 Nicole Simms
Nicole Simms John R. P. Knight
John R. P. Knight
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. RNA Res. , 06 September 2023
Sec. RNA Processing and Regulation
Volume 1 - 2023 | https://doi.org/10.3389/frnar.2023.1248236
Unlike DNA, RNA can be found in every sub-cellular compartment, where it is used to impart the genetic code or perform essential catalytic activities. As a result, damage to RNA is more spatially pervasive than damage to DNA and can have profound effects on gene expression and RNA-dependent activities. The past decade has seen the pathways involved in detecting and responding to damage of specific RNAs defined. These studies largely used high concentrations of tool compounds or deletion of essential factors for the response to RNA damage to study its effects. RNA is damaged by both endogenous and exogenous agents, with the effect of exogenous agents administered as therapeutics the focus of this review. In an effort to formalise studies into clinical RNA damage biology we propose 4 types of RNA damaging drug that we divide into 2 broad classes. Class 1 drugs result from synthesis using non-canonical nucleotides, which are incorporated into RNA in place of the canonical nucleotides. This class is subdivided depending on the outcome of this misincorporation on the nascent transcript. Class 2 drugs result in covalent ligation of moieties that alter RNA structure. This class is subdivided according to the functionality of the covalent ligation—class 2a are monovalent while class 2b are divalent. We discuss the evidence for and mechanisms of RNA damage as well as highlighting the unknown factors that require further investigation to determine the molecular mechanisms of these drugs.
RNA is the nucleic acid intermediate between the genes encoded in DNA and their functional output as protein. RNAs are also functional, carrying out essential and highly conserved catalytic activities across the cell, not least within the ribosome where the genetic code is translated into protein. Many of the agents we will discuss have broad effects on RNA, DNA, and metabolic processes, with the latter two mechanisms receiving the greatest interest and producing a dogma for many compounds. We highlight seminal work performed decades ago that define the effects these compounds have on RNA as well as emerging stories that are causing the field to revisit RNA as a forgotten target. These exciting developments are being fuelled by amazing advances in our abilities to study RNA in greater detail, which we comment on throughout.
There is much still to learn about how current clinical compounds damage RNA. First, let’s define the scope of this review. We will focus on small molecules that are approved for use in the clinic that cause covalent changes in the structures of RNA. As such, the application of therapeutic RNA species for genetic manipulation is out of our remit as these are not small molecules and rely on host cell machinery to alter host RNAs (Khorkova et al., 2023; Lightfoot and Smith, 2023). Similarly, the stunning advances in the design and application of small molecule RNA binding drugs is related but outside of our focus as they do not covalently alter RNAs (Tong et al., 2023). The recent advance made in targeted RNA has drawn the attention of big pharmaceutical companies, who have invested at least $1.5billion in this area at the time of press (Garber, 2023). This interest in RNA as a target for refined therapies should act as a catalyst to revisit the RNA-based mechanisms of action (MOAs) of non-targeted therapies, which has the potential to identify RNA targets with exceptional disease positioning. For example, if we understood the crucial RNA targets of compounds such as 5-fluorouracil to enable the design of targeted therapies against these RNAs there is a ready market of over 2million patients who already receive 5-FU each year worldwide.
What are the advantages of targeting RNA that may have contributed to the success of the compounds discussed here? As mentioned above, RNAs can have either coding or catalytic functions, meaning that its targeting can have a wide range of effects. Specific examples of this are included below. From a drug targeting perspective, RNA ticks a lot of boxes as an excellent approach to therapy. Firstly, it is abundant, with specific RNAs of interest individually also very abundant. The DNA and RNA content within a cell is about equal (Darzynkiewicz et al., 1979), but the impact of targeting RNA will have far greater instant impact. Only 3% of a cellular DNA is protein coding, whereas it is assumed that all of the RNA that is synthesised has a purpose in one way or another (Bellacosa and Moss, 2003). This is a particularly provocative point to consider when assessing the relative importance of RNA and DNA damage by non-targeting therapies. Does equal incidence of damage mean equal impact of damage? Another important consideration is the subcellular localisation of RNA. RNA is everywhere, present in almost all organelles and subcellular compartments. This may make it more amenable to damage than DNA which reside in the nucleus, plus a small amount in mitochondria. Given the complex metabolism required to activate many of the drugs to be discussed, we comment on the localisation of these metabolic enzymes and expression of organelle transporters that will also impact which nucleic acids are damaged. Obviously, restricted flux of drug metabolites to the nucleus could innately favour RNA damage due to its pervasive expression within cells.
Class 1 compounds, or their metabolites, are incorporated into nascent RNA strands during transcription by the endogenous DNA-dependent RNA transcriptase (Figure 1A). Many of the first drugs approved for chemotherapy fall into this category, with RNA damage a component of their multifaceted MOAs. We further subdivide these drugs into Classes 1a and 1b to differentiate the outcome of drug incorporation into RNA; class 1a do not halt RNA transcription, class 1b on the other hand induces early termination of transcription.
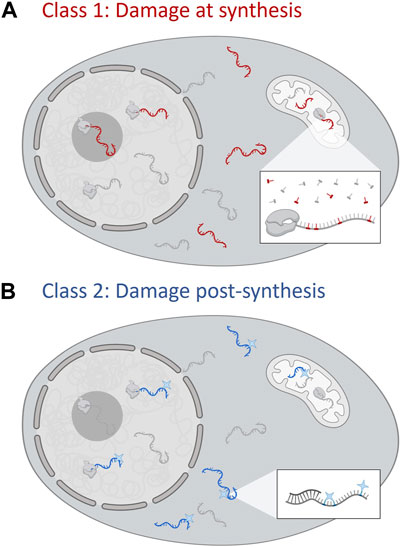
FIGURE 1. The two classes of RNA damage-inducing small molecules. (A) Class 1 RNA damaging agents produce covalent changes in RNA structure at the site of synthesis. Damaged RNAs, shown in red, contain individual nucleotides that are misincorporated and thus different to the canonical nucleotides. These can be made by any of the four mammalian cell RNA polymerases. Inset shows the mitochondrial RNA polymerase, POLRMT, utilising non-canonical nucleotides to synthesise damaged RNA. Class 1a compounds permit further RNA synthesis on the same chain. Class 1b compounds are chain terminators. (B) Class 2 RNA damage occurs post-synthesis by the covalent ligation of small molecules (blue stars) on to specific moeities of RNA. This is dependent on the association of the compound and RNA, which need to colocalise. Factors such as base pairing of nucleotides and occlusion by proteins reduces the propensity for damage (inset). Created with Biorender.com.
Class 1a drugs are nucleobase, nucleoside or nucleotide analogues that are erroneously incorporated in place of canonical purine or pyrimidine bases into RNA, usually having similar effect on DNA. For a summary of the canonical nucleobases, nucleoside and nucleotides see Figure 2. These compounds are subjected to processing by cellular metabolic enzymes to generate multiple active compounds that impinge nucleotide biology, earning them the broad name of antimetabolites. As well as being incorporated into RNA or DNA, these compounds can inhibit metabolic enzymes to prevent synthesis of new nucleic acid strands by depleting nucleotide pools.
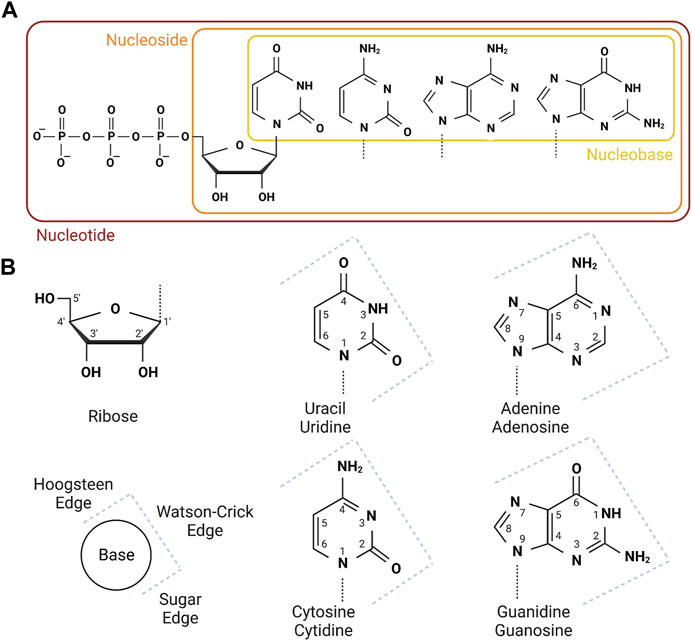
FIGURE 2. Nomenclature and numbering of nucleobases, nucleosides and nucleotides. (A) The four canonical nucleobases are shown boxed in yellow. These join with ribose to form nucleosides, boxed in orange. Dashed lines on the nucleobases shows the conjugation point to ribose. Nucleosides are sequentially phophorylated to mono-, di- and tri-phosphate nucleotides. Nucleotide triphosphates are the substrates for the RNA polymerases. (B) The ribose and named nucleobase (top) nucleoside/nucleotide (bottom) structures are shown, annotated with numbers for each carbon or nitrogen. This will assist with interpretation of small compound analouges where the nomenclature includes reference to the modified position. The key in the bottom left shows the three nucleotide edges as annotated on the four structures on the right. Created with Biorender.com.
5-flurouracil (5-FU), first synthesised as an anti-cancer compound by C. Heidelberger in 1957 (Duschinsky et al., 1957), is a classic example in which the nature of the drug is being re-explored in relation to RNA alterations and RNA damage. 5-FU is an uracil analogue with fluorine in place of a hydrogen at ring position 5. This gives 5-FU the ability to disrupt metabolism modifications that occur at position 5, and also alter the Hoogsteen edge when incorporated into nucleic acids. Throughout the late 1950s to early 2000s, research boomed in the study of 5-FU’s MOA, revealing profound RNA damaging effects of 5-FU in strains of E. coli, viruses and various forms of cancer (Horowitz and Chargaff, 1959; Andoh and Chargaff, 1965; Graham and Kirk, 1965; Kadowaki et al., 1965; Wilkinson and Crumley, 1977; Spiegelman et al., 1980a; Spiegelman et al., 1980b; Ghoshal and Jacob, 1997). Subsequently, the concept that 5-FU targets RNA was surpassed by the rise in interest of the role 5-FU plays in limiting DNA synthesis, which is engrained as the current dogma. In truth, compounds such as 5-FU have multiple MOAs, which we still do not fully understand (Figure 3A). This is despite 5-FU being given to around 2 million patients worldwide each year to treat diseases such as colorectal, breast and pancreatic cancer. The dogmatic MOA is that 5-FU inhibits thymidylate synthase (TS); an enzyme essential for the conversion of deoxyuridine monophosphate to deoxythymidine monophosphate (dTMP) via methylation of uridine at position 5. This reduces the pool of deoxythymidine triphosphate (dTTP) for use by DNA polymerases, stopping the synthesis of new DNA strands. At the same time 5-FU leads to incorporation of fluorodeoxyuridine triphosphate (FdUTP) into DNA hence replacing dTTP. It was shown that FdUTP in DNA is recognised as an error, stalling DNA synthesis and promoting cancer cell death (reviewed in Miura et al., 2010). Although this is the widely accepted view of 5-FU, the 5-FU metabolite fluorouridine triphosphate (FUTP) is in fact incorporated 3,000-15,000 fold more into RNA than FdUTP is into DNA, and knockdown of DNA repair proteins in colon cancer cell lines, fails to alter cancer cell sensitivity to 5-FU (Pettersen et al., 2011). Furthermore, a recent study showed that resistance to 5-FU is driven by functional changes in RNA binding proteins and not by changes in TS activity (Liang et al., 2022). Consistent with this, 5-FU blockade of TS activity has also been challenged; supplementing 5-FU treated cancer cells with dTMP, which is metabolically downstream of TS, did not significantly reverse 5-FU cytotoxicity (Pettersen et al., 2011), suggesting that 5-FU is not dependent on blocking TS activity. An intriguing recent study showed that this TS blockade can in fact act as a molecular trap for the RNA precursor metabolite FdUMP (Kurasaka et al., 2022). TS in 5-FU resistant colon cancer cells trap more FdUMP than cells naïve to treatment, resulting in reduced cell death due to reduced active 5-FU metabolites. How this relates to 5-FU incorporation into RNA is of great interest.
