 Yang Ouyang
Yang Ouyang Jennifer R. Reeve
Jennifer R. Reeve Jeanette M. Norton
Jeanette M. Norton- 1Department of Plants, Soils and Climate, Utah State University, Logan, UT, United States
- 2Department of Microbiology and Plant Biology, Institute for Environmental Genomics, University of Oklahoma, Norman, OK, United States
Organic amendments are applied in organic farming systems to provide nutrients for crop uptake and to improve soil health. Compost is often favored over fresh manure for food safety reasons, while fresh manure can be a valuable source of readily available nitrogen (N). However, the potential for fresh versus composted manure to differentially affect soil microbial and N-cycling functional communities over multiple seasons remains unknown. We compared the effect of composted vs. fresh cattle manure on soil microbial communities using taxonomic and functional approaches. Soils were collected from field plots with three organic N treatments: control (no amendment), composted manure (compost, 224 kg/ha total N), and fresh manure (manure, 224 kg/ha total N) in an organic production system. Illumina amplicon sequencing was used to comprehensively assess the bacterial community (16S rRNA genes), fungal community (ITS), ureolytic community (ureC), chitinolytic community (chiA), bacterial ammonia oxidizers (AOB amoA), and nitrite oxidizers (Nitrospira nxrB). The results showed that both compost and manure treatment significantly changed the soil microbial communities. Manure had a stronger effect than compost on soil bacterial and fungal community composition, as well as on the ureolytic and chitinolytic communities, while compost treated soils had higher microbial richness than manure treated soils. Both taxonomic and functional approaches showed that the microbial community was more responsive to fresh manure than to compost. Manure treated soil also had more complex microbial interactions than compost treated soil. The abundance and community composition of N-cycling functional groups often played more limited roles than soil chemical properties (soil organic carbon, extractable organic carbon, and pH) in driving N-cycling processes. Results from our study may guide strategies for the management of organic amendments in organic farming systems and provide insights into the linkages between soil microbial communities and soil function.
Introduction
Organic amendments are widely used in organic farming systems to supply nutrients for crop uptake and to improve soil health. Compost is often favored over fresh manure for food safety reasons as well as for ease of transportation and application due to its reduced moisture content and lower vitality of weed seeds. However, fresh manure can be a valuable source of readily available N in organic farming systems and hence is often valued by farmers especially when applied to nutrient demanding crops such as corn. The release of nutrients from organic amendments is largely mediated by soil microbes and their enzymes. A number of studies have found that compost and/or manure have altered soil microbial communities (1–5), while others have shown no or little effect (6, 7). This variability in findings is likely due to the type, rate and frequency of the compost and manure that were applied (5, 8). However, few studies have directly compared the effect of fresh manure versus compost side by side, especially with the same total nitrogen (N) application rate, on the soil microbial communities in organic farming systems over multiple years. For the same total N application rate, manure often has higher N availability, but less overall material is applied than with compost. Differences between manure and compost in carbon (C) and N content and their availability may result in contrasting impacts on soil microbial communities. Improving management of organic amendments for a desirable response in soil microbiomes and their nutrient cycling processes is a long-term goal for sustainable agriculture.
The transformation of organic polymers to biologically available N forms is largely controlled by N mineralization and nitrification. N mineralization converts organic polymers to monomers or ammonium (9), while nitrification transforms ammonium to nitrite or nitrate (10). Numerous microbial enzymes are involved in N mineralization (11). Previous studies have consistently showed that organic amendments increased the activities of soil enzymes involved in N mineralization (reviewed in Luo et al., 2018 (12)). However, few studies have examined the effect of organic amendments on the diversity of genes encoding these key N mineralization enzymes (2, 13, 14). Several studies have assessed the effect of organic fertilization on the communities of nitrifiers, but the results are controversial (15–18). This is probably due to the differences in rates, quality, and duration of organic fertilization. The current study examines the effect of fresh steer manure versus composted cattle manure on select N mineralizers and nitrifiers.
Microbial co-occurrence networks provide critical insights into microbial associations (19–21). Network analysis has been widely used to examine the response of microbial associations to agricultural management and to identify putative keystone species (22–25). Several studies have revealed that organic fertilization increased the complexity of microbial interaction compared with mineral fertilization (22, 26). Compared with bacteria and fungi, it is less understood how different soil N-cycling functional groups interact and respond to the quality of organic fertilizers. Our study seeks to use an existing organic long-term rotational experiment with a robust experimental design to examine microbial community changes in response to organic fertilizer amendments.
The use of manures and composts is common on organic farms yet knowledge gaps in their microbial characteristics remain for their safe and sustainable use (27). Previous research at our site indicated fresh manure released more available N for plant uptake while composted manure increased soil organic C, with no differences in soil enzymatic activity involved in nutrient cycling detected (28). In this study, we measure bacterial and fungal community composition in soils treated with fresh manure and composted manure at the same total N application rate. We expected that soil microbial community compositions and their co-occurrence networks would be different between manure and compost treated soils. To complement our previous study, which demonstrated organic N fertilizers increased soil functions and functional gene abundances (28). we examined the community composition using four functional genes involved in N mineralization and nitrification. We hypothesized that the community compositions of these N-cycling functional groups would be changed by organic N fertilizers and that the community composition might play a role in prediction of the corresponding microbial process rates.
Methods
Site history and soil sampling
The field site and experiment design is a certified organic farming systems trial that was started in 2007 and has been previously described (28). In brief, the experiment consisted of three different rotations incorporating three different cover crops with or without fresh manure or composted manure in a completely randomized block with split split plot design. Cover crop (buckwheat (Fagopyrum esculentum, Moench), millet (Pennisetum glaucum L.), or black turtle bean (Phaseolus vulgaris L.)) was the main plot factor, crop rotation the sub plot factor and fertility treatment (compost, manure, nothing as control) the sub-sub plot factor, each with three replicates. In this study, we used the most intensive rotation: field corn (in 2007), potatoes (2008), dry beans (2009), sweet corn (2011), potatoes (2012), cover crops (2013), dry beans (2014), sweet corn (in 2015). In total, 27 plots were selected with three organic N amendment treatments, three cover crop treatments, and three replicates. A cover crop mixture of winter wheat (Triticum aestivum L), and hairy vetch (Vicia villosa L.) was grown every year across all treatments over the winter and mowed and incorporated immediately prior to applying composted or fresh manure. Organic fertilizers were applied and immediately incorporated two weeks before planting in mid-June. Organic fertilizers were not applied to cover crops in 2013 and beans in 2014. For each fertilization, field plots received applications of 224 kg/ha total N either in the form of a commercial steer manure compost (compost), fresh cattle manure (manure) or no amendment (control). The basic characteristics of the compost and manure were previously described (28). Each year, manure was selected with a higher total N content than the compost so that manure plots received less overall material for the same total N application rate. The C:N ratio of the compost (1:12 to 1:16) was higher than the manure three years out of six but otherwise similar (see Supplementary Figure 1. for rotations and treatment timing).
Soil samples (0-15 cm) were collected in late Sep 2011 (after corn harvest), in Jul 2015 (six weeks after fertilization), and in early Oct 2015 (after corn harvest). Six soil cores were randomly taken from each plot, composited and thoroughly mixed, and about 10 g sample of soil was stored at -80°C immediately after soils were brought to the laboratory. Soil chemical and microbiological properties were measured and described in Ouyang et al. (28). In this study, we used Illumina amplicon sequencing to evaluate the overall prokaryotic and fungal communities in soils sampled in Sep 2011 and Oct 2015. Illumina amplicon sequencing was performed for four N-cycling genes in soils sampled in Jul 2015 (See Supplementary Figure 1).
Illumina sequencing and data processing for 16S rRNA genes and fungal ITS
Soil DNA was extracted using a MoBio PowerSoil DNA isolation kit (MoBio Laboratories Inc, Carlsbad, USA). DNA extracts were quantified by using the Quant-iT™ PicoGreen dsDNA BR Assay Kit (Invitrogen) according to the manufacturer’s protocol. The variable V4-V5 region of the 16S ribosomal rRNA gene (16S) was amplified with 515F-Y (GTGYCAGCMGCCGCGGTAA) and 926R (CCGYCAATTYMTTTRAGTTT) universal primers for the bacterial and archaeal community (29). The fungal internal transcribed spacer (ITS) was amplified with the primer pair ITS9F (GAACGCAGCRAAIIGYGA) and ITS948R (TCCTCCGCTTATTGATATGC). The amplicon sequencing was performed on an Illumina MiSeq instrument (Illumina Inc., San Diego, CA, USA). The Illumina raw reads were processed using a custom pipeline developed at the Joint Genome Institute (JGI) (https://bitbucket.org/berkeleylab/jgi_itagger). Briefly, raw reads were first quality-filtered, and then the high quality sequences were clustered into amplicon sequence variants (ASV) based on zero-radius operational taxonomic units (OTU) using the USEARCH pipeline (30). Singletons were removed. Taxonomies were assigned to each ASV using the RDP Classifier with a confidence threshold of 0.60 (31). Maximum-likelihood phylogenetic trees were constructed from representative sequences using FastTree with default parameters (32). All data files were then organized using R package phyloseq (33). Sequence data can be accessed from JGI data portal at https://data.jgi.doe.gov/ and searching for Utah Georgia Soil iTags (plate 1 and plate 2 are available).
