
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Soil Sci. , 16 November 2022
Sec. Soil Biology, Ecosystems and Biodiversity
Volume 2 - 2022 | https://doi.org/10.3389/fsoil.2022.1020869
 Expedito Olimi1*
Expedito Olimi1* Samuel Bickel1
Samuel Bickel1 Wisnu Adi Wicaksono1
Wisnu Adi Wicaksono1 Peter Kusstatscher1
Peter Kusstatscher1 Robert Matzer4
Robert Matzer4 Tomislav Cernava1
Tomislav Cernava1 Gabriele Berg1,2,3*
Gabriele Berg1,2,3*Soil microbial communities are crucial for plant growth and are already depleted by anthropogenic activities. The application of microbial transplants provides a strategy to restore beneficial soil traits, but less is known about the microbiota of traditional inoculants used in biodynamic agriculture. In this study, we used amplicon sequencing and quantitative PCR to decipher microbial communities of composts, biodynamic manures, and plant preparations from Austria and France. In addition, we investigated the effect of extracts derived from biodynamic manure and compost on the rhizosphere microbiome of apple trees. Microbiota abundance, composition, and diversity of biodynamic manures, plant preparations, and composts were distinct. Microbial abundances ranged between 1010-1011 (bacterial 16S rRNA genes) and 109-1011 (fungal ITS genes). The bacterial diversity was significantly higher in biodynamic manures compared to compost without discernible differences in abundance. Fungal diversity was not significantly different while abundance was increased in biodynamic manures. The microbial communities of biodynamic manures and plant preparations were specific for each production site, but all contain potentially plant-beneficial bacterial genera. When applied in apple orchards, biodynamic preparations (extracts) had the non-significant effect of reducing bacterial and fungal abundance in apple rhizosphere (4 months post-application), while increasing fungal and lowering bacterial Shannon diversity. One to four months after inoculation, individual taxa indicated differential abundance. We observed the reduction of the pathogenic fungus Alternaria, and the enrichment of potentially beneficial bacterial genera such as Pseudomonas. Our study paves way for the science-based adaptation of empirically developed biodynamic formulations under different farming practices to restore the vitality of agricultural soils.
The soil microbiome plays an essential role in crop production, but is a strongly influenced component in the Anthropocene (1, 2). Therefore, it is critical to identify threats that cause microbiome disturbances, as well as to find sustainable restoration strategies for affected microbiomes (2). Microbial diversity is crucial for soil and plant health (3) and for human health as well, which can be measured in taxonomic and functional diversity as well as by total gene count within the microbiota (4). However, enriching microbial diversity in the soil is a challenge, which can be managed directly by applying (i) microbiome transplants, (ii) microbes with beneficial properties, (iii) microbiota-active metabolites, or indirectly by changing environmental conditions in a way that microbiomes also shift their structure and function from dysbiosis into a healthy state (1, 5). The use of compost in field fertilization, a classical microbiome transplant, can be traced back to the advent of human civilization, marked by the agricultural revolution (6). Numerous benefits for enhancing soil ecosystem functions, soil quality, and plant health, are attributed to composting in arable and natural ecosystems (7–9). However, many extensive farming systems from the past have been replaced by what is nowadays known as intense, conventional agriculture. It is characterized by high-yield cultivars, which need high inputs of synthetic fertilizer, and pesticides and commonly result in reduced soil vitality, emerging and multi-resistant soil-borne pathogens and an overall yield decline, which prompts a re-evaluation of agricultural practices (10–12).
An ongoing evaluation of current agricultural practices has shifted attention towards alternative and environmentally sustainable systems, including biodynamic farming to restore the “lost soil microbiome” (13–15). Furthermore, the globally increasing demand for foods and beverages produced under organic management practices (16–18) motivates a deeper investigation of microbial constituents that are present in the utilized organic amendments. Biodynamic (BD) farming was first conceptualized by the Austrian philosopher Rudolf Steiner (1861-1925) (19) and is considered the first systematic form of ecological or organic farming (15, 19, 20). Although organic and BD farming are related, the latter considers specific practices which are intended to influence the biological metaphysical aspects including natural rhythms concerning the sun and moon, weather, and seasons of the farm (10, 15, 21). A special feature of BD agriculture is the use of specific BD manure and fermented plant preparations (20). These products are applied as solid manure or liquid extracts in form of field sprays (i.e., soil or foliar application) to enhance soil quality and stimulate plant health (22–24). Recent studies highlighted the role of BD preparations, especially in enhancing soil diversity (15, 18, 25), and have also found application in viticulture (17), especially in enhancing winegrape quality (26). In addition, long-term field trials indicated differences in the soil microbiome between BD, integrated, and organic farming systems (27, 28) even under desert farming conditions (29).
BD farming is rooted in anthroposophy, which views humans as the primary link between the cycles of the earth and the cosmos, with humans bridging a gap between the spiritual and material worlds (17). Special natural biodynamic preparations (dubbed “Steiner preparations”) were formulated and utilized in this system to replace synthetic products (17). The procedure to obtain BD formulations involves fermenting cow manure in cow horns to create the so called biodynamic extracts (compost teas), which are then applied according to Steiner (30). While the reason for fermentation in cow horns may still be unknown, preparations from this method have been proven to have special qualities including soil fertility improvement and enhancing plant physiological responses to light radiation (25). Moreover, (26) found a substantial impact of BD preparations in the enhancement of wine quality, winegrape canopy and chemistry, despite contradicting results regarding effects on soil quality. Yet, the microbiome of BD formulations (manures and plant preparations), the differences between BD manures and non-biodynamic composts as well as their impact on the rhizosphere microbiota remain unexplored.
Therefore, we analysed bacterial and fungal communities of BD formulations (manures and plant preparations) and composts (standard-, horse-, and apple composts). Extracts from the BD manures and composts are commonly applied as field sprays; thus, we also investigated the microbiome of liquid extracts and their precursor materials. The two analysed extracts were applied in differently managed (organic and integrated) apple orchards and samples were taken at bloom (one month), and post-harvest (four months) to investigate the temporal variability in the rhizosphere microbiome. The study was based on the hypotheses that: (i) BD manures and composts have a different microbiome; (ii) the microbiome of BD manures is affected by the amendment with biodynamic plant preparations; (iii) the microbiome of BD manures differs between regions and the year of production; (iv) the microbiomes of extracts obtained from BD manure and compost are different from the precursor materials, and (v) the microbiome of the apple rhizosphere is affected by the application of extracts.
We sampled four BD manure products and six biodynamic plant preparations for amplicon sequence analysis. BD horn manures were obtained from Demeter (Vienna, Austria) and BioDynamie Services sarl (Château, France), while all plant preparations were obtained from France. In the BD farming community, the plant preparations are referred to as P-502, P-503, P-504, P-505, P-506, and P-507. In the production process, BD manure (500) is prepared from cow manure that is fermented in cow horns and buried in the soil at a depth of 50 cm for six months (i.e., during autumn and winter). After the horns are recovered from the soil, the retrieved horn manure (500) can be amended with six different plant preparations (P-502 to P-507) and referred to as 500P.
The BD formulations used in this study originated from two different regions of production: France (FR-500/FR-500P) and Austria (AT-500/AT-500P). Apart from differences in the region of origin, we included BD products from different production years (Table 1). According to the producers, BD preparations were stored in ceramic pots and placed in a double-walled wooden box covered with a wooden lid. The boxes were kept in cellars exclusively dedicated for storage of preparations at constant environmental conditions. We analysed BD manure samples without preparations (AT/FR-500) produced in the years 2019, 2020, and 2021 and BD manures with preparations (AT/FR-500P) from the years 2012, 2016, and 2020 (Table 1). The BD manure FR-500P produced in 2016 was amended with four plant preparations coded F, T, S, and V, according to the producer in France.
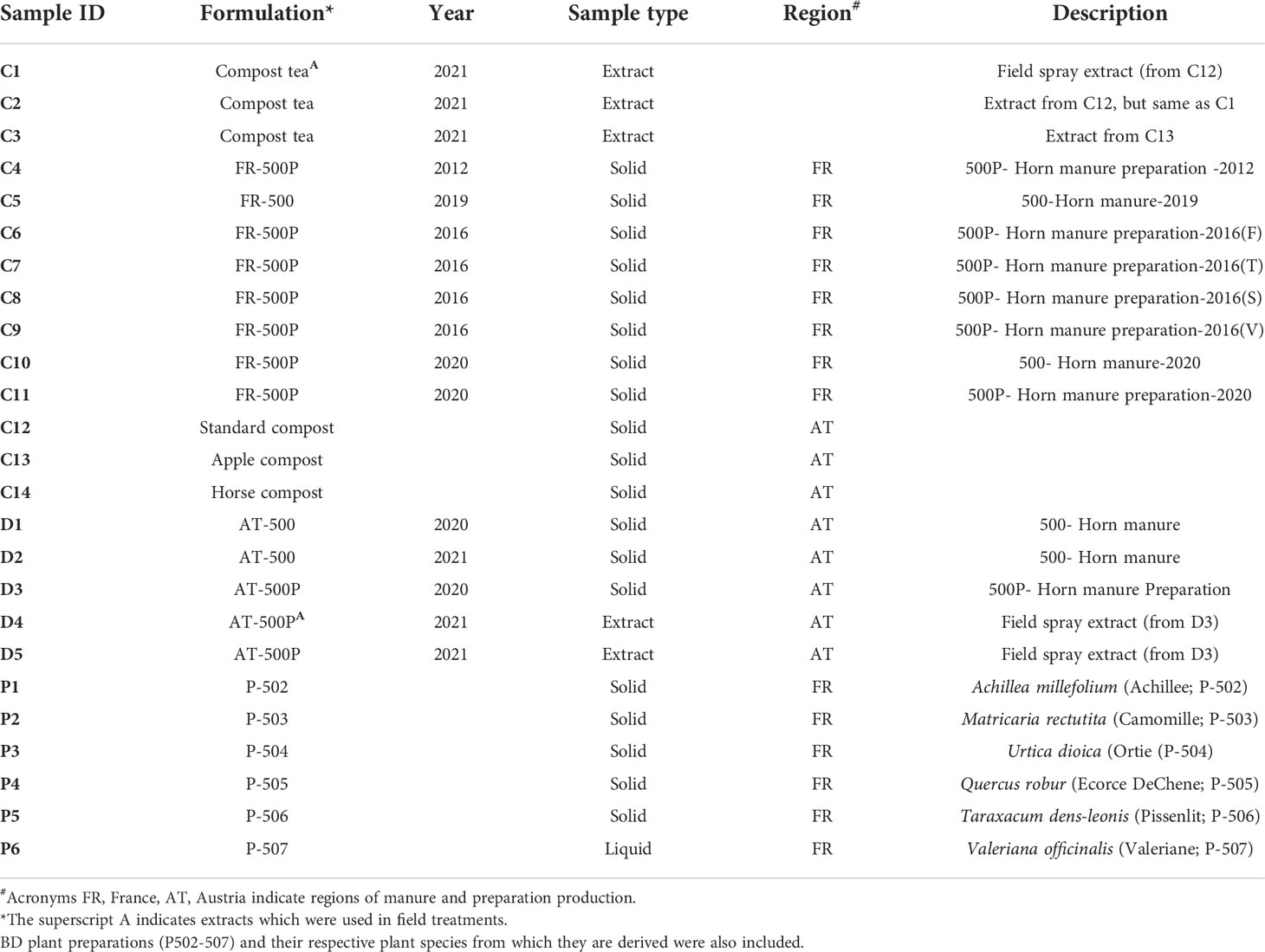
Table 1 Overview of the studied composts (i.e., apple-, horse-, and standard compost) and biodynamic manures (with/without plant preparations).
For comparison between biodynamic manures and compost, three composts (standard-, apple, and horse compost) were included in the analysis (Table 1). Briefly, the different composts were constituted as follows: (i) Standard compost (70% shrub cuttings, 5% soil, and 25% organic waste); (ii) Horse compost (60% shrub cuttings, 5% soil, and 35% horse manure with straw); and (iii) Apple compost (75% shrub cuttings, 5% soil, and 20% pressed organic apple fruits).