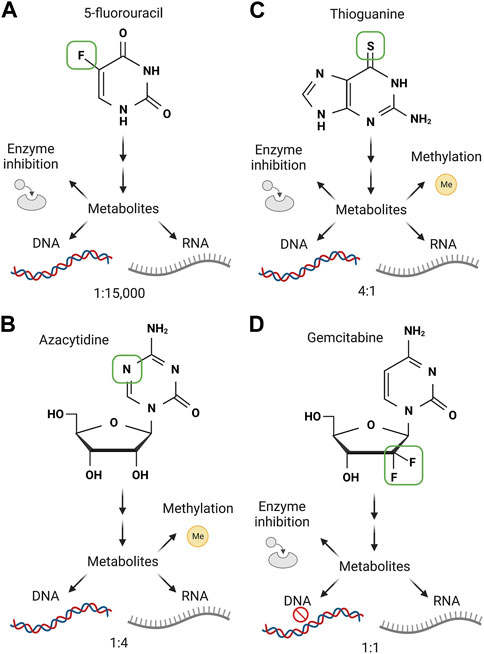
FIGURE 3. The multiple mechanisms of action of four class 1a compounds. The important mechanisms of action for 5-fluorouracil (A), azacytidine (B), thioguanine (C) and gemcitabine (D) are shown. The green box on each nucleobase/nucleoside analogue shows the divergent structure from the canonical structure. Each small molecule is metabolised by host cell enzymes into active compounds which have multiple effects. These are listed and enzyme inhibition, effects via nucleotide methylation, incorporation into DNA and incorporation into RNA. Below the DNA and RNA for each is an annotation for the relative incorporation rate between the two nucleic acids. Note, gemcitabine causes DNA chain termination, shown by stop sign. Created with Biorender.com.
In support of RNA damage as the determinant of the response to 5FU, mice administered uridine to compete with 5-FU metabolites for RNA incorporation 2 h after 5-FU dosing showed a reduction of intestinal epithelial cell apoptosis compared to 5-FU alone (Pritchard et al., 1997). Furthermore, the level of RNA incorporation of 5-FU correlates with cytotoxicity in human breast and colon cancer cell lines (Kufe and Major, 1981; Glazer and Lloyd, 1982). In agreement, measuring bulk incorporation of 5-FU metabolites into RNA identified that the presence of RNA damage correlates with drug efficacy in a panel of responsive and non-responsive model cell lines (Chen et al., 2023). In this study, it was also found that in cells from tumours that respond to 5-FU in the clinic it was the active incorporation of 5-FU metabolites into RNA that determined drug response, not any effect on overall RNA or DNA synthesis. Overall, these studies point to a considerable role for RNA damage in the cytotoxicity of 5-FU, but also highlights heterogeneity in the molecular effects of the drug. Understanding all of 5-FU’s molecular impacts, and how these vary between cell types, will be the first step towards directing 5-FU towards specific mechanisms and cell types. Previous research has shown that pairing other anti-cancer drugs or biological compounds with 5-FU can indeed impact how 5-FU is utilised in cells. In the colon cancer cell line HCT-8 pre-treatment with thymidine sensitised the cells to 5-FU and reduced viability in comparison to single treatments (Greenhalgh and Parish, 1990). Although the study used 5-FU concentrations above clinically achievable levels it is important to note that pre-treatment with thymidine increased the quantity of 5-FU incorporated into RNA by 1.5-fold in preference to DNA. Furthermore 5-FU increased overall nucleolar surface area and cell size, likely indicating an accumulation of pre-ribosomes containing 5-FU (Greenhalgh and Parish, 1990).
How pervasive is 5-FU incorporation into RNA? With new technological advances in the way we isolate and detect RNA, 5-FU-damaged RNA species such as mRNAs, rRNAs, microRNAs, tRNAs can readily be identified. Therizols et al. (2022), adapted a liquid chromatography method coupled to high resolution mass spectrometry and quantified the number of 5-FU containing ribosomes across multiple colorectal cancer cell lines as well as breast cancer and pancreatic cancer cell lines. The group found between 7 and 14 5-FU molecules were present in cytoplasmic ribosomes, confirming that these fluorinated ribosomes can escape quality control processes. These results were reflected in colorectal cancer xenografts derived from mice and tissue from patients treated with 5-FU. Interestingly, fluorinated ribosomes had altered activity and favoured the translation of specific mRNAs. Isolation of fluorinated ribosomes by sucrose gradient revealed increased translation of the cancer promoting mRNA insulin growth factor-1 receptor (IGF-1R) compared to non-fluorinated ribosomes (Therizols et al., 2022). Due to the multifaceted nature of 5-FU it is difficult to attribute these changes to alteration solely in rRNA, but the link to 5-FU damaged RNA in turn providing a resistance mechanism is of incredible interest. Of note the relative abundance of 5FUTP to canonical UTP after a 24 h incubation with 5-FU in HCT116 was quantified in a metabolomic analysis at ∼1:300 (Ser et al., 2016). Therizols et al. (2022) performed the exact same experiment (time, drug concentration, cell line) and observed a relative incorporation of 5FUTP:UTP into rRNA of ∼1:200. The convergence of this data is startling and indicates the extent of 5-FU damage to RNA is likely governed largely by drug exposure. Consistent with high levels of 5-FU incorporation in ribosomes, a genome wide screen in yeast found that 5-FU induced haploinsufficiency in only 7 genes, all of which are related to the processing of the ribosome subunits (Lum et al., 2004). This included four of the ten components of the RNA degrading exosome. These data imply that 5FU perturbs exosome activity in degrading unwanted RNA species, and furthermore implies this is the major pathway linked to cell fitness, at least in this yeast model. The biophysical changes in nucleic acids that result from 5-FU incorporation are not fully understood. Due to presumably random incorporation of 5-FU into RNA species it is difficult to study specific effects. Structurally, 5-FU does not disrupt Watson-Crick base pairing, instead sitting on the Hoogsteen edge. It has been suggested that Hoogsteen base pairing is more stable between 5-fluorouridine:adenosine than the canonical uridine:adenosine, but whether this has functional consequences is unknown (Abdulnur, 1976).
As well as rRNA, 5-FU incorporation has functional consequences in other RNAs. 5-FU stops the formation of pseudouridine, which is essential for the activity of U2 snRNA in splicing. 5-FU was found to inhibit U2 pseudouridylation and thus stop the splicing of pre-mRNAs in a Xenopus model where endogenous U2 snRNA was depleted (Zhao and Yu, 2006). Using both in vitro transcribed 5-FU RNAs and in vivo synthesised 5-FU RNA this work demonstrated how 5-FU metabolites have profound and specific effects on essential RNA biology. Similarly, 5-FU inhibits the post-transcriptional methylation of uridine at position 5 of most tRNAs, which is an essential modification for their activity. Using 5-FU as a chemical crosslinking probe in a novel FICC-seq methodology, Carter et al. defined the mammalian methyl-transferase responsible for uridine methylation due to the pervasive incorporation of 5-FU within tRNAs (Carter et al., 2019). Expanding this approach to identify more binding partners of 5-FU-damaged RNA should allow the molecular response to be defined in detail. As a final remarkable example of 5-FU incorporation having functional consequences, 5-FU in the mRNA synthesised from mutant TP53 alleles was able to promote readthrough of the mutation, restore p53 protein expression and induce apoptosis (Palomar-Siles et al., 2022). More studies need to be conducted to elucidate other RNA targets of 5-FU and how this can influence multiple biological factors such as cancer progression, resistance, known side effects and most importantly patient outcome.
Azacytidine is an antimetabolite developed in the late 1960s and is well documented for its effectiveness against leukaemia (Von Hoff et al., 1976; Veselý and Čihák, 1977). It is approved for use in patients with myelodysplastic syndrome (MDS) and acute myeloid leukaemia (AML). Azacytidine is a cytidine nucleoside analogue with a nitrogen substituted for carbon at position 5. This substitution leaves Watson-Crick base pairing unaffected and has been reported to have little effect on Hoogsteen pairing (Oellerich et al., 2019). Azacytidine has a similar MOA to 5-FU in terms of RNA and DNA incorporation (Figure 3B). Early studies showed that administration of azacytidine to leukemic mice depleted endogenous 2′deoxy-cytidine uptake into cells and blocked incorporation into DNA, but did not affect cytidine uptake into RNA. In addition, a heavy labelled radioactive azadeoxycytidine confirmed efficient integration into DNA (Von Hoff et al., 1976). In vitro investigations demonstrated cells arrested in G1 phase when treated with azacytidine did not synthesise DNA however if cells had already entered the S phase at the time of treatment, continued to synthesise DNA but failed to undergo mitosis (Tobey, 1972), suggesting azacytidine’s cytotoxic effects can be delayed and halts cell cycle progression. A well-documented MOA of azacytidine is the inhibition of cytidine methylation in de novo DNA strands. Since methylation in cancer can lead to selective gene expression, e.g., hypermethylation of tumour suppressor genes such as retinoblastoma (RB1) (Greger et al., 1989) and cell cycle checkpoint genes p16INK4A and p14(ARF) (Baur et al., 1999) and hence their repression, or the hypomethylation of mismatch repair (MMR) genes such as human-mut-L-homologue (hMLH1) (Fleisher et al., 2001), reactivation of cancer suppressive genes by azacytidine is a mechanism of interest (Christman, 2002). Although reports demonstrate the DNA incorporation and damaging effects by azacytidine, it is known that the drug is not limited to incorporating into DNA. In fact, 80%–90% of azacytidine is incorporated into RNA, with only 10%–20% incorporated into DNA (Li et al., 1970). Like its effects on DNA, mounting evidence confirms azacytidine inhibition of RNA methylation. The RNA methyltransferase DNMT2 enzyme methylates tRNAAsp at cytidine 38 (C38). tRNAAsp charging and delivery of aspartate to translating ribosomes is dependent on C38 methylation by DNMT2 (Shanmugam et al., 2015). Treating the colorectal cancer cell line HCT116 and the MCF7 breast cancer cell line with azacytidine significantly reduced the number of C38 methylated residues in tRNAAsp (Schaefer et al., 2009). Azacytidine also reduced the viability of myeloid leukemic cell lines and was associated with reduced methylation of C38 and C39 in tRNAAsp. Therefore, the cytotoxic effects of azacytidine may stem from failure to load tRNAs with amino acids, which would cause mistranslation and ribosome pausing. The MOA of azacytidine is broad and has been shown that when incorporated into newly synthesised RNA strands triggers endoplasmic reticulum stress response and cell death (Pawlak et al., 2022). Despite the success achieved with azacytidine treatment of leukemic cancers, in which the 12-month overall survival rate is 75% in MDS, 60% AML and 75% chronic myelomonocytic leukaemia (CMML), more than 60% of patients do not respond to the drug and over 80% do not experience complete disease remission (Helbig et al., 2019). It is interesting to speculate on how patient response is linked to RNA damage. Understanding the specifics of what dictates the levels of and response to azacytidine damaged RNA may be central to improving patient outcomes.
Other nucleobase/nucleoside analogues that cause Class 1a RNA damage are the thiopurines; azathioprine, 6-mercaptopurine and 6-thioguanine. These are commonly used to treat AML but have also proved effective as immunosuppressants and are prescribed to patients with chronic bowel diseases, psoriasis, and rheumatoid arthritis. The known MOA of thiopurines is their misincorporation in place of canonical guanosine bases during DNA synthesis while thiodeoxyguanosine triphosphate (TdGTP), also leads to the depletion of purine pools by inhibiting metabolic enzymes (Figure 3C). Thioguanines incorporated into DNA are subsequently heavily methylated to form S6-methylthioguanine in nascent DNA strands. Erroneous pairing of S6-methylthioguanine to thymidine is recognised as an error by the MMR system, further contributing to cytotoxicity (Swann et al., 1996). The azathioprine metabolite 6-thioguanosine can also inhibit GTP-dependent proteins such as the GTPase Rac1 (Tiede et al., 2003), highlighting the multiple mechanisms of thiopurine compounds (Coulthard et al., 2018).