Illumina sequencing and data processing for four N-cycling genes
Amplicon sequencing of four N-cycling genes was accomplished for soils sampled in July 2015 (six weeks after fertilization). Four N-cycling genes were ureC (encoding urease), chiA (encoding chitinase), bacterial amoA (encoding ammonia monooxygenase) and Nitrospira nxrB (encoding nitrite oxidoreductase). The primer sets and library preparation of Miseq sequencing were the same as our previous studies (2, 16). We tried to design new archaeal amoA primers with amplicon length less than 500bp, but the primer sets failed to amplify archaeal amoA in our soils. The sequencing data was processed using RDPTools (34) and USEARCH (30). Briefly, raw forward and reverse reads were merged using the USEARCH workflow (30). High quality sequences were extracted from merged reads in each sample using the RDP SeqFilters with a read Q score cutoff of 20 (34). Chimera sequences were detected and removed using UCHIME (35). The obtained sequences were further processed using the FrameBot tool (36) to fix frame shifts. Sequences were dereplicated and clustered at 90% nucleotide similarity using USEARCH. Singletons were removed. To obtain the phylum-level classification of representative sequences for ureC and chiA, the taxonomy from the closet matches to the protein reference downloaded from the FunGene were used (37). If the percent identity to the reference sequences was less than 80%, we defined the phylum as unclassified. A maximum-likelihood phylogenetic tree was constructed from representative sequences using FastTree with default parameters (32). OTU table and taxonomy files were further organized for diversity analysis using R package phyloseq (33). The Sequence Read Archive for the amplicon sequencing is available through Bioproject PRJNA804976 at https://www.ncbi.nlm.nih.gov//bioproject/PRJNA804976.
Network analysis
We aimed to examine the effect of organic fertilizers on microbial network properties. Three networks were constructed for each organic N treatment. Cover crop treatments and samples in 2011 and 2015 were pooled together in each organic N treatment. Therefore, there were 18 replicates in each N treatment. To ensure network reliability, mitigate temporal variance, and compare organic fertilizer, only bacterial and fungal ASVs detected in all 54 samples were used for each network construction. Finally, 854 bacterial and 25 fungal ASVs were used for network construction. The networks were built using the pipeline of Molecular Ecological Network Analyses (MENA) (http://ieg4.rccc.ou.edu/mena/) (38). Briefly, pairwise similarities of ASVs were calculated based on Spearman correlation coefficients. The random matrix theory was applied to identify the similarity threshold before network construction (19, 38). Network topological features were calculated by the MENA pipeline, including the total node number, total link number, average degree, average cluster coefficient, average path distance, and modularity. Network modules were identified by the fast-greedy modularity optimization. Detailed definitions of these network features were described previously (39, 40). To randomize network in each organic N treatment, a total 100 randomly rewired networks were generated (19). To test if there were differences in network topological characteristics among the three organic N treatments, one-way ANOVA was performed by using averages and standard deviations of their corresponding 100 random networks. Putative keystone taxa were also identified based on network hubs, module hubs, and connectors (38). All networks were visualized using Gephi (41).
To evaluate the effect of organic N fertilizers on the network properties of N-cycling genes, OTUs presented in all soil samples were obtained to build three networks for each organic N treatment. There were 595 OTUs, 40 OTUs, ten OTUs, and seven OTUs for ureC, chiA, amoA, and nxrB, respectively. Nine replicates regardless of the cover crop treatments were pooled together to construct the network in each organic N treatment, since cover crop type had no effect on the microbial communities of N-cycling functional groups. The same procedure as above was used to generate networks. All networks with a focus on the linkage among four N-cycling genes were visualized using Cytoscape (42).
Statistical analysis
All statistical analyses were conducted in R software (https://www.R-project.org). For 16S and ITS sequencing data, we had a very high sequencing depth. The retained high-quality sequences were randomly resampled to a depth of 141,248 and 108,584 reads per sample for 16S and ITS, respectively. For four N-cycling genes, the retained high-quality sequences were randomly resampled to a depth of 20000, 6000, 540, and 1400 reads per sample for ureC, chiA, bacterial amoA, and Nitrospira nxrB, respectively. The rarefaction curves showed that these normalized reads were sufficient to capture the diversity of these four N-cycling genes. Alpha diversity and beta diversity of the microbial communities were then calculated. Nonmetric multidimensional scaling (NMDS) and PERMANOVA were conducted to visualize and assess the Weighted UniFrac distance matrices using the R package vegan version 2.5-6 (43). Canonic correspondence analysis (CCA) was used to evaluate relationships between chemical properties and soil structural and functional microbial communities. Soil chemical properties included soil organic carbon (SOC), total N, pH, extractable organic C (EOC), extractable organic N (EON), amino acids, ammonium, and nitrate. Forward selection of soil chemical properties was performed to determine a parsimonious set of explanatory variables. To evaluate the effect of organic N fertilization on the relative abundances of ASVs or OTUs, the fold changes of ASVs or OTUs in compost or manure versus the control was calculated using the R package DESeq2 (44). The p values were adjusted using the Benjamini-Hochberg correlation method (44). The analysis of variance (ANOVA) and Tukey’s honestly significant difference (HSD) were used to characterize the statistical significance of the differences among organic N treatments.
Several analyses were performed to evaluate the relative importance of soil chemical properties and functional genes in controlling the rates of selected soil N-cycling processes in soil. Four selected N-cycling processes were urease activity (i.e. urea hydrolysis), N-acetyl β-glucosaminidase (NAGase) activity, octyne-sensitive nitrification potential (nitrification potential contributed by AOB, NPAOB), and potential nitrite oxidation (PNO). Firstly, the Mantel test was used to evaluate the relationships between the community composition of N-cycling functional groups and their corresponding process rates. Secondly, Random Forest analysis was used to explore the importance of each soil chemical parameter and the abundance and community of N-cycling functional groups for explaining selected N-cycling process rates. We used NMDS1 to represent the community composition of N-cycling functional groups. The Random Forest analysis was conducted using the R package randomForest. The significance of each predictor on N-cycling process rates was assessed with the R package rfPermute. Lastly, structural equation modeling (SEM) was used to evaluate the direct or indirect effects of soil chemical properties and the abundance and diversity of N-cycling groups on selected N-cycling process rates. To simplify the SEM, only three soil chemical factors (SOC, EOC, and pH) were included in the model, since the Random Forest analysis showed that these three factors were all significant for predicting the four N-cycling process rates. We first generated a full model that included all reasonable pathways, and then sequentially eliminated nonsignificant pathways. The χ2 test and the root mean square error of approximation were used to evaluate the fit of our model. The SEM analysis was performed using the R package lavaan.
Results
Similar to the results for soil basic properties and soil function in our previous study (Supplementary Table 1; 28), we found that cover crop type had no effect on the compositions of bacterial and fungal communities, and selected N-cycling functional groups (Supplementary Tables 2, 3). Therefore, we focused on the effects of organic N treatment only for this study.
Bacterial and fungal community compositions
Both organic N treatment and year significantly changed soil bacterial (R2N = 0.21, p < 0.001; R2year=0.30, p < 0.001) and fungal (R2N = 0.14, p < 0.001; R2year=0.10, p < 0.001) community composition (Figure 1 and Supplementary Table 2). Pairwise comparison indicated that the three organic N treatments were different from each other in the soil bacterial and fungal community composition. Overall, manure had a stronger impact than compost on the soil bacterial and fungal community composition, based on their dissimilar distances to the control soil. Organic N treatment (p < 0.001) and year (p < 0.001) also significantly influenced the alpha diversity of the soil bacterial community (Supplementary Figure 2A). Specifically, compost treatment increased richness, Chao1, and Shannon diversity in both 2011 and 2015. For the soil fungal community, compost treatment increased the alpha diversity in 2011, but organic N treatment had no effect on the alpha diversity in 2015 (Supplementary Figure 2B).
