 Nayely Vélez-Cruz
Nayely Vélez-Cruz Antonia Papandreou-Suppappola
Antonia Papandreou-Suppappola- School of Electrical, Computer and Energy Engineering, Arizona State University, Tempe, AZ, United States
Introduction: Gene regulatory networks (GRNs) are characterized by their dynamism, meaning that the regulatory interactions which constitute these networks evolve with time. Identifying when changes in the GRN architecture occur can inform our understanding of fundamental biological processes, such as disease manifestation, development, and evolution. However, it is usually not possible to know a priori when a change in the network architecture will occur. Furthermore, an architectural shift may alter the underlying noise characteristics, such as the process noise covariance.
Methods: We develop a fully Bayesian hierarchical model to address the following: a) sudden changes in the network architecture; b) unknown process noise covariance which may change along with the network structure; and c) unknown measurement noise covariance. We exploit the use of conjugate priors to develop an analytically tractable inference scheme using Bayesian sequential Monte Carlo (SMC) with a local Gibbs sampler.
Results: Our Bayesian learning algorithm effectively estimates time-varying gene expression levels and architectural model indicators under varying noise conditions. It accurately captures sudden changes in network architecture and accounts for time-evolving process and measurement noise characteristics. Our algorithm performs well even under high noise conditions. By incorporating conjugate priors, we achieve analytical tractability, enabling robust inference despite the inherent complexities of the system. Furthermore, our method outperforms the standard particle filter in all test scenarios.
Discussion: The results underscore our method’s efficacy in capturing architectural changes in GRNs. Its ability to adapt to a range of time-evolving noise conditions emphasizes its practical relevance for real-world biological data, where noise presents a significant challenge. Overall, our method provides a powerful tool for studying the dynamics of GRNs and has the potential to advance our understanding of fundamental biological processes.
1 Introduction
Gene regulatory networks (GRNs) are complex systems consisting of genes and a toolkit of molecular elements responsible for coordinating the spatiotemporal allocation of gene expression in every cell of the body. They are involved in the execution of essential biological processes, such as development, metabolism, and responses to environmental signals. One of their key characteristics is dynamicity. The regulatory interactions constituting these networks are not static; rather, they evolve, giving rise to time-dependent GRN architectures. These temporal alterations in regulatory structures have consequences for the biological processes which are encoded by GRNs across both short and long time scales and can affect all major biological processes, including development, disease, and phenotypic evolution. GRNs also exhibit a complex hierarchical architecture comprised of interlinked subunits called “subcircuits.” Ranging from two to eight genes, these subcircuits consist of specific regulatory interactions that execute specific biological functions (Peter and Davidson, 2011). Subcircuits thus intimately link structure and function GRNs (Hinman and Cheatle Jarvela, 2014). Research shows that these subcircuits are evolutionarily conserved across Metazoa, reinforcing their critical role (Peter and Davidson, 2015). Various subcircuit types, such as positive-feedback loops, feed-forward connections, and reciprocal repression mechanisms, have been identified (Alon, 2019). During development, different subcircuits are involved in different temporal stages of the developmental process, such as pattern formation, state stabilization, and cellular differentiation (Peter and Davidson, 2015). As development proceeds, the sets of active genes and their associated regulatory interactions change. In regard to disease, the rewiring of regulatory networks can cause disruptions in essential biological functions, giving rise to diseases such as cancer or disorders such as schizophrenia (Potkin et al., 2010). These rewirings can manifest at the level of subcircuits, which emphasizes their importance in understanding disease mechanisms (Saunders and McClay, 2014). Such alterations may involve transitions from one subcircuit type to another—for example, a shift from a positive-feedback loop to a feed forward cascade (Saunders and McClay, 2014). Similarly, evolutionary changes in GRN configurations contribute to the emergence of novel phenotypes within populations (Ha et al., 2022). Identifying these shifts in GRN interactions, especially within subcircuits, is thus an imperative task for understanding essential biological processes. This is fundamentally a change point detection problem.
To this extent, state-space models provide a mathematical framework that captures the dynamical behavior of GRNs, including their subcircuits, over time. GRN estimation using a state space approach has been extensively studied (Noor et al., 2012; Bugallo et al., 2015; Ancherbak et al., 2016; Pirgazi and Khanteymoori, 2018; Amor et al., 2019). However, these approaches assume that the network structure is static across all time. To this extent, several works have considered the problem of estimating GRNs with time-varying structures using linear models. Specifically, the authors in (Xiong and Zhou, 2013; Pirgazi and Khanteymoori, 2018) use Kalman filtering for inference by assuming a linear state-space model. In (Noor et al., 2012; Bugallo et al., 2015; Ancherbak et al., 2016), particle filtering is used to infer the dynamic network and the process and measurement noise are assumed to be known and constant. This may not capture changes in the noise statistics that can arise when a change in the regulatory network structure occurs. In (Dondelinger et al., 2013), the network structure assumed to change slowly across time. However, environmental stressors, disease, or mutations may substantially alter the network structure in a more abrupt manner. Furthermore, gene regulation is a nonlinear process due to the feedback loops present in the architectures, threshold effects, and combinatorial binding of transcription factors on DNA. Linear models may therefore obscure these phenomena. Alternatively, nonlinear system models based on Hill kinetics, Michaelis-Menten kinetics, or the S-system are able to more accurately capture the molecular mechanisms of gene regulation (Wang et al., 2007; Youseph et al., 2015; 2019; Elahi and Hasan, 2018). Nonlinear Bayesian filtering inference methods, such as extended Kalman filtering and particle filtering, were used with these nonlinear models (Wang et al., 2009; Zhang et al., 2014; Bugallo et al., 2015). In (Lee et al., 2013), a nonlinear chemical kinetics model is considered, but the transition probabilities for switching between various network structures (or modes) are assumed to be known, which is usually not the case in practice.
Given the limitations of these works, we propose a fully Bayesian hierarchical model with sequential learning to account for the following: (a) sudden changes in the network architecture, (b) unknown modeling error process covariance, and (c) unknown measurement noise covariance. Using Bayesian hierarchical modeling allows for uncertainty at all levels and the use of prior information and inference techniques. It can also be integrated with sequential Monte Carlo (SMC) methods, offering the ability to handle nonlinear and non-Gaussian processes, which are often the case in biological systems. Within this framework, we use Bayesian learning for the predictive modeling of dynamic transitions between GRN subcircuits. This approach not only allows us to build upon existing domain knowledge but also to develop models with the complexity necessary for identifying temporal changes in the GRN interactions. In our model, the regulatory network interactions are encoded by kinetic order parameters, which we assume change at unknown times. Our proposed method exploits the use of conjugate priors to enable tractable and efficient inference and SMC with local Gibbs sampling Gelfand et al. (1990); Gelman et al. (1995) to choose among the network configurations and estimate the model and trajectories of the gene expression values.
This paper is organized as follows. In Section 2.1, we review the dynamic nonlinear state space model for GRNs and extend it to allow for time-variations in the direction and type of gene regulation in Section 2.2. In Section 2.3, we present our proposed approach to learning time-variations in GRNs; and in Section 2.4, we discuss our implementation using SMC inference with local Gibbs sampling. Simulations are presented in Section 3 to demonstrate the efficacy of our Bayesian hierarchical learning and tracking.
2 Materials and methods
2.1 Modeling gene regulatory networks
In (Vélez-Cruz et al., 2021), we developed a stochastic nonlinear GRN model to capture the molecular mechanisms of gene regulation and account for inherent variability in biological processes. The model includes information on binding affinity, expression and self-decay rates. It is based on Michaelis-Menten kinetics that describe different types of regulation processes between a gene and multiple other genes (Youseph et al., 2015; Youseph et al., 2019; Krishnan et al., 2020). It also incorporates the Hill coefficient that represents the effect of binding affinity between genes (Alon, 2019). We represent the GRN model by a dynamics state space formulation for N genes Xi, i = 1, …, N as.
