 Ryan Johnston
Ryan Johnston Anne Aldrich
Anne Aldrich Shawn M. Lyons
Shawn M. Lyons
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. RNA Res., 19 January 2024
Sec. Non-coding RNA
Volume 1 - 2023 | https://doi.org/10.3389/frnar.2023.1331185
This article is part of the Research TopicNon-coding RNAs in disease development and progressionView all 4 articles
Ribosomes are amongst the most ancient molecular machines in cells, showing conservation from the simplest prokaryotes to humans. Ribosomes are an assembly of ribosomal (r)RNA and ribosomal proteins, but the rRNA comprises most of the mass of the ribosome and performs key enzymatic tasks. In humans, rRNA undergoes a laborious maturation that involves multiple processing steps and the deposition of chemical modifications. The correct processing and modification of rRNA ensures the proper function of the mature ribosome. Disturbance of these processes may lead to human disease. Understanding the role of rRNA in protein synthesis and the consequences of its dysregulation is key to deciphering and mitigating the emergence of pathological states in human biology.
De novo protein synthesis is a requirement of all forms of life. Cellular proliferation increases the demand for new proteins. Ribosomes are the macromolecular machines tasked with synthesizing new proteins; ironically, their synthesis represents a major source of new protein production. Ribosome biogenesis accounts for more than 60% of energy consumption in a growing cell (Warner et al., 2001) due to both their complexity and size.
The human 80S ribosome is 4.3 megadaltons and is composed of 80 ribosomal proteins and 4 non-coding RNAs. Owing to the centrality of protein synthesis in life, both ribosomes and tRNAs are conserved throughout evolution, despite the stark differences in translation regulation—particularly at the initiation stage. Some 33 of the 80 human ribosomal proteins are conserved in Escherichia coli, which diverged from humans 3–4 billion years ago. Bacteria have 55, rather than 80, ribosomal proteins, meaning that 60% of their ribosomal proteins are conserved in humans (Table 1).
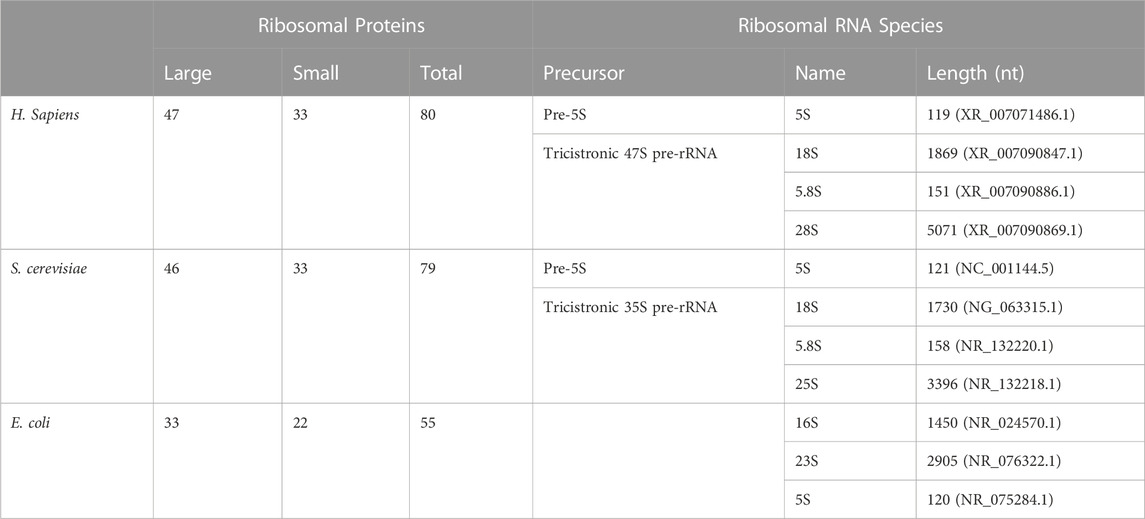
TABLE 1. Evolutionary conservation of ribosomal components. Ribosomal proteins and rRNAs found in Homo sapiens, S. cerevisiae, and E. coli. Note that the lengths of each mature rRNA are an approximation as there is heterogeneity in rRNA length amongst individuals or between strains. Numbers included here are derived from indicated GenBank or RefSeq submissions.
From prokaryotes to eukaryotes, rRNA plays a disproportionate role in the activity of ribosomes. rRNA coordinates accessory factor binding, plays a key role in discriminating amongst incoming charged tRNAs to ensure proper mRNA decoding, and, perhaps most importantly, rRNA plays a critical role in catalyzing peptide bond synthesis. This role increases the rate of peptide bond formation by a factor of 2 × 107 (Sievers et al., 2004). Emerging evidence also suggests that rRNA is a source of heterogeneity amongst ribosomes and hints at specialized ribosomes.
In this review, we will discuss the maturation of rRNA and its role in protein synthesis. We will also highlight disorders that arise from defects in these processes, with an emphasis on human disease.
To better understand how ribosome malfunctions related to rRNA contribute to disease, it is first necessary to understand the canonical roles that rRNA plays in protein synthesis. Following translation initiation, the joined 80S ribosome translocates along an mRNA, catalyzing new polypeptides as it goes. The large 60S subunit contains three sites through which tRNAs and mRNAs traverse to facilitate this activity: the aminoacyl (A), peptidyl (P), and exit (E) sites. During translocation, an individual tRNA will move from the A to the P site, from the P to the E site, or from the E site out of the ribosome. The A site contains aminoacylated tRNAs still conjugated to their cognate amino acid, the P site contains a peptidyl tRNA conjugated to the growing polypeptide chain, and the E site contains a deacylated tRNA ready for expulsion from the ribosome. As they enter, the tRNA and mRNA complex first encounters the decoding center (DC) before being transferred to the catalytic site, known as the peptidyl transferase center (PTC). The solution of the crystal structures of the large subunits, initially from prokaryotes and later in eukaryotes, reveals that the functional core of the PTC is rRNA and that rRNA plays a critical role in interrogating and proofreading tRNAs in the DC.
Many of the mechanistic studies regarding the molecular interactions of mRNA, tRNA, and the ribosome were initially conducted in prokaryotic systems. Subsequent data have revealed key similarities and distinctions between prokaryotic and eukaryotic ribosomes. An aminoacyl (aa)-tRNA is transported to the ribosome by EF-Tu, the bacterial homolog of mammalian eEF1a, where it is sampled at the DC. The ribosome must function as a “gatekeeper” in which non-cognate tRNAs are distinguished from cognate tRNAs. This is initially monitored by base-pairing between the mRNA codon and the anti-codon of the tRNA, although this is not sufficient for codon recognition.
Pioneering work by the Noller Lab showed that chemical modification of the 16S rRNA—the bacterial homolog of mammalian 18S—by kethoxal abolished tRNA binding to the A site of the ribosome (Noller and Chaires, 1972). Further chemical probing used bound tRNAs to protect against chemical modification and identified specific nucleotides of the 16S rRNA that mediated tRNA binding: G530, A1492, and A1493 (Moazed and Noller, 1990). Homologous nucleotides in the 18S rRNA of humans—G626, A1824, and A1825—serve similar roles (Table 2). When a cognate tRNA binds to a codon through its anticodon, this interaction is validated by flipping of A1492/1824 and A1493/1825, and conversion of G530/626 from a syn- to an anti-conformation. This facilitates an “A-minor interaction” with the first two base pairs in the codon–anticodon helix (Ogle et al., 2001). The mutational analysis further demonstrates the essential nature of these universally conserved bases (Cochella et al., 2007). This triggers large-scale movements of the small ribosome subunit (Ogle et al., 2002). In this conformation, there is evidence in prokaryotes that the 23S rRNA plays some role in stabilizing cognate tRNAs within the A site. A1913 of the 23S rRNA interacts with the A site tRNA within the DC directly and via Mg2+-mediated contacts (Selmer et al., 2006). Mammalian ribosomes similarly interact with tRNAs (Lomakin and Steitz, 2013). These movements prime EF-Tu/eEF1a for GTP hydrolysis to complete the first step of tRNA selection. In both prokaryotes and eukaryotes, EF-Tu/eEF1a bind the sarcin–ricin loop (SRL) of the 23S/28S rRNA, indicating the role of rRNA in the recruitment of elongation factors during translation (Correll et al., 1998; Lancaster et al., 2008; Budkevich et al., 2014). In prokaryotes, A2662 of the 23S rRNA interacts with His-84 of EF-Tu to stimulate GTP hydrolysis and, ultimately, EF-Tu dissociation. In eukaryotes, this mechanism is coordinated by the interaction of A4607 of the 28S rRNA and catalytic His-95 of eEF1a (Shao et al., 2016). At this point, interactions within the DC between rRNA and tRNA are a major driving force in cognate tRNA stability within the ribosome. Thermodynamic properties make non-cognate tRNAs more likely to disassociate at this point. The conformational changes made by cognate tRNA recognition instead drive the tRNA toward the P site and the PTC. Recent data demonstrate that the decoding mechanism in humans is ten times slower than in prokaryotes and may be functionally distinct (Holm et al., 2023). Despite the great strides gained from the study of the prokaryotic system, these results highlight the need for additional analysis of the mechanism of human ribosomal function.
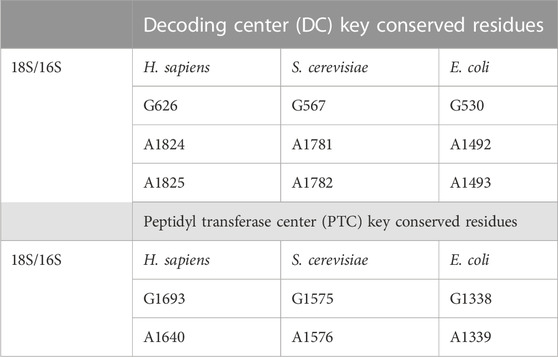
TABLE 2. Universally conserved residues in rRNA. Positions of residues within the decoding center and peptidyl transferase domains in H. sapiens, S. cerevisiae, and E. coli. As with the lengths, note that the exact location of these sites may be different in an individual molecule due to sequence heterogeneity. However, the identity of these residues does not vary.
The role of rRNA within the PTC and its role in catalyzing peptide bond synthesis cannot be overstated (Steitz and Moore, 2003). Ultimately, the mechanism of peptide bond formation was validated by crystallographic analysis of ribosomes; the first evidence that rRNA, rather than proteins, was responsible for catalysis was gained through biochemical analysis.
In hindsight, in vitro reconstitution experiments using E. coli components initially provided the first evidence that rRNA played a key role in peptidyl transferase activity. The 23S rRNA was identified as one of the essential factors required for reconstituting peptidyl transferase activity in vitro (Hampl et al., 1981; Schulze and Nierhaus, 1982). However, these studies also identified six ribosomal proteins that were essential, so activity could not be attributed to the rRNA.
The prokaryotic 23S rRNA has six major domains (Noller et al., 1981). Due to its increased photoreactivity when irradiated with 320 nm light and crosslinking to both RNA and protein, 3-(4′-benzoylphenyl)propionyl (BP)-[3H]-tRNAPhe was used as a chemical probe. The tRNAs crosslinked directly to the central single-stranded loop of domain V of the 23S rRNA in the PTC (Barta et al., 1984; Steiner et al., 1988). Primer extension determined that the tRNA crosslinked to U2584 and U2525 in the A site and to A2451 and C2452 in the P site—early evidence that RNA, not protein, was at the catalytic heart of the ribosome.
Chemical probing of tRNA-bound 70S ribosomes also showed that tRNAs bound in the P site would protect the PTC from chemical modification (Moazed and Noller, 1986). The protective effect was contingent upon the tRNA’s “CCA end”. This is critical because the CCA end is the site of amino acid conjugation, suggesting the 23S rRNA is near the site of catalysis. Antibiotic studies also provided key insights into the role of rRNA in peptide bond formation, as chemical probing showed that several antibiotics which were known to inhibit peptidyl transferase activity would protect nucleotides near A2451 (Moazed and Noller, 1987). These studies suggested that the antibiotics target the same region of 23S rRNA, in domain V, to block protein synthesis. Strong biochemical evidence was also presented by demonstrating that purified ribosomes treated with proteinase K, or partially extracting proteins, did not abolish peptidyl transferase activity. However, treatment with RNase T1 was sufficient to inhibit this activity (Noller et al., 1992).
While these experiments provided evidence that peptide bond formation was an rRNA-catalyzed event, the solution of the crystal structure of the 50S ribosome gave the clearest evidence that this was so (Ban et al., 2000; Nissen et al., 2000). This structure from the archaeon Haloarcula marismortui showed that A2451 was positioned precisely to mediate peptide bond synthesis. This suggested a model through which the N3 of A2451 could act to catalyze the reaction through general acid-base catalysis (Nissen et al., 2000). Later work refined this model to show that the 2′-OH of this nucleotide was the critical functional group (Erlacher et al., 2005). If A2451 were replaced with deoxyA2541, catalysis was severely compromised. Follow-up work demonstrated that the 2′-OH group of A2541 could act as a hydrogen donor during catalysis (Lang et al., 2008). This model was further refined using a higher resolution structure of the ribosome that revealed an additional water molecule present in the PTC (Polikanov et al., 2014). It has been proposed that this water molecule acts as a proton wire between the 2′-OH group of A2451 and A76 of the tRNA.
Finally, regions of rRNA beyond the core catalytic domains have been implicated in ribosome function. As the rRNA lengths have increased throughout evolution, much of the added length can be found in the aptly named “expansion segments (ES).” Analysis of some of the first structures of eukaryotic ribosomes revealed that they play many important roles. Most notably, these extensions form many eukaryotic-specific inter-subunit bridges between the 60S and 40S ribosomes (Ben-Shem et al., 2011). Expansion segments have also been shown to be directly involved in protein synthesis. In yeast, ES27Lb coordinates an interaction with NatA—a complex of proteins that acetylates the N-terminus of nascent peptides (Knorr et al., 2019). Binding at this ES positions NatA at the peptide exit tunnel so that it is in the opportune position to acetylate newly synthesized proteins. Deletion of ES27Lb sensitizes cells to reductive stresses by causing an increase in protein aggregates (Shankar et al., 2020). While ΔES27Lb cells show no difference in global translation levels, there are transcript-specific defects. This highlights what may be a theme for eukaryotic expansion segments in which they function as binding sites for accessory proteins. ES7L of the 28S rRNA interacts with SBP2, a protein involved in selenoprotein synthesis (Kossinova et al., 2014) and with aminoacyl-tRNA synthetases (Krauer et al., 2021), although the consequence of these interactions is unclear. ES7 has also been shown to contain sequences that assemble into G-quadruplexes (G4s) (Mestre-Fos et al., 2019). G4s are non-canonical nucleic acid structures that form following Hoogsteen base-pairing of four guanosines into a G-quartet (Fay et al., 2017). If the sequence of a nucleic acid permits, several G-quartets may stack to form a G4. This analysis shows that the ES7 G4s can mediate interaction with several proteins; however, the function of these interactions also remains elusive. Another striking example of transcript-specific translation regulation by expansion segments was proposed during development (Leppek et al., 2020). In this model, ES9S was proposed to have a role in directing the translation of Hox gene mRNAs. However, these data have recently been called into question based on misannotation of 5′-UTR start sequences (Akirtava et al., 2022).
Decades of work have revealed the roles of rRNA in protein synthesis; however, our knowledge regarding the functions of ribosomes still largely relies on work conducted in prokaryotic systems. In recent years, there have been giant leaps in the ability to resolve ribosome structures in different states (Ben-Shem et al., 2010; Ben-Shem et al., 2011; Anger et al., 2013; Khatter et al., 2015). The coming years can be expected to reveal important distinctions and similarities between prokaryotic and human ribosomes.
Despite similarities in ribosome function, the assembly of ribosomes is dramatically distinct between humans and prokaryotes. Prokaryotic ribosomes can be assembled in vitro using purified components (Traub and Nomura, 1968; Nierhaus and Dohme, 1974) and contain three rRNAs: the 23S and 5S in the large subunit and the 16S in the small subunit. Mammalian ribosomes contain four rRNAs: the 28S, 5.8S, and 5S in the large 60S subunit and the 18S rRNA in the small 40S subunit. Additionally, in vitro reconstitution from purified components is not possible for any eukaryotic ribosome. It is also worth noting that while the yeast ribosome also contains four rRNAs, the largest rRNA is referred to as the 25S, not the 28S, rRNA.
Mammalian ribosome biogenesis predominantly unfolds within the nucleolus, a specialized compartment within the nucleus (Lafontaine et al., 2021). Constituting approximately 80–90% of the total cellular RNA mass, rRNA synthesis primarily relies on RNA polymerase I (Pol I), which synthesizes the tricistronic 47S rRNA precursor, encoding 18S, 5.8S, and 28S rRNAs (Figure 1A). This precursor’s processing is essential for releasing mature rRNAs. The 5S rRNA, produced from monocistronic genes by RNA polymerase III, requires only 5′-end modifications to attain maturity, in stark contrast to the intricate processing of the 47S rRNA.
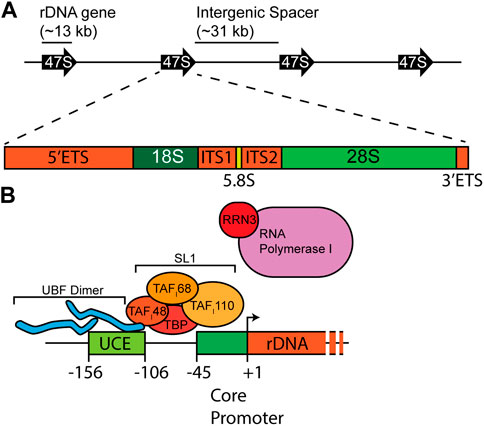
FIGURE 1. Schema of the rDNA locus and promoter. (A) rDNA genes are organized in a head-to-tail configuration. Each gene contains the mature 18S, 5.8S, and 28S rRNA flanked by 5′-and 3′-external transcribed spacers (ETS) and separated by internal transcribed spacers 1 and 2 (ITS1/2). (B) Two important sequence elements, the upstream control element (UCE) and the core promoter, drive efficient transcription of rRNA. UBF binds the UCE, while SL1, a multi-subunit complex of TATA-binding protein (TBP) and TAFs, assembles on the core promoter. These proteins work to recruit RNA polymerase I bound by RRN3 for active transcription.
The synthesis of rRNA emerges as a pivotal bottleneck in the intricate process of ribosome biogenesis (Maden et al., 1969; Emerson, 1971; Liebhaber et al., 1978). Each cell may contain 107 ribosomes that necessitate duplication during cell division to ensure an ample supply for the daughter cells. Failing to achieve this duplication would compromise the cell’s ability to engage in robust protein synthesis. Moreover, this challenge extends beyond duplication as it does not account for the replacement of ribosomes lost due to their natural half-life. Cells adopt a dual-pronged strategy to address the demand for new rRNA synthesis. Actively growing cells exhibit heightened rates of rRNA gene synthesis—a straightforward and effective response. Additionally, cells throughout evolution have expanded their repertoire of rRNA genes. In the case of humans, more than 300 individual copies of the 47S rDNA gene are distributed on the short arms of five acrocentric chromosomes at 13p12, 14p12, 15p12, 21p12, and 22p12 (Henderson et al., 1972; Gaubatz et al., 1976). Traditionally organized in a “head-to-tail” orientation within 43 kb repeated elements, each repeat encompasses a 13 kb rDNA gene and an intergenic spacer (IGS) of approximately 30 kb in length (Figure 1A) (Wellauer and Dawid, 1979). Newer imaging and sequencing approaches have revealed significant variations in rDNA structure and rDNA gene sequences in humans (Caburet et al., 2005; Kim et al., 2018; Nurk et al., 2022). The functional consequences of rRNA variation are still being analyzed.
In humans, the primary cluster for 5S rRNA is located at a specific genomic locus on chromosome 1 at 1q42 (Steffensen et al., 1974; Sorensen et al., 1991). While this cluster boasts over 2000 individual genes, stringent dot-blotting using placenta DNA has indicated that the majority of these may be pseudogenes (Sorensen and Frederiksen, 1991). A more refined assessment revealed that only approximately 400 genes are potentially active. This results in the number of 5S genes roughly aligning with the count of 47S genes. Given that 5S rRNA is equimolarly abundant in ribosomes, this unique genomic organization may serve as a strategic mechanism to balance 5S production with the synthesis of other rRNAs. However, this genomic arrangement is distinctive to higher eukaryotes. In organisms like Saccharomyces cerevisiae and Dictyostelium discoideum, the 5S rRNA genes are intricately integrated within clusters responsible for transcribing the 18S, 5.8S, and 28S rRNAs (Maizels, 1976; Petes et al., 1978), positioned on the opposite strand of the tricistronic rRNA genes. In contrast, Schizosaccharomyces pombe and Neurospora crassa exhibit a dispersion of 5S genes throughout their genomes (Selker et al., 1981; Mao et al., 1982).
Despite this expansion, only approximately half of the rDNA repeats appear to be active as the remaining are epigenetically silenced (reviewed in Srivastava et al. (2016)). However, a percentage of active and inactive genes can change in response to stimuli, such as metabolic and environmental signals or stages of the cell cycle. There are three distinct rDNA silencing complexes in humans: the nucleolar remodeling complex (NoRC), the nucleosome remodeling and deacetylation (NuRD) complex, and the energy-dependent nucleolar silencing complex (eNoSC). The NoRC establishes a constitutively silenced rDNA (Strohner et al., 2001). The NuRD complex leaves rDNA promoters in an inactive state but remains accessible for transcription factors, allowing genes to stay “poised” for transcription (Xie et al., 2012). As its name suggests, eNoSC regulates rDNA silencing in response to energy deprivation (Murayama et al., 2008). In mammals, effective silencing is facilitated by the concerted effort of the methylation of histone tails (e.g., H3K4 and H3K79) and by DNA methylation. The repetitive nature of rDNA repeats has historically posed challenges for analyzing the transcriptional status of individual genes, causing a lag in understanding which rDNA genes are selected for silencing and why. However, innovative data analysis approaches and longer read lengths may help bridge these knowledge gaps. Active and silenced genes are interspersed, rather than entire loci being uniformly silenced (Sharp et al., 1984; Dammann et al., 1995; Zillner et al., 2015).