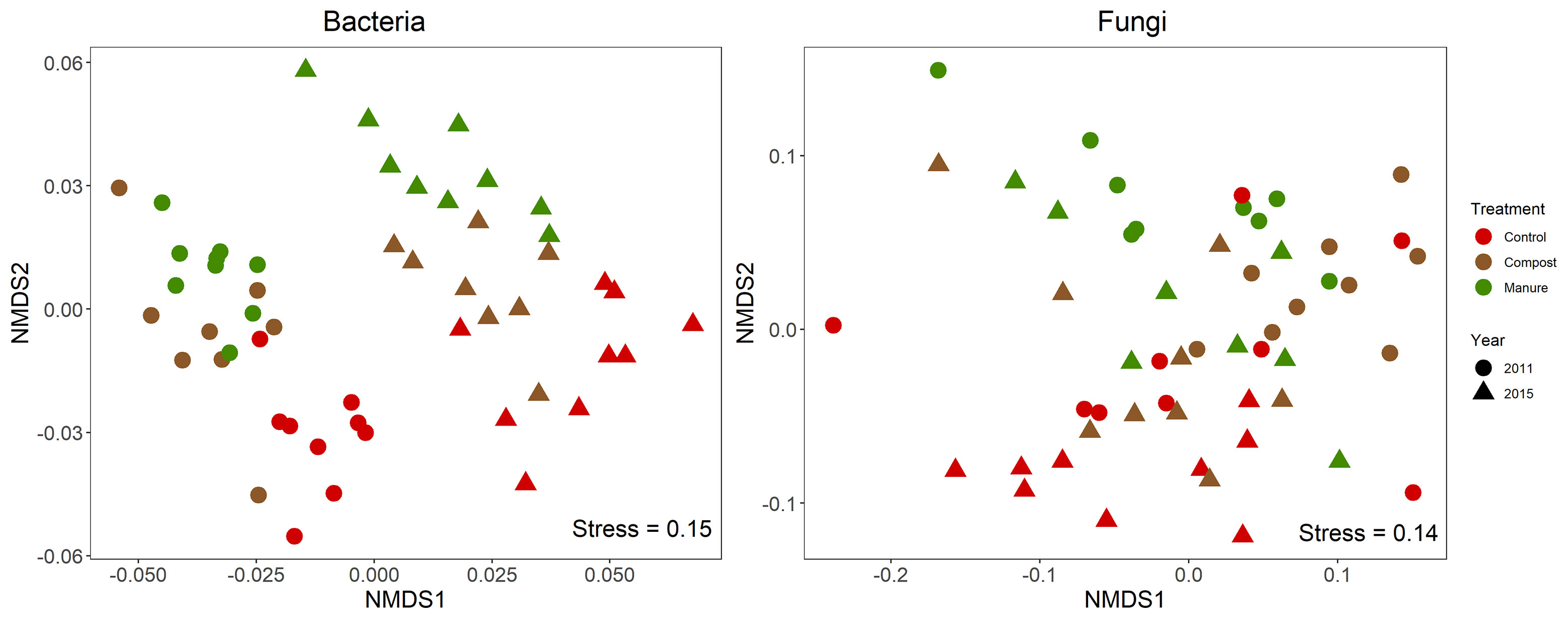
Figure 1 Nonmetric multidimensional scaling (NMDS) ordination of the weighted UniFrac distance for soil bacterial and fungal communities. PERMANOVA indicates significant effect of organic N treatment and Year on both bacterial and fungal community compositions (p < 0.001).
Responsive bacterial and fungal taxa to organic N fertilizers
Organic N fertilizers significantly changed many bacterial taxa. Five of 12 abundant bacterial phyla (> 1%) were significantly changed by organic N treatment. Organic N fertilizers increased Proteobacteria and Bacteroidetes, but decreased Acidobacteria, Chloroflexi, and Gemmatimonadetes in both 2011 and 2015 (Figure 2A). Overall, these five responsive phyla showed no difference between compost and manure treatments. Only Chloroflexi in the manure treatment was significantly lower than the compost treatment in 2015. Numerous bacterial ASVs were enriched (16.19%) or depleted (12.68%) by organic N fertilizers (Figure 2B). Most of these responsive ASVs belonged to Proteobacteria, Bacteroidetes, Planctomycetes, Chloroflexi, Actinobacteria, and Acidobacteria. Manure treated soil had more numbers of responsive ASVs than compost treated soil in both 2011 and 2015 (Supplementary Figure 3A). In both compost and manure treated soils, there were higher numbers of responsive ASVs in 2015 than in 2011. Many responsive ASVs were shared between compost and manure treated soils (50-61%), or between 2011 and 2015 (36-42%). Both the numbers and log2-fold change values of responsive ASVs responded more positively than negatively to compost and manure treatment (Supplementary Figure 3B).
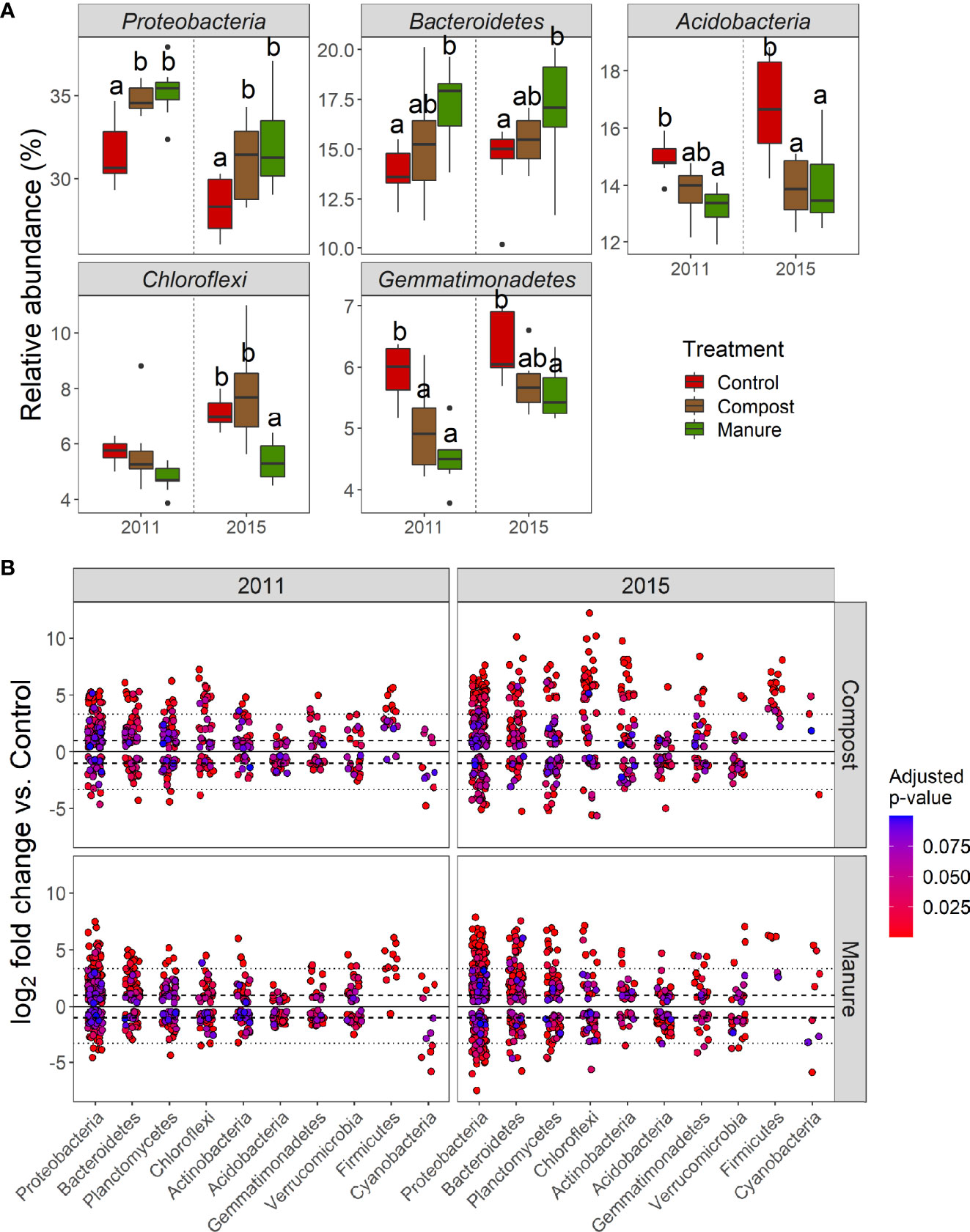
Figure 2 (A) Relative abundance of several bacteria phyla (> 1%) are significantly different among organic N treatments (p < 0.05, ANOVA). Lowercase letters indicate significant differences among organic N treatment in a specific year. (B) Relative abundance changes (log2-fold change) of bacterial ASVs in soils treated with compost and manure compared with the control soil. Each circle represents a single bacterial ASV with an adjusted p value of < 0.1. Dashed line: 2-fold change. Dotted lines: 10-fold change.