Where
where the terms
And
All model parameters are described in Table 1. We specifically note the following parameters for Gene Xi and Gene Xj, i ≠ j. The Hill coefficient qij in Eq. 4 represents how the binding affinity of Xj increases the binding affinity of Xi. The Michaelis-Menten constant Kij accounts for the binding affinity between the product of Xj and the binding site of a target Xi; large values of Kij indicate a low binding affinity. The kinetic order parameters hij and gij in Eq. 5 take values of 0 or 1. Different combinations of these parameters indicate the type of regulation, as summarized in Table 2. Note that parameters Kii, qii, hii and gii specify autoregulatory interactions.
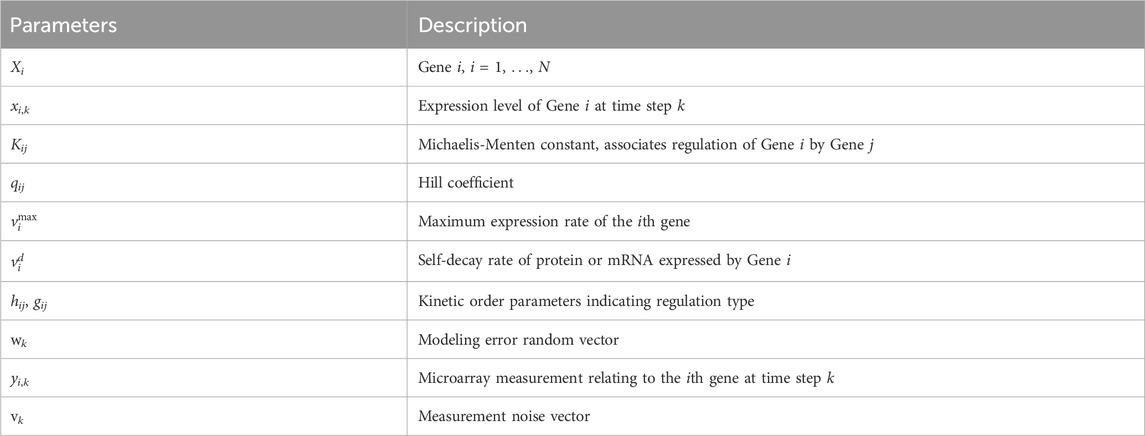
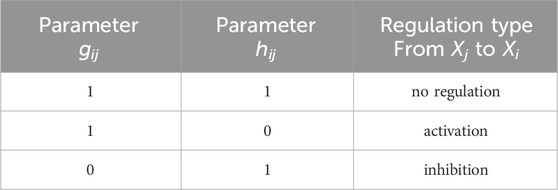
Table 2. GRN kinetic order parameters indicating type of regulation from Gene Xj to Gene Xi, i = j; autoregulation is indicated by gii = hii = 1.
2.2 Formulation of time-varying GRN model
We consider a TV GRN model where both the direction and type of regulation vary with time. This time variation is reflected in the kinetic order parameters in Eq. 5.
and thus in the transition function ϕk (⋅) in Eqs. (1) and (3). With this TV model, we can describe interactions between any number of genes and capture any changes in the GRN configuration with varying degrees of complexity.
As a simple illustration, we consider the transition from time step k − 1 to k of the simple GRN in Figure 1A to the one in Figure 1B. In Figure 1A, Genes X1 and X2 activate each other’s expression in a positive feedback loop at k − 1. From Table 2, activation of Gene X1 by Gene X2 is indicated by h12 = 0 and g12 = 1 and activation of Gene X2 by Gene X1 is indicated by h21 = 0 and g21 = 1. Thus, the kinetic order parameter vector at time step k − 1 is given by
where notation X1 ← X2 denotes the interaction from Gene X2 to Gene X1. In Figure 1B, the interaction from Gene X2 to Gene X1 switches from activation to inhibitory at time k. Whereas a21,k = a21,k−1, since inhibition of Gene X1 by Gene X2 is indicated by h12 = 1 and g12 = 0, then

Figure 1. Simple feedback loops demonstrating different types of regulatory interactions: (A) Gene X1 activates the expression of gene X2 and gene X2 activates the expression of gene X1. (B) Gene X1 activates the expression of gene X2 and gene X2 inhibits the expression of gene X1. The arrows denote activating regulatory interactions, whereas the line with the vertical bar denotes an inhibitory regulatory interaction.
For a realistic scenario, we consider the transcriptional activator Gcn4 of several amino acid biosynthesis genes in Saccharomyces cerevisiae or yeast (Mittal et al., 2017) and the transcription factor LexA responsible for repressing several genes involved in DNA repair in bacteria (McKenzie et al., 2000). Under stressful conditions such as nutrient deprivation, the network may switch from a single-input module configuration to a state which requires the coordination between several genes such as in a feed forward loop. For N = 5 genes, this configuration switch is depicted as changing from the subcircuit in Figure 2A to the subcircuit in Figure 2B. For example, when yeast are subject to osmotic stress, the high-osmolarity glycerol pathway is activated; this involves the subsequent activation of several genes involved in metabolism regulation, temporary arrest of cell cycle progression, or many other processes that are required for cellular adaptation in a feed forward architecture (Nadal and Posas, 2022). More complex subcircuits are found, for example, in the hypothalamic-pituitary-adrenal axis, which is responsible for regulating the stress response in humans (Tsigos and Chrousos, 2002).
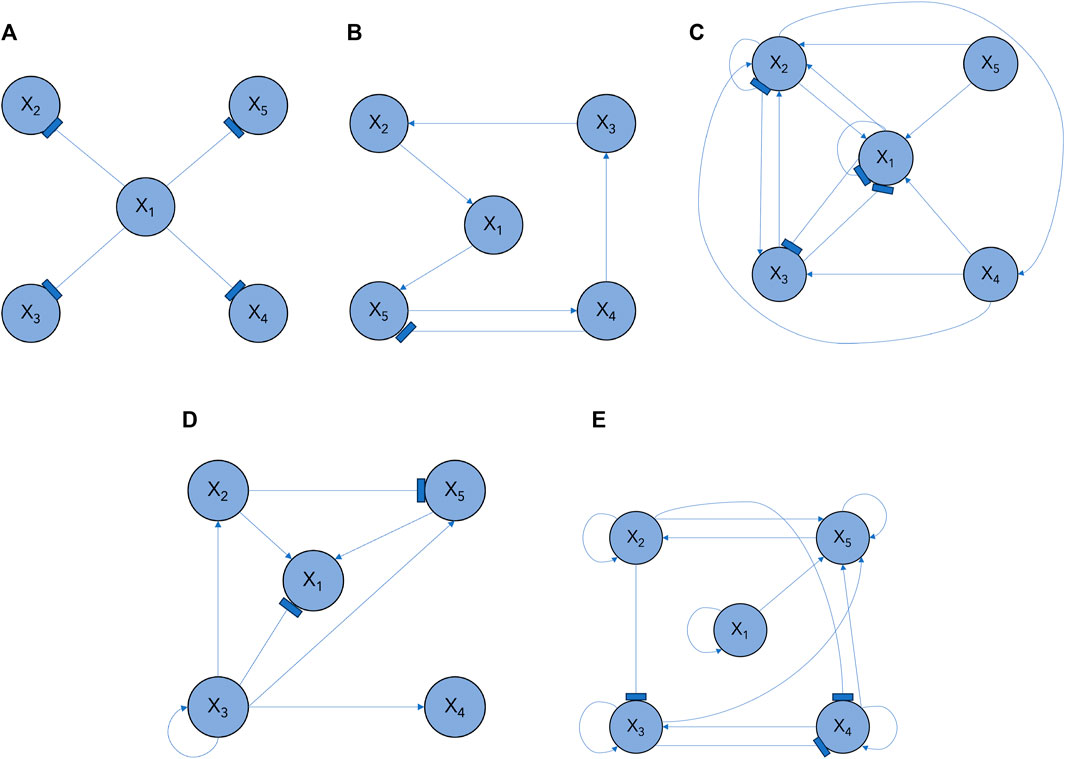
Figure 2. GRN subcircuit models in Section 3.1: (A) M1, (B) M2, (C) M3, (D) M4, (E) M5.
