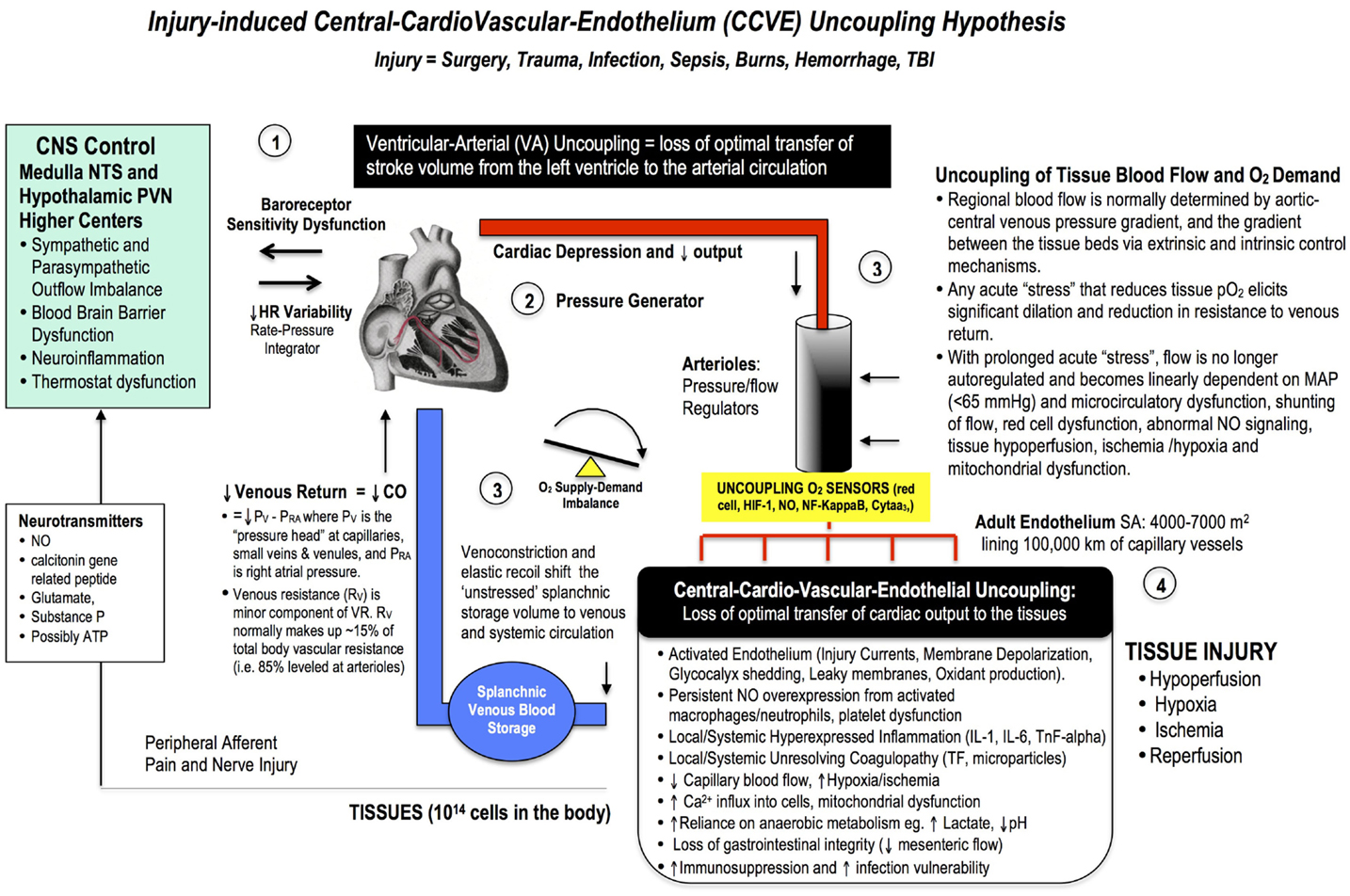
Figure 4. A broad schematic of the Central-CardioVascular-Endothelium (CCVE) “uncoupling” hypothesis that may be responsible for the high mortality and morbidity after major surgery. Loss of whole body homeostatic control during surgical trauma may be leveled at: (1) the CNS, (2) the heart, (3) the vascular tree, and (4) the endothelium. There is an urgent need to develop a pharmacological therapy that supports a high flow (maintained cardiac output), hypotensive, vasodilatory state with endothelial protection and tissue oxygenation (287). If central and local control of cardiac output and ventricular-arterial coupling are improved, endothelial and micro-vascular function will be improved and tissue O2 delivery will be maintained. An uncoupling is reflected in increased stress hormones, sympathetic discharge, loss of baroreceptor sensitivity, and loss of heart rate variability (229, 230). Impaired sympathetic control and a loss of heart rate variability are two of the strongest predictors of death in critically ill patients (188), and promote a pro-inflammatory state with higher IL-1, IL-6, TnF-alpha, and CRP levels, and coagulopathy. A new whole body therapy is required to bolster the patient’s defense against the trauma of surgery and prevent “secondary hit” complications from ischemic and inflammatory cascades, coagulopathy, multiple organ failure, and immunosuppression.
Maintaining the health of the vascular endothelium is a key to reduce surgical stress because this “organ” is the master integrator and regulator of vascular tone, inflammation and coagulation, vascular permeability, blood fluidity, and lymphatic function (Figures 1–4) (289). Its vast surface area of up to 7000 m2 is lined with negatively charged 0.1–1 uM thick glycocalyx mesh of fibril projections made of proteoglycans and glycoproteins (290–294). When injured, the glycocalyx releases syndecan-1, hyaluronic acid, and heparan sulfate into the circulation and the endothelium becomes leaky and damage occurs to underlying tissues (291, 293, 295, 296). Once injured, there is evidence that the glycocalyx can repair itself quickly under the right conditions, which has great significance to surgery and recovery (110, 297). An underlying assumption of the CCVE hypothesis is that if the stress of surgery is controlled, the patient will do the recovery since every cell in the body is programed and working hard in that direction already.
Perioperative Innovation: From Natural Hibernators to Heart Surgery
For a large number of problems there will be some animal of choice on which it can be most conveniently studied.
Krogh (298), p. 202
Cardiac Surgery
In 1998, the author (GPD) utilized the August Krogh principle and asked: “Could the human heart in cardiac surgery be pharmacologically manipulated to operate more like a heart of a natural hibernator?” (25). Natural hibernators are extraordinary animals and can become profoundly hypotensive and hypothermic with up to 98% reductions in body metabolism (25). In this “pilot-light” state, the hibernator does not flood its heart with high potassium, as is standard practice in cardiac surgery today. The objective was to arrest the human heart at its natural resting “polarized” potential of −80 mV by: (1) inhibiting the voltage-dependent Na+ fast channels responsible for the phase O upstroke of the action potential (AP) (lidocaine), and (2) simultaneously decreasing the AP duration assisted by opening K+ATP channels (adenosine) (191). Magnesium was included to reduce Ca2+ entry and protect the heart from ischemia-reperfusion injury and post-operative arrhythmias.
Theoretically, this drug strategy should “flat-line” the heart at its natural “diastolic” membrane potential and confer protection by having fewer channels open, less Na2+ and Ca2+ loading, less inflammation (from a polarized potential), and less arrhythmias during reanimation (299). What emerged was the world’s first low potassium polarizing adenosine and lidocaine with Mg2+ (ALM) cardioplegia (299). Recently an Italian prospective, randomized trial showed that the new cardioplegia was superior to a Buckberg high potassium solution by demonstrating significantly lower perioperative troponin levels, improved post-operative cardiac function (arterial ventricular coupling), 50% less blood transfusions, one full day less in ICU, and two days less in hospital (300). The key to this “polarizing” concept was that ALM at high concentrations arrests the heart, and at lower concentrations it resuscitates the heart. What follows are studies involving the lower, non-arrest ALM levels to rescue the heart following MI, hemorrhagic shock, cardiac arrest and sepsis, which may have applications to major surgery administered as an IV drip after anesthesia but before the first incision.
Possible Applications to Major Surgery
Our first set of in vivo rat studies showed that AL infusion administered 5 min before severe regional myocardial ischemia from tying off the left anterior descending coronary artery for 30 min led to 100% survival and a 92% reduction in ventricular arrhythmias compared to 60% deaths in controls (301). We also showed using 31P NMR that AL led to improved ATP supply by lowering myocardial demand during insult (302). Our second set of studies showed that ultra-small volumes of an IV bolus of 7.5% NaCl ALM (~3% of shed volume) resuscitated the heart and raised mean arterial pressure (MAP) into the hypotensive range after severe 40–60% blood loss and shock, and it corrected coagulopathy at 60 min in the rat model (303, 304) (305). In 2015, we showed that coagulation correction occurred in 5 min indicating that the coagulopathy was not consumptive because the clotting factors, post-shock platelets, and coagulation pathways were fully operational compared with controls (110, 306). We proposed that small-volume ALM assisted the heart and the body to recover with the blood left in the circulation after severe loss without large volume fluid therapy. In addition, ALM has potent anti-inflammatory properties by reducing the priming and activation of neutrophils (307), reduces TnF-alpha (308) and reduces endothelial damage (306). We have also shown similar protection in the rat model of 8 min asphyxial hypoxia where a small bolus of 0.9% NaCl ALM improved return of spontaneous circulation (ROSC), hemodynamics, coagulation status and survival (108, 309).
Small-volume 7.5% NaCl ALM resuscitation has translated from rat to pig after 75% blood loss (310). Around 2 l of blood was removed from 40 kg pigs and only ~140 ml IV bolus of 7.5% NaCl ALM (~7% return of shed volume) was administered (310). During 60 min hypotensive phase (MAP ~50 mmHg), the ALM group had a 1.8-fold increase in stroke volume, a 34% fall in blood lactate, and a 43% higher O2 delivery compared to controls which began decompensate (310). How can 140 ml of ALM fluid increase stroke volume by 1.8 times when added to only ~25% of the animal’s normal circulating blood volume? One possible explanation is that ALM improved the coupling between the heart and arterial supply venous capacitance system, and increased the mean systemic pressure (PMS) that was sufficient to increase venous return by 1.8-fold (see Figure 4). After 60 min, shed blood was returned and whole body O2 consumption fell, systemic vascular resistance increased 30%, and urine output in the ALM group increased threefold compared with controls (310). Lastly, the hypotensive cardiac rescue potential of 7.5% NaCl ALM in the pig model of 75% blood loss was further demonstrated by Granfeldt and colleagues who showed that a 20 ml bolus (0.5 ml/kg) significantly reduced fluid requirement by 40% to reach a target MAP of 50 mmHg (311). Interestingly, when shed blood was returned a 10 ml bolus of 0.9% NaCl AL (no Mg2+) there was a significant drop in whole body O2 consumption (27% fall) and improved cardiac and renal function (311).