Thiopurines have been known to incorporate into RNA for decades with 6-thioguanosine seen pervasively in rRNA and mRNA after exposure in cultured cells (MELVIN et al., 1978). Furthermore, high levels of 6-thioguanosine in RNA significantly reduced overall protein synthesis and protein half-life, which was attributed to an erroneous decoding of damaged RNA. Similar effects were seen on RNA and protein synthesis in vivo (Kwan et al., 1973). Consistent with deleterious effects of 6-thioguanosine upon incorporation into RNA, leukemic cells treated with 6-thioguanine exhibited greater adenosine-to-inosine RNA editing via induction of the editing enzyme ADAR2 (You et al., 2023). Furthermore, this work showed that 6-thioguanosine incorporation was biased towards shorter RNAs than longer RNAs, giving an intriguing insight into the unknown RNA damage biases of class 1a compounds. In terms of relative effects on DNA and RNA, 6-mercaptopurine metabolites were shown to be incorporated into DNA around 4 times more prevalently than RNA in a mouse lymphoma cell line (Tidd and Paterson, 1974).
Although thiopurines have proved highly successful in the treatment of malignant disease including patients undergoing organ transplant there is cause for concern of drug-induced secondary cancer, particularly after long term use (Karran et al., 2003; Karran, 2007). Further investigation is needed into the outcomes of thiopurines incorporation into RNA. Understanding the consequences of thiopurine damaged RNA may significantly change how the drugs are used and increase their safety in long term use.
The cytidine nucleoside analogue gemcitabine is used for the treatment of pancreatic tumours, as well in some cases of ovarian and bladder cancer. Structurally it differs from cytidine due to the inclusion of two fluorines in place of the 2′hydroxyl group and its adjacent hydrogen atom in the ribose sugar. Thus, unlike the other drugs in this class listed above, the base of gemcitabine is identical to its orthologue, with the difference in the sugar. Gemcitabine undergoes processing to impinge on multiple aspects of pyrimidine metabolism (Bergman et al., 2002) (Figure 3D). Deamination of gemcitabine and its metabolites generates uridine analogues containing the same difluororibose modification that inhibit thymidylate synthase much like 5-FU metabolites. However, gemcitabine is also metabolised into phosphorylated forms that can be incorporated into DNA and RNA. The relative incorporation into RNA or DNA was seen to be comparable for some cell lines, but divergent in others, with either a preference for RNA or DNA noted (Ruiz van Haperen et al., 1993). The cell line with the highest incorporation of gemcitabine into RNA was the most sensitive to drug treatment. Gemcitabine causes termination of DNA synthesis after the next nucleotide has been incorporated, through a well described mechanism that is difficult for cells to resolve (reviewed by Plunkett et al., 1995). In contrast, gemcitabine appears to have a less of an inhibitory effect on RNA synthesis, which persists after exposure, giving a strong time dependent increase in gemcitabine incorporation into RNA (Ruiz van Haperen et al., 1993).
Gemcitabine can be incorporated into RNA (and indeed DNA) as 2′,2′-difluorinated cytidine, but also as 2′,2′-difluorinated uridine, due to the action if cytidine deaminase (Bergman et al., 2002). Indeed, exposing cells to 2′,2′-difluorinated uridine resulted in incorporation into RNA and DNA, indicating that it can be phosphorylated and is an acceptable substrate for RNA and DNA polymerases (Veltkamp et al., 2008). Derissen et al. (2018) analysed the pharmacokinetics of the cytidine and uridine metabolites derived from gemcitabine in patient plasma and peripheral blood cells. They found higher levels of the cytidine analogues than the uridine analogues, but observed triphosphate forms of both nucleotides at levels where nucleotide incorporation occurs in vitro. Importantly, this work only quantified free nucleotides, with the gemcitabine metabolites incorporated into RNA and DNA removed by precipitation with perchloric acid prior to analysis. Determining the extent of RNA damage by these antimetabolites is now a priority to shed new light on their pharmacokinetics and target engagement.
Class 1b agents are also incorporated into cellular RNA, but in doing so terminate the transcription process, resulting in truncated RNAs. This termination occurs due to either i) the lack of a 3’ hydroxyl group for conjugation of the subsequent nucleotide or ii) the presence of other moieties that block the addition of the next nucleotide. They have primarily been designed and applied as antivirals. Here we focus on targeting the two types of retroviruses, whose RNA genomes are either reverse transcribed into DNA or simply duplicated directly into new RNA copies. Both activities rely on viral RNA dependent DNA/RNA polymerase enzymes, with these enzymes the target of the nucleoside antivirals. Compound design has aimed to specifically inhibit the synthesis of these DNA or RNA genomes by the viral DNA- or RNA-dependent RNA polymerases (Ami and Ohrui, 2021). But, given the structural similarity of the modified nucleotide antiviral metabolites to the canonical nucleotides there are significant off-target effects on the host cell polymerases.
Nucleoside antivirals were rapidly approved to combat the human immunodeficiency virus (HIV) pandemic due to their ability to halt reverse transcription of the HIV RNA genome (Feng, 2018). Combinations of these compounds remain the standard of care for people infected with HIV, who require constant suppression of reverse transcription to reduce further genomic incorporation of the viral genome. The first nucleotide antiviral approved to treat HIV was zidovudine (also called azidothymidine) which has an azide group at the 3′ position, causing termination of reverse transcription (Figure 4). Over 20 similar compounds with changes at the 3’ (as well as 1′, 2′ and 4′) positions have also been approved for treatment of HIV, with these and other compounds also approved for viruses such as hepatitis C virus (HCV) and SARS-CoV2 (Ami and Ohrui, 2021). Different modifications at each position change the efficacy of these compounds against specific viruses, while also altering the side effects associated with them (Figure 4). This is a clear indication that the molecular targets behind the side effects are the host (human) nucleotide polymerases, which will utilise the different antivirals to differing extents (Moyle, 2000). Fortunately, the human polymerases appear to be more selective than the viral polymerases, generating a therapeutic window for viral targeting, with the toxicity profile of these compounds reduced by increasing divergence from the canonical nucleotides (Ami and Ohrui, 2021) (Figure 4). An excellent example of this in action is the antiviral stavudine, which is thymidine analogue lacking both the 2′ and 3′ hydroxyl groups—i.e., the minimal divergence from the canonical nucleoside possible (Figure 4). Stavudine is effective as an anti-HIV agent, but patients suffer from severe side effects, leading to its removal from the market in 2020.
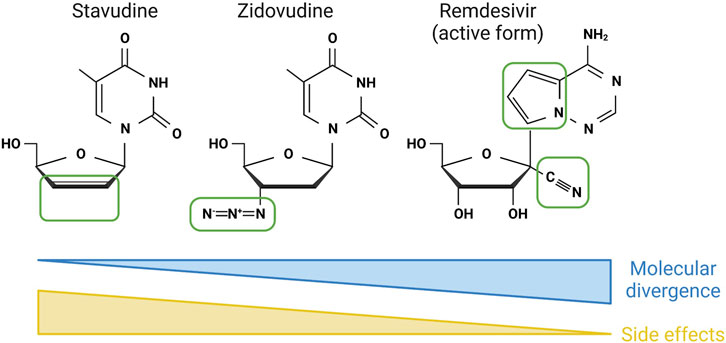
FIGURE 4. Molecular divergence inversely correlates with side-effects in class 1b compounds. The structures of three class 1b compounds with increasing molecular divergence from their canonical nucleoside—shown by areas in green boxes. As a general rule, the increase in divergence inversely correlates with the ability of the mammalian cell RNA polymerases to utilise these as substrates, and as a result the number of side effects. Created with Biorender.com.
The toxicities arising from these antivirals range from peripheral neuropathy, cardiomyopathy, lactic acidosis and cytopenia (Feng, 2018). Many of these side effects stem from changes in post-mitotic cell function, leading to an investigation of the mitochondrial DNA polymerase, rather than the nuclear DNA polymerases, as the molecular mechanism behind the side effects. Mitochondrial toxicity was first attributed to the inhibition of the mitochondrial DNA polymerase, but in some cases toxicities were observed in the absence of changes in mitochondrial DNA levels (Moyle, 2000). This led to an interest in the mitochondrial RNA polymerase (POLRMT) as an alternative source of toxicity. Arnold et al. (2012) used recombinant POLRMT and purified eukaryotic RNA polymerase II to screen for their ability to utilise 12 nucleoside antiviral analogues that cause termination of HCV RNA genome synthesis. The authors found that all the analogues were viable substrates for POLRMT but were less effectively used in their RNA polymerase II assays. Of note, the compounds which retain their 3′ hydroxyl groups did not cause chain termination by the host cell RNA polymerases. Likewise, a cell-based screen of compounds that had failed in trials against HCV due to toxicity found that they selectively inhibit POLRMT over host cell DNA polymerases, with little effect on the nuclear host RNA polymerases (Feng et al., 2016). Expanding this further, additional nucleoside antivirals were found to be substrates for POLRMT in vitro, with some resulting in chain termination despite possessing a 3′ hydroxyl while others did not terminate elongation (Jin et al., 2017; Lu et al., 2018). These methodologies open the possibility to define the substrate repertoire of nucleosides that POLRMT can use, which should now be applied to the obligate chain terminating antivirals lacking 3′ hydroxyls. Remdesivir, a prodrug that is metabolised to 2′cyano-modified adenosine analogue, is the only approved antiviral for patients with SARS-CoV2 infection and has shown remarkable tolerance. Consistent with this, molecular profiling of how remdesivir interacts with host cell polymerases showed very little impact (Xu et al., 2021) (Figure 4). Going forward, performing in silico simulations of POLRMT binding to antiviral nucleosides is an exciting development (Freedman et al., 2018), which may also be possible for the recently solved structures of the human nuclear RNA polymerases (Ramsay et al., 2020; Aibara et al., 2021; Misiaszek et al., 2021).
The success of antiviral nucleosides is an excellent demonstration of structure-function relationships between the analogues and the polymerases that require their canonical analogues for function. It remains to be seen if these compounds designed to be antivirals may be effective at targeting genetic human diseases due to their ability to target human polymerases. Resistance to nucleoside antivirals occur rapidly through mutations in the viral polymerases (Seley-Radtke and Yates, 2018), and it will be interesting to see if human polymerases are also able to become resistant under the same selection pressure.
Class 2 drugs are defined by their effects on RNA post-synthesis. For class 1 drugs that are incorporated at synthesis the final product is restrained by the input molecule. For example, following 5-FU treatment RNAs will have uridines incorporated that contained fluorine at position 5. However, for class 2 damage a variety of final products are possible, determined by which chemical component of the nucleic acid is altered and the nature of the drug. The most reactive elements of RNA, and indeed DNA, are the bases themselves, but damage can occur in several positions. The agents below have long been studied as damaging agents for DNA, but their ability to damage the same bases in RNA has been less studied and largely forgotten in recent years. Drawing on the expertise from DNA damaging agents we align our stratification of class 2 small molecules according to their damaging functionality (Fu et al., 2012). Class 2a compounds are monofunctional, causing damage at a single site on a given RNA, while class 2b compounds are bifunctional, causing damage to two sites, resulting in a molecular bridge between the two.
Alkylating agents are compounds that form direct covalent bonds with biological molecules. They are termed alkylating for their ability to deposit alkyl groups on DNA, as well as RNA and protein. These alkyl groups vary in size from the simplest methyl linkage through to large branched organic compounds. Hoogsteen base paired DNA is more susceptible to damage by alkylating agents (Xu et al., 2020), although it is not known if this is the same for RNA. Alkylating agents were the first compounds recognised as having anti-tumour effects, since the serendipitous (yet unfortunate) finding that the mustard gas used as a biological weapon during the World Wars, caused leukopenia in soldiers exposed to the toxin. This discovery gave the impetus to study the anti-neoplastic properties of nitrogen mustards (Colvin, 2003). The MOA of these drugs has been primarily attributed to the induction of DNA damage. This is despite the evident ability of these agents to damage the identical chemical structures also present in RNA. The importance of RNA damage is perhaps best demonstrated by the enzymatic removal of alkylation using the same conserved enzymes for both DNA and RNA repair (Aas et al., 2003). Thus, RNA repair is an evolutionarily conserved activity showing that nucleic acid methylation by cellular factors needs to be controlled and that this is perhaps equally important for damage to RNA or DNA. Consistent with this, methylation of specific mRNAs and tRNAs had a profound effect on translation, which could be readily reversed by the human dealkylating enzyme hABH3 (Ougland et al., 2004). The importance of this repair mechanism has been studied in relation to drug-induced DNA alkylation, but as yet no formal analysis of how RNA alkylation has been performed.