2.3 Bayesian learning for tracking
In this paper, we propose a new method for estimating the TV gene expressions under the realistic conditions of sudden changes in the GRN architecture and unknown statistics in the state space formulation. In particular, we assume that the GRN circuit features on developmental function can be selected from M subcircuit models. The mth model at time step k, denoted by the indicator sk = m, m = 1, …, M, is associated with the mth TV kinematic order parameter vector
Where the modeling error process
The new method, Bayesian Learning for Tracking or BLT, sequentially estimates the gene expressions while learning the probability of selecting one of the M models at each time step k as well as the unknown covariance matrices. From Eq. 8, the model indicator is drawn from a categorical distribution with parameter vector
where
The overall learning model is summarized by the following hierarchy
Following a sequential Bayesian Monte Carlo approach, the model first predicts xk using
where
Before using measurement yk to update the state xk at time step k, the noise covariance matrix Σv ∼IWD (Ψv) is learned using an IWD prior with hyperparameter Ψv = {Λv, νv}. This results in
where p (yk ∣ xk, Σv) is given in Eq. 9 and p (Σv ∣ Ψv) is the IWD prior. Using Eqs. (11) and (12), the joint posterior distribution at each time step k is given by
The last distribution in Eq. 13 is the posterior from the previous time step, obtained using model sk−1, where
Note that the derivation of the BLT is provided in Supplementary Appendix A.
2.4 BLT implementation using particle filtering and local Gibbs sampling
The dynamic state space model in Eqs. (1)-(6) is highly nonlinear, so we implement the BLT using particle filtering (Arulampalam et al., 2002; Elvira et al., 2019). As Eq. 11 is not explicitly known, the PF also estimates both the unknown TV model indicator sk and unknown constant covariance matrix
The BLT implementation steps are summarized in Algorithm 1. The algorithm uses the joint prior in Eq. 11 as the PF proposal distribution and assumes Ns state particles. At each time step and particle, we perform L Gibbs sampling iterations. In what follows,
Algorithm 1.Sequential Monte Carlo with Local Gibbs Sampling for Subcircuit Detection.
1: Initialization at t = 1
2: for i = 1, …, N particles do
3: Sample
4: Sample
5: Sample
6: Sample
7: end for
8: Sequential Updates for t ≥ 2
9: for ℓ = 1, …, L Gibbs iterations do
10: Sample a new model indicator
11: Sample
12: Sample
13: end for
14: Update measurement
15: Update hyperparameters and sample
16: Compute particle weights
17: Normalize weights
18: Resample particles
(i) Samples probability
(ii) uses
where Σv,prev is the modeling error covariance from the previous time step.
(iii) samples state
(iv) Samples
At the end of the L iterations, the predicted state particle is given by
where the Gaussian likelihood, using N measurements, is given as
The covariance matrix Σv,i is estimated using the IWD hyperparameters that are updated from Ψv,prev = {Λv,prev, νv,prev} using
3 Results and discussion
3.1 Simulation settings
We demonstrate the efficacy of our proposed Bayesian learning approach using N = 5 genes that transition between multiple GRN configurations over time. We consider M = 5 subcircuits, of varying degrees of complexity, as shown in Figure 2. Their configurations vary from a simple single-input module (SIM) configuration, which regulates several genes and is present across a range of systems, to more complex ones; these include compositions of different canonical subcircuits, such as feedforward and mutual repression loops. Switching between these specific network structures highlights the versatility of our model as it demonstrates the adaptive capacities of our Bayesian learning approach in identifying architectural changes. The configurations are described as follows.
M1 SIM subcircuit (Figure 2A): Gene X1 inhibits the expression of genes X2, X3, X4, and X5.
M2 Subcircuit with feed forward loop and an inhibitory interaction (Figure 2B): Gene X1 activates a cascade of activating regulatory interactions proliferating throughout the network; Gene X5 activates Gene X4, which in turn inhibits the expression of Gene X5.
M3 Complex subcircuit with negative autoinhibitory interactions on genes X1 and X2 and a feed forward loop (Figure 2C): X2 activates the expression of X4, which then activates the expression of X3 and X2; Gene X5 activates the expression of Gene X1, which inhibits the expression of X3; and Gene X3 inhibits the expression of X1, forming a mutually repressive loop.
M4 Complex subcircuit (Figure 2D): Gene X1 is activated by genes X2 and X5; when X3 is activated via an autoregulatory interaction, it inhibits Gene X1 and activates genes X4 and X2; in turn, Gene X2 activates X1 and inhibits X5, which activates the expression of X1.
M5 Complex subcircuit characterized by positive feedback loops (Figure 2E): each gene activates its own expression; Genes X2 and X5 form a positive feedback loop; Genes X2 and X3 inhibit X4; Genes X1 and X3 activate X5.
All configurations use the same constant model parameters provided in Table 3. The TV kinetic order parameters for each configuration are given in Table 4.
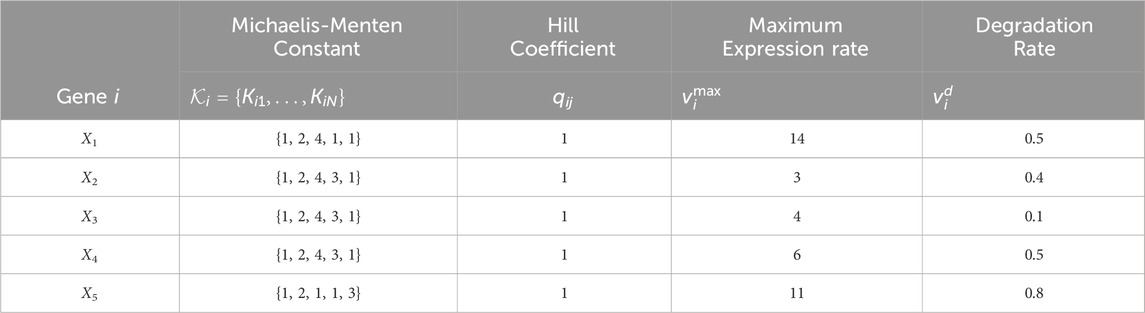
Table 3. Constant parameter values for the GRN models with N = 5 genes in Figure 2. The parameter values correspond to those in the nonlinear dynamic GRN equation in Eq. 4.