The ALM drug therapy also appears to be protective against infection. We showed that a bolus and infusion of 0.9% NaCl ALM in the rat model of polymicrobial sepsis elicited a stable, hypotensive state and reduced lung edema compared to controls (312). The therapy also corrected coagulopathy due to the laparotomy itself which may have clinical implications (312). Importantly, IV infusion rates were kept low (1.0 ml/kg/h) to avoid “secondary hit” complications from fluid overload such as cardiovascular and endothelial dysfunction, inflammation and coagulopathy. These low volumes are consistent with human studies by Lamke and colleagues who showed that the basal evaporation rate and typical fluid losses in humans undergoing major abdominal surgery were ~0.5 ml/kg/h (313, 314). Today, high fluid volumes up to 3–4 l are common in major surgery and may amplify the stress response by shocking the body a second time (315–317). In the pig endotoxin model, we also showed that a bolus and infusion of 0.9% NaCl ALM induced a profound hypotensive, vasodilatory state (MAP ~47 mmHg) with maintained CO and tissue oxygenation for 5 h. This state was accompanied by improved ventricular-arterial coupling, a significant reduction in TnF alpha and reduced lung edema compared to LPS controls which began to decompensate (308).
To summarize, our work using a small-volume ALM bolus and infusion “drip” appears to induce a mild hibernating-like state in rat and pig models, and may find clinical utility in protecting the patient against the stress of major surgery. It is possible that an ALM bolus/drip may blunt the sympathetic discharge that accompanies surgical stress and improve the coupling between the CNS support of the cardiovascular system and endothelium to reduce the inflammatory and coagulopathy responses with reduced mortality and morbidity. Clinical safety trials are required to examine the effect of the ALM “drip” (0.25–0.5 ml/kg/h) administered after anesthesia but before the first incision to reduce the stress response of major surgery.
Concluding Remarks
Major surgery elicits profound changes in the neuroendocrine, metabolic, inflammatory, and immune systems, which collectively constitutes the “stress response.” The stress response is normally self-limiting and resolving. However, during surgical stress, the system can quickly “overshoot” and result in potentially harmful outcomes such as cognitive and cardiac dysfunction, vascular instability, endothelial activation, inflammation, coagulopathy and possibly immunosuppression. Since cardiovascular function is key to a healthy endothelium, it is proposed frontline drugs that improve CNS control of CO and arterial supply venous return functions will help to maintain tissue oxygenation and improve perioperative outcomes. Improved endothelium function may reduce an overexpression of inflammatory, immune and complement discharges and reduce “secondary hit” complications such as SIRS, MODS, and MOF. A systems-based approach to perioperative protection may also find wide utility in treating the critically ill or casualties in prehospital and military environments, and help stabilize the patient during transport to definitive care.
Author Contributions
GD is the sole contributor to the design, implementation, literature analysis and writing of the manuscript.
Conflict of Interest Statement
Geoffrey P. Dobson is the sole inventor of the ALM concept for cardioplegia, organ preservation, surgery, infection, and trauma.
Funding
Internal Research Grant funds (to Geoffrey P. Dobson).
Acknowledgments
The author would like to thank his research team Hayley Letson and Maddison Griffin and Dr Rajiv Sharma for many discussions on the topic of surgical trauma. This study was supported by internal research funds from AITHM, and the College of Medicine and Dentistry, James Cook University.
References
1. Moore FD. Bodily changes in surgical convelescence. 1.The normal sequence-observations and interpretations. Ann Surg (1953) 137:289–315. doi:10.1097/00000658-195303000-00001
2. Weiser TG, Regenbogen SE, Thompson KD, Haynes AB, Lipsitz SR, Berry WR, et al. An estimation of the global volume of surgery: a modelling strategy based on available data. Lancet (2008) 372:139–44. doi:10.1016/S0140-6736(08)60878-8
3. Wilmore DW. From Cuthbertson to fast-track surgery: 70 years of progress in reducing stress in surgical patients. Ann Surg (2002) 236:643–8. doi:10.1097/00000658-200211000-00015
4. Giannoudis PV, Dinopoulos H, Chalidis B, Hall GM. Surgical stress response. Injury (2006) 37:S3–9. doi:10.1016/S0020-1383(07)70005-0
5. Li L, Messina JL. Acute insulin resistance following surgery. Trends Endocrinol Metab (2009) 20:429–35. doi:10.1016/j.tem.2009.06.004
6. Abdelmalak BB, Bonilla AM, Yang D, Chowdary HT, Gottlieb A, Lyden SP, et al. The hyperglycemic response to major noncardiac surgery and the added effect of steroid administration in patients with and without diabetes. Anesth Analg (2013) 116:1116–22. doi:10.1213/ANE.0b013e318288416d
7. Desborough JP. The stress response to trauma and surgery. Br J Anaesth (2000) 85:109–17. doi:10.1093/bja/85.1.109
8. Jin F, Chung F. Minimizing peroperative adverse events in the elderly. Br J Anaesth (2001) 87:608–24. doi:10.1093/bja/87.4.608
9. Cecconi M, Corredor C, Arulkumaran N, Abuella G, Ball J, Grounds RM, et al. Clinical review: goal-directed therapy – what is the evidence in surgical patients? The effect on different risk groups. Crit Care (2013) 17:209. doi:10.1186/cc11823
10. Reddy SV. Effect of general anesthetics on the developing brain. J Anaesthesiol Clin Pharmacol (2012) 28:6–10. doi:10.4103/0970-9185.92426
11. Coggins M, Rosenzweig A. The fire within: cardiac inflammatory signalling in health and disease. Circ Res (2012) 110:116–25. doi:10.1161/CIRCRESAHA.111.243196
12. Farmer PE, Kim JY. Surgery and global health: a view from beyond the OR. World J Surg (2008) 32:533–6. doi:10.1007/s00268-008-9525-9
13. Rose J, Chang DC, Weiser TG, Kassebaum NJ, Bickler SW. The role of surgery in global health: analysis of united states inpatient procedure frequency by condition using the global burden of disease 2010 framework. PLoS One (2014) 9:e9693. doi:10.1371/journal.pone.0089693
14. Maggio PM, Taheri PA. Perioperative issues: myocardial ischemia and protection – beta-blockade. Surg Clin North Am (2005) 85:1091–102. doi:10.1016/j.suc.2005.09.016
15. Kristensen SD, Knuuti J, Saraste A, Anker S, Bøtker HE, Hert SD, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management: the joint task force on non-cardiac surgery: cardiovascular assessment and management of the European society of cardiology (ESC) and the European society of anaesthesiology (ESA). Eur Heart J (2014) 35:2383–431. doi:10.1093/eurheartj/ehu282
16. Leape LL. Hospital readmissions following surgery. Turning complications into ‘treasures’. JAMA (2015) 313:467–8. doi:10.1001/jama.2014.18666
17. Dimick JB, Ghaferi AA. Hospital readmission as a quality measure in surgery. JAMA (2015) 313:512–3. doi:10.1001/jama.2014.14179
18. Bartels K, Sullivan BL, Holger K, Elzschig HK. Blowing the cover from perioperative myocardial injury. Anesthesiology (2014) 120(120):533–5. doi:10.1097/ALN.0000000000000112
19. Pearse RM, Moreno RP, Bauer P, Pelosi P, Metnitz P, Spies C, et al. Mortality after surgery in Europe: a 7 day cohort study. Lancet (2012) 380:1059–65. doi:10.1016/S0140-6736(12)61148-9
20. Ozgediz D, Jamison D, Cherian M, Mcqueen K. The burden of surgical conditions and access to surgical care in low- and middle-income countries. Bull World Health Organ (2008) 86(8):646–7.
23. Moore FD. The growth of surgical biology. Ann Surg (1953) 138:807–22. doi:10.1097/00000658-195311000-00023
24. Cooper SJ. From Claude Bernard to Walter Cannon. Emergence of the concept of homeostasis. Appetite (2008) 51:419–27. doi:10.1016/j.appet.2008.06.005
25. Dobson GP. Organ arrest, protection and preservation: natural hibernation to cardiac surgery: a review. Comp Biochem Physiol B Biochem Mol Biol (2004) 139:469–85. doi:10.1016/j.cbpc.2004.06.002
26. Nathoo N, Lautzenheiser FK, Barnett GH. George W. Crile, Ohio’s first neurosurgeon, and his relationship with Harvey cushing. J Neurosurg (2005) 103:378–86. doi:10.3171/jns.2005.103.2.0378
27. Crile G. Nitrous oxide anaesthesia and a note on anoci-association, a new principle in operative surgery. Surg Gynecol Obstet (1911) 13:170–3.