The simplest alkylating agents covalently conjugate methyl groups to specific reactive positions on the bases of DNA and RNA. Most of the analyses in this area have focused on the relative damage of DNA base positions and their consequences. This can guide assumptions about how RNA damage may mirror that of DNA but highlights the need to study this in more detail. The methylating agent temozolomide is approved for treatment of malignant glioma; it is a prodrug that undergoes non-enzymatic activation to form a highly reactive methyldiazonium ion (Denny et al., 1994) (Figure 5). This ion methylates a range of positions on DNA, and presumably RNA, with a preference for N7 of guanosine (70%), N3 of adenosine (9%) and O6 of guanosine (5%) (reviewed in Barciszewska et al., 2015). The relative preferences in RNA are not known but are likely different to those in DNA due to the higher solvent accessibility of RNA’s bases. Consistent with this, endogenous RNA in human cells in culture receives around 20-fold more oxidative damage per base than DNA (Hofer et al., 2005). Furthermore, the methylating tool compound methyl methanesulfonate, often used to mimic temozolomide damage in laboratory experiments, results in over 10-fold more RNA damage than DNA damage in E. coli (Vågbø et al., 2013) (Figure 5). Intriguingly, in this study an enrichment for RNA over DNA methylation was seen at the N1 position of adenosine, potentially due to this position participating in base pairing in helical DNA while being more readily solvent exposed in RNA. The overwhelmingly higher incidence of RNA damage over DNA damage by these monovalent methylating agents lead to the interesting dual hypothesis that RNA acts both as a sponge for damage to protect DNA and also as an early warning system of potential genotoxicity (Vågbø and Slupphaug, 2020). Understanding what responds to this early warning is of great interest, and if inhibited could enhance the effects of these RNA/DNA damaging agents in the future.
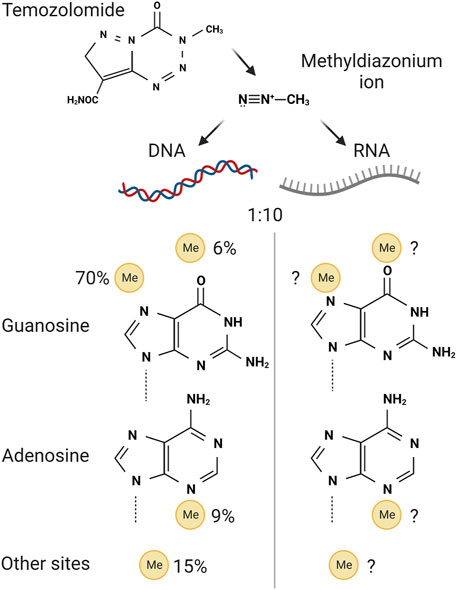
FIGURE 5. Activation and mechanism of action of temozolomide. Top, the class 2a prodrug temozolomide forms methyldiazonium ions through non-enzymatic processing in cells. These ions are highly reactive towards positions on the purine bases guanosine and adenosine, bottom. Only the bases are shown for simplicity, but these are damaged while present within DNA or RNA. The relative propensity for temozolomide-derived damage in DNA is shown on the left. The split in the propensity for damage in RNA is unknown, despite RNA damage being up to 10 times more prevalent than DNA damage. Created with Biorender.com.
The weaponised mustard gas that lead to the analysis of similar compounds for anti-cancer therapy was a bifunctional sulphur mustard called bis-chloroethyl sulphide (Singh et al., 2018). Using similar chemicals to treat patients brought success with the nitrogen mustards, with compounds such as chlorambucil used to treat leukemic patients. Subsequent improvements of this same bifunctional mustard moiety resulted in the widely used compound cyclophosphamide (Figure 6). Cyclophosphamide is used to treat several neoplasms, including lymphomas, myelomas, breast cancer, ovarian cancers, and neuroblastoma. Due to cyclophosphamide’s immunosuppressive properties, it is also used for treatment of autoimmune diseases such as lupus and multiple sclerosis (Ogino and Tadi, 2022). As a bifunctional alkylating agent, cyclophosphamide can form cross links between nucleic acid strands which induce apoptotic events. Surprisingly, the DNA adducts of the major active component of cyclophosphamide, phosphoramide mustard, primarily form monofunctional adducts on guanosines or the phosphate backbone with a maximum of only 12% bifunctional crosslinks observed (Povirk and Shuker, 1994) (Figure 6). This low incidence of crosslinking is consistently seen across all similar bifunctional alkylating compounds.
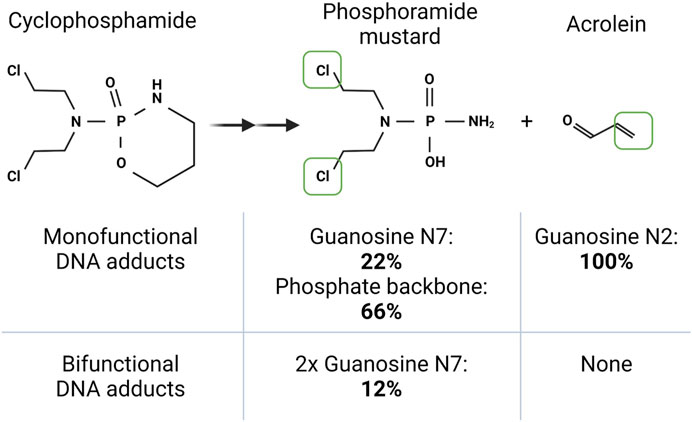
FIGURE 6. The bi- and mono-functional damaging agents of cyclophosphamide. Cyclophosphamide is metabolised in the liver to form two nucleic acid damaging agents. The class 2b bifunctional phosphoramide mustard, which covalently links to bases within DNA or RNA via its two chlorines (boxed in green). A second metabolite, acrolein, is a class 2a monofunctional (functional group in green) damaging agent able to damage DNA or RNA. The table shows the propensity of the two compounds to cause damage to specific positions in DNA. Note that for this bifunctional class 2b metabolite, bifunctional adducts are only formed 12% of the time. The specific site propensity for RNA damage by cyclophosphamide metabolites is not known. Created with Biorender.com.
Cyclophosphamide metabolic activation occurs in the liver with the first event being hydroxylation by the enzyme cytochrome P450 to non-toxic products 4-hydroxycyclophosphamide followed by aldophosphamide. Aldophosphamide is further processed by phosphodiesterases to acrolein and phosphamide mustard. Both products are capable of DNA alkylation and inducing cell death (reviewed by Voelcker, 2020) (Figure 6). Studies in mid to late 20th century evidence the ability of cyclophosphamide to also exert cytotoxic effects through RNA damage. Neo-natal mice treated with cyclophosphamide demonstrated a significant decrease in the incorporation of 14C uridine into newly synthesised RNA strands. Cyclophosphamide treatment reduced RNA synthesis in the livers and brains of these mice by 64% and 36% respectively. (Short and Gibson, 1973). In addition, studies using E. coli strains showed that the cyclophosphamide metabolite 4-hydroxyperoxycyclophosphamide (4-OOH-CP) could be used to determine the relative effects of cyclophosphamide on RNA and DNA. RNA was predominantly targeted by 4-OOH-CP, in a dose-dependent manner (Frank et al., 1981). Furthermore 4-OOH-CP targeted specific RNA species and prevented charging of tRNALeu with leucine amino acids. These RNA effects resulted in cyclophosphamide stalling protein synthesis (Frank et al., 1981), most likely through interrupting RNA synthesis and regulatory processes such as amino acid loading of tRNAs. Given that this work is 50 years old, these mechanisms should be revisited using newer technologies to elucidate other RNA targets that could be linked to cytotoxicity. The toxic by-product of cyclophosphamide metabolism, acrolein, can also directly damage DNA and RNA (Figure 6). Acrolein is released in the formation of aldophosphamide and is known to have mutagenic effects (Singh et al., 2014). Patients taking cyclophosphamide for prolonged times, for example, to treat vasculitis disease, have a higher risk of developing lymphoma or bladder cancer (Shang et al., 2015). Using A549 cancer cells a study showed the RNAse and DNase sensitive accumulation of acrolein within RNA-rich nucleoli (Wang et al., 2016). Functionally, this perturbed rRNA synthesis, as shown by reduced nascent RNA synthesis within nucleoli and a decrease in pre-rRNA 45S transcript levels. Consistent with defective ribosome synthesis, acrolein reduced global protein synthesis and the abundance of translating polysomes, while also causing structural changes to the nucleolus. How much the acrolein by-product contributes to cyclophosphamide cytotoxicity in cancer patients is an area that requires further investigation. Since acrolein does have devastating side effects, cyclophosphamide is paired with the acrolein neutralising medicine sodium-2-mercaptoethane-sulfonate (MESNA). MESNA however also induces adverse effects (Links and Lewis, 1999), therefore quality of life for the patient may be heavily reduced. The multimodal effects of cyclophosphamide’s metabolic products on nucleic acid biology offer a fascinating opportunity to study the relative roles of RNA and DNA damage, with huge scope of using this knowledge to improve patient treatment options.
Although not strictly alkylating agents, platinum-based compounds have very similar MOAs by bifunctionally linking to nucleic acids, usually via guanosine N7 but also via adenosine N7. The bifunctional nature of these agents is almost always engaged, usually through intrastrand linkage of eitherdirectly proximal bases. These compounds were discovered by chance in 1965 when Barnett Rosenberg found that products derived from platinum electrodes inhibited bacterial cell division (Weber, 2015). Cisplatin was the first platinum based drug and is frequently used to treat breast, bladder, ovarian and testicular cancers, but also proved effective against head and neck carcinomas, lymphomas and lung cancer (Eastman, 1987; Weber, 2015). The limitations of cisplatin are the multitude of adverse reactions, which brought about a 2nd generation of platinum drug called carboplatin. Unfortunately, both platinum drugs cause neurotoxicity and in addition patients become resistant to therapy after long term use. In effort to limit neurotoxicity and overcome resistance, the 3rd generation platinum drug oxaliplatin was developed. Oxaliplatin is commonly used to treat colorectal cancer in combination with 5-FU (Giacchetti et al., 2000). The MOA of platinum compounds is similar to nitrogen mustards; they form intra-strand and inter-strand cross links of DNA strands. Cisplatin and oxaliplatin form cross-links between guanine bases creating bulky adducts that alter DNA and RNA structure (Eastman, 1987; Chaney et al., 2005). What differs between the drugs are the DNA repair proteins that recognise cisplatin or oxaliplatin induced adducts (Wang and Lippard, 2005). Ultimately platinum drugs are lethal to cancer cells through removal of bulky adducts by MMR and damage recognition proteins. Breaks in DNA strands occur because of these repair processes, with the accumulation of DNA breaks leading to programmed cell death.
The current view that platinum drugs exert cytotoxic effects solely through DNA damage has recently been challenged. A mammalian RNA interference strategy revealed that oxaliplatin’s MOA differs significantly from those of cisplatin and carboplatin (Bruno et al., 2017). Short hairpin RNAs were designed to target genes involved in regulating cell cycle checkpoint and cell death and transfected cells were exposed to platinum drugs, resulting in a gene signature for each compound. Subsequent analysis revealed that oxaliplatin clustered with transcription and translation inhibitors such as actinomycin D, puromycin and rapamycin, while cisplatin and carboplatin showed similar activity to compounds that induce DNA damage repair. The DT40 avian cell line often used in gene manipulation studies due to its high targeted integration efficiency, was used to target DNA damage repair proteins. Forty DT40 cell lines each missing a specific repair protein revealed treatment with cisplatin enhanced cell sensitivity, specifically knock out of homologous recombination repair genes such as BRCA2. In contrast, BRCA2 knockout cells were significantly less sensitive to oxaliplatin (Bruno et al., 2017). This implies that oxaliplatin cytotoxicity is not predominantly occurring through platinum-based DNA lesions. In fact, high-performance liquid chromatography (HPLC) of DNA isolated from the ovarian cancer cell line A2780, confirmed that out of the expected 60% of platinum G-G adducts, 33% was retrieved in cisplatin treated cells while only 7% of adducts could be retrieved in cells dosed with oxaliplatin. Cisplatin is more reactive than oxaliplatin, producing a higher amount of G-G and A-G adducts, ultimately demonstrating that cisplatin binds DNA faster than oxaliplatin. Oxaliplatin adducts do occur, but at a much slower rate and lower frequency (Saris et al., 1996). In support of these findings, assessment of γH2AX expression; a common marker used to confirm the presence of DNA damage repair events, showed oxaliplatin treated cells lacked γH2AX foci compared to cisplatin, indicative of failure to initiate a DNA damage response (Bruno et al., 2017). Furthermore, the same work showed that oxaliplatin did not result in DNA damage measured by the comet assay, whereas cisplatin produced archetype comet tails indicative of damage to genomic DNA. In truth this will not be black and white, both cisplatin and oxaliplatin will cause damage to both DNA and RNA. For example, there is strong evidence that cisplatin damages RNA within ribosomes and arrests elongation along mRNAs in in vitro translation experiments (Heminger et al., 1997; Melnikov et al., 2016). However, the consensus now is that there is a preference for cisplatin to damage DNA and for oxaliplatin to damage RNA (Figure 7).