As a common practice in BD farming, sprays for field application are extracted from BD formulations. Similarly, compost can be applied in liquid form as “compost tea”; it is prepared by mixing liquid extracts from compost with molasses and stone dust. We applied two extracts from the BD manure (AT-500P) and compost (standard compost) in two apple orchards, which were maintained following organic and integrated management practices. The organic and integrated orchards were located at 47.1289°N, 15.7807°E and 47.2125°N, 15.8569°E, respectively. Apple trees were grown for approximately ten years in each orchard. Treatments were performed at the beginning of spring (May 2021). The BD preparations were applied by sprinkling the extracts in the vicinity of the plant roots, using the “Demeter” recommended application rate of 1:10 (i.e. liquid extract: water) liters per acre. For each orchard, a randomized plot design of 4 plots per treatment and 8-10 trees per plot was chosen. To account for spatial variations, plots were randomly distributed across six neighbouring rows. Rhizosphere samples were randomly taken from each plot using an auger of 5 cm diameter. Ten soil cores from each plot were pooled to comprise a biological replicate. Rhizosphere (soil associated with roots) sampling was performed one- and four- months after treatment and corresponded with the spring and autumn seasons. Samples were kept cooled and transported within six hours to the laboratory at the Institute of Environmental Biotechnology (Graz University of Technology, Graz, Austria).
For compost, BD manures, and BD plant preparations, four grams of sample were stored at -70°C until DNA was extracted. Before DNA extraction from liquid extracts (“compost tea”), the extracts from compost and BD manure were centrifuged at 16000g for 16 minutes, and the microbial pellet was frozen at -70°C until DNA extraction. For orchard samples, approx. four grams of rhizosphere soil were stored at -70°C. For all samples, total microbial genomic DNA was extracted using the E.Z.N.A.® Soil DNA Kit (Omega Bio-tek, Inc.; Norcross-Georgia, United States) following the manufacturer’s instructions. DNA was quality checked using a Nanodrop 2000 (Thermo Scientific, Wilmington, DE, USA) and stored at -20°C before performing PCR and amplicon sequencing.
The primer pairs Unibac-II-515f/Unibac-II-806r for bacteria as previously used by (29) and ITS1f/ITS2r for fungi (31) were used for real-time qPCR quantifications to determine the copy numbers of 16S rRNA and ITS genes in the different samples. Reactions were performed in a total volume of 10 μL in a reaction mix composed of 5 μL of KAPA SYBR Green (Bio-Rad, Hercules, CA, U.S.A.), 0.5 μL of each primer (10 µM), 3 μL of PCR grade water and 1 μL template DNA (samples were diluted 1:10 in PCR grade water). Amplifications were performed in triplicates for each sample using a Rotor-GeneTM 6000 series thermal cycler (Corbett Research, Sydney, Australia) with the following program settings: initial denaturation (95°C,5 min) followed by 35 cycles of denaturation (95°C,10 s); annealing (54°C, 15 s); extension (72°C, 10 s); then melt down from 72 to 96°C. Serial dilutions of standards containing defined copy numbers were generated according to (29) and used to calculate gene copy numbers in different samples. As standards, known concentrations of 16S rRNA gene fragments from a Bacillus sp., and the ITS region from a Penicillium sp. were used.
The extracted DNA was further used for amplicon library preparation based on the hypervariable V4 region of the bacterial 16S rRNA gene and the ITS1 regions of fungal DNA. We employed one-step PCR using the primer pair 515F (5’-GTGCCAGCMGCCGCGGTAA-3’), and 806R (5’-GGACTACHVGGGTWTCTAAT-3’) for the bacterial library preparation (32). Both forward and reverse primers contained sample-specific barcodes to facilitate multiplexed sequencing. For each PCR, 1 µL of extracted DNA was used as a template in a 30 µL reaction. Peptide nucleic acid (PNA) PCR clamps were used to block the amplification of plant plastid and mitochondrial 16S rRNA genes during the PCR amplification of the bacterial community (33, 34). The reaction mixture contained 6 µL (5xTaq &GO, PCR pre-mix, MP Biomedicals), 0.6 µL (10 µM 515F/806R) primers, 0.45 µL (50 µM mPNA and pPNA), and 20.9 µL of PCR grade water. All reactions were performed in triplicates in a thermocycler (Bio-metra GmbH, Jena, Germany). The PCR program included an initial denaturation (96°C, 5 min), followed by 30 cycles (94°C for 60 s, 78°C PNA step for 5 s, 54°C for 1 min, 74°C for 60 s), followed by 74°C for 10 min and then a cool down to 10°C.
For the fungal community, we used the primer pair ITS1f (5’-CTTGGTCATTTAGAGGAAGTAA-3’) and ITS2r (5’-GCTGCGTTCTTCATCGATGC-3’) (31, 35) for library preparation. All amplifications were performed in triplicates. One µL of extracted DNA was used in each 30 µL reaction. The reaction mixture contained 6 µL (5xTaq &GO, PCR pre-mix, MP Biomedicals), 0.6 µL (10 µM ITS1F/ITS2R) primers, 21.8 µL of PCR grade water. The PCR program included the first step of initial denaturation (96°C, 5 min), followed by 30 cycles (denaturation: 96°C for 60 s, annealing: 58°C for 60 s, and extension: 74°C for 60 s), followed by 74°C for 10 min (final extension) and a cool-down step to 10°C. Successful PCR amplifications at the correct amplicon size were confirmed by gel electrophoresis. PCR amplicons were purified using Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI) following the manufacturer’s instructions. Purified PCR amplicons were quantified using a Nanodrop 2000 (Thermo Scientific, Wilmington, DE, USA) and pooled in equimolar concentrations. Paired-end Illumina MiSeq 2x 250 sequencing of the amplicon library was performed at Eurofin (Berlin, Germany). All raw reads obtained from the sequencing company were deposited at the European Nucleotide Archive (ENA) under study accession number PRJEB53400.
Paired-end reads from the sequencing facility were quality-checked and demultiplexed using Cutadapt (36). Demultiplexed reads were analysed using QIIME2 version 2021.11.0 (37). Primer sequences were removed, and the DADA2 algorithm in the QIIME2 environment was employed to quality filter, denoise, and remove chimeric sequences (38); thus generating amplicon sequence variants (ASVs), and a feature table of microbial counts. Taxonomic assignment of denoised reads was performed using the VSEARCH (39) tool in QIIME2 by comparing the reads against reference databases: SILVA132 for 16S rRNA (40, 41) and UNITE v7 (42) for ITS sequence reads.
All data were analysed with R statistical software (version 4.0.3) using RStudio (Version 1.1.423) (43), supplemented by the web-based microbiome analyst tools (44). The obtained microbial ASV tables and taxonomic assignment were processed using phyloseq (45) and vegan 2.5.7 (46) packages.
For alpha and beta diversity analyses, the statistics were performed on datasets rarefied to minimum sampling depths of 1000 and 700 reads per sample for 16S and ITS analyses, respectively. The sample-specific rarefaction curves for bacterial and fungal communities were visualized (Figure S1). One sample of a biodynamic plant preparation (P-507) was excluded from the analysis due to insufficient replicates (one sample was retained).
Microbial alpha diversity was calculated using the Shannon diversity index (H’). The non-parametric Kruskal-Wallis test (47), followed by Dunn’s test (corrected for multiple comparisons using the Bonferroni method) was performed on Shannon diversity index and abundance data to test for differences between the samples (e.g., BD manures vs. composts, extracts vs. precursor materials); and treatment comparisons (extract from standard compost and AT-500P) post-inoculation in the different orchards. Also, the effect of treatment on the rhizosphere microbiome at different sampling times was tested. Permutational analysis of variance (PERMANOVA, 999 permutations) based on Bray-Curtis dissimilarity was used to test for differences in community structure between sample groups. For significant factors (and combinations), pairwise comparisons were performed using the ‘pairwise.adonis2’ function in vegan (46) with p-values adjusted using the Bonferroni method. The differences in microbial community structure between the sample groups were visualized using dendrograms (generated by complete clustering of Bray-Curtis distances), and non-metric multidimensional scaling (NMDS).
Microbial taxonomic composition based on the percentage relative abundance of top20 taxa at class, order, and family levels was represented using stacked bar plots. Linear discriminant analysis Effect Size (LEfSE) (48) implemented in microbiome analyst (44) was used to identify the taxa underlying the observed microbiome differences between the composts and biodynamic preparations, as well as revealing treatment effects in the different orchards and sampling times. The common microbiome (defined by a prevalence of 50%, and detection threshold of 0.001% relative abundance) was used to obtain unique and shared ASVs associated with the different samples. The list of ASVs was visualized using a Venn diagram generated by the systemPipeR package (49) in R studio.
We used the SourceTracker R script (50) to estimate the proportion of microbiota in the apple rhizosphere that potentially originated from the field sprays extracted from compost and BD manures after soil treatment. SourceTracker estimates the proportion of microbiota that originates from a set of source and sink environments as previously described (50). In our case, the extracts from BD manure (AT-500P) and standard compost which were used for orchard treatment were considered as the source environment and the rhizosphere soil as the sink environment. By considering control soil as the microbial source, we also tracked microbiota in the treated rhizosphere soil which was not of extract origin, but likely originated from the soil.
Amplicon sequencing of bacterial and fungal communities yielded 2,213,856 and 4,534,963 high-quality reads, assigned to 9,387 bacterial and 2,742 fungal ASVs, respectively. Generally, we observed significant differences in microbiome structures between BD manures (AT-500/P or FR-500/P) and composts (i.e. apple-, horse-, standard composts), especially for the bacterial community (Figure 1). Moreover, a separation based on the country of origin of BD manures was observed (Figures 1A, B). Significant differences (P=0.008) in Shannon diversity between compost and BD manures were observed for bacterial communities (Table S3A), while no significant differences (P=0.19) were observed for the fungal community (Table S4A). Microbial abundance was significantly different for bacterial (P=0.04; Table S1A) and fungal communities (P=0.009; Table S2A).
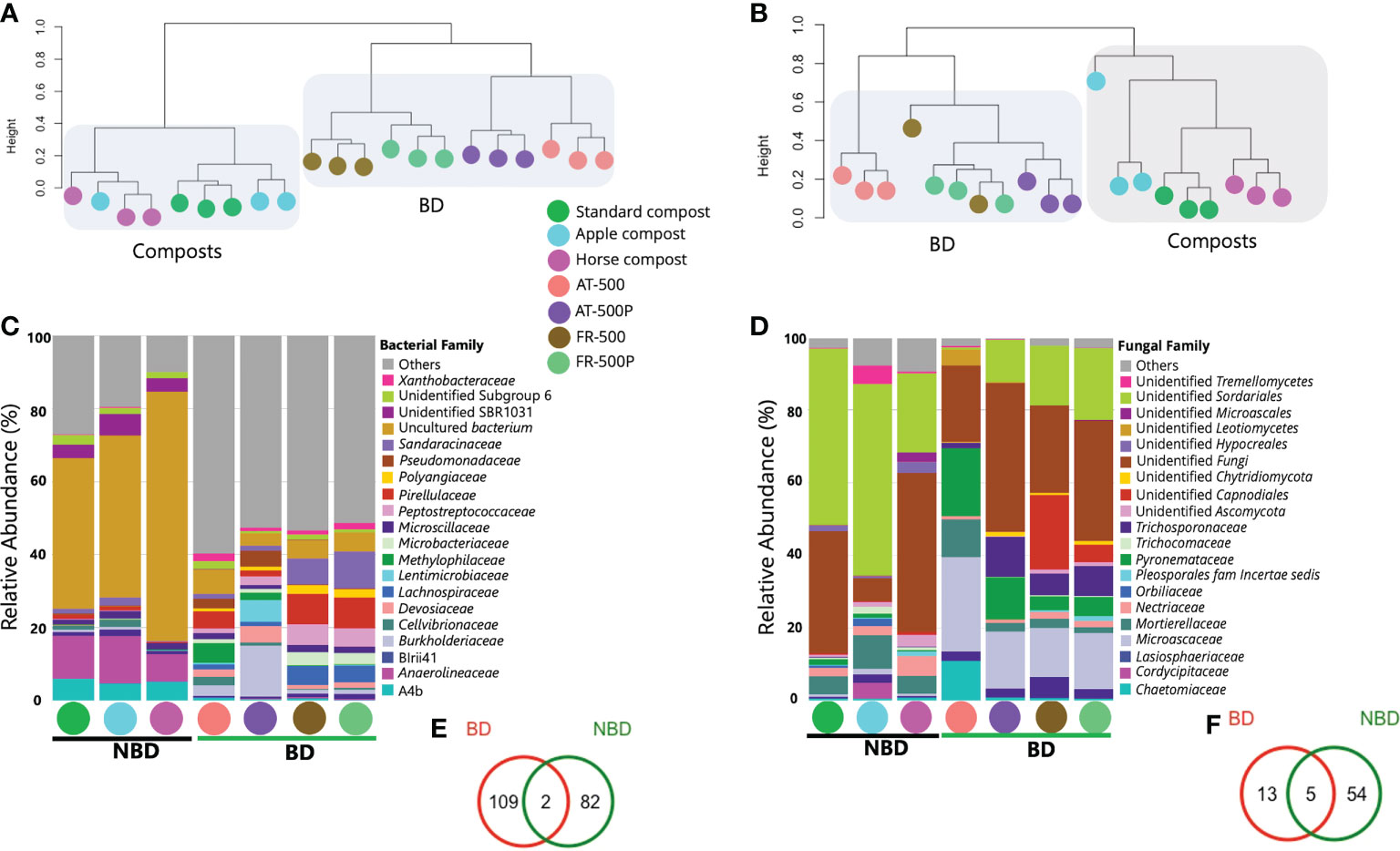
Figure 1 Microbial community structure and composition of BD manures and composts. Coloured circles represent the different BD manures (500/P) and non-biodynamic (NBD) composts. Figures (A, B) are dendrogram representation of the bacterial and fungal community obtained by complete clustering of the Bray Curtis distance matrix. (C, D) are stacked bar plots showing the bacterial, and fungal taxonomic composition in the different BD and compost samples; while (E, F) represent shared and unique ASVs between BD manures and composts for bacterial and fungal communities, respectively. The common microbiome was defined as ASVs with prevalence >50% and relative abundance >0.001%. AT and FR indicate countries Austria and France, respectively.