RNA Pol I is specialized for transcribing only the 47S rDNA genes. It is recruited to rDNA promoters which contain two critical regulatory elements: the upstream control element (UCE) and the core promoter (Figure 1B). Initial cell-free transcription experiments from mouse Ehrlich ascites tumor-cell extracts and cloned mouse DNA templates demonstrated that base pairs immediately upstream of the transcription start site, which defined the core promoter, were sufficient to drive transcription (Grummt, 1982). However, these same experiments showed that nucleotides further upstream were important in stimulating the transcription of rRNA when competing with templates containing only core promoters; this upstream region came to be known as the UCE. Later experiments using HeLa extract and human DNA templates validated that human Pol I transcription was also governed by a core promoter and UCE (Learned et al., 1983). Despite the conserved promoter structure across species, there is little promoter sequence homology—a contrast with mature rRNA sequences. In fact, mouse extract is incapable of driving transcription from the human rDNA promoter and vice versa (Grummt et al., 1982).
Identification of these elements initiated the search for the protein factors that acted as trans factors. The first of these factors to be identified was termed selectivity factor 1 (SL1) (Learned et al., 1985). SL1 is a multi-subunit complex containing TATA-binding protein (TBP) and multiple transcription activating factors (TAFs) (Comai et al., 1992; Comai et al., 1994; Zomerdijk et al., 1994; Heix et al., 1997; Gorski et al., 2007). However, SL1 has no specificity for the rDNA promoters and no DNA binding activity within the promoter (Bell et al., 1988). DNase I foot-printing experiments showed that SL1 alone would not bind to the rDNA promoters, but partially purified RNA Pol I would associate with the UCE (Learned et al., 1986). The DNA binding activity was further purified from crude RNA Pol I preparations and was termed upstream binding factor 1 (UBF) (Bell et al., 1988). Importantly, UBF and SL1 cooperate to mediate rDNA transcription. The TAFI48 subunit of SL1 interacts with UBF (Beckmann et al., 1995), at which point the UBF1 DNA binding targets the complex at rDNA promoters (Bell et al., 1988). Conversely, binding to SL1 stabilizes UBF on DNA (Friedrich et al., 2005).
Another key factor for efficient rRNA transcription initiation is RRN3/TIF-IA. Biochemical fractionation partially purified this protein from mouse cells, based on its ability to stimulate rRNA transcription in growing cells, and it was named TIF-IA (Buttgereit et al., 1985). Later work demonstrated that TIF-IA is regulated by mTOR activity, which is consistent with its loss of activity during reduced growth phases (Mayer et al., 2004). Other work identified the same protein as being able to stimulate rRNA synthesis in response to hormonal stimulation but termed the protein the name TFIC (Mahajan and Thompson, 1990). Yet another group identified the same activity and gave this protein a different name: Factor C* (Brun et al., 1994). It was not until the homologous protein was identified in yeast that this gene was cloned (Milkereit and Tschochner, 1998), and shortly thereafter it was discovered that TIF-IA, TFIC, and Factor C* were the same protein and were the mammalian homolog of yeast Rrn3 (Bodem et al., 2000; Moorefield et al., 2000).
Work from yeast and mammalian systems showed that RRN3/TIF-IA is the limiting factor for rRNA synthesis. Pol I that is unbound to DNA can exist freely or associated with RRN3/TIF-IA. However, only RRN3/TIF-IA-bound Pol I is recruited to DNA to initiate transcription (Peyroche et al., 2000; Blattner et al., 2011), although RRN3/TIF-IA is inactivated and may dissociate following transcription (Brun et al., 1994; Hirschler-Laszkiewicz et al., 2003). Thus, it is likely needed only for DNA recruitment, melting of DNA, and promoter escape (Sadian et al., 2019). Given its key role in regulating rRNA synthesis, RRN3/TIF-IA is a target of many pathways that regulate stress response and growth. In addition to being regulated by mTOR activity, RRN3/TIF-IA is a target of JNK, ERK, RSK, and AMPK (Zhao et al., 2003; Mayer et al., 2005; Hoppe et al., 2009).
Treacher Collins syndrome (TCS) is the most common cause of mandibulofacial dysostosis, occurring in 1 in 50,000 live births (Gorlin et al., 1990; Ulhaq et al., 2023). Prior to the identification of the causative gene for TCS, it was noted that this disorder followed an autosomal dominant inheritance pattern but that most cases were sporadic, likely arising from novel private mutations (Jones et al., 1975). Through laborious genetic mapping using short tandem repeat polymorphisms and the generation of yeast artificial chromosomes (YACs) containing the affected genomic region, the major causative gene was identified and named TCOF1 (Dixon et al., 1996). A wide array of seemingly sporadic mutations in TCOF1 have been associated with or are causative of the development of TCS (Wise et al., 1997). Eventually, a mouse model generated heterozygous knockouts of TCOF1, mimicking the disease conditions (Dixon et al., 2006). In this model, there was a defect in neural crest cell migration during development, which led to defects in facial bones, cleft palate, and ear defects consistent with clinical presentations (Jones et al., 2008). The complete analysis of the TCOF1 amino acid sequence led to the hypothesis that it was a possible nucleolar phosphoprotein, based on its similarity to other resident nucleolar proteins (e.g., NOLC1 and NPM) (Wise et al., 1997). This work also noted that TCOF1 displayed particularly low complexity throughout its amino acid sequence, a feature that is now known to aid in the condensation of non-membranous organelles such as the nucleolus (Lafontaine et al., 2021). Experiments formally demonstrated that TCOF1 localized to the nucleolus, and this required sequences in its C-terminus (Winokur and Shiang, 1998; Isaac et al., 2000). A subset of TCS patients do not have mutations in TCOF1. The second and third most common mutations are found in POLR1D and POLR1C, which are subunits of both Pol I and Pol III (Dauwerse et al., 2011). Disease-associated mutations destabilize the function of these enzymes which are critical for the synthesis of rRNAs, emphasizing that TCS arises from a defect in this process (Walker-Kopp et al., 2017).
The first clues about the mechanistic function of TCOF1 emerged as its role in rRNA transcription was revealed. Co-immunoprecipitation and yeast two-hybrid experiments demonstrated that TCOF1 formed a direct complex with UBF (Valdez et al., 2004). Consistent with these data, siRNA-mediated depletion of TCOF1 resulted in a reduced rate of rRNA synthesis. However, data from Xenopus laevis also support the hypothesis that TCOF1 may be involved in rRNA methylation, another form of dysfunction found in rRNA synthesis (see below) (Gonzales et al., 2005). However, there is mounting evidence that the rate of rRNA synthesis is linked to the effectiveness of rRNA methylation, so it is unclear whether the effect on rRNA methylation is direct or is a secondary effect from altered rRNA synthesis. However, proteomic analysis of Nop56, a key factor in rRNA methylation, has identified TCOF1 as an interacting factor (Hayano et al., 2003).
Cockayne syndrome (CS) is an autosomal recessive disorder that results in diverse clinical presentations. The most common presentations are neonatal failure of growth and neurological dysfunction that worsens with age. However, microcephaly, dwarfism, cardiac defects, increased sensitivity to UV light, and ophthalmologic dysfunction, including retinal degeneration and the increased propensity for cataract formation, are also common (Nance and Berry, 1992). Life expectancy for afflicted individuals is 12 years (Laugel et al., 2008). Fibroblasts from CS patients display reduced proliferation after exposure to UV light based on colony-forming assays (Schmickel et al., 1977). Curiously, these cells appear to have no defect in the rate of DNA repair but have a reduced rate of recovery based on RNA synthesis (Mayne and Lehmann, 1982; Lehmann et al., 1993). In these studies, RNA synthesis was monitored by the uptake of 3H-uridine into acid-soluble RNA. Since rRNA accounts for the majority of all cellular RNA synthesis, it can be inferred that these studies monitored the resumption of rRNA synthesis after UV irradiation. Genetic analysis revealed that CS patients fell into at least two distinct complementation groups: CSA or CSB (Tanaka et al., 1981). Clarity regarding the complementation groups and the effect on nucleic acid synthesis came with the isolation of the relevant genes, which were termed CSA (ERCC8) and CSB (ERCC6) (Troelstra et al., 1990; Henning et al., 1995). Both CSA and CSB were determined to associate with transcription factor IIH (TFIIH), which originally implicated a defect in RNA Pol II transcription in CS. However, later data demonstrated that TFIIH was also crucial for Pol I transcription initiation and elongation (Bradsher et al., 2002; Iben et al., 2002; Assfalg et al., 2012). These data, and the dramatic effect on bulk rRNA synthesis, implicated rRNA synthesis as a component of CS pathology.
To date, multiple mechanisms have been proposed for how mutations to CSA and CSB alter rRNA synthesis rates. These mechanisms are not mutually exclusive, and some interplay between proposed mechanisms is possible. TFIIH, which interacts with CSA and CSB, is essential for Pol I transcription initiation in humans and yeast, and it interacts with Pol I and SL1 (Iben et al., 2002; Koch et al., 2014). CSB and TFIIH are also necessary for efficient Pol I elongation (Lebedev et al., 2008; Assfalg et al., 2012). The elongation-promoting activity found in these studies may be related to recent data showing that CSA and CSB can resolve G4 structures (Scheibye-Knudsen et al., 2016; Liano et al., 2021). These are highly stable structures that can block a polymerase if not resolved. Reduction of CSA or CSB leads to the stalling of polymerases at sequences capable of forming G4s. Further analysis shows that CSB can melt these structures, which would allow for more efficient transcription elongation. Surprisingly, chemical stabilization of G4 leads to decreased rRNA synthesis and an increased rate of aging (Scheibye-Knudsen et al., 2016; Liano et al., 2021). These results could tie the rRNA synthesis seen in CS mutations to the pathological aging symptoms in CS patients. CS has also been proposed to function through interaction with nucleolin (NCL) (Okur et al., 2020), which is noteworthy, as NCL has also been implicated in the regulation of G4 structures in rDNA (Drygin et al., 2009; Xu and Hurley, 2022).
Other work has hinted at an alteration of the epigenetic landscape as a mechanism for reduced rRNA transcription. Co-immunoprecipitation studies established that CSB interacts with G9a, a histone methyltransferase that deposits mono- and di-methyl groups on H3K9 (Yuan et al., 2007). The authors propose that these methylation marks aid the recruitment of HP1γ which promotes Pol I elongation. CSB may also interact with the NuRD complex, which establishes a chromatin state that poises individual rDNA genes for transcription (Xie et al., 2012).
Finally, CS is also associated with aberrant ribosome function, although it has not been formally established that this is a consequence of altered rRNA synthesis rates. Nevertheless, cells harboring CS mutations have reduced translation fidelity (Alupei et al., 2018). In these conditions, proteins with misincorporated amino acids have an increased propensity to misfold, which activates the unfolded protein response. Mutations in TFIIH have similar effects on proteostasis and translation fidelity (Khalid et al., 2023). These mutations are associated with trichothiodystrophy (TTD) developmental disorder, which shares common pathologies with CS.
The rDNA repeats are highly recombinogenic (Gaubatz et al., 1976; Kobayashi, 2011). In humans, the average number of rDNA genes is 300, but the absolute number can differ between individuals and within an individual (Johnson and Strehler, 1972; Medvedev, 1972; Johnson et al., 1975; Wang and Lemos, 2017; Malinovskaya et al., 2018). Early work suggested an age-dependent loss of rDNA repeats in brain and heart tissue (Johnson and Strehler, 1972; Medvedev, 1972; Johnson et al., 1975). Follow-up studies have found similar reductions in rDNA number during aging (Zafiropoulos et al., 2005; Ren et al., 2017). This suggested an intriguing hypothesis that an age-related decrease in fitness may be partly due to the loss of proliferative potential by reducing the number of rDNA transcription units. In turn, this would reduce the rate of rRNA synthesis and ultimately ribosome production. In fact, earlier work using Drosophila melanogaster genetics had demonstrated that to be true (Ritossa et al., 1966). Correlative work has shown that a low rDNA copy number is associated with mild cognitive impairment and dementia, although these studies could not demonstrate a causative link (Hallgren et al., 2014; Veiko et al., 2022). Expansion and loss of rDNA repeats are associated with human cancers (Wang and Lemos, 2017). In these cases, there is a trend for expansion of the 5S rDNA genes and a decrease in the 47S rDNA genes, leading to a general imbalance. Loss of rDNA repeats is also common in Werner’s syndrome (Caburet et al., 2005), a progeroid syndrome characterized by adult onset rapid aging (Oshima et al., 2017). These are coupled with a wealth of data from S. cerevisiae demonstrating an age-dependent loss of rDNA on linear chromosomes and the generation of extrachromosomal rDNA circles (Sinclair and Guarente, 1997). Age in yeast can be measured by replicative lifespan, which is the number of daughter cells a mother cell may produce. Recent data have demonstrated that the loss of rDNA repeats is a major driver of replicative senescence (Hotz et al., 2022).
However, the relevance of this phenotype in humans and its causal effect on aging has been challenged (Gaubatz et al., 1976). A Russian population study demonstrated a narrowing of the range of rDNA copy number with age, but the mean and median number of repeats were unchanged (Malinovskaya et al., 2018).
As the maintenance of rDNA repeats has been implicated in regulating longevity and aging, so has the maintenance of epigenetic rDNA silencing. Overexpression of SIRT1, a component of the eNoSC, has been shown to extend lifespan in D. melanogaster, Caenorhabditis elegans, and mice (Tissenbaum and Guarente, 2001; Satoh et al., 2013; Whitaker et al., 2013). In contrast, patients with Alzheimer’s disease also show an increase in epigenetic silencing of rDNA repeats, which would result in a reduced transcriptional potential (Pietrzak et al., 2011).
As loss of rRNA synthesis, either due to epigenetic silencing or rDNA loss, is linked to reduced fitness, increased rRNA synthesis is intrinsically tied to hyperproliferation. The ability to duplicate the cell’s complement of ribosomes is a key factor in its ability to divide, and rRNA synthesis is a rate-limiting step in this process. Some of the earliest histological analyses of cancer cells revealed enlarged nucleoli—the site of rRNA transcription and ribosome biogenesis (Maccarty, 1936). Nearly a century of work has now shown that this is due to an increase in rRNA synthesis.
Stabilization of c-Myc, a protooncogenic transcription factor, is a key feature of carcinogenesis. Transcriptional regulation of RNA Pol II and RNA Pol III genes had been under intense investigation in regulating cell transformation. Data showed that ribosomal protein genes were stimulated by c-Myc (Kim et al., 2000). However, in 2005, three groups independently revealed that c-Myc is also a major driver of rRNA (Arabi et al., 2005; Grandori et al., 2005; Grewal et al., 2005). c-Myc was shown to localize to the nucleus, and its overexpression triggered a dramatic increase in transcription. Conversely, siRNA knockdown of c-Myc reduced rRNA synthesis rates. E-boxes are the DNA element to which c-Myc binds. Analysis of rDNA repeats demonstrates a wide distribution of these sites throughout each repeat, which ChIP experiments demonstrated were bound by c-Myc. Later work showed that c-Myc may re-organize the chromatin to better allow access to SL1 and UBF (Shiue et al., 2009). Recent data also point to UBF mutations as playing a role in acute myeloid leukemia (Stratmann et al., 2021; Umeda et al., 2022; Duployez et al., 2023). Screening of adult and pediatric cases showed internal tandem duplication of exons in UBF. However, the effect of these tandem duplications on rRNA expression is not known.
The increased requirement for de novo ribosome production in cancer and metastasis has made inhibition of RNA Pol I an attractive target for anti-cancer therapies. CX-3543 is a small molecule that has been shown to inhibit rRNA synthesis. A proposed mechanism of action is to disrupt nucleolin (NCL) from G-rich sequences in rDNA (Drygin et al., 2009). Under normal conditions, NCL has been proposed to prevent G4 structures from forming, suggesting a mechanism in which CX-3543 displaces NCL from rDNA, leading to an increase in G4 formation and impaired Pol I elongation. This is similar to a proposed cause of Cockayne syndrome. A derivative of CX-3543—CX-5461—was shown to target RNA Pol I more directly (Mars et al., 2020). This drug allows the recruitment of Pol I/RRN3 complexes to promoters but prevents promoter escape and transition to an elongation phase. However, polymerases that have begun elongation are not inhibited, and the validity of this proposed mechanism is a matter of debate. Other work has suggested that the primary mode of action is through the “poisoning” of topoisomerase II (Bruno et al., 2020).
In mammals, the synthesis of four mature rRNAs involves the transcription of two precursor rRNAs. The pre-5S rRNA is transcribed by RNA polymerase III, while the tricistronic 47S pre-rRNA (equivalent to 35S in yeast) is transcribed by RNA polymerase I. Minimal 5′-end processing is required to convert pre-5S rRNA into mature 5S rRNA, whereas the 47S precursor undergoes an intricate maturation process that leads to the release of 18S, 5.8S, and 28S rRNAs (Figure 2). Significant strides in understanding rRNA maturation have been achieved using S. cerevisiae, yielding foundational discoveries. However, distinctions between yeast and mammals underscore our incomplete understanding of the maturation process in humans. This review will primarily focus on the maturation of mammalian rRNAs, with a conscientious acknowledgment of pivotal findings derived from yeast studies.
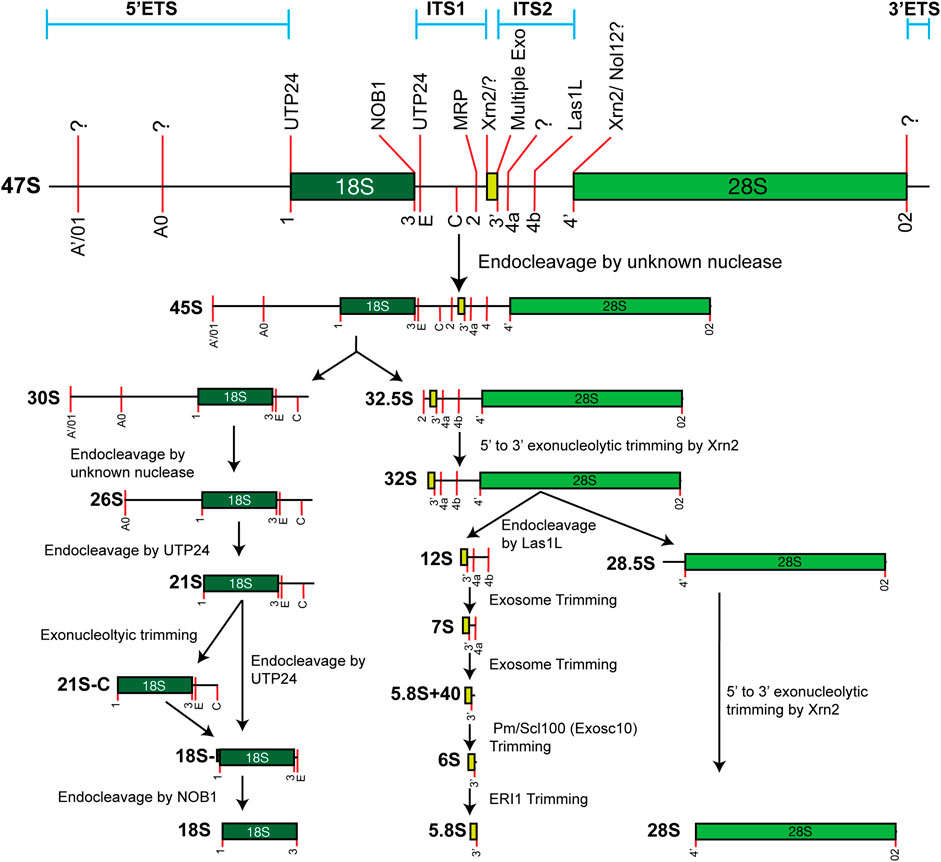
FIGURE 2. Simplified model of the major 47S rRNA processing pathway. Processing sites and identified processing enzymes are noted on the 47S rRNA, when known. This only schematizes the major pathway. Note that alternative minor maturation pathways exist.
The mature 5S rRNA is the smallest of the four rRNAs. The human 5S is homologous to the prokaryotic 5S rRNA and shares a similar secondary structure. The establishment of in vitro transcription systems using isolated human or rat nuclei revealed that the transcription product for the 5S genes was longer than the mature 5S rRNA, suggesting the possibility of a longer precursor (Yamamoto and Seifart, 1978; Hamada et al., 1979). However, these apparent precursors contained extended uridine residues at their 5′-end, so researchers acknowledged the possibility that these arose because of the artificial nature of in vitro transcription.
Later work confirmed that this 3′-extended precursor was a natural transcription product. These experiments took advantage of antisera from patients with systemic lupus erythematosus (Rinke and Steitz, 1982). The antisera recognized that the La protein and immunoprecipitation effectively precipitated most Pol III transcripts, including the 5S rRNA. These transcripts were shown to contain similar U-rich 3′-extensions, as had been seen in in vitro transcription experiments. Shortly thereafter, mutagenesis experiments established the existence of 5S precursor molecules in yeast (Piper et al., 1983). Similarly, it was discovered that the initial transcripts contained short uridine-rich 3′-extensions. Further work on the mechanism of Pol III termination has shown that these U-rich sequences function as a termination signal, like that seen in prokaryotic transcription (reviewed in Arimbasseri et al. (2013)).
Despite the relatively simplistic processing required to mature the pre-5S to 5S rRNA, the mechanism remains unclear in humans. Work from yeast has shown that the 3′-end of the pre-5S is exonucleolytically processed by Rex1p to generate the mature rRNA (van Hoof et al., 2000). A distantly related protein, Rexo5, appears to be involved in D. melanogaster (Gerstberger et al., 2017). However, there are important differences that have yet to be resolved in species-specific processing. In yeast, mutants of Rex1p are viable, whereas mutations to fly Rexo5 are lethal. It is unclear if this is due to added functions of Rexo5 or a reduced stringency in yeast for 3′-extensions. Further complicating this is the unclear role of the mammalian homolog of Rexo5, known as NEF-sp (Silva et al., 2017). In mice, this protein is restricted to gonads, and its loss does not alter viability. The maturation of human 5S rRNA clearly warrants additional research to determine its unique process relative to yeast.