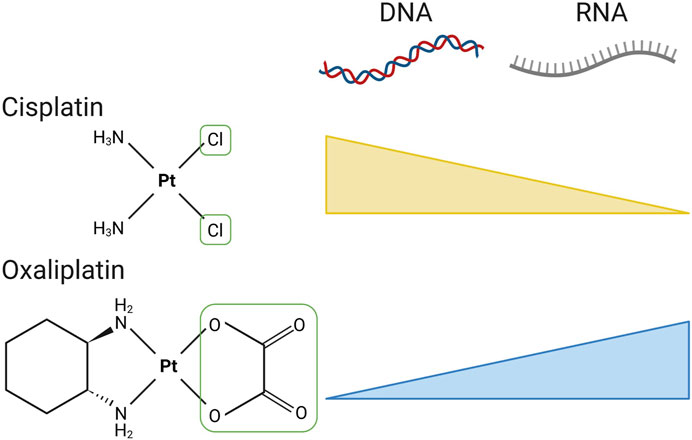
FIGURE 7. The divergent molecular damage caused by cisplatin and oxaliplatin. The chemical structures of cisplatin and oxaliplatin are shown, with the reactive leaving group(s) that permit bifunctional conjugation to purines boxed in green. Both compounds are able to form adducts with either DNA or RNA, with cisplatin showing a preference for DNA and oxaliplatin a preference for RNA. Created with Biorender.com.
Evidence also supports a role for oxaliplatin in inducing ribosomal stress, an effect likely linked to damage to rRNA. Ribosomal stress results in activation of the tumour suppressor protein p53, through the elevation of non-ribosome associated ribosomal proteins. Instead, these ribosomal proteins bind and sequester the E3 ubiquitin-protein ligase HDM2 (human double mutant minute 2 homolog) thereby relieving the HDM2 block on p53 and permitting p53 activation (Knight et al., 2016). Following this mechanism, silencing of ribosome proteins such as RPL11 conferred resistance to oxaliplatin, while no effect was seen with RPL11 knockdown in cisplatin treated cells (Bruno et al., 2017). To further support the role of oxaliplatin cytotoxicity mediated through the disruption of ribosome biogenesis, studies have explored redistribution of nucleolar markers and disruption of nucleolar structure in response to drug induced nucleolar stress. The nucleolus is structured into liquid subphases; the fibrillar centre (FC), dense fibrillar centre (DFC) and granular component (GC) (reviewed by Boisvert et al., 2007). Each phase is essential for the processing of rRNA transcripts and assembly of ribosome subunits. Transcription of rDNA occurs in the FC, while pre-rRNA is processed and modified to 18S, 5.8S and 28S rRNA transcripts in the DFC. Within the GC the pre-RNA transcripts mature, the 5.8S is combined with the 28S pre-RNA. Mature rRNAs form ribosome subunits that can then be exported to the cytoplasm (Boisvert et al., 2007). Nucleolar structures can be identified by the abundance of proteins located at each liquid subphase; nucleophosmin (NPM1) and SURF6 are distributed in the GC and fibrillarin (FBL) is abundant in the DFC. NPM1 is of particular interest as it is a chaperone protein that translocates from the nucleolus to the nucleoplasm and has a broad role in the cell including in ribosome biogenesis, DNA repair mechanisms to apoptosis (Box et al., 2016). NPM1 redistribution was demonstrated in A549 cells treated with oxaliplatin, using concentrations below that do not induce excessive cell death (Sutton et al., 2019). No change in NPM1 positioning in the nucleolus was evident in carboplatin treated cells while a small change was seen with cisplatin although only at concentrations with significant cell death. This re-localisation of NPM1 coincides with reduced rRNA transcription. After pulse labelling A549 with radiolabelled 32P followed by chasing with oxaliplatin, extensive NPM1 re-localisation was observed just 3 h after drug treatment, simultaneously to reductions in the 47S and 28S rRNA transcripts (Sutton and DeRose, 2021). This recent work has given cause to reanalyse oxaliplatin as an RNA-trophic drug. In agreement, a subsequent independent study also found oxaliplatin to result in disintergation of nucleolar liquid subphases, as a consequnce disrupting the rRNA assembly line (Schmidt et al., 2022). Although, as mentioned above, the molecular preferences of the platinum compounds are by no means black and white. For example, cisplatin is able to damage rRNA in yeast, a method developed based on the ability of platinum adducts to inhibit reverse transcription (Plakos and DeRose, 2017). Specific sites are seen to be enriched for platination in a dose responsive manner, which correspond to the solvent exposed sections of rRNA, inviting the speculation that the more RNA-rich human ribosome may present more target sites for platination (Petrov et al., 2014).
We have described the mechanisms and extent of RNA damage resulting directly from small molecules used in the clinic. Throughout we have also commented on some of the current topics in the field that need to be addressed to understand this RNA damage. Chief amongst these is the simple question of whether all damage is made equal. Is the incorporation of class 1 compounds even across the synthesis of all RNAs? Do class 2 RNAs have equal propensity to damage any RNA within a cell? Using the existing class 1 compounds it is not possible to target RNA damage to specific RNA molecules or to specific RNA sequences. However, there is evidence for a degree of specificity dependent on fundamental biology. On the face of it there seems a very high chance that RNA damage is not equal. Taking a class 1 compound as an example. These can be incorporated into RNA by one of four cellular RNA polymerases—RNA polymerase I, II and III in the nucleus and POLRMT in the mitochondria. These polymerases have very different properties, such as their localisation, speed, fidelity and perhaps most importantly in their products (Barba-Aliaga et al., 2021) (Figure 8). For example, mitochondrial RNA is enriched for uridine and unenriched for guanosine. Would this make POLRMT more sensitive to uridine analogues and less sensitive to guanosine analogues? Adding further to this is the observation that the two thymidine kinases (TK1 and TK2) that are required for activation of uridine and thymidine analogues are temporally and spatially divergent. Cytosolic TK1 is only expressed during S phase to fuel DNA synthesis, while mitochondrial TK2 is expressed uniformly across the cell cycle (Moyle, 2000). This leads to the assumption that mitochondrial activation of class 1 uracil analogues, such as 5-FU, could occur more readily than activation within the cytosol for the nuclear polymerases. There is also a lower ratio of pyrimidine triphosphates (uridine and cytosine) compared to purine triphosphates (adenosine and guanosine) within the mitochondria compared to the cytosol, suggesting a high flux of pyrimidine metabolism and usage within this organelle (Wheeler and Mathews, 2011). Together this demonstrates the potential for mitochondrial RNAs to be particularly sensitive to pyrimidine analogue drugs, but other means to drive specific incorporation biases likely exist and are yet to be discovered.
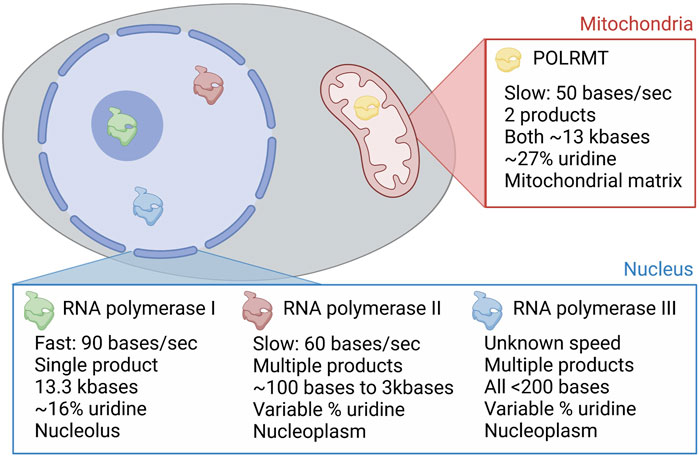
FIGURE 8. Molecular and spatial factors that influence class 1 compound RNA selectivity. The four RNA polymerases in mammalian cells are shown in different colours in the image of a cell. These are named in the pop out boxes, with POLRMT the only polymerase in the mitochondria, and RNA polymerases I, II and III found in the nucleus. RNA polymerase I is restricted to the nucleolus and RNA polymerases II and III to the nucleoplasm. These three different locations are shown, with molecular divergence annotated in the pop out boxes. Uridine percentages in the polymerase transcriptome are included as an example of the different decoding requirements of the polymerases, to accompany the discussion in the text. Created with Biorender.com.
As mentioned above, there is a stark coincident ratio of 5-FU incorporation into rRNA (∼1:200) to the observed 5FUTP:UTP nucleotide pool (∼1:300) (Ser et al., 2016; Therizols et al., 2022). These numbers perhaps mask an enriched incorporation into rRNA as Therizols and colleagues analysed 5-FU content across all rRNA after only 24 h of treatment. With the half-life of the ribosome on the order of days it is likely that the nascent 5-FU containing ribosomes have been diluted by older rRNA (Therizols et al., 2022). However, disagreeing with this is the use of direct 5-FU tracing where only a 50% reduction in the rate of incorporation was seen after inhibition of rRNA synthesis using a selective RNA polymerase I inhibitor (Chen et al., 2023).
For class 2 compounds, the limiting factor for causing damage to RNA is the accessibility of reactive sites in the RNA chain. This will be determined by several factors, i) the secondary structure of the RNA itself, ii) the binding and occlusion of areas of the RNA by protein factors and iii) the subcellular localisation of the RNA and the active metabolite of the drug. As an example of this in action, the ability of oxaliplatin to damage rRNA would seem to be higher than that of other RNAs due to its apparent tropism for nucleoli, where rRNAs are synthesised (Sutton and DeRose, 2021; Schmidt et al., 2022). Indeed, is it possible that this tropism is driven by the innate reactivity that oxaliplatin has for RNA, with nucleoli being one of the most RNA rich parts of the cell? Further work is required to define the RNA damage transcriptome of both class 1 and class 2 drugs. This potential for specificity in the RNA repertoire would provide a strong rationale for targeting the specifically damaged RNAs with RNA therapeutics. Understanding this could lead to patient benefit, either by enhancing RNA-linked cytotoxicity or minimising RNA-linked side-effects.
Technological developments mean that revisiting the roles of RNA damaging agents can now be done in exquisite detail. The ability to define the RNAs that are most at risk of damage by each of the agents described herein is within reach. Furthermore, doing this at nucleotide resolution will reveal the exact positions of RNA damage and how this relates to the MOA of each compound. The most promising methodologies available are sequencing based approaches. Platinum-seq, or Pt-seq, builds on the success of Mod-seq which uses small molecule probes to analyse RNA structure (Talkish et al., 2014). By using platinating agents as these small molecule probes it has been possible to define the locations of cisplatin induced RNA damage at nucleotide resolution. Applied to a yeast model system, this revealed specific sites within ribosomal RNA as targets for cisplatin adduct formation (Plakos and DeRose, 2017). The emergence of native RNA sequencing methods may also allow for the direct detection of drug-induced RNA modifications, similar to the analysis of endogenous RNA modifications such as methylation of adenosine at position 6 (Liu et al., 2019). Oxford Nanopore Technology’s direct RNA sequencing platform is a highly accurate method that will allow these analyses to be performed on single molecules at nucleotide resolution across the whole transcriptome. This is one of the reasons this technology was named Nature’s method of the year for 2022 (Nature Methods, 2023). This method will be useful for the analysis of class 1 compounds that incorporate into nascent RNA chains as well as class 2 compounds where damage occurs post-synthesis. Though the protein-based motor proteins and nanopores may struggle to accommodate the larger class 2 adducts.