The bacterial abundance (16S rRNA gene fragments per gram soil) was higher in BD manure than in compost and ranged as follows and ranged as follows: (BD manure: 2.52×1011 to 4.17×1011 vs compost: 8.89.16×1010 to 1.46×1011). Meanwhile, the fungal community (ITS region copies per gram soil) were: (BD manure: 1.34×109 to 1.18×1011 vs compost: 8.89×1010 to 1.46×1011). However, a significant difference (P<0.05) was observed only for the fungal abundance between BD manure (AT-500) and horse compost (Figure S2B), while no significant differences (P>0.05) were observed for the bacterial community (Figure S2A).
The bacterial Shannon index was higher in BD manures than in composts and ranged as follows: BD manure (5.28-5.63) versus compost (1.81-3.54). Significantly higher bacterial diversity (P=0.02) in BD manures (AT-500 and FR-500) compared with horse compost was observed (Figure S2C). In contrast, the fungal Shannon diversity ranged between (2.63-3.13) for BD manures as compared to compost (2.41-2.60), and significant differences (P>0.05) in Shannon diversity were observed for the fungal community (Figure S2D).
PERMANOVA between BD manures and composts revealed significant differences in community compositions between these organic formulations for bacteria (R2 = 0.85, P=0.001) and fungi (R2 = 0.85, P=0.001). In addition, BD manures amended with plant preparations (AT/FR-500P) could be distinguished from those without preparations (AT/FR-500), especially for the bacterial community (Figures 1A, B).
Comparing higher-level taxonomy of BD manures and composts revealed differences in microbial composition between samples. Dominant phyla included Chloroflexi (average percentage abundance: 35.0%), Proteobacteria (29.5%), and Firmicutes (8.9%). The phylum Chloroflexi was dominant in composts (standard-, apple-, and horse), with abundance (69.8%, 72.4%, and 87.7%, respectively). Meanwhile, Proteobacteria, Firmicutes, and Actinobacteria were highest in BD manures with relative abundances of 43.7%, 14.8%, and 9.5%, respectively. For the fungal community, the phyla Ascomycota (59.7%), Basidiomycota (5.7%), and Mortierellomycota (5.1%) were predominant. We observed differences among compost microbiomes, especially for the bacterial community. The bacterial family Anaerolineaceae was present only in composts; however, with a dominance of uncultured bacteria (Figure 1C). Meanwhile, BD manures were seen to contain members of families Pseudomonadaceae, Peptostreptococcaceae, Microbacteriaceae, Lentimicrobiaceae, Lachnospiraceae, Devosiaceae, and Burkholderiaceae (Figure 1C). The fungal families including unidentified Microascales, unidentified Hypocreales, and Cladosporiaceae were observed in composts (Figure 1D). Trichosporonaceae, Microascaceae, Lasiosphaeriaceae, Mortierellaceae, and Chaetomiaceae were observed in BD manures, while the fungal family Cordycipitacea was only found in the apple compost (Figure 1D).
Moreover, bacterial genera including Bacillus, Devosia, and Pseudomonas were mainly associated with BD manures (AT-500/P and FR-500/P). The fungal genera Lecanicillium, and Arthrobotrys were mainly present in composts, while Trichosporon, Scutellinia, and Sporobolomyces, were associated with BD manures.
Analysis of the common microbiome analysis revealed that 109, and 82 bacterial ASVs were unique to BD manures and composts, respectively, while only two ASVs were shared (Figure 1E). Some bacterial genera in the common microbiome of BD manure were Paenibacillus, Mesorhizobium, Mycobacterium, Cellulomonas, and Clostridium. On the other hand, 13 and 54 unique fungal ASVs were detected in BD manures and compost, while five ASVs were shared between both microbiomes (Figure 1F). The shared fungal ASVs included unidentified Capnodiales and unidentified Microascacea.
We compared BD manures that differed in production year and region of production (Austria or France). Differences (P<0.05) in fungal diversity were observed between FR-500P [2012] and AT-500 [2021] as shown (Figure S3D), while the fungal abundance was substantially higher in AT-500 [2020/2021] as compared to FR-500P [2016: S, T, and V] (Figure S3B). No significant differences (P>0.05) in bacterial abundance and diversity were observed among the biodynamic manures, for the two regions and different years of production (Figures S3A, C).
In contrast to microbial diversity and abundance, we observed substantial differences in microbial composition between BD manures based on the country of production as follows: bacterial community (BD manure: R2 = 0.78, P=0.001, country: R2 = 0.24, P=0.001), and fungal community (BD manure: R2 = 0.78, P=0.001, country: R2 = 0.26, P=0.001). These differences were also shown by NMDS in which clear separation of samples according to the region and BD manure type was observed (Figure S4).
The dominant bacterial classes were Gammaproteobacteria (19.3 average percentage relative abundance), Clostridia (14.5%) and Alphaproteobacteria (12.1%). Predominant fungal classes included Sordariomycetes (47.9%), Tremellomycetes (6.7%), Pezizomycetes (5.9%) and Dothideomycetes (5.2%). Differences between regions were visible, especially for the bacterial community. For instance, the order Actinomarinales was only found in BD manures produced in France (Figure S3C). Meanwhile, Sphingobacteriales were observed exclusively in Austrian BD manure samples (Figure S4C). The fungal community between the two regions was separated by the presence of orders Capnodiales, Ascosphaerales, and Pleosporales were associated with the BD manures produced in France. Independent of the region of production, BD manures were found to contain bacterial families such as Bacillaceae, Burkholderiaceae, Clostridiaceae, Flavobacteriaceae, and Pseudomonadaceae.
The assessment of the common microbiome of BD manures from the two countries revealed that 30 bacterial and 13 fungal ASVs were shared. Most bacterial ASVs were unique (120 and 123 for Austria and France, respectively). In contrast, a total of 16 and 11 fungal ASVs were unique for Austria and France (Figures S4E, F). Some of the shared bacterial ASVs included Devosia, Azotobacter, Acidibacter, Clostridium, Romboutsia, Cellulomonas, Sorangium, Solirubrobacter, and Turicibacter. The shared fungal genera were Trichosporon, Mortierella, unidentified Microascaceae, unidentified Lasiosphaeriaceae and unidentified Sordariales.
To establish whether BD plant preparations altered the microbiome of BD manures (FR/AT-500P), the community structure and diversity of BD preparations (manure and plant preparations) were analysed. Significant differences in microbiome composition were seen between amended BD manures and the different plant preparations (bacterial: R2 = 0.83, P=0.001; fungal community: R2 = 0.94, P=0.001. NMDS visualization showed a clear separation between BD manures and plant preparations. Microbiome separation between plant preparations was observed for bacterial and fungal communities, respectively (Figures S5A, C).
There was a high microbiota load both in BD plant preparations and manures (bacterial: 8.12 × 1010 to 2.65 × 1011), and fungal (9.70 × 109 to 1.34 × 1010). A significant difference (P<0.05) was observed for the fungal abundance between FR-500 and P-505 (Figure S6B), and no differences (P>0.05) were observed for the bacterial community (Figure S6A). On the other hand, the Shannon diversity index of BD plant preparations and amended manure (i.e. FR-500P) ranged between 4.38-5.28 and 2.20-3.18, both for bacterial and fungal communities, respectively. Significant differences (Kruskal-Wallis, P<0.05) in bacterial diversity were observed between BD manure (FR-500P) and plant preparation (P-505), as well as between P-504 with P-505 and P-506 (Figure S5C). The fungal diversity was substantially higher (P<0.05) in preparations (P-505 and P-506) as compared to P-503 (Figure S6D).
The bacterial order Bacillales was present both in BD manures and plant preparations, except in preparation P-505, and was highest in P-503 and P-504 with average relative abundances of 11.8% and 36.0%, respectively. The bacterial orders Rhizobiales, Pseudomonadales, and Flavobacteriales were observed in both plant preparations and BD manures (Figure 5B). Meanwhile, Bacteroidales was especially present in AT-500/P and plant preparations (P-503 and P-505). For the fungal community, BD manures were distinguished from plant preparations by orders Capnodiales, Hypocreales, Ascosphaerales, Eurotiales and Unidentified fungi. The fungal orders Microascales, and Pezizales were associated with BD manures and plant preparations (Figure S4D).
A comparison between preparation amended and non-amended manures revealed that 120 and 155 bacterial ASVs were shared between (AT-500 and AT-500P), as well as (FR-500 and FR-500P), respectively (Figures S7B, D). Similarly, for the fungal community 25 and 23 ASVs were shared (Figures S8B, D). There was a common microbiome shared between BD manures and plant preparations, both for bacterial (Figures S7A, C) and fungal communities (Figures S8A, C), respectively. More unique than shared members of the bacterial (Figures S7A, C) and fungal (Figures S8A, C) ASVs were observed for plant preparations and BD manures. The shared part of the common microbiome between BD manures and plant preparations was composed of bacterial genera Romboutsia, Azotobacter, Pedomicrobium, and Bacillus. Meanwhile, shared fungal genera were Fusarium, Arthrographis, Unidentified Microascaceae, Unidentified Chaetomiaceae, and Unidentified Nectriaceae.
LEfSe analysis identified the bacterial and fungal genera explaining the differences between BD manures (FR-500P) and plant preparations (Figures 2A, B, respectively). The BD plant preparations (P-502, P-503, P-504, P-505, and P-506) were differentiated by the bacterial genera Luteibacter, Oceanobacillus, Virgibacillus, Mycobacterium, and Staphylococcus (Figure 2A). Lentimicrobium, Romboutsia, and Devosia were associated with BD manures (AT/FR-500P). However, the genus Bacillus was shared between BD manures (AT/FR-500P) and plant preparations. Meanwhile, fungal genera Scutellinia, Trichosporon, and Sporobolomyces were mainly associated with BD manures, while Fusarium, Ascosphaera, Pseudallescheria, Arthrographis, Chaetomium, Cyberlindnera, and Scedosporium were associated with BD plant preparations (Figure 2B).