Mammalian tricistronic precursor maturation has been better characterized, although important gaps in knowledge remain. In humans, the most commonly used reference 47S rRNA sequence is 13,304 nucleotides. This sequence is known to contain errors, and increasing evidence supports heterogeneity amongst 47S sequences. The initial transcript contains the 18S, 5.8S, and 28S rRNAs, which are flanked by 5′- and 3′-external transcribed spacers (ETSs). The 18S and 5.8S sequences are separated from each other by internal transcribed spacer 1 (ITS1), while the 5.8S and 28S sequences are separated by ITS2. This general structure is conserved in metazoans, but the spacer regions have greatly expanded in mammals. As with 5S, the presence of precursor molecules from which the 18S, 5.8S, and 28S originated was not a foregone conclusion. The 18S and 28S can be analyzed by radiolabeling RNA and purification by sucrose gradient ultracentrifugation (Philipson, 1961; Scherrer and Darnell, 1962). Brief labeling of RNA identified larger intermediates that are dependent upon active transcription, suggesting that these were precursors of the mature rRNAs (Scherrer and Darnell, 1962; Scherrer et al., 1963; Tiollais et al., 1971).
The first processing events occur nearly simultaneously at the extreme 5′- and 3′-ends to convert the 47S to the 45S pre-rRNA. The 5′-end processing occurs at two closely related sites: A′ and 01 (A’/01). In humans, this site is located approximately 415 nucleotides from the 5′-end of the 47S, although the precise cleavage site is not known. S1 nuclease protection assays and primer extension assays suggest a heterogeneous distribution of cleavage events (Kass et al., 1987). This processing site is not present in yeast, making this system unable to provide further insights into the mechanism of cleavage in humans. Despite these challenges, a pivotal contributor to A’/01 processing is the U3 small nucleolar RNA (snoRNA) (Bachellerie et al., 1983; Crouch et al., 1983; Tague and Gerbi, 1984; Parker and Steitz, 1987; Kass et al., 1990). Notably, this RNA molecule assumes multiple roles in the intricate process of maturing the 18S rRNA. The U3 snoRNA is a member of the Box C/D snoRNA family that primarily guides the 2′-O methylation of rRNA (see below). However, the U3 snoRNA assembles with a host of protein co-factors to then base-pair with the pre-rRNA to direct the processing of the 18S rRNA (Prestayko et al., 1970; Calvet and Pederson, 1981). Evidence suggests that A’/01 processing occurs within minutes (Popov et al., 2013).
Processing at the 3′-end of the 47S is less clear in humans. A processing site known as 02 is found at the 3′-end of the mature 28S. However, it is not known if this is generated by an exo- or endo-nucleolytic processing event. In yeast, the concerted efforts of an endonuclease—Rnt1p—and exonuclease—Rex1p—are required (Kempers-Veenstra et al., 1986; Kufel et al., 1999). As with 5′-end processing, a snoRNA has been implicated in this processing reaction in vertebrates and mammals. Work in Xenopus oocytes demonstrated that the U8 snoRNA was involved in this process (Peculis and Steitz, 1993), and later work has confirmed that this is conserved in mammals (Srivastava et al., 2010; Langhendries et al., 2016).
Although minor processing pathways exist in metazoans, the next major processing event is endo-nucleolytic cleavage in ITS1, which separates the small and large subunit rRNAs. This cleavage event at Site 2 is mediated by the MRP complex, a ribonucleoprotein that contains the RMRP catalytic RNA (see below for additional details).
Maturation of the 18S requires removal of the 5′-ETS and the 5′-portion of ITS1 after cleavage. Like cleavage at A’/01, the discrete processing requirements for the removal of 5′-ETS still require clarification. However, they are dependent upon the U3 snoRNP, which aids in the assembly of the small-subunit (SSU) processome. There are two identified processing sites remaining in the 5′-ETS after A’/01 cleavage—A0 and 1—which may be functionally related to the A0 and A1 sites in yeast. This supposition is strengthened by the fact that Site 1 in humans and A1 in yeast is cleaved by homologous protein Utp24, a PIN (PilT N terminus) domain-containing endonuclease. Utp24 and other U3 proteins (Utp) are associated with the U3 snoRNP (Bleichert et al., 2006). Genetic evidence, crosslinking, and in vitro cleavage experiments in yeast and humans have established that Utp24 performs this processing (Bleichert et al., 2006; Wells et al., 2016). Some evidence has suggested that another UTP, Utp23, may be involved in both A0 and A0 cleavage in humans and yeast, respectively. However, in yeast, the catalytic nucleotides are not required for efficient cleavage, suggesting that Utp23 serves a structural role (Bleichert et al., 2006; Hoareau-Aveilla et al., 2012). In humans, the catalytic PIN domain does appear to have a role in promoting cleavage, though it is yet to be validated if Utp23 is directly responsible (Wells et al., 2017).
Utp24 also has a role in generating the mature 3′-end of the 18S rRNA by cleaving at Site E to generate the 18S-E precursor (Bleichert et al., 2006; Wells et al., 2016). However, the ITS1 fragment may be exonucleolytically trimmed by the exosome before Utp23 cleavage to generate the 21S-C precursor (Carron et al., 2011). It is not clear if this is an obligatory step in processing, although the 21S-C precursor is stabilized in patients with Diamond–Blackfan anemia, a ribosomopathy that arises due to mutations in ribosomal proteins. 18S-E contains approximately 78 extra nucleotides at its 3′-end relative to the mature rRNA. Unlike other steps in this process, the final maturation is completed in the cytoplasm by Nob1, another PIN-domain-containing protein (Udem and Warner, 1973; Rouquette et al., 2005). Various proteins, including PNO1, a binding partner of Nob1, ensure that pre-40S ribosomes do not prematurely engage with the translation machinery before final maturation (Tone and Toh-e, 2002; Lebaron et al., 2012). Phosphorylation of PNO1 by RIOK1 triggers its displacement from pre-40S ribosome and allows for final 18S maturation by Nob1 (Ameismeier et al., 2020).
Maturation of the large subunit rRNAs requires separation of the 28S from the 5.8S rRNA via cleavage in ITS2 and final 5′- and 3′-end processing of each mature rRNA. The endonuclease Las1L cleaves within ITS2 to separate the two immature rRNAs, requiring the polynucleotide kinase activity of Grc3/Nol9 (Gasse et al., 2015; Fromm et al., 2017; Pillon et al., 2017; Pillon et al., 2018; Pillon et al., 2019). The majority of these analyses have been conducted in yeast, but it is presumed that their function is conserved in humans. Yeast Grc3 recruits Las1 to the site of cleavage and “programs” it by promoting its proper conformation. In humans, RNAi-mediated depletion of these proteins blocks ITS2 cleavage (Castle et al., 2010; Heindl and Martinez, 2010). Las1L and Nol9 also assemble into a complex whose components are necessary for ITS2 processing (Castle et al., 2012).
The 5′-end of the 5.8S rRNA is likely matured primarily through the action of exo-nucleolytic trimming. This generates two distinct 5′-ends, resulting in the longer 5.8SL or the 5.8SS, which differ by seven nucleotides (Henry et al., 1994). While most mechanistic details were discovered in yeast, the basic processes appear to be conserved in humans. The 5′- to 3′-exonuclease Rat1 is responsible for 5.8S 5′-end maturation in yeast (Amberg et al., 1992; Xue et al., 2000). Structurally, human Xrn2 is homologous to yeast Rat1 (Shobuike et al., 1995), and its depletion impairs 5′-end maturation of the 5.8S rRNA (Wang and Pestov, 2011). These same proteins are responsible for 5′-end maturation of the 28S rRNA after ITS2 cleavage (Geerlings et al., 2000; Wang and Pestov, 2011).
Finally, the maturation of the 3′-end of the 5.8S rRNA is a concerted effort of several exonucleases that generate discrete intermediates. The major player in this process is a multi-subunit complex known as the exosome (Mitchell et al., 1997). Human exosome was first characterized using autoantisera from patients with scleroderma, while the yeast exosome was identified genetically (Reimer et al., 1986; Allmang et al., 1999; Brouwer et al., 2001). The Exo9 complex is the structural unit of the exosome that contains nine polypeptides in a barrel-like structure. Each polypeptide has features of an exonuclease, but catalytic activity is granted by adding one or two additional proteins—Exosc10 (Pm/Scl100) and Dis3—to form the Exo10 or Exo11 complex. The exosome trims the largest 5.8S precursor, known as the 12S, to generate the 7S precursor. This processing may also involve a second exonuclease called interferon-stimulated 20-kDa exonuclease-like 2 (ISG20L2) (Coute et al., 2008). The exosome then completes the next processing reaction to generate the 5.8S + 40 precursor. As the name implies, this precursor contains an additional 40 nucleotides at the 3′-end of the 5.8S rRNA. This length is dictated by the distance an RNA must traverse through the Exo9 barrel before reaching the catalytic subunits (Schuller et al., 2018). At this point, Exosc10 can function independently of the exosome complex to trim the 5.8S + 40 to generate the final precursor, known as the 6S. As with the final 3′-end-maturation of the 18S, the final processing event occurs in the cytoplasm. The multifunctional exonuclease Eri1 (3′-hExo) removes the final nucleotides in the cytoplasm (see below for additional details).
Cartilage–hair hypoplasia (CHH) is a type of dwarfism first described by McKusick in Old Order Amish populations (McKusick et al., 1965). CHH is a pleiotropic disease causing a variation of clinical presentations that include immune dysfunction and increased cancer risk (Ridanpaa et al., 2001). The etiology of CHH was unknown until the late 1990s, when a candidate genomic region was mapped to chromosome 9 using linkage analysis on genomic data from affected Finnish families (Sulisalo et al., 1993). This region was further narrowed down to Chr9p13 using both linkage and physical mapping techniques (Sulisalo et al., 1994; Vakkilainen et al., 1999). The causative gene remained elusive until 2001, when an insertion in RMRP was discovered in a patient suffering from CHH. The RMRP gene encodes an untranslated RNA that acts as the RNA component of the ribonuclease RNase MRP (Ridanpaa et al., 2001).
Ribonuclease mitochondrial RNA processing (RNase MRP) was first identified in 1987 as a ribonuclease responsible for the cleavage of mitochondrial RNA transcribed during replication in mice and humans. It was subsequently discovered that RNase MRP has an RNA component encoded in the nucleus that is required for activity (Chang and Clayton, 1987a; Chang and Clayton, 1987b). Despite having mitochondrial processing functions, RNase MRP was predominantly localized to the nucleolus, suggesting an alternative role outside of mitochondria (Yuan et al., 1989; Topper and Clayton, 1990; Karwan et al., 1991; Kiss et al., 1992; Schmitt and Clayton, 1992). Due to its known processing roles, it warranted further investigation as an rRNA processing factor. RMRP is also referred to as 7-2 RNA and NME1 (Kiss et al., 1992; Schmitt and Clayton, 1992). Interestingly, RNase MRP has a conserved secondary structure similar to that of RNase P, which plays a role in tRNA processing. Taken together, its nucleolar localization and RNA cleavage activity suggested that RNase MRP was acting as an rRNA processing enzyme (López et al., 2009).
This supposition was strengthened by extensive studies in S. cerevisiae. Two temperature-sensitive (ts) mutants, coined rrp2-1 and rrp2-2, were found to display impaired rRNA processing (Shuai and Warner, 1991; Lindahl et al., 1992). Northern blotting studies revealed an abnormal processing of yeast 35S rRNA. Further characterization of ts-mutants revealed misprocessing of the 5.8S rRNA, with the major effect being a non-canonical product that was extended by 149 nucleotides. They also determined there was an alteration to the stoichiometry between 5.8SL and 5.8SS forms. (Lindahl et al., 1992). Two years later, the NME1 gene was shown to complement the rrp2-2 mutants. By deleting portions of the NME1 gene at various lengths (14, 22, and 65 nucleotides), it was found that longer deletions prevent the ability of NME1 to complement rrp2-2 mutations. These data provided evidence that rrp2-2 mutants were actually NME1 mutants (Chu et al., 1994). Studies in S. cerevisiae revealed that NME1, the gene homologous to human RMRP, was required for viability (Schmitt and Clayton, 1992). Schmitt and Clayton (1993) later confirmed that RNase MRP/NME1 in the nucleus is required for appropriate processing of yeast 5.8S rRNA. The complete processing activity of RNase MRP in S. cerevisiae was proposed shortly after these studies. In vitro experiments revealed that yeast RNase MRP-mediated processing of pre-rRNA is achieved through cleavage at the A3 site, which is located in ITS1 (Henry et al., 1994; Lygerou et al., 1996). It was also shown that mutations in POP1, a protein component of the RNase MRP RNP, cause defects in pre-rRNA processing similar to RNase MRP mutants, as well inhibiting A3 cleavage (Lygerou et al., 1994).
It took significantly longer to clarify the role of RMRP in humans and the connection to CHH, but this has now been validated to direct the analogous cleavage event at site 2 in ITS1 of the human pre-rRNA. Earlier studies initially shed doubt on whether RMRP served a similar role as yeast NME1. RNAi-depletion of POP1, RPP38, or RPP40 protein cofactors of RNase MRP and RNase P did not show alterations in rRNA processing (Sloan et al., 2013). However, the Cech group later concluded that siRNA or antisense oligonucleotides were not sufficient to disrupt RMRP and used CRISPR-Cas9 to edit RMRP (Goldfarb and Cech, 2017). In agreement with yeast studies, complete ablation of RMRP is lethal. An increase in rRNA precursors, specifically on the 5′-end of the 5.8S, was observed in HeLa clones that did accumulate RMRP mutations.
In CHH, two main classes of mutation have been described; the first consists of either insertions or duplications that occur between the promoter and the transcription initiation site. It is thought that these mutations prevent the effective transcription of RMRP. The second category consists of single-nucleotide substitutions that are generally found in conserved regions of RMRP. These mutations, primarily A70G and G262T, are thought to have functional consequences (Ridanpaa et al., 2001). The authors also found that all patients with mutations in RMRP express RNase MRP to some degree, but patients who are heterozygous for 5′-end insertions have one allele silenced, resulting in lower expression of RNase MRP RNA; this indicates that the 5′-insertion mutations do, in fact, suppress transcription. Co-immunoprecipitation studies show that proteinaceous subunits still interact with mutant RMRP (Ridanpaa et al., 2001). In contrast with yeast rrp2-2 mutants, CHH patients do not present with the 149 nt extended 5.8S. Stabilization of such an rRNA may be inviable outside of the context of ts-mutants.
Recent studies have described A70G mutations resulting in a decreased abundance of cytosolic ribosomes, as well as a reduction in rRNA on a per-cell basis (Robertson et al., 2022). While previous studies found abnormalities in pre-rRNA processing, effects on ribosomal biogenesis of abundance were not previously described. Robertson et al. (2022) has cemented CHH as a bone fide ribosomopathy. A previously uncharacterized mutation in the gene NEPRO was recently described as causing a CHH-like phenotype. While NEPRO is thought to interact with RMRP components, these studies are still in their infancy and warrant further investigation (Narayanan et al., 2019; Remmelzwaal et al., 2023).
CHH has been primarily investigated on the level of pre-rRNA processing, although only recently has it been studied at the level of ribosome abundance and biogenesis. It would be interesting to explore with new biochemical tools whether these ribosomes are translation competent and, if so, what their translation capacity and efficiency is. While the pre-rRNA processing role in CHH has been well established, understanding downstream effects on translation would be a significant continuation of research.
North American Indian childhood cirrhosis (NAIC) is a familial disease displaying an autosomal recessive inheritance that specifically affects the Oji-Cree people of Northwestern Quebec (Betard et al., 2000). The disease is characterized by neonatal intrahepatic cholestasis, jaundice, and progressive liver damage (Weber et al., 1981).
Using genome-wide scanning techniques and linkage analysis on samples from affected and unaffected individuals, the locus responsible for this condition was mapped to chromosome 16q22 (Betard et al., 2000). Just 2 years after identifying the locus, the gene implicated in NAIC was found, which encoded an uncharacterized protein referred to as FLJ14728—now called Cirhin/CIRH1A. This protein is homologous to yeast UTP4, which is a component of the SSU processome. Homozygous individuals who had the R565W mutation (c.1741C>T), located in the C-terminal end of the protein, were identified. Conservation of the Cirhin gene has also been shown across vertebrates and specifically the presence of a dibasic residue in the 565 position (Chagnon et al., 2002).
As Cirhin was previously uncharacterized, its normal function was largely unknown. It was found that both wildtype and R565W mutant Cirhin localize to the nucleolus. Interestingly, the nucleolus is not generally implicated in diseases of intrahepatic cholestasis, suggesting a potential function for Cirhin in the nucleolus (Yu et al., 2005). In S. cerevisiae, U3 snoRNA and associated proteins (t-Utps) are required for processing of 18S pre-rRNA. In the absence of t-Utps, production of mature 18S rRNA is abrogated (Dragon et al., 2002). While the function of t-Utps has been well established in pre-18S rRNA processing, it has also been characterized as linking its ribosomal RNA processing function with the transcription of rDNA (Gallagher et al., 2004). Through TBLASTN searches, potential human orthologs of several yeast t-Utps were identified: hUTP4, hUTP5, hUTP10, hUTP15, and hUTP17, with Cirhin being hUTP4. These UTPs comprise the SSU processome (Prieto and McStay, 2007). Given that CIRHIN/Utp4 are part of the SSU processome, this may indicate potential processing defects of rRNA in the small ribosomal subunit.
To further elucidate the functions of CIRHIN and its role in the pathogenesis of NAIC, yeast two-hybrid (Y2H) screens and mass spectrometry (MS) were performed to identify potential binding partners for CIRHIN. NOL11, another member of the human t-Utp complex, was found to be a binding partner with CIRHIN. To further parse this, co-immunoprecipitation revealed that NOL11 is part of the SSU processome based on its co-immunoprecipitation with FBL. Interestingly, NAIC mutations (R565W) in CIRHIN ablate interaction with NOL11 (Freed et al., 2012). In addition to association with CIRHIN, siRNA knockdown of NOL11 indicates that it is required to generate mature 18S rRNA (Freed et al., 2012). While it has been shown that hUTP4 with the R565W mutation can localize to the nucleolus normally (Yu et al., 2005), it is reported that CIRHIN interaction with NOL11 may be required for appropriate localization, as C-terminal truncations of CIRHIN prevent nucleolar localization (Freed et al., 2012). While the R565W mutation in hUtp4 appears to interfere with NOL11 interaction, it is curious that nucleolar localization still occurs despite the potential weakening of this interaction. While biochemical data are beginning to reveal the function of this protein, the precise effect on rRNA production and ribosome activity in NAIC awaits clarification.
ERI1 (3′-hExo) is a multifunctional member of the DEDDh family of exoribonucleases that possesses 3′- to 5′-exonuclease activity. Amongst other activities, it conducts the final processing reaction to generate the mature 3′-end of the 5.8S rRNA. Originally identified via genetic screening of C. elegans mutants with a sensitivity to dsRNA, the ERI1 gene was identified as a potentially important regulator of siRNA. It was found that mutations in eri-1 result in an accumulation of siRNAs, potentially pointing toward regulatory role for ERI1 in siRNA levels. In fact, ERI1 acts as a 3′- to 5′-nuclease and can degrade siRNAs at the 3′-end (Kennedy et al., 2004). This nuclease function against siRNAs is found to be conserved in humans and the fission yeast S. pombe (Kennedy et al., 2004; Iida et al., 2006). In addition to negatively regulating siRNAs, ERI1 has also been shown to degrade the 3′-end of histone mRNAs, as well as 3′-end processing of the 5.8S rRNA (Gabel and Ruvkun, 2008). Despite its low sequence homology, structural studies using crystallography and characterization of its nuclease activity confirm its identity as a DEDDh nuclease (Dominski et al., 2003; Yang et al., 2006).
Given the characterized function of ERI1 as a 3′-end processing enzyme of the 5.8S rRNA in C. elegans, (Gabel and Ruvkun, 2008), it appears that this function is conserved to mouse ERI1, as ERI1-deficient mice exhibit a longer 5.8S rRNA and can be rescued by reintroducing wild-type Eri1. Biochemical in vitro processing studies indicate that recombinant ERI1 can catalyze the final 3′-end processing required for the appropriate formation of the 3′-end of 5.8S. ERI1 performs its 3′-end processing activities in a duplex formed by a 5.8S and 28S rRNA molecule, and subsequently chewing back unpaired nucleotides at the 3′-end of the 5.8S rRNA (Ansel et al., 2008). In addition to carrying out processing activities of the 5.8S, ERI1 also appears to associate with ribosomal proteins and co-sediments with the 40S, 60S, and 80S peaks, but is depleted from actively translating polysomes. While the rRNA processing function has been characterized, the significance of sustained association with the ribosome after processing is unclear (Ansel et al., 2008).
Aside from rRNA processing functions, ERI1 is also necessary for the efficient decay of histone mRNAs at the end of the S-phase (Dominski et al., 2003). Histone mRNAs are the only cellular mRNAs that are not polyadenylated, instead ending in a conserved stem-loop. At the end of the S-phase, when bulk histone production is no longer necessary, they are rapidly degraded. ERI1 binds to the histone stem-loop and participates in the early stages of degradation.