The targets of platinum containing compounds have also been revealed in spatial and molecular detail using agents that have been modified to contain a click chemistry molecular handle (Moghaddam et al., 2015; White et al., 2016). It will be fascinating to apply a similar approach to other clinically important compounds. This will likely be most applicable to other class 2 compounds such as cyclophosphamide. The addition of clickable elements may restrict the incorporation of class 1 drugs into nascent RNA. One exception is the anti-viral compound zidovudine, which has an azide group as part of its structure. This azide group has previously been used for click chemistry conjugation to a fluorescent tag to demonstrate zidovudine incorporation into DNA in vitro and in cells (Koh et al., 2011).
Most of the drugs discussed here are used in combination with each other or other therapeutic agents. For example, oxaliplatin is used in combination with 5-fluorouracil as part of the extensively used FOLFOX chemotherapy regimen for cancers such as colorectal and pancreatic cancer and HIV antiviral nucleosides are only ever prescribed in combination. These therapies likely have greater efficacy in combination than as single agents due to augmented mechanisms of the combination, the additive effect of the drugs or a reduced likelihood of resistance developing. Modelling these combinations in cell lines, the Peters group showed in the 1990s that combination of gemcitabine with either cisplatin or paclitaxel increased the incorporation of gemcitabine metabolites into both RNA and to a lesser extent DNA (Moorsel et al., 1999; Kroep et al., 2000). Indeed, cisplatin could induce a 70-fold increase in gemcitabine found in RNA, while paclitaxel induced at least a 6-fold induction. Whether this contributes to the increased efficacy of gemcitabine is not fully understood. The FOLFOX combination treatment of oxaliplatin plus leucovorin followed by 5-FU significantly improved the treatment of patients with colorectal cancers compared to leucovorin and 5-FU treatment alone and has been the gold standard first line therapy since the turn of the millennium (Giacchetti et al., 2000). However, with concurrent treatment of oxaliplatin and 5-FU, oxaliplatin was found to reduce the incorporation of 5-FU into nascent RNA (Chen et al., 2023). This was attributed to the suppression of rRNA synthesis by oxaliplatin, reducing 5-FU usage by RNA polymerase I. Relating this to the above findings for gemcitabine combinations, it would be interesting to investigate the scheduled dosing of the two compounds, both in cell lines and in vivo models, to see if patient mimicking treatments also showed changes in RNA damage. Treatment with 5-FU prior to oxaliplatin may result in more RNA damage, given that 5-FU is able to stabilise Hoogsteen base pairing (Abdulnur, 1976), which makes nucleic acids more sensitive to platination (Xu et al., 2020).
Many of the compounds discussed herein are used in the treatment and management of the same diseases. The prime example being the antiviral nucleoside analogues used in combination against HIV, HCV, and other viral infections. Similarly, the standard of care therapies for some tumour types, but not others, rely on combination treatments with the compounds discussed here. Breast cancer regimens are based on 5-FU and cyclophosphamide, colorectal and other digestive tract tumours use 5-FU and oxaliplatin while leukaemias rely heavily on nucleoside analogues such as azacytidine and historically the thioguanines. Why do these drugs have efficacy in these specific indications, while they presumably do not in other tumours where different targeting modalities are used? A clue comes from Bruno et al. (2017), who identified a translation addiction signature that correlated with oxaliplatin response across multiple tumour types. Consistent with this, targeting of translation through alterations in the canonical translation machinery is more effective in certain tumours types, such as colorectal cancer, than others (Knight and Sansom, 2021; Smith et al., 2021). Given that RNA is essential for translation this translation addiction likely equates to RNA addiction. Providing further evidence for certain tumours being more susceptible to RNA targeting, promoting 5-FU incorporation into RNA was more cytotoxic in cell lines from tumours that are treated with 5-FU in the clinic than in cell lines from tumours that are not responsive to 5-FU in the clinic (Chen et al., 2023). This gives a clear indication that innate differences in susceptibility to RNA damage underly the response to RNA damaging agents and provides robust evidence that RNA damage is a strong determinate of clinical utility. The true measure of this will be observing that tumours that suffer higher RNA damage correlate with better outcomes in patients. Neoadjuvant trials of solid tumours or serial blood samples from liquid tumour patients may provide the opportunity to demonstrate exactly this.
RNA damage caused by clinical small molecules is a common phenomenon. Here we define the types of damage that result from these agents to provide a framework for the future study of clinical RNA damage biology. Studying the MOAs of current chemotherapies such as those described here was commonplace up until the time the human genome was published in 2001. In the early post-genomic era research moved towards developing targeted therapies (Sun et al., 2017) based on the improved knowledge of the genetics of human disease. Sadly, this was to the detriment of further analysis of RNA-based mechanisms of the contemporary therapeutics. Despite the change in research focus and development of alternative targeted therapies, many of the same chemotherapies are still used to this day. Advances in our understanding of how these RNA damaging agents function have focused on their effects on DNA or metabolic process for the past 20 years. Revisiting the forgotten RNA-based mechanisms of these chemotherapies using modern technologies will provide a fuller appreciation of their multiple effects and has the potential to revolutionise our understanding of what gives these drugs their clinical impact.
NS and JK contributed to the conception and scope of the review. NS and JK wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Funding was provided by the Division of Cancer Sciences, University of Manchester.
The author JK declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aas, P. A., Otterlei, M., Falnes, P. Ø., Vågbø, C. B., Skorpen, F., Akbari, M., et al. (2003). Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 421 (6925), 859–863. doi:10.1038/nature01363
Abdulnur, S. (1976). Why is 5-fluorouracil a mutagen? J. Theor. Biol. 58 (1), 165–175. doi:10.1016/0022-5193(76)90145-4
Aibara, S., Schilbach, S., and Cramer, P. (2021). Structures of mammalian RNA polymerase II pre-initiation complexes. Nature 594 (7861), 124–128. doi:10.1038/s41586-021-03554-8
Ami, E.-i., and Ohrui, H. (2021). Intriguing antiviral modified nucleosides: A retrospective view into the future treatment of COVID-19. ACS Med. Chem. Lett. 12 (4), 510–517. doi:10.1021/acsmedchemlett.1c00070
Andoh, T., and Chargaff, E. (1965). Formation and fate of abnormal ribosomes of E. coli cells treated with 5-fluorouracil. Proc. Natl. Acad. Sci. 54(4), 1181–1189. doi:10.1073/pnas.54.4.1181
Arnold, J. J., Sharma, S. D., Feng, J. Y., Ray, A. S., Smidansky, E. D., Kireeva, M. L., et al. (2012). Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. PLOS Pathog. 8 (11), e1003030. doi:10.1371/journal.ppat.1003030
Barba-Aliaga, M., Alepuz, P., and Pérez-Ortín, J. E. (2021). Eukaryotic RNA polymerases: the many ways to transcribe a gene. Front. Mol. Biosci. 8, 663209. doi:10.3389/fmolb.2021.663209
Barciszewska, A.-M., Gurda, D., Głodowicz, P., Nowak, S., and Naskręt-Barciszewska, M. Z. (2015). A new epigenetic mechanism of temozolomide action in glioma cells. PLOS ONE 10 (8), e0136669. doi:10.1371/journal.pone.0136669
Baur, A. S., Shaw, P., Burri, N., Delacretaz, F., Bosman, F. T., and Chaubert, P. (1999). Frequent methylation silencing of p15INK4b(MTS2) and p16INK4a (MTS1) in B-cell and T-cell lymphomas. Blood 94 (5), 1773–1781. doi:10.1182/blood.V94.5.1773
Bellacosa, A., and Moss, E. G. (2003). RNA repair: damage control. Curr. Biol. 13 (12), R482–R484. doi:10.1016/S0960-9822(03)00408-1
Bergman, A. M., Pinedo, H. M., and Peters, G. J. (2002). Determinants of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine). Drug Resist. Updat. 5 (1), 19–33. doi:10.1016/S1368-7646(02)00002-X
Boisvert, F.-M., van Koningsbruggen, S., Navascués, J., and Lamond, A. I. (2007). The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 8 (7), 574–585. doi:10.1038/nrm2184
Box, J. K., Paquet, N., Adams, M. N., Boucher, D., Bolderson, E., O’Byrne, K. J., et al. (2016). Nucleophosmin: from structure and function to disease development. BMC Mol. Biol. 17 (1), 19. doi:10.1186/s12867-016-0073-9
Bruno, P. M., Liu, Y., Park, G. Y., Murai, J., Koch, C. E., Eisen, T. J., et al. (2017). A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 23 (4), 461–471. doi:10.1038/nm.4291
Carter, J.-M., Emmett, W., Mozos, I. R., Kotter, A., Helm, M., Ule, J., et al. (2019). FICC-seq: a method for enzyme-specified profiling of methyl-5-uridine in cellular RNA. Nucleic Acids Res. 47 (19), e113. doi:10.1093/nar/gkz658
Chaney, S. G., Campbell, S. L., Bassett, E., and Wu, Y. (2005). Recognition and processing of cisplatin- and oxaliplatin-DNA adducts. Crit. Rev. Oncology/Hematology 53 (1), 3–11. doi:10.1016/j.critrevonc.2004.08.008
Chen, J.-K., Merrick, K. A., Kong, Y. W., Izrael-Tomasevic, A., Eng, G., Handly, E. D., et al. (2023). An RNA damage response network mediates the lethality of 5-FU in clinically relevant tumor types. bioRxiv. doi:10.1101/2023.04.28.538590
Christman, J. K. (2002). 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21 (35), 5483–5495. doi:10.1038/sj.onc.1205699
Coulthard, S. A., McGarrity, S., Sahota, K., Berry, P., and Redfern, C. P. F. (2018). Three faces of mercaptopurine cytotoxicity in vitro: methylation, nucleotide homeostasis, and deoxythioguanosine in DNA. Drug Metabolism Dispos. 46 (8), 1191–1199. doi:10.1124/dmd.118.081844
Darzynkiewicz, Z., Evenson, D. P., Staiano-Coico, L., Sharpless, T. K., and Melamed, M. L. (1979). Correlation between cell cycle duration and RNA content. J. Cell. Physiology 100 (3), 425–438. doi:10.1002/jcp.1041000306
Denny, B. J., Wheelhouse, R. T., Stevens, M. F. G., Tsang, L. L. H., and Slack, J. A. (1994). NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 33 (31), 9045–9051. doi:10.1021/bi00197a003
Derissen, E. J. B., Huitema, A. D. R., Rosing, H., Schellens, J. H. M., and Beijnen, J. H. (2018). Intracellular pharmacokinetics of gemcitabine, its deaminated metabolite 2′,2′-difluorodeoxyuridine and their nucleotides. Br. J. Clin. Pharmacol. 84 (6), 1279–1289. doi:10.1111/bcp.13557
Duschinsky, R., Pleven, E., and Heidelberger, C. (1957). The synthesis of 5-fluoropyrimidines. J. Am. Chem. Soc. 79 (16), 4559–4560. doi:10.1021/ja01573a087
Eastman, A. (1987). The formation, isolation and characterization of DNA adducts produced by anticancer platinum complexes. Pharmacol. Ther. 34 (2), 155–166. doi:10.1016/0163-7258(87)90009-X
Feng, J. Y. (2018). Addressing the selectivity and toxicity of antiviral nucleosides. Antivir. Chem. Chemother. 26, 204020661875852. doi:10.1177/2040206618758524
Feng, J. Y., Xu, Y., Barauskas, O., Perry, J. K., Ahmadyar, S., Stepan, G., et al. (2016). Role of mitochondrial RNA polymerase in the toxicity of nucleotide inhibitors of hepatitis C virus. Antimicrob. Agents Chemother. 60(2), 806–817. doi:10.1128/aac.01922-15
Fleisher, A. S., Esteller, M., Tamura, G., Rashid, A., Stine, O. C., Yin, J., et al. (2001). Hypermethylation of the hMLH1 gene promoter is associated with microsatellite instability in early human gastric neoplasia. Oncogene 20 (3), 329–335. doi:10.1038/sj.onc.1204104
Frank, S., Eck, W., Kersten, W., and Ogilvie, A. (1981). On the cytostatic mechanism of cyclophosphamide. Inhibition of aminoacylation of transfer RNA and induction of stringent control in Escherichia coli by 4-hydroperoxycyclophosphamide. Hoppe-Seyler´s Z. für Physiol. Chem., 362(2), 1059–1068. doi:10.1515/bchm2.1981.362.2.1059
Freedman, H., Winter, P., Tuszynski, J., Tyrrell, D. L., and Houghton, M. (2018). A computational approach for predicting off-target toxicity of antiviral ribonucleoside analogues to mitochondrial RNA polymerase. J. Biol. Chem. 293 (25), 9696–9705. doi:10.1074/jbc.RA118.002588
Fu, D., Calvo, J. A., and Samson, L. D. (2012). Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 12 (2), 104–120. doi:10.1038/nrc3185
Ghoshal, K., and Jacob, S. T. (1997). An alternative molecular mechanism of action of 5-fluorouracil, a potent anticancer drug. Biochem. Pharmacol. 53 (11), 1569–1575. doi:10.1016/S0006-2952(97)00040-3
Giacchetti, S., Perpoint, B., Zidani, R., Le Bail, N., Faggiuolo, R., Focan, C., et al. (2000). Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil–leucovorin as first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 18 (1), 136. doi:10.1200/jco.2000.18.1.136
Glazer, R. I., and Lloyd, L. S. (1982). Association of cell lethality with incorporation of 5-fluorouracil and 5-fluorouridine into nuclear RNA in human colon carcinoma cells in culture. Mol. Pharmacol. 21 (2), 468–473.