Compared with bacteria, fungal taxa were less responsive to organic N fertilizers. At the phylum level, we found that Ascomycota and Zygomycota were significantly affected by organic N treatment (Supplementary Figure 4A). Specifically, manure treatment significantly decreased Ascomycota in 2011, and compost treatment significantly increased Zygomycota in 2015 compared with control. There were also fewer fungal responsive ASVs than bacterial responsive ASVs (Supplementary Figure 4B). In 2011, there were only 24 and 35 responsive ASVs in compost and manure treatments, respectively. In 2015, there were 45 and 57 responsive ASVs in compost and manure treatments, respectively. Most of these responsive ASVs were enriched by organic N treatment.
Microbial network
Using bacterial and fungal ASVs present in all soil samples, we built networks for each organic N treatment. These three networks were non-random and scale-free with power-law R2 ranging from 0.84 to 0.88 (Table 1). Compared with the control, networks in compost and manure treatments had higher total nodes, total links, numbers of modules, and average degree (Table 1 and Figure 3). Based on 100 randomized networks, average path distance was lowest in the manure treatment, medium in compost treatment and highest in control treatment (p < 0.001). Compost and manure treatments also had significant higher average clustering coefficients than the control (p < 0.001). We also calculated the percentage of negative correlations and found that compost and manure treatments had higher negative interactions than the control (Table 1).
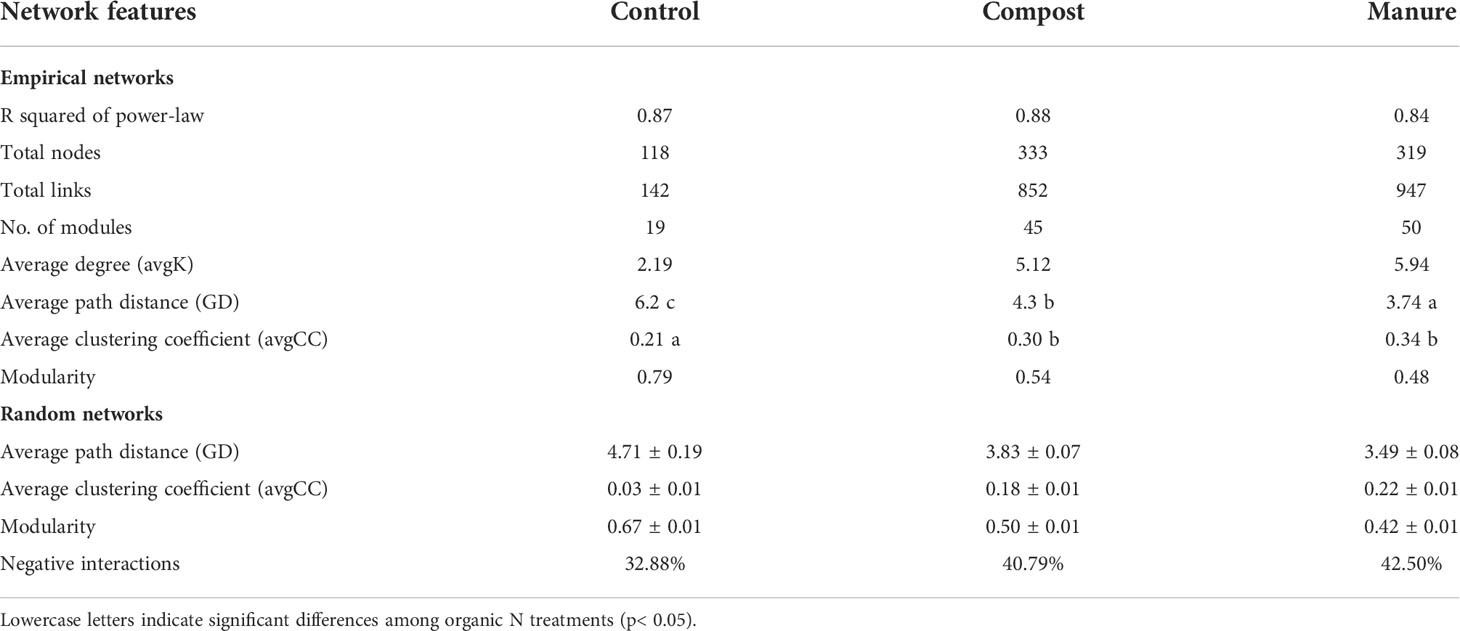
Table 1 Topological features of empirical networks and random networks for combined bacterial and fungal communities.

Figure 3 Microbial networks under three organic N treatments. Networks are built using the random matrix theory. Nodes represent ASVs and links indicate significant correlation. Modules are randomly colored. Detailed topological features of networks are shown in Table 1.
Putative keystone taxa were also identified by the network analysis (Supplementary Table 4). We only identified one connector in the control network, belonging to Verrucomicrobia. There were six connectors and five module hubs in the compost network. Most of these connectors and module hubs were in Acidobacteria. In the manure network, there were 22 connectors and three module hubs, mainly belonging to Protobacteria, Acidobacteria, and Planctomycetes. Module hubs and connectors were largely from bacteria, and we identified only one fungal connector (Chytridiomycota) in the compost network. The relative abundance of module hubs and connectors ranged from 0.02% to 0.9%. Notably, most of module hubs and connectors were unclassified at the genus level. We also evaluated the response of module hubs and connectors to organic N fertilization using the log2-fold change approach, but no clear pattern was identified.
Community compositions of N-cycling functional groups
Four N-cycling genes were sequenced for soils sampled in July 2015, which was about six weeks after compost and manure application. Based on 90% nucleotide cutoff, there were 7001, 3658, 34, and 91 OTUs for ureC, chiA, bacterial amoA, and Nitrospira nxrB, respectively. Organic amendments significantly changed the community composition for N-cycling functional groups as indicated by ureC, chiA and AOB amoA but not for nxrB amplicon sequencing, (Figure 4; Supplementary Table 3). Pairwise PERMANOVA further showed that the community compositions of ureC (p<0.001) and chiA (p=0.018) were significantly different between manure and compost treatments. The CCA indicated that SOC, nitrate, EOC, and pH were often significantly associated with the community composition of N-cycling functional guilds (Supplementary Figure 5). We also built networks for N-cycling functional groups in each N treatment (Supplementary Figure 6 and Supplementary Table 5). Overall, compost treatment had the most complex network, mainly due to the higher interaction in the ureolytic community. Interestingly, both compost and manure treatments increased links among the four different N-cycling functional groups compared with control.
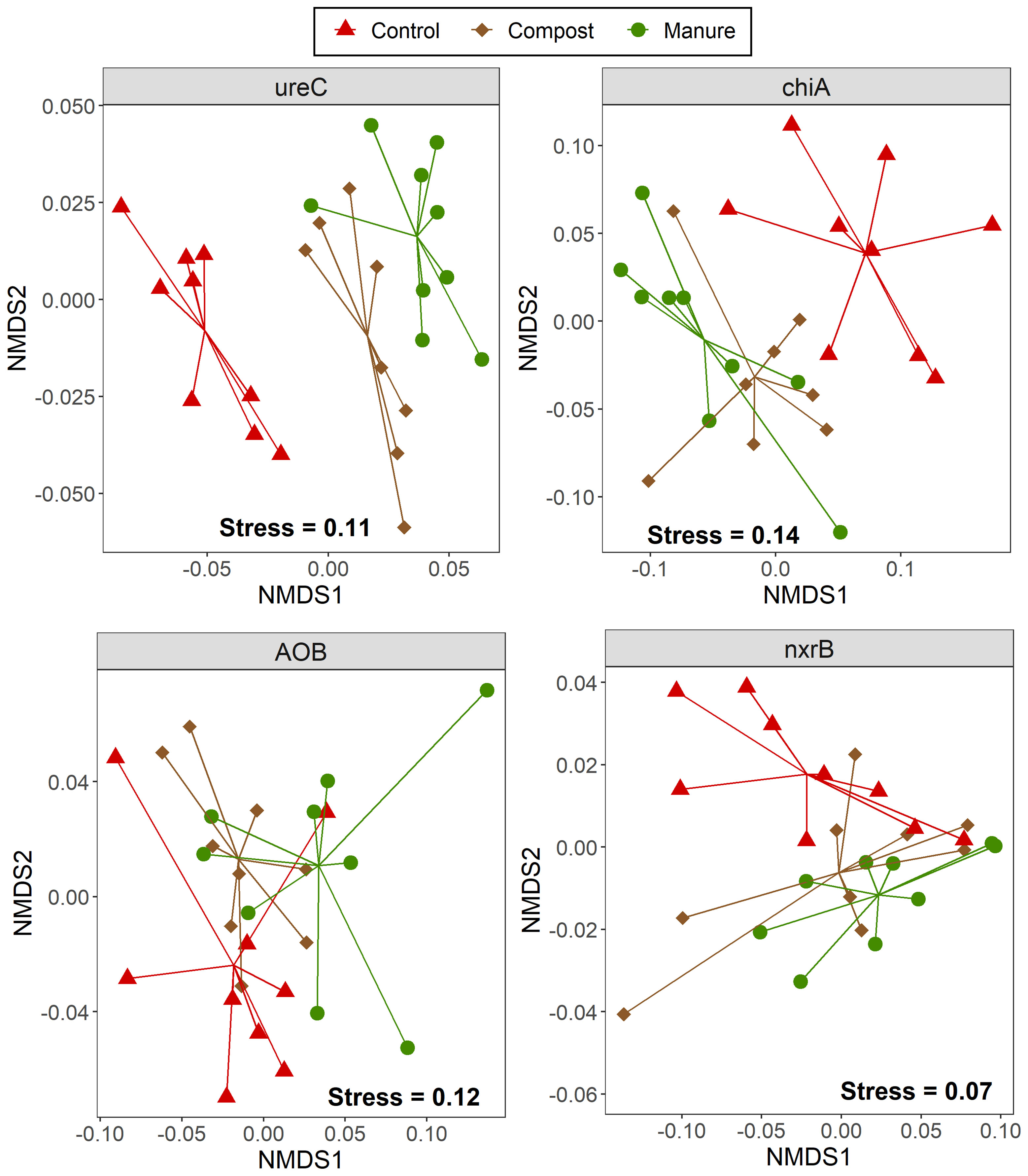
Figure 4 NMDS ordination of the weighted UniFrac distance for four N-cycling functional groups. Four N-cycling genes were ureC (encoding urease), chiA (encoding chitinase), bacterial amoA (encoding ammonia monooxygenase) and Nitrospira nxrB (encoding nitrite oxidoreductase). Except for Nitrospira nxrB, organic N treatment significantly changed the community composition of other N-cycling functional groups (p < 0.01).