28. Katz J. George Washington Crile, anoci-association, and pre-emptive analgesia. Pain (1993) 53:243–5. doi:10.1016/0304-3959(93)90219-F
29. Crile GW. The kinetic theory of shock and its prevention through anoci-association {shockless operation). Lancet (1913) 185:7–16. doi:10.1016/S0140-6736(01)65552-1
30. Cushing H. On the avoidance of shock in major amputations by cocainization of large nerve-trunks preliminary to their division. Ann Surg (1902) 36:321–45. doi:10.1097/00000658-190209000-00001
31. Soto-Ruiz KM, Varon J. Resuscitation great. George W. Crile: a visionary mind in resuscitation. Resuscitation (2009) 80:6–8. doi:10.1016/j.resuscitation.2008.09.008
32. Cuthbertson DP. Symposium on ‘surgery and nutrition’. Historical approach. Proc Nutr Soc (1980) 39:101–5. doi:10.1079/PNS19800017
33. Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev (2010) 90:513–57. doi:10.1152/physrev.00007.2009
34. Goldstein DS, Kopin IJ. Evolution of concepts of stress. Stress (2007) 10:109–20. doi:10.1080/10253890701288935
35. Minto G, Biccard B. Assessment of the high-risk perioperative patient. Contin Educ Anaesth Crit Care Pain (2014) 14(1):12–7. doi:10.1093/bjaceaccp/mkt020
37. O’Connor TM, O’halloran DJ, Shanahan F. The stress response and the hypothalamic-pituitary-adrenal axis: from molecule to melancholia. Q J Med (2000) 93:323–33. doi:10.1093/qjmed/93.6.323
38. Gelman S, Mushlin PS. Catecholamine-induced changes in the splanchnic circulation affecting systemic hemodynamics. Anesthesiology (2004) 11:434–9. doi:10.1097/00000542-200402000-00036
39. Deitch EA. Multiple organ failure. Pathophysiology and potential future therapy. Ann Surg (1992) 216:117–34. doi:10.1097/00000658-199208000-00002
40. Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res (2010) 107:1170–84. doi:10.1161/CIRCRESAHA.110.223883
41. Baue AE. Multiple, progressive, or sequential systems failure. Arch Surg (1975) 110:779–81. doi:10.1001/archsurg.1975.01360130011001
43. Faist E, Baue AE, Dittmer H, Heberer G. Multiple organ failure in polytrauma patients. J Trauma (1983) 23:775–87. doi:10.1097/00005373-198309000-00002
44. Goris RJ, Boekhorst TP, Nuytinck JK, Gimbrère JS. Multiple-organ failure. Generalized autodestructive inflammation? Arch Surg (1985) 120:1109–15. doi:10.1001/archsurg.1985.01390340007001
45. Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am (1995) 75:25–77.
46. Bone RC. Toward an epidemiology and natural history of SIRS (systemic inflammatory response syndrome). JAMA (1992) 268:3452–5. doi:10.1001/jama.1992.03490240060037
47. Dewar DC, Butcher NE, King KL, Balogh ZJ. Post injury multiple organ failure. Trauma (2011) 13:81–91. doi:10.1177/1460408610386657
48. Moore FA. The role of the gastrointestinal tract in postinjury multiple organ failure. Am J Surg (1999) 178:449–53. doi:10.1016/S0002-9610(99)00231-7
49. Saadia R, Saadia M. Multiple organ failure. How valid is the “two hit” model? J Accid Emerg Med (1999) 16:163–7. doi:10.1136/emj.16.3.163
50. Moore EE, Moore FA, Harken AH, Johnson JL, Ciesla D, Banerjee A. The two-event construct of postinjury multiple organ failure. Shock (2005) 24:71–4. doi:10.1097/01.shk.0000191336.01036.fe
51. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi:10.1038/nature08780
52. Mikhail J. The trauma triad of death: hypothermia, acidosis, and coagulopathy. AACN Clin Issues (1999) 10:85–94. doi:10.1097/00044067-199902000-00008
53. Prêtre R. Minimal invasive surgery in congenital heart defects: keeping sight of our priority. Eur J Surg Cardiothorac Surg (2012) 42:980. doi:10.1093/ejcts/ezs235
54. Oka Y, Murata A, Nishijima J, Yasuda T, Hiraoka N, Ohmachi Y, et al. Circulating interleukin 6 as a useful marker for predicting postoperative complications. Cytokine (1992) 4:298–304. doi:10.1016/1043-4666(92)90070-8
55. Helmy SAK, Wahby MAM, El-Nawaway M. The effect of anaesthesia and surgery on plasma cytokine production. Anaesthesia (1999) 54:733–8. doi:10.1046/j.1365-2044.1999.00947.x
56. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10:826–37. doi:10.1038/nri2873
57. Gutierrez T, Hornigold R, Pearce A. The systemic response to surgery. Surgery (2011) 29(2):93–6. doi:10.1016/j.mpsur.2010.11.010
58. Kimura F, Shimizu H, Yoshidome H, Ohtsuka M, Miyazaki M. Immunosuppression following surgical and traumatic injury. Surg Today (2010) 40:793–808. doi:10.1007/s00595-010-4323-z
59. Fitzpatrick SF, Tambuwala MM, Bruning U, Schaible B, Scholz CC, Byrne A, et al. An intact canonical NF-κB pathway is required for inflammatory gene expression in response to hypoxia. J Immunol (2011) 186:1091–6. doi:10.4049/jimmunol.1002256
60. Hirsiger S, Simmen H-P, Werner CML, Wanner GA, Rittirsch D. Danger signals activating the immune response after trauma. Mediators Inflamm (2012) 2012:1–10. doi:10.1155/2012/315941
61. Lawrence T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol (2009) 1:a001651. doi:10.1101/cshperspect.a001651
62. Ueda A, Turner P, Gagner M. Stress response to laparoscopic liver resection. HPB (Oxford) (2004) 6:247–52. doi:10.1080/13651820410023987
63. Kehlet H. Fast-track colorectal surgery. Lancet (2008) 371:791–3. doi:10.1016/S0140-6736(08)60357-8
64. Myers JL, Marks LJ, O’hara ET, Heizer JW, Liberman H. Regulatory function of circulating corticosteroids in the pituitary-adrenal response to surgical operation. Ann Surg (1961) 154:859–63.
65. Swanson LW, Sawchenko PE. Paraventricular nucleus: a site for the integration of neuroendocrine and autonomic mechanisms. Neuroendocrinology (1980) 31:410–7. doi:10.1159/000123111
66. Hall GM. The anesthetic modification of the endocrine and metabolic response to surgery. Ann R Coll Surg Engl (1985) 67:25–9.
67. Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med (1995) 332:1351–62. doi:10.1056/NEJM199505183322008
68. Burton D, Nicholson G, Hall G. Endocrine and metabolic response to surgery. Contin Educ Anesth Crit Care Pain (2004) 4:144–7. doi:10.1093/bjaceaccp/mkh040
69. Ferri CC, Yuill EA, Ferguson AV. Interleukin-1h depolarizes magnocellular neurons in the paraventricular nucleus of the hypothalamus through prostaglandin-mediated activation of a non selective cationic conductance. Regul Pept (2005) 129:63–71. doi:10.1016/j.regpep.2005.01.004
70. Toft P, Tonnesen E. The systemic inflammatory response to anesthesia and surgery. Curr Anesth Crit Care (2008) 19:349–53. doi:10.1016/j.cacc.2008.07.009
71. Gadek-Michalska A, Tadeusz J, Rachwalska P, Bugajski J. Cytokines, prostaglandins and nitric oxide in the regulation of stress-response systems. Pharmacol Rep (2013) 65:1655–62. doi:10.1016/S1734-1140(13)71527-5
72. Tao J, Youtan L. Impact of anesthesia on systemic and cerebral glucose metabolism in diabetes patients undergoing neurosurgery “updates of diabetes and neurosurgical anesthesia”. J Diabetes Metab (2013) 4:297. doi:10.4172/2155-6156.1000297
73. Marshall JW, Jawdat DM. Mast cells in innate immunity. J Allergy Clin Immunol (2004) 114:21–7. doi:10.1016/j.jaci.2004.04.045
74. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi:10.1038/nri2448
75. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11:723–37. doi:10.1038/nri3073
76. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol (2012) 4:a006049. doi:10.1101/cshperspect.a006049
77. Vacas S, Degos V, Feng X, Maze M. The neuroinflammatory response of postoperative cognitive decline. Br Med Bull (2013) 106:161–78. doi:10.1093/bmb/ldt006
78. Weidenbusch M, Anders HJ. Tissue microenvironments define and get reinforced by macrophage phenotypes in homeostasis or during inflammation, repair and fibrosis. J Innate Immun (2012) 4:463–77. doi:10.1159/000336717
79. Lech M, Anders HJ. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta (2013) 1832:989–97. doi:10.1016/j.bbadis.2012.12.001
80. Portou MJ, Baker D, Abraham D, Tsui J. The innate immune system, toll-like receptors and dermal wound healing: a review. Vascul Pharmacol (2015) 11:S1537–891. doi:10.1016/j.vph.2015.02.007
81. Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev (2006) 51:240–64. doi:10.1016/j.brainresrev.2005.11.004
82. Walker ME, Hatfield JK, Brown MA. New insights into the role of mast cells in autoimmunity: evidence for a common mechanism of action? Biochim Biophys Acta (2012) 1822:57–65. doi:10.1016/j.bbadis.2011.02.009
83. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi:10.1038/nri3452
84. Delvaeye M, Conway EM. Coagulation and innate immune responses: can we view them separately? Blood (2009) 114:2367–74. doi:10.1182/blood-2009-05-199208
85. Semple JW, Italiano JEJ, Freedman J. Platelets and the immune continuum. Nat Rev Immunol (2011) 11:264–74. doi:10.1038/nri2956
86. Langer HF, Chavakis T. Platelets and neurovascular inflammation. Thromb Haemost (2013) 110:888–93. doi:10.1160/TH13-02-0096
87. Menger MD, Vollmar B. Surgical trauma: hyperinflammation versus immunosuppression? Langenbecks Arch Surg (2004) 389:475–84. doi:10.1007/s00423-004-0472-0
88. Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett (2004) 361:184–7. doi:10.1016/j.neulet.2003.12.007
89. Angele MK, Chaudry IH. Surgical trauma and immunosupression: pathophysiology and potential immumodulatory responses. Langenbecks Arch Surg (2005) 390:333–41. doi:10.1007/s00423-005-0557-4
91. Berda-Haddad Y, Robert S, Salers P, Zekraoui L, Farnarier C, Dinarello CA, et al. Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1α. Proc Natl Acad Sci U S A (2011) 108:20684–9. doi:10.1073/pnas.1116848108
92. Ridker PM, Lüscher TF. Anti-inflammatory therapies for cardiovascular disease. Eur Heart J (2014) 35:1782–91. doi:10.1093/eurheartj/eht398
93. Turnbull AV, Rivier C. Regulation of the HPA axis by cytokines. Brain Behav Immun (1995) 9:253–75. doi:10.1006/brbi.1995.1026
94. Cruickshank AM, Fraser WD, Burns HJ, Van Damme J, Shenki NA. Response of serum interleukin-6 in patients undergoing elective surgery of varying severity. Clin Sci (Lond) (1990) 79:161–5. doi:10.1042/cs0790161
95. Ishibashi S, Takeuchi H, Fujii K, Shiraishi N, Adachi Y, Kitano S. Length of laparotomy incision and surgical stress assessed by serum IL-6 level. Injury (2006) 37:247–51. doi:10.1016/j.injury.2005.08.008
96. Lin MT, Yeh SL, Wu MS, Lin JT, Lee PH, Liaw KY, et al. Impact of surgery on local and systemic responses of cytokines and adhesion molecules. Hepatogastroenterology (2009) 56:1341–5.