Graham, A. F., and Kirk, C. (1965). Effect of 5-fluorouracil on the growth of bacteriophage R17. J. Bacteriol. 90(4), 928–935. doi:10.1128/jb.90.4.928-935.1965
Greenhalgh, D. A., and Parish, J. H. (1990). Effect of 5-fluorouracil combination therapy on RNA processing in human colonic carcinoma cells. Br. J. Cancer 61 (3), 415–419. doi:10.1038/bjc.1990.91
Greger, V., Passarge, E., Höpping, W., Messmer, E., and Horsthemke, B. (1989). Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet. 83 (2), 155–158. doi:10.1007/BF00286709
Helbig, G., Chromik, K., Woźniczka, K., Kopińska, A. J., Boral, K., Dworaczek, M., et al. (2019). Real life data on efficacy and safety of azacitidine therapy for myelodysplastic syndrome, chronic myelomonocytic leukemia and acute myeloid leukemia. Pathology Oncol. Res. 25 (3), 1175–1180. doi:10.1007/s12253-018-00574-0
Heminger, K. A., Hartson, S. D., Rogers, J., and Matts, R. L. (1997). Cisplatin inhibits protein synthesis in rabbit reticulocyte lysate by causing an arrest in elongation. Archives Biochem. Biophysics 344 (1), 200–207. doi:10.1006/abbi.1997.0198
Hofer, T., Badouard, C., Bajak, E., Ravanat, J.-L., Mattsson, Å., and Cotgreave, I. A. (2005). Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol. Chem., 386(4), 333–337. doi:10.1515/BC.2005.040
Horowitz, J., and Chargaff, E. (1959). Massive incorporation of 5-fluorouracil into a bacterial ribonucleic acid. Nature 184 (4694), 1213–1215. doi:10.1038/1841213a0
Jin, Z., Kinkade, A., Behera, I., Chaudhuri, S., Tucker, K., Dyatkina, N., et al. (2017). Structure-activity relationship analysis of mitochondrial toxicity caused by antiviral ribonucleoside analogs. Antivir. Res. 143, 151–161. doi:10.1016/j.antiviral.2017.04.005
Kadowaki, K., Hosoda, J., and Maruo, B. (1965). Effects of actinomycin D and 5-fluorouracil on the formation of enzymes in Bacillus subtilis. Biochimica Biophysica Acta (BBA) - Nucleic Acids Protein Synthesis 103 (2), 311–318. doi:10.1016/0005-2787(65)90170-X
Karran, P., Offman, J., and Bignami, M. (2003). Human mismatch repair, drug-induced DNA damage, and secondary cancer. Biochimie 85 (11), 1149–1160. doi:10.1016/j.biochi.2003.10.007
Karran, P. (2006). Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br. Med. Bull. 79-80 (1), 153–170. doi:10.1093/bmb/ldl020
Khorkova, O., Stahl, J., Joji, A., Volmar, C.-H., and Wahlestedt, C. (2023). Amplifying gene expression with RNA-targeted therapeutics. Nat. Rev. Drug Discov. 22, 539–561. doi:10.1038/s41573-023-00704-7
Knight, J. R. P., Sbarrato, T., Stoneley, M., and Willis, A. E. (2016). in Ribosomes and stress – linked from birth to death. Editors R. A. Bradshaw, and P. D. B. T. E. C. B. Stahl (Waltham: Academic Press), 270–280.
Knight, J., and Sansom, O. (2021). Tuning protein synthesis for cancer therapy. Mol. Cell. Oncol. 8, 1884034. doi:10.1080/23723556.2021.1884034
Koh, M., Park, J., An, H., and Park, S. B. (2011). Ratiometric analysis of zidovudine (ZDV) incorporation by reverse transcriptases or polymerases via bio-orthogonal click chemistry. Chem. Commun. 47 (27), 7614–7616. doi:10.1039/C1CC12518D
Kroep, J. R., Giaccone, G., Tolis, C., Voorn, D. A., Loves, W. J. P., Groeningen, C. J. v., et al. (2000). Sequence dependent effect of paclitaxel on gemcitabine metabolism in relation to cell cycle and cytotoxicity in non-small-cell lung cancer cell lines. Br. J. Cancer 83 (8), 1069–1076. doi:10.1054/bjoc.2000.1399
Kufe, D. W., and Major, P. P. (1981). 5-Fluorouracil incorporation into human breast carcinoma RNA correlates with cytotoxicity. J. Biol. Chem. 256 (19), 9802–9805. doi:10.1016/S0021-9258(19)68695-3
Kurasaka, C., Nishizawa, N., Ogino, Y., and Sato, A. (2022). Trapping of 5-fluorodeoxyuridine monophosphate by thymidylate synthase confers resistance to 5-fluorouracil. ACS Omega 7 (7), 6046–6052. doi:10.1021/acsomega.1c06394
Kwan, S.-W., Kwan, S.-P., and Mandel, H. G. (1973). The incorporation of 6-thioguanine into RNA fractions and its effect on RNA and protein biosynthesis in mouse sarcoma 180 ascites cells. Cancer Res. 33 (5), 950–955.
Li, L. H., Olin, E. J., Buskirk, H. H., and Reineke, L. M. (1970). Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res. 30 (11), 2760–2769.
Liang, Y. Y., Bacanu, S., Sreekumar, L., Ramos, A. D., Dai, L., Michaelis, M., et al. (2022). CETSA interaction proteomics define specific RNA-modification pathways as key components of fluorouracil-based cancer drug cytotoxicity. Cell Chem. Biol. 29 (4), 572–585.e8. doi:10.1016/j.chembiol.2021.06.007
Lightfoot, H. L., and Smith, G. F. (2023). Targeting RNA with small molecules—a safety perspective. Br. J. Pharmacol. 2023. doi:10.1111/bph.16027
Links, M., and Lewis, C. (1999). Chemoprotectants. Drugs 57 (3), 293–308. doi:10.2165/00003495-199957030-00003
Liu, H., Begik, O., Lucas, M. C., Ramirez, J. M., Mason, C. E., Wiener, D., et al. (2019). Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun. 10 (1), 4079. doi:10.1038/s41467-019-11713-9
Lu, G., Bluemling, G. R., Mao, S., Hager, M., Gurale, B. P., Collop, P., et al. (2018). Simple in vitro assay to evaluate the incorporation efficiency of ribonucleotide analog 5′-triphosphates into RNA by human mitochondrial DNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 62 (2), e01830. doi:10.1128/aac.01830-17
Lum, P. Y., Armour, C. D., Stepaniants, S. B., Cavet, G., Wolf, M. K., Butler, J. S., et al. (2004). Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell 116 (1), 121–137. doi:10.1016/S0092-8674(03)01035-3
Melnikov, S. V., Söll, D., Steitz, T. A., and Polikanov, Y. S. (2016). Insights into RNA binding by the anticancer drug cisplatin from the crystal structure of cisplatin-modified ribosome. Nucleic Acids Res. 44 (10), 4978–4987. doi:10.1093/nar/gkw246
Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., and Keir, H. M. (1978). Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J. Biochem. 92 (2), 373–379. doi:10.1111/j.1432-1033.1978.tb12756.x
Misiaszek, A. D., Girbig, M., Grötsch, H., Baudin, F., Murciano, B., Lafita, A., et al. (2021). Cryo-EM structures of human RNA polymerase I. Nat. Struct. Mol. Biol. 28 (12), 997–1008. doi:10.1038/s41594-021-00693-4
Miura, K., Kinouchi, M., Ishida, K., Fujibuchi, W., Naitoh, T., Ogawa, H., et al. (2010). 5-FU metabolism in cancer and orally-administrable 5-FU drugs. Cancers 2 (3), 1717–1730. doi:10.3390/cancers2031717
Moghaddam, A. D., White, J. D., Cunningham, R. M., Loes, A. N., Haley, M. M., and DeRose, V. J. (2015). Convenient detection of metal–DNA, metal–RNA, and metal–protein adducts with a click-modified Pt(ii) complex. Dalton Trans. 44 (8), 3536–3539. doi:10.1039/C4DT02649G
Moorsel, C. J. A. v., Pinedo, H. M., Veerman, G., Bergman, A. M., Kuiper, C. M., Vermorken, J. B., et al. (1999). Mechanisms of synergism between cisplatin and gemcitabine in ovarian and non-small-cell lung cancer cell lines. Br. J. Cancer 80 (7), 981–990. doi:10.1038/sj.bjc.6690452
Moyle, G. (2000). Toxicity of antiretroviral nucleoside and nucleotide analogues. Drug Saf. 23 (6), 467–481. doi:10.2165/00002018-200023060-00001
Nature Methods (2023). Method of the year 2022: long-read sequencing. Nat. Methods 20 (1), 1. doi:10.1038/s41592-022-01759-x
Oellerich, T., Schneider, C., Thomas, D., Knecht, K. M., Buzovetsky, O., Kaderali, L., et al. (2019). Selective inactivation of hypomethylating agents by SAMHD1 provides a rationale for therapeutic stratification in AML. Nat. Commun. 10 (1), 3475. doi:10.1038/s41467-019-11413-4
Ogino, M. H., and Tadi, P. (2022). “Cyclophosphamide,” in StatPearls (Florida, USA: Treasure Island).
Ougland, R., Zhang, C.-M., Liiv, A., Johansen, R. F., Seeberg, E., Hou, Y.-M., et al. (2004). AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol. Cell 16 (1), 107–116. doi:10.1016/j.molcel.2004.09.002
Palomar-Siles, M., Heldin, A., Zhang, M., Strandgren, C., Yurevych, V., van Dinter, J. T., et al. (2022). Translational readthrough of nonsense mutant TP53 by mRNA incorporation of 5-Fluorouridine. Cell Death Dis. 13 (11), 997. doi:10.1038/s41419-022-05431-2
Pawlak, A., Chybicka, K., Zioło, E., Strządała, L., and Kałas, W. (2022). The contrasting delayed effects of transient exposure of colorectal cancer cells to decitabine or azacitidine. Cancers 14 (6), 1530. doi:10.3390/cancers14061530
Petrov, A. S., Bernier, C. R., Hsiao, C., Norris, A. M., Kovacs, N. A., Waterbury, C. C., et al. (2014). Evolution of the ribosome at atomic resolution. Proc. Natl. Acad. Sci. 111(28), 10251–10256. doi:10.1073/pnas.1407205111
Pettersen, H. S., Visnes, T., Vågbø, C. B., Svaasand, E. K., Doseth, B., Slupphaug, G., et al. (2011). UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 39 (19), 8430–8444. doi:10.1093/nar/gkr563
Plakos, K., and DeRose, V. J. (2017). Mapping platinum adducts on yeast ribosomal RNA using high-throughput sequencing. Chem. Commun. 53 (95), 12746–12749. doi:10.1039/C7CC06708A
Plunkett, W., Huang, P., Xu, Y. Z., Heinemann, V., Grunewald, R., and Gandhi, V. (1995). Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Seminars Oncol. 22, 3–10.