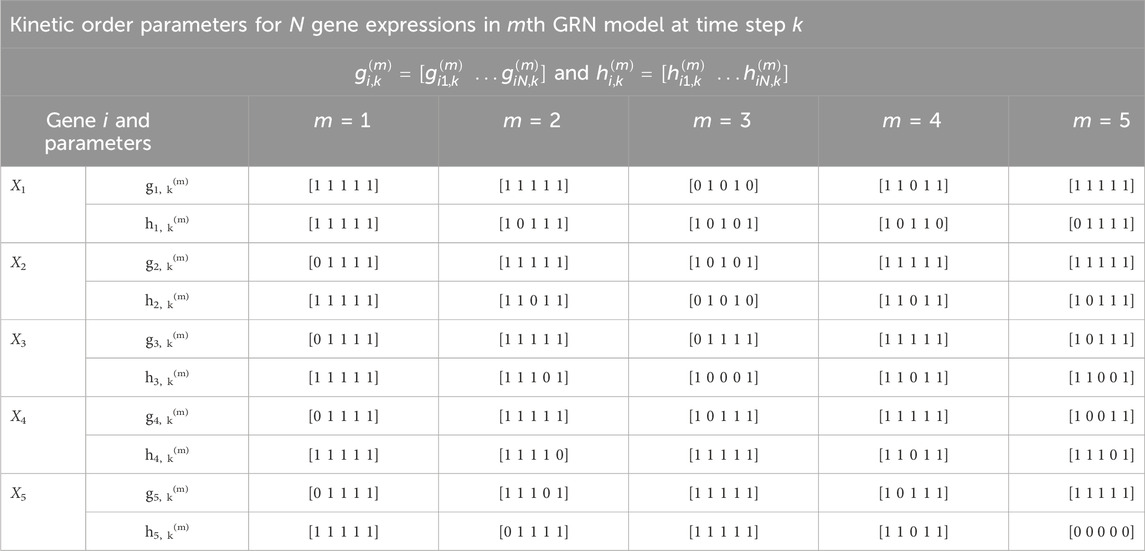
Table 4. Time-varying kinetic order parameters for M = 5 GRN models with N = 5 genes in Figure 2. Each m denotes a specific GRN configuration which is parameterized by g and h. These parameters correspond to those in Eq. 7.
The BLT implementation using the PF and Gibbs sampling follows Algorithm 1. In the simulations, the modeling error process and measurement noise process are both assumed Gaussian with zero-mean and uncorrelated samples; their corresponding unknown covariance matrices are
3.2 Simulation results
We consider three different simulation scenarios with a varying number of configuration models during different time step segments, as summarized in Table 5. In the table,
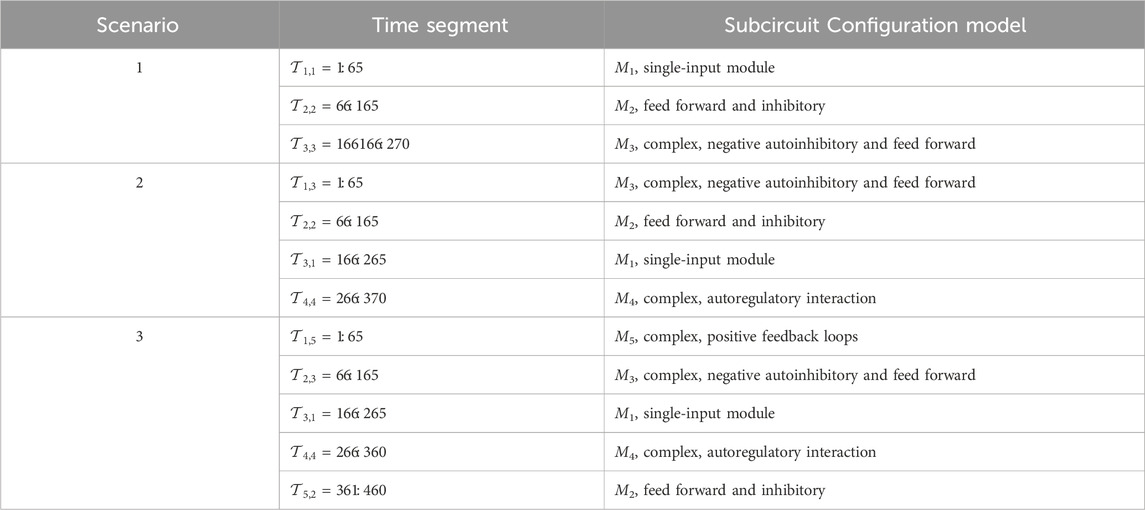
Table 5. GRN configuration models as they vary with time in the simulation scenarios. Time segment
Scenario 1: Three-model transition.
The BLT considers an unknown number of TV transitions between M = 3 models: M1 in Figure 2A during
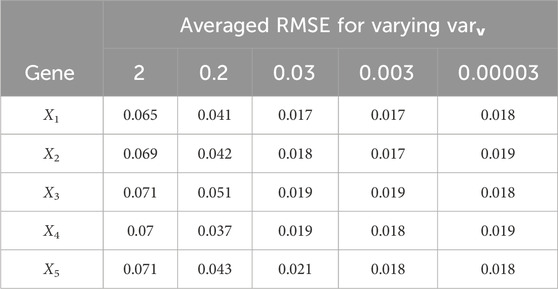
Table 6. Scenario 1: Averaged gene expression estimation RMSE for varying measurement noise variance varv; the modeling error variances were varw,1 = 0.00002, varw,2 = 0.0005, varw,3 = 0.00004.
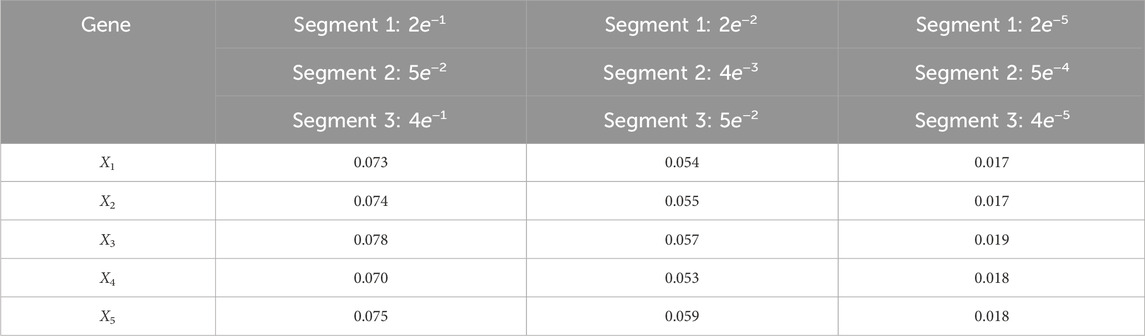
Table 7. Averaged RMSE for varying process noise intensities. For time steps k = 1: 65, the dynamics are described by model 1; for time steps k = 66: 165, the dynamics are described by model 2; and for time steps k = 166: 270, the dynamics are described by model 3. We use 1,000 particles, 5,000 Monte Carlo runs and 10,000 Gibbs iterations were used.
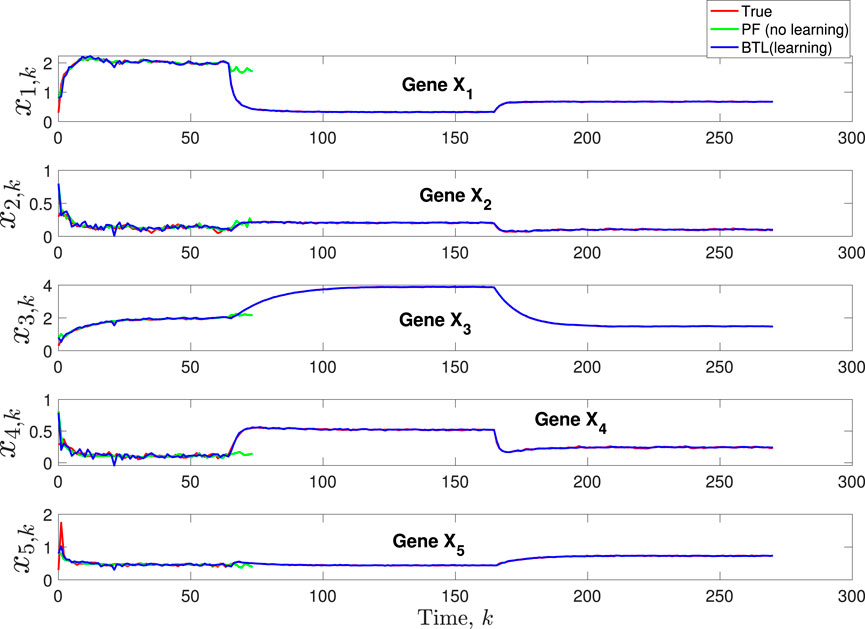
Figure 3. Scenario 1: Comparison of true, PF estimated and BLT estimated expression level xi,k of Gene Xi, i = 1, …, 5 using modeling error variances varw,1 = 0.00002, varw,2 = 0.0005, varw,3 = 0.00004 and measurement noise variance varv = 0.003.