97. Volk HD. Immunodepression in the surgical patient and increased susceptibility to infection. Crit Care (2002) 6:279–81. doi:10.1186/cc1507
98. Norman MU, Lister KJ, Yan YH, Issekutz A, Hickey MJ. TNF regulates leukocyte–endothelial cell interactions and microvascular dysfunction during immune complex-mediated inflammation. Br J Pharmacol (2005) 144:265–74. doi:10.1038/sj.bjp.0706081
99. Bradley JR. TNF-mediated inflammatory disease. J Pathol (2008) 214:149–60. doi:10.1002/path.2287
100. Zelová H, Hošek J. TNF-α signalling and inflammation: interactions between old acquaintances. Inflamm Res (2013) 62:641–51. doi:10.1007/s00011-013-0633-0
101. Masli S, Turpie B. Anti-inflammatory effects of tumour necrosis factor (TNF)-alpha are mediated via TNF-R2 (p75) in tolerogenic transforming growth factor-beta-treated antigen-presenting cells. Immunology (2009) 127:62–72. doi:10.1111/j.1365-2567.2008.02933.x
102. Rao LV, Pendurthi UR. Tissue factor-factor VIIa signaling. Arterioscler Thromb Vasc Biol (2005) 25(1):47–56.
103. Amara U, Rittirsch D, Flierl MA, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol (2008) 632:71–9.
104. Rittenberger JC, Hostler DP, Tobin T, Gaines J, Callaway CW. Predictors of ROSC in witnessed aeromedical cardiac arrest. Resuscitation (2008) 76:43–6. doi:10.1016/j.resuscitation.2007.06.009
105. Mackman N. Role of tissue factor in hemostasis and thrombosis. Blood Cells Mol Dis (2006) 36:104–7. doi:10.1016/j.bcmd.2005.12.008
106. Østerud B. Tissue factor expression in blood cells. Thromb Res (2010) 125:S31–4. doi:10.1016/j.thromres.2010.01.032
107. Rao LV, Pendurthi UR. Regulation of tissue factor coagulant activity on cell surfaces. J Thromb Haemost (2012) 10:2242–53. doi:10.1111/jth.12003
108. Djabir Y, Letson HL, Dobson GP. Adenosine, lidocaine and Mg2+ (ALM) increases survival and corrects coagulopathy after 8 min asphyxial cardiac arrest in the rat. Shock (2013) 40:222–32. doi:10.1097/SHK.0b013e3182a03566
109. Iwam IT, Kitamura T, Kawamura T, Mitamura H, Nagao K, Takayama M, et al. Chest compression-only cardiopulmonary resuscitation for out-of-hospital cardiac arrest with public-access defibrillation: a nationwide cohort study. Circulation (2012) 126:2844–51. doi:10.1161/CIRCULATIONAHA.112.109504
110. Dobson GP, Letson HL, Sharma R, Sheppard F, Cap AP. Mechanisms of early traumatic-induced coagulopathy (TIC): the clot thickens or not? J Trauma Acute Care Surg (2015) 79:301–9. doi:10.1097/TA.0000000000000729
111. Gronchi F, Perret A, Ferrari E, Marcucci CM, Flèche J, Crosset M, et al. Validation of rotational thromboelastometry during cardiopulmonary bypass: a prospective, observational in-vivo study. Eur J Anaesthesiol (2014) 31:68–75. doi:10.1097/EJA.0b013e328363171a
112. Raja SC. The response of the coagulation system to cardiac surgery is not similar with or without cardiopulmonary bypass. J Cardiothorac Vasc Anesth (2005) 19:700–4. doi:10.1053/j.jvca.2005.02.014
113. Yavari M, Becker RC. Coagulation and fibrinolytic protein kinetics in cardiopulmonary bypass. J Thromb Thrombolysis (2009) 29:95–104. doi:10.1007/s11239-007-0187-5
114. Putzu M, Casati A, Berti M, Pagliarini G, Fanelli G. Clinical complications, monitoring and management of perioperative mild hypothermia: anesthesiological features. Acta Biomed (2007) 78(3):163–9.
115. Albaladejo P, Charbonneau H, Samama CM, Collet JP, Marret E, Piriou V, et al. Bleeding complications in patients with coronary stents during non-cardiac surgery. Thromb Res (2014) 15:S0049–3848. doi:10.1016/j.thromres.2014.05.015
116. Devine EB, Chan LN, Babigumira J, Kao H, Drysdale T, Reilly D, et al. Postoperative acquired coagulopathy: a pilot study to determine the impact on clinical and economic outcomes. Pharmacotherapy (2010) 30:994–1003. doi:10.1592/phco.30.10.994
118. Fleming SD, Tsokos GC. Complement, natural antibodies, autoantibodies and tissue injury. Autoimmun Rev (2006) 5:89–92. doi:10.1016/j.autrev.2005.09.006
119. Markiewski MM, Nilaaon B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol (2007) 28:184–92. doi:10.1016/j.it.2007.02.006
120. Ioannou A, Dalle Lucca J, Tsokos GC. Immunopathogenesis of ischemia/reperfusion-associated tissue damage. Clin Immunol (2011) 141:3–14. doi:10.1016/j.clim.2011.07.001
121. Burk AM, Martin M, Flierl MA, Rittirsch D, Helm M, Lampl L, et al. Early complementopathy after multiple injuries in humans. Shock (2012) 37:348–54. doi:10.1097/SHK.0b013e3182471795
122. Rittirsch D, Redl H, Huber-Lang M. Role of complement in multiorgan failure. Clin Dev Immunol (2012) 2012:1–10. doi:10.1155/2012/962927
123. Gorsuch WB, Chrysanthou E, Schwaeble WJ, Stahl GL. The complement system in ischemia-reperfusion injuries. Immunobiology (2012) 217:1026–33. doi:10.1016/j.imbio.2012.07.024
124. Lu X, Li Y, Simovic MO, Peckham R, Wang Y, Tsokos GC, et al. Decay-accelerating factor attenuates C-reactive protein-potentiated tissue injury after mesenteric ischemia/reperfusion. J Surg Res (2011) 167:e103–15. doi:10.1016/j.jss.2009.10.021
125. Kirklin JK, Westaby S, Blackstone EH, Kirklin JW, Chenoweth DE, Pacifico AD. Complement and the damaging effects of cardiopulmonary bypass. J Thorac Cardiovasc Surg (1983) 86:845–57.
126. Stahl GL, Shernan SK, Smith PK, Levy JH. Complement activation and cardiac surgery: a novel target for improving outcomes. Anesth Analg (2012) 115:759–71. doi:10.1213/ANE.0b013e3182652b7d
127. Gu YJ, Mariani MA, Boonstra PW, Grandjean JG, Van Oeveren W. Complement activation in coronary artery bypass grafting patients without cardiopulmonary bypass: the role of tissue injury by surgical incision. Chest (1999) 116:892–8. doi:10.1378/chest.116.4.892
128. Watkins LR, Maier S, Goehler LE. Immune activation: the role of pro-inflammatory cytokines in inflammation, illness responses and pathological pain states. Pain (1995) 63:289–302. doi:10.1016/0304-3959(95)00186-7
129. Lorenz W, Duda D, Dick W, Sitter H, Doenicke A, Black A, et al. Incidence and clinical importance of perioperative histamine release: randomised study of volume loading and antihistamines after induction of anaesthesia. Trial group Mainz/Marburg. Lancet (1994) 343:933–40. doi:10.1016/S0140-6736(94)90063-9
130. Rittner HL, Brack A, Stein C. Pro-algesic versus analgesic actions of immune cells. Curr Opin Anaesthesiol (2003) 16:527–33. doi:10.1097/00001503-200310000-00014
131. Rittner HL, Brack A. Leukocytes as mediators of pain and analgesia. Curr Rheumatol Rep (2007) 9:503–10. doi:10.1007/s11926-007-0081-3
132. Borsook D, George E, Kussman B, Becerra L. Anesthesia and perioperative stress: consequences on neural networks and postoperative behaviors. Prog Neurobiol (2010) 92:601–12. doi:10.1016/j.pneurobio.2010.08.006
133. Tobias JD, Leder M. Procedural sedation: a review of sedative agents, monitoring, and management of complications. Saudi J Anaesth (2011) 5:395–410. doi:10.4103/1658-354X.87270
134. Moore RA, Allen MC, Wood PJ, Rees LH, Sear JW. Peri-operative endocrine effects of etomidate. Anaesthesia (1985) 40:124–30. doi:10.1111/j.1365-2044.1985.tb10702.x
135. Citerio G, Pesenti A, Latini R, Masson S, Barlera S, Gaspari F, et al. A multicentre, randomised, open-label, controlled trial evaluating equivalence of inhalational and intravenous anaesthesia during elective craniotomy. Eur J Anaesthesiol (2012) 29:371–9. doi:10.1097/EJA.0b013e32835422db
136. Roth-Isigkeit A, Brechmann J, Dibbelt L, Sievers HH, Raasch W, Schmucker P. Persistent endocrine stress response in patients undergoing cardiac surgery. J Endocrinol Invest (1998) 21:12–9. doi:10.1007/BF03347280
137. Apostolakis E, Akinosoglou K. The methodologies of hypothermic circulatory arrest and of antegrade and retrograde cerebral perfusion for aortic arch surgery. Ann Thorac Cardiovasc Surg (2008) 14(3):138–48. doi:10.1097/EJA.0b013e328334c017
138. Kunst G. From coronary steal to myocardial, renal, and cerebral protection: more questions than answers in anaesthetic preconditioning? Br J Anaesth (2014) 112:958–60. doi:10.1093/bja/aeu007
139. Alsina E, Matute E, Ruiz-Huerta AD, Gilsanz F. The effects of sevoflurane or remifentanil on the stress response to surgical stimulus. Curr Pharm Des (2014) 20(34):5449–68.