In recent years, an emerging set of data implicates ERI1 in several disease types. Skeletal and cardiac malformations and intellectual disability were reported in an individual with a homozygous deletion of ERI1, MFHAS1, and MIR4660 (miR-4660). While ERI1 deletion likely plays a role in this pathology, it cannot solely be attributed to it (Choucair et al., 2017). Another patient was also identified recently as having a homozygous nonsense mutation (K188Stop) (Hoxha and Aliu, 2023). Similarly, this patient presented with limb abnormalities and intellectual disabilities, strengthening the case that ERI1 loss was the disease-causing mutation in the previous case. Finally, a recent study contrasts clinical manifestations in patients with either missense variants or bi-allelic null mutations in ERI1 (Guo et al., 2023). Patients with missense variants tend to have major skeletal defects known as severe spondyloepimetaphyseal dysplasia (SEMD), while those with biallelic null mutations suffer from much milder skeletal malformations and intellectual disabilities. Patients with ERI1 mutations exhibit defective processing of the 3′-end of 5.8S rRNA, as well as accumulating replication-dependent histone mRNAs (Guo et al., 2023). While these recent cases strongly implicate ERI1 in disease, the cause of these diseases is not known. Since ERI1 has many RNA targets, it is unclear whether these conditions arise from the misprocessing of rRNA, a defect in histone mRNA decay, or the stabilization of other uncharacterized RNAs. Future work is needed to determine the mechanistic causes of these disease states.
Production of mature rRNAs requires both efficient processing and elaborate chemical modifications. rRNAs are second only to tRNAs in the number and diversity of chemical modifications deposited on the RNA (Figure 3). Most of these modifications are 2′-O methylations (2′-Ome) and pseudouridylations (Ψ), although other base modifications, such as methylations and acetylations, are also present. While these modifications can be found throughout an rRNA, they are clustered around active sites on the ribosome, such as the decoding center and the peptidyl transferase center. The majority of the functional experimental data are derived from yeast and prokaryotic work, but data from various human disease states strongly suggest that these results also hold true for humans. It has been shown for 2′-Ome and Ψ modifications that loss of one or two modifications often has little impact on ribosome structure or function. However, defects begin to emerge as increasing modifications are lost. A seminal study from yeast analyzed modifications found in helix 69 of the 25S rRNA (Liang et al., 2007); helix 69 interacts with helix 44 of the 18S rRNA to form an inter-subunit bridge between the 40S and 60S ribosome and is well conserved from prokaryotes to humans (Yusupov et al., 2001). This helix contains several modifications over an 11-nucleotide stretch (AmCΨAΨGACCΨCΨ). Preventing the deposition of the modifications in this region has a severe effect on protein synthesis and growth (Liang et al., 2007). Yeast ribosomes lacking these modifications have a loss of translational fidelity, a decreased rate of protein synthesis, an increased rate of turnover, and a decreased rate of rRNA processing. Data from prokaryotes demonstrate that the Ψ modifications are important for the proper structure of helix 69 (Jiang et al., 2014). Other work analyzed 18S rRNA modifications present within the decoding center and found similar perturbations to ribosome function and maturation (Liang et al., 2009). More recent data analyzing only 2′-Ome and Ψ modifications highlight the global role of these modifications in accurate translation and in mediating proper ribosome dynamics (Khoshnevis et al., 2022; Zhao et al., 2023).
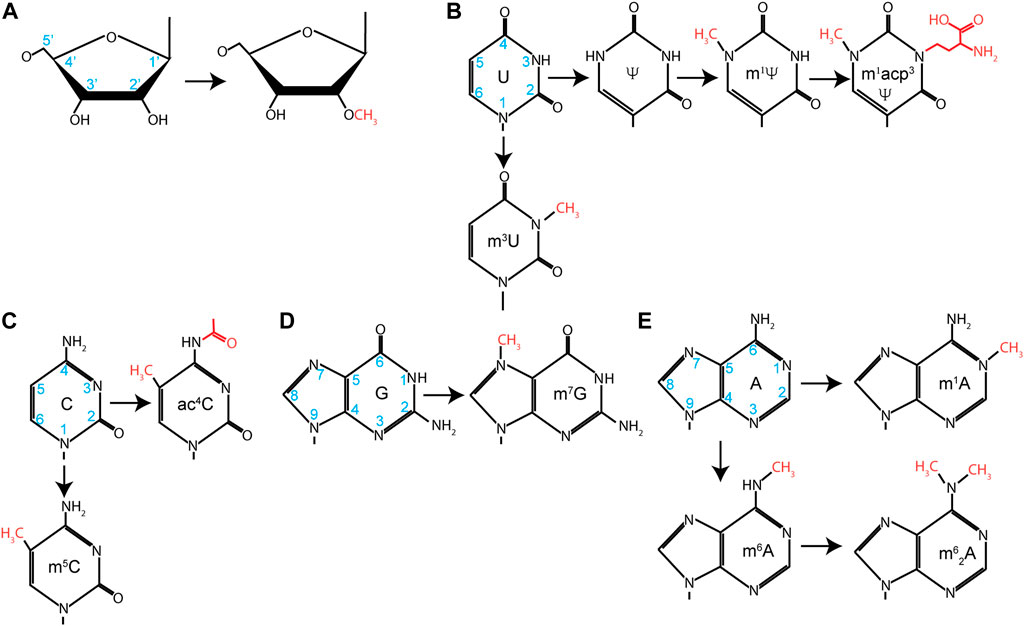
FIGURE 3. Nucleotide modifications found in rRNA. Chemical modification found on (A) ribose, (B) uridine, (C) cytosine, (D) guanosine, or (E) adenosine. Carbon number is noted on unmodified nucleotides in blue, and additional groups added during modification are identified in red.
Functionally, we are still gaining new knowledge about how these modifications regulate translation, although their chemical role is broadly clear. The 2′-OH groups of the rRNA are hydrophilic, which encourages preferential orientation away from the more hydrophobic core of the ribosome. Methylation of this group increases the hydrophobicity of the nucleotides, allowing them to be buried within the ribosome, and stabilizes RNA helices. Isomerization of uridine by 180° rotation around C6 converts U to Ψ. This generates a carbon–carbon glycosidic bond and frees the N1 to take part in additional hydrogen bonding interactions, further stabilizing the ribosome structure.
rRNA modifications are a significant source of ribosome heterogeneity. This is a hypothesis that not all ribosomes are identical and that the cellular pool of ribosomes is more heterogeneous than previously thought. In recent years, this hypothesis has gained widespread acceptance (Xue and Barna, 2012; Genuth and Barna, 2018). Further speculation has proposed that some ribosomes may have specialized activities in cells by actively translating distinct subsets of mRNAs. The existence of such specialized ribosomes is still a matter of debate. While some studies strongly suggest the existence of specialized ribosomes, the field awaits definitive proof (Ferretti and Karbstein, 2019). Much of these data have focused on alterations of the protein complement of ribosomes, either through differences in core r-proteins or through association with different ribosome-associated factors, although alterations in rRNA modifications have now been seen in various organisms. Ribo-meth seq revealed that several sites of 2′-O methylation are sub-stoichiometric and that the stoichiometry of modification changes depending upon cell type, leading to the proposal that it may provide the basis for differential regulation through methylation (Birkedal et al., 2015; Krogh et al., 2016). This has been given further credence by a quantitative mass-spectrometry approach showing that other modifications, such as Ψ and base methylations (e.g., m1A), are also sub-stoichiometric at some positions (Taoka et al., 2018). Furthermore, there are developmentally regulated changes in rRNA methylation in mice (Hebras et al., 2020). Recent work has even shown that these developmental changes are important for proper cellular differentiation in mouse embryos (Hafner et al., 2023). Loss of U3904 2′-O methylation on the 28S rRNA drives mouse embryonic stem cells toward neuroectodermal cell fate.
In metazoans, 2′-Ome and Ψ modification are catalyzed by small nucleolar ribonucleoproteins (snoRNPs) (Ojha et al., 2020). SnoRNPs fall into two classes based on their structure, associated proteins, and which chemical modifications they catalyze: Box C/D snoRNPs and Box H/ACA snoRNPs. Box C/D snoRNPs catalyze 2′-Ome, while Box H/ACA catalyzes Ψ deposition. SnoRNPs are assemblages of catalytic protein subunits and snoRNA subunits that guide chemical modification through Watson–Crick base-pairing. Fibrillarin (FBL) assembles onto Box C/D snoRNAs along with SNU13, NOP58, and NOP56. FBL is a methyltransferase that uses S-adenosylmethionine as a donor to deposit methyl groups. Alternatively, dyskerin (DKC), a pseudouridine synthetase, assembles on Box H/ACA snoRNAs with NHP2, NOP10, and GAR1 to form the active Box H/ACA snoRNP. The assembly of these snoRNPs and their functional regulation have undergone intense study, initially in archaea and yeast systems and eventually in mammals (Ojha et al., 2020). However, new discoveries are still being made. N4-acetylation of C1773 (ac4C1773) is required for ribosome biogenesis in yeast and is deposited by KRE33 (Ito et al., 2014a). In humans, NAT10 was identified as the acetyltransferase required for this event at C1337 and C1842 of the 18S (Ito et al., 2014b; Sharma et al., 2015). Surprisingly, the U13 snoRNP was shown to guide this modification. The U13 snoRNA is a Box C/D snoRNA, but it has not yet been shown to guide 2′-O methylation of any other nucleotide. Thus, it is likely that this snoRNA has evolved to direct the acetylation rather than the methylation of rRNA.
This highlights the fact that, while the majority of modifications are 2′-Ome and Ψ, others, including ac4C, are present. Simple base modifications such as 7-methylguanosine (m7G) and 1-methyladenosine (m1A) are typically carried out by standalone methyltransferases. More elaborate modifications, such as 1-methyl-3-(3-amino-3-caboxypropyl) pseudouridine (m1acp3Ψ), take the concerted efforts of several modification enzymes. In the case of m1acp3Ψ, uridine is first isomerized to Ψ, at which point a methyl group is added to the N1 position by EMG1 (Wurm et al., 2010; Meyer et al., 2011). Finally, a 3-amino-3-carboxypropyl moiety is added by TSR3 (Meyer et al., 2016). Recent evidence points to this modification as being important in the development of cancer in addition to previous evidence highlighting this modification in Bowen–Conradi syndrome (see below).
X-linked dyskeratosis congenita (XDC), also known as Zinsser–Cole–Engman syndrome, is a rare multisystem disease presenting clinical features such as reticulate skin hyperpigmentation, nail dystrophy, bone marrow failure, oral leukoplakia, and an increased risk of cancer (Savage et al., 1993). The original name for the syndrome suggests an X-linked pattern of disease inheritance, although a haplo-insufficient autosomal dominant and autosomal recessive mode of transmittance has since been demonstrated. A second disorder, Hoyeraal–Hreidarsson (HH) syndrome, is considered a severe form of XDC that is associated with cerebellar hypoplasia, intrauterine growth retardation (IUGR), and immunodeficiency (Hoyeraal et al., 1970; Aalfs et al., 1995).
Earlier genetic analysis linked the causative gene for XDC to Xp28 (Connor et al., 1986). Later analysis determined that the vast majority of XDC and HH patients have mutations in the DKC gene (Heiss et al., 1998; Knight et al., 1999). DKC is the catalytic subunit of Box H/ACA snoRNP that catalyzes the isomerization of uridine to pseudouridine in rRNA and is the human homolog of yeast S. cerevisiae Cbf5 (Meier and Blobel, 1994; Cadwell et al., 1997). However, mutations in other Box H/ACA-associated factors—NOP10 and NHP2—have been shown to cause disease with similar clinical presentations (Walne et al., 2007; Vulliamy et al., 2008; Benyelles et al., 2020).
Despite the clear role of DKC in ribosome biogenesis, the cause of symptoms in XDC patients has been a matter of debate due to the assembly of DKC in the non-canonical snoRNP telomerase RNA component (TERC). Importantly, TERC is necessary for the maintenance of telomeres (Heiss et al., 1998); patients with XDC also have defects in telomere maintenance (Mitchell et al., 1999). However, XDC patients also have a marked decrease in rRNA pseudouridylation and defects in translation. Model system data have reported conflicting data, with mouse models recapitulating disease while primarily altering ribosome dynamics with no immediate loss of telomere length (Ruggero et al., 2003); however, work on iPSCs demonstrates the opposite (Gu et al., 2015). Furthermore, mutations in the RNA component of telomerase (hTR) cause disease similar to XDC (Vulliamy et al., 2001), which would implicate telomerase maintenance as the major culprit in XDC etiology. Mutations in nucleophosmin, another ribosome biogenesis factor, perturb 2′-Ome rather than Ψ and cause XDC (Nachmani et al., 2019). These data support the model in which alterations to rRNA modifications are the major driver of disease. Recent data have also implicated mutation of DKC and NOP10 in nephrotic syndrome (Balogh et al., 2020). Like XDC, these mutations result in a defect in telomere maintenance and in ribosome activity. However, the clinical presentations are distinct, further highlighting the pleiotropic role of DKC mutations. Thus, it is likely that the clinical symptoms arise from an additive effect of telomere and translational dysfunction.
The global reduction of Ψ in rRNA, as seen in DKC, is intrinsically linked with translational dysfunction as are site-specific losses of Ψ (Liang et al., 2007; Penzo et al., 2015).
A mouse model of XDC demonstrates diverse defects in ribosome function, demonstrating a loss of Ψ deposition and a reduced rate of rRNA maturation (Ruggero et al., 2003). This same model system reveals a defect in the translation of p27, BCL-xL, and XIAP mRNAs and a concomitant decrease in p27, BCL-xL, and XIAP protein levels in DKC-mutant cells (Yoon et al., 2006; Bellodi et al., 2010a). Further work also implicated alterations in p53 mRNA translation as having a role in XDC (Bellodi et al., 2010b). In each case, the defect in translating these mRNAs appears connected with non-canonical modes of translation initiation. In particular, these mRNAs have been proposed to contain internal ribosome entry sites (IRES) that may recruit the ribosome to each mRNA independently of a 5′-cap. While the concept of IRES is well established in viral mRNAs, the existence of cellular IRES remains controversial (Jackson, 2013; Akirtava et al., 2022). However, defects in the synthesis of these mRNAs also explain the predisposition toward cancer observed in XDC patients.
Williams–Beuren syndrome (WBS) is a rare condition that affects approximately 1 in 20,000 live births and is characterized genetically by a of 1.5–1.8 Mb deletion at chromosomal position 7q11.23 (Pober, 2010). This deletion results in the loss of 28 individual genes. Patients with WBS have mild to moderate neurocognitive defects, hypersensitivity to sounds, and difficulty with visual–spatial abilities in addition to connective tissue abnormalities and premature skin aging. Because of the many genes deleted in WBS, disease symptoms are likely an accumulated effect of many insufficiencies. However, one of the 28 deleted genes encodes Nop2/Sun RNA methyltransferase 5 (NSUN5). This is a member of a family of methyltransferases that deposits methyl groups on specific RNAs, including tRNAs and rRNAs. Defects in these proteins have been associated with other neurological disorders, including WBS (Blanco et al., 2014; Paramasivam et al., 2020; Mattioli et al., 2023; PerezGrovas-Saltijeral et al., 2023).
NSUN5 is responsible for m5C3782 modification of the 28S rRNA and is the homolog of yeast Rcm1, which similarly modified m5C2278 in the yeast 25S rRNA (Sharma et al., 2013; Heissenberger et al., 2019). Loss of Rcm1 in yeast induces structural changes to the ribosome which results in a defect in translational fidelity (Schosserer et al., 2015), but also confers increased lifespan and expression of stress-responsive genes. This seeming paradox between disease state and presumed beneficial gene expression is also seen in cancer at this site (see below). Mouse knockout studies have also shown that this gene is necessary for proper cerebral cortex development, which may link the cognitive defects in WBS to the loss of this gene (Chen et al., 2019; Zhang et al., 2019). In contrast to studies in yeast, CRISPR/Cas9-mediated knockout of NSUN5 in HeLa cells decreased rates of global translation but did not show defects in translational fidelity (Heissenberger et al., 2019). Importantly, this same study formally demonstrated that the loss of one copy in WBS patients was sufficient to reduce m5C3782 levels on patient 28S rRNAs. It is currently unclear how much of the symptoms present in WBS are directly related to the haploinsufficiency of NSUN5 or a reduction of m5C3782. However, given the defects in model systems, it is worthy of further investigation and highlights the role of a single nucleotide modification in ribosome function.
Bowen–Conradi syndrome (BCS) is a rare lethal autosomal recessive disorder that was first described in a pair of brothers from a Hutterite family in 1976 (Bowen and Conradi, 1976). Despite its rarity in the larger population, its incidence in Hutterite communities is greater than 1 in every 400 live births. Patients present with low birth weight, microcephaly, and anatomical malformations. Failure to thrive typically results in death within the first year of life (Hunter et al., 1979). Lineage and linkage analysis isolated the gene to a 1.9 Mbp region of chromosome 12p13.3 (Lamont et al., 2005). This region contained 59 genes. Of these, 35 were sequenced and revealed a single nucleotide change (NM_006331.6:c.400A>G) in the EMG1 gene as segregating with disease (Armistead et al., 2009). This mutation results in a D86G mutation in the EMG1 protein.
EMG1 is the human homolog of yeast Nep1, which had previously been shown to be a methyltransferase that acted upon the 18S rRNA (Eschrich et al., 2002; Buchhaupt et al., 2006). Crystallographic analysis of archaeal Nep1 had shown that D86 interacted with R84, which is essential for RNA binding (Taylor et al., 2008). Further studies of archaeal Nep1 and human EMG1 revealed that it is the N1-pseudouridine methyltransferase that aids in the conversion of U1248 to m1acp3Ψ1248 in the human 18S rRNA. It is also important to note that N1-methylation is a prerequisite for 3-amino-3-carboxypropyl modification at this location. Later, the yeast homolog of EMG1 was identified and validated to be a pseudouridine synthetase acting upon a homologous site in the yeast 18S rRNA (Meyer et al., 2011). Surprisingly, this study revealed that mutation of this aspartic acid residue did not diminish methyltransferase activity. These data strongly suggest that BCS is not a defect in rRNA modification but that EMG1 plays additional roles in the cell. Importantly, this is coupled with the initial study demonstrating that D86G results in EMG1 aggregation and a dramatic decrease in EMG1 protein in patients (Armistead et al., 2009).
Indeed, further analysis revealed that EMG1 also plays a key structural role in ribosome biogenesis. Patient samples displayed a decreased rate in 18S rRNA synthesis, although, basal levels of protein synthesis are unaffected (Armistead et al., 2014). The major phenotype thus appears to be a defect in ribosome assembly rather than in ribosome function. Indeed, siRNA-mediated depletion of EMG1 results in an accumulation of 30S precursors and a relative depletion in 18S-E precursors (Warda et al., 2016). Therefore, it is likely that BCS arises from a failure to properly form the SSU processome and efficiently generate 18S rRNA, despite the relevant mutation being found in an enzyme required for chemical modification of the rRNA.
The burgeoning pool of data consistently highlights significant variation in rRNA modification, particularly 2′-Ome and Ψ, during oncogenesis. Nevertheless, the prevailing trend is toward correlative connections rather than establishing clear causative effects in the majority of these instances. Misregulation of either FBL or DKC is commonly seen in cancers (Montanaro et al., 2006; Marcel et al., 2013). Unsurprisingly, this correlates with alterations in the Ψ and 2′-Ome profiles of rRNAs (Marcel et al., 2020; Barozzi et al., 2023; Zhou et al., 2023). The modulation of snoRNA expression has long been seen as a biomarker of tumorigenesis (Williams and Farzaneh, 2012; Krishnan et al., 2016; Gong et al., 2017; Barros-Silva et al., 2021). In various tumor types, decreases or increases in specific snoRNAs are common, but specific effects from gain or loss of modification guided by these snoRNAs have been more difficult to validate. Some data suggest that specific snoRNAs and the modifications they guide may promote tumor development. A form of acute myeloid leukemia (AML) undergoes genetic alterations that culminate in the enhanced formation of snoRNPs (Zhou et al., 2017). In particular, SNORD14D and SNORD35A have been shown in vivo and in vitro to be necessary for AML cells’ oncogenic properties. These two snoRNAs direct the 2′-Ome of C1708 in the 18S rRNA and G1328 in the 28S, respectively.
In contrast to Ψ and 2′-Ome modifications, there appears to be a stronger causative link between oncogenesis and base methylations. METTL5 is a methyltransferase that installs m6A modifications at A1832 of the 18S rRNA (m6A1832) (van Tran et al., 2019). Studies using mouse genetic models implicated METTL5 and m6A1832 in transcript-specific translation regulation, suggesting that METTL5 misregulation could be important in reshaping the translatome in specific disease contexts (Sepich-Poore et al., 2022). Indeed, METTL5 was shown to be upregulated in breast cancer cell lines, with its loss reducing their growth rate in vitro (Rong et al., 2020). Research suggests that m6A1832 localization to the decoding center is important for efficient translation initiation. More recently, both METTL5 and m6A1832 were found to be elevated in nasopharyngeal carcinoma (NPC) and intrahepatic cholangiocarcinoma (ICC) (Chen et al., 2023; Dai et al., 2023). Similarly, loss of METTL5 has reduced tumorigenesis in vitro and in vivo for these cancers.
Surprisingly, not all alterations to the rRNA modification profile are negative. NSUN5, which is also implicated in WBS, was identified as being alternatively regulated in a subset of gliomas (Janin et al., 2019). This gene becomes epigenetically silenced but was shown to be associated with better prognosis in patients. This same work paradoxically suggested that loss of m5C3782 reprograms the ribosomes to better translate stress-responsive mRNAs, which aids in cancer cell survival in response to tumor-associated stresses. This seeming contradiction may be reconciled by the fact that NSUN5-depleted cells have lower rates of protein synthesis. Thus, while these cells can better survive stress, they are less prolific, leading to a better prognosis for patients.
One intriguing case regarding a potential causative link occurs, again, with the case of m1acp3Ψ. Analysis of colorectal carcinoma (CRC) cells revealed downregulation of m1acp3Ψ at 1,248 of the 18S rRNA (Babaian et al., 2020). Given its location in the P-site of the ribosome, the authors suggest that the loss of this site may be key to loss of translational control seen during oncogenesis. Indeed, loss of the acp3 moiety following TSR3 knockout revealed an increase in ribosomal protein synthesis, an important driver in cancer development. While more work is needed to confirm loss of m1acp3Ψ as an oncogenic driver, these data support the hypothesis that alterations to individual modifications can promote cancer development.