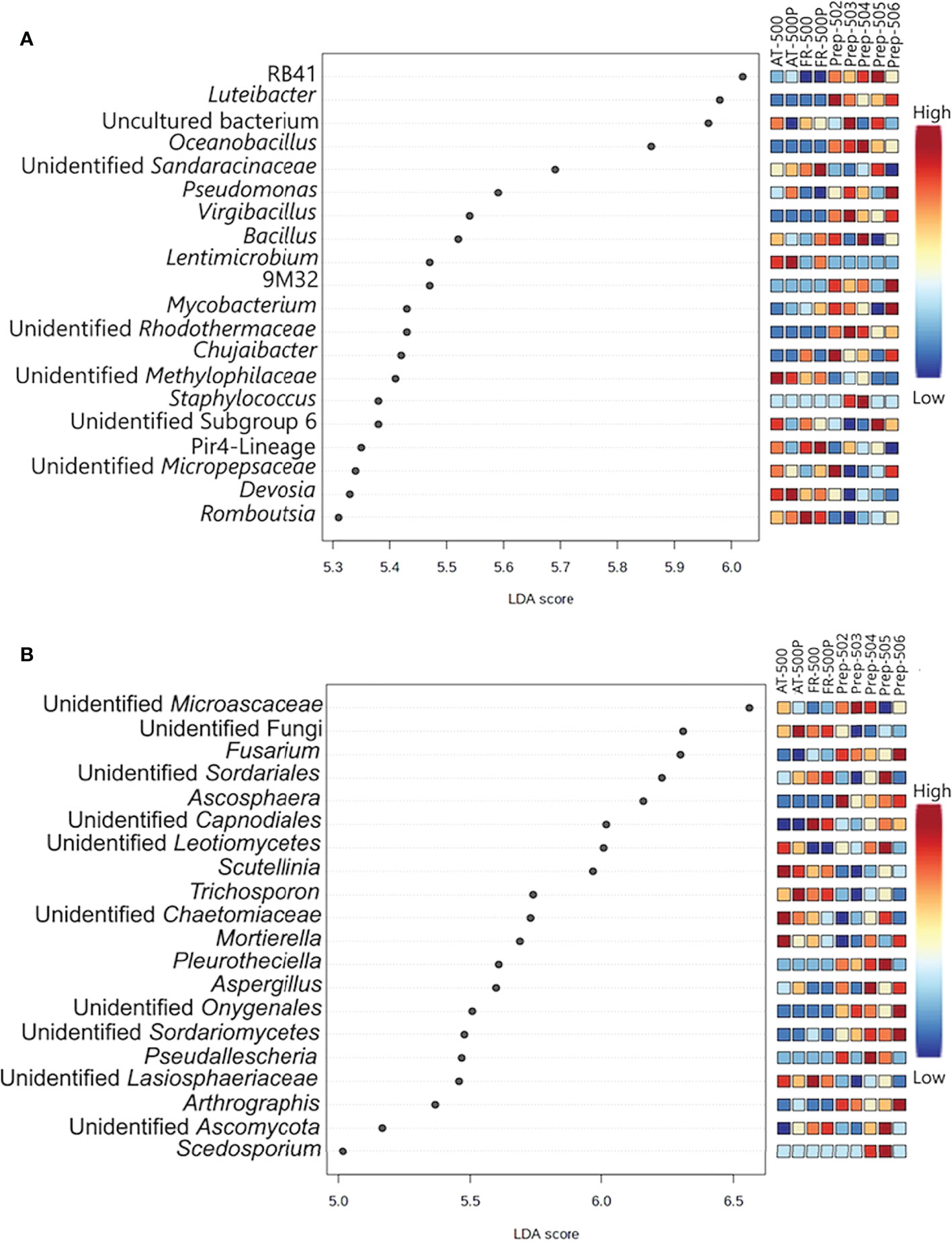
Figure 2 Dot plot representation of differentially abundant bacterial (A) and fungal (B) genera between BD manures and BD plant preparations. Plant preparations are shown as P-502, P-503, P-504, P-505, and P-506; while biodynamic manures were represented as AT -500/P and FR-500/P. Acronym P- represent preparation, while AT and FR represent the countries Austria and France where the BD manures were produced. The differentially abundant genera with LDA scores between 5 and 7 and were based on FDR adjusted P-value. The coloured bar represents the relative abundances of the differentially abundant genera, with the lowest indicated as blue and highest as red.
The microbial diversity and abundance comparison between extracts and precursor materials of BD manure (AT-500P) and standard compost revealed that extracts generally contained a higher microbial diversity than precursor materials, except for AT-500P (Figures S9C, D); and vice versa for microbial abundance (Figures S9A, B). Significantly higher (t-test: P<0.05) microbial abundance was observed in the precursor of BD manure (AT-500P) in comparison to the respective extract, both for bacterial and fungal communities (Figures S9A, B). No significant differences (P>0.05) in microbial abundance were observed between the precursor and extract of standard compost (Figures S9A, B). The bacterial diversity in the standard compost extract was significantly lower (t-test, P=0.004) than in the extract derived from the compost materials (Figure S9C). Contrarily, the fungal diversity in extract derived from standard compost and BD manure (AT-500P) was significantly higher (t-test, P<0.05) in comparison to precursor material (Figure S9D).
The microbial community structure between extract and precursor materials of BD manure (AT-500P) and standard compost was distinct. This was confirmed by PERMANOVA analysis in which we observed significant differences between the extracts and their derivative materials (i.e., bacteria: R2 = 0.65, P=0.001 and fungi: R2 = 0.72, P=0.001). In addition, pairwise community comparison revealed significant differences (P<0.05) in community structure between precursor materials (standard compost and AT-500P) and their respective extracts, both for bacterial and fungal communities (Tables S7 and S8, respectively).
The extracts and precursor materials of standard compost were distinct in microbial composition. BD manure and their respective extract displayed similar microbiome compositions (Figures 3A, B). The bacterial classes Anaerolineae (61.6%), and Chloroflexia (8.2%) predominated in standard compost, while Gammaproteobacteria (52.3%) and Bacilli (34.0%) were mainly present in the extract of standard compost (Figure 3A). The BD manure (AT-500P) and extract were mainly associated with the bacterial classes Gammaproteobacteria (precursor: 30.8% and extract: 20.6%), Bacteroidia (18.0% and 11.3%), Alphaproteobacteria (11.8% and 18.1%), Clostridia (11.8% and 11.5%)), and Deltaproteobacteria (5.2% and 8.2%). For the fungal community, extracts of standard compost and BD manure as compared to the precursor materials contained a higher abundance of Dothideomycetes (10.0% versus 1.2%, and 2.6% versus 0.1%, respectively), Eurotiomycetes (7.7% versus 1.2%, and 4.1% versus 0.3%), as well as Leotiomycetes (2.0% versus 0.7%, and 3.1% versus 0.5%) (Figure 3B).
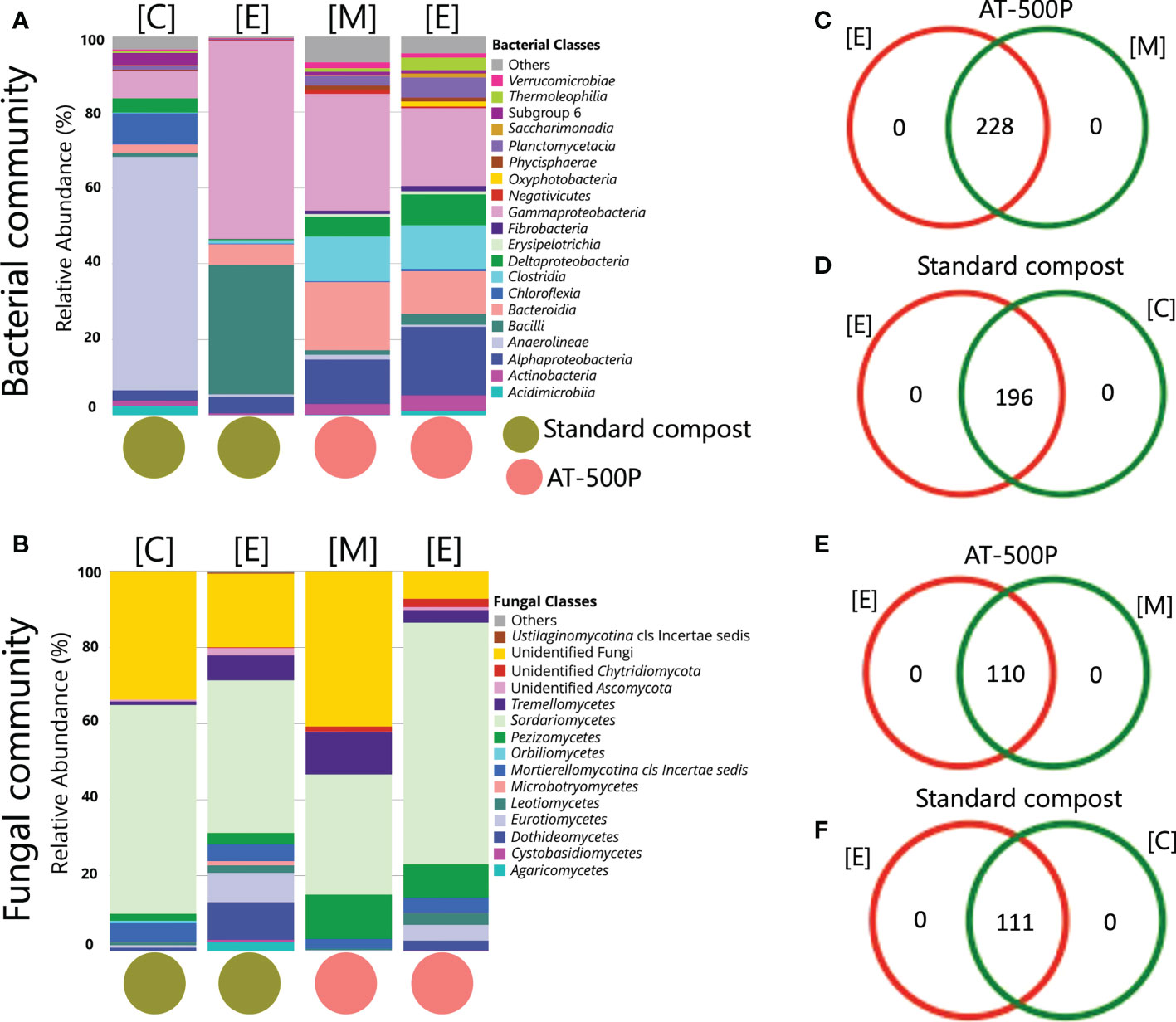
Figure 3 Comparison of microbial structure composition between AT-500P and standard compost as well as their respective extract. Panels (A, B) are stacked barplot representations of the bacterial and fungal community composition in precursors and extracts of compost and BD manure. Panels (C, D) are Venn diagrams representing the common bacterial ASVs shared between extract and precursor materials of compost and BD manure, while (E, F) represent the same for the fungal community. The acronyms [E], [C], and [M] represent extract, compost, and BD manure, respectively.
Interestingly, BD manure (AT-500P) and the associated extract shared 228 and 110 ASVs of the common microbiome, both for bacterial and fungal communities, respectively (Figures 3C, E). Conversely, standard compost and extract were found to share 196 and 111 ASVs (Figures 3D, F). Differential abundance analysis revealed the enrichment of bacterial genera like Bacillus, Pseudomonas, Paenibacillus, Flavobacterium, and Azotobacter in the extract of standard compost as compared to precursor materials (Figure S10A). Meanwhile, in extract (AT-500P) as compared to precursor material, an enrichment of bacterial genera including Roseobacter and Polynucleobacter was observed, while Lentimicrobium, Devosia, and Simplicispira were enriched in AT-500P (precursors) Figure S10B.
For the fungal community, genus-level differences between extracts and precursor materials of standard compost and BD manure (AT-500P) were marked by the enrichment of fungal genera Fusarium, Trichosporon and Aspergillus in the extract as compared to standard compost and AT-500P precursors (Figures S10C, D). In addition, Penicillium, and Scutellinia comprised the extracts of standard compost (Figure S10C), while Arthrographis was enriched in AT-500P extract (Figure S10D).
The microbial structure and composition, as well as diversity and abundance analyses, were performed with rhizosphere samples from two apple orchards managed using organic and integrated management systems. Samples were obtained at two sampling times (spring and autumn) from both orchards. There were no significant differences (P>0.05) in microbial abundance between treatments and controls, both for organic and integrated orchards at one- and four-months post-inoculation (Figure S11). Generally, after four months, bacterial diversity was lower in treatments in comparison to controls. In contrast, the fungal diversity was higher in treatments as compared to controls (Figure S12). At four months post-inoculation, the bacterial diversity was significantly lower (P<0.05) in standard compost (extract treatment) compared to the control in the organic orchard, as well as between AT-500P and the control for the integrated orchard (Figure S12B). Furthermore, the fungal diversity was significantly higher in the AT-500P treatment compared to the control in the integrated orchard four-month post-inoculation (Figure S12D).