140. Hofbauer R, Frass M, Salfinger H, Moser D, Hornykewycz S, Gmeiner B, et al. Propofol reduces the migration of human leukocytes through endothelial cell monolayers. Crit Care Med (1999) 27:1843–7. doi:10.1097/00003246-199901001-00347
141. De Hert SG. Cardioprotection by volatile anesthetics: what about noncardiac surgery? J Cardiothorac Vasc Anesth (2011) 25:899–901. doi:10.1053/j.jvca.2011.08.004
142. Campiglia L, Consales G, De Gaudio AR. Pre-emptive analgesia for postoperative pain control: a review. Clin Drug Investig (2010) 30:15–26. doi:10.2165/1158411-S0-000000000-00000
143. Yardeni IZ, Beilin B, Mayburd E, Levinson Y, Bessler H. The effect of perioperative intravenous lidocaine on postoperative pain and immune function. Anesth Analg (2009) 109:1464–9. doi:10.1213/ANE.0b013e3181bab1bd
144. Biber K, Boddeke E. Neuronal CC chemokines: the distinct roles of CCL21 and CCL2 in neuropathic pain. Front Cell Neurosci (2014) 8:210. doi:10.3389/fncel.2014.00210
145. Tønnesen E, Wahlgreen C. Influence of extradural and general anaesthesia on natural killer cell activity and lymphocyte subpopulations in patients undergoing hysterectomy. Br J Anaesth (1988) 60:500–7. doi:10.1093/bja/60.5.500
146. Beilin B, Shavit Y, Trabekin E, Mordashev B, Mayburd E, Zeidel A, et al. The effects of postoperative pain management on immune response to surgery. Anesth Analg (2003) 97:822–7. doi:10.1213/01.ANE.0000078586.82810.3B
147. Turan A, Sessler DI. Steroids to ameliorate postoperative pain. Anesthesiology (2011) 115:457–9. doi:10.1097/ALN.0b013e31822a2871
148. De Oliveira GSJ, Fitzgerald P, Streicher LF, Marcus RJ, Mccarthy RJ. Systemic lidocaine to improve postoperative quality of recovery after ambulatory laparoscopic surgery. Anesth Analg (2012) 115:262–7. doi:10.1213/ANE.0b013e318257a380
149. Sun Y, Li T, Wang N, Yun Y, Gan TJ. Perioperative systemic lidocaine for postoperative analgesia and recovery after abdominal surgery: a meta-analysis of randomized controlled trials. Dis Colon Rectum (2012) 55:1183–94. doi:10.1097/DCR.0b013e318259bcd8
150. Joshi GP, Schug SA, Kehlet H. Procedure-specific pain management and outcome strategies. Best Pract Res Clin Anaesthesiol (2014) 28:191–201. doi:10.1016/j.bpa.2014.03.005
151. Thorell A, Efendic S, Giutniak M, Haggmark T, Ljungqvist O. Development of postoperative insulin resistance is associated with the magnitude of operation. Eur J Surg (1993) 159:593–9.
152. McCowen KC, Malhotra A, Bistrian BR. Stress-induced hyperglycemia. Crit Care Clin (2001) 17:107–24. doi:10.1016/S0749-0704(05)70154-8
153. Losser MR, Damoisel C, Payen D. Bench-to-bedside review: glucose and stress conditions in the intensive care unit. Crit Care (2010) 14:231. doi:10.1186/cc9100
154. Baban B, Thorell A, Nygren J, Bratt A, Ljungqvist O. Determination of insulin resistance in surgery: the choice of method is crucial. Clin Nutr (2015) 34(1):123–8. doi:10.1016/j.clnu.2014.02.002
155. Whang W, Bigger JTJ. Diabetes and outcomes of coronary artery bypass graft surgery in patients with severe left ventricular dysfunction: results from the CABG patch trial database. J Am Coll Cardiol (2000) 36:1166–72. doi:10.1016/S0735-1097(00)00823-8
156. Krenk L, Rasmussen LS, Kehlet H. New insights into the pathophysiology of postoperative cognitive dysfunction. Acta Anaesthesiol Scand (2010) 54:951–6. doi:10.1111/j.1399-6576.2010.02268.x
157. Bakker RC, Osse RJ, Tulen JH, Kappetein AP, Bogers AJ. Preoperative and operative predictors of delirium after cardiac surgery in elderly patients. Eur J Cardiothorac Surg (2012) 41:544–9. doi:10.1093/ejcts/ezr031
158. Newman MF, Kirchner JL, Phillips-Bute B, Gaver V, Grocott H, Jones RH, et al. Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. N Engl J Med (2001) 344:395–402. doi:10.1056/NEJM200102083440601
159. Rudolph JL, Jones RN, Levkoff SE, Rockett C, Inouye SK, Sellke FW, et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation (2009) 119:229–36. doi:10.1161/CIRCULATIONAHA.108.795260
160. Price CC, Garvan CW, Monk TG. Type and severity of cognitive decline in older adults after noncardiac surgery. Anesthesiology (2008) 108:8–17. doi:10.1097/01.anes.0000296072.02527.18
161. Newman S, Stygall J, Hirani S, Shaefi S, Maze M. Postoperative cognitive dysfunction after noncardiac surgery: a systematic review. Anesthesiology (2007) 106:572–90. doi:10.1097/00000542-200703000-00023
162. Chen J, Zimmerman RA, Jarvik GP, Nord AS, Clancy RR, Wernovsky G, et al. Perioperative stroke in infants undergoing open heart operations for congenital heart disease. Ann Thorac Surg (2009) 88:823–9. doi:10.1016/j.athoracsur.2009.03.030
163. Morriss FH Jr, Saha S, Bell EF, Colaizy TT, Stoll BJ, Hintz SR, et al. Surgery and neurodevelopmental outcome of very low-birth-weight infants. JAMA Pediatr (2014) 168:746–54. doi:10.1001/jamapediatrics.2014.307
164. Harrison MJG. Neurologic complications of coronary artery bypass grafting: diffuse or focal ischemia? Ann Thorac Surg (1995) 59:1356–8. doi:10.1016/0003-4975(95)00056-Q
165. Schoen J, Meyerrose J, Paarmann H, Heringlake M, Hueppe M, Berger KU. Preoperative regional cerebral oxygen saturation is a predictor of postoperative delirium in on-pump cardiac surgery patients: a prospective observational trial. Crit Care (2011) 15:R218. doi:10.1186/cc10454
166. Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab (2012) 32:598–611. doi:10.1038/jcbfm.2012.6
167. Bartels K, Mcdonagh DL, Newman MF, Mathew JP. Neurocognitive outcomes after cardiac surgery. Curr Opin Anaesthesiol (2013) 26:91–7. doi:10.1097/ACO.0b013e32835bf24c
168. Rasmussen LS. Postoperative cognitive dysfunction: incidence and prevention. Best Pract Res Clin Anaesthesiol (2006) 20:315–30. doi:10.1016/j.bpa.2005.10.011
169. Fines DP, Severn AM. Anaesthesia and cognitive disturbance in the elderly. Contin Educ Anaesth Crit Care Pain (2006) 6(1):37–40. doi:10.1093/bjaceaccp/mki066
170. Kellermann K, Jungwirth B. Avoiding stroke during cardiac surgery. Semin Cardiothorac Vasc Anesth (2010) 14:95–101. doi:10.1177/1089253210370902
171. Baufreton C. Role of surgical factors in strokes after cardiac surgery. Arch Cardiovasc Dis (2010) 103:326–32. doi:10.1016/j.acvd.2009.12.007
172. Streit WJ, Mrak RE, Griffin WST. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation (2004) 1:14. doi:10.1186/1742-2094-1-14
173. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci (2007) 8:57–69. doi:10.1038/nrn2038
174. Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun (2011) 25:181–213. doi:10.1016/j.bbi.2010.10.015
175. Hovens IB, Schoemaker RG, Van Der Zee EA, Absalom AR, Heineman E, Van Leeuwen BL. Postoperative cognitive dysfunction: involvement of neuroinflammation and neuronal functioning. Brain Behav Immun (2014) 38:202–10. doi:10.1016/j.bbi.2014.02.002
176. Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol (2010) 41:242–7. doi:10.1007/s12035-010-8105-9
177. Sohrabji F, Lewis DK. Estrogen-BDNF interactions: implications for neurodegenerative diseases. Front Neuroendocrinol (2006) 27:404–14. doi:10.1016/j.yfrne.2006.09.003
178. Guyon A, Massa F, Rovere C, Nahon J-L. How cytokines can influence the brain: a role for chemokines? J Neuroimmunol (2008) 198:46–55. doi:10.1016/j.jneuroim.2008.04.009
179. Harris DN, Bailey SM, Smith PL, Taylor KM, Oatridge A, Bydder GM. Brain swelling in first hour after coronary artery bypass surgery. Lancet (1993) 342:586–7. doi:10.1016/0140-6736(93)91412-F
180. Merino JG, Latour LL, Tso A, Lee KY, Kang DW, Davis LA, et al. Blood-brain barrier disruption after cardiac surgery. Am J Neuroradiol (2013) 34:518–23. doi:10.3174/ajnr.A3251
181. Oakley R, Tharakan B. Vascular hyperpermeability and aging. Aging Dis (2014) 5(2):114–25. doi:10.14336/AD.2014.0500114
182. Singh SP, Kapoor PM, Chowdhury U, Kiran U. Comparison of S100β levels, and their correlation with hemodynamic indices in patients undergoing coronary artery bypass grafting with three different anesthetic techniques. Ann Card Anaesth (2011) 14:197–202. doi:10.4103/0971-9784.83998
183. He HJ, Wang Y, Le Y, Duan KM, Yan XB, Liao Q, et al. Surgery upregulates high mobility group box-1 and disrupts the blood-brain barrier causing cognitive dysfunction in aged rats. CNS Neurosci Ther (2012) 18:994–1002. doi:10.1111/cns.12018
184. Lin GX, Wang T, Chen MH, Hu ZH, Ouyang W. Serum high-mobility group box 1 protein correlates with cognitive decline after gastrointestinal surgery. Acta Anaesthesiol Scand (2014) 58(6):668–74. doi:10.1111/aas.12320
185. Botto F, Alonso-Coello P, Chan MT, Villar JC, Xavier D, Srinathan S, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology (2014) 120:564–78. doi:10.1097/ALN.0000000000000113
186. Biccard BM, Rodseth RN. The pathophysiology of peri-operative myocardial infarction. Anaesthesia (2010) 65:733–41. doi:10.1111/j.1365-2044.2010.06338.x
187. Landesberg G, Beattie S, Mosseri M, Jaffe AS, Alpert JS. Perioperative myocardial infarction. Circulation (2009) 119:2936–44. doi:10.1161/CIRCULATIONAHA.108.828228
188. Dunser MW, Hasibeder WR. Sympathetic overstimulation during critical illness: adverse effects of adrenergic stress. J Intensive Care Med (2009) 24:293–316. doi:10.1177/0885066609340519
189. Devereaux PJ, Sessler DI, Leslie K, Kurz A, Mrkobrada M, Alonso-Coello P, et al. Clonidine in patients undergoing noncardiac surgery. N Engl J Med (2014) 370:1504–13. doi:10.1056/NEJMoa1401105
190. Hausenloy DJ, Kunst G, Boston-Griffiths E, Kolvekar S, Chaubey S, John L, et al. The effect of cyclosporin-A on peri-operative myocardial injury in adult patients undergoing coronary artery bypass graft surgery: a randomised controlled clinical trial. Heart (2014) 100:544–9. doi:10.1136/heartjnl-2013-304845
191. Dobson GP, Faggian G, Onorati F, Vinten-Johansen J. Hyperkalemic cardioplegia in adult and pediatric cardiac surgery: end of an era? Front Clin Transl Physiol (2013) 4:228. doi:10.3389/fphys.2013.00228
192. Lee MA, Yatani A, Sambol JT, Deitch EA. Role of gut-lymph factors in the induction of burn-induced and trauma-shock-induced acute heart failure. Int J Clin Exp Med (2008) 1(2):171–80.