RJ: writing–original draft, writing–review and editing. AA: writing–original draft, writing–review and editing. SL: conceptualization, writing–original draft, writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by funding from the NIH to SML (GM146769).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aalfs, C. M., van den Berg, H., Barth, P. G., and Hennekam, R. C. (1995). The Hoyeraal-Hreidarsson syndrome: the fourth case of a separate entity with prenatal growth retardation, progressive pancytopenia and cerebellar hypoplasia. Eur. J. Pediatr. 154, 304–308. doi:10.1007/bf01957367
Akirtava, C., May, G. E., and McManus, C. J. (2022). False-positive IRESes from Hoxa9 and other genes resulting from errors in mammalian 5' UTR annotations. Proc. Natl. Acad. Sci. U. S. A. 119, e2122170119. doi:10.1073/pnas.2122170119
Allmang, C., Petfalski, E., Podtelejnikov, A., Mann, M., Tollervey, D., and Mitchell, P. (1999). The yeast exosome and human PM-Scl are related complexes of 3' right-arrow 5' exonucleases. Genes & Dev. 13, 2148–2158. doi:10.1101/gad.13.16.2148
Alupei, M. C., Maity, P., Esser, P. R., Krikki, I., Tuorto, F., Parlato, R., et al. (2018). Loss of proteostasis is a pathomechanism in cockayne syndrome. Cell Rep. 23, 1612–1619. doi:10.1016/j.celrep.2018.04.041
Amberg, D. C., Goldstein, A. L., and Cole, C. N. (1992). Isolation and characterization of RAT1: an essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev. 6, 1173–1189. doi:10.1101/gad.6.7.1173
Ameismeier, M., Zemp, I., van den Heuvel, J., Thoms, M., Berninghausen, O., Kutay, U., et al. (2020). Structural basis for the final steps of human 40S ribosome maturation. Nature 587, 683–687. doi:10.1038/s41586-020-2929-x
Anger, A. M., Armache, J. P., Berninghausen, O., Habeck, M., Subklewe, M., Wilson, D. N., et al. (2013). Structures of the human and Drosophila 80S ribosome. Nature 497, 80–85. doi:10.1038/nature12104
Ansel, K. M., Pastor, W. A., Rath, N., Lapan, A. D., Glasmacher, E., Wolf, C., et al. (2008). Mouse Eri1 interacts with the ribosome and catalyzes 5.8S rRNA processing. Nat. Struct. Mol. Biol. 15, 523–530. doi:10.1038/nsmb.1417
Arabi, A., Wu, S., Ridderstrale, K., Bierhoff, H., Shiue, C., Fatyol, K., et al. (2005). c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 7, 303–310. doi:10.1038/ncb1225
Arimbasseri, A. G., Rijal, K., and Maraia, R. J. (2013). Transcription termination by the eukaryotic RNA polymerase III. Biochimica Biophysica Acta (BBA) - Gene Regul. Mech. 1829, 318–330. doi:10.1016/j.bbagrm.2012.10.006
Armistead, J., Hemming, R., Patel, N., and Triggs-Raine, B. (2014). Mutation of EMG1 causing Bowen-Conradi syndrome results in reduced cell proliferation rates concomitant with G2/M arrest and 18S rRNA processing delay. BBA Clin. 1, 33–43. doi:10.1016/j.bbacli.2014.05.002
Armistead, J., Khatkar, S., Meyer, B., Mark, B. L., Patel, N., Coghlan, G., et al. (2009). Mutation of a gene essential for ribosome biogenesis, EMG1, causes Bowen-Conradi syndrome. Am. J. Hum. Genet. 84, 728–739. doi:10.1016/j.ajhg.2009.04.017
Assfalg, R., Lebedev, A., Gonzalez, O. G., Schelling, A., Koch, S., and Iben, S. (2012). TFIIH is an elongation factor of RNA polymerase I. Nucleic Acids Res. 40, 650–659. doi:10.1093/nar/gkr746
Babaian, A., Rothe, K., Girodat, D., Minia, I., Djondovic, S., Milek, M., et al. (2020). Loss of m1acp3Ψ ribosomal RNA modification is a major feature of cancer. Cell Rep. 31, 107611. doi:10.1016/j.celrep.2020.107611
Bachellerie, J. P., Michot, B., and Raynal, F. (1983). Recognition signals for mouse pre-rRNA processing. A potential role for U3 nucleolar RNA. Mol. Biol. Rep. 9, 79–86. doi:10.1007/bf00777477
Balogh, E., Chandler, J. C., Varga, M., Tahoun, M., Menyhard, D. K., Schay, G., et al. (2020). Pseudouridylation defect due to DKC1 and NOP10 mutations causes nephrotic syndrome with cataracts, hearing impairment, and enterocolitis. Proc. Natl. Acad. Sci. U. S. A. 117, 15137–15147. doi:10.1073/pnas.2002328117
Ban, N., Nissen, P., Hansen, J., Moore, P. B., and Steitz, T. A. (2000). The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 289, 905–920. doi:10.1126/science.289.5481.905
Barozzi, C., Zacchini, F., Corradini, A. G., Morara, M., Serra, M., De Sanctis, V., et al. (2023). Alterations of ribosomal RNA pseudouridylation in human breast cancer. Nar. Cancer 5, zcad026. doi:10.1093/narcan/zcad026
Barros-Silva, D., Klavert, J., Jenster, G., Jeronimo, C., Lafontaine, D. L. J., and Martens-Uzunova, E. S. (2021). The role of OncoSnoRNAs and ribosomal RNA 2'-O-methylation in cancer. RNA Biol. 18, 61–74. doi:10.1080/15476286.2021.1991167
Barta, A., Steiner, G., Brosius, J., Noller, H. F., and Kuechler, E. (1984). Identification of a site on 23S ribosomal RNA located at the peptidyl transferase center. Proc. Natl. Acad. Sci. U. S. A. 81, 3607–3611. doi:10.1073/pnas.81.12.3607
Beckmann, H., Chen, J. L., O'Brien, T., and Tjian, R. (1995). Coactivator and promoter-selective properties of RNA polymerase I TAFs. Science 270, 1506–1509. doi:10.1126/science.270.5241.1506
Bell, S. P., Learned, R. M., Jantzen, H. M., and Tjian, R. (1988). Functional cooperativity between transcription factors UBF1 and SL1 mediates human ribosomal RNA synthesis. Science 241, 1192–1197. doi:10.1126/science.3413483
Bellodi, C., Kopmar, N., and Ruggero, D. (2010b). Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J. 29, 1865–1876. doi:10.1038/emboj.2010.83
Bellodi, C., Krasnykh, O., Haynes, N., Theodoropoulou, M., Peng, G., Montanaro, L., et al. (2010a). Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res. 70, 6026–6035. doi:10.1158/0008-5472.can-09-4730
Ben-Shem, A., Garreau de Loubresse, N., Melnikov, S., Jenner, L., Yusupova, G., and Yusupov, M. (2011). The structure of the eukaryotic ribosome at 3.0 A resolution. Science 334, 1524–1529. doi:10.1126/science.1212642
Ben-Shem, A., Jenner, L., Yusupova, G., and Yusupov, M. (2010). Crystal structure of the eukaryotic ribosome. Science 330, 1203–1209. doi:10.1126/science.1194294
Benyelles, M., O'Donohue, M. F., Kermasson, L., Lainey, E., Borie, R., Lagresle-Peyrou, C., et al. (2020). NHP2 deficiency impairs rRNA biogenesis and causes pulmonary fibrosis and Hoyeraal-Hreidarsson syndrome. Hum. Mol. Genet. 29, 907–922. doi:10.1093/hmg/ddaa011
Betard, C., Rasquin-Weber, A., Brewer, C., Drouin, E., Clark, S., Verner, A., et al. (2000). Localization of a recessive gene for North American Indian childhood cirrhosis to chromosome region 16q22-and identification of a shared haplotype. Am. J. Hum. Genet. 67, 222–228. doi:10.1086/302993
Birkedal, U., Christensen-Dalsgaard, M., Krogh, N., Sabarinathan, R., Gorodkin, J., and Nielsen, H. (2015). Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. Int. Ed. 54, 451–455. doi:10.1002/anie.201408362
Blanco, S., Dietmann, S., Flores, J. V., Hussain, S., Kutter, C., Humphreys, P., et al. (2014). Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 33, 2020–2039. doi:10.15252/embj.201489282
Blattner, C., Jennebach, S., Herzog, F., Mayer, A., Cheung, A. C., Witte, G., et al. (2011). Molecular basis of Rrn3-regulated RNA polymerase I initiation and cell growth. Genes Dev. 25, 2093–2105. doi:10.1101/gad.17363311
Bleichert, F., Granneman, S., Osheim, Y. N., Beyer, A. L., and Baserga, S. J. (2006). The PINc domain protein Utp24, a putative nuclease, is required for the early cleavage steps in 18S rRNA maturation. Proc. Natl. Acad. Sci. U. S. A. 103, 9464–9469. doi:10.1073/pnas.0603673103
Bodem, J., Dobreva, G., Hoffmann-Rohrer, U., Iben, S., Zentgraf, H., Delius, H., et al. (2000). TIF-IA, the factor mediating growth-dependent control of ribosomal RNA synthesis, is the mammalian homolog of yeast Rrn3p. EMBO Rep. 1, 171–175. doi:10.1093/embo-reports/kvd032
Bowen, P., and Conradi, G. J. (1976). Syndrome of skeletal and genitourinary anomalies with unusual facies and failure to thrive in Hutterite sibs. Birth Defects Orig. Artic. Ser. 12, 101–108.
Bradsher, J., Auriol, J., de Santis, L. P., Iben, S., Vonesch, J. L., Grummt, I., et al. (2002). CSB is a component of RNA pol I transcription. Mol. Cell 10, 819–829. doi:10.1016/s1097-2765(02)00678-0
Brouwer, R., Allmang, C., Raijmakers, R., van Aarssen, Y., Egberts, W. V., Petfalski, E., et al. (2001). Three novel components of the human exosome. J. Biol. Chem. 276, 6177–6184. doi:10.1074/jbc.m007603200
Brun, R. P., Ryan, K., and Sollner-Webb, B. (1994). Factor C*, the specific initiation component of the mouse RNA polymerase I holoenzyme, is inactivated early in the transcription process. Mol. Cell. Biol. 14, 5010–5021. doi:10.1128/mcb.14.7.5010-5021.1994
Bruno, P. M., Lu, M., Dennis, K. A., Inam, H., Moore, C. J., Sheehe, J., et al. (2020). The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. U. S. A. 117, 4053–4060. doi:10.1073/pnas.1921649117
Buchhaupt, M., Meyer, B., Kotter, P., and Entian, K. D. (2006). Genetic evidence for 18S rRNA binding and an Rps19p assembly function of yeast nucleolar protein Nep1p. Mol. Genet. Genomics 276, 273–284. doi:10.1007/s00438-006-0132-x
Budkevich, T. V., Giesebrecht, J., Behrmann, E., Loerke, J., Ramrath, D. J., Mielke, T., et al. (2014). Regulation of the mammalian elongation cycle by subunit rolling: a eukaryotic-specific ribosome rearrangement. Cell 158, 121–131. doi:10.1016/j.cell.2014.04.044
Buttgereit, D., Pflugfelder, G., and Grummt, I. (1985). Growth-dependent regulation of rRNA synthesis is mediated by a transcription initiation factor (TTF-IA). Nucleic Acids Res. 13, 8165–8180. doi:10.1093/nar/13.22.8165
Caburet, S., Conti, C., Schurra, C., Lebofsky, R., Edelstein, S. J., and Bensimon, A. (2005). Human ribosomal RNA gene arrays display a broad range of palindromic structures. Genome Res. 15, 1079–1085. doi:10.1101/gr.3970105
Cadwell, C., Yoon, H. J., Zebarjadian, Y., and Carbon, J. (1997). The yeast nucleolar protein Cbf5p is involved in rRNA biosynthesis and interacts genetically with the RNA polymerase I transcription factor RRN3. Mol. Cell. Biol. 17, 6175–6183. doi:10.1128/mcb.17.10.6175
Calvet, J. P., and Pederson, T. (1981). Base-pairing interactions between small nuclear RNAs and nuclear RNA precursors as revealed by psoralen cross-linking in vivo. Cell 26, 363–370. doi:10.1016/0092-8674(81)90205-1
Carron, C., O'Donohue, M. F., Choesmel, V., Faubladier, M., and Gleizes, P. E. (2011). Analysis of two human pre-ribosomal factors, bystin and hTsr1, highlights differences in evolution of ribosome biogenesis between yeast and mammals. Nucleic Acids Res. 39, 280–291. doi:10.1093/nar/gkq734
Castle, C. D., Cassimere, E. K., and Denicourt, C. (2012). LAS1L interacts with the mammalian Rix1 complex to regulate ribosome biogenesis. Mol. Biol. Cell 23, 716–728. doi:10.1091/mbc.e11-06-0530
Castle, C. D., Cassimere, E. K., Lee, J., and Denicourt, C. (2010). Las1L is a nucleolar protein required for cell proliferation and ribosome biogenesis. Mol. Cell. Biol. 30, 4404–4414. doi:10.1128/mcb.00358-10
Chagnon, P., Michaud, J., Mitchell, G., Mercier, J., Marion, J. F., Drouin, E., et al. (2002). A missense mutation (R565W) in cirhin (FLJ14728) in North American Indian childhood cirrhosis. Am. J. Hum. Genet. 71, 1443–1449. doi:10.1086/344580
Chang, D. D., and Clayton, D. A. (1987a). A novel endoribonuclease cleaves at a priming site of mouse mitochondrial DNA replication. EMBO J. 6, 409–417. doi:10.1002/j.1460-2075.1987.tb04770.x
Chang, D. D., and Clayton, D. A. (1987b). A mammalian mitochondrial RNA processing activity contains nucleus-encoded RNA. Science 235, 1178–1184. doi:10.1126/science.2434997
Chen, B., Huang, Y., He, S., Yu, P., Wu, L., and Peng, H. (2023). N(6)-methyladenosine modification in 18S rRNA promotes tumorigenesis and chemoresistance via HSF4b/HSP90B1/mutant p53 axis. Cell Chem. Biol. 30, 144–158.e10. doi:10.1016/j.chembiol.2023.01.006
Chen, P., Zhang, T., Yuan, Z., Shen, B., and Chen, L. (2019). Expression of the RNA methyltransferase Nsun5 is essential for developing cerebral cortex. Mol. Brain 12, 74. doi:10.1186/s13041-019-0496-6
Choucair, N., Rajab, M., Megarbane, A., and Chouery, E. (2017). Homozygous microdeletion of the ERI1 and MFHAS1 genes in a patient with intellectual disability, limb abnormalities, and cardiac malformation. Am. J. Med. Genet. Part A 173, 1955–1960. doi:10.1002/ajmg.a.38271
Chu, S., Archer, R. H., Zengel, J. M., and Lindahl, L. (1994). The RNA of RNase MRP is required for normal processing of ribosomal RNA. Proc. Natl. Acad. Sci. U. S. A. 91, 659–663. doi:10.1073/pnas.91.2.659
Cochella, L., Brunelle, J. L., and Green, R. (2007). Mutational analysis reveals two independent molecular requirements during transfer RNA selection on the ribosome. Nat. Struct. Mol. Biol. 14, 30–36. doi:10.1038/nsmb1183
Comai, L., Tanese, N., and Tjian, R. (1992). The TATA-binding protein and associated factors are integral components of the RNA polymerase I transcription factor, SL1. Cell 68, 965–976. doi:10.1016/0092-8674(92)90039-f
Comai, L., Zomerdijk, J. C., Beckmann, H., Zhou, S., Admon, A., and Tjian, R. (1994). Reconstitution of transcription factor SL1: exclusive binding of TBP by SL1 or TFIID subunits. Science 266, 1966–1972. doi:10.1126/science.7801123
Connor, J. M., Gatherer, D., Gray, F. C., Pirrit, L. A., and Affara, N. A. (1986). Assignment of the gene for dyskeratosis congenita to Xq28. Hum. Genet. 72, 348–351. doi:10.1007/bf00290963
Correll, C. C., Munishkin, A., Chan, Y. L., Ren, Z., Wool, I. G., and Steitz, T. A. (1998). Crystal structure of the ribosomal RNA domain essential for binding elongation factors. Proc. Natl. Acad. Sci. U. S. A. 95, 13436–13441. doi:10.1073/pnas.95.23.13436
Coute, Y., Kindbeiter, K., Belin, S., Dieckmann, R., Duret, L., Bezin, L., et al. (2008). ISG20L2, a novel vertebrate nucleolar exoribonuclease involved in ribosome biogenesis. Mol. Cell. Proteomics 7, 546–559. doi:10.1074/mcp.m700510-mcp200
Crouch, R. J., Kanaya, S., and Earl, P. L. (1983). A model for the involvement of the small nucleolar RNA (U3) in processing eukaryotic ribosomal RNA. Mol. Biol. Rep. 9, 75–78. doi:10.1007/bf00777476
Dai, Z., Zhu, W., Hou, Y., Zhang, X., Ren, X., Lei, K., et al. (2023). METTL5-mediated 18S rRNA m(6)A modification promotes oncogenic mRNA translation and intrahepatic cholangiocarcinoma progression. Mol. Ther. 31, 3225–3242. doi:10.1016/j.ymthe.2023.09.014
Dammann, R., Lucchini, R., Koller, T., and Sogo, J. M. (1995). Transcription in the yeast rRNA gene locus: distribution of the active gene copies and chromatin structure of their flanking regulatory sequences. Mol. Cell. Biol. 15, 5294–5303. doi:10.1128/mcb.15.10.5294
Dauwerse, J. G., Dixon, J., Seland, S., Ruivenkamp, C. A., van Haeringen, A., Hoefsloot, L. H., et al. (2011). Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 43, 20–22. doi:10.1038/ng.724
Dixon, J., Edwards, S. J., Gladwin, A. J., Dixon, M. J., Loftus, S. K., Bonner, C. A., et al. (1996). Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat. Genet. 12, 130–136. doi:10.1038/ng0296-130
Dixon, J., Jones, N. C., Sandell, L. L., Jayasinghe, S. M., Crane, J., Rey, J. P., et al. (2006). Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. U. S. A. 103, 13403–13408. doi:10.1073/pnas.0603730103
Dominski, Z., Yang, X. C., Kaygun, H., Dadlez, M., and Marzluff, W. F. (2003). A 3' exonuclease that specifically interacts with the 3' end of histone mRNA. Mol. Cell 12, 295–305. doi:10.1016/s1097-2765(03)00278-8
Dragon, F., Gallagher, J. E., Compagnone-Post, P. A., Mitchell, B. M., Porwancher, K. A., Wehner, K. A., et al. (2002). A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417, 967–970. doi:10.1038/nature00769
Drygin, D., Siddiqui-Jain, A., O'Brien, S., Schwaebe, M., Lin, A., Bliesath, J., et al. (2009). Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 69, 7653–7661. doi:10.1158/0008-5472.can-09-1304
Duployez, N., Vasseur, L., Kim, R., Largeaud, L., Passet, M., L'Haridon, A., et al. (2023). UBTF tandem duplications define a distinct subtype of adult de novo acute myeloid leukemia. Leukemia 37, 1245–1253. doi:10.1038/s41375-023-01906-z
Emerson, C. P. (1971). Regulation of the synthesis and the stability of ribosomal RNA during contact inhibition of growth. Nat. New Biol. 232, 101–106. doi:10.1038/newbio232101a0
Erlacher, M. D., Lang, K., Shankaran, N., Wotzel, B., Huttenhofer, A., Micura, R., et al. (2005). Chemical engineering of the peptidyl transferase center reveals an important role of the 2'-hydroxyl group of A2451. Nucleic Acids Res. 33, 1618–1627. doi:10.1093/nar/gki308
Eschrich, D., Buchhaupt, M., Kotter, P., and Entian, K. D. (2002). Nep1p (Emg1p), a novel protein conserved in eukaryotes and archaea, is involved in ribosome biogenesis. Curr. Genet. 40, 326–338. doi:10.1007/s00294-001-0269-4
Fay, M. M., Lyons, S. M., and Ivanov, P. (2017). RNA G-quadruplexes in biology: principles and molecular mechanisms. J. Mol. Biol. 429, 2127–2147. doi:10.1016/j.jmb.2017.05.017
Ferretti, M. B., and Karbstein, K. (2019). Does functional specialization of ribosomes really exist? RNA 25, 521–538. doi:10.1261/rna.069823.118
Freed, E. F., Prieto, J. L., McCann, K. L., McStay, B., and Baserga, S. J. (2012). NOL11, implicated in the pathogenesis of North American Indian childhood cirrhosis, is required for pre-rRNA transcription and processing. PLoS Genet. 8, e1002892. doi:10.1371/journal.pgen.1002892
Friedrich, J. K., Panov, K. I., Cabart, P., Russell, J., and Zomerdijk, J. C. (2005). TBP-TAF complex SL1 directs RNA polymerase I pre-initiation complex formation and stabilizes upstream binding factor at the rDNA promoter. J. Biol. Chem. 280, 29551–29558. doi:10.1074/jbc.m501595200
Fromm, L., Falk, S., Flemming, D., Schuller, J. M., Thoms, M., Conti, E., et al. (2017). Reconstitution of the complete pathway of ITS2 processing at the pre-ribosome. Nat. Commun. 8, 1787. doi:10.1038/s41467-017-01786-9
Gabel, H. W., and Ruvkun, G. (2008). The exonuclease ERI-1 has a conserved dual role in 5.8S rRNA processing and RNAi. Nat. Struct. Mol. Biol. 15, 531–533. doi:10.1038/nsmb.1411
Gallagher, J. E., Dunbar, D. A., Granneman, S., Mitchell, B. M., Osheim, Y., Beyer, A. L., et al. (2004). RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev. 18, 2506–2517. doi:10.1101/gad.1226604
Gasse, L., Flemming, D., and Hurt, E. (2015). Coordinated ribosomal ITS2 RNA processing by the Las1 complex integrating endonuclease, polynucleotide kinase, and exonuclease activities. Mol. Cell 60, 808–815. doi:10.1016/j.molcel.2015.10.021
Gaubatz, J., Prashad, N., and Cutler, R. G. (1976). Ribosomal RNA gene dosage as a function of tissue and age for mouse and human. Biochimica Biophysica Acta (BBA) - Nucleic Acids Protein Synthesis 418, 358–375. doi:10.1016/0005-2787(76)90297-5
Geerlings, T. H., Vos, J. C., and Raue, H. A. (2000). The final step in the formation of 25S rRNA in Saccharomyces cerevisiae is performed by 5'-->3' exonucleases. RNA 6, 1698–1703. doi:10.1017/s1355838200001540
Genuth, N. R., and Barna, M. (2018). The discovery of ribosome heterogeneity and its implications for gene regulation and organismal life. Mol. Cell 71, 364–374. doi:10.1016/j.molcel.2018.07.018
Gerstberger, S., Meyer, C., Benjamin-Hong, S., Rodriguez, J., Briskin, D., Bognanni, C., et al. (2017). The conserved RNA exonuclease Rexo5 is required for 3' end maturation of 28S rRNA, 5S rRNA, and snoRNAs. Cell Rep. 21, 758–772. doi:10.1016/j.celrep.2017.09.067
Goldfarb, K. C., and Cech, T. R. (2017). Targeted CRISPR disruption reveals a role for RNase MRP RNA in human preribosomal RNA processing. Genes Dev. 31, 59–71. doi:10.1101/gad.286963.116
Gong, J., Li, Y., Liu, C. J., Xiang, Y., Li, C., Ye, Y., et al. (2017). A pan-cancer analysis of the expression and clinical relevance of small nucleolar RNAs in human cancer. Cell Rep. 21, 1968–1981. doi:10.1016/j.celrep.2017.10.070
Gonzales, B., Henning, D., So, R. B., Dixon, J., Dixon, M. J., and Valdez, B. C. (2005). The Treacher Collins syndrome (TCOF1) gene product is involved in pre-rRNA methylation. Hum. Mol. Genet. 14, 2035–2043. doi:10.1093/hmg/ddi208
Gorlin, R. J., Cohen, M. M., and Levin, L. S. (1990). Syndromes of the head and neck. New York: Oxford University Press.