Organic N fertilizers also changed the N-cycling functional groups at both phylum and OTU level. In the ureolytic community, the majority of OTUs were assigned to Proteobacteria and Actinobacteria (Supplementary Figure 7A). Proteobacteria and Verrucomicrobia were increased, but Nitrospirae were decreased by compost and manure treatments. There was no difference between compost and manure treatments at the phylum level. Using the log2 fold-change approach, we identified 707 and 858 responsive OTUs in the compost and manure treatments (475 OTUs were shared), respectively (Supplementary Figure 7B). Most of these responsive OTUs were from Proteobacteria (Supplementary Figure 7C). In the chitinolytic community, the majority of OTUs were assigned to Actinobacteria or unclassified (Supplementary Figure 8). Compared with control and manure treatments, compost treatment significantly increased Actinobacteria. We also identified more responsive OTUs in the manure treatment (103 OTUs) than compost treatment (65 OTUs). In the AOB community, we identified three and seven responsive OTUs in the compost and manure treatments, respectively. The Nitrospira community was less responsive to organic N treatment at the OTU level, compared with other N cycling groups. We only detected three responsive OTUs (one in compost treatment and two in manure treatment).
Several approaches were used to evaluate the relative importance of soil geochemical properties and functional genes on soil processes. First, the Mantel test indicated that the community composition of three N-cycling functional groups were significantly correlated with its corresponding process (RureC=0.60, p < 0.001; RchiA = 0.27, p = 0.002; RAOB = 0.28, p = 0.001) but not that of nxrB;(RnxrB = 0.04, p = 0.26). However, the Random Forest analysis indicated that SOC, pH, EOC, and nitrate pools played significant roles in the N-cycling processes (Table 2). The abundance and community composition of N-cycling functional groups were often not important in explaining the N-cycling processes. We only found that the ureolytic community was significantly associated with urease activity. Similarly, the SEM analysis showed that EOC and pH often directly impacted the abundance of N-cycling genes and N-cycling processes (Figure 5). We found that the AOB community significantly influenced the activity of AOB (p = 0.02) while the abundance and community composition of the other three N-cycling functional groups often played limited roles in explaining their associated N-cycling processes.
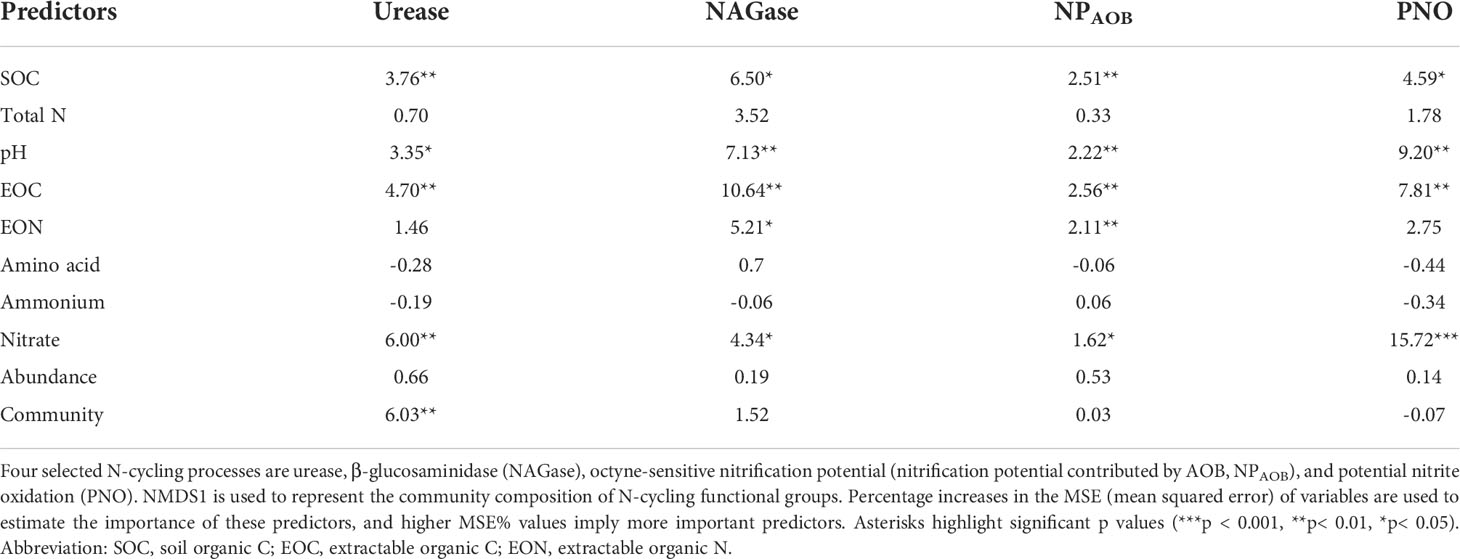
Table 2 Random forest mean predictor importance (percentage of increase of mean square error) of soil chemical properties and the abundance and community of N-cycling functional groups as drivers of their corresponding N process rates.
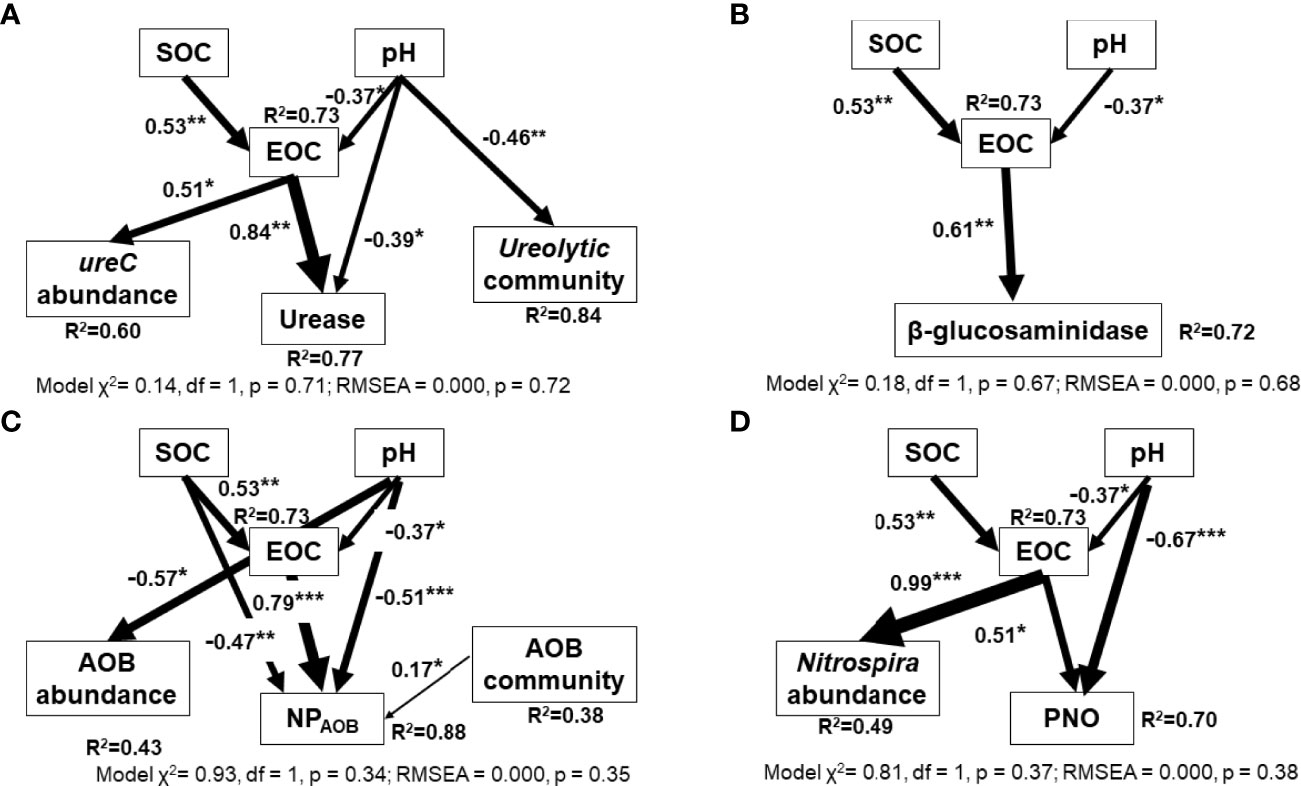
Figure 5 Structural equation models (SEM) showing the effect of soil chemical properties (SOC, EOC, and pH) and the abundance and community of N-cycling functional groups on their corresponding N process rates. Four selected N-cycling processes are (A) urease, (B) β-glucosaminidase (NAGase), (C) octyne-sensitive nitrification potential (nitrification potential contributed by AOB, NPAOB), and (D) potential nitrite oxidation (PNO). NMDS1 is used to represent the community composition of N-cycling functional groups. The arrow width is proportional to the strength of the relationship. R2 represents the proportion of variance explained for every dependent variable. SOC, soil organic carbon; EOC, extractable organic C. Asterisks indicate significant p values (***p < 0.001, **p < 0.01, *p < 0.05)..