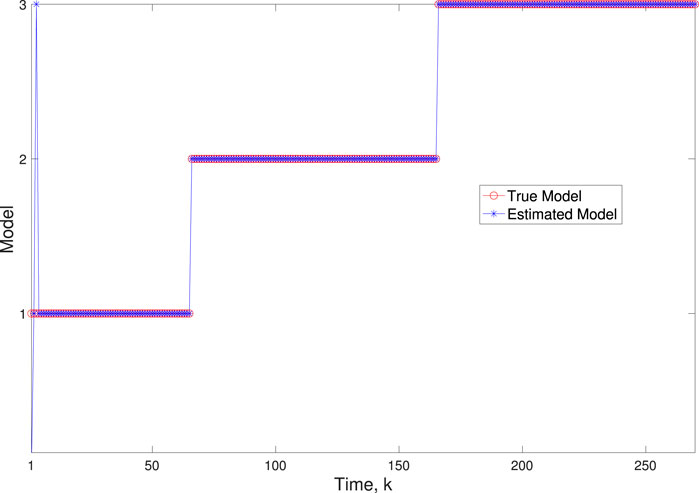
Figure 4. Scenario 1: Comparison of true and BLT estimated labels using varw,1 = 0.00002, varw,2 = 0.0005, varw,3 = 0.00004, and varv = 0.003.
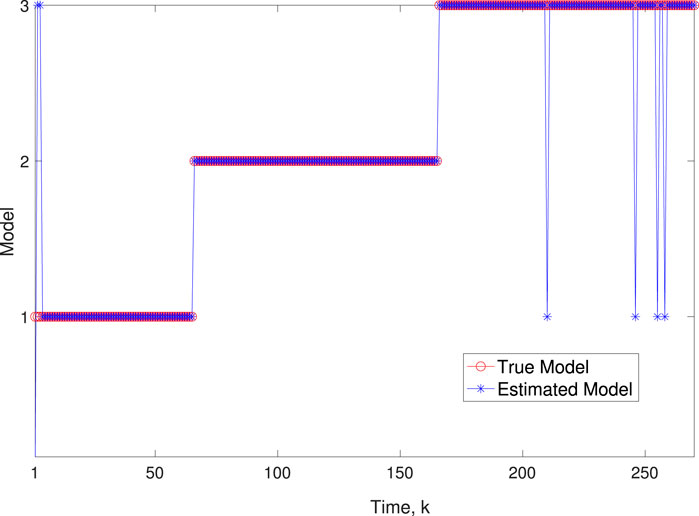
Figure 5. Scenario 1: Comparison of true and BLT estimated labels using varw,1 = 0.00002, varw,2 = 0.0005, varw,3 = 0.00004 and varv = 0.2.
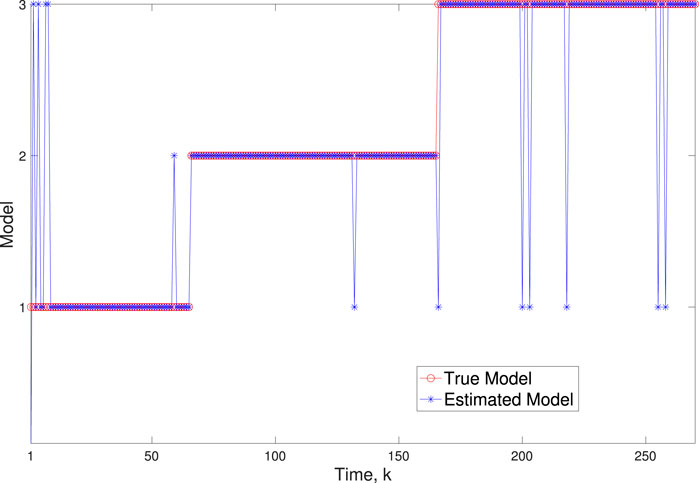
Figure 6. Scenario 1: Comparison of true and BLT estimated labels using varw,1 = 0.02, varw,2 = 0.004, varw,3 = 0.005 and varv = 0.003.
Scenario 2: Four-model transition.
In this scenario, the BLT considers transitions between M = 4 models: M3 in Figure 2C during
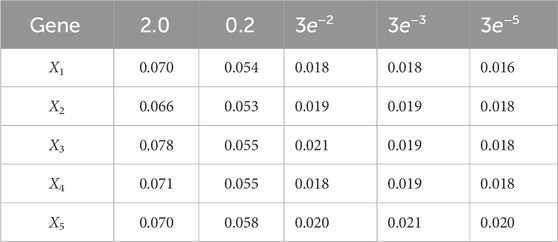
Table 8. Root mean-square error (RMSE) averaged across all time steps for varying measurement noise intensities. For time steps k = 1: 65, the dynamics are described by model 3; for time steps k = 66: 165, the dynamics are described by model 2; for time steps k = 166: 265, the dynamics are described by model 1; and for k = 265: 370, the dynamics are described by model 4. We use 1,000 particles, 5,000 Monte Carlo runs and 10,000 Gibbs iterations were used.

Table 9. Root mean-square error (RMSE) averaged across all time steps for varying process noise intensities. For time steps k = 1: 65, the dynamics are described by model 3; for time steps k = 66: 165, the dynamics are described by model 2; for time steps k = 166: 265, the dynamics are described by model 1; and for k = 265: 370, the dynamics are described by model 4. We use 1,000 particles, 5,000 Monte Carlo runs and 10,000 Gibbs iterations were used.
A comparison of our learning algorithm to the standard PF (no learning) is shown in Figure 7 for varv = 0.003. The standard PF (no learning method) assumes that the dynamics are only described by M3. As in scenario 1, the standard particle filter (no learning) can only provide estimates of the gene expressions whose trajectories are described by M3. Until the network architecture switches. Then it is no longer able to estimate the trajectory. In contrast, the BLT is able to estimate the trajectory at each of the change points. The true versus estimated model for different noise values are shown in Figure 8, Figure 9, Figure 10, which demonstrates that the BLT effectively estimates the correct model. The RMSE across different measurement noise intensity values is shown in Table 8. We also vary the modeling error variance and the RMSE values are shown in Table 9. Figure 9 further demonstrates the performance of the proposed BLT approach under high measurement noise conditions. Figure 10 demonstrates the BLT performance under increased modeling error variance.