Gorski, J. J., Pathak, S., Panov, K., Kasciukovic, T., Panova, T., Russell, J., et al. (2007). A novel TBP-associated factor of SL1 functions in RNA polymerase I transcription. EMBO J. 26, 1560–1568. doi:10.1038/sj.emboj.7601601
Grandori, C., Gomez-Roman, N., Felton-Edkins, Z. A., Ngouenet, C., Galloway, D. A., Eisenman, R. N., et al. (2005). c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 7, 311–318. doi:10.1038/ncb1224
Grewal, S. S., Li, L., Orian, A., Eisenman, R. N., and Edgar, B. A. (2005). Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat. Cell Biol. 7, 295–302. doi:10.1038/ncb1223
Grummt, I. (1982). Nucleotide sequence requirements for specific initiation of transcription by RNA polymerase I. Proc. Natl. Acad. Sci. U. S. A. 79, 6908–6911. doi:10.1073/pnas.79.22.6908
Grummt, I., Roth, E., and Paule, M. R. (1982). Ribosomal RNA transcription in vitro is species specific. Nature 296, 173–174. doi:10.1038/296173a0
Gu, B. W., Apicella, M., Mills, J., Fan, J. M., Reeves, D. A., French, D., et al. (2015). Impaired telomere maintenance and decreased canonical WNT signaling but normal ribosome biogenesis in induced pluripotent stem cells from X-linked dyskeratosis congenita patients. PLoS One 10, e0127414. doi:10.1371/journal.pone.0127414
Guo, L., Salian, S., Xue, J. Y., Rath, N., Rousseau, J., Kim, H., et al. (2023). Null and missense mutations of ERI1 cause a recessive phenotypic dichotomy in humans. Am. J. Hum. Genet. 110, 1068–1085. doi:10.1016/j.ajhg.2023.06.001
Hafner, S. J., Jansson, M. D., Altinel, K., Andersen, K. L., Abay-Norgaard, Z., Menard, P., et al. (2023). Ribosomal RNA 2'-O-methylation dynamics impact cell fate decisions. Dev. Cell 58, 1593–1609.e9. doi:10.1016/j.devcel.2023.06.007
Hallgren, J., Pietrzak, M., Rempala, G., Nelson, P. T., and Hetman, M. (2014). Neurodegeneration-associated instability of ribosomal DNA. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1842, 860–868. doi:10.1016/j.bbadis.2013.12.012
Hamada, H., Muramatsu, M., Urano, Y., Onishi, T., and Kominami, R. (1979). In vitro synthesis of a 5S RNA precursor by isolated nuclei of rat liver and HeLa cells. Cell 17, 163–173. doi:10.1016/0092-8674(79)90304-0
Hampl, H., Schulze, H., and Nierhaus, K. H. (1981). Ribosomal components from Escherichia coli 50 S subunits involved in the reconstitution of peptidyltransferase activity. J. Biol. Chem. 256, 2284–2288. doi:10.1016/s0021-9258(19)69775-9
Hayano, T., Yanagida, M., Yamauchi, Y., Shinkawa, T., Isobe, T., and Takahashi, N. (2003). Proteomic analysis of human nop56p-associated pre-ribosomal ribonucleoprotein complexes. J. Biol. Chem. 278, 34309–34319. doi:10.1074/jbc.m304304200
Hebras, J., Krogh, N., Marty, V., Nielsen, H., and Cavaille, J. (2020). Developmental changes of rRNA ribose methylations in the mouse. RNA Biol. 17, 150–164. doi:10.1080/15476286.2019.1670598
Heindl, K., and Martinez, J. (2010). Nol9 is a novel polynucleotide 5'-kinase involved in ribosomal RNA processing. EMBO J. 29, 4161–4171. doi:10.1038/emboj.2010.275
Heiss, N. S., Knight, S. W., Vulliamy, T. J., Klauck, S. M., Wiemann, S., Mason, P. J., et al. (1998). X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 19, 32–38. doi:10.1038/ng0598-32
Heissenberger, C., Liendl, L., Nagelreiter, F., Gonskikh, Y., Yang, G., Stelzer, E. M., et al. (2019). Loss of the ribosomal RNA methyltransferase NSUN5 impairs global protein synthesis and normal growth. Nucleic Acids Res. 47, 11807–11825. doi:10.1093/nar/gkz1043
Heix, J., Zomerdijk, J. C., Ravanpay, A., Tjian, R., and Grummt, I. (1997). Cloning of murine RNA polymerase I-specific TAF factors: conserved interactions between the subunits of the species-specific transcription initiation factor TIF-IB/SL1. Proc. Natl. Acad. Sci. U. S. A. 94, 1733–1738. doi:10.1073/pnas.94.5.1733
Henderson, A. S., Warburton, D., and Atwood, K. C. (1972). Location of ribosomal DNA in the human chromosome complement. Proc. Natl. Acad. Sci. U. S. A. 69, 3394–3398. doi:10.1073/pnas.69.11.3394
Henning, K. A., Li, L., Iyer, N., McDaniel, L. D., Reagan, M. S., Legerski, R., et al. (1995). The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 82, 555–564. doi:10.1016/0092-8674(95)90028-4
Henry, Y., Wood, H., Morrissey, J. P., Petfalski, E., Kearsey, S., and Tollervey, D. (1994). The 5' end of yeast 5.8S rRNA is generated by exonucleases from an upstream cleavage site. EMBO J. 13, 2452–2463. doi:10.1002/j.1460-2075.1994.tb06530.x
Hirschler-Laszkiewicz, I., Cavanaugh, A. H., Mirza, A., Lun, M., Hu, Q., Smink, T., et al. (2003). Rrn3 becomes inactivated in the process of ribosomal DNA transcription. J. Biol. Chem. 278, 18953–18959. doi:10.1074/jbc.m301093200
Hoareau-Aveilla, C., Fayet-Lebaron, E., Jady, B. E., Henras, A. K., and Kiss, T. (2012). Utp23p is required for dissociation of snR30 small nucleolar RNP from preribosomal particles. Nucleic Acids Res. 40, 3641–3652. doi:10.1093/nar/gkr1213
Holm, M., Natchiar, S. K., Rundlet, E. J., Myasnikov, A. G., Watson, Z. L., Altman, R. B., et al. (2023). mRNA decoding in human is kinetically and structurally distinct from bacteria. Nature 617, 200–207. doi:10.1038/s41586-023-05908-w
Hoppe, S., Bierhoff, H., Cado, I., Weber, A., Tiebe, M., Grummt, I., et al. (2009). AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc. Natl. Acad. Sci. U. S. A. 106, 17781–17786. doi:10.1073/pnas.0909873106
Hotz, M., Thayer, N. H., Hendrickson, D. G., Schinski, E. L., Xu, J., and Gottschling, D. E. (2022). rDNA array length is a major determinant of replicative lifespan in budding yeast. Proc. Natl. Acad. Sci. U. S. A. 119, e2119593119. doi:10.1073/pnas.2119593119
Hoxha, V., and Aliu, E. (2023). ERI1: a case report of an autosomal recessive syndrome associated with developmental delay and distal limb abnormalities. Am. J. Med. Genet. Part A 191, 64–69. doi:10.1002/ajmg.a.62987
Hoyeraal, H. M., Lamvik, J., and Moe, P. J. (1970). Congenital hypoplastic thrombocytopenia and cerebral malformations in two brothers. Acta Paediatr. 59, 185–191. doi:10.1111/j.1651-2227.1970.tb08986.x
Hunter, A. G., Woerner, S. J., Montalvo-Hicks, L. D., Fowlow, S. B., Haslam, R. H., Metcalf, P. J., et al. (1979). The Bowen-Conradi syndrome – a highly lethal autosomal recessive syndrome of microcephaly, micrognathia, low birth weight, and joint deformities. Am. J. Med. Genet. 3, 269–279. doi:10.1002/ajmg.1320030305
Iben, S., Tschochner, H., Bier, M., Hoogstraten, D., Hozak, P., Egly, J. M., et al. (2002). TFIIH plays an essential role in RNA polymerase I transcription. Cell 109, 297–306. doi:10.1016/s0092-8674(02)00729-8
Iida, T., Kawaguchi, R., and Nakayama, J. (2006). Conserved ribonuclease, Eri1, negatively regulates heterochromatin assembly in fission yeast. Curr. Biol. 16, 1459–1464. doi:10.1016/j.cub.2006.05.061
Isaac, C., Marsh, K. L., Paznekas, W. A., Dixon, J., Dixon, M. J., Jabs, E. W., et al. (2000). Characterization of the nucleolar gene product, treacle, in Treacher Collins syndrome. Mol. Biol. Cell 11, 3061–3071. doi:10.1091/mbc.11.9.3061
Ito, S., Akamatsu, Y., Noma, A., Kimura, S., Miyauchi, K., Ikeuchi, Y., et al. (2014a). A single acetylation of 18 S rRNA is essential for biogenesis of the small ribosomal subunit in Saccharomyces cerevisiae. J. Biol. Chem. 289, 26201–26212. doi:10.1074/jbc.m114.593996
Ito, S., Horikawa, S., Suzuki, T., Kawauchi, H., Tanaka, Y., Suzuki, T., et al. (2014b). Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J. Biol. Chem. 289, 35724–35730. doi:10.1074/jbc.c114.602698
Jackson, R. J. (2013). The current status of vertebrate cellular mRNA IRESs. Cold Spring Harb. Perspect. Biol. 5, a011569. doi:10.1101/cshperspect.a011569
Janin, M., Ortiz-Barahona, V., de Moura, M. C., Martinez-Cardus, A., Llinas-Arias, P., Soler, M., et al. (2019). Epigenetic loss of RNA-methyltransferase NSUN5 in glioma targets ribosomes to drive a stress adaptive translational program. Acta Neuropathol. 138, 1053–1074. doi:10.1007/s00401-019-02062-4
Jiang, J., Aduri, R., Chow, C. S., and SantaLucia, J. (2014). Structure modulation of helix 69 from Escherichia coli 23S ribosomal RNA by pseudouridylations. Nucleic Acids Res. 42, 3971–3981. doi:10.1093/nar/gkt1329
Johnson, L. K., Johnson, R. W., and Strehler, B. L. (1975). Cardiac hypertrophy, aging and changes in cardiac ribosomal RNA gene dosage in man. J. Mol. Cell. Cardiol. 7, 125–133. doi:10.1016/0022-2828(75)90014-0
Johnson, R., and Strehler, B. L. (1972). Loss of genes coding for ribosomal RNA in ageing brain cells. Nature 240, 412–414. doi:10.1038/240412a0
Jones, K. L., Smith, D. W., Harvey, M. A., Hall, B. D., and Quan, L. (1975). Older paternal age and fresh gene mutation: data on additional disorders. J. Pediatr. 86, 84–88. doi:10.1016/s0022-3476(75)80709-8
Jones, N. C., Lynn, M. L., Gaudenz, K., Sakai, D., Aoto, K., Rey, J. P., et al. (2008). Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 14, 125–133. doi:10.1038/nm1725
Karwan, R., Bennett, J. L., and Clayton, D. A. (1991). Nuclear RNase MRP processes RNA at multiple discrete sites: interaction with an upstream G box is required for subsequent downstream cleavages. Genes Dev. 5, 1264–1276. doi:10.1101/gad.5.7.1264
Kass, S., Craig, N., and Sollner-Webb, B. (1987). Primary processing of mammalian rRNA involves two adjacent cleavages and is not species specific. Mol. Cell. Biol. 7, 2891–2898. doi:10.1128/mcb.7.8.2891
Kass, S., Tyc, K., Steitz, J. A., and Sollner-Webb, B. (1990). The U3 small nucleolar ribonucleoprotein functions in the first step of preribosomal RNA processing. Cell 60, 897–908. doi:10.1016/0092-8674(90)90338-f
Kempers-Veenstra, A. E., Oliemans, J., Offenberg, H., Dekker, A. F., Piper, P. W., Planta, R. J., et al. (1986). 3'-End formation of transcripts from the yeast rRNA operon. EMBO J. 5, 2703–2710. doi:10.1002/j.1460-2075.1986.tb04554.x
Kennedy, S., Wang, D., and Ruvkun, G. (2004). A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature 427, 645–649. doi:10.1038/nature02302
Khalid, F., Phan, T., Qiang, M., Maity, P., Lasser, T., Wiese, S., et al. (2023). TFIIH mutations can impact on translational fidelity of the ribosome. Hum. Mol. Genet. 32, 1102–1113. doi:10.1093/hmg/ddac268
Khatter, H., Myasnikov, A. G., Natchiar, S. K., and Klaholz, B. P. (2015). Structure of the human 80S ribosome. Nature 520, 640–645. doi:10.1038/nature14427
Khoshnevis, S., Dreggors-Walker, R. E., Marchand, V., Motorin, Y., and Ghalei, H. (2022). Ribosomal RNA 2'-O-methylations regulate translation by impacting ribosome dynamics. Proc. Natl. Acad. Sci. U. S. A. 119, e2117334119. doi:10.1073/pnas.2117334119
Kim, J. H., Dilthey, A. T., Nagaraja, R., Lee, H. S., Koren, S., Dudekula, D., et al. (2018). Variation in human chromosome 21 ribosomal RNA genes characterized by TAR cloning and long-read sequencing. Nucleic Acids Res. 46, 6712–6725. doi:10.1093/nar/gky442
Kim, S., Li, Q., Dang, C. V., and Lee, L. A. (2000). Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc. Natl. Acad. Sci. U. S. A. 97, 11198–11202. doi:10.1073/pnas.200372597
Kiss, T., Marshallsay, C., and Filipowicz, W. (1992). 7-2/MRP RNAs in plant and mammalian cells: association with higher order structures in the nucleolus. EMBO J. 11, 3737–3746. doi:10.1002/j.1460-2075.1992.tb05459.x
Knight, S. W., Heiss, N. S., Vulliamy, T. J., Greschner, S., Stavrides, G., Pai, G. S., et al. (1999). X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am. J. Hum. Genet. 65, 50–58. doi:10.1086/302446
Knorr, A. G., Schmidt, C., Tesina, P., Berninghausen, O., Becker, T., Beatrix, B., et al. (2019). Ribosome-NatA architecture reveals that rRNA expansion segments coordinate N-terminal acetylation. Nat. Struct. Mol. Biol. 26, 35–39. doi:10.1038/s41594-018-0165-y
Kobayashi, T. (2011). Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell Mol. Life Sci. 68, 1395–1403. doi:10.1007/s00018-010-0613-2
Koch, S., Garcia Gonzalez, O., Assfalg, R., Schelling, A., Schafer, P., Scharffetter-Kochanek, K., et al. (2014). Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth. Cell Cycle 13, 2029–2037. doi:10.4161/cc.29018
Kossinova, O., Malygin, A., Krol, A., and Karpova, G. (2014). The SBP2 protein central to selenoprotein synthesis contacts the human ribosome at expansion segment 7L of the 28S rRNA. RNA 20, 1046–1056. doi:10.1261/rna.044917.114
Krauer, N., Rauscher, R., and Polacek, N. (2021). tRNA synthetases are recruited to yeast ribosomes by rRNA expansion segment 7L but do not require association for functionality. Noncoding RNA 7, 73. doi:10.3390/ncrna7040073
Krishnan, P., Ghosh, S., Wang, B., Heyns, M., Graham, K., Mackey, J. R., et al. (2016). Profiling of small nucleolar RNAs by next generation sequencing: potential new players for breast cancer prognosis. PLoS One 11, e0162622. doi:10.1371/journal.pone.0162622
Krogh, N., Jansson, M. D., Hafner, S. J., Tehler, D., Birkedal, U., Christensen-Dalsgaard, M., et al. (2016). Profiling of 2'-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 44, 7884–7895. doi:10.1093/nar/gkw482
Kufel, J., Dichtl, B., and Tollervey, D. (1999). Yeast Rnt1p is required for cleavage of the pre-ribosomal RNA in the 3' ETS but not the 5' ETS. RNA 5, 909–917. doi:10.1017/s135583829999026x
Lafontaine, D. L. J., Riback, J. A., Bascetin, R., and Brangwynne, C. P. (2021). The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol. 22, 165–182. doi:10.1038/s41580-020-0272-6
Lamont, R. E., Loredo-Osti, J., Roslin, N. M., Mauthe, J., Coghlan, G., Nylen, E., et al. (2005). A locus for Bowen-Conradi syndrome maps to chromosome region 12p13.3. Am. J. Med. Genet. Part A 132A, 136–143. doi:10.1002/ajmg.a.30420
Lancaster, L., Lambert, N. J., Maklan, E. J., Horan, L. H., and Noller, H. F. (2008). The sarcin-ricin loop of 23S rRNA is essential for assembly of the functional core of the 50S ribosomal subunit. RNA 14, 1999–2012. doi:10.1261/rna.1202108
Lang, K., Erlacher, M., Wilson, D. N., Micura, R., and Polacek, N. (2008). The role of 23S ribosomal RNA residue A2451 in peptide bond synthesis revealed by atomic mutagenesis. Chem. Biol. 15, 485–492. doi:10.1016/j.chembiol.2008.03.014
Langhendries, J. L., Nicolas, E., Doumont, G., Goldman, S., and Lafontaine, D. L. (2016). The human box C/D snoRNAs U3 and U8 are required for pre-rRNA processing and tumorigenesis. Oncotarget 7, 59519–59534. doi:10.18632/oncotarget.11148
Laugel, V., Dalloz, C., Stary, A., Cormier-Daire, V., Desguerre, I., Renouil, M., et al. (2008). Deletion of 5' sequences of the CSB gene provides insight into the pathophysiology of Cockayne syndrome. Eur. J. Hum. Genet. 16, 320–327. doi:10.1038/sj.ejhg.5201991
Learned, R. M., Cordes, S., and Tjian, R. (1985). Purification and characterization of a transcription factor that confers promoter specificity to human RNA polymerase I. Mol. Cell. Biol. 5, 1358–1369. doi:10.1128/mcb.5.6.1358
Learned, R. M., Learned, T. K., Haltiner, M. M., and Tjian, R. T. (1986). Human rRNA transcription is modulated by the coordinate binding of two factors to an upstream control element. Cell 45, 847–857. doi:10.1016/0092-8674(86)90559-3
Learned, R. M., Smale, S. T., Haltiner, M. M., and Tjian, R. (1983). Regulation of human ribosomal RNA transcription. Proc. Natl. Acad. Sci. U. S. A. 80, 3558–3562. doi:10.1073/pnas.80.12.3558
Lebaron, S., Schneider, C., van Nues, R. W., Swiatkowska, A., Walsh, D., Bottcher, B., et al. (2012). Proofreading of pre-40S ribosome maturation by a translation initiation factor and 60S subunits. Nat. Struct. Mol. Biol. 19, 744–753. doi:10.1038/nsmb.2308
Lebedev, A., Scharffetter-Kochanek, K., and Iben, S. (2008). Truncated Cockayne syndrome B protein represses elongation by RNA polymerase I. J. Mol. Biol. 382, 266–274. doi:10.1016/j.jmb.2008.07.018
Lehmann, A. R., Thompson, A. F., Harcourt, S. A., Stefanini, M., and Norris, P. G. (1993). Cockayne's syndrome: correlation of clinical features with cellular sensitivity of RNA synthesis to UV irradiation. J. Med. Genet. 30, 679–682. doi:10.1136/jmg.30.8.679
Leppek, K., Fujii, K., Quade, N., Susanto, T. T., Boehringer, D., Lenarčič, T., et al. (2020). Gene- and species-specific Hox mRNA translation by ribosome expansion segments. Mol. Cell 80, 980–995.e13. doi:10.1016/j.molcel.2020.10.023
Liang, X. H., Liu, Q., and Fournier, M. J. (2007). rRNA modifications in an intersubunit bridge of the ribosome strongly affect both ribosome biogenesis and activity. Mol. Cell 28, 965–977. doi:10.1016/j.molcel.2007.10.012
Liang, X. H., Liu, Q., and Fournier, M. J. (2009). Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA 15, 1716–1728. doi:10.1261/rna.1724409
Liano, D., Chowdhury, S., and Di Antonio, M. (2021). Cockayne syndrome B protein selectively resolves and interact with intermolecular DNA G-quadruplex structures. J. Am. Chem. Soc. 143, 20988–21002. doi:10.1021/jacs.1c10745
Liebhaber, S. A., Wolf, S., and Schlessinger, D. (1978). Differences in rRNA metabolism of primary and SV40-transformed human fibroblasts. Cell 13, 121–127. doi:10.1016/0092-8674(78)90143-5
Lindahl, L., Archer, R. H., and Zengel, J. M. (1992). A new rRNA processing mutant of Saccharomyces cerevisiae. Nucleic Acids Res. 20, 295–301. doi:10.1093/nar/20.2.295
Lomakin, I. B., and Steitz, T. A. (2013). The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature 500, 307–311. doi:10.1038/nature12355
López, M. D., Rosenblad, M. A., and Samuelsson, T. (2009). Conserved and variable domains of RNase MRP RNA. RNA Biol. 6, 208–221. doi:10.4161/rna.6.3.8584
Lygerou, Z., Allmang, C., Tollervey, D., and Seraphin, B. (1996). Accurate processing of a eukaryotic precursor ribosomal RNA by ribonuclease MRP in vitro. Science 272, 268–270. doi:10.1126/science.272.5259.268
Lygerou, Z., Mitchell, P., Petfalski, E., Seraphin, B., and Tollervey, D. (1994). The POP1 gene encodes a protein component common to the RNase MRP and RNase P ribonucleoproteins. Genes Dev. 8, 1423–1433. doi:10.1101/gad.8.12.1423
Maccarty, W. C. (1936). The value of the macronucleolus in the cancer problem. Am. J. Cancer 26, 529–532. doi:10.1158/ajc.1936.529
Maden, B. E., Vaughan, M. H., Warner, J. R., and Darnell, J. E. (1969). Effects of valine deprivation on ribosome formation in HeLa cells. J. Mol. Biol. 45, 265–275. doi:10.1016/0022-2836(69)90104-1
Mahajan, P. B., and Thompson, E. A. (1990). Hormonal regulation of transcription of rDNA. Purification and characterization of the hormone-regulated transcription factor IC. J. Biol. Chem. 265, 16225–16233. doi:10.1016/s0021-9258(17)46212-0
Maizels, N. (1976). Dictyostelium 17S, 25S, and 5S rDNAs lie within a 38,000 base pair repeated unit. Cell 9, 431–438. doi:10.1016/0092-8674(76)90088-x
Malinovskaya, E. M., Ershova, E. S., Golimbet, V. E., Porokhovnik, L. N., Lyapunova, N. A., Kutsev, S. I., et al. (2018). Copy number of human ribosomal genes with aging: unchanged mean, but narrowed range and decreased variance in elderly group. Front. Genet. 9, 306. doi:10.3389/fgene.2018.00306
Mao, J., Appel, B., Schaack, J., Sharp, S., Yamada, H., and Soll, D. (1982). The 5S RNA genes of Schizosaccharomyces pombe. Nucleic Acids Res. 10, 487–500. doi:10.1093/nar/10.2.487
Marcel, V., Ghayad, S. E., Belin, S., Therizols, G., Morel, A. P., Solano-Gonzalez, E., et al. (2013). p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 24, 318–330. doi:10.1016/j.ccr.2013.08.013
Marcel, V., Kielbassa, J., Marchand, V., Natchiar, K. S., Paraqindes, H., Nguyen Van Long, F., et al. (2020). Ribosomal RNA 2'O-methylation as a novel layer of inter-tumour heterogeneity in breast cancer. Nar. Cancer 2, zcaa036. doi:10.1093/narcan/zcaa036
Mars, J. C., Tremblay, M. G., Valere, M., Sibai, D. S., Sabourin-Felix, M., Lessard, F., et al. (2020). The chemotherapeutic agent CX-5461 irreversibly blocks RNA polymerase I initiation and promoter release to cause nucleolar disruption, DNA damage and cell inviability. Nar. Cancer 2, zcaa032. doi:10.1093/narcan/zcaa032
Mattioli, F., Worpenberg, L., Li, C. T., Ibrahim, N., Naz, S., Sharif, S., et al. (2023). Biallelic variants in NSUN6 cause an autosomal recessive neurodevelopmental disorder. Genet. Med. 25, 100900. doi:10.1016/j.gim.2023.100900
Mayer, C., Bierhoff, H., and Grummt, I. (2005). The nucleolus as a stress sensor: JNK2 inactivates the transcription factor TIF-IA and down-regulates rRNA synthesis. Genes Dev. 19, 933–941. doi:10.1101/gad.333205
Mayer, C., Zhao, J., Yuan, X., and Grummt, I. (2004). mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 18, 423–434. doi:10.1101/gad.285504
Mayne, L. V., and Lehmann, A. R. (1982). Failure of RNA synthesis to recover after UV irradiation: an early defect in cells from individuals with Cockayne's syndrome and xeroderma pigmentosum. Cancer Res. 42, 1473–1478. doi:10.1016/0027-5107(82)90047-1
McKusick, V. A., Eldridge, R., Hostetler, J. A., Ruangwit, U., and Egeland, J. A. (1965). Dwarfism in the amish. Ii. Cartilage-hair hypoplasia. Bull. Johns Hopkins Hosp. 116, 285–326.