Povirk, L. F., and Shuker, D. E. (1994). DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Research/Reviews Genet. Toxicol. 318 (3), 205–226. doi:10.1016/0165-1110(94)90015-9
Pritchard, D. M., Watson, A. J. M., Potten, C. S., Jackman, A. L., and Hickman, J. A. (1997). Inhibition by uridine but not thymidine of p53-dependent intestinal apoptosis initiated by 5-fluorouracil: evidence for the involvement of RNA perturbation. Proc. Natl. Acad. Sci. 94(5), 1795–1799. doi:10.1073/pnas.94.5.1795
Ramsay, E. P., Abascal-Palacios, G., Daiß, J. L., King, H., Gouge, J., Pilsl, M., et al. (2020). Structure of human RNA polymerase III. Nat. Commun. 11 (1), 6409. doi:10.1038/s41467-020-20262-5
Ruiz van Haperen, V. W. T., Veerman, G., Vermorken, J. B., and Peters, G. J. (1993). 2′,2′-Difluoro-deoxycytidine (gemcitabine) incorporation into RNA and DNA of tumour cell lines. Biochem. Pharmacol. 46 (4), 762–766. doi:10.1016/0006-2952(93)90566-F
Saris, C. P., van de Vaart, P. M., Rietbroek, R. C., and Bloramaert, F. (1996). In vitro formation of DNA adducts by cisplatin, lobaplatin and oxaliplatin in calf thymus DNA in solution and in cultured human cells. Carcinogenesis 17 (12), 2763–2769. doi:10.1093/carcin/17.12.2763
Schaefer, M., Hagemann, S., Hanna, K., and Lyko, F. (2009). Azacytidine inhibits RNA methylation at DNMT2 target sites in human cancer cell lines. Cancer Res. 69 (20), 8127–8132. doi:10.1158/0008-5472.Can-09-0458
Schmidt, H. B., Jaafar, Z. A., Wulff, B. E., Rodencal, J. J., Hong, K., Aziz-Zanjani, M. O., et al. (2022). Oxaliplatin disrupts nucleolar function through biophysical disintegration. Cell Rep. 41 (6), 111629. doi:10.1016/j.celrep.2022.111629
Seley-Radtke, K. L., and Yates, M. K. (2018). The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antivir. Res. 154, 66–86. doi:10.1016/j.antiviral.2018.04.004
Ser, Z., Gao, X., Johnson, C., Mehrmohamadi, M., Liu, X., Li, S., et al. (2016). Targeting one carbon metabolism with an antimetabolite disrupts pyrimidine homeostasis and induces nucleotide overflow. Cell Rep. 15 (11), 2367–2376. doi:10.1016/j.celrep.2016.05.035
Shang, W., Ning, Y., Xu, X., Li, M., Guo, S., Han, M., et al. (2015). Incidence of cancer in ANCA-associated vasculitis: A meta-analysis of observational studies. PLOS ONE 10 (5), e0126016. doi:10.1371/journal.pone.0126016
Shanmugam, R., Fierer, J., Kaiser, S., Helm, M., Jurkowski, T. P., and Jeltsch, A. (2015). Cytosine methylation of tRNA-Asp by DNMT2 has a role in translation of proteins containing poly-Asp sequences. Cell Discov. 1 (1), 15010. doi:10.1038/celldisc.2015.10
Short, R. D., and Gibson, J. E. (1973). Biosynthesis of deoxyribonucleic acid, ribonucleic acid and protein in vivo by neonatal mice after a toxic dose of cyclophosphamide. Biochem. Pharmacol. 22 (24), 3181–3188. doi:10.1016/0006-2952(73)90092-0
Singh, R. K., Kumar, S., Prasad, D. N., and Bhardwaj, T. R. (2018). Therapeutic journery of nitrogen mustard as alkylating anticancer agents: historic to future perspectives. Eur. J. Med. Chem. 151, 401–433. doi:10.1016/j.ejmech.2018.04.001
Singh, V., Fedeles, B. I., Li, D., Delaney, J. C., Kozekov, I. D., Kozekova, A., et al. (2014). Mechanism of repair of acrolein- and malondialdehyde-derived exocyclic guanine adducts by the α-ketoglutarate/Fe(II) dioxygenase AlkB. Chem. Res. Toxicol. 27 (9), 1619–1631. doi:10.1021/tx5002817
Smith, R. C. L., Kanellos, G., Vlahov, N., Alexandrou, C., Willis, A. E., Knight, J. R. P., et al. (2021). Translation initiation in cancer at a glance. J. Cell Sci. 134 (1), jcs248476. doi:10.1242/jcs.248476
Spiegelman, S., Nayak, R., Sawyer, R., Stolfi, R., and Martin, D. (1980a). Potentiation of the anti-tumor activity of 5FU by thymidine and its correlation with the formation of (5FU) RNA. Cancer 45 (S5), 1129–1134. doi:10.1002/1097-0142(19800315)45:5+<1129::AID-CNCR2820451317>3.0.CO;2-I
Spiegelman, S., Sawyer, R., Nayak, R., Ritzi, E., Stolfi, R., and Martin, D. (1980b). Improving the anti-tumor activity of 5-fluorouracil by increasing its incorporation into RNA via metabolic modulation. Proc. Natl. Acad. Sci. 77(8), 4966–4970. doi:10.1073/pnas.77.8.4966
Sun, J., Wei, Q., Zhou, Y., Wang, J., Liu, Q., and Xu, H. (2017). A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 11 (5), 87. doi:10.1186/s12918-017-0464-7
Sutton, E. C., and DeRose, V. J. (2021). Early nucleolar responses differentiate mechanisms of cell death induced by oxaliplatin and cisplatin. J. Biol. Chem. 296, 100633. doi:10.1016/j.jbc.2021.100633
Sutton, E. C., McDevitt, C. E., Prochnau, J. Y., Yglesias, M. V., Mroz, A. M., Yang, M. C., et al. (2019). Nucleolar stress induction by oxaliplatin and derivatives. J. Am. Chem. Soc. 141 (46), 18411–18415. doi:10.1021/jacs.9b10319
Swann, P. F., Waters, T. R., Moulton, D. C., Xu, Y.-Z., Zheng, Q., Edwards, M., et al. (1996). Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 273(5278), 1109–1111. doi:10.1126/science.273.5278.1109
Talkish, J., May, G., Lin, Y., Woolford, J. L., and McManus, C. J. (2014). Mod-seq: high-throughput sequencing for chemical probing of RNA structure. RNA 20 (5), 713–720. doi:10.1261/rna.042218.113
Therizols, G., Bash-Imam, Z., Panthu, B., Machon, C., Vincent, A., Ripoll, J., et al. (2022). Alteration of ribosome function upon 5-fluorouracil treatment favors cancer cell drug-tolerance. Nat. Commun. 13 (1), 173. doi:10.1038/s41467-021-27847-8
Tidd, D. M., and Paterson, A. R. P. (1974). A biochemical mechanism for the delayed cytotoxic reaction of 6-mercaptopurine. Cancer Res. 34 (4), 738–746.
Tiede, I., Fritz, G., Strand, S., Poppe, D., Dvorsky, R., Strand, D., et al. (2003). CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Investigation 111 (8), 1133–1145. doi:10.1172/JCI16432
Tobey, R. A. (1972). Effects of cytosine arabinoside, daunomycin, mithramycin, azacytidine, adriamycin, and camptothecin on mammalian cell cycle traverse. Cancer Res. 32 (12), 2720–2725.
Tong, Y., Lee, Y., Liu, X., Childs-Disney, J. L., Suresh, B. M., Benhamou, R. I., et al. (2023). Programming inactive RNA-binding small molecules into bioactive degraders. Nature 618 (7963), 169–179. doi:10.1038/s41586-023-06091-8
Vågbø, C. B., and Slupphaug, G. (2020). RNA in DNA repair. DNA Repair 95, 102927. doi:10.1016/j.dnarep.2020.102927
Vågbø, C. B., Svaasand, E. K., Aas, P. A., and Krokan, H. E. (2013). Methylation damage to RNA induced in vivo in Escherichia coli is repaired by endogenous AlkB as part of the adaptive response. DNA Repair 12 (3), 188–195. doi:10.1016/j.dnarep.2012.11.010
Veltkamp, S., Pluim, D., van Eijndhoven, M., Bolijn, M., Ong, F., Govindarajan, R., et al. (2008). New insights into the pharmacology and cytotoxicity of gemcitabine and 2',2'-difluorodeoxyuridine. Mol. Cancer Ther. 7 (8), 2415–2425. doi:10.1158/1535-7163.MCT-08-0137
Veselý, J., and Čihák, A. (1977). Incorporation of a potent antileukemic agent, 5-Aza-2′-deoxycytidine, into DNA of cells from leukemic mice. Cancer Res. 37 (10), 3684–3689.
Voelcker, G. (2020). The mechanism of action of cyclophosphamide and its consequences for the development of a new generation of oxazaphosphorine cytostatics. Sci. Pharm. 88 (4), 42. doi:10.3390/scipharm88040042
Von Hoff, D. D., Slavik, M., and Muggia, F. M. (1976). 5-Azacytidine: A new anticancer drug with effectiveness in acute myelogenous leukemia. Ann. Intern. Med. 85 (2), 237–245. doi:10.7326/0003-4819-85-2-237
Wang, D., and Lippard, S. J. (2005). Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 4 (4), 307–320. doi:10.1038/nrd1691
Wang, H.-t., Chen, T.-y., Weng, C.-w., Yang, C.-h., and Tang, M.-s. (2016). Acrolein preferentially damages nucleolus eliciting ribosomal stress and apoptosis in human cancer cells. Oncotarget 7 (49), 80450–80464. doi:10.18632/oncotarget.12608
Weber, G. F. (2015). “DNA damaging drugs,” in Molecular therapies of cancer. Editor G. F. Weber (Cham: Springer International Publishing), 9–112.
Wheeler, L. J., and Mathews, C. K. (2011). Nucleoside triphosphate pool asymmetry in mammalian mitochondria *. J. Biol. Chem. 286 (19), 16992–16996. doi:10.1074/jbc.M111.236968
White, J. D., Haley, M. M., and DeRose, V. J. (2016). Multifunctional Pt(II) reagents: covalent modifications of Pt complexes enable diverse structural variation and in-cell detection. Accounts Chem. Res. 49 (1), 56–66. doi:10.1021/acs.accounts.5b00322
Wilkinson, D. S., and Crumley, J. (1977). Metabolism of 5-fluorouracil in sensitive and resistant Novikoff hepatoma cells. J. Biol. Chem. 252 (3), 1051–1056. doi:10.1016/S0021-9258(19)75204-1
Xu, Y., Barauskas, O., Kim, C., Babusis, D., Murakami, E., Kornyeyev, D., et al. (2020). Off-target in vitro profiling demonstrates that remdesivir is a highly selective antiviral agent. Antimicrob. Agents Chemother. 65 (2). doi:10.1128/aac.02237-20
Xu, Y., Manghrani, A., Liu, B., Shi, H., Pham, U., Liu, A., et al. (2020). Hoogsteen base pairs increase the susceptibility of double-stranded DNA to cytotoxic damage. J. Biol. Chem. 295 (47), 15933–15947. doi:10.1074/jbc.RA120.014530
You, X.-J., Li, L., Ji, T.-T., Xie, N.-B., Yuan, B.-F., and Feng, Y.-Q. (2023). 6-Thioguanine incorporates into RNA and induces adenosine-to-inosine editing in acute lymphoblastic leukemia cells. Chin. Chem. Lett. 34 (1), 107181. doi:10.1016/j.cclet.2022.01.074
Keywords: RNA damage, antimetabolites, alkylating agents, chemotherapies, antivirals
Citation: Simms N and Knight JRP (2023) RNA damage: the forgotten target of clinical compounds. Front. RNA Res. 1:1248236. doi: 10.3389/frnar.2023.1248236
Received: 26 June 2023; Accepted: 23 August 2023;
Published: 06 September 2023.
Edited by:
Nikolay Shirokikh, Australian National University, AustraliaReviewed by:
Sarah Walker, University at Buffalo, United StatesCopyright © 2023 Simms and Knight. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John R. P. Knight, am9obi5rbmlnaHRAbWFuY2hlc3Rlci5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.