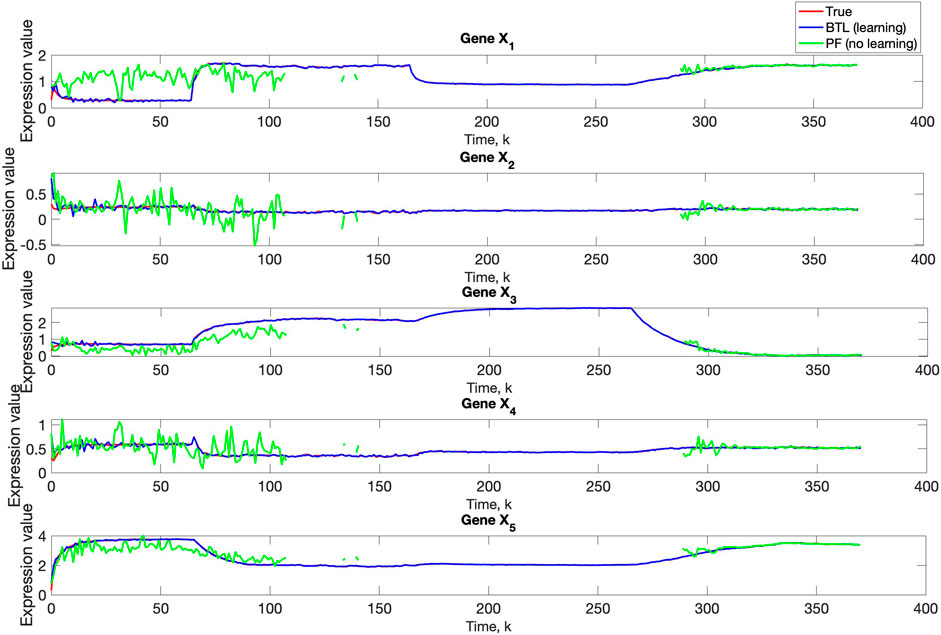
Figure 7. Comparison of true, PF estimated and BLT estimated expression level xi,k of Gene Xi, i = 1, …, 5 in Scenario 2. The TV model configuration is provided in Table 5.
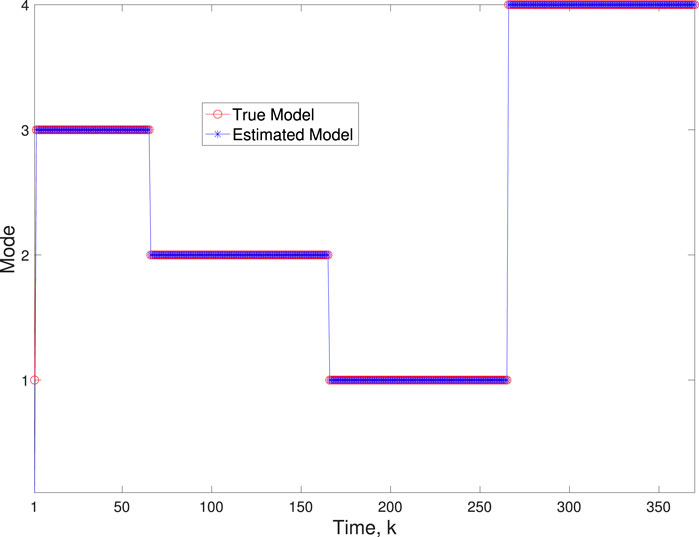
Figure 8. Scenario 2: Comparison of true and BLT estimated labels using varw,1 = 0.00002, varw,2 = 0.0005, varw,3 = 0.00004, varw,3 = 0.0002, and varv = 0.003.
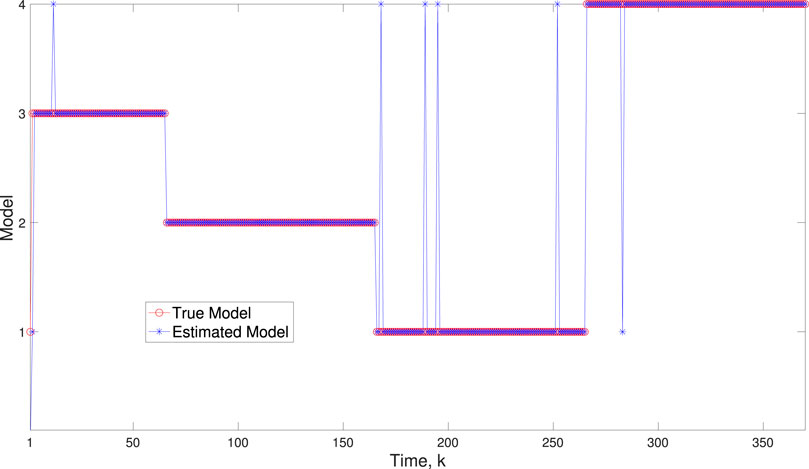
Figure 9. Scenario 2: Comparison of true and BLT estimated labels using varw,1 = 0.00002, varw,2 = 0.0005, varw,3 = 0.00004, varw,3 = 0.0002, varv = 2.
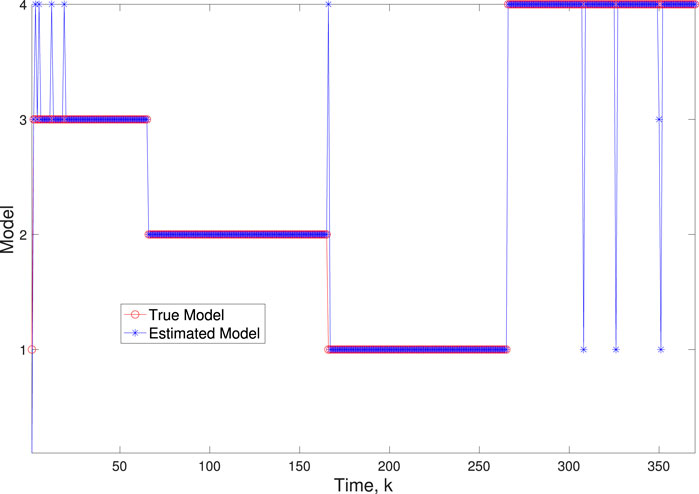
Figure 10. Scenario 2: Comparison of true and BLT estimated labels using varw,1 = 0.02, varw,2 = 0.004, varw,3 = 0.05, varw,3 = 0.002, varv = 0.003.
Scenario 3: Transitions Among Five Models.
In this scenario, the BLT considers transitions between M = 5 models: M3 in Figure 2C during
Using varv = 0.02, Figure 11 and Figure 12 further demonstrate the performance of the proposed BLT approach. Figure 11 compares the true expression levels of each of the 5 genes to those estimated by the BLT and PF methods. As in the previous scenarios, the standard particle filter (no learning) is only able to estimate the trajectory during the third segment where model M1 is assumed. Then it is no longer able to estimate the trajectory. In contrast, the BLT method is able to estimate the trajectory at each of the change points. Figure 12 compares the actual model labels with those learned by the BLT. The averaged RMSE for varying measurement and process noise intensities are shown in Tables 10 and 11, respectively. Note that the BLT algorithm exhibits similar performance to the previous scenarios despite increasing the number of models to M = 5.
Figure 11. Comparison of true, PF estimated and BLT estimated expression level xi,k of Gene Xi, i = 1, …, 5 in Scenario 3. The TV model configuration is provided in Table 5.