Discussion
Organic amendments improve nutrient cycling and soil health in organic farming systems (45, 46). In this study, we compare the effect of organic amendments with different qualities (e.g. composted vs. fresh manure) on soil function and microbial communities in an organic farming systems. Our current and previous studies have comprehensively examined soil chemical properties, soil enzyme activities and process rates, soil bacterial and fungal communities, and the abundance and diversity of N-cycling genes under organic amendments with different qualities (28). We found that organic amendments improved these soil chemical and microbial properties, compared with the control soil. Compost and manure treatments often had similar enzyme activities and abundance of N-cycling genes, while manure treatment had higher N transformation rates than compost (28). This current study further demonstrates that manure treatment had a stronger effect than compost treatment on bacterial and fungal communities, as well as on ureolytic and chitinolytic communities. Although the compost treated soils had higher microbial richness than manure treated soils, both taxonomic and functional microbial groups were more responsive to manure than compost. Manure treated soils also had more complex microbial interactions than compost treated soils. Overall, our study suggests that manure treatment has a stronger influence than compost treatment on structural and functional community composition in this organic farming system.
Organic fertilization affected bacterial and fungal community compositions
Interestingly, we found that compost and manure treatments exerted a contrasting impact on the alpha and beta diversity of soil microbial communities. Compost treated soils had a higher microbial alpha diversity than manure treated soils, while manure treatment had a stronger effect on the bacterial and fungal community compositions. In our study, fresh cattle manure was selected with a higher total N content than the compost so that manure plots received less overall material for the same total N application rate. Therefore, the compost treatment had higher organic C, while the manure treatment had a higher available N in soils (28). Higher organic C may increase the microbial richness in the compost treatment. In addition, although both compost and manure could introduce exogenous species to soils, microbes from compost may be more adaptive to the soil environment than those from manure, which originate from the cattle’s gut microbiome. Thus, microbes from compost have a higher chance to survive in soils. In contrast, the changes in soil bacterial and fungal community composition may be more driven by the N availability, which was higher in manure treatment. Furthermore, we found more ASVs were responsive to manure than compost. The sensitivity of microbes to organic fertilizers may result in a difference in microbial community composition at finer taxonomic levels.
Soil microbial communities often exhibit strong temporal dynamics (47). We collected soils in the corn phase at the end of two cropping cycles in 2011 and 2015. Organic fertilizers were applied twice from 2011 to 2015. We expected that soil microbial communities would be different between 2011 and 2015, and that there would be a stronger organic fertilization effect in 2015 due to repeated application of organic fertilizers. Bacterial and fungal communities did significantly vary between 2011 and 2015 in our study. More interestingly, both bacterial and fungal communities had a higher number of responsive ASVs and a higher magnitude of their response to compost and manure in 2015 than 2011. These results suggest that both bacterial and fungal communities could be gradually manipulated by agricultural management practices, such as crop rotation and organic fertilization. Organic N fertilizers increased Proteobacteria and Bacteroidetes, but decreased Acidobacteria, Chloroflexi, and Gemmatimonadetes. Similar changes in the relative abundances of these phyla have been identified through on-farm surveys of manure or compost treated soils (48).
Organic fertilization affected the microbial network
Co-occurrence networks reveal shared ecological niches and potential biological interactions among organisms (38). In this study, several topological properties of the networks (e.g. average path distance, average clustering coefficient, and modularity) suggested that organic fertilizer intensified soil microbial co-occurrence networks. Previous studies also showed that organic amendment supported more interactions within the soil microbial community (22, 23, 25, 26). The probable mechanism is that the degradation of organic amendments, mainly complex and polymeric organic compounds, was mediated by many specialized microbes. Microbes interacted to each other closely to efficiently degrade these polymeric compounds. To support this assumption, we found that organic fertilizers increased the interaction among four N-cycling genes, especially the links between the ureolytic and chitinolytic communities. Furthermore, we also found that manure treated soils had a more intensive microbial co-occurrence network than compost treated soils. This may be associated with higher N transformation rates in manured soils.
Negative interactions may indicate competition and niche separation between organisms (49). We found that compost and manure treated soils had higher negative associations than the control soils, indicative of increased microbial competition in soil treated with organic fertilizers. Although organic fertilizers increase soil organic C and nutrient availability, microbial biomass and its diversity are also increased. The application of organic fertilizers is a pulse event. Fresh labile substrates thus may be consumed quickly. Thereafter the reduced available C and nutrients may lead to an intense competition among microbes. However, it is notable that most network analyses cannot reveal specific ecological interactions based on positive or negative correlations between nodes.
Organic fertilization affected N-cycling functional groups
The ureolytic community composition was significantly changed by organic fertilizers in our soil, which is consistent with other studies in agricultural soils (12, 13). However, our study further demonstrated that manure had a stronger effect than compost on the ureolytic community composition. The shift of the ureolytic community between compost and manure treatments were closely associated with total N and nitrate suggesting that N availability plays a key role in shaping ureolytic communities. Additionally, there were more responsive OTUs to manure than to compost additions. Urea turnover or concentration may be higher with manure than compost treatment, therefore exerting a stronger effect on urea degraders at both the community and species levels. Supporting this observation, we also found manured soils had higher urease activity than compost treated soils (28).
Numerous studies have focused on the effect of chitin amendment on the chitinolytic communities (14, 50–52). It is largely unknown how organic fertilizers directly mediate the chitinolytic community. In our study, both compost and manure treatment significantly changed the chitinolytic community composition. However, in our previous study on a conventional farm, we found that compost treatment effect was not significant on the chitinolytic community, largely due to the heterogeneity of the microbiome in compost-treated soil (2). The change in the chitinolytic community was associated with pH in this study. Previous studies have also demonstrated that pH was a key factor for shaping the chitinolytic community (14, 53). Furthermore, we reveal that the compost and manure treated soils harbored distinct chitinolytic communities. This difference was correlated with SOC, which was higher in compost treated soils. Compost treatment also had higher proportions of Actinobacteria than the manure treated soils. These outcomes suggest that organic C availability may play a more important role than N availability in shaping the chitinolytic community composition.
Organic fertilizers significantly changed the community of AOB, but not the Nitrospira community in this study. There was no distinction between manure and compost treatments on the community of AOB, though we found manured soils had higher nitrate, octyne-sensitive nitrification potential, and AOB abundance than compost treated soils (28). Ammonium was slowly released by N mineralization in both compost and manure treated soils, which may result in similar selective forces on the community of AOB. At the nearby site under conventional management, the same compost was applied at the same rate and we found that compost had no effect on the community of AOB with three consecutive years of application in a continuous corn cropping system (15). This contrasting result suggests that the duration of compost application or agricultural management may have an important role in shaping the AOB community, since the organic site reported in this current study received six applications of compost with organic management in a diverse rotation. In contrast, the community composition of Nitrospira was not changed by the quality and duration of organic fertilizers (17). The response of the Nitrospira community to organic fertilizer may be masked by their physiologically diverse life-strategies (54). Collectively, our results suggest the quality of organic fertilizers has relatively limited effect on the composition of AOB and Nitrospira communities particularly when compared to the effect of conventional ammonia based fertilizers.