193. Sambol JT, Lee MA, Caputo FJ, Kawai K, Badami C, Kawai T, et al. Mesenteric lymph duct ligation prevents trauma/hemorrhage shock-induced cardiac contractile dysfunction. J Appl Physiol (2009) 106:57–65. doi:10.1152/japplphysiol.90937.2008
194. Schmittinger CA, Wurzinger B, Deutinger M, Wohlmuth C, Knotzer H, Torgersen C, et al. How to protect the heart in septic shock: a hypothesis on the pathophysiology and treatment of septic heart failure. Med Hypotheses (2010) 74:460–5. doi:10.1016/j.mehy.2009.10.012
195. Cain BS, Harken AH, Meldrum DR. Therapeutic strategies to reduce TNF-alpha mediated cardiac contractile depression following ischemia and reperfusion. J Mol Cell Cardiol (1999) 31:931–47. doi:10.1006/jmcc.1999.0924
196. McGhan LJ, Jaroszewski DE. The role of toll-like receptor-4 in the development of multi-organ failure following traumatic haemorrhagic shock and resuscitation. Injury (2012) 43:129–36. doi:10.1016/j.injury.2011.05.032
197. Hassoun HT, Kone BC, Mercer DW, Moody FG, Weisbrodt NW, Moore FA. Post-injury multiple organ failure: the role of the gut. Shock (2001) 15:1–10. doi:10.1097/00024382-200115010-00001
198. Clark JA, Coopersmith CM. Intestinal crosstalk: a new paradigm for understanding the gut as the “motor” of critical illness. Shock (2007) 28:384–93. doi:10.1097/shk.0b013e31805569df
200. London MJ. Cardiovascular problems in noncardiac surgery. Curr Opin Crit Care (2009) 15:333–41. doi:10.1097/MCC.0b013e32832e4795
201. Brienza N, Giglio MT, Marucci M, Fiore T. Does perioperative hemodynamic optimization protect renal function in surgical patients? A meta-analytic study. Crit Care Med (2009) 37:2079–90. doi:10.1097/CCM.0b013e3181a00a43
202. Alsabbagh MM, Asmar A, Ejaz NI, Aiyer RK, Kambhampati G, Ejaz AA. Update on clinical trials for the prevention of acute kidney injury in patients undergoing cardiac surgery. Am J Surg (2013) 206:86–95. doi:10.1016/j.amjsurg.2012.08.007
203. Coleman MD, Shaefi S, Sladen RN. Preventing acute kidney injury after cardiac surgery. Curr Opin Anaesthesiol (2011) 24:70–6. doi:10.1097/ACO.0b013e3283422ebc
204. Vives M, Wijeysundera D, Marczin N, Monedero P, Rao V. Cardiac surgery-associated acute kidney injury. Interact Cardiovasc Thorac Surg (2014) 18:637–45. doi:10.1093/icvts/ivu014
205. Brienza N, Giglio MT, Marucci M. Preventing acute kidney injury after noncardiac surgery. Curr Opin Crit Care (2010) 16:353–8. doi:10.1097/MCC.0b013e32833a9ef5
206. Johnson DC, Kaplan LJ. Perioperative pulmonary complications. Curr Opin Crit Care (2011) 17:362–9. doi:10.1097/MCC.0b013e328348bfc8
207. Lawrence VA, Dhanda R, Hilsenbeck SG, Page CP. Risk of pulmonary complications after elective abdominal surgery. Chest (1996) 110:744–50. doi:10.1378/chest.110.3.744
208. Singer P, Kilpatrick B. Perioperative lung protection strategies in cardiothoracic anesthesia. Anesth Clin (2012) 30:607–28. doi:10.1016/j.anclin.2012.07.001
209. Hedenstierna G, Edmark L. The effects of anesthesia and muscle paralysis on the respiratory system. Intensive Care Med (2005) 31:1327–35. doi:10.1007/s00134-005-2761-7
210. Eischenbaum KD, Neustein SM. Acute lung injury after thoracic surgery. J Cardiothorac Vasc Anesth (2010) 24:681–90. doi:10.1053/j.jvca.2009.10.032
211. Groeneveld ABJ, Jansen EK, Verheij J. Mechanisms of pulmonary dysfunction after on-pump and off-pump cardiac surgery: a prospective cohort study. J Cardiothorac Surg (2007) 2:11. doi:10.1186/1749-8090-2-11
212. Hedenstierna G, Edmark L. Mechanisms of atelectasis in the perioperative period. Best Pract Res Clin Anaesthesiol (2010) 24:157–69. doi:10.1016/j.bpa.2009.12.002
213. Della Rocca G, Coccia C. Acute lung injury in thoracic surgery. Curr Opin Anaesthesiol (2013) 26:40–6. doi:10.1097/ACO.0b013e32835c4ea2
214. Stephens RS, Shah AS, Whitman GJ. Lung injury and acute respiratory distress syndrome after cardiac surgery. Ann Thorac Surg (2013) 95:1122–9. doi:10.1016/j.athoracsur.2012.10.024
215. Dushianthan A, Grocott MP, Postle AD, Cusack R. Acute respiratory distress syndrome and acute lung injury. Postgrad Med J (2011) 87:612–22. doi:10.1136/pgmj.2011.118398
216. Moore EE. Claude H. Organ, Jr. memorial lecture: splanchnic hypoperfusion provokes acute lung injury via a 5-lipoxygenase-dependent mechanism. Am J Surg (2010) 200:681–9. doi:10.1016/j.amjsurg.2010.05.010
217. Adeniji K, Steel AC. The pathophysiology of perioperative lung injury. Anesth Clin (2012) 30:573–90. doi:10.1016/j.anclin.2012.08.011
218. Clifford L, Jia Q, Subramanian A, Yadav H, Wilson GA, Murphy SP, et al. Characterizing the epidemiology of postoperative transfusion-related acute lung injury. Anesthesiology (2015) 122:12–20. doi:10.1097/ALN.0000000000000514
219. Clifford L, Jia Q, Yadav H, Subramanian A, Wilson GA, Murphy SP, et al. Characterizing the epidemiology of perioperative transfusion-associated circulatory overload. Anesthesiology (2015) 122:21–8. doi:10.1097/ALN.0000000000000513
220. Giglio MT, Marucci M, Testini M, Brienza N. Goal-directed haemodynamic therapy and gastrointestinal complications in major surgery: a meta-analysis of randomized controlled trials. Br J Anaesth (2009) 103:637–46. doi:10.1093/bja/aep279
221. Vollmar B, Menger MD. Intestinal ischemia/reperfusion: microcirculatory pathology and functional consequences. Langenbecks Arch Surg (2011) 396:13–29. doi:10.1007/s00423-010-0727-x
222. Diaz GC, Moitra V, Sladen RN. Hepatic and renal protection during cardiac surgery. Anesthesiol Clin (2008) 26:565–90. doi:10.1016/j.anclin.2008.05.001
223. Puleo F, Arvanitakis M, Van Gossum A, Preiser JC. Gut failure in the ICU. Semin Respir Crit Care Med (2011) 32:626–38. doi:10.1055/s-0031-1287871
224. Caricilli AM, Castoldi A, Câmara NO. Intestinal barrier: a gentlemen’s agreement between microbiota and immunity. World J Gastrointest Pathophysiol (2014) 5:18–32. doi:10.4291/wjgp.v5.i1.18
225. Koboziev L, Reinoso C, Furr KL, Grisham MB. Role of the enteric microbiota in intestinal homeostasis. Free Radic Biol Med (2014) 68:122–33. doi:10.1016/j.freeradbiomed.2013.11.008
226. Wullaert A, Bonnet MC, Pasparakis M. NF-κ B in the regulation of epithelial homeostasis and inflammation. Cell Res (2011) 21:146–58. doi:10.1038/cr.2010.175
227. Takahata R, Ono S, Tsujimoto H, Hiraki S, Kimura A, Kinoshita M, et al. Postoperative serum concentrations of high mobility group box chromosomal protein-1 correlates to the duration of SIRS and pulmonary dysfunction following gastrointestinal surgery. J Surg Res (2011) 170:e135–40. doi:10.1016/j.jss.2011.04.040
228. Marik PE, Flemmer M. The immune response to surgery and trauma: implications for treatment. J Trauma Acute Care Surg (2012) 73:801–8. doi:10.1097/TA.0b013e318265cf87
229. Huston JM, Tracey KJ. The pulse of inflammation: heart rate variability, the cholinergic anti-inflammatory pathway and implications for therapy. J Intern Med (2011) 269:45–53. doi:10.1111/j.1365-2796.2010.02321.x
230. Matteoli G, Boeckxstaens GE. The vagal innervation of the gut and immune homeostasis. Gut (2013) 62:1214–22. doi:10.1136/gutjnl-2012-302550
232. Beck C, Schwartges I, Picker O. Perioperative liver protection. Curr Opin Crit Care (2010) 16:142–7. doi:10.1097/MCC.0b013e328336ea83
233. Friedman LS. Surgery in the patient with liver disease. Trans Am Clin Climatol Assoc (2010) 121:192–204.
234. Lautt WW. Intrinsic regulation of hepatic blood flow. Can J Physiol Pharmacol (1996) 74:223–33. doi:10.1139/y96-029
235. Greenway CV. Mechanisms and quantitative assessment of drug effects on cardiac output with a new model of the circulation. Pharmacol Rev (1981) 33:213–51.
236. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroeneterology (2012) 143:1158–72. doi:10.1053/j.gastro.2012.09.008
237. Weeder PD, Porte RJ, Lisman T. Hemostasis in liver disease: implications of new concepts for perioperative management. Transfus Med Rev (2014) 28:107–13. doi:10.1016/j.tmrv.2014.03.002
238. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology (2006) 43:S54–62. doi:10.1002/hep.21060
239. Elenkov IJ. Neurohormonal-cytokine interactions: implications for inflammation, common human diseases and well-being. Neurochem Int (2008) 52:40–51. doi:10.1016/j.neuint.2007.06.037
240. Kurosawa S, Kato M. Anesthetics, immune cells, and immune responses. J Anesth (2008) 22:263–77. doi:10.1007/s00540-008-0626-2
241. Angele MK, Faist E. Clinical review: immunodepression in the surgical patient and increased susceptibility to infection. Crit Care (2002) 6:298–305. doi:10.1186/cc1514
242. Hogan BV, Peter MB, Shenoy HG, Horgan K, Hughes TA. Surgery induced immunosuppression. Surgeon (2011) 9:38–43. doi:10.1016/j.surge.2010.07.011
243. Heaney A, Buggy DJ. Can anesthetic and analgesic techniques affect cancer recurrence or metastasis? Br J Anaesth (2012) 109:i17–28. doi:10.1093/bja/aes421
244. Walz CR, Zedler S, Schneider CP, Albertsmeier M, Loehe F, Bruns CJ, et al. The potential role of T-cells and their interaction with antigen-presenting cells in mediating immunosuppression following trauma-hemorrhage. Innate Immun (2009) 15:233–41. doi:10.1177/1753425909104679
245. Elenkov IJ, Chrousos GP. Stress system – organization, physiology and immunoregulation. Neuroimmunomodulation (2006) 13:257–67. doi:10.1159/000104853
246. Martino M, Rocchi G, Escelsior A, Fornaro M. Immunomodulation mechanism of antidepressants: interactions between serotonin and norepinephrine balance and Th1/Th2 balance. Curr Neuropharmacol (2012) 10:97–123. doi:10.2174/157015912800604542
247. Carrico CJ, Meakins JL, Marshall JC, Fry D, Maier RV. Multiple-organ-failure syndrome. Arch Surg (1986) 121:196–208. doi:10.1001/archsurg.1986.01400020082010
248. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med (2011) 208:2581–90. doi:10.1084/jem.20111354
249. Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Annu Rev Immunol (2012) 30:313–35. doi:10.1146/annurev-immunol-020711-075015
250. Nowbar AN, Cole GD, Shun-Shin MJ, Finegold JA, Francis DP. International RCT-based guidelines for use of preoperative stress testing and perioperative beta-blockers and statins in non-cardiac surgery. Int J Cardiol (2014) 171:138–43. doi:10.1016/j.ijcard.2013.12.309
251. Poldermans D, Bax JJ, Boersma E, De Hert S, Eeckhout E, Fowkes G, et al. Guidelines for preoperative cardiac risk assessment and perioperative cardiac management in non-cardiac surgery: the task force for preoperative cardiac risk assessment and perioperative cardiac management in non-cardiac surgery of the European society of cardiology and endorsed by the European society of anaesthesiology. Eur J Anaesthesiol (2010) 27(2):92–137. doi:10.1097/EJA.0b013e328334c017
252. Skrlin S, Hou V. A review of perioperative statin therapy for noncardiac surgery. Semin Cardiothorac Vasc Anesth (2010) 14:283–90. doi:10.1177/1089253210386387
253. Singh N, Patel P, Wyckoff T, Augoustides JG. Progress in perioperative medicine: focus on statins. J Cardiothorac Vasc Anesth (2010) 24:892–6. doi:10.1053/j.jvca.2010.06.015
254. Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov (2005) 4:977–87. doi:10.1038/nrd1901
255. Group HPSC. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet (2002) 360:7–22. doi:10.1016/S0140-6736(02)09327-3
256. Gurm HS, Hoogwerf B. The heart protection study: high-risk patients benefit from statins, regardless of LDL-C level. Cleve Clin J Med (2003) 70:991–7. doi:10.3949/ccjm.70.11.991
257. Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol (2005) 46:1855–62. doi:10.1016/j.jacc.2005.05.085
258. de Waal BA, Buise MP, Van Zundert AA. Perioperative statin therapy in patients at high risk for cardiovascular morbidity undergoing surgery: a review. Br J Anaesth (2015) 114:44–52. doi:10.1093/bja/aeu295
259. Paraskevas KI, Veith FJ, Liapis CD, Mikhailidis DP. Perioperative/periprocedural effects of statin treatment for patients undergoing vascular surgery or endovascular procedures: an update. Curr Vasc Pharmacol (2013) 11:112–20. doi:10.2174/1570161111309010112
260. Liakopoulos OJ, Kuhn EW, Slottosch I, Wassmer G, Wahlers T. Preoperative statin therapy for patients undergoing cardiac surgery. Cochrane Database Syst Rev (2012) 4:CD008493. doi:10.1002/14651858.CD008493.pub2
261. Sanders RD, Nicholson A, Lewis SR, Smith AF, Alderson P. Perioperative statin therapy for improving outcomes during and after noncardiac vascular surgery. Cochrane Database Syst Rev (2013) 7:CD009971. doi:10.1002/14651858.CD009971.pub2
262. Ouattara A, Benhaoua H, Le Manach Y, Mabrouk-Zerguini N, Itani O, Osman A, et al. Perioperative statin therapy is associated with a significant and dose-dependent reduction of adverse cardiovascular outcomes after coronary artery bypass graft surgery. J Cardiothorac Vasc Anesth (2009) 23:633–8. doi:10.1053/j.jvca.2009.02.008
263. Kulik A, Ruel M. Statins and coronary artery bypass graft surgery: preoperative and postoperative efficacy and safety. Expert Opin Drug Saf (2009) 8:559–71. doi:10.1517/14740330903188413
264. Chopra V, Wesorick DH, Sussman JB, Greene T, Rogers M, Froehlich JB, et al. Effect of perioperative statins on death, myocardial infarction, atrial fibrillation, and length of stay: a systematic review and meta-analysis. Arch Surg (2012) 147:181–9. doi:10.1001/archsurg.2011.897
265. Koh KK, Sakuma I, Quon MJ. Differential metabolic effects of distinct statins. Atherosclerosis (2011) 215:1–8. doi:10.1016/j.atherosclerosis.2010.10.036
266. Williams TM, Harken AH. Statins for surgical patients. Ann Surg (2008) 247:30–7. doi:10.1097/SLA.0b013e3181492c0d
267. Poldermans D, Devereaux PJ. The experts debate: perioperative beta-blockade for noncardiac surgery – proven safe or not? Cleve Clin J Med (2009) 76:S84–92. doi:10.3949/ccjm.76.s4.14
268. Plosker GL. Landiolol: a review of its use in intraoperative and postoperative tachyarrhythmias. Drugs (2013) 73:959–77. doi:10.1007/s40265-013-0077-4
269. Shahian DM. Preoperative β-blockade in coronary artery bypass grafting surgery. JAMA Intern Med (2014) 174(8):1328–9. doi:10.1001/jamainternmed.2014.155
270. Kim Y, Hwang W, Cho ML, Her YM, Ahn S, Lee J. The effects of intraoperative esmolol administration on perioperative inflammatory responses in patients undergoing laparoscopic gastrectomy: a dose-response study. Surg Innov (2015) 22(2):177–82. doi:10.1177/1553350614532534
271. Brinkman W, Herbert MA, O’brien S, Filardo G, Prince S, Dewey T, et al. Preoperative β-blocker use in coronary artery bypass grafting surgery: national database analysis. JAMA Intern Med (2014) 174(8):1320–7. doi:10.1001/jamainternmed.2014.2356
272. Wiesbauer F, Schlager O, Domanovits H, Wildner B, Maurer G, Muellner M, et al. Perioperative beta-blockers for preventing surgery-related mortality and morbidity: a systematic review and meta-analysis. Anesth Analg (2007) 104:27–41. doi:10.1213/01.ane.0000247805.00342.21
273. Bangalore S, Wetterslev J, Pranesh S, Sawhney S, Gluud C, Messerli FH. Perioperative beta blockers in patients having non-cardiac surgery: a meta-analysis. Lancet (2008) 372:1962–76. doi:10.1016/S0140-6736(08)61560-3
274. Devereaux PJ, Yang H, Yusuf S, Et A. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet (2008) 371:1839–47. doi:10.1016/S0140-6736(08)60601-7
275. Angeli F, Verdecchia P, Karthikeyan G, Mazzotta G, Gentile G, Reboldi G. ß-Blockers reduce mortality in patients undergoing high-risk non-cardiac surgery. Am J Cardiovasc Drugs (2010) 10:247–59. doi:10.2165/11539510-000000000-00000
276. Blessberger H, Kammler J, Domanovits H, Schlager O, Wildner B, Azar D, et al. Perioperative beta-blockers for preventing surgery-related mortality and morbidity. Cochrane Database Syst Rev (2014) 18:CD004476. doi:10.1002/14651858.CD004476.pub2
277. Cole GD, Francis DP. Perioperative β blockade: guidelines do not reflect the problems with the evidence from the DECREASE trials. BMJ (2014) 349:g5210. doi:10.1136/bmj.g5210
278. Bouri S, Shun-Shin MJ, Cole GD, Mayet J, Francis DP. Meta-analysis of secure randomised controlled trials of β-blockade to prevent perioperative death in non-cardiac surgery. Heart (2014) 100:456–64. doi:10.1136/heartjnl-2013-304262
279. Viljoen JF, Estafanous FG, Kellner GA. Propranolol and cardiac surgery. J Thorac Cardiovasc Surg (1972) 64:826–30.
280. Ogawa S, Okawa Y, Goto Y, Aoki M, Baba H. Perioperative use of a beta blocker in coronary artery bypass grafting. Asian Cardiovasc Thorac Ann (2013) 21:265–9. doi:10.1177/0218492312451166
281. Blessberger H, Kammler J, Steinwender C. Perioperative use of β-blockers in cardiac and noncardiac surgery. JAMA (2015) 313:2070–1. doi:10.1001/jama.2015.1883
282. Bangalore S, Makani H, Radford M, Thakur K, Toklu B, Katz SD, et al. Clinical outcomes with β-blockers for myocardial infarction a meta-analysis of randomized trials. Am J Med (2014) 127(10):939–53. doi:10.1016/j.amjmed.2014.05.032
283. Wijeysundera DN, Beattie WS, Rao V, Ivanov J, Karkouti K. Calcium antagonists are associated with reduced mortality after cardiac surgery: a propensity analysis. J Thorac Cardiovasc Surg (2004) 127:755–62. doi:10.1016/S0022-5223(03)00695-0
284. Biccard BM, Howell SJ. Calcium channel blockers, beta-blockers, and perioperative cardiovascular stability. Br J Anaesth (2008) 101:439–41. doi:10.1093/bja/aen247
285. Kertai MD, Westerhout CM, Varga KS, Acsady G, Gal J. Dihydropiridine calcium-channel blockers and perioperative mortality in aortic aneurysm surgery. Br J Anaesth (2008) 101:458–65. doi:10.1093/bja/aen173
286. Kluger MT, Gale S, Plummer JL, Owen H. Peri-operative drug prescribing pattern and manufacturers’ guidelines. An audit. Anaesthesia (1991) 46:456–9. doi:10.1111/j.1365-2044.1991.tb11682.x
287. Dobson GP. Addressing the global burden of sepsis: importance of a systems-based approach. Crit Care Med (2014) 42:e797–8. doi:10.1097/CCM.0000000000000595
288. Shoemaker WC, Beez M. Pathophysiology, monitoring, and therapy of shock with organ failure. Appl Cardiopulm Pathophyiol (2010) 14:5–15.
289. Aird WC. Spatial and temporal dynamics of the endothelium. J Thromb Haemost (2005) 3:1392–406. doi:10.1111/j.1538-7836.2005.01328.x
290. Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev (2004) 84:869–901. doi:10.1152/physrev.00035.2003
291. Reitsma S, Slaaf DW, Vink H, Van Zandvoort MA, Oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch (2007) 454:345–59. doi:10.1007/s00424-007-0212-8
292. Woodcock TE, Woodcock TM. Revised starling equation and the glycocalyx model of transvascular fluid exchange: an improved paradigm for prescribing intravenous fluid therapy. Br J Anaesth (2012) 108:384–94. doi:10.1093/bja/aer515
293. Aditianingsih D, George YWH. Guiding principles of fluid and volume therapy. Best Pract Res Clin Anaesthesiol (2014) 28:249–60. doi:10.1016/j.bpa.2014.07.002
294. Chappell D, Jacob M. Role of the glycocalyx in fluid management: small things matter. Best Pract Res Clin Anaesthesiol (2014) 28:227–34. doi:10.1016/j.bpa.2014.06.003
295. Chappell D, Westphal M, Jacob M. The impact of the glycocalyx on microcirculatory oxygen distribution in critical illness. Curr Opin Anaesthesiol (2009) 22:155–62. doi:10.1097/ACO.0b013e328328d1b6
296. Biddle C. Like a slippery fish, a little slime is a good thing: the glycocalyx revealed. AANA J (2013) 81(6):473–80.
297. Zeng Y, Tarbell JM. The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS One (2014) 9:e86249. doi:10.1371/journal.pone.0086249
299. Dobson GP. Membrane polarity: a target for myocardial protection and reduced inflammation in adult and pediatric cardiothoracic surgery (editorial – free standing). J Thorac Cardiovasc Surg (2010) 140:1213–7. doi:10.1016/j.jtcvs.2010.05.040
300. Onorati F, Santini F, Dandale R, Ucci G, Pechlivanidis K, Menon T, et al. “Polarizing” microplegia improves cardiac cycle efficiency after CABG for unstable angina. Int J Cardiol (2013) 167:2739–46. doi:10.1016/j.ijcard.2012.06.099
301. Canyon SJ, Dobson GP. Protection against ventricular arrhythmias and cardiac death using adenosine and lidocaine during regional ischemia in the in vivo rat. Am J Physiol Heart Circ Physiol (2004) 287:H1286–95. doi:10.1152/ajpheart.00273.2004
302. Canyon SJ, Dobson GP. The effect of adenosine and lidocaine infusion on myocardial high energy phosphates and pH during regional ischemia in the rat model in vivo. Can J Physiol Pharmacol (2006) 84:903–12. doi:10.1139/Y06-035
303. Letson HL, Dobson GP. Small-volume 7.5% NaCl with 6% dextran-70 or 6% and 10% Hetastarch are associated with arrhythmias and death following 60 min of severe hemorrhagic shock in the rat in vivo. J Trauma (2011) 70:1444–52. doi:10.1097/TA.0b013e3181e99c63
304. Letson HL, Dobson GP. Unexpected 100% survival following 60% blood loss using small-volume 7.5% NaCl with adenocaine and Mg2+ in the rat model of extreme hemorrhagic shock. Shock (2011) 36:586–94. doi:10.1097/SHK.0b013e318237eb0c
305. Letson HL, Pecheniuk NM, Mhango LP, Dobson GP. Reversal of acute coagulopathy during hypotensive resuscitation using small-volume 7.5% NaCl with adenocaine and Mg2+ in the rat model of severe hemorrhagic shock. Crit Care Med (2012) 40:2417–22. doi:10.1097/CCM.0b013e31825334c3
306. Letson HL, Dobson GP. Correction of acute traumatic coagulopathy with small-volume 7.5% NaCl adenosine, lidocaine and Mg2+ (ALM) occurs within 5 min: a ROTEM Analysis. J Trauma Acute Care Surg (2015) 78:773–83. doi:10.1097/TA.0000000000000587
307. Shi W, Jiang R, Dobson GP, Granfeldt A, Vinten-Johansen J. The non-depolarizing, normokalemic cardioplegia formulation adenosine-lidocaine (adenocaine) exerts anti-neutrophil effects by synergistic actions of its components. J Thorac Cardiovasc Surg (2012) 143:1167–75. doi:10.1016/j.jtcvs.2011.06.045
308. Granfeldt A, Letson HL, Dobson GP, Shi W, Vinten-Johansen J, Tønnesen E. Adenosine, lidocaine and Mg2+ improves cardiac and pulmonary function, induces reversible hypotension and exerts anti-inflammatory effects in an endotoxemic porcine model. Crit Care (2015) 18:682. doi:10.1186/s13054-014-0682-y [Epub ahead of print].,
309. Djabir Y, Dobson GP. Hemodynamic rescue and ECG stability during chest compressions using adenocaine following 8 min asphyxial hypoxia in the rat. Am J Emerg Med (2013) 31:1539–45. doi:10.1016/j.ajem.2013.05.044
310. Granfeldt A, Letson HL, Hyldebrandt JA, Wang ER, Salcedo PA, Nielson TK, et al. Small-Volume 7.5% NaCl adenosine, lidocaine and Mg2+ has multiple benefits during hypotensive and blood resuscitation in the pig following severe blood loss: rat to pig translation. Crit Care Med (2014) 42:e329–44. doi:10.1097/CCM.0000000000000225
311. Granfeldt A, Nielsen TK, Solling C, Hyldebrandt JA, Frøkiær J, Wogensen L, et al. Adenocaine and Mg2+ reduces fluid requirement to maintain hypotensive resuscitation and improves cardiac and renal function in a porcine model of severe hemorrhagic shock. Crit Care Med (2012) 40:3013–25. doi:10.1097/CCM.0b013e31825fd4ed
312. Griffin MJ, Letson HL, Dobson GP. Adenosine, lidocaine and Mg2+ (ALM) induces a reversible hypotensive state, reduces lung edema and prevents coagulopathy in the rat model of polymicrobial sepsis. J Trauma Acute Care Surg (2014) 77:471–8. doi:10.1097/TA.0000000000000361
313. Lamke LO, Nilsson GE, Reithner HL. Water loss by evaporation from the abdominal cavity during surgery. Acta Chir Scand (1977) 143:279–84.
314. Miller TE, Raghunathan K, Gan TJ. State-of-the-art fluid management in the operating room. Best Pract Res Clin Anaesthesiol (2014) 28:261–73. doi:10.1016/j.bpa.2014.07.003
315. Maitland K, George EC, Evans JA, Kiguli S, Olupot-Olupot P, Akech SO, et al. Exploring mechanisms of excess mortality with early fluid resuscitation: insights from the FEAST trial. BMC Med (2013) 11:68. doi:10.1186/1741-7015-11-68
316. Myburgh JA, Finfer S. Causes of death after fluid bolus resuscitation: new insights from FEAST. BMC Med (2013) 11:67. doi:10.1186/1741-7015-11-67
Keywords: perioperative, injury, trauma, surgery, inflammation, coagulopathy, endothelium, sepsis
Citation: Dobson GP (2015) Addressing the global burden of trauma in major surgery. Front. Surg. 2:43. doi: 10.3389/fsurg.2015.00043
Received: 08 July 2015; Accepted: 17 August 2015;
Published: 03 September 2015
Edited by:
Sarah Longnus, University of Berne, SwitzerlandReviewed by:
Antonio Miceli, Istituto Clinico Sant’Ambrogio, ItalyPradeep Narayan, Rabindranath Tagore International Institute of Cardiac Sciences, India
Copyright: © 2015 Dobson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geoffrey P. Dobson, Heart, Trauma and Sepsis Research Laboratory, Australian Institute of Tropical Health and Medicine, College of Medicine and Dentistry, James Cook University, 1 James Cook University Drive, Townsville, QLD 4811, Australia,Z2VvZmZyZXkuZG9ic29uQGpjdS5lZHUuYXU=