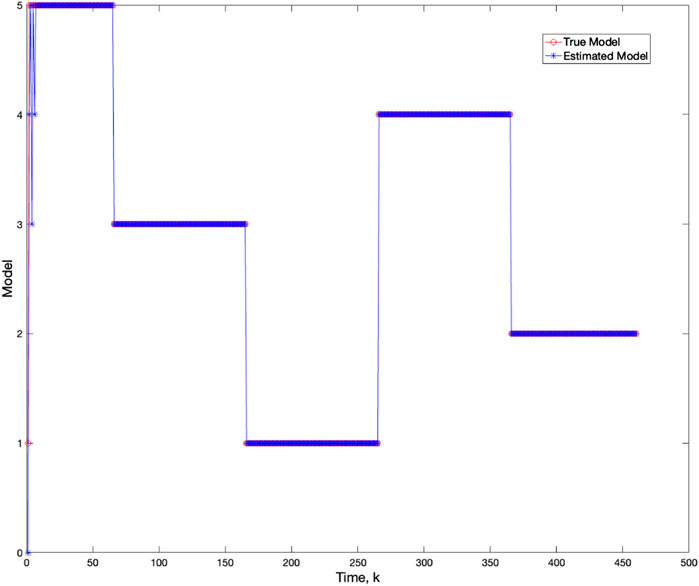
Figure 12. Comparison of true and BLT estimated labels of the configuration models in Scenario 3.
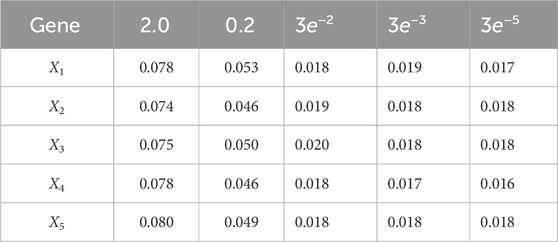
Table 10. Root mean-square error (RMSE) averaged across all time steps for varying measurement noise intensities. For time steps k = 1: 65, the dynamics are described by model 5; for time steps k = 66: 165, the dynamics are described by model 3; for time steps k = 166: 265, the dynamics are described by model 1; for k = 265: 360, the dynamics are described by model 4; and for k = 360: 460 the dynamics are described by model 5. We use 1,000 particles, 5,000 Monte Carlo runs and 10,000 Gibbs iterations were used.
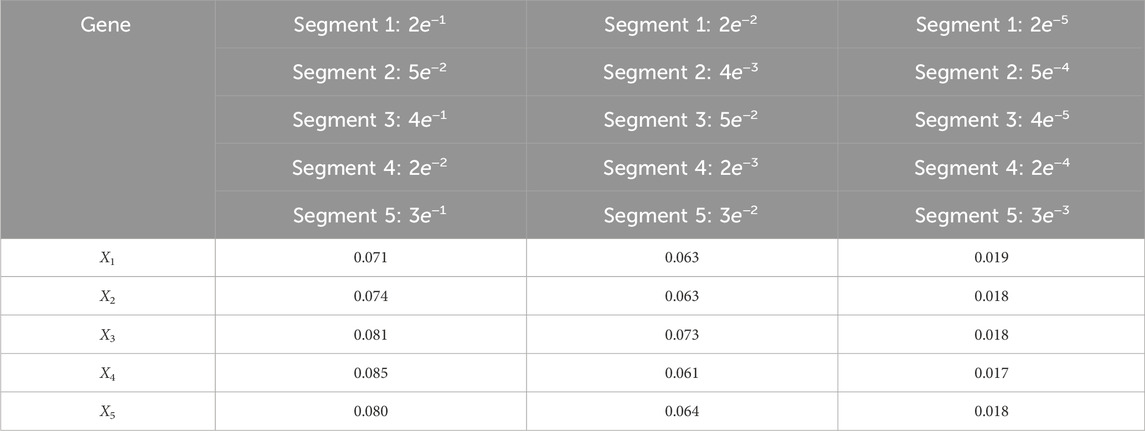
Table 11. Root mean-square error (RMSE) averaged across all time steps for varying process noise intensities. For time steps k = 1: 65, the dynamics are described by model 5; for time steps k = 66: 165, the dynamics are described by model 3; for time steps k = 166: 265, the dynamics are described by model 1; for k = 265: 370, the dynamics are described by model 4; and for k = 371: 460, the dynamics are described by model 2. We use 1,000 particles, 5,000 Monte Carlo runs and 10,000 Gibbs iterations were used.
4 Conclusion and future directions
In this work, we introduced a fully Bayesian hierarchical model for learning nonlinear gene regulatory networks with dynamically switching subcircuit architectures. We showed that our algorithm, which employs sequential Monte Carlo (SMC) with a local Gibbs step, effectively estimates the correct model corresponding to the subcircuit architecture as well as the unknown state under varying measurement and process noise conditions with a high degree of accuracy, though with greater sensitivity to variations in the process noise. Through the use of conjugate priors, we have formulated an analytically tractable inference scheme which effectively addresses the challenges posed by the inherent complexities of the system. By learning the unknown transition probabilities, which describe changes between different network configurations and by learning the unknown measurement and process noise covariances using Bayesian updating of the Inverse-Wishart distribution, we were able to account for uncertainty at all levels of the hierarchy. Our approach has demonstrated robustness and versatility in identifying changes in subcircuit architectures of varying degrees of complexity, ranging from a simple single-input-module (SIM) to subcircuits consisting of a composition of different types. To showcase this robustness, we applied our algorithm to different scenarios where the architecture switches between three, four, and five types.
It is worth noting that the methodology developed in this work is not confined to the analysis of gene regulatory networks but can also be generalized to other domains which involve switching dynamics in nonlinear systems. For example, our methodology can be applied to change point detection problems in financial markets, where a sudden shift in the market dynamics could be caused by changes in nonlinear interactions between different economic factors. While our Bayesian hierarchical model offers an effective and robust framework for learning switching dynamics in nonlinear gene regulatory networks, the complexity of the SMC algorithm with a local Gibbs step poses computational challenges. The drawbacks of incorporating Gibbs within SMC include potential inefficiencies, particularly in large-scale systems, where the iterative Gibbs sampling step at each stage of the sequential updating process may lead to increased computational burden. This challenge is accentuated when dealing with multi-omics data. As such, future research aims to extend our methodology to large-scale systems through the use of stochastic gradient Markov Chain Monte Carlo (SGMCMC), leveraging its efficiency in handling high-dimensional data and providing a computationally scalable approach for the analysis of switching gene regulatory networks.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author contributions
NV-C: Conceptualization, Formal Analysis, Investigation, Methodology, Writing–original draft, Writing–review and editing. AP-S: Conceptualization, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was partially funded by AFOSR grant FA9550-23-1-0328.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frsip.2024.1323538/full#supplementary-material
References
Alon, U. (2019) An introduction to systems biology: design principles of biological circuits. United States: CRC Press.
Amor, N., Meddeb, A., Marrouchi, S., and Chebbi, S. (2019). A comparative study of nonlinear bayesian filtering algorithms for estimation of gene expression time series data. Turkish J. Electr. Eng. Comput. Sci. 27, 2648–2665. doi:10.3906/elk-1809-187
Ancherbak, S., Kuruoglu, E. E., and Vingron, M. (2016). Time-dependent gene network modelling by sequential Monte Carlo. IEEE/ACM Trans. Comput. Biol. Bioinforma. 13, 1183–1193. doi:10.1109/TCBB.2015.2496301
Arulampalam, M. S., Maskell, S., Gordon, N., and Clapp, T. (2002). A tutorial on particle filters for online nonlinear/non-Gaussian Bayesian tracking. IEEE Trans. Signal Process. 50, 174–188. doi:10.1109/78.978374
Bugallo, M. F., Taşdemir, c., and Djurić, P. M. (2015). “Estimation of gene expression by a bank of particle filters,” in 2015 23rd European Signal Processing Conference (EUSIPCO), Nice, France, 31 August - 4 September 2015, 494–498.