For beta diversity, significant microbial structure differences between orchard types and sampling time were observed: orchard type (bacterial: R2 = 0.14, P=0.001; fungal: R2 = 0.30, P=0.001) and sampling time (bacterial: R2 = 0.06, P=0.001; fungal: R2 = 0.06, P=0.004). An overall effect of treatment on the microbiome was observed in the organic orchard, both after one month (PERMANOVA; bacterial: R2 = 0.32, P=0.02; fungal: R2 = 0.26, P=0.001) and four months (bacterial: R2 = 0.27, P=0.002; fungal: R2 = 0.23, P=0.05) as shown (Tables S7, S8 for the bacterial and fungal community, respectively). However, in the integrated orchard, a significant effect of treatment on bacterial community structure was observed four months after treatment (R2 = 0.32, P=0.002), while no significant effect (P>0.05) was seen for the fungal community (Tables S7, S8). Pairwise significant differences (P<0.05) between treatment and control were evidenced for the fungal community at one-month post-inoculation (Table S8), and for the bacterial community in both orchards at four months post-inoculation (Table S7). The microbial community clustering by NMDS was shown for different treatments and orchards, at one month (Figures S13A, C), and four months (Figures S14A, C) post inoculation, both for bacterial and fungal community, respectively.
The apple rhizosphere of the two orchards was dominated by the bacterial phyla Proteobacteria (Organic: Integrated; 26.1%: 25.8%), Firmicutes (30.0%: 3.4%), Actinobacteria (18.8%: 26.9%), Chloroflexi (13.1%: 15.5%), and Acidobacteria (17.2%; 10.4%). On the other hand, the fungal community was predominated by Ascomycota (70.1%: 68.1%), Mortierellomycota (21.8%: 21.8%), and Basidiomycota (6.5%: 8.3%). For both orchards, the bacterial composition was similar at one-month post-inoculation, but a higher average relative abundance of less abundant taxa categorized into (“others”) was seen in rhizosphere soil treated with extract of BD manure (AT-500P) and standard compost as compared to the control (Figure S13B). For the fungal community, we observed that families such as Microascaceae, and Chaetomiaceae were higher in treatments as compared to the control (Figure S13D).
At four months post-inoculation, the family-level taxonomic composition was similar, but differences were observed in the less abundant taxa (“others”), whose abundance was high in treatments as compared to the control (Figures S14B, D). In contrast, a high abundance of the family Chaetomiaceae was observed in treatments as compared to controls, both for organic and integrated orchards (Figure S14D).
All microbial genera which were responsible for significant pairwise differences in community composition between treatments and controls were further explored. At one-month post-inoculation, enrichment in the control of the fungal genera Alternaria, Thelonectria, and Solicoccozyma was observed in the organic orchard (Figure 4A). Meanwhile, the genus Chaetomium was highly abundant in the rhizosphere which was treated with the extract from BD manure (AT-500P). At four months post-inoculation, bacterial genera including Bacillus, Bradyrhizobium, Pseudolabrys, and unidentified Burkholderiacea were enriched in the controls, compared to the treatments, while Chthniobacter and Candidatus Xiphinemebacter were enriched in the rhizosphere after treatments in the organic orchard (Figure 4B). For the integrated orchard, the bacterial genera Pseudomonas, Mycobacterium, and Candidatus Udeobacter were enriched in the rhizosphere that had been treated with AT-500P (extract), while Candidatus Xiphinemebacter, and uncultured Pirellulaceae were enriched in rhizosphere treated with the extract from standard compost (Figure 4B).
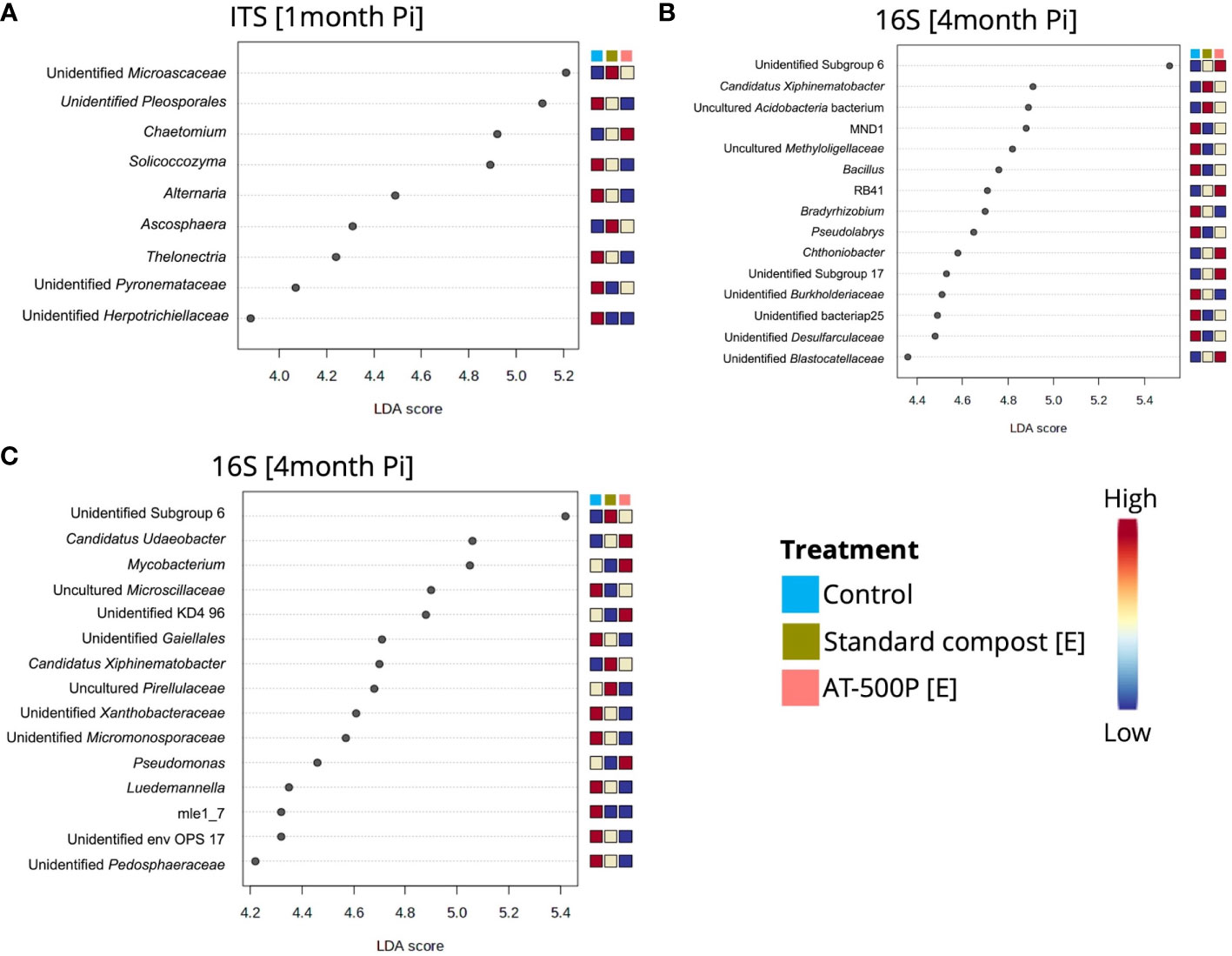
Figure 4 Dot plot representation of differentially abundant genera in treatments which showed significant pairwise differences to the control for the different orchards and sampling times. (A) shows differentially abundant fungal genera at one-month post inoculation in organic orchard, while (B, C) represent significant differentially abundant bacterial genera between treatment and control in organic and integrated orchard at four-months post inoculation, respectively. The significant (P<0.05) differentially abundant genera were obtained by LEfSe, and the genera with high LDA scores (3 to 6) were shown. The [E] in the legend represents extract, and the coloured scale bar represents relative abundances, with the lowest indicated as blue, and highest as red.
SourceTracker was employed to reveal the portion of the rhizosphere microbiome, which was potentially introduced by the treatments that included extracts from standard compost and AT-500P for organic and integrated orchards (Figure 5) at two sampling times. We observed time-dependent differences in the proportion of the rhizosphere microbiome that was introduced by extracts. A higher proportion of the microbiota at one month post-inoculation, than at four months post-inoculation could be traced back to the treatments, especially for the integrated orchard.
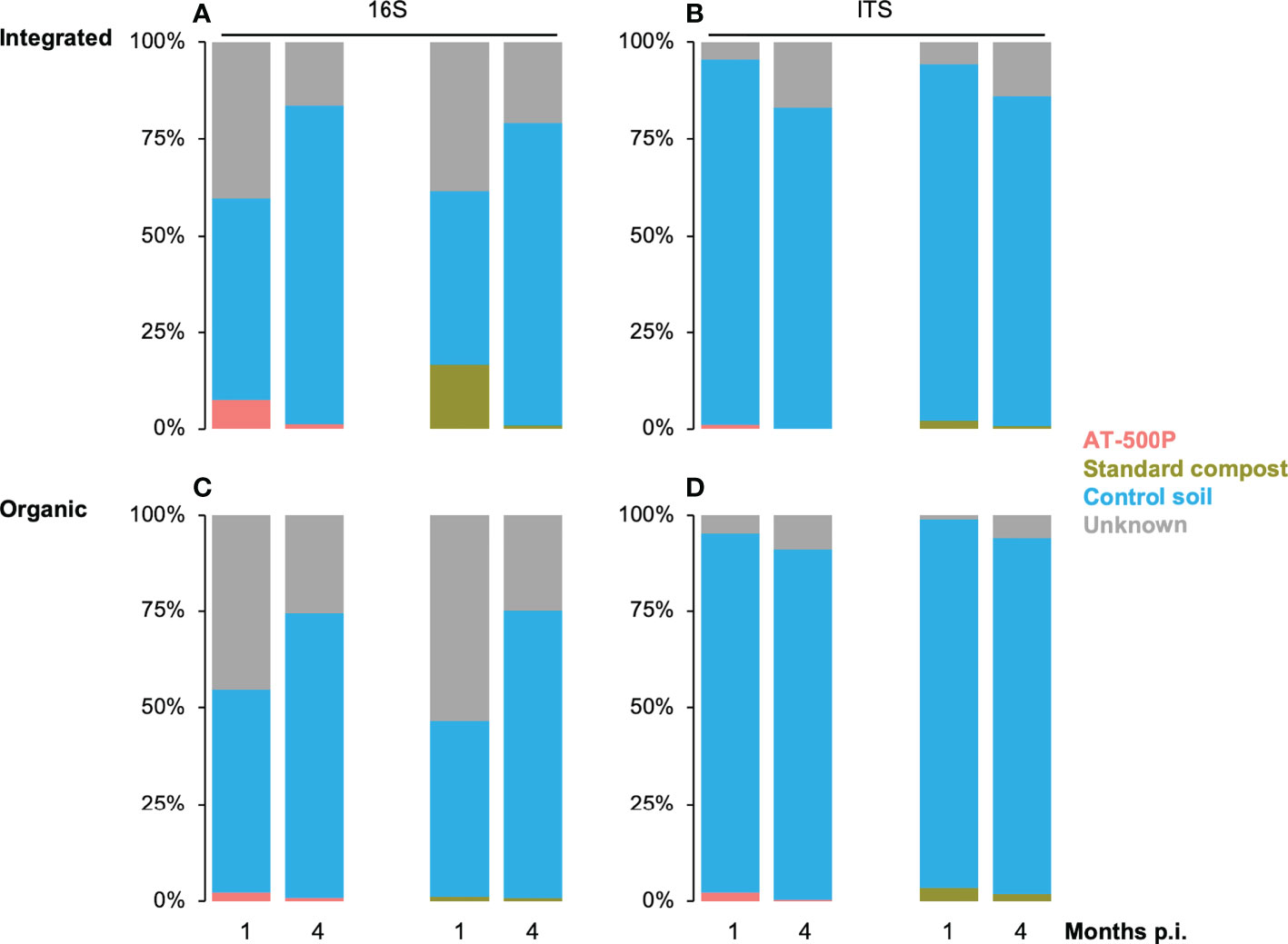
Figure 5 Tracking the microbiome transfers from standard compost and BD manure (AT-500P) extracts (i.e. microbiome source) into the treated rhizosphere soil under different management systems and sampling times. Panel (A) and (B) shows the proportion of microbiota tracked from treatment into rhizosphere in the integrated orchard for bacterial and fungal community, respectively, while (C) and (D) represent the same for the organic orchard. The proportion of the microbiome in the treated rhizosphere which was shared with the untreated soil (i.e. control soil) is shown both for organic and integrated orchards. The unknown part of the microbiome includes the percentage proportion of microbiota in treatments which could not be traced in the rhizosphere. Coloured stacked bars represent proportions of microbiome in treated rhizosphere (i.e. AT-500P and standard compost extracts), shared microbiome between control and treated rhizosphere soil, as well as the unknown microbiome (i.e. the unassigned part of treatment microbiome). The x-axis represents time in months post inoculation. The y-axis shows the proportion of the community originating from the different sources.