Medvedev, Z. A. (1972). Repetition of molecular-genetic information as a possible factor in evolutionary changes of life span. Exp. Gerontol. 7, 227–238. doi:10.1016/0531-5565(72)90012-5
Meier, U. T., and Blobel, G. (1994). NAP57, a mammalian nucleolar protein with a putative homolog in yeast and bacteria. J. Cell Biol. 127, 1505–1514. doi:10.1083/jcb.127.6.1505
Mestre-Fos, S., Penev, P. I., Suttapitugsakul, S., Hu, M., Ito, C., Petrov, A. S., et al. (2019). G-quadruplexes in human ribosomal RNA. J. Mol. Biol. 431, 1940–1955. doi:10.1016/j.jmb.2019.03.010
Meyer, B., Wurm, J. P., Kotter, P., Leisegang, M. S., Schilling, V., Buchhaupt, M., et al. (2011). The Bowen–Conradi syndrome protein Nep1 (Emg1) has a dual role in eukaryotic ribosome biogenesis, as an essential assembly factor and in the methylation of Ψ1191 in yeast 18S rRNA. Nucleic Acids Res. 39, 1526–1537. doi:10.1093/nar/gkq931
Meyer, B., Wurm, J. P., Sharma, S., Immer, C., Pogoryelov, D., Kotter, P., et al. (2016). Ribosome biogenesis factor Tsr3 is the aminocarboxypropyl transferase responsible for 18S rRNA hypermodification in yeast and humans. Nucleic Acids Res. 44, 4304–4316. doi:10.1093/nar/gkw244
Milkereit, P., and Tschochner, H. (1998). A specialized form of RNA polymerase I, essential for initiation and growth-dependent regulation of rRNA synthesis, is disrupted during transcription. EMBO J. 17, 3692–3703. doi:10.1093/emboj/17.13.3692
Mitchell, J. R., Wood, E., and Collins, K. (1999). A telomerase component is defective in the human disease dyskeratosis congenita. Nature 402, 551–555. doi:10.1038/990141
Mitchell, P., Petfalski, E., Shevchenko, A., Mann, M., and Tollervey, D. (1997). The exosome: a conserved eukaryotic RNA processing complex containing multiple 3'-->5' exoribonucleases. Cell 91, 457–466. doi:10.1016/s0092-8674(00)80432-8
Moazed, D., and Noller, H. F. (1986). Transfer RNA shields specific nucleotides in 16S ribosomal RNA from attack by chemical probes. Cell 47, 985–994. doi:10.1016/0092-8674(86)90813-5
Moazed, D., and Noller, H. F. (1987). Chloramphenicol, erythromycin, carbomycin and vernamycin B protect overlapping sites in the peptidyl transferase region of 23S ribosomal RNA. Biochimie 69, 879–884. doi:10.1016/0300-9084(87)90215-x
Moazed, D., and Noller, H. F. (1990). Binding of tRNA to the ribosomal A and P sites protects two distinct sets of nucleotides in 16 S rRNA. J. Mol. Biol. 211, 135–145. doi:10.1016/0022-2836(90)90016-f
Montanaro, L., Brigotti, M., Clohessy, J., Barbieri, S., Ceccarelli, C., Santini, D., et al. (2006). Dyskerin expression influences the level of ribosomal RNA pseudo-uridylation and telomerase RNA component in human breast cancer. J. Pathology 210, 10–18. doi:10.1002/path.2023
Moorefield, B., Greene, E. A., and Reeder, R. H. (2000). RNA polymerase I transcription factor Rrn3 is functionally conserved between yeast and human. Proc. Natl. Acad. Sci. U. S. A. 97, 4724–4729. doi:10.1073/pnas.080063997
Murayama, A., Ohmori, K., Fujimura, A., Minami, H., Yasuzawa-Tanaka, K., Kuroda, T., et al. (2008). Epigenetic control of rDNA loci in response to intracellular energy status. Cell 133, 627–639. doi:10.1016/j.cell.2008.03.030
Nachmani, D., Bothmer, A. H., Grisendi, S., Mele, A., Bothmer, D., Lee, J. D., et al. (2019). Germline NPM1 mutations lead to altered rRNA 2'-O-methylation and cause dyskeratosis congenita. Nat. Genet. 51, 1518–1529. doi:10.1038/s41588-019-0502-z
Nance, M. A., and Berry, S. A. (1992). Cockayne syndrome: review of 140 cases. Am. J. Med. Genet. 42, 68–84. doi:10.1002/ajmg.1320420115
Narayanan, D. L., Shukla, A., Kausthubham, N., Bhavani, G. S., Shah, H., Mortier, G., et al. (2019). An emerging ribosomopathy affecting the skeleton due to biallelic variations in NEPRO. Am. J. Med. Genet. Part A 179, 1709–1717. doi:10.1002/ajmg.a.61267
Nierhaus, K. H., and Dohme, F. (1974). Total reconstitution of functionally active 50S ribosomal subunits from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 71, 4713–4717. doi:10.1073/pnas.71.12.4713
Nissen, P., Hansen, J., Ban, N., Moore, P. B., and Steitz, T. A. (2000). The structural basis of ribosome activity in peptide bond synthesis. Science 289, 920–930. doi:10.1126/science.289.5481.920
Noller, H. F., and Chaires, J. B. (1972). Functional modification of 16S ribosomal RNA by kethoxal. Proc. Natl. Acad. Sci. U. S. A. 69, 3115–3118. doi:10.1073/pnas.69.11.3115
Noller, H. F., Hoffarth, V., and Zimniak, L. (1992). Unusual resistance of peptidyl transferase to protein extraction procedures. Science 256, 1416–1419. doi:10.1126/science.1604315
Noller, H. F., Kop, J., Wheaton, V., Brosius, J., Gutell, R. R., Kopylov, A. M., et al. (1981). Secondary structure model for 23S ribosomal RNA. Nucleic Acids Res. 9, 6167–6189. doi:10.1093/nar/9.22.6167
Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., et al. (2022). The complete sequence of a human genome. Science 376, 44–53. doi:10.1126/science.abj6987
Ogle, J. M., Brodersen, D. E., Clemons, W. M., Tarry, M. J., Carter, A. P., and Ramakrishnan, V. (2001). Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292, 897–902. doi:10.1126/science.1060612
Ogle, J. M., Murphy, F. V., Tarry, M. J., and Ramakrishnan, V. (2002). Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111, 721–732. doi:10.1016/s0092-8674(02)01086-3
Ojha, S., Malla, S., and Lyons, S. M. (2020). snoRNPs: functions in ribosome biogenesis. Biomolecules 10, 783. doi:10.3390/biom10050783
Okur, M. N., Lee, J. H., Osmani, W., Kimura, R., Demarest, T. G., Croteau, D. L., et al. (2020). Cockayne syndrome group A and B proteins function in rRNA transcription through nucleolin regulation. Nucleic Acids Res. 48, 2473–2485. doi:10.1093/nar/gkz1242
Oshima, J., Sidorova, J. M., and Monnat, R. J. W. (2017). Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 33, 105–114. doi:10.1016/j.arr.2016.03.002
Paramasivam, A., Meena, A. K., Venkatapathi, C., Pitceathly, R. D. S., and Thangaraj, K. (2020). Novel biallelic NSUN3 variants cause early-onset mitochondrial encephalomyopathy and seizures. J. Mol. Neurosci. 70, 1962–1965. doi:10.1007/s12031-020-01595-8
Parker, K. A., and Steitz, J. A. (1987). Structural analysis of the human U3 ribonucleoprotein particle reveal a conserved sequence available for base pairing with pre-rRNA. Mol. Cell. Biol. 7, 2899–2913. doi:10.1128/mcb.7.8.2899-2913.1987
Peculis, B. A., and Steitz, J. A. (1993). Disruption of U8 nucleolar snRNA inhibits 5.8S and 28S rRNA processing in the Xenopus oocyte. Cell 73, 1233–1245. doi:10.1016/0092-8674(93)90651-6
Penzo, M., Rocchi, L., Brugiere, S., Carnicelli, D., Onofrillo, C., Coute, Y., et al. (2015). Human ribosomes from cells with reduced dyskerin levels are intrinsically altered in translation. FASEB J. 29, 3472–3482. doi:10.1096/fj.15-270991
PerezGrovas-Saltijeral, A., Rajkumar, A. P., and Knight, H. M. (2023). Differential expression of m(5)C RNA methyltransferase genes NSUN6 and NSUN7 in Alzheimer's disease and traumatic brain injury. Mol. Neurobiol. 60, 2223–2235. doi:10.1007/s12035-022-03195-6
Petes, T. D., Hereford, L. M., and Skryabin, K. G. (1978). Characterization of two types of yeast ribosomal DNA genes. J. Bacteriol. 134, 295–305. doi:10.1128/jb.134.1.295-305.1978
Peyroche, G., Milkereit, P., Bischler, N., Tschochner, H., Schultz, P., Sentenac, A., et al. (2000). The recruitment of RNA polymerase I on rDNA is mediated by the interaction of the A43 subunit with Rrn3. EMBO J. 19, 5473–5482. doi:10.1093/emboj/19.20.5473
Philipson, L. (1961). Chromatographic separation, and characteristics of nucleic acids from HeLa cells. J. General Physiology 44, 899–910. doi:10.1085/jgp.44.5.899
Pietrzak, M., Rempala, G., Nelson, P. T., Zheng, J. J., and Hetman, M. (2011). Epigenetic silencing of nucleolar rRNA genes in Alzheimer's disease. PLoS One 6, e22585. doi:10.1371/journal.pone.0022585
Pillon, M. C., Hsu, A. L., Krahn, J. M., Williams, J. G., Goslen, K. H., Sobhany, M., et al. (2019). Cryo-EM reveals active site coordination within a multienzyme pre-rRNA processing complex. Nat. Struct. Mol. Biol. 26, 830–839. doi:10.1038/s41594-019-0289-8
Pillon, M. C., Sobhany, M., Borgnia, M. J., Williams, J. G., and Stanley, R. E. (2017). Grc3 programs the essential endoribonuclease Las1 for specific RNA cleavage. Proc. Natl. Acad. Sci. U. S. A. 114, E5530–E5538. doi:10.1073/pnas.1703133114
Pillon, M. C., Sobhany, M., and Stanley, R. E. (2018). Characterization of the molecular crosstalk within the essential Grc3/Las1 pre-rRNA processing complex. RNA 24, 721–738. doi:10.1261/rna.065037.117
Piper, P. W., Bellatin, J. A., and Lockheart, A. (1983). Altered maturation of sequences at the 3' terminus of 5S gene transcripts in a Saccharomyces cerevisiae mutant that lacks a RNA processing endonuclease. EMBO J. 2, 353–359. doi:10.1002/j.1460-2075.1983.tb01430.x
Pober, B. R. (2010). Williams–beuren syndrome. N. Engl. J. Med. 362, 239–252. doi:10.1056/nejmra0903074
Polikanov, Y. S., Steitz, T. A., and Innis, C. A. (2014). A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nat. Struct. Mol. Biol. 21, 787–793. doi:10.1038/nsmb.2871
Popov, A., Smirnov, E., Kováčik, L., Raška, O., Hagen, G., Stixova, L., et al. (2013). Duration of the first steps of the human rRNA processing. Nucleus 4, 134–141. doi:10.4161/nucl.23985
Prestayko, A. W., Tonato, M., and Busch, H. (1970). Low molecular weight RNA associated with 28 s nucleolar RNA. J. Mol. Biol. 47, 505–515. doi:10.1016/0022-2836(70)90318-9
Prieto, J. L., and McStay, B. (2007). Recruitment of factors linking transcription and processing of pre-rRNA to NOR chromatin is UBF-dependent and occurs independent of transcription in human cells. Genes Dev. 21, 2041–2054. doi:10.1101/gad.436707
Reimer, G., Scheer, U., Peters, J. M., and Tan, E. M. (1986). Immunolocalization and partial characterization of a nucleolar autoantigen (PM-Scl) associated with polymyositis/scleroderma overlap syndromes. J. Immunol. 137, 3802–3808. doi:10.4049/jimmunol.137.12.3802
Remmelzwaal, P. C., Verhagen, M. V., Jongbloed, J. D. H., van den Akker, P. C., Veenstra-Knol, H. E., and Hitzert, M. M. (2023). Expanding the phenotype of anauxetic dysplasia caused by biallelic <i>NEPRO</i> mutations: a case report. Am. J. Med. Genet. Part A 191, 2440–2445. doi:10.1002/ajmg.a.63316
Ren, R., Deng, L., Xue, Y., Suzuki, K., Zhang, W., Yu, Y., et al. (2017). Visualization of aging-associated chromatin alterations with an engineered TALE system. Cell Res. 27, 483–504. doi:10.1038/cr.2017.18
Ridanpaa, M., van Eenennaam, H., Pelin, K., Chadwick, R., Johnson, C., Yuan, B., et al. (2001). Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell 104, 195–203. doi:10.1016/s0092-8674(01)00205-7
Rinke, J., and Steitz, J. A. (1982). Precursor molecules of both human 5S ribosomal RNA and transfer RNAs are bound by a cellular protein reactive with anti-La lupus antibodies. Cell 29, 149–159. doi:10.1016/0092-8674(82)90099-x
Ritossa, F. M., Atwood, K. C., and Spiegelman, S. (1966). A molecular explanation of the bobbed mutants of Drosophila as partial deficiencies of "ribosomal" DNA. Genetics 54, 819–834. doi:10.1093/genetics/54.3.819
Robertson, N., Shchepachev, V., Wright, D., Turowski, T. W., Spanos, C., Helwak, A., et al. (2022). A disease-linked lncRNA mutation in RNase MRP inhibits ribosome synthesis. Nat. Commun. 13, 649. doi:10.1038/s41467-022-28295-8
Rong, B., Zhang, Q., Wan, J., Xing, S., Dai, R., Li, Y., et al. (2020). Ribosome 18S m(6)A methyltransferase METTL5 promotes translation initiation and breast cancer cell growth. Cell Rep. 33, 108544. doi:10.1016/j.celrep.2020.108544
Rouquette, J., Choesmel, V., and Gleizes, P. E. (2005). Nuclear export and cytoplasmic processing of precursors to the 40S ribosomal subunits in mammalian cells. EMBO J. 24, 2862–2872. doi:10.1038/sj.emboj.7600752
Ruggero, D., Grisendi, S., Piazza, F., Rego, E., Mari, F., Rao, P. H., et al. (2003). Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science 299, 259–262. doi:10.1126/science.1079447
Sadian, Y., Baudin, F., Tafur, L., Murciano, B., Wetzel, R., Weis, F., et al. (2019). Molecular insight into RNA polymerase I promoter recognition and promoter melting. Nat. Commun. 10, 5543. doi:10.1038/s41467-019-13510-w
Satoh, A., Brace, C. S., Rensing, N., Cliften, P., Wozniak, D. F., Herzog, E. D., et al. (2013). Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 18, 416–430. doi:10.1016/j.cmet.2013.07.013
Savage, S. A., and Niewisch, M. R. (1993). “Dyskeratosis congenita and related telomere biology disorders,” in GeneReviews((R)). Editors M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Beanet al. (Seattle (WA): National Library of Medicine).