Chopin, N. (2002). A sequential particle filter method for static models. Biometrika 89, 539–552. doi:10.1093/biomet/89.3.539
Dondelinger, F., Lebre, S., and Husmeier, D. (2013). Non-homogeneous dynamic bayesian networks with bayesian regularization for inferring gene regulatory networks with gradually time-varying structure. Mach. Learn. 90, 191–230. doi:10.1007/s10994-012-5311-x
Elahi, F., and Hasan, A. (2018). A method for estimating hill function-based dynamic models of gene regulatory networks. R. Soc. Open Sci. 5, 171226. doi:10.1098/rsos.171226
Elvira, V., Martino, L., Bugallo, M. F., and Djuric, P. M. (2019). Elucidating the auxiliary particle filter via multiple importance sampling [lecture notes]. IEEE Signal Process. Mag. 36, 145–152. doi:10.1109/MSP.2019.2938026
Gelfand, A. E., Hills, S. E., Racine-Poon, A., and Smith, A. F. M. (1990). Illustration of Bayesian inference in normal data models using Gibbs sampling. J. Am. Stat. Assoc. 85, 972–985. doi:10.1080/01621459.1990.10474968
Gelman, A., Carlin, J. B., Stern, H. S., and Rubin, D. B. (1995) Bayesian data analysis. United States: Chapman and Hall/CRC.
Ha, D., Kim, D., Kim, I., Oh, Y., Kong, J., Han, S., et al. (2022). Evolutionary rewiring of regulatory networks contributes to phenotypic differences between human and mouse orthologous genes. Nucleic Acids Res. 50, 1849–1863. doi:10.1093/nar/gkac050
Hinman, V. F., and Cheatle Jarvela, A. M. (2014). Developmental gene regulatory network evolution: insights from comparative studies in echinoderms. genesis 52, 193–207. doi:10.1002/dvg.22757
Krishnan, M., Small, M., Bosco, A., and Stemler, T. (2020). Network using Michaelis-Menten kinetics: constructing an algorithm to find target genes from expression data. J. Complex Netw. 8. doi:10.1093/comnet/cnz016
Lee, D. S., and Chia, N. K. (2002). A particle algorithm for sequential Bayesian parameter estimation and model selection. IEEE Transaction Signal Process. 50, 326–336. doi:10.1109/78.978387
Lee, T. H., Lakshmanan, S., Park, J. H., and Balasubramaniam, P. (2013). State estimation for genetic regulatory networks with mode-dependent leakage delays, time-varying delays, and markovian jumping parameters. IEEE Trans. NanoBioscience 12, 363–375. doi:10.1109/TNB.2013.2294478
Li, Y., Papandreou-Suppappola, A., and Morrell, D. (2006). “Instantaneous frequency estimation using sequential Bayesian techniques,” in Asilomar Conference on Signals, Systems, and Computers, Oct. 27th - Oct. 30th, 2024, 569–573.
Martino, L., Elvira, V., and Camps-Valls, G. (2018). The recycling gibbs sampler for efficient learning. Digit. Signal Process. 74, 1–13. doi:10.1016/j.dsp.2017.11.012
McKenzie, G. J., Harris, R. S., Lee, P. L., and Rosenberg, S. M. (2000). The sos response regulates adaptive mutation. Proc. Natl. Acad. Sci. U. S. A. 97, 6646–6651. doi:10.1073/pnas.120161797
Mittal, N., Guimaraes, J., Gross, T., Schmidt, A., Vina, A., Nedialkova, D., et al. (2017). The gcn4 transcription factor reduces protein synthesis capacity and extends yeast lifespan. Nat. Commun. 8, 457. doi:10.1038/s41467-017-00539-y
Nadal, E., and Posas, F. (2022). The hog pathway and the regulation of osmoadaptive responses in yeast. FEMS Yeast Res. 22, foac013. doi:10.1093/femsyr/foac013
Noor, A., Serpedin, E., Nounou, M., and Nounou, H. (2012). Inferring gene regulatory networks via nonlinear state-space models and exploiting sparsity. IEEE/ACM Trans. Comput. Biol. Bioinforma. 9, 1203–1211. doi:10.1109/TCBB.2012.32
Peter, I. S., and Davidson, E. H. (2011). Evolution of gene regulatory networks controlling body plan development. Cell 144, 970–985. doi:10.1016/j.cell.2011.02.017
Peter, I. S., and Davidson, E. H. (2015) Genomic control process: development and evolution. Massachusetts, United States: Academic Press.
Pirgazi, J., and Khanteymoori, A. R. (2018). A robust gene regulatory network inference method base on kalman filter and linear regression. PLOS ONE 13, 02000944–e200117. doi:10.1371/journal.pone.0200094
Potkin, S. G., Macciardi, F., Guffanti, G., Fallon, J. H., Wang, Q., Turner, J. A., et al. (2010). Identifying gene regulatory networks in schizophrenia. NeuroImage 53, 839–847. doi:10.1016/j.neuroimage.2010.06.036
Saunders, L., and McClay, D. (2014). Sub-circuits of a gene regulatory network control a developmental epithelial-mesenchymal transition. Dev. Camb. Engl. 141, 1503–1513. doi:10.1242/dev.101436
Tsigos, C., and Chrousos, G. P. (2002). Hypothalamic–pituitary–adrenal axis, neuroendocrine factors and stress. J. Psychosomatic Res. 53, 865–871. doi:10.1016/S0022-3999(02)00429-4
Vélez-Cruz, N., Moraffah, B., and Papandreou-Suppappola, A. (2021). “Sequential Bayesian inference using stochastic models of gene regulatory networks,” in Asilomar Conference on Signals, Systems, and Computers, Oct. 27th - Oct. 30th, 2024, 568–572.
Wang, H., Qian, L., and Dougherty, E. (2007). “Inference of gene regulatory networks using s-system: a unified approach,” in 2007 IEEE Symposium on Computational Intelligence and Bioinformatics and Computational Biology, Hawaii, USA, April 1-5, 2007, 82–89.
Wang, Z., Liu, X., Liu, Y., Liang, J., and Vinciotti, V. (2009). An extended kalman filtering approach to modeling nonlinear dynamic gene regulatory networks via short gene expression time series. IEEE/ACM Trans. Comput. Biol. Bioinforma. 6, 410–419. doi:10.1109/TCBB.2009.5
Xiong, J., and Zhou, T. (2013). A kalman-filter based approach to identification of time-varying gene regulatory networks. PLOS ONE 8, e74571–e74578. doi:10.1371/journal.pone.0074571
Youseph, A. S. K., Chetty, M., and Karmakar, G. (2015). “Gene regulatory network inference using michaelis-menten kinetics,” in 2015 IEEE Congress on Evolutionary Computation (CEC), Sendai, Japan, 25-28 May 2015, 2392–2397.
Youseph, A. S. K., Chetty, M., and Karmakar, G. (2019). Reverse engineering genetic networks using nonlinear saturation kinetics. Biosystems 182, 30–41. doi:10.1016/j.biosystems.2019.103977
Keywords: gene regulatory networks, Bayesian hierarchical modeling, dynamic model learning, Gibbs sampler, sequential Monte Carlo, Bayesian learning, nonlinear state-space model
Citation: Vélez-Cruz N and Papandreou-Suppappola A (2024) Bayesian learning of nonlinear gene regulatory networks with switching architectures. Front. Sig. Proc. 4:1323538. doi: 10.3389/frsip.2024.1323538
Received: 18 October 2023; Accepted: 18 April 2024;
Published: 22 May 2024.
Edited by:
Dirk Slock, EURECOM, FranceReviewed by:
Shuo Cao, Mayo Clinic Arizona, United StatesLuca Martino, Rey Juan Carlos University, Spain
Copyright © 2024 Vélez-Cruz and Papandreou-Suppappola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nayely Vélez-Cruz, bnZlbGV6Y3JAYXN1LmVkdQ==