We observed that treatments introduced a higher proportion of bacteria in integrated than in the organic orchard, especially at one-month post-inoculation, and the average percentage proportions ranged as follows: organic (1%-2%) and integrated management (1%-20%). However, a high proportion of bacteria (10% to 60%), and fungi (0%- 20%) from the extracts (i.e. labelled as “unknown”) could not be traced in the rhizosphere (Figure 5). For the fungal community, both in the organic and integrated orchard, the extract from standard compost as compared to AT-500P was found to potentially introduce more fungi into the rhizosphere: organic (AT-500P: 0%-2% vs. standard compost: 0%-3%), and integrated management (0%-1% vs. 0%-2%). Considering control soil as the microbial source, we observed that a high proportion of the microbiome (bacteria: 40% to 90%, and fungi: 80% to 95%) was shared with the treated rhizosphere soil.
Biodynamic manures, composts, and plant preparations carried distinct microbiomes. We observed a higher microbial diversity in BD manures than in BD plant preparations and composts (apple-, horse-, and standard composts). While the impact of type and quantity of organic material on compost microbiomes was previously reported (51), our study reveals for the first time specific microbiome differences between BD manures and composts. These variations could be attributed to various factors such as the type of starting material and the composting technology used (52). Moreover, the role of physical and chemical parameters such as oxygen, temperature, moisture content, pH, and nutrient availability shape the compost microbiome (53).
Specific to BD farming, the fermentation of animal waste and the subsequent amendment with plant preparations form a critical part of BD products (20). This study revealed that plant preparations and amended BD manures contain a distinct microbiome. In addition, the microbiome varied among the different plant preparations. Such variation among preparations might be attributed to plant materials from which they are derived. Moreover, the effect of the plant genotype on the microbiome has been reviewed elsewhere (54, 55). Our study provides a link between the plant microbiome and the microbiome of plant-derived organic amendments. We propose that the specific procedures used in making BD manures might have contributed significantly to the uniqueness of BD products. This is apparent from the clear differences between composts and their BD counterparts (Figures 1A, B for bacterial and fungal communities, respectively). However, a common microbiome comprised of Paenibacillus, Cellulomonas, and Clostridium was shared between all BD manures and composts. These taxa play essential roles as proficient degraders of plant biomass (56). The presence of members under the family Clostridiaceae could be attributed to the use of cow manure in BD preparations. Clostridium is associated with severe bacillary hemoglobinuria in cattle and sheep, and it was suggested that its incidence can be reduced by the annual vaccination of herds (57). Clostridia in traditional BD preparations could fulfil similar functions. Additionally, Nitrogen fixation by Clostridia (58, 59), underpins the potential effect of organic amendments in promoting beneficial soil microbial traits.
Biodynamic manures amended with preparations contained a high abundance of Actinobacteria, a phylum known for its active role in the composting process (60, 61). Evidence linking Actinobacteria in a consortium of Firmicutes, Proteobacteria, and Bacteroidetes to the breakdown of lignocellulose has recently been revealed (56). Potential plant-beneficial bacterial families like Pseudomonadaceae, Burkholderiaceae, and Bacillaceae (62) were observed to dominate the BD formulations and plant preparations. This suggests a potential contribution of the microbiome associated with BD manure and plant preparations in farming systems.
Interestingly, a common microbiome of the bacterial genus Azotobacter, and the fungal genera Fusarium and Arthrographis was observed between BD manures and plant preparations and further emphasizes the potential soil enrichment with beneficial bacteria. The genus Fusarium mainly contains plant pathogenic species (63), and was observed to be mainly associated with BD plant preparations. However, non-pathogenic and beneficial Fusarium species are well known (64) supporting the idea that intraspecific microbial diversity can supress pathogenicity (3). The fungal family Ascosphaeracea was observed in some of the BD plant preparations and has previously been found associated with the microbiome of bees (65). We suggest that this could have originated from the flowers of the plants used as starting materials in the production of BD plant preparations.
Independent of the production year (storage effect), the microbiome of BD manures was distinct between the regions of production (Austria and France). We suggest that various factors including plant and animal raw materials, as well as product handling in the two regions, have contributed significantly to the observed microbiome differences. Our study extends this insight to explain the likely cause of the region-specific difference observed in BD manures. Furthermore, the microbiome consistency across years suggests that BD manures can be reproduced in a production line. The specific procedures to make BD preparations have been practised for decades by the BD farming communities and were also standardized (10, 15, 20), which provides an explanation for the reproducibility.
The use of liquid extracts from BD manures as field sprays is a common practice in BD farming (15). While the biochemical characteristics of such extracts were already deciphered (66, 67), our study serves as a primer to further understand the microbiome of BD extracts. The microbiomes of extracts and precursor materials of composts and BD manures were distinct but shared a common microbiome. We likely attribute the shared microbiome to the material from which the extracts are derived; also the procedure that materials are subjected during the fermentation. Moreover, the common microbiome of compost, mainly composed of Firmicutes and Actinobacteria was recently revealed (68). Bacterial genera including Pseudomonas, Paenibacillus, Azotobacter, and unidentified Sphingomonadaceae were associated with standard compost extract Microbially enhanced compost extracts were previously found to be essential in improving plant growth and nutrition (69). Thus, their microbiome composition could potentially explain the effects attributed to extract application on plant health.
Several fungal genera such as Fusarium, Aspergillus Penicillium, Mortierella, and Lecanicillium were observed in extracts of the different compost and BD manures. Interestingly, Lecanicillium has been found to have entomopathogenic properties, which led to its use as a biocontrol agent against aphids (70), as well as phytoparasitic nematodes and fungi (71, 72). The fungal genus Penicillium is diverse and widely distributed in the soil environment (73–75). It can exert positive effects through the production of secondary products (76), as well as play critical roles in plant growth and defence (77–79). High microbial diversity was found in extracts, as compared to the precursor standard compost and BD manure. This could be attributed to additives such as molasses and stone dust which were used to make the extracts. In addition, water as a solvent used in the extract production process may have contributed to its microbiome. Recent studies often found high microbial diversity in water microbiomes (80–82).
Overall, a positive impact of BD manure and compost amendments on soil fungal diversity was found, especially at four months post-inoculation, both for organic and integrated orchards. Enrichment effects of BD formulations on the soil microbiome were reported in a forgoing study (27), and further confirmed by the current study. Previous studies indicated that the use of BD manures and associated extracts in viticulture affects soil biodiversity and ecosystem functions (13, 18, 83, 84), and plays important roles in improving soil quality (23); as well as their potential role in biodynamic viticulture including the enhancement of wine quality (17, 25, 26). In the present study, treatments increased Acidobacteria, both in organic and integrated orchards. Acidobacteria is a rather underrepresented phylum, yet ubiquitous in the soil environment (85, 86), and involved in various processes in biogeochemical cycles, decomposition of biopolymers, exopolysaccharide secretion, and plant growth promotion (85, 87, 88). The fungal genus Alternaria was found to be decreased in treatments as compared to the untreated rhizosphere at one-month post-inoculation in the organic orchard. Thus, we suggest that one potential role of BD treatments is the reduction of plant pathogenic fungi, which also includes Alternaria. This genus is ubiquitous with saprophytic, endophytic, and pathogenic species which are commonly found associated with various plants, including fruits (89–91). Moreover, some Alternaria species were linked with allergies in humans (92, 93). Interestingly, strains of Fusarium and Alternaria have recently been found to be strongly associated with seeds of numerous crop plants, where they are vertically transmitted to the root and rhizosphere (94). On the other hand, the bacteria genera Pseudomonas and Mycobacterium were found to be enriched in the apple rhizosphere of the integrated orchard which had been treated with extracts from BD manure. Thus, in the context of BD farming, our findings provide insights into the potential role of BD products especially in increasing the abundance of beneficial bacterial genera. Moreover, the determination of various soil quality indicators suggests larger response in the BD system, than in conventional management systems (95, 96).
Our attempt to track the microbiome from extracts to the rhizosphere revealed that a specific proportion of the extract microbiome was transferable. In contrast to a previous study by (27), we observed that a higher proportion of the bacterial and fungal community was transferred to the rhizosphere in the integrated management orchard when compared the organic orchard. This implies that the use of BD extracts differently affects local soil microbiomes. This might be due to competition with indigenous microorganisms that are present in these soils. However, microbial signatures of organic amendment origin can survive in the soil microbial biomass (97, 98). Interestingly, even with the low applied rates for BD preparations (i.e. 1L:10L; extract: water) as shown by Koepf etal. (99) and the international “Demeter” guidelines for biodynamic farming (https://demeter.net/), we observed a significant effect of treatment on the microbial community structure and diversity, particularly an increase in fungal diversity during autumn for the integrated orchard. Thus, we suggest that an increase in the frequency of treatment application could more effectively modulate the local soil microbiome. Moreover, the application of small quantities (i.e. ≥ 5%) of suppressive soil was found to induce suppressiveness to disease conducive soils (100, 101). The low proportion of microbes transferred to the apple rhizosphere of the organic as compared to the integrated management orchard could be associated with high competition from the resident microbiota. Limitations for the establishment of bioinoculant strains in soil environments were previously described (102, 103), and may include competition, as well as limitation in colonization due to different soil physico-chemical parameters.
The findings of this study provide first insights into the microbiome of biodynamic manures and their applied liquid extracts. Overall, our findings provide knowledge on approaches to enhance microbial diversity in agricultural soils, especially in intensively managed fields. Moreover, the in-depth assessment of biodynamic products in orchards subjected to different field management systems provides valuable information related to their contribution to plant rhizosphere microbiomes. Interestingly, the low standardization of compost amendments criticized in the past (104), can be an advantage in future agriculture, which needs more diversification at all levels. We detected potentially beneficial microbes originating from biodynamic treatments that may be established in different farming systems upon their introduction; although, consistent application will be needed to provide expected benefits in the long term.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
GB, PK, EO, and RM designed the study. EO, PK, and RM performed the experiment and took the samples. EO performed library preparation and other laboratory work. EO, PK and WW analysed the data and interpreted it together with TC, SB, and GB. EO, SB, and PK wrote the manuscript. All authors read, corrected, and approved the final version.
We gratefully acknowledge funding by EXCALIBUR research project with funding from the European Union’s Horizon 2020 Research and Innovation Program under grant agreement No. 817946 (to GB).
We sincerely thank Robert Matzer (Gleisdorf, Steirmark-Austria) and Josef Singer (Gleisdorf Steirmark-Austria) for allocating us experimental orchards to apply our treatments, perform sampling, as well as in maintaining the experimental plots. The cooperation and hospitality from their families is also highly appreciated.
Author RM was employed by the company BODENmanagement.net.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsoil.2022.1020869/full#supplementary-material
1. Berg G, Rybakova D, Fischer D, Cernava T, Vergès MCC, Charles T, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome (2020) 8, 103. doi: 10.1186/S40168-020-00875-0
2. Berg G, Cernava T. The plant microbiota signature of the anthropocene as a challenge for microbiome research. Microbiome (2022) 10:1–12. doi: 10.1186/S40168-021-01224-5/FIGURES/1
3. Berg G, Köberl M, Rybakova D, Müller H, Grosch R, Smalla K. Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol Ecol (2017) 93:50. doi: 10.1093/FEMSEC/FIX050
4. Malard F, Dore J, Gaugler B, Mohty M. Introduction to host microbiome symbiosis in health and disease. Mucosal Immunol (2020) 14:547–54. doi: 10.1038/s41385-020-00365-4
5. Malusà E, Berg G, Biere A, Bohr A, Canfora L, Jungblut AD, et al. A holistic approach for enhancing the efficacy of soil microbial inoculants in agriculture:: From Lab to field scale. Glob J Agric Innov Res Dev (2021) 8:176–90. doi: 10.15377/2409-9813.2021.08.14
6. Ozores-Hampton M. Past, present and future of compost in horticulture crop production. Compost Util. Prod Hortic Crop (2021), 1–8. doi: 10.1201/9781003140412-1
7. De Corato U. Agricultural waste recycling in horticultural intensive farming systems by on-farm composting and compost-based tea application improves soil quality and plant health: A review under the perspective of a circular economy. Sci Total Environ (2020) 738:139840. doi: 10.1016/J.SCITOTENV.2020.139840
8. Lutz S, Thuerig B, Oberhaensli T, Mayerhofer J, Fuchs JG, Widmer F, et al. Harnessing the microbiomes of suppressive composts for plant protection: From metagenomes to beneficial microorganisms and reliable diagnostics. Front Microbiol (2020) 11:1810/BIBTEX. doi: 10.3389/FMICB.2020.01810/BIBTEX
9. Heisey S, Ryals R, Maaz TM, Nguyen NH. A single application of compost can leave lasting impacts on soil microbial community structure and alter cross-domain interaction networks. Front Soil Sci (2022) 0:749212. doi: 10.3389/FSOIL.2022.749212
10. Diver S. Biodynamic farming and compost preparation (1999). Available at: www.attra.ncat.org (Accessed February 17, 2022).
11. Raaijmakers JM, Mazzola M. Soil immune responses soil microbiomes may be harnessed for plant health. Sci (80-. ) (2016) 352:1392–3. doi: 10.1126/SCIENCE.AAF3252/ASSET/D65C8EED-FD4D-4B9F-8F8E-AB5BFACED03C/ASSETS/GRAPHIC/352_1392_F1.JPEG
12. Lupatini M, Korthals GW, de Hollander M, Janssens TKS, Kuramae EE. Soil microbiome is more heterogeneous in organic than in conventional farming system. Front Microbiol (2017) 7:2064/BIBTEX. doi: 10.3389/FMICB.2016.02064/BIBTEX
13. Villanueva-Rey P, Vázquez-Rowe I, Moreira MT, Feijoo G. Comparative life cycle assessment in the wine sector: Biodynamic vs. conventional viticulture activities in NW Spain. J Clean Prod (2014) 65:330–41. doi: 10.1016/J.JCLEPRO.2013.08.026
14. Pergola M, Persiani A, Pastore V, Palese AM, Arous A, Celano G. A comprehensive life cycle assessment (LCA) of three apricot orchard systems located in metapontino area (Southern Italy). J Clean. Prod (2017) 142:4059–71. doi: 10.1016/J.JCLEPRO.2016.10.030
15. Brock C, Geier U, Greiner R, Olbrich-Majer M, Fritz J. Research in biodynamic food and farming- a review. Open Agric (2019) 4:743–57. doi: 10.1515/opag-2019-0064
16. Yiridoe EK, Bonti-Ankomah S, Martin RC. Comparison of consumer perceptions and preference toward organic versus conventionally produced foods: A review and update of the literature. Renew. Agric Food Syst (2005) 20:193–205. doi: 10.1079/RAF2005113
17. Castellini A, Mauracher C, Troiano S. An overview of the biodynamic wine sector. Int J Wine Res (2017) 9:1–11. doi: 10.2147/IJWR.S69126
18. Döring J, Collins C, Frisch M, Kauer R. Organic and biodynamic viticulture affect biodiversity and properties of vine and wine: A systematic quantitative review. Am J Enol Vitic (2019) 70:221–42. doi: 10.5344/AJEV.2019.18047
19. Paull J, Kolisko L, Vreede E, Wachsmuth G, Pfeiffer E, Kaufmann GA. Attending the first organic agriculture course: Rudolf steiner’s agriculture course at koberwitz 1924. Eur J Soc Sci (2011) 21, 64–70. Available at: https://www.biographien.kulturimpuls.org [Accessed May 12, 2022]
20. Leiber F, Fuchs N, Spiess H. ‘Biodynamic agriculture today’, in Organic agriculture: a global perspective (CABI), 141–9. doi: 10.1079/9781845931698.0141
21. Nabi A, Narayan S, Afroza B, Mushtaq F, Mufti S, Hm U, et al. Biodynamic farming in vegetables. J Pharmacogn. Phytochem. (2017) 6, 212–9. Available at: https://www.phytojournal.com/archives/2017/vol6issue6/PartD/6-5-383-396.pdf [Accessed November 7, 2022].
22. Ponzio C, Gangatharan R, N. D. Organic and biodynamic agriculture: A review in relation to sustainability (2013). Available at: https://asianarchive.co.in/index.php/IJPSS/article/view/1432 (Accessed February 19, 2022). Asian Arch.
23. Reganold JP. Soil quality and profitability of biodynamic and conventional farming systems: A review. Am J Altern. Agric (1995) 10:36–45. doi: 10.1017/S088918930000610X
24. Scheuerell S, Mahaffee W. Compost tea: Principles and prospects for plant disease control. Compost Sci Util (2002) 10:313–38. doi: 10.1080/1065657X.2002.10702095
25. Spaccini R, Mazzei P, Squartini A, Giannattasio M, Piccolo A. Molecular properties of a fermented manure preparation used as field spray in biodynamic agriculture. Environ Sci pollut Res (2012) 19:4214–25. doi: 10.1007/s11356-012-1022-x
26. Reeve JR, Carpenter-Boggs L, Reganold JP, York AL, McGourty G, McCloskey LP. Soil and winegrape quality in biodynamically and organically managed vineyards. Am J Enol Vitic (2005) 56:367–76. Available at: https://www.ajevonline.org/content/56/4/367.short [Accessed October 10, 2022].
27. Hartmann M, Frey B, Mayer J, Mäder P, Widmer F. Distinct soil microbial diversity under long-term organic and conventional farming. ISME J (2015) 9:1177–94. doi: 10.1038/ismej.2014.210
28. Fritz J, Athmann M, Meissner G, Kauer R, Köpke U. Quality characterisation via image forming methods differentiates grape juice produced from integrated, organic or biodynamic vineyards in the first year after conversion. Biol Agric Hortic (2017) 33:195–213. doi: 10.1080/01448765.2017.1322003
29. Köberl M, Müller H, Ramadan EM, Berg G. Desert farming benefits from microbial potential in arid soils and promotes diversity and plant health. PLoS One (2011) 6:e24452. doi: 10.1371/JOURNAL.PONE.0024452
30. Steiner R. Agriculture: a course of eight lectures. Bio-Dynamic Agric Assoc (1958) 327. Available at: https://rsarchive.org/Lectures/GA327/English/BDA1958/Ag1958_index.html [Accessed October 10, 2022].
31. White TJ, Bruns T, Lee S, Taylor J. ‘Amplification and direct sequencing of fungal ribosomal rna genes for phylogenetics’, in PCR Protocols (Elsevier), 315–22. doi: 10.1016/b978-0-12-372180-8.50042-1
32. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A (2011) 108:4516–22. doi: 10.1073/pnas.1000080107
33. Lundberg DS, Yourstone S, Mieczkowski P, Jones CD, Dangl JL. Practical innovations for high-throughput amplicon sequencing. Nat Methods (2013) 10:999–1002. doi: 10.1038/nmeth.2634
34. Fitzpatrick CR, Lu-Irving P, Copeland J, Guttman DS, Wang PW, Baltrus DA, et al. Chloroplast sequence variation and the efficacy of peptide nucleic acids for blocking host amplification in plant microbiome studies. Microbiome (2018) 6:144. doi: 10.1186/s40168-018-0534-0
35. Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol Ecol (1993) 2:113–8. doi: 10.1111/j.1365-294X.1993.tb00005.x
36. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal (1994) 17:10–2. doi: 10.14806/EJ.17.1.200.
37. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol (2019) 37:852–7. doi: 10.1038/s41587-019-0209-9
38. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from illumina amplicon data. Nat Methods (2016) 13:581–3. doi: 10.1038/nmeth.3869
39. Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: A versatile open source tool for metagenomics. PeerJ (2016) 2016:e2584. doi: 10.7717/PEERJ.2584/FIG-7
40. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res (2013) 41:590–6. doi: 10.1093/nar/gks1219
41. Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, et al. The SILVA and ‘all-species living tree project (LTP)’ taxonomic frameworks. Nucleic Acids Res (2014) 42:D643–8. doi: 10.1093/nar/gkt1209
42. Abarenkov K, Nilsson RH, Larsson K-H, Alexander IJ, Eberhardt U, Erland S, et al. The UNITE database for molecular identification of fungi: recent updates and future perspectives. New Phytol (2010) 186:281–5. doi: 10.1111/J.1469-8137.2009.03160.X
43. R Core Team. R: A language and environment for statistical computing (2020). Vienna, Austria: R Foundation for Statistical Computing. Available at: https://stat.ethz.ch/R-manual/R-devel/library/utils/html/citation.html (Accessed May 8, 2021).
44. Chong J, Liu P, Zhou G, Xia J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat Protoc (2020) 15:799–821. doi: 10.1038/s41596-019-0264-1
45. McMurdie PJ, Holmes S. Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PLoS One (2013) 8, e61217. doi: 10.1371/journal.pone.0061217
46. Oksanen AJ, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, et al. vegan: Community Ecology Package. R package version 2.5-7. (2020) Available at: https://cran.r-project.org/package=vegan [Accessed November 5, 2022].
47. McKight PE, Najab J. ‘Kruskal-Wallis Test’, in The Corsini Encyclopedia of Psychology (John Wiley & Sons, Inc.). doi: 10.1002/9780470479216.CORPSY0491
48. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol (2011) 12:R60. doi: 10.1186/gb-2011-12-6-r60
49. Tyler TW, Girke T. systemPipeR: NGS workflow and report generation environment. BMC Bioinf (2016) 17:1–8. doi: 10.1186/S12859-016-1241-0/TABLES/1
50. Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, et al. Bayesian Community-wide culture-independent microbial source tracking. Nat Methods (2011) 8:761–5. doi: 10.1038/NMETH.1650
51. Franke-Whittle IH, Confalonieri A, Insam H, Schlegelmilch M, Körner I. Changes in the microbial communities during co-composting of digestates. Waste Manage (2014) 34:632–41. doi: 10.1016/J.WASMAN.2013.12.009
52. Palaniveloo K, Amran MA, Norhashim NA, Mohamad-Fauzi N, Peng-Hui F, Hui-Wen L, et al. Food waste composting and microbial community structure profiling. Processes (2020) 8:1–30. doi: 10.3390/pr8060723
53. Bohacz J. Changes in mineral forms of nitrogen and sulfur and enzymatic activities during composting of lignocellulosic waste and chicken feathers. Environ Sci pollut Res (2019) 26:10333–42. doi: 10.1007/S11356-019-04453-2/TABLES/1
54. Berg G, Smalla K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol (2009) 68:1–13. doi: 10.1111/J.1574-6941.2009.00654.X
55. Dastogeer KMG, Tumpa FH, Sultana A, Akter MA, Chakraborty A. Plant microbiome–an account of the factors that shape community composition and diversity. Curr Plant Biol (2020) 23:100161. doi: 10.1016/J.CPB.2020.100161
56. Wang C, Dong D, Wang H, Müller K, Qin Y, Wang H, et al. Metagenomic analysis of microbial consortia enriched from compost: New insights into the role of actinobacteria in lignocellulose decomposition. Biotechnol Biofuels (2016) 9:1–17. doi: 10.1186/S13068-016-0440-2/FIGURES/5
57. Alves MLF, Ferreira MRA, Rodrigues RR, Conceição FR. Clostridium haemolyticum, a review of beta toxin and insights into the antigen design for vaccine development. Mol Immunol (2022) 148:45–53. doi: 10.1016/J.MOLIMM.2022.05.007
58. Minamisawa K, Nishioka K, Miyaki T, Ye B, Miyamoto T, You M, et al. Anaerobic nitrogen-fixing consortia consisting of clostridia isolated from gramineous plants. Appl Environ Microbiol (2004) 70:3096–102. doi: 10.1128/AEM.70.5.3096-3102.2004/ASSET/1E5A8199-933F-4E18-8F22-E119E7C73F56/ASSETS/GRAPHIC/ZAM0050421490004.JPEG
59. Miyamoto T, Kawahara M, Minamisawa K. Novel endophytic nitrogen-fixing clostridia from the grass Miscanthus sinensis as revealed by terminal restriction fragment length polymorphism analysis. Appl Environ Microbiol (2004) 70:6580–6. doi: 10.1128/AEM.70.11.6580-6586.2004/ASSET/41CCA7D1-5612-46F6-A7C8-E6E5FCEBDA9F/ASSETS/GRAPHIC/ZAM0110449120004.JPEG
60. Partanen P, Hultman J, Paulin L, Auvinen P, Romantschuk M. Bacterial diversity at different stages of the composting process. BMC Microbiol (2010) 10:1–11. doi: 10.1186/1471-2180-10-94/TABLES/2
61. Dhanasekaran D, Jiang Y. Actinobacteria - basics and biotechnological applications. Actinobacteria - Basics Biotechnol Appl (2016). doi: 10.5772/60457
62. Berg G, Roskot N, Steidle A, Eberl L, Zock A, Smalla K. Plant-dependent genotypic and phenotypic diversity of antagonistic rhizobacteria isolated from different verticillium host plants. Appl Environ Microbiol (2002) 68:3328–38. doi: 10.1128/AEM.68.7.3328-3338.2002
63. Summerell BA. Resolving fusarium: Current status of the genus. Annu Rev Phytopathol (2019) 57:323–39. doi: 10.1146/ANNUREV-PHYTO-082718-100204
64. Mulero-Aparicio A, Cernava T, Turrà D, Schaefer A, Di Pietro A, López-Escudero FJ, et al. The role of volatile organic compounds and rhizosphere competence in mode of action of the non-pathogenic Fusarium oxysporum fo12 toward verticillium wilt. Front Microbiol (2019) 10:1808/BIBTEX. doi: 10.3389/FMICB.2019.01808/BIBTEX
65. Brudzynski K. Honey as an ecological reservoir of antibacterial compounds produced by antagonistic microbial interactions in plant nectars, honey and honey bee. Antibiotics (2021) 10, 551. doi: 10.3390/ANTIBIOTICS10050551
66. Pant AP, Radovich TJK, Hue NV, Paull RE. Biochemical properties of compost tea associated with compost quality and effects on pak choi growth. Sci Hortic (Amsterdam) (2012) 148:138–46. doi: 10.1016/J.SCIENTA.2012.09.019
67. Andreu V, Levert A, Amiot A, Cousin A, Aveline N, Bertrand C. Chemical composition and antifungal activity of plant extracts traditionally used in organic and biodynamic farming. Environ Sci pollut Res (2018) 25:29971–82. doi: 10.1007/S11356-018-1320-Z/TABLES/8
68. Wang Y, Gong J, Li J, Xin Y, Hao Z, Chen C, et al. Insights into bacterial diversity in compost: Core microbiome and prevalence of potential pathogenic bacteria. Sci Total Environ (2020) 718:137304. doi: 10.1016/J.SCITOTENV.2020.137304
69. Shrestha K, Walsh KB, Midmore DJ. Microbially enhanced compost extract: Does it increase solubilisation of minerals and mineralisation of organic matter and thus improve plant nutrition? J Bioremediation Biodegrad (2012) 3:1–9. doi: 10.4172/2155-6199.1000149
70. Vu VH, Hong SI, Kim K. Production of aerial conidia of lecanicillium lecanii 41185 by solid-state fermentation for use as a mycoinsecticide. Mycobiology (2008) 36:183. doi: 10.4489/MYCO.2008.36.3.183
71. Goettel MS, Koike M, Kim JJ, Aiuchi D, Shinya R, Brodeur J. Potential of Lecanicillium spp. for management of insects, nematodes and plant diseases. J Invertebr Pathol (2008) 98:256–61. doi: 10.1016/J.JIP.2008.01.009
72. Bakthavatsalam N. Chapter 19, in: Semiochemicals (2016). Elsevier Inc. Available at: http://dx.doi.org/10.1016/B978-0-12-803265-7.00018-X (Accessed April 25, 2022).
73. Visagie CM, Houbraken J, Frisvad JC, Hong SB, Klaassen CHW, Perrone G, et al. Identification and nomenclature of the genus penicillium. Stud Mycol (2014) 78:343. doi: 10.1016/J.SIMYCO.2014.09.001
74. Yadav AN, Verma P, Kumar V, Sangwan P, Mishra S, Panjiar N, et al. ‘Biodiversity of the genus penicillium in different habitats’, in New and Future Developments in Microbial Biotechnology and Bioengineering: Penicillium System Properties and Applications (Elsevier) (2017), 3–18. doi: 10.1016/B978-0-444-63501-3.00001-6
75. Patel TK. ‘Metarhizium’, in Beneficial Microbes in Agro-Ecology: Bacteria and Fungi (Elsevier) (2020), 593–610. doi: 10.1016/B978-0-12-823414-3.00029-0
76. Guzmán-Chávez F, Zwahlen RD, Bovenberg RAL, Driessen AJM. Engineering of the filamentous fungus Penicillium chrysogenumas cell factory for natural products. Front Microbiol (2018) 9:2768/BIBTEX. doi: 10.3389/FMICB.2018.02768/BIBTEX
77. Hossain MM, Sultana F, Kubota M, Koyama H, Hyakumachi M. The plant growth-promoting fungus Penicillium simplicissimum GP17-2 induces resistance in Arabidopsis thaliana by activation of multiple defense signals. Plant Cell Physiol (2007) 48:1724–36. doi: 10.1093/PCP/PCM144
78. Hossain MM, Sultana F, Kubota M, Hyakumachi M. Differential inducible defense mechanisms against bacterial speck pathogen in Arabidopsis thaliana by plant-growth-promoting-fungus Penicillium sp. GP16-2 and its cell free filtrate. Plant Soil (2008) 304:227–39. doi: 10.1007/S11104-008-9542-3/FIGURES/4
79. Radhakrishnan R, Kang SM, Baek IY, Lee IJ. Characterization of plant growth-promoting traits of penicillium species against the effects of high soil salinity and root disease. J Plant Interact (2014) 9:754–62. doi: 10.1080/17429145.2014.930524
80. Pinto AJ, Xi C, Raskin L. Bacterial community structure in the drinking water microbiome is governed by filtration processes. Environ Sci Technol (2012) 46:8851–9. doi: 10.1021/ES302042T/SUPPL_FILE/ES302042T_SI_001.PDF
81. Ling F, Whitaker R, LeChevallier MW, Liu WT. Drinking water microbiome assembly induced by water stagnation. ISME J (2018) 12:1520–31. doi: 10.1038/s41396-018-0101-5
82. Hull NM, Ling F, Pinto AJ, Albertsen M, Jang HG, Hong PY, et al. Drinking water microbiome project: Is it time? Trends Microbiol (2019) 27:670–7. doi: 10.1016/J.TIM.2019.03.011
83. Zaller JG, Köpke U. Effects of traditional and biodynamic farmyard manure amendment on yields, soil chemical, biochemical and biological properties in a long-term field experiment. Biol Fertil. Soils (2004) 40:222–9. doi: 10.1007/S00374-004-0772-0/TABLES/3
84. Birkhofer K, Bezemer TM, Bloem J, Bonkowski M, Christensen S, Dubois D, et al. Long-term organic farming fosters below and aboveground biota: Implications for soil quality, biological control and productivity. Soil Biol Biochem (2008) 40:2297–308. doi: 10.1016/J.SOILBIO.2008.05.007
85. Kalam S, Basu A, Ahmad I, Sayyed RZ, El-Enshasy HA, Dailin DJ, et al. Recent understanding of soil acidobacteria and their ecological significance: A critical review. Front Microbiol (2020) 11:580024. doi: 10.3389/fmicb.2020.580024
86. Sikorski J, Baumgartner V, Birkhofer K, Boeddinghaus RS, Bunk B, Fischer M, et al. The evolution of ecological diversity in acidobacteria. Front Microbiol (2022) 13:715637. doi: 10.3389/fmicb.2022.715637
87. Ward NL, Challacombe JF, Janssen PH, Henrissat B, Coutinho PM, Wu M, et al. Three genomes from the phylum acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl Environ Microbiol (2009) 75:2046–56. doi: 10.1128/AEM.02294-08/SUPPL_FILE/COMMON_GENES_1_24_09.ZIP
88. Belova SE, Ravin NV, Pankratov TA, Rakitin AL, Ivanova AA, Beletsky AV, et al. Hydrolytic capabilities as a key to environmental success: Chitinolytic and cellulolytic acidobacteriafrom acidic sub-arctic soils and boreal peatlands. Front Microbiol (2018) 9:2775/BIBTEX. doi: 10.3389/FMICB.2018.02775/BIBTEX
89. Woudenberg JHC, Groenewald JZ, Binder M, Crous PW. Alternaria redefined. Stud Mycol (2013) 75:171–212. doi: 10.3114/SIM0015
90. Matić S, Tabone G, Garibaldi A, Gullino ML. Alternaria leaf spot caused by alternaria species: An emerging problem on ornamental plants in Italy. Plant Dis (2020) 104:2275–87. doi: 10.1094/PDIS-02-20-0399-RE/ASSET/IMAGES/LARGE/PDIS-02-20-0399-RE_F4.JPEG
91. Olimi E, Kusstatscher P, Wicaksono WA, Abdelfattah A, Cernava T, Berg G. Insights into the microbiome assembly during different growth stages and storage of strawberry plants. Environ Microbiomes (2022) 17:1–15. doi: 10.1186/S40793-022-00415-3/FIGURES/5
92. Dang HX, Pryor B, Peever T, Lawrence CB. The Alternaria genomes database: A comprehensive resource for a fungal genus comprised of saprophytes, plant pathogens, and allergenic species. BMC Genomics (2015) 16:1–9. doi: 10.1186/S12864-015-1430-7/FIGURES/5
93. Hernandez-ramirez G, Barber D, Tome-amat J, Garrido-arandia M, Diaz-perales A. Alternaria as an inducer of allergic sensitization. J Fungi (2021) 7:1–14. doi: 10.3390/jof7100838
94. Johnston-Monje D, Gutiérrez JP, Lopez-Lavalle LAB. Seed-transmitted bacteria and fungi dominate juvenile plant microbiomes. Front Microbiol (2021) 12:737616/BIBTEX. doi: 10.3389/FMICB.2021.737616/BIBTEX
95. Ryan M, Ash J. Effects of phosphorus and nitrogen on growth of pasture plants and VAM fungi in SE Australian soils with contrasting fertiliser histories (conventional and biodynamic). Agric Ecosyst Environ (1999) 73:51–62. doi: 10.1016/S0167-8809(99)00014-6
96. Mäder P, Fließbach A, Dubois D, Gunst L, Fried P, Niggli U. Soil fertility and biodiversity in organic farming. Science (2002) 296:1694–7. doi: 10.1126/SCIENCE.1071148/SUPPL_FILE/MAEDERSUPPL.PDF
97. Knapp BA, Ros M, Insam H. Do composts affect the soil microbial community? Microbes Work From Wastes to Resources (Springer-Verlag Berlin Heidelberg) (2010), 271–91. doi: 10.1007/978-3-642-04043-6_14/FIGURES/2_14
98. Doan TT, Bouvier C, Bettarel Y, Bouvier T, Henry-des-Tureaux T, Janeau JL, et al. Influence of buffalo manure, compost, vermicompost and biochar amendments on bacterial and viral communities in soil and adjacent aquatic systems. Appl Soil Ecol (2014) 73:78–86. doi: 10.1016/J.APSOIL.2013.08.016
99. Koepf HH, Pettersson BD, Schaumann W. Bio-dynamic agriculture: an introduction. (1976) 429. Available at: https://biblio.co.uk/book/bio-dynamic-agriculture-introduction-herbert-h/d/952570183 [Accessed November 6, 2022].
100. Jayaraman S, Naorem AK, Lal R, Dalal RC, Sinha NK, Patra AK, et al. Disease-suppressive soils-beyond food production: a critical review. J Soil Sci Plant Nutr (2021) 21:1437–65. doi: 10.1007/S42729-021-00451-X
101. Stutz EW. Naturally occurring fluorescent pseudomonads involved in suppression of black root rot of tobacco. Phytopathology (1986) 76:181. doi: 10.1094/PHYTO-76-181
102. Compant S, Samad A, Faist H, Sessitsch A. A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J Adv Res (2019) 19:29–37. doi: 10.1016/J.JARE.2019.03.004
103. Berg G, Kusstatscher P, Abdelfattah A, Cernava T, Smalla K. Microbiome modulation–toward a better understanding of plant microbiome response to microbial inoculants. Front Microbiol (2021) 12:650610/BIBTEX. doi: 10.3389/FMICB.2021.650610/BIBTEX
Keywords: biodynamic farming, compost microbiome, biodynamic manures, biodynamic preparations, rhizosphere microbiome, 16S rRNA/ITS amplicon sequencing, organic soil amendments
Citation: Olimi E, Bickel S, Wicaksono WA, Kusstatscher P, Matzer R, Cernava T and Berg G (2022) Deciphering the microbial composition of biodynamic preparations and their effects on the apple rhizosphere microbiome. Front. Soil Sci. 2:1020869. doi: 10.3389/fsoil.2022.1020869
Received: 16 August 2022; Accepted: 31 October 2022;
Published: 16 November 2022.
Edited by:
David Johnston-Monje, University of Valle, ColombiaReviewed by:
Oluwadara Pelumi Omotayo, North-West University, South AfricaCopyright © 2022 Olimi, Bickel, Wicaksono, Kusstatscher, Matzer, Cernava and Berg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Expedito Olimi, ZXhwZWRpdG8ub2xpbWlAdHVncmF6LmF0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.