Scheibye-Knudsen, M., Tseng, A., Borch Jensen, M., Scheibye-Alsing, K., Fang, E. F., Iyama, T., et al. (2016). Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. U. S. A. 113, 12502–12507. doi:10.1073/pnas.1610198113
Scherrer, K., and Darnell, J. E. (1962). Sedimentation characteristics of rapidly labelled RNA from HeLa cells. Biochem. Biophysical Res. Commun. 7, 486–490. doi:10.1016/0006-291x(62)90341-8
Scherrer, K., Latham, H., and Darnell, J. E. (1963). Demonstration of an unstable RNA and of a precursor to ribosomal RNA in HeLa cells. Proc. Natl. Acad. Sci. U. S. A. 49, 240–248. doi:10.1073/pnas.49.2.240
Schmickel, R. D., Chu, E. H., Trosko, J. E., and Chang, C. C. (1977). Cockayne syndrome: a cellular sensitivity to ultraviolet light. Pediatrics 60, 135–139. doi:10.1542/peds.60.2.135
Schmitt, M. E., and Clayton, D. A. (1992). Yeast site-specific ribonucleoprotein endoribonuclease MRP contains an RNA component homologous to mammalian RNase MRP RNA and essential for cell viability. Genes Dev. 6, 1975–1985. doi:10.1101/gad.6.10.1975
Schmitt, M. E., and Clayton, D. A. (1993). Nuclear RNase MRP is required for correct processing of pre-5.8S rRNA in Saccharomyces cerevisiae. Mol. Cell Biol. 13, 7935–7941. doi:10.1128/mcb.13.12.7935
Schosserer, M., Minois, N., Angerer, T. B., Amring, M., Dellago, H., Harreither, E., et al. (2015). Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat. Commun. 6, 6158. doi:10.1038/ncomms7158
Schuller, J. M., Falk, S., Fromm, L., Hurt, E., and Conti, E. (2018). Structure of the nuclear exosome captured on a maturing preribosome. Science 360, 219–222. doi:10.1126/science.aar5428
Schulze, H., and Nierhaus, K. H. (1982). Minimal set of ribosomal components for reconstitution of the peptidyltransferase activity. EMBO J. 1, 609–613. doi:10.1002/j.1460-2075.1982.tb01216.x
Selker, E. U., Yanofsky, C., Driftmier, K., Metzenberg, R. L., Alzner-DeWeerd, B., and RajBhandary, U. L. (1981). Dispersed 5S RNA genes in N. crassa: structure, expression and evolution. Cell 24, 819–828. doi:10.1016/0092-8674(81)90107-0
Selmer, M., Dunham, C. M., Murphy, F. V. t., Weixlbaumer, A., Petry, S., Kelley, A. C., et al. (2006). Structure of the 70S ribosome complexed with mRNA and tRNA. Science 313, 1935–1942. doi:10.1126/science.1131127
Sepich-Poore, C., Zheng, Z., Schmitt, E., Wen, K., Zhang, Z. S., Cui, X. L., et al. (2022). The METTL5-TRMT112 N(6)-methyladenosine methyltransferase complex regulates mRNA translation via 18S rRNA methylation. J. Biol. Chem. 298, 101590. doi:10.1016/j.jbc.2022.101590
Shankar, V., Rauscher, R., Reuther, J., Gharib, W. H., Koch, M., and Polacek, N. (2020). rRNA expansion segment 27Lb modulates the factor recruitment capacity of the yeast ribosome and shapes the proteome. Nucleic Acids Res. 48, 3244–3256. doi:10.1093/nar/gkaa003
Shao, S., Murray, J., Brown, A., Taunton, J., Ramakrishnan, V., and Hegde, R. S. (2016). Decoding mammalian ribosome-mRNA states by translational GTPase complexes. Cell 167, 1229–1240.e15. doi:10.1016/j.cell.2016.10.046
Sharma, S., Langhendries, J. L., Watzinger, P., Kotter, P., Entian, K. D., and Lafontaine, D. L. (2015). Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 43, 2242–2258. doi:10.1093/nar/gkv075
Sharma, S., Yang, J., Watzinger, P., Kotter, P., and Entian, K. D. (2013). Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 41, 9062–9076. doi:10.1093/nar/gkt679
Sharp, S., Garcia, A., Cooley, L., and Soll, D. (1984). Transcriptionally active and inactive gene repeats within theD. meianogaster5S RNA gene cluster. Nucleic Acids Res. 12, 7617–7632. doi:10.1093/nar/12.20.7617
Shiue, C. N., Berkson, R. G., and Wright, A. P. (2009). c-Myc induces changes in higher order rDNA structure on stimulation of quiescent cells. Oncogene 28, 1833–1842. doi:10.1038/onc.2009.21
Shobuike, T., Sugano, S., Yamashita, T., and Ikeda, H. (1995). Characterization of cDNA encoding mouse homolog of fission yeast dhp1+ gene: structural and functional conservation. Nucleic Acids Res. 23, 357–361. doi:10.1093/nar/23.3.357
Shuai, K., and Warner, J. R. (1991). A temperature sensitive mutant of Saccharomyces cerevisiae defective in pre-rRNA processing. Nucleic Acids Res. 19, 5059–5064. doi:10.1093/nar/19.18.5059
Sievers, A., Beringer, M., Rodnina, M. V., and Wolfenden, R. (2004). The ribosome as an entropy trap. Proc. Natl. Acad. Sci. U. S. A. 101, 7897–7901. doi:10.1073/pnas.0402488101
Silva, S., Homolka, D., and Pillai, R. S. (2017). Characterization of the mammalian RNA exonuclease 5/NEF-sp as a testis-specific nuclear 3'--> 5' exoribonuclease. RNA 23, 1385–1392. doi:10.1261/rna.060723.117
Sinclair, D. A., and Guarente, L. (1997). Extrachromosomal rDNA circles–a cause of aging in yeast. Cell 91, 1033–1042. doi:10.1016/s0092-8674(00)80493-6
Sloan, K. E., Mattijssen, S., Lebaron, S., Tollervey, D., Pruijn, G. J., and Watkins, N. J. (2013). Both endonucleolytic and exonucleolytic cleavage mediate ITS1 removal during human ribosomal RNA processing. J. Cell Biol. 200, 577–588. doi:10.1083/jcb.201207131
Sorensen, P. D., and Frederiksen, S. (1991). Characterization of human 5S rRNA genes. Nucleic Acids Res. 19, 4147–4151. doi:10.1093/nar/19.15.4147
Sorensen, P. D., Lomholt, B., Frederiksen, S., and Tommerup, N. (1991). Fine mapping of human 5S rRNA genes to chromosome 1q42.11–--q42.13. Cytogenet. Genome Res. 57, 26–29. doi:10.1159/000133107
Srivastava, L., Lapik, Y. R., Wang, M., and Pestov, D. G. (2010). Mammalian DEAD box protein Ddx51 acts in 3' end maturation of 28S rRNA by promoting the release of U8 snoRNA. Mol. Cell. Biol. 30, 2947–2956. doi:10.1128/mcb.00226-10
Srivastava, R., Srivastava, R., and Ahn, S. H. (2016). The epigenetic pathways to ribosomal DNA silencing. Microbiol. Mol. Biol. Rev. 80, 545–563. doi:10.1128/mmbr.00005-16
Steffensen, D. M., Duffey, P., and Prensky, W. (1974). Localisation of 5S ribosomal RNA genes on human chromosome 1. Nature 252, 741–743. doi:10.1038/252741a0
Steiner, G., Kuechler, E., and Barta, A. (1988). Photo-affinity labelling at the peptidyl transferase centre reveals two different positions for the A- and P-sites in domain V of 23S rRNA. EMBO J. 7, 3949–3955. doi:10.1002/j.1460-2075.1988.tb03281.x
Steitz, T. A., and Moore, P. B. (2003). RNA, the first macromolecular catalyst: the ribosome is a ribozyme. Trends Biochem. Sci. 28, 411–418. doi:10.1016/s0968-0004(03)00169-5
Stratmann, S., Yones, S. A., Mayrhofer, M., Norgren, N., Skaftason, A., Sun, J., et al. (2021). Genomic characterization of relapsed acute myeloid leukemia reveals novel putative therapeutic targets. Blood Adv. 5, 900–912. doi:10.1182/bloodadvances.2020003709
Strohner, R., Nemeth, A., Jansa, P., Hofmann-Rohrer, U., Santoro, R., Langst, G., et al. (2001). NoRC–a novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J. 20, 4892–4900. doi:10.1093/emboj/20.17.4892
Sulisalo, T., Francomano, C. A., Sistonen, P., Maher, J. F., McKusick, V. A., de la Chapelle, A., et al. (1994). High-resolution genetic mapping of the cartilage-hair hypoplasia (CHH) gene in amish and Finnish families. Genomics 20, 347–353. doi:10.1006/geno.1994.1187
Sulisalo, T., Sistonen, P., Hastbacka, J., Wadelius, C., Makitie, O., de la Chapelle, A., et al. (1993). Cartilage-hair hypoplasia gene assigned to chromosome 9 by linkage analysis. Nat. Genet. 3, 338–341. doi:10.1038/ng0493-338
Tague, B. W., and Gerbi, S. A. (1984). Processing of the large rRNA precursor: two proposed categories of RNA-RNA interactions in eukaryotes. J. Mol. Evol. 20, 362–367. doi:10.1007/bf02104742
Tanaka, K., Kawai, K., Kumahara, Y., Ikenaga, M., and Okada, Y. (1981). Genetic complementation groups in cockayne syndrome. Somat. Cell Genet. 7, 445–455. doi:10.1007/bf01542989
Taoka, M., Nobe, Y., Yamaki, Y., Sato, K., Ishikawa, H., Izumikawa, K., et al. (2018). Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 46, 9289–9298. doi:10.1093/nar/gky811
Taylor, A. B., Meyer, B., Leal, B. Z., Kotter, P., Schirf, V., Demeler, B., et al. (2008). The crystal structure of Nep1 reveals an extended SPOUT-class methyltransferase fold and a pre-organized SAM-binding site. Nucleic Acids Res. 36, 1542–1554. doi:10.1093/nar/gkm1172
Tiollais, P., Galibert, F., and Boiron, M. (1971). Evidence for the existence of several molecular species in the "45S fraction" of mammalian ribosomal precursor RNA. Proc. Natl. Acad. Sci. U. S. A. 68, 1117–1120. doi:10.1073/pnas.68.6.1117
Tissenbaum, H. A., and Guarente, L. (2001). Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410, 227–230. doi:10.1038/35065638
Tone, Y., and Toh-e, A. (2002). Nob1p is required for biogenesis of the 26S proteasome and degraded upon its maturation in Saccharomyces cerevisiae. Genes Dev. 16, 3142–3157. doi:10.1101/gad.1025602
Topper, J. N., and Clayton, D. A. (1990). Characterization of human MRP/Th RNA and its nuclear gene: full length MRP/Th RNA is an active endoribonuclease when assembled as an RNP. Nucleic Acids Res. 18, 793–799. doi:10.1093/nar/18.4.793
Traub, P., and Nomura, M. (1968). Structure and function of E. coli ribosomes. V. Reconstitution of functionally active 30S ribosomal particles from RNA and proteins. Proc. Natl. Acad. Sci. U. S. A. 59, 777–784. doi:10.1073/pnas.59.3.777
Troelstra, C., Odijk, H., de Wit, J., Westerveld, A., Thompson, L. H., Bootsma, D., et al. (1990). Molecular cloning of the human DNA excision repair gene ERCC-6. Mol. Cell. Biol. 10, 5806–5813. doi:10.1128/mcb.10.11.5806
Udem, S. A., and Warner, J. R. (1973). The cytoplasmic maturation of a ribosomal precursor ribonucleic acid in yeast. J. Biol. Chem. 248, 1412–1416. doi:10.1016/s0021-9258(19)44314-7
Ulhaq, Z. S., Nurputra, D. K., Soraya, G. V., Kurniawati, S., Istifiani, L. A., Pamungkas, S. A., et al. (2023). A systematic review on Treacher Collins syndrome: correlation between molecular genetic findings and clinical severity. Clin. Genet. 103, 146–155. doi:10.1111/cge.14243
Umeda, M., Ma, J., Huang, B. J., Hagiwara, K., Westover, T., Abdelhamed, S., et al. (2022). Integrated genomic analysis identifies UBTF tandem duplications as a recurrent lesion in pediatric acute myeloid leukemia. Blood Cancer Discov. 3, 194–207. doi:10.1158/2643-3230.bcd-21-0160
Vakkilainen, T., Kivipensas, P., Kaitila, I., de le Chapelle, A., and Ridanpaa, M. (1999). Integrated high-resolution BAC, P1, and transcript map of the CHH region in chromosome 9p13. Genomics 59, 319–325. doi:10.1006/geno.1999.5883
Valdez, B. C., Henning, D., So, R. B., Dixon, J., and Dixon, M. J. (2004). The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc. Natl. Acad. Sci. U. S. A. 101, 10709–10714. doi:10.1073/pnas.0402492101
van Hoof, A., Lennertz, P., and Parker, R. (2000). Three conserved members of the RNase D family have unique and overlapping functions in the processing of 5S U5, RNase MRP and RNase P RNAs in yeast. EMBO J. U4, 1357–1365. doi:10.1093/emboj/19.6.1357
van Tran, N., Ernst, F. G. M., Hawley, B. R., Zorbas, C., Ulryck, N., Hackert, P., et al. (2019). The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 47, 7719–7733. doi:10.1093/nar/gkz619
Veiko, N. N., Ershova, E. S., Veiko, R. V., Umriukhin, P. E., Kurmyshev, M. V., Kostyuk, G. P., et al. (2022). Mild cognitive impairment is associated with low copy number of ribosomal genes in the genomes of elderly people. Front. Genet. 13, 967448. doi:10.3389/fgene.2022.967448
Vulliamy, T., Beswick, R., Kirwan, M., Marrone, A., Digweed, M., Walne, A., et al. (2008). Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc. Natl. Acad. Sci. U. S. A. 105, 8073–8078. doi:10.1073/pnas.0800042105
Vulliamy, T., Marrone, A., Goldman, F., Dearlove, A., Bessler, M., Mason, P. J., et al. (2001). The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413, 432–435. doi:10.1038/35096585
Walker-Kopp, N., Jackobel, A. J., Pannafino, G. N., Morocho, P. A., Xu, X., and Knutson, B. A. (2017). Treacher Collins syndrome mutations in Saccharomyces cerevisiae destabilize RNA polymerase I and III complex integrity. Hum. Mol. Genet. 26, 4290–4300. doi:10.1093/hmg/ddx317
Walne, A. J., Vulliamy, T., Marrone, A., Beswick, R., Kirwan, M., Masunari, Y., et al. (2007). Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum. Mol. Genet. 16, 1619–1629. doi:10.1093/hmg/ddm111
Wang, M., and Lemos, B. (2017). Ribosomal DNA copy number amplification and loss in human cancers is linked to tumor genetic context, nucleolus activity, and proliferation. PLoS Genet. 13, e1006994. doi:10.1371/journal.pgen.1006994
Wang, M., and Pestov, D. G. (2011). 5'-end surveillance by Xrn2 acts as a shared mechanism for mammalian pre-rRNA maturation and decay. Nucleic Acids Res. 39, 1811–1822. doi:10.1093/nar/gkq1050
Warda, A. S., Freytag, B., Haag, S., Sloan, K. E., Gorlich, D., and Bohnsack, M. T. (2016). Effects of the Bowen-Conradi syndrome mutation in EMG1 on its nuclear import, stability and nucleolar recruitment. Hum. Mol. Genet. 25, 5353–5364. doi:10.1093/hmg/ddw351
Warner, J. R., Vilardell, J., and Sohn, J. H. (2001). Economics of ribosome biosynthesis. Cold Spring Harb. Symposia Quantitative Biol. 66, 567–574. doi:10.1101/sqb.2001.66.567
Weber, A. M., Tuchweber, B., Yousef, I., Brochu, P., Turgeon, C., Gabbiani, G., et al. (1981). Severe familial cholestasis in North American Indian children: a clinical model of microfilament dysfunction? Gastroenterology 81, 653–662. doi:10.1016/0016-5085(81)90487-x
Wellauer, P. K., and Dawid, I. B. (1979). Isolation and sequence organization of human ribosomal DNA. J. Mol. Biol. 128, 289–303. doi:10.1016/0022-2836(79)90089-5
Wells, G. R., Weichmann, F., Colvin, D., Sloan, K. E., Kudla, G., Tollervey, D., et al. (2016). The PIN domain endonuclease Utp24 cleaves pre-ribosomal RNA at two coupled sites in yeast and humans. Nucleic Acids Res. 44, 5399–5409. doi:10.1093/nar/gkw213
Wells, G. R., Weichmann, F., Sloan, K. E., Colvin, D., Watkins, N. J., and Schneider, C. (2017). The ribosome biogenesis factor yUtp23/hUTP23 coordinates key interactions in the yeast and human pre-40S particle and hUTP23 contains an essential PIN domain. Nucleic Acids Res. 45, 4796–4809. doi:10.1093/nar/gkw1344
Whitaker, R., Faulkner, S., Miyokawa, R., Burhenn, L., Henriksen, M., Wood, J. G., et al. (2013). Increased expression of Drosophila Sir2 extends life span in a dose-dependent manner. Aging (Albany NY) 5, 682–691. doi:10.18632/aging.100599
Williams, G. T., and Farzaneh, F. (2012). Are snoRNAs and snoRNA host genes new players in cancer? Nat. Rev. Cancer 12, 84–88. doi:10.1038/nrc3195
Winokur, S. T., and Shiang, R. (1998). The Treacher Collins syndrome (TCOF1) gene product, treacle, is targeted to the nucleolus by signals in its C-terminus. Hum. Mol. Genet. 7, 1947–1952. doi:10.1093/hmg/7.12.1947
Wise, C. A., Chiang, L. C., Paznekas, W. A., Sharma, M., Musy, M. M., Ashley, J. A., et al. (1997). TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc. Natl. Acad. Sci. U. S. A. 94, 3110–3115. doi:10.1073/pnas.94.7.3110
Wurm, J. P., Meyer, B., Bahr, U., Held, M., Frolow, O., Kotter, P., et al. (2010). The ribosome assembly factor Nep1 responsible for Bowen-Conradi syndrome is a pseudouridine-N1-specific methyltransferase. Nucleic Acids Res. 38, 2387–2398. doi:10.1093/nar/gkp1189
Xie, W., Ling, T., Zhou, Y., Feng, W., Zhu, Q., Stunnenberg, H. G., et al. (2012). The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc. Natl. Acad. Sci. U. S. A. 109, 8161–8166. doi:10.1073/pnas.1201262109
Xu, H., and Hurley, L. H. (2022). A first-in-class clinical G-quadruplex-targeting drug. The bench-to-bedside translation of the fluoroquinolone QQ58 to CX-5461 (Pidnarulex). Bioorg. Med. Chem. Lett. 77, 129016. doi:10.1016/j.bmcl.2022.129016
Xue, S., and Barna, M. (2012). Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 13, 355–369. doi:10.1038/nrm3359
Xue, Y., Bai, X., Lee, I., Kallstrom, G., Ho, J., Brown, J., et al. (2000). Saccharomyces cerevisiae RAI1 (YGL246c) is homologous to human DOM3Z and encodes a protein that binds the nuclear exoribonuclease Rat1p. Mol. Cell. Biol. 20, 4006–4015. doi:10.1128/mcb.20.11.4006-4015.2000
Yamamoto, M., and Seifart, K. H. (1978). Heterogeneity in the 3'-terminal sequence of ribosomal 5S RNA synthesized by isolated HeLa cell nuclei in vitro. Biochemistry 17, 457–461. doi:10.1021/bi00596a013
Yang, X. C., Purdy, M., Marzluff, W. F., and Dominski, Z. (2006). Characterization of 3'hExo, a 3' exonuclease specifically interacting with the 3' end of histone mRNA. J. Biol. Chem. 281, 30447–30454. doi:10.1074/jbc.m602947200
Yoon, A., Peng, G., Brandenburg, Y., Zollo, O., Xu, W., Rego, E., et al. (2006). Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 312, 902–906. doi:10.1126/science.1123835
Yu, B., Mitchell, G. A., and Richter, A. (2005). Nucleolar localization of cirhin, the protein mutated in North American Indian childhood cirrhosis. Exp. Cell Res. 311, 218–228. doi:10.1016/j.yexcr.2005.08.012
Yuan, X., Feng, W., Imhof, A., Grummt, I., and Zhou, Y. (2007). Activation of RNA polymerase I transcription by cockayne syndrome group B protein and histone methyltransferase G9a. Mol. Cell 27, 585–595. doi:10.1016/j.molcel.2007.06.021
Yuan, Y., Singh, R., and Reddy, R. (1989). Rat nucleolar 7-2 RNA is homologous to mouse mitochondrial RNase mitochondrial RNA-processing RNA. J. Biol. Chem. 264, 14835–14839. doi:10.1016/s0021-9258(18)63776-7
Yusupov, M. M., Yusupova, G. Z., Baucom, A., Lieberman, K., Earnest, T. N., Cate, J. H., et al. (2001). Crystal structure of the ribosome at 5.5 A resolution. Science 292, 883–896. doi:10.1126/science.1060089
Zafiropoulos, A., Tsentelierou, E., Linardakis, M., Kafatos, A., and Spandidos, D. A. (2005). Preferential loss of 5S and 28S rDNA genes in human adipose tissue during ageing. Int. J. Biochem. Cell Biol. 37, 409–415. doi:10.1016/j.biocel.2004.07.007
Zhang, T., Chen, P., Li, W., Sha, S., Wang, Y., Yuan, Z., et al. (2019). Cognitive deficits in mice lacking Nsun5, a cytosine-5 RNA methyltransferase, with impairment of oligodendrocyte precursor cells. Glia 67, 688–702. doi:10.1002/glia.23565
Zhao, J., Yuan, X., Frodin, M., and Grummt, I. (2003). ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol. Cell 11, 405–413. doi:10.1016/s1097-2765(03)00036-4
Zhao, Y., Rai, J., and Li, H. (2023). Regulation of translation by ribosomal RNA pseudouridylation. Sci. Adv. 9, eadg8190. doi:10.1126/sciadv.adg8190
Zhou, F., Aroua, N., Liu, Y., Rohde, C., Cheng, J., Wirth, A. K., et al. (2023). A dynamic rRNA ribomethylome drives stemness in acute myeloid leukemia. Cancer Discov. 13, 332–347. doi:10.1158/2159-8290.cd-22-0210
Zhou, F., Liu, Y., Rohde, C., Pauli, C., Gerloff, D., Kohn, M., et al. (2017). AML1-ETO requires enhanced C/D box snoRNA/RNP formation to induce self-renewal and leukaemia. Nat. Cell Biol. 19, 844–855. doi:10.1038/ncb3563
Zillner, K., Komatsu, J., Filarsky, K., Kalepu, R., Bensimon, A., and Nemeth, A. (2015). Active human nucleolar organizer regions are interspersed with inactive rDNA repeats in normal and tumor cells. Epigenomics 7, 363–378. doi:10.2217/epi.14.93
Keywords: ribosome, translation, rRNA, ribosomopathy, protein synthesis
Citation: Johnston R, Aldrich A and Lyons SM (2024) Roles of ribosomal RNA in health and disease. Front. RNA Res. 1:1331185. doi: 10.3389/frnar.2023.1331185
Received: 31 October 2023; Accepted: 11 December 2023;
Published: 19 January 2024.
Edited by:
Chandrasekhar Kanduri, University of Gothenburg, SwedenReviewed by:
Franck Martin, Université de Strasbourg (IBMC), FranceCopyright © 2024 Johnston, Aldrich and Lyons. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shawn M. Lyons, c21seW9uczFAYnUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.