Relative importance of soil chemical properties and functional genes on soil processes
In a previous study, we demonstrated that soil edaphic properties rather than functional gene abundance dominantly impacted N transformation processes (28). In this study, we complemented these findings by including the community composition of four selected N-cycling groups. We expected that the community composition would be strongly linked to N-cycling processes. We did find that community composition was often significantly correlated with the corresponding process rates based on Mantel test. However, our SEM results showed that the direct effects of community composition on soil N-cycling process rates were not maintained after considering multiple biotic and abiotic factors together. Soil pH and EOC often directly affected selected N-cycling activities and rates. This result was contradicted by Trivedi et al. (55), who found that several C-cycling functional genes directly drove the corresponding soil enzyme activities. This discrepancy may be partly explained by the low coverage of some N-cycling groups by amplicon sequencing. We only targeted bacterial ureC and chiA, but did not include fungi, which are also involved in urea and chitin degradation (56, 57). We only targeted Nitrospira in our study, but both Nitrospira and Nitrobacter may be important nitrite oxidizers in soils (58, 59). However, we found the AOB community with a high coverage of AOB amoA by amplicon sequencing directly controlled the octyne-sensitive nitrification potential (contributed by AOB). Collectively, our study suggests that improved primer coverage of N-cycling groups may be necessary to link functional genes and their corresponding process rates especially when the level of functional redundancy in communities is unknown.
Conclusion
We investigated the community compositions of soil microbiome and N-cycling bacteria under fresh and composted manure treatments in a long-term organic farming system. We found that fresh manure treatment had a stronger impact than compost treatment on the composition of bacterial and fungal communities, as well as on the ureolytic and chitinolytic community. Both taxonomic and functional microbial groups were more responsive to fresh manure than compost. Fresh manure treated soils also had more complex microbial interactions than compost treated soils. Collectively, our study suggests that fresh manure selects for a microbial community with an increased functional capacity of supplying available N including nitrate. Further investigation should include crop yield, soil C storage, disease suppression, N loss and greenhouse gas emissions to assess holistically the advantages and disadvantages of using fresh manure versus compost for organic farming systems.
Data availability statement
The data presented in this study are deposited in the NCBI Sequence Read Archive, accession PRJNA804976 (https://www.ncbi.nlm.nih.gov//bioproject/PRJNA804976).
Author contribution
JN and JR designed the study and revised the manuscript. YO conducted the experiment, analyzed the data, and wrote the draft of the manuscript. All authors have read and approved the final manuscript.
Funding
The USDA National Institute of Food and Agriculture funded this work under awards 2011- 67019-30178 and 2016-35100-25091. The research was supported by the Utah Agricultural Experiment Station, Utah State University and approved as journal paper number 9392. ITS and 16S Illumina sequencing was conducted by the U.S. Department of Energy Joint Genome Institute, an Office of Science User Facility, and was supported by the Office of Science of the U.S. Department of Energy under contract DE-AC02-05CH11231.
Acknowledgments
We would like to thank Craig Marlen Rice, Jeremiah Moore and Henry Linford for soil sampling and laboratory assistance. Davie Olsen and Kareem Adeleke performed organic farm plot maintenance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsoil.2022.869136/full#supplementary-material
References
1. Bernard E, Larkin RP, Tavantzis S, Erich MS, Alyokhin A, Sewell G, et al. Compost, rapeseed rotation, and biocontrol agents significantly impact soil microbial communities in organic and conventional potato production systems. Appl Soil Ecol (2012) 52:29–41. doi: 10.1016/j.apsoil.2011.10.002
2. Ouyang Y, Norton JM. Short-term nitrogen fertilization affects microbial community composition and nitrogen mineralization functions in an agricultural soil. Appl Environ Microbiol (2020) 86:e03378-19. doi: 10.1128/AEM.02278-19
3. Wang J, Song Y, Ma T, Raza W, Li J, Howland JG, et al. Impacts of inorganic and organic fertilization treatments on bacterial and fungal communities in a paddy soil. Appl Soil Ecol (2017) 112:42–50. doi: 10.1016/j.apsoil.2017.01.005
4. Sun R, Zhang X-X, Guo X, Wang D, Chu H. Bacterial diversity in soils subjected to long-term chemical fertilization can be more stably maintained with the addition of livestock manure than wheat straw. Soil Biol Biochem (2015) 88:9–18. doi: 10.1016/j.soilbio.2015.05.007
5. Zhang Y, Hao X, Alexander TW, Thomas BW, Shi X, Lupwayi NZ. Long-term and legacy effects of manure application on soil microbial community composition. Biol Fertil Soils (2018) 54:269–83. doi: 10.1007/s00374-017-1257-2
6. Carrera LM, Buyer JS, Vinyard B, Abdul-Baki AA, Sikora LJ, Teasdale JR. Effects of cover crops, compost, and manure amendments on soil microbial community structure in tomato production systems. Appl Soil Ecol (2007) 37:247–55. doi: 10.1016/J.APSOIL.2007.08.003
7. Marschner P, Kandeler E, Marschner B. Structure and function of the soil microbial community in a long-term fertilizer experiment. Soil Biol Biochem (2003) 35:453–61. doi: 10.1016/S0038-0717(02)00297-3
8. Pérez-Piqueres A, Edel-Hermann V, Alabouvette C, Steinberg C. Response of soil microbial communities to compost amendments. Soil Biol Biochem (2006) 38:460–70. doi: 10.1016/j.soilbio.2005.05.025
9. Schimel JP, Bennett J. Nitrogen mineralization: challenges of a changing paradigm. Ecology (2004) 85:591–602. doi: 10.1890/03-8002
10. Norton J, Ouyang Y. Controls and adaptive management of nitrification in agricultural soils. Front Microbiol (2019) 10:1931. doi: 10.3389/fmicb.2019.01931
11. Norton JM, Schimel JP. “Nitrogen mineralization immobilization turnover,”. In: Huang PM, Li Y, Summers ME, editors. Handbook of soil science, 2nd ed. Boca Raton, FL USA: CRC Press (2011).
12. Luo G, Li L, Friman VP, Guo J, Guo S, Shen Q, et al. Organic amendments increase crop yields by improving microbe-mediated soil functioning of agroecosystems: A meta-analysis. Soil Biol Biochem (2018) 124:105–15. doi: 10.1016/j.soilbio.2018.06.002
13. Wang L, Luo X, Liao H, Chen W, Wei D, Cai P, et al. Ureolytic microbial community is modulated by fertilization regimes and particle-size fractions in a black soil of northeastern China. Soil Biol Biochem (2018) 116:171–8. doi: 10.1016/j.soilbio.2017.10.012
14. Kielak AM, Cretoiu MS, Semenov AV, Sørensen SJ, van Elsas JD. Bacterial chitinolytic communities respond to chitin and pH alteration in soil. Appl Environ Microbiol (2013) 79:263–72. doi: 10.1128/AEM.02546-12
15. Ouyang Y, Norton JM, Stark JM, Reeve JR, Habteselassie MY. Ammonia-oxidizing bacteria are more responsive than archaea to nitrogen source in an agricultural soil. Soil Biol Biochem (2016) 96:4–15. doi: 10.1016/j.soilbio.2016.01.012
16. Ouyang Y, Norton JM. Nitrite oxidizer activity and community are more responsive than their abundance to ammonium-based fertilizer in an agricultural soil. Front Microbiol (2020) 11:1736. doi: 10.3389/fmicb.2020.01736
17. Han S, Zeng L, Luo X, Xiong X, Wen S, Wang B, et al. Shifts in nitrobacter- and nitrospira-like nitrite-oxidizing bacterial communities under long-term fertilization practices. Soil Biol Biochem (2018) 124:118–25. doi: 10.1016/j.soilbio.2018.05.033
18. He J, Shen J, Zhang L, Zhu Y, Zheng Y, Xu M, et al. Quantitative analyses of the abundance and composition of ammonia-oxidizing bacteria and ammonia-oxidizing archaea of a Chinese upland red soil under long-term fertilization practices. Environ Microbiol (2007) 9:2364–74. doi: 10.1111/j.1462-2920.2007.01358.x
19. Zhou J, Deng Y, Luo F, He Z, Tu Q, Zhi X. Functional molecular ecological networks. MBio (2010) 1. doi: 10.1128/mBio.00169-10
20. de Vries FT, Griffiths RI, Bailey M, Craig H, Girlanda M, Gweon HS, et al. Soil bacterial networks are less stable under drought than fungal networks. Nat Commun (2018) 9:1–12. doi: 10.1038/s41467-018-05516-7
21. Banerjee S, Schlaeppi K, van der Heijden MGA. Keystone taxa as drivers of microbiome structure and functioning. Nat Rev Microbiol (2018) 16:567–76. doi: 10.1038/s41579-018-0024-1
22. Hartmann M, Frey B, Mayer J, Mäder P, Widmer F. Distinct soil microbial diversity under long-term organic and conventional farming. ISME J (2015) 9:1177–94. doi: 10.1038/ismej.2014.210
23. Banerjee S, Walder F, Büchi L, Meyer M, Held AY, Gattinger A, et al. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J (2019) 13:1722–36. doi: 10.1038/s41396-019-0383-2
24. Hartman K, van der Heijden MGA, Wittwer RA, Banerjee S, Walser J-C, Schlaeppi K. Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome (2018) 6:14. doi: 10.1186/s40168-017-0389-9
25. Schmid CAO, Schröder P, Armbruster M, Schloter M. Organic amendments in a long-term field trial–consequences for the bulk soil bacterial community as revealed by network analysis. Microb Ecol (2018) 76:226–39. doi: 10.1007/s00248-017-1110-z
26. Ling N, Zhu C, Xue C, Chen H, Duan Y, Peng C, et al. Insight into how organic amendments can shape the soil microbiome in long-term field experiments as revealed by network analysis. Soil Biol Biochem (2016) 99:137–49. doi: 10.1016/j.soilbio.2016.05.005
27. Ramos TM, Jay-Russell MT, Millner PD, Shade J, Misiewicz T, Sorge US, et al. Assessment of biological soil amendments of animal origin use, research needs, and extension opportunities in organic production. Front Sustain Food Syst (2019) 3:73. doi: 10.3389/fsufs.2019.00073
28. Ouyang Y, Reeve JR, Norton JM. Soil enzyme activities and abundance of microbial functional genes involved in nitrogen transformations in an organic farming system. Biol Fertil Soils (2018) 54:437–50. doi: 10.1007/s00374-018-1272-y
29. Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol (2016) 18:1403–14. doi: 10.1111/1462-2920.13023
30. Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods (2013) 10:996–8. doi: 10.1038/nmeth.2604
31. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol (2007) 73:5261–7. doi: 10.1128/AEM.00062-07
32. Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for Large alignments. PloS One (2010) 5:e9490. doi: 10.1371/journal.pone.0009490
33. McMurdie PJ, Holmes S. Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PloS One (2013) 8:e61217. doi: 10.1371/journal.pone.0061217
34. Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res (2014) 42:D633–42. doi: 10.1093/nar/gkt1244
35. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics (2011) 27:2194–200. doi: 10.1093/bioinformatics/btr381
36. Wang Q, Quensen JF, Fish JA, Lee TK, Sun Y, Tiedje JM, et al. Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using FrameBot, a new informatics tool. MBio (2013) 4:e00592–13. doi: 10.1128/mBio.00592-13
37. Fish JA, Chai B, Wang Q, Sun Y, Brown CT, Tiedje JM, et al. FunGene: the functional gene pipeline and repository. Front Microbiol (2013) 4:291. doi: 10.3389/fmicb.2013.00291
38. Deng Y, Jiang YH, Yang Y, He Z, Luo F, Zhou J. Molecular ecological network analyses. BMC Bioinf (2012) 13:1–20. doi: 10.1186/1471-2105-13-113
39. Wu L, Yang Y, Chen S, Zhao M, Zhu Z, Yang S, et al. Long-term successional dynamics of microbial association networks in anaerobic digestion processes. Water Res (2016) 104:1–10. doi: 10.1016/j.watres.2016.07.072
40. Shi S, Nuccio EE, Shi ZJ, He Z, Zhou J, Firestone MK. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol Lett (2016) 19:926–36. doi: 10.1111/ele.12630
41. Bastian M, Heymann S, Jacomy M. Gephi : An open source software for exploring and manipulating networks. Proceedings of the International AAAI Conference on Web and Social Media (2009) 3(1):361–2. Retrieved from https://ojs.aaai.org/index.php/ICWSM/article/view/13937.
42. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res (2003) 13:2498–504. doi: 10.1101/gr.1239303
43. Oksanen J, Blanchet FG, Kindt R, Legendre P, McGlinn D, Wagner H, et al. Vegan: Community ecology package. (version 2.5-6) The Comprehensive R Archive Network (CRAN) (2019). https://cran.r-project.org/web/packages/vegan/vegan.pdf
44. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15:550. doi: 10.1186/s13059-014-0550-8
45. Lori M, Symnaczik S, Mäder P, De Deyn G, Gattinger A. Organic farming enhances soil microbial abundance and activity–a meta-analysis and meta-regression. PloS One (2017) 12:e0180442. doi: 10.1371/journal.pone.0180442
46. Reeve JR, Hoagland LA, Villalba JJ, Carr PM, Atucha A, Cambardella C, et al. Organic farming, soil health, and food quality: Considering possible links. Adv Agron (2016) 137:319–67. doi: 10.1016/bs.agron.2015.12.003
47. Shade A, Gregory Caporaso J, Handelsman J, Knight R, Fierer N. A meta-analysis of changes in bacterial and archaeal communities with time. ISME J (2013) 7:1493–506. doi: 10.1038/ismej.2013.54
48. Peng M, Tabashsum Z, Millner P, Parveen S, Biswas D. Influence of manure application on the soil bacterial microbiome in integrated crop-livestock farms in Maryland. Microorganisms (2021) 9:2586. doi: 10.3390/microorganisms9122586
49. Ghoul M, Mitri S. The ecology and evolution of microbial competition. Trends Microbiol (2016) 24:833–45. doi: 10.1016/j.tim.2016.06.011
50. Hui C, Jiang H, Liu B, Wei R, Zhang Y, Zhang Q, et al. Chitin degradation and the temporary response of bacterial chitinolytic communities to chitin amendment in soil under different fertilization regimes. Sci Total Environ (2020) 705:136003. doi: 10.1016/j.scitotenv.2019.136003
51. Jacquiod S, Franqueville L, Cécillon S, M. Vogel T, Simonet P. Soil bacterial community shifts after chitin enrichment: An integrative metagenomic approach. PloS One (2013) 8:e79699. doi: 10.1371/journal.pone.0079699
52. Cretoiu MS, Kielak AM, Schluter A, Van Elsas JD. Bacterial communities in chitin-amended soil as revealed by 16S rRNA gene based pyrosequencing. Soil Biol Biochem (2014) 76:5–11. doi: 10.1016/j.soilbio.2014.04.027
53. Terahara T, Ikeda S, Noritake C, Minamisawa K, Ando K, Tsuneda S, et al. Molecular diversity of bacterial chitinases in arable soils and the effects of environmental factors on the chitinolytic bacterial community. Soil Biol Biochem (2009) 41:473–80. doi: 10.1016/J.SOILBIO.2008.11.024
54. Daims H, Lücker S, Wagner M. A new perspective on microbes formerly known as nitrite-oxidizing bacteria. Trends Microbiol (2016) 24:699–712. doi: 10.1016/j.tim.2016.05.004
55. Trivedi P, Delgado-Baquerizo M, Trivedi C, Hu H, Anderson IC, Jeffries TC, et al. Microbial regulation of the soil carbon cycle: evidence from gene–enzyme relationships. ISME J (2016) 10:2593–604. doi: 10.1038/ismej.2016.65
56. Mobley HLT, Island MD, Hausinger RP. Molecular biology of microbial ureases. Microbiol Rev (1995) 59:451–80. doi: 10.1128/mr.59.3.451-480.1995
57. Treseder KK, Lennon JT. Fungal traits that drive ecosystem dynamics on land. Microbiol Mol Biol Rev (2015) 79:243–62. doi: 10.1128/mmbr.00001-15
58. Poly F, Wertz S, Brothier E, Degrange V. First exploration of nitrobacter diversity in soils by a PCR cloning-sequencing approach targeting functional gene nxrA. FEMS Microbiol Ecol (2008) 63:132–40. doi: 10.1111/j.1574-6941.2007.00404.x
Keywords: organic nitrogen management, compost, manure, microbial community network analysis, ureC, chiA, amoA, nxrB
Citation: Ouyang Y, Reeve JR and Norton JM (2022) The quality of organic amendments affects soil microbiome and nitrogen-cycling bacteria in an organic farming system. Front. Soil Sci. 2:869136. doi: 10.3389/fsoil.2022.869136
Received: 03 February 2022; Accepted: 28 June 2022;
Published: 02 August 2022.
Edited by:
Nigel Victor Gale, University of Toronto, CanadaReviewed by:
Carlos Garcia, Spanish National Research Council (CSIC), SpainStephanie Ann Yarwood, University of Maryland, United States
Paulo Roger Lopes Alves, Universidade Federal da Fronteira Sul, Brazil
Leandro Nascimento Lemos, National Laboratory for Scientific Computing (LNCC), Brazil
Copyright © 2022 Ouyang, Reeve and Norton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeanette M. Norton, amVhbmV0dGUubm9ydG9uQHVzdS5lZHU=