 Haiyang Zhang
Haiyang Zhang Maoting Tang
Maoting Tang Deyuan Li1,2
Deyuan Li1,2 Liangkang Lin
Liangkang Lin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Public Health , 19 June 2024
Sec. Infectious Diseases: Epidemiology and Prevention
Volume 12 - 2024 | https://doi.org/10.3389/fpubh.2024.1410672
This article is part of the Research Topic Nontuberculous Mycobacterial Infections in Animals And Humans: Pathogenesis, Diagnosis, Prevention, Treatment, And Epidemiology View all 13 articles
Non-tuberculous mycobacteria (NTM) infections pose a significant public health challenge worldwide, affecting individuals across a wide spectrum of immune statuses. Recent epidemiological studies indicate rising incidence rates in both immunocompromised and immunocompetent populations, underscoring the need for enhanced diagnostic and therapeutic approaches. NTM infections often present with symptoms similar to those of tuberculosis, yet with less specificity, increasing the risk of misdiagnosis and potentially adverse outcomes for patients. Consequently, rapid and accurate identification of the pathogen is crucial for precise diagnosis and treatment. Traditional detection methods, notably microbiological culture, are hampered by lengthy incubation periods and a limited capacity to differentiate closely related NTM subtypes, thereby delaying diagnosis and the initiation of targeted therapies. Emerging diagnostic technologies offer new possibilities for the swift detection and accurate identification of NTM infections, playing a critical role in early diagnosis and providing more accurate and comprehensive information. This review delineates the current molecular methodologies for NTM species and subspecies identification. We critically assess the limitations and challenges inherent in these technologies for diagnosing NTM and explore potential future directions for their advancement. It aims to provide valuable insights into advancing the application of molecular diagnostic techniques in NTM infection identification.
Non-tuberculous mycobacteria (NTM) encompass a diverse assembly of species distinct from the Mycobacterium tuberculosis complex (MTBC) and Mycobacterium leprae (M. leprae). Identified initially in the late 19th century, NTM are characterized as Gram-positive, rod-shaped bacteria, often exhibiting branching or curved formations. The cell wall structure of NTM, rich in lipids, phenolic glycolipids, and mycolic acids, is a key biological marker, rendering these bacteria acid-fast positive, similar to Mycobacterium tuberculosis (MTB), as demonstrated by staining techniques like the Ziehl-Neelsen staining method (1, 2). However, while useful, acid-fast staining alone cannot distinguish between MTB and NTM, necessitating further molecular diagnostic interventions for accurate identification. NTM is classified based on their growth rate in subculture as either rapidly growing mycobacteria (RGM), with a growth cycle of less than 7 days, or slowly growing mycobacteria (SGM), requiring 7 days or more to grow. To date, approximately 200 species of NTM have been identified, which can be further differentiated into various subspecies. The distinct clinical features and treatment responses of different subspecies play a crucial role in understanding the clinical management and therapeutic strategies for diseases associated with NTM (3, 4). The species diversity of NTM significantly varies across global regions, with the pathogenic potential of NTM species being geographically distinct. Additionally, in developed nations, the disease burden attributable to NTM infections now exceeds that of MTB (5–8).
NTM infections can manifest across multiple tissue and organ systems, such as the respiratory, central nervous, lymphatic, articular, and dermal systems. Notably, pulmonary infections account for roughly 90% of these cases (9, 10). The clinical manifestations of pulmonary infections caused by NTM are characterized by their nonspecific nature. Although chronic or recurrent coughing is almost universally reported among patients, there is variability in symptoms such as expectoration, fatigue, malaise, dyspnea, fever, hemoptysis, chest pain, and weight loss. In children aged 1 to 5 years, NTM primarily infects submandibular, cervical, or preauricular lymph nodes, indicating its capacity to also target tissues beyond the commonly affected systems (11, 12). Currently, approximately 80% of culture-confirmed NTM lymphadenitis cases are caused by the Mycobacterium avium complex (MAC) (13). Recent studies emphasize the evolving landscape of NTM species affecting lymph nodes, highlighting the need for advanced molecular diagnostics to accurately identify and tailor treatment strategies. Furthermore, studies reported a steady increase in the variety of NTM species isolated from lymph nodes, including Mycobacterium lentiflavum (M. lentiflavum) (14).
Accurate diagnosis and treatment of NTM infections necessitate a nuanced and individualized assessment of therapeutic risks versus benefits. The widespread environmental presence of NTM poses substantial challenges for clinicians in accurately identifying infections. Detecting NTM in non-sterile respiratory specimens does not necessarily indicate pathogenic involvement in pulmonary conditions, potentially indicating colonization, transient infection without clinical disease, or sample contamination. Furthermore, the pathogenic potential of NTM significantly depends on the host’s immune status and the specific anatomical site of culture extraction. When NTM is isolated from sterile sites, including blood, tissues, cerebrospinal fluid, pleural fluid, or the brain, its presence almost always indicates clinical significance (15).
Traditionally, the identification of species within the NTM complex has relied on time-consuming biochemical tests and methods based on phenotypic characteristics, which often fail to provide accurate identification results. Pathogenic culture, despite its limitations, has historically been the gold standard for NTM identification and remains critical for drug sensitivity testing (DST) (16). While culture is indispensable for DST, molecular diagnostics play an increasingly crucial role in genotypic identification, offering more rapid and accurate results (4). The effectiveness of culture-based identification is markedly impacted by the growth kinetics of mycobacteria. Specifically, slow-growing strains of NTM often necessitate upwards of 7 days to yield discernible colonies. This contrasts sharply with molecular techniques, which can often identify NTM species within hours. Additionally, specific NTM species and subspecies, such as Mycobacterium haemophilum (M. haemophilum), Mycobacterium marinum (M. marinum), Mycobacterium ulcerans (M. ulcerans), and MAC, often cannot be identified through conventional culturing methods due to their stringent dependency on certain nutrients or specific culturing conditions (17–19).
To address these challenges, advancements in molecular diagnostics have revolutionized the identification of NTM infections, offering substantial improvements over traditional methods in terms of accuracy, diagnostic sensitivity, speed, and cost-effectiveness, especially in differentiating NTM species and genotyping. Several detection methods for NTM have been developed by adapting techniques originally designed for mycobacterial identification. While targeted single-gene sequencing effectively identifies most NTM species, the distinction at the subspecies level often requires multigene sequencing approaches (20, 21). Table 1 summarizes the comparison of DNA-based molecular techniques for identification of genetically closa NTM species, underscoring the critical role of these diagnostics in the precise detection of NTM (22–54).
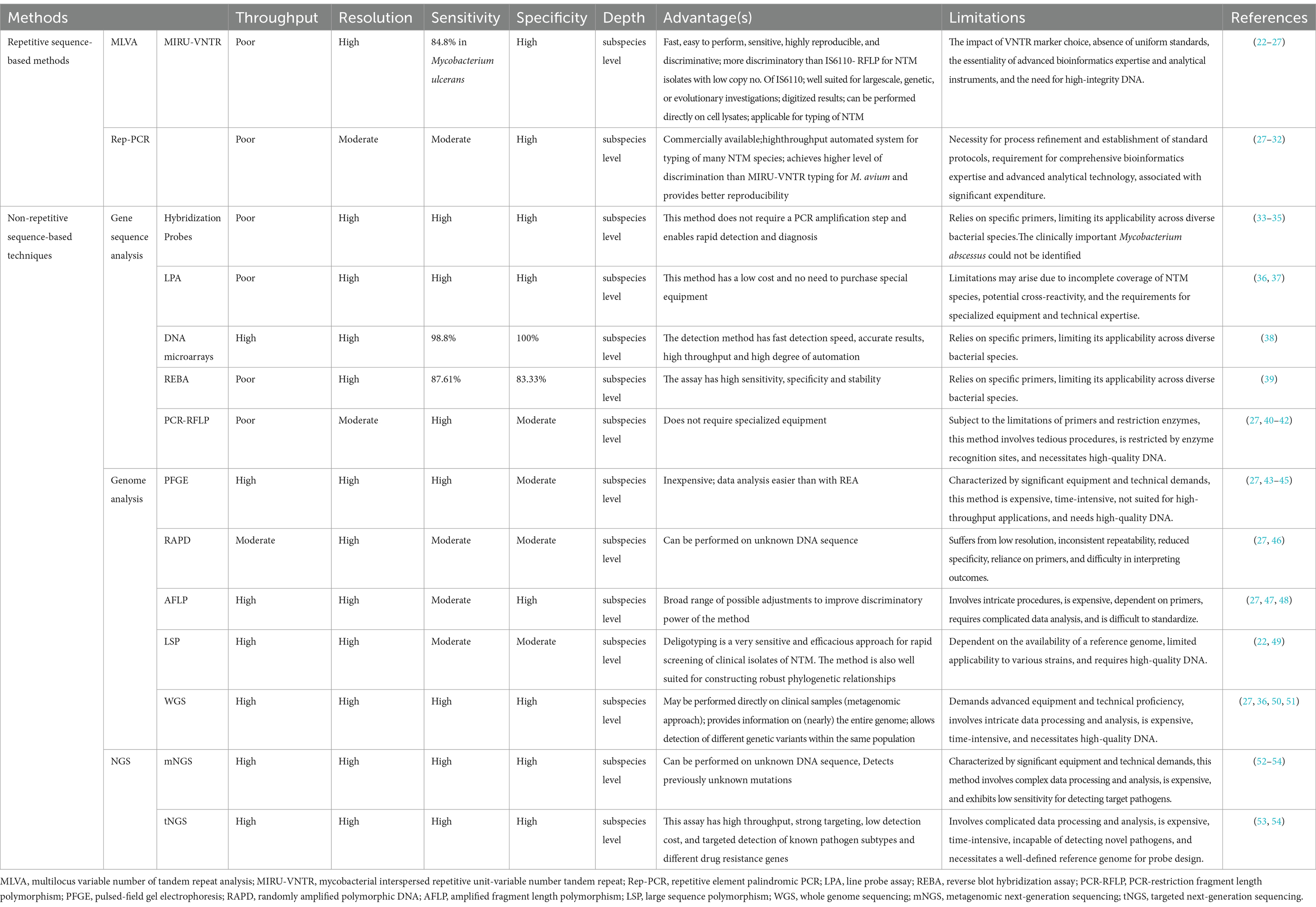
Table 1. Comparison of DNA-based molecular techniques for identification of genetically closa NTM species.
Repetitive sequence-based methods are widely utilized in molecular biology for the accurate identification of NTM. Leveraging the substantial variability inherent in the repetitive regions of bacterial genomes, these techniques distinguish between strains through comparative analysis of sequence differences.
Insertion sequence (IS) is a compact, mobile genetic element that predominantly encodes for transposition and regulatory functions. Its integration into specific genomic loci can disrupt gene functionality and alter the expression of nearby genes. Investigations across various NTM species have revealed multiple distinct IS types, underscoring their pivotal role in molecular diagnostics and contributing to genomic diversity. Among these IS elements, IS1245 and IS1311 are closely associated with the molecular typing of MAC strains (55). In addition to IS1245 and IS1311, the MAC genome includes other insertion sequences. These insertion elements include IS900, present in M. avium subsp. paratuberculosis (MAP), IS901 in M. avium subsp. avium (MAA), IS902 in M. avium subsp. silvaticum (MAS), and those yet to be thoroughly investigated in M. avium isolates (21, 56, 57). IS900, IS901, and IS902, of the reported IS elements thus far, have been widely used to identify and differentiate various MAC strains (58–60). Extending beyond MAC, there are numerous IS used for NTM epidemiological studies, highlighting the expansive utility of IS in the broader context of NTM research. These IS include IS1395 in M. xenopi, IS1511/IS1512 in Mycobacterium gordonae (M. gordonae), IS1407 in M. celatum, IS6120 and IS2404 in Mycobacterium smegmatis (M. smegmatis), IS2606 in M. ulcerans, M. lentiflavum, and Mycobacterium kansasii (M. kansasii), as well as IS1652 in M. kansasii (61–66).
Trinucleotide repeat sequence (TRS) is ubiquitously present across bacterial genomes, varying in quantity, and serve as a pivotal tool for bacterial genotyping. Within the genomes of mycobacteria, the majority of these TRS are located within the genes of the Pro-Glu (PE) and PPE families (67). The PGRS-restriction fragment length polymorphism (RFLP) typing technique, which relies on the PGRS sequences, involves cloning these sequences into the recombinant plasmid pTBN12 to serve as probes for species identification. This approach has identified PGRS sequences across various NTM species, thereby facilitating the differentiation of strains such as M. kansasii and M. ulcerans through similar PGRS-RFLP typing methods. This advancement underscores the utility of TRS in the molecular diagnostics landscape, significantly enhancing the resolution of mycobacterial genotyping and contributing to our understanding of NTM infection identification (21).
Enterobacterial repetitive intergenic consensus (ERIC), characterized as imperfect palindromic sequences of approximately 126 bp, are predominantly distributed within the genomes of Gram-negative bacteria. The variability of ERIC sequences has facilitated the development of a novel genotyping approach, known as enterobacterial repetitive intergenic consensus-polymerase chain reaction (ERIC-PCR) typing (68). ERIC, a derivative of the RAPD technique, proves to be informative when utilized for targets beyond Enterobacteria (69). This method has been extensively applied to assess the genetic diversity among various species of Mycobacteria, including MTB, M. gordonae, Mycobacterium intracellulare (M. intracellulare), Mycobacterium szulgai (M. szulgai), Mycobacterium fortuitum (M. fortuitum), Mycobacterium chelonae (M. chelonae), and Mycobacterium abscessus (M. abscessus) (70–73). ERIC-PCR typing has significantly enhanced insights into the molecular epidemiology and phylogenetic relationships of these species, improving our comprehension of their transmission dynamics and pathogenic potential (74).
Multilocus variable number of tandem repeat analysis (MLVA) characterizes tandem repeat regions dispersed across the NTM genome. These regions mirror the polymorphic minisatellites found in eukaryotic genomes. The variable number of tandem repeat (VNTR) loci encompasses five major polymorphic tandem repeat (MPTR) sequences (MPTR-A to E) and six exact tandem repeat (ETR) sequences (ETR-A to F) (75). MPTR sequences, comprising unique 10 bp sequences separated by 5 bp intervals, are identified across various NTM species, including M. gordonae, M. kansasii, and M. szulgai (76). MPTR sequences have been demonstrated to facilitate RFLP typing of M. kansasii, utilizing MPTR sequences as probes (66, 77). Supply et al. (78) introduced an optimized 24-locus mycobacterial interspersed repetitive units-variable number tandem repeats (MIRU-VNTR) typing scheme, incorporating 12 loci previously identified. This expanded 24-locus format offers enhanced phylogenetic insight, establishing it as a standard method for typing the MTBC. MIRU-VNTR analysis is extensively applied in NTM typing, notably within M. avium, where the identification of multiple loci has revealed substantial discriminatory capability. In the case of M. intracellulare, up to 45 potential loci have been identified, with 7 exhibiting high variability, making them suitable for differentiation purposes. Additionally, 13 loci have been employed in the study of isolated M. ulcerans, further evidencing the utility of MIRU-VNTR analysis in nuanced differentiation and identification (24, 79).
Repetitive element palindromic PCR (Rep-PCR) represents a commercialized, high-throughput, automated system designed to genotype various species of mycobacteria by leveraging the variability in repetitive sequences scattered across bacterial genomes. This method amplifies repetitive fragments within non-coding sequences and separates them using a microfluidic electrophoresis chip (28). The NTM genome is characterized by various repetitive elements, including IS6110. The variability in sequence copy numbers and configurations yields unique genomic fingerprints. Rep-PCR leverages this to generate distinct profiles, differentiating closely related species or strains via subtle pattern shifts. Although some NTM species conserve specific repeating elements, others exhibit significant variations. This combination of conservation and variability aids in identifying NTM at both the species and strain levels with precision. Rep-PCR has been applied to the genotyping of M. abscessus, and its typing patterns have been compared with those obtained via pulsed-field gel electrophoresis (PFGE). The results demonstrated a 90% concordance in the typing of identical strains between Rep-PCR and PFGE analyses, suggesting that Rep-PCR may offer superior discriminatory power over PFGE (80). In the typing of various NTM species, Rep-PCR demonstrates enhanced repeatability relative to random amplified polymorphic DNA (RAPD) analysis. Furthermore, Rep-PCR offers superior discriminatory power compared to MIRU-VNTR typing for the MAC, underscoring its potential to improve resolution in NTM infection molecular diagnostics (72, 81).
Non-repetitive sequence-based methods, while serving as a cornerstone in genetics, evolutionary biology, and biological research, have also been increasingly adopted in the molecular diagnostics of infectious diseases, including the identification of NTM infections. These methods facilitate the precise differentiation of NTM species, which are crucial for tailored therapeutic strategies. Contrary to repetitive sequences, non-repetitive sequences encompass gene-coding regions, regulatory elements, and functional sequences that, although not as prevalent as repetitive elements in the genome, play pivotal roles in genetic diversity and function. The analysis of non-repetitive sequences, through sequence alignment and specific gene or fragment sequencing, is pivotal for detecting genetic variations within NTM species.
Gene sequence analysis stands as a pivotal bioinformatics approach for examining and decoding the DNA sequences within genomes, playing an essential role in the identification and study of NTM infections. By leveraging computational algorithms and bioinformatics tools for DNA sequence alignment, assembly, annotation, and analysis, this approach elucidates the NTM genome’s structure, function, and evolution. It is instrumental in accurately distinguishing NTM species and subspecies, essential for precise treatment and management. Furthermore, genomic insights not only enhance our understanding of NTM biodiversity but also illuminate its public health implications, including the emerging patterns of antibiotic resistance and the geographical distribution of infections. This knowledge is imperative for informing public health strategies and interventions aimed at preventing and managing NTM outbreaks. Single nucleotide polymorphism (SNP) typing has shown low levels of homogeneity, making it a valuable tool for differentiating between species and thus has been employed in the identification of mycobacterial species. Technological advancements now enable the use of molecular beacons to identify single nucleotide substitutions, facilitating simultaneous analysis of multiple SNP loci. Gene sequence analysis leveraging SNP typing facilitates the identification of a broad spectrum of mycobacterial species, encompassing members within the MTBC, and enables the detection of various resistance markers (21, 82). Gene sequence analysis methods yield better results with the analysis of multiple loci. Multilocus sequence typing (MLST), which involves sequencing allele groups to differentiate species, is extended significantly in mycobacterial identification. Tools like PubMLST and mlstverse cater to the nuanced demand for subspecies-level identification (83, 84). PubMLST, combining conventional and ribosomal MLST, facilitates comprehensive bacterial identification. By accumulating and integrating genomic sequences from MLST, enhanced typing can be linked to prognosis and treatment resistance in emerging subspecies. This approach has effectively distinguished members of the MAC and fast-growing mycobacteria, although some studies indicate that MLST alone may not suffice for precise strain differentiation (43, 55, 85).
Initially, the utilization of DNA probe-based techniques to identify partial gene fragments represented a common technique in molecular biology. Depending on the type of target nucleic acid, a variety of amplification methods are employed based on their specific advantages, including conventional PCR for its simplicity and cost-effectiveness, real-time PCR for its quantitative capabilities, nucleic acid sequence-based amplification for high sensitivity, loop-mediated isothermal amplification for rapid and equipment-free amplification, and transcription-mediated amplification for its high amplification efficiency (86–90). Gene probe assays are available in two primary variants. Direct nucleic acid testing (NAT) traditionally required a substantial amount of bacterial material, although recent advancements in sensitivity allow for effective analysis with reduced bacterial loads. Incorporating an amplification step in nucleic acid amplification tests (NAATs) allows for the direct detection of bacterial DNA in clinical samples. The market offers a diverse array of commercial nucleic acid probes, which, when combined with the aforementioned amplification methods, enhance the detection and differentiation capabilities for a wide range of mycobacteria (encompassing both MTBC and NTM), with target sequences often including rRNA, 16S rRNA, and the 16S-23S rRNA spacer region (65, 91, 92). The line probe assay (LPA) employs a technique where target DNA is amplified using biotin-labeled specific primers. Subsequently, the amplified product is denatured and hybridized with specific oligonucleotides affixed to a nylon membrane. The results are then visualized through enzyme-linked immunochromatography. Notable LPA kits include the INNO-LiPA Mycobacteria v2 (Fujirebio Europe, Belgium), GenoType Mycobacteria CM, GenoType Mycobacteria AS, and GenoType NTM-DR kits (Hain Life Sciences, Germany), along with the Speed-oligo Mycobacteria (Vircell, Spain). The GenoType NTM-DR kits not only facilitates the identification of commonly encountered NTM to the species level, such as MAC, Mycobacterium abscessus complex (MABC), and M. chelonae, but also enables the detection of resistance to macrolides. This is achieved through the identification of mutations in the erm41 gene and rrl gene, as well as resistance to aminoglycosides by detecting mutations in the rrs gene (37). However, in clinical mycobacterial identification laboratories, many still rely on commercial single-stranded DNA nucleic acid probe technology. This approach typically offers high species identification accuracy, with results available in a short timeframe (less than 24 h), facilitating rapid identification (93). The reverse blot hybridization assay (REBA) operates on the principle of affixing numbered oligonucleotide probes to a nitrocellulose or nylon membrane. These probes are then hybridized with biotin-labeled PCR amplification products. The presence of a colored signal at specific positions on the membrane strip indicates successful hybridization of the probe to the DNA fragment. Wang HY et al. conducted a comparative analysis of PCR-REBA with real-time PCR and RFLP techniques (94). Their findings demonstrated that PCR-REBA delivers highly sensitive and specific results in identifying NTM and distinguishing between NTM species from mycobacterial liquid cultures. DNA microarrays present a compelling alternative for conducting high-throughput analyses of multiple genetic markers concurrently, distinguishing themselves from traditional methods reliant on predefined probes. These chips can be tailored to target specific regions of interest within bacterial genomes, including conserved sequences typically associated with NTM species. This customization enables a more exhaustive analysis, facilitating the detection of genetic variations and the differentiation of closely related species. Despite the potential for elevated costs, the versatility and efficiency of DNA microarrays are increasingly appealing to clinical laboratories specializing in mycobacterium identification. Research has explored the utility of DNA microarrays in NTM identification (95, 96). With ongoing technological advancements driving down costs and enhancing performance, DNA microarrays are poised to become indispensable tools for NTM monitoring and research, addressing critical challenges in clinical settings and aiding in the development of more effective management strategies.
Currently, PCR-RFLP has been effectively employed in the identification of NTM, marking a significant advancement in gene sequence analysis. This method integrates PCR amplification, restriction enzyme digestion, and electrophoresis to generate species or strain-specific profiles. For the identification of NTM species using this approach, the process begins with the PCR amplification of the rpoB gene and the hsp65 gene, resulting in PCR products. These are then subjected to digestion with specific restriction enzymes, such as MspI, HaeIII, and BstEII, followed by analysis using agarose gel electrophoresis. By employing additional restriction enzymes, this technique can further differentiate members of the MABC into subspecies levels (97).
Genome analysis transcends basic gene sequencing, offering a detailed exploration of an organism’s full genome, including gene organization, structure, and overall genomic context. This inclusive strategy provides intricate, high-resolution insights into both coding and non-coding regions and regulatory elements, shedding light on genetic variations, evolutionary links, and functional attributes. Consequently, it delivers a nuanced understanding of strain characteristics and their interconnections.
PFGE uses restriction enzyme digestion to fragment chromosomal DNA from different mycobacterial strains, producing unique fingerprint patterns that are visible under ultraviolet light after gel electrophoresis. The technique applies periodically alternating electric fields to direct DNA fragments, facilitating the separation of large molecules more effectively. A principal advantage is its capacity to separate larger DNA fragments, beyond the 50 kb limit of conventional unidirectional electrophoresis, by employing rare-cutting restriction enzymes. PFGE is characterized by its high repeatability and discriminatory capacity. Despite subtle strain variabilities, it proves highly effective in typing diverse NTM species, demonstrating particular success with slow-growing strains such as M. kansasii, MAC, and M. avium (98). However, it’s important to acknowledge that PFGE involves significant costs, demands extensive technical expertise, and has a lengthy turnaround time, sometimes extending to 5 days. Like RFLP, PFGE also necessitates access to a comprehensive database of high-quality DNA sequences for reference (99).
Randomly amplified polymorphic DNA (RAPD), or arbitrary primed PCR (AP-PCR), is a technique independent of prior DNA sequence knowledge. Utilizing a single, arbitrarily chosen primer of 5 to 50 bp, it generates strain-specific DNA profiles by binding at sites with full or partial template DNA matches (100). Despite its high discriminative power, RAPD is limited by its poor reproducibility. Furthermore, it is generally believed that the observed differences between strains are more attributable to technical and procedural variations inherent to the method rather than true genetic polymorphism (101). Despite its limitations, RAPD-especially with multiple primer combinations-has been effectively used for NTM strain analysis. The polymorphism of DNA profiles from these combinations matches or exceeds those from PFGE. Moreover, RAPD’s application extends to genotyping M. abscessus and M. chelonae, which are prone to spontaneous fragmentation during gel electrophoresis, thus complicating PFGE assessment (102, 103). Similarly, this method has been used for typing strains of Mycobacterium phocaicum (M. phocaicum), M. gordonae, M. szulgai, and Mycobacterium malmoense (M. malmoense) (104–107).
Amplified fragment length polymorphism (AFLP) analysis represents a PCR-based genotyping technique that utilizes dual restriction enzymes for DNA digestion: one rare cutter and one frequent cutter, recognizing sites of 6 bp and 4 bp, respectively. Following digestion, the resultant DNA fragments are ligated to double-stranded adaptors ranging from 10 to 30 bp, which are complementary to PCR primers. This setup facilitates the selective amplification of fragment subsets. AFLP analysis has emerged as a novel approach for typing NTM species, including members of the MAC and M. hemophilum (27). Additionally, AFLP has proven effective in distinguishing between closely related species, such as M. marinum and M. ulcerans, offering a refined tool for microbial identification and strain differentiation within the context of molecular diagnostics (21).
Large sequence polymorphism (LSP) analysis, a key molecular marker for examining genetic diversity in mycobacteria, depends on pre-existing sequence knowledge and often demands significant DNA quantities. It utilizes targeted PCR genotyping for focused studies and microarray technology for extensive genomic screenings. While microarray technology provides thorough genomic insights, it has discernible limitations. For instance, some platforms cannot detect deletions smaller than 350 bp, potentially compromising the sensitivity of NTM infection identification (108). This limitation underscores the need for integrating or developing more refined molecular diagnostic tools capable of detecting smaller genetic variations. Furthermore, the challenge of cross-hybridization among similar sequences necessitates the optimization of probe design and hybridization conditions, particularly for the non-repetitive segments of bacterial genomes, to enhance the method’s applicability and accuracy in distinguishing between NTM subtypes. LSP analysis has been employed to identify differences between various subtypes within the MAC and M. abscessus (109).
Whole genome sequencing (WGS) represents a novel and highly precise method for identifying and characterizing various species of mycobacteria. Offering superior resolution compared to techniques such as targeted PCR genotyping and microarray analysis, WGS enables the differentiation of NTM subspecies down to their specific evolutionary branches, while also providing comprehensive genomic information. WGS provides unparalleled accuracy and precision in identifying genetic variances between strains by detecting almost all markers utilized in the genotyping methods mentioned previously. Its utility spans from offering comprehensive data at the global (population-wide), local (community), and individual (single patient) levels to yielding profound insights into the pathogens. It facilitates the precise identification of new NTM species and aids in predicting the virulence genes of NTM, laying a foundation for targeted therapy (50, 51). For example, WGS has successfully distinguished between closely related strains of M. ulcerans and M. marinum, providing insights into their evolutionary paths and virulence factors. Furthermore, its application in the study of MABC outbreaks has revealed specific transmission chains, highlighting its potential in public health surveillance and response (110, 111). Large-scale studies have validated the efficacy of WGS in mapping outbreaks and elucidating transmission pathways (112, 113). Compared with WGS, traditional methods such as PFGE and RFLP analysis often result in imprecise clustering, potentially missing fine-scale genetic variations critical for accurate pathogen identification and outbreak tracking. Furthermore, WGS has the capability to forecast undetected NTM species, thus facilitating the provision of timely and suitable interventions for emerging infections (114–117).
In recent years, NGS has significantly advanced our ability to harness comprehensive genetic information, especially via WGS. These technologies excel in pathogen detection with notable specificity, even amidst complex or sparse datasets, as demonstrated by metagenomic DNA analysis. Metagenomic NGS (mNGS) emerges as a superior, high-throughput method for DNA/RNA sequencing, outperforming traditional Sanger sequencing by offering quicker processing, reduced costs, and enhanced sensitivity. NGS markedly accelerates the breadth of genomic or transcriptomic analyses, substantially boosting sequencing speed and efficiency (118). This enhanced depth and coverage are particularly valuable in NTM infection identification, as they facilitate the detection of low-frequency mutations or variants that might be missed by other methods (119). In bacterial evolution analysis, NGS shows superior advantages, including applications in phylogenetic analysis, speculation on species origins, and predicting microbial communities (120–122). NGS encompasses diverse approaches, including mNGS and targeted NGS (tNGS), each offering distinct advantages and applications across various contexts.
mNGS represents a broadly unbiased sequencing technique, capable of sequencing the genomes of a wide array of microbes present in a sample, although its efficacy can be influenced by sample quality and sequencing depth. This approach offers significant advantages for organisms like NTM, which are notoriously difficult to culture and identify. Crucially, mNGS eliminates the need for prior knowledge of the microbial composition within a sample, allowing for direct DNA extraction and sequencing from the specimen. While circumventing the limitations and biases inherent to traditional culturing methods, it’s important to acknowledge scenarios where traditional methods may still offer valuable insights, particularly in understanding microbial growth characteristics and antibiotic susceptibility. mNGS facilitates the detection of microbial nucleic acids across a variety of specimen types and enables rapid identification of multiple pathogens, with numerous cases of NTM infections being diagnosed through this method (50, 52, 53).
Moreover, mNGS is adept at detecting low-abundance microbes, including NTM, making it essential for a comprehensive understanding of microbial communities within samples. mNGS exhibits the capability to uncover novel or hitherto unrecognized microbial species. In a study by Dougherty et al. (123), the utility of metagenomic analysis in identifying mixed infections was further corroborated. Their study underscored the prowess of shotgun metagenomics, not only for sequencing DNA from uncultured samples, but also for elevating mixed infection detection via target-specific amplification. This highlights its capability to refine microbial identification techniques. Thus, metagenomic analysis could signify a breakthrough in diagnostics, especially for multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains.
tNGS is a high-throughput sequencing method focused on specific genes or genomic regions, playing a pivotal role in the identification and study of NTM. This technique begins with the alignment of sequenced DNA against NTM sequences from curated reference databases, a critical step that enables the precise identification of specific microbial species and subspecies within the sample. Subsequently, by analyzing sequence variations, tNGS can elucidate critical information such as the NTM subtype, virulence factors, and resistance genes (124, 125). By focusing sequencing efforts on targeted genes or genomic regions of interest, tNGS not only reduces costs and turnaround times compared to WGS but also enables the rapid identification of resistance genes and virulence factors, crucial for tailoring treatment strategies in NTM infections. This streamlined focus makes tNGS an invaluable tool in the nuanced field of NTM research, offering precise insights into microbial genetics with efficiency (126, 127). While tNGS offers significant advantages in terms of cost and speed, it’s important to acknowledge that by focusing only on predetermined genomic regions, it may miss novel or unexpected variants outside these regions, potentially limiting the comprehensive understanding of microbial genome complexity.
Merely identifying the pathogen is insufficient for clinical satisfaction. Once the species of mycobacteria in a clinical sample is identified, the crucial next step is to determine its drug susceptibility to devise an appropriate treatment strategy. Inherent resistance, characteristic of certain NTM species, guides initial antibiotic choices, whereas acquired resistance, emerging during treatment, necessitates ongoing monitoring and potentially adjusting therapeutic strategies. While studies on MTB have laid the foundation for understanding antibiotic resistance mechanisms, the genetic and phenotypic diversity among NTM species necessitates targeted research to uncover specific resistance patterns and develop effective treatments. Increasing reports indicate that antibiotics previously effective, including macrolides, fluoroquinolones, and aminoglycosides, are now facing resistance, an issue particularly prevalent and posing a higher risk in slow-growing NTM, exacerbated by the extensive use of these antibiotics in both clinical and animal husbandry. Settings, leading to increased selection pressure for resistant strains (128, 129).
Managing NTM infections is notably challenging, largely owing to two main factors. First, the relationship between in vitro DST outcomes and clinical success is not consistently direct. This inconsistency can be attributed to factors such as the complex biology of NTM species and variability in patient responses to treatment. For example, DST results for M. abscessus and Mycobacterium simiae (M. simiae) have a weak correlation with clinical efficacy, in contrast to species such as M. kansasii, M. marinum, and M. fortuitum, where there is a stronger alignment with treatment success (130, 131). This variation in correlation may be due to differences in the mechanisms of antibiotic resistance and pathogenicity among these species, affecting the predictability of treatment outcomes based on DST alone. Secondly, clinical case management ranges from no treatment to requiring a multidrug regimen. The decision to opt for no treatment, monitor, or pursue aggressive multidrug regimens is guided by a comprehensive evaluation of the specific NTM species involved, infection severity, and individual patient health considerations, highlighting the need for a personalized approach to treatment. Thus, DST, aligned with Clinical and Laboratory Standards Institute (CLSI) guidelines, is critical for crafting optimal treatment strategies. Adhering to CLSI guidelines, which are regularly updated to reflect emerging data on NTM behavior and antibiotic resistance, ensures that DST is applied effectively to inform treatment strategies, underscoring the dynamic nature of NTM management. Identification of clinically significant antibiotic resistance genes (ARGs) enables the prediction of pathogen resistance.
The CLSI guidelines were revised and expanded in 2018, establishing broth microdilution as the most common laboratory method for DST to determine the minimum inhibitory concentration (MIC) (132). Traditional culture-dependent approaches for DST, prevalent in clinical microbiology laboratories, suffer from drawbacks including lengthy processing times, bias towards certain microbial communities, and contamination risks from overgrowth. Recent studies have elucidated the intrinsic and acquired resistance mechanisms of mycobacteria to classes of antibiotics including macrolides, aminoglycosides, oxazolidinones (such as linezolid), riminophenazines (like clofazimine), and di-substituted diazepanes (such as bedaquiline), paving the way for the application of molecular diagnostic techniques in DST (133). Notably, molecular diagnostics can also detect resistance heterogeneity, identifying resistant subpopulations within drug-sensitive mycobacterial communities (134, 135). The Comprehensive Antibiotic Resistance Database (CARD) (https://card.mcmaster.ca) aggregates well-documented, peer-reviewed resistance determinants alongside their corresponding antibiotics. Utilizing CARD facilitates the identification of genes and mutations associated with NTM resistance, enabling comparison of these sequences against WGS and NGS data. Regrettably, no existing database perfectly correlates NTM resistance predictions from WGS data with DST outcomes, reinforcing the need for refined bioinformatic tools and databases. Table 2 showcases a meticulously compiled inventory of NTM-relevant mutations and genes, including their reference data, sourced from the CARD (136–150). Table 3 outlines the comparison of DST and WGS in antibiotic susceptibility identification for NTM.
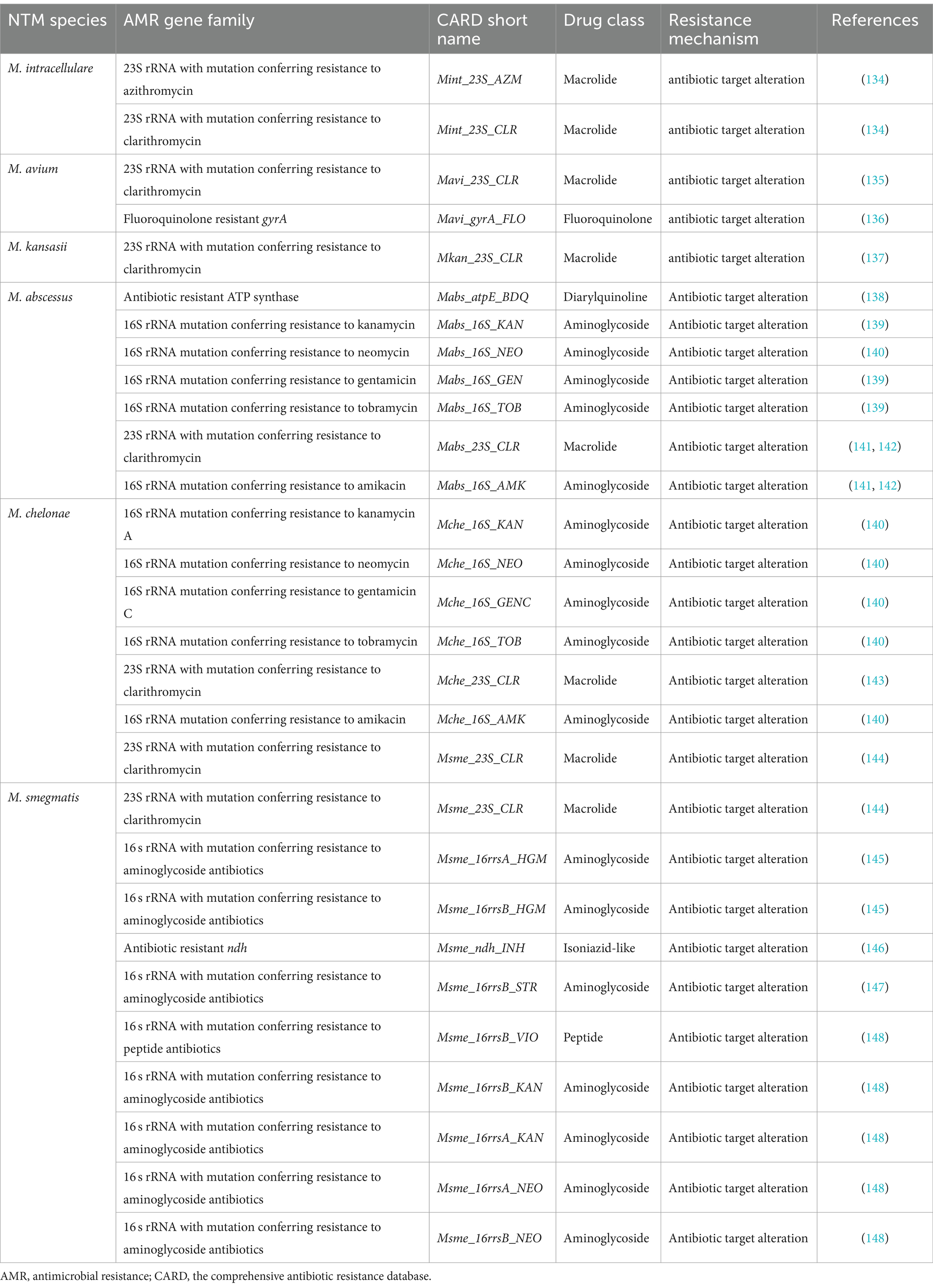
Table 2. Mutated genes associated with NTM and their mutation details retrieved from the CARD database.
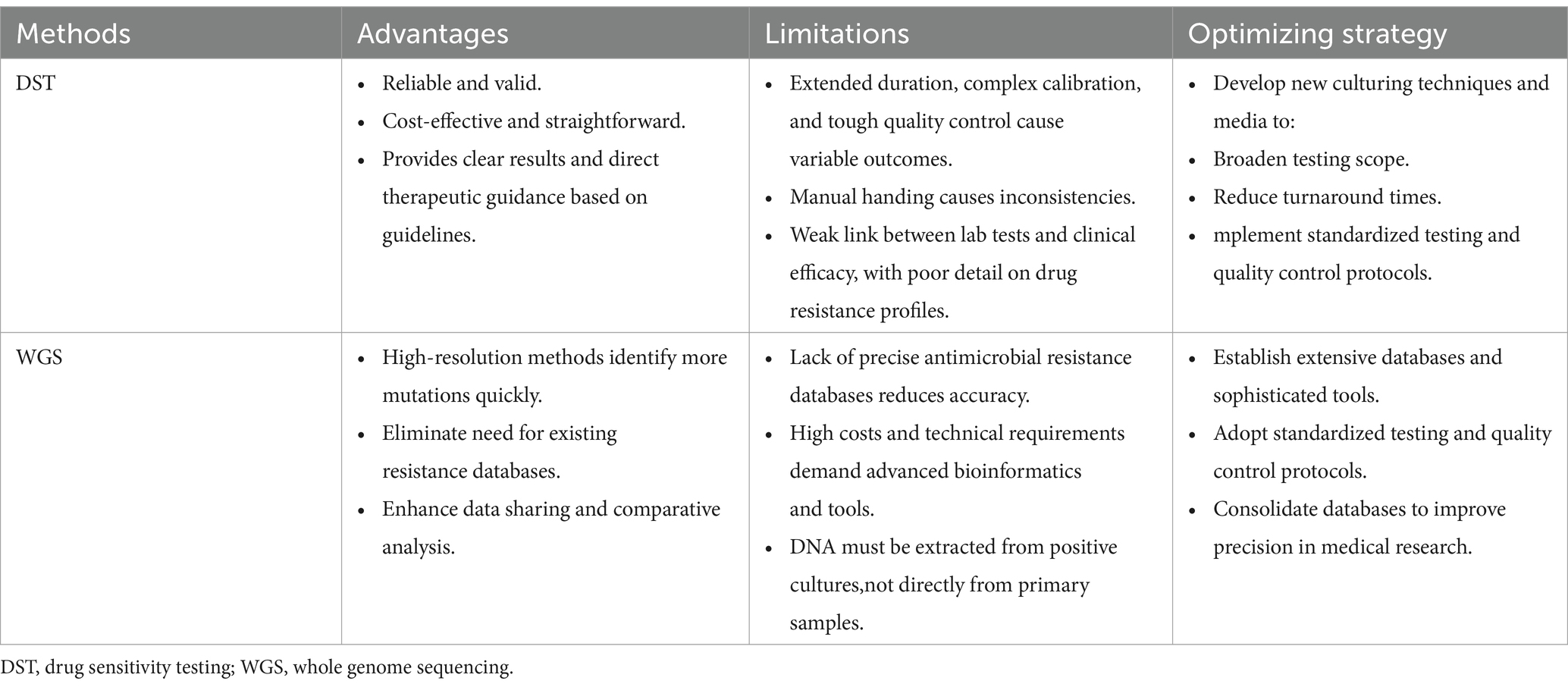
Table 3. Comparison of DST and WGS in antibiotic susceptibility identification for NTM.
Given the predominance of macrolide antibiotics as the first-line treatment for most NTM infections, the detection of resistance to these drugs is particularly critical. Macrolide resistance mechanisms involve the methylation of 23S rRNA by the erythromycin ribosome methylation (erm) gene, preventing macrolide binding to its ribosomal target. Additionally, mutations in the 23S rRNA itself can render macrolides ineffective, a scenario relatively common in mycobacteria due to their limited number of rRNA operons. A single mutation in any of these operons can significantly alter the ribosomal structure, inhibiting macrolide binding (151–153). Rifampin remains among the preferred treatments for most NTM infections, generally linked to mutations in its target, the rpoB gene, which encodes the β-subunit of RNA polymerase. Studies suggested rifampin preferentially inhibits one of the rpoB promoters (Promoter I), leading to increased expression from the second promoter (Promoter II) and promoting the growth of resistant strains (154). Recent research indicates additional mechanisms might be involved. There are reported cases of rifampin resistance in both MAC and M. kansasii (155, 156). M. abscessus resistance constitutes a formidable clinical hurdle, as evidenced by multiple DST studies demonstrating resistance across several antibiotic classes, including macrolides, quinolones, and aminoglycosides. Notably, intrinsic high-level resistance has been predominantly associated with established mutations in the embB gene, conferring resistance to ethambutol, and the gyrA gene, conferring resistance to fluoroquinolones (157). Additionally, intrinsic resistance to rifampin in M. abscessus is due to mutations in the rpoB gene and the presence of the MAB_0591 gene, which is known to contribute to rifampin resistance processes (158). Another mechanism of intrinsic resistance is the overexpression of efflux pumps, contributing to resistance against bedaquiline and clofazimine (159). Chen et al. (160) conducted WGS on clofazimine-resistant M. abscessus strains, revealing several significant mutations in the MAB_2299c, MAB_1483, and MAB_0540 genes, which were found to play crucial roles in conferring drug resistance. The identification of the erm41 gene and its pivotal role in macrolide resistance has facilitated the critical differentiation of three distinct species within the MABC: M. abscessus subsp. abscessus (MABSa), M. abscessus subsp. massiliense (MABSm), and M. abscessus subsp. bolletii (MABSb). Prior to this discovery, the American Thoracic Society/Infectious Disease Society of America (ATS/IDSA) guidelines on NTM infections did not reflect the central influence of the erm41 gene on macrolide susceptibility. This oversight has significant clinical implications, as the majority of MABC isolates exhibit a functional erm41 gene, correlating with poor treatment outcomes in patients (161). In MABSa, a T/C polymorphism at position 28 of the erm41 gene determines inducible macrolide resistance (28 T) or susceptibility (T28C). While most MABSa and MABSb strains exhibit inducible macrolide resistance, those with the T28C substitution are sensitive due to the loss of erm41 functionality. Conversely, MABSm contains a large deletion in the erm41 gene, resulting in nonfunctional erm41 and macrolide susceptibility (162, 163). The commercialized GenoType NTM-DR kits (Hain Life science, Germany) enables rapid identification of M. abscessus subspecies and concurrent detection of resistance to macrolides and aminoglycosides. Studies have demonstrated the test’s high sensitivity in detecting acquired resistance, underscoring its utility in the molecular diagnostics landscape for NTM infections (164, 165).
The adoption of WGS has markedly advanced our comprehension of NTM antibiotic resistance, thus improving treatment strategies. In a significant validation of WGS’s precision, Wetzstein et al. (166) demonstrated complete concordance between WGS forecasting of M. abscessus resistance to macrolides and aminoglycosides and results from both GenoType NTM-DR kits and phenotypic DST. Lipworth et al. (167) leveraged sequencing data from M. abscessus to identify novel mutations in the erm gene and rrs gene linked to macrolide resistance, mutations not covered by traditional genotyping, raising the possibility of false-negative outcomes. Yoshida et al. (168) developed a WGS-based diagnostic approach, incorporating DNA chromatography and PCR, to distinguish macrolide-resistant and macrolide-sensitive M. abscessus subspecies, including M. massiliense, M. abscessus, and M. bolletii, achieving a 99.7% concordance with conventional DST findings. With a promising start, Realegeno et al. (169) have formulated a WGS assay aimed at accurately predicting M. abscessus resistance to clarithromycin and amikacin, achieving an impressive 100% accuracy when compared to phenotypic results. Yet, it should be noted that before wide clinical application of such a WGS-based prediction method, extensive validation against an array of drugs and broader NTM species is imperative. NGS, encompassing both mNGS and tNGS, is increasingly valued for its potential in detecting drug resistance genes in NTM. tNGS, in particular, sequences specific genes or genomic regions, offering higher specificity in detecting target genes compared to mNGS (126). Due to the highly conserved DNA sequences within the Mycobacterium and limited nucleotide sequence data, mNGS often fails to accurately identify species within the Mycobacterium or detect multiple drug-resistant mutations (170). tNGS combines multiplex PCR amplification with sequencing to rapidly capture diverse drug resistance gene sequences, thus advancing the molecular diagnosis of NTM resistance (124, 125).
In contemporary clinical practice, advances in molecular biology have revolutionized the diagnosis of NTM infections. These methodologies offer rapid, accurate, and comprehensive detection capabilities for NTM species identification and genotyping, along with critical insights into antimicrobial resistance and susceptibility. The systematic standardization, which ensures consistency and reliability across different laboratories, and a modular approach that allows for the tailored combination of diagnostic tests, together with rigorous quality control measures, significantly enhance the efficacy and reliability of clinical diagnostics and thereby inform more precise therapeutic strategies.
Numerous molecular techniques are applicable for species identification and genotyping of NTM, each offering distinct advantages and specific applications in clinical settings. Techniques such as MLVA, MIRU-VNTR, Rep-PCR, RFLP, PFGE, RAPD, AFLP, and LSP are primarily employed for NTM subspecies in clinical diagnostics. Conversely, methods such as hybridization probes, WGS, and NGS provide significant advantages in identifying drug resistance genes. This capability is particularly critical in anticipating NTM drug resistance, especially in the absence of available DST. Figure 1 depicts the application and workflow of molecular diagnostic techniques in NTM species and subspecies identification.
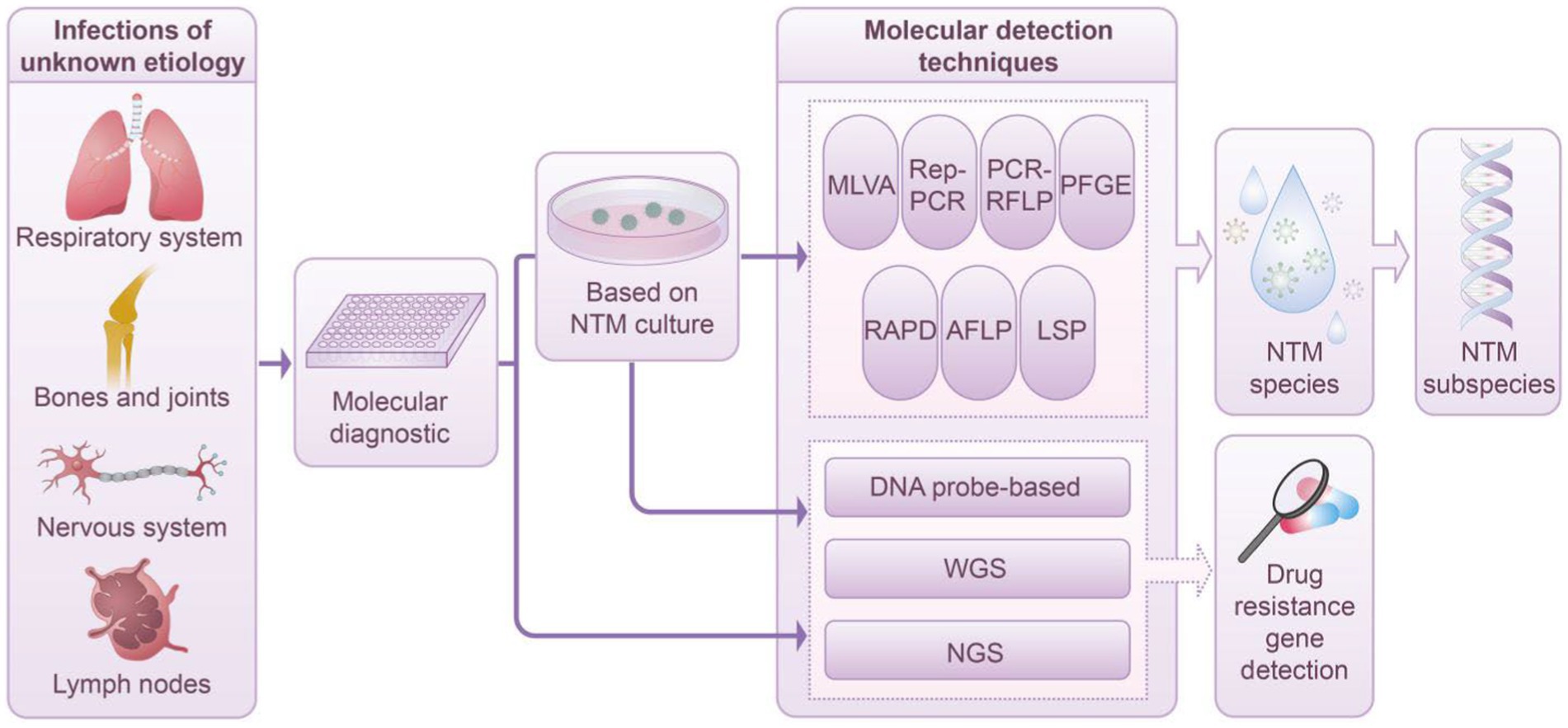
Figure 1. Diagnostic workflow based on molecular technology in NTM infection management. This schematic depicts the integration of advanced molecular techniques within the clinical framework for the diagnosis of NTM infections. Techniques such as MLVA, Rep-PCR, PCR-RFLP, PFGE, RAPD, AFLP, LSP, and DNA probe-based techniques, along with WGS and NGS, facilitate the detailed characterization of NTM species and their subspecies. The process entails isolating and culturing a singular NTM colony from a clinical specimen, followed by its purification. Notably, cutting-edge methodologies such as DNA probe-based techniques, WGS, and NGS enable direct application to clinical samples, thereby circumventing the conventional culture step. Upon completing the identification of NTM species and subspecies, tools such as DNA probe-based techniques, WGS, and NGS are employed to comprehensively evaluate NTM resistance genes. This approach significantly enriches our understanding of the mechanisms underlying microbial resistance.
The delineation and phylogenetic affiliations of NTM remain critical challenges due to their diverse genetic backgrounds and widespread environmental presence, which complicate clinical identification and treatment. Conventional methodologies, primarily relying on phenotypic characteristics and limited genetic markers, often lead to inconsistent and conflicting taxonomic identifications when different tools analyze the same environmental samples. The NTM taxonomy is dynamic, with phylogenetic updates substantially influencing the terminology across various fields. Advancements in molecular diagnostics, particularly through 16S rRNA gene amplification and sequencing, have unveiled extensive microbial diversity and intricate interrelations (171, 172). Nonetheless, exclusive reliance on 16S rRNA gene-based taxonomy presents significant shortcomings, such as inadequate resolution at and below the genus level, primer mismatches, and the potential distortion of phylogenetic relationships owing to chimeric sequence formation during PCR, which may impact the integrity of phylogenetic trees (173, 174). Thus, there’s a necessity for genomic data, beyond the 16S rRNA gene sequences, for accurate species classification and matching (175). Analyses of metagenomes and the construction of phylogenetic trees using single-copy, vertically transmitted protein sequences provide enhanced resolution compared to those derived from a single phylogenetic marker gene, such as 16S rRNA. These approaches are increasingly recommended for taxonomic reference (176, 177). The advent of the molecular diagnostics era, marked by the application of technologies such as NGS, including WGS and mNGS, has not only clarified NTM genomes but also provided a more accurate and comprehensive understanding of their phylogenetic relationships, thereby overcoming the limitations of previous methodologies. Genomic data enables the construction of more robust, comprehensive phylogenetic trees with higher resolution. A comprehensive review of the literature was undertaken, focusing on instances where molecular methodologies have been utilized to construct phylogenetic and evolutionary analyses of NTM, as detailed in Table 4 (178–185).
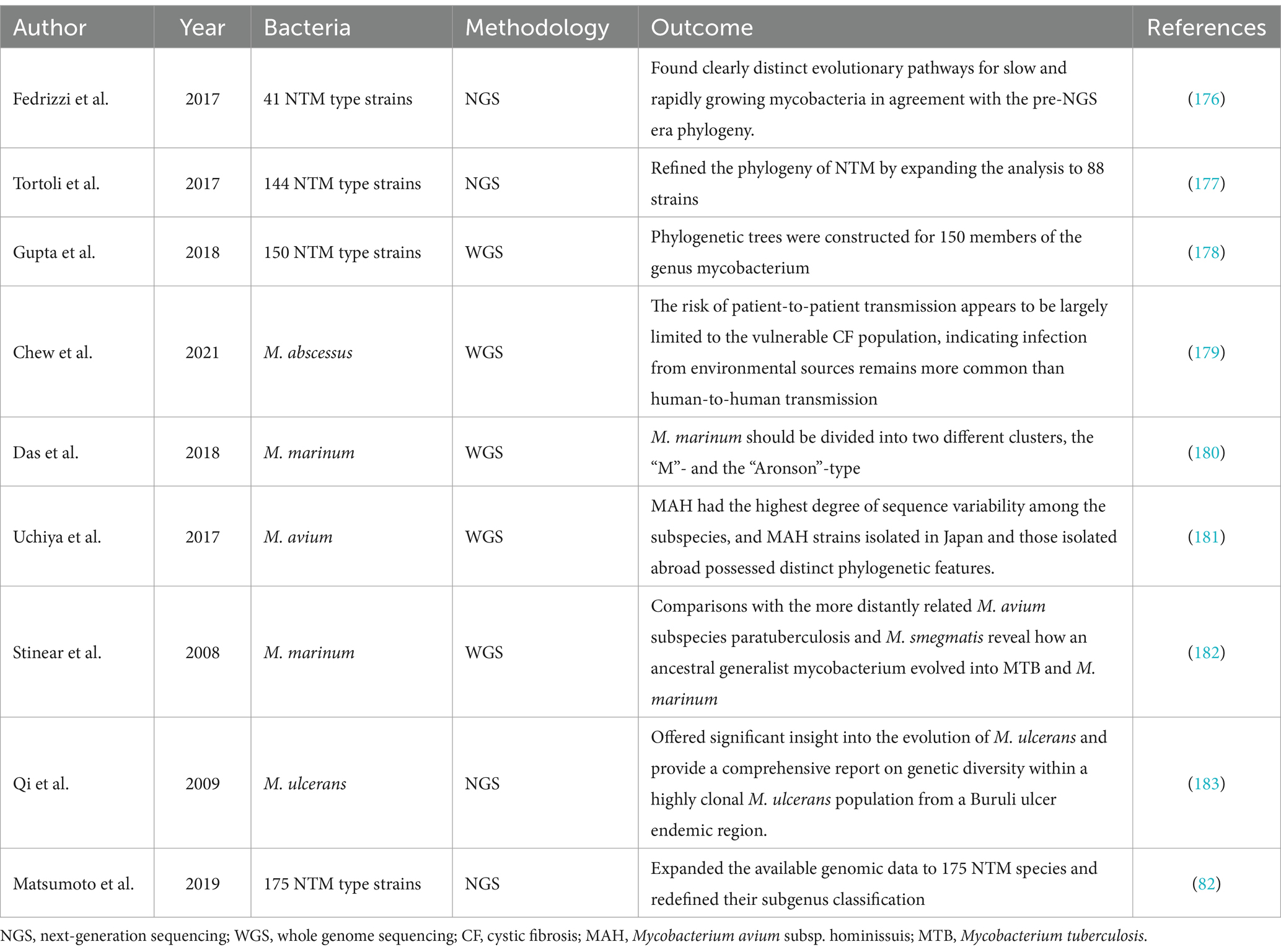
Table 4. Published data on studies involving molecular diagnostic techniques in constructing phylogenetic and evolutionary of NTM.
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) offers significant advantages for biomolecular analysis (186). It has proven effective for identifying pathogenic microorganisms and their resistance patterns, demonstrating specificity and sensitivity superior to Sanger sequencing, with detection limits on par with next-generation sequencing (187–191). While the identification of bacterial isolates, including NTM, is well established using MALDI-TOF MS, the technology is optimized for pure cultures. Challenges arise with mixed Mycobacterium cultures, where molecular methods are necessary to identify multiple NTM species in respiratory specimens. Despite its utility, differentiation of closely related species, such as subspecies of M. abscessus, poses difficulties. Literature reports occasional misidentifications by MALDI-TOF MS, primarily among closely related species or those within the same mycobacterial complex (192). Although some studies have explored subspecies identification through protein peak analysis via MALDI-TOF MS, a consensus on the optimal approach remains elusive (193–195). Recent years have seen the adoption of advanced data analysis techniques, including machine learning, which extend beyond simple species identification to provide more comprehensive insights (196). While MALDI-TOF MS has demonstrated efficacy in DST, it has yet to gain global consensus (197). In summary, MALDI-TOF MS offers rapid, sensitive, accurate, and cost-effective detection with high resolution and minimal sample requirements, enabling precise identification of NTM. Nevertheless, for complex NTM infections, integration with newer molecular diagnostic technologies is essential for accurate identification.
High-performance liquid chromatography (HPLC), developed in the mid-1960s within the chemical sciences, has been extensively applied across inorganic, organic, and biochemical disciplines (198). In the realm of NTM species identification, HPLC facilitates the analysis of mycobacterial components like mycolic acids, offering a faster alternative to traditional, time-intensive identification methods (199). While HPLC enhances species-level differentiation over biochemical tests, its effectiveness is limited by its inability to consistently discern clinically significant NTM species (200). Moreover, the proliferation of similar HPLC chromatograms among various species has diminished its utility as a standalone diagnostic tool (201). Consequently, for robust and accurate NTM species identification and typing, HPLC should be integrated with other molecular diagnostic techniques.
Despite the benefits of molecular diagnostics for NTM infection identification, the literature identifies key limitations and challenges. The lack of standardized protocols results in operational and data processing variability, impacting comparability and reproducibility across labs. Additionally, the necessity for high-quality samples and the variability in sample sources complicate standardizing pretreatment procedures, affecting detection accuracy and reliability. Furthermore, the elevated costs and complexity of certain molecular methods limit their broad clinical adoption, underscoring the need for more affordable and straightforward technologies to enhance their practicality and acceptance in clinical settings.
In upcoming research, molecular diagnostics will focus on key areas. Microfluidic technologies, known for their high throughput, sensitivity, and cost-effectiveness, are expected to revolutionize NTM detection by enabling automated sample processing and analysis. Additionally, integrating artificial intelligence (AI) and machine learning (ML) shows promise in extracting insights from extensive molecular data, aiding in precise diagnostic and treatment decisions. Nucleic acid mass spectrometry (NAMS) has gained prominence, with clinical laboratories increasingly adopting these techniques, poised to become standard in routine lab settings, offering potential for NTM infection detection advancements (202). Advances in genome editing technologies, like CRISPR/Cas9, offer opportunities for precise NTM genome editing, enhancing understanding of pathogenic mechanisms, drug responses, and resistance (203). These technological advancements will deepen comprehension of NTM diseases and drive diagnostic and therapeutic methodologies forward.
Molecular diagnostics, notably through WGS and NGS, have revolutionized NTM infection research, providing efficient, precise tools for species identification and drug sensitivity prediction. These methods have exceeded traditional limitations, offering fresh insights into NTM treatment approaches. Yet, challenges remain in studying less common NTM species, highlighting the need for technique refinement and a deeper understanding of NTM diversity.
In summary, the significant influence of molecular diagnostics on NTM identification is undeniable. As research progresses and technological refinement continues, there exists a potential for pioneering discoveries that will elucidate the complex biology of NTM and enable the development of more precise and effective diagnostic and therapeutic strategies for conditions related to NTM.
HZ: Funding acquisition, Methodology, Resources, Writing – original draft. TM: Conceptualization, Writing – original draft. DL: Supervision, Validation, Writing – review & editing. MX: Conceptualization, Writing – original draft. YA: Investigation, Writing – review & editing. LL: Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the key research and development project of the Sichuan Science and Technology Department (No. 2022YFS0268).
The authors thank all staff involved in the implementation of this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Falkinham, JO 3rd. Nontuberculous mycobacteria in the environment. Tuberculosis. (2022) 137:102267. doi: 10.1016/j.tube.2022.102267
2. Falkinham, JO 3rd. Nontuberculous mycobacteria in the environment. Clin Chest Med. (2002) 23:529–51. doi: 10.1016/S0272-5231(02)00014-X
3. Haworth, CS, Banks, J, Capstick, T, Fisher, AJ, Gorsuch, T, Laurenson, IF, et al. British Thoracic Society guidelines for the management of NTMl pulmonary disease (NTM-PD). Thorax. (2017) 72:ii1–ii64. doi: 10.1136/thoraxjnl-2017-210927
4. Cowman, S, Wilson, R, and Loebinger, MR. Opportunistic mycobacterial diseases. Medicine. (2016) 44:390–2. doi: 10.1016/j.mpmed.2016.03.010
5. Furuuchi, K, Morimoto, K, Yoshiyama, T, Tanaka, Y, Fujiwara, K, Okumura, M, et al. Interrelational changes in the epidemiology and clinical features of nontuberculous mycobacterial pulmonary disease and tuberculosis in a referral hospital in Japan. Respir Med. (2019) 152:74–80. doi: 10.1016/j.rmed.2019.05.001
6. Cowman, S, van Ingen, J, Griffith, DE, and Loebinger, MR. NTMl pulmonary disease. Eur Respir J. (2019) 54:1900250. doi: 10.1183/13993003.00250-2019
7. Kumar, K, and Loebinger, MR. Nontuberculous mycobacterial pulmonary disease: clinical epidemiologic features, risk factors, and diagnosis: the nontuberculous mycobacterial series. Chest. (2022) 161:637–46. doi: 10.1016/j.chest.2021.10.003
8. Ym, L, Mj, K, and Yj, K. Increasing trend of nontuberculous mycobacteria isolation in a referral clinical Laboratory in South Korea. Medicina. (2021) 57:720. doi: 10.3390/medicina57070720
9. Gopalaswamy, R, Shanmugam, S, Mondal, R, and Subbian, S. Of tuberculosis and NTMl infections - a comparative analysis of epidemiology, diagnosis and treatment. J Biomed Sci. (2020) 27:74. doi: 10.1186/s12929-020-00667-6
10. Donohue, MJ, and Wymer, L. Increasing prevalence rate of nontuberculous mycobacteria infections in five states, 2008-2013. Ann Am Thorac Soc. (2016) 13:2143–50. doi: 10.1513/AnnalsATS.201605-353OC
11. Domenech, P, Jimenez, MS, Menendez, MC, Bull, TJ, Samper, S, Manrique, A, et al. Mycobacterium mageritense sp. nov. Int J Syst Bacteriol. (1997) 47:535–40. doi: 10.1099/00207713-47-2-535
12. Miqueleiz-Zapatero, A, Santa Olalla-Peralta, C, Guerrero-Torres, MD, Cardeñoso-Domingo, L, Hernández-Milán, B, and Domingo-García, D. Mycobacterium lentiflavum as the main cause of lymphadenitis in pediatric population. Enferm Infecc Microbiol Clin. (2018) 36:640–3. doi: 10.1016/j.eimc.2017.12.006
13. Martínez-Planas, A, Baquero-Artigao, F, Santiago, B, Fortuny, C, Méndez-Echevarría, A, Del Rosal, T, et al. Interferon-gamma release assays differentiate between Mycobacterium avium complex and tuberculous lymphadenitis in children. J Pediatr. (2021) 236:211–218.e2. doi: 10.1016/j.jpeds.2021.05.008
14. Garcia-Marcos, PW, Plaza-Fornieles, M, Menasalvas-Ruiz, A, Ruiz-Pruneda, R, Paredes-Reyes, P, and Miguelez, SA. Risk factors of non-tuberculous mycobacterial lymphadenitis in children: a case-control study. Eur J Pediatr. (2017) 176:607–13. doi: 10.1007/s00431-017-2882-3
15. Tortoli, E. Clinical manifestations of nontuberculous mycobacteria infections. Clin Microbiol Infect. (2009) 15:906–10. doi: 10.1111/j.1469-0691.2009.03014.x
16. Morimoto, K, Hasegawa, N, Izumi, K, Namkoong, H, Uchimura, K, Yoshiyama, T, et al. A laboratory-based analysis of nontuberculous mycobacterial lung disease in Japan from 2012 to 2013. Ann Am Thorac Soc. (2017) 14:49–56. doi: 10.1513/AnnalsATS.201607-573OC
17. Sun, Y, and Li, H. Mycobacterium haemophilum infection in immunocompetent adults: a literature review and case report. Int J Dermatol. (2024) 63:169–76. doi: 10.1111/ijd.16874
18. Medel-Plaza, M, and Esteban, J. Current treatment options for Mycobacterium marinum cutaneous infections. Expert Opin Pharmacother. (2023) 24:1113–23. doi: 10.1080/14656566.2023.2211258
19. Peng, M, Li, W, Li, F, Tang, B, Deng, Y, Peng, S, et al. Mycobacterium xenopi related spine infections: a case report and systematic literature review. One Health. (2023) 16:100502. doi: 10.1016/j.onehlt.2023.100502
20. Jarzembowski, JA, and Young, MB. Nontuberculous mycobacterial infections. Arch Pathol Lab Med. (2008) 132:1333–41. doi: 10.5858/2008-132-1333-NMI
21. Huh, HJ, Kim, SY, Jhun, BW, Shin, SJ, and Koh, WJ. Recent advances in molecular diagnostics and understanding mechanisms of drug resistance in nontuberculous mycobacterial diseases. Infect Genet Evol. (2019) 72:169–82. doi: 10.1016/j.meegid.2018.10.003
22. Jagielski, T, van Ingen, J, Rastogi, N, Dziadek, J, Mazur, PK, and Bielecki, J. Current methods in the molecular typing of Mycobacterium tuberculosis and other mycobacteria. Biomed Res Int. (2014) 2014:645802. doi: 10.1155/2014/645802
23. Reynaud, Y, Millet, J, Couvin, D, Rastogi, N, Brown, C, Couppié, P, et al. Heterogeneity among Mycobacterium ulcerans from French Guiana revealed by multilocus variable number tandem repeat analysis (MLVA). PLoS One. (2015) 10:e0118597. doi: 10.1371/journal.pone.0118597
24. IMPERIALE, BR, MOYANO, RD, di GIULIO, AB, ROMERO, MA, ALVARADO PINEDO, MF, SANTANGELO, MP, et al. Genetic diversity of Mycobacterium avium complex strains isolated in Argentina by MIRU-VNTR. Epidemiol Infect. (2017) 145:1382–91. doi: 10.1017/S0950268817000139
25. Mansoori, N, Yaseri, M, Vaziri, F, and Douraghi, M. Genetic diversity of Mycobacterium tuberculosis complex isolates circulating in an area with high tuberculosis incidence: using 24-locus MIRU-VNTR method. Tuberculosis. (2018) 112:89–97. doi: 10.1016/j.tube.2018.08.003
26. Sohal, JS, Arsenault, J, Labrecque, O, Fairbrother, JH, Roy, JP, Fecteau, G, et al. Genetic structure of Mycobacterium avium subsp. paratuberculosis population in cattle herds in Quebec as revealed by using a combination of multilocus genomic analyses. J Clin Microbiol. (2014) 52:2764–75. doi: 10.1128/JCM.00386-14
27. Jagielski, T, Minias, A, van Ingen, J, Rastogi, N, Brzostek, A, Żaczek, A, et al. Methodological and clinical aspects of the molecular epidemiology of Mycobacterium tuberculosis and other mycobacteria. Clin Microbiol Rev. (2016) 29:239–90. doi: 10.1128/CMR.00055-15
28. Shin, JI, Ha, JH, Kim, KM, Choi, JG, Park, SR, Park, HE, et al. A novel repeat sequence-based PCR (rep-PCR) using specific repeat sequences of Mycobacterium intracellulare as a DNA fingerprinting. Front Microbiol. (2023) 14:1161194. doi: 10.3389/fmicb.2023.1161194
29. Karauz, M, Koç, A, and Atalay, A. Identification and molecular epidemiology of mycobacterial isolates in Kayseri/Turkey. Int J Respir Pulm Med. (2018) 5:092. doi: 10.23937/2378-3516/1410092
30. Gilchrist, FJ, Bright-Thomas, RJ, Jones, AM, Smith, D, Spaněl, P, Webb, AK, et al. Hydrogen cyanide concentrations in the breath of adult cystic fibrosis patients with and without Pseudomonas aeruginosa infection. J Breath Res. (2013) 7:026010. doi: 10.1088/1752-7155/7/2/026010
31. Halstrom, S, Price, P, and Thomson, R. Review: environmental mycobacteria as a cause of human infection. Int J Mycobacteriol. (2015) 4:81–91. doi: 10.1016/j.ijmyco.2015.03.002
32. Shah, J, Weltman, H, Narciso, P, Murphy, C, Poruri, A, Baliga, S, et al. Dual color fluorescence in situ hybridization (FISH) assays for detecting Mycobacterium tuberculosis and Mycobacterium avium complexes and related pathogens in cultures. PLoS One. (2017) 12:e0174989. doi: 10.1371/journal.pone.0174989
33. Shankar, H, Singh, SV, Singh, PK, Singh, AV, Sohal, JS, and Greenstein, RJ. Presence, characterization, and genotype profiles of Mycobacterium avium subspecies paratuberculosis from unpasteurized individual and pooled milk, commercial pasteurized milk, and milk products in India by culture, PCR, and PCR-REA methods. Int J Infect Dis. (2010) 14:e121–6. doi: 10.1016/j.ijid.2009.03.031
34. Singh, AV, Singh, SV, Makharia, GK, Singh, PK, and Sohal, JS. Presence and characterization of Mycobacterium avium subspecies paratuberculosis from clinical and suspected cases of Crohn's disease and in the healthy human population in India. Int J Infect Dis. (2008) 12:190–7. doi: 10.1016/j.ijid.2007.06.008
35. Whittington, RJ, Hope, AF, Marshall, DJ, Taragel, CA, and Marsh, I. Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis: IS900 restriction fragment length polymorphism and IS1311 polymorphism analyses of isolates from animals and a human in Australia. J Clin Microbiol. (2000) 38:3240–8. doi: 10.1128/JCM.38.9.3240-3248.2000
36. Khieu, V, Ananta, P, Kaewprasert, O, Laohaviroj, M, Namwat, W, and Faksri, K. Whole-genome sequencing analysis to identify infection with multiple species of nontuberculous mycobacteria. Pathogens. (2021) 10:879. doi: 10.3390/pathogens10070879
37. Huh, HJ, Kim, SY, Shim, HJ, Kim, DH, Yoo, IY, Kang, OK, et al. GenoType NTM-DR performance evaluation for identification of Mycobacterium avium complex and Mycobacterium abscessus and determination of clarithromycin and amikacin resistance. J Clin Microbiol. (2019) 57:e00516–9. doi: 10.1128/JCM.00516-19
38. Fang, H, Shangguan, Y, Wang, H, Ji, Z, Shao, J, Zhao, R, et al. Multicenter evaluationof the biochip assay for rapid detection of mycobacteriaisolates in smear positive specimens. Int J Infect Dis. (2019) 81:46–51. doi: 10.1016/j.ijid.2019.01.036
39. Zhang, H, Luo, M, Zhang, K, Yang, X, Hu, K, Fu, Z, et al. Species identification andantimicrobial susceptibility testing of non tuberculous mycobacteria isolated in Chongqing. Southwest China Epidemiol Infect. (2020) 149:e7. doi: 10.1017/S0950268820003088
40. Nour-Neamatollahie, A, Ebrahimzadeh, N, Siadat, SD, Vaziri, F, Eslami, M, Akhavan Sepahi, A, et al. Distribution of non-tuberculosis mycobacteria strains from suspected tuberculosis patients by heat shock protein 65 PCR-RFLP. Saudi J Biol Sci. (2017) 24:1380–6. doi: 10.1016/j.sjbs.2016.02.001
41. Saifi, M, Jabbarzadeh, E, Bahrmand, AR, Karimi, A, Pourazar, S, Fateh, A, et al. HSP65-PRA identification of non-tuberculosis mycobacteria from 4892 samples suspicious for mycobacterial infections. Clin Microbiol Infect. (2013) 19:723–8. doi: 10.1111/j.1469-0691.2012.04005.x
42. Nasr-Esfahani, B, Sarikhani, E, Moghim, S, Faghri, J, Fazeli, H, Hoseini, N, et al. Molecular characterization of environmental non-tuberculous mycobacteria using PCR-RFLP analysis of 441 Bp heat shock protein 65 fragments. Iran J Public Health. (2012) 41:108–14. doi: 10.1016/j.ijheh.2011.08.018
43. Jeon, S, Lim, N, Kwon, S, Shim, T, Park, M, Kim, BJ, et al. Molecular typing of Mycobacterium intracellulare using pulsed-field gel electrophoresis, variable-number tandem-repeat analysis, mycobacteria interspersed repetitive-unit-variable-number tandem repeat typing, and multilocus sequence typing: molecular characterization and comparison of each typing methods. Osong Public Health Res Perspect. (2014) 5:119–30. doi: 10.1016/j.phrp.2014.04.003
44. Kröner, C, Ganster, B, Kappler, M, Grimmelt, A, Belohradsky, BH, Hogardt, M, et al. Molecular epidemiology of nontuberculous mycobacteria in a German CF Center and clinical course of NTM positive patients. Open J Med Microbiol. (2013) 3:39–47. doi: 10.4236/ojmm.2013.31006
45. Scorzolini, L, Mengoni, F, Mastroianni, CM, Baldan, R, Cirillo, DM, De Giusti, M, et al. Pseudo-outbreak of Mycobacterium gordonae in a teaching hospital: importance of strictly following decontamination procedures and emerging issues concerning sterilization. New Microbiol. (2016) 39:25–34.
46. Khosravi, AD, Mehrabzadeh, RS, Farahani, A, and Jamali, H. Molecular identification of clinical isolates of Mycobacterium fortuitum by random amplified polymorphic DNA (RAPD) polymerase chain reaction and ERIC PCR. J Clin Diagn Res. (2015) 9:DC01–5. doi: 10.7860/JCDR/2015/15504.6909
47. Blanco-Conde, S, González-Cortés, C, López-Medrano, R, Palacios-Gutiérrez, JJ, Diez-Tascón, C, Nebreda-Mayoral, T, et al. A strategy based on amplified fragment length polymorphism (AFLP) for routine genotyping of nontuberculous mycobacteria at the clinical laboratory. Mol Biol Rep. (2020) 47:3397–405. doi: 10.1007/s11033-020-05420-8
48. Buijtels, PC, Petit, PL, Verbrugh, HA, van Belkum, A, and van Soolingen, D. Isolation of nontuberculous mycobacteria in Zambia: eight case reports. J Clin Microbiol. (2005) 43:6020–6. doi: 10.1128/JCM.43.12.6020-6026.2005
49. Streit, ES. Mycobactérium tuberculosis and non tuberculous mycobacteria in the French Departments of the Americas and in the Caribbean: Studying epidemiological aspects and transmission using molecular tools and database comparison. (2015). [dissertation/doctor’s thesis]. (Guadeloupe: Université des Antilles).
50. Dohál, M, Porvazník, I, Solovič, I, and Mokrý, J. Whole genome sequencing in the management of NTMl infections. Microorganisms. (2021) 9:2237. doi: 10.3390/microorganisms9112237
51. Turenne, CY. Nontuberculous mycobacteria: insights on taxonomy and evolution. Infect Genet Evol. (2019) 72:159–68. doi: 10.1016/j.meegid.2019.01.017
52. Liu, Y, Ma, X, Chen, J, Wang, H, and Yu, Z. Nontuberculous mycobacteria by metagenomic next-generation sequencing: three cases reports and literature review. Front Public Health. (2022) 10:972280. doi: 10.3389/fpubh.2022.972280
53. Wang, S, and Xing, L. Metagenomic next-generation sequencing assistance in identifying NTMl infections. Front Cell Infect Microbiol. (2023) 13:1253020. doi: 10.3389/fcimb.2023.1253020
54. Zhu, H, Zhu, M, Lei, JH, Xiao, YL, and Zhao, LM. Metagenomic next-generation sequencing can clinch diagnosis of NTMl infections: a case report. Front Med. (2021) 8:679755. doi: 10.3389/fmed.2021.679755
55. Kolb, J, Hillemann, D, Möbius, P, Reetz, J, Lahiri, A, Lewin, A, et al. Genetic characterization of German Mycobacterium avium strains isolated from different hosts and specimens by multilocus sequence typing. Int J Med Microbiol. (2014) 304:941–8. doi: 10.1016/j.ijmm.2014.06.001
56. Mundo, SL, Gilardoni, LR, Hoffman, FJ, and Lopez, OJ. Rapid and sensitive method to identify Mycobacterium avium subsp. paratuberculosis in cow's milk by DNA methylase genotyping. Appl Environ Microbiol. (2013) 79:1612–8. doi: 10.1128/AEM.02719-12
57. Rindi, L, and Garzelli, C. Genetic diversity and phylogeny of Mycobacterium avium. Infect Genet Evol. (2014) 21:375–83. doi: 10.1016/j.meegid.2013.12.007
58. Taheri, MM, Mosavari, N, Feizabadi, MM, Tadayon, K, Keshavarz, R, Pajoohi, RA, et al. Rapid identification of Mycobacterium avium ssp paratuberculosis laboratory strains by IS900-nested polymerase chain reaction. Int J Mycobacteriol. (2016) 5:S232–3. doi: 10.1016/j.ijmyco.2016.10.018
59. Orłowska, B, Majchrzak, M, Didkowska, A, Anusz, K, Krajewska-Wędzina, M, Zabost, A, et al. Mycobacterial interspersed repeat unit-variable number tandem repeat typing of Mycobacterium avium strains isolated from the lymph nodes of free-living carnivorous animals in Poland. Pathogens. (2023). 12:1184. doi: 10.3390/pathogens12091184
60. Starkova, DA, Otten, TF, Mokrousov, IV, Viazovaia, AA, Vishnevskiĭ, BI, and Narvskaia, OV. Genotypic characteristics of Mycobacterium avium subsp. hominissuis strains. J Genet. (2013) 49:1048–54. doi: 10.1134/s1022795413090093
61. Picardeau, M, Varnerot, A, Rauzier, J, Gicquel, B, and Vincent, V. Mycobacterium xenopi IS1395, a novel insertion sequence expanding the IS256 family. Microbiology. (1996) 142:2453–61. doi: 10.1099/00221287-142-9-2453
62. Picardeau, M, Bull, TJ, and Vincent, V. Identification and characterization of IS-like elements in Mycobacterium gordonae. FEMS Microbiol Lett. (1997) 154:95–102. doi: 10.1111/j.1574-6968.1997.tb12629.x
63. Picardeau, M, Bull, TJ, Prod'hom, G, Pozniak, AL, Shanson, DC, and Vincent, V. Comparison of a new insertion element, IS1407, with established molecular markers for the characterization of Mycobacterium celatum. Int J Syst Bacteriol. (1997) 47:640–4. doi: 10.1099/00207713-47-3-640
64. Guilhot, C, Gicquel, B, Davies, J, and Martín, C. Isolation and analysis of IS6120, a new insertion sequence from Mycobacterium smegmatis. Mol Microbiol. (1992) 6:107–13. doi: 10.1111/j.1365-2958.1992.tb00842.x
65. Stinear, T, Ross, BC, Davies, JK, Marino, L, Robins-Browne, RM, Oppedisano, F, et al. Identification and characterization of IS2404 and IS2606: two distinct repeated sequences for detection of Mycobacterium ulcerans by PCR. J Clin Microbiol. (1999) 37:1018–23. doi: 10.1128/JCM.37.4.1018-1023.1999
66. Picardeau, M, Prod'Hom, G, Raskine, L, LePennec, MP, and Vincent, V. Genotypic characterization of five subspecies of Mycobacterium kansasii. J Clin Microbiol. (1997) 35:25–32. doi: 10.1128/jcm.35.1.25-32.1997
67. Otsuka, Y, Parniewski, P, Zwolska, Z, Kai, M, Fujino, T, Kirikae, F, et al. Characterization of a trinucleotide repeat sequence (CGG)5 and potential use in restriction fragment length polymorphism typing of Mycobacterium tuberculosis. J Clin Microbiol. (2004) 42:3538–48. doi: 10.1128/JCM.42.8.3538-3548.2004
68. Seifi, K, Kazemian, H, Heidari, H, Rezagholizadeh, F, Saee, Y, Shirvani, F, et al. Evaluation of biofilm formation among Klebsiella pneumoniae isolates and molecular characterization by ERIC-PCR. Jundishapur J Microbiol. (2016) 9:e30682. doi: 10.5812/jjm.30682
69. Gillings, M, and Holley, M. Repetitive element PCR fingerprinting (rep-PCR) using enterobacterial repetitive intergenic consensus (ERIC) primers is not necessarily directed at ERIC elements. Lett Appl Microbiol. (1997) 25:17–21. doi: 10.1046/j.1472-765x.1997.00162.x
70. Sechi, LA, Zanetti, S, Dupré, I, Delogu, G, and Fadda, G. Enterobacterial repetitive intergenic consensus sequences as molecular targets for typing of Mycobacterium tuberculosis strains. J Clin Microbiol. (1998) 36:128–32. doi: 10.1128/JCM.36.1.128-132.1998
71. Asiimwe, BB, Bagyenzi, GB, Ssengooba, W, Mumbowa, F, Mboowa, G, Wajja, A, et al. Species and genotypic diversity of non-tuberculous mycobacteria isolated from children investigated for pulmonary tuberculosis in rural Uganda. BMC Infect Dis. (2013) 13:88. doi: 10.1186/1471-2334-13-88
72. Sampaio, JL, Chimara, E, Ferrazoli, L, da Silva Telles, MA, Del Guercio, VM, Jericó, ZV, et al. Application of four molecular typing methods for analysis of Mycobacterium fortuitum group strains causing post-mammaplasty infections. Clin Microbiol Infect. (2006) 12:142–9. doi: 10.1111/j.1469-0691.2005.01312.x
73. Sampaio, JL, Viana-Niero, C, de Freitas, D, Höfling-Lima, AL, and Leão, SC. Enterobacterial repetitive intergenic consensus PCR is a useful tool for typing Mycobacterium chelonae and Mycobacterium abscessus isolates. Diagn Microbiol Infect Dis. (2006) 55:107–18. doi: 10.1016/j.diagmicrobio.2006.01.006
74. Da Mata, JO, Hernández-Pérez, R, Corrales, H, Cardoso-Leao, S, and de Waard, JH. Seguimiento de un brote de infección en tejido blando causado por Mycobacterium abscessus posterior a la mesoterapia en Venezuela [follow-up on an outbreak in Venezuela of soft-tissue infection due to Mycobacterium abscessus associated with Mesotherapy]. Enferm Infecc Microbiol Clin. (2010) 28:596–601. Spanish. doi: 10.1016/j.eimc.2009.08.003
75. Fawzy, A, Zschöck, M, Ewers, C, and Eisenberg, T. New polymorphisms within the variable number tandem repeat (VNTR) 7 locus of Mycobacterium avium subsp. paratuberculosis. Mol Cell Probes. (2016) 30:132–7. doi: 10.1016/j.mcp.2016.02.002
76. Hermans, PW, van Soolingen, D, and van Embden, JD. Characterization of a major polymorphic tandem repeat in Mycobacterium tuberculosis and its potential use in the epidemiology of Mycobacterium kansasii and Mycobacterium gordonae. J Bacteriol. (1992) 174:4157–65. doi: 10.1128/jb.174.12.4157-4165.1992
77. Taillard, C, Greub, G, Weber, R, Pfyffer, GE, Bodmer, T, Zimmerli, S, et al. Clinical implications of Mycobacterium kansasii species heterogeneity: Swiss National Survey. J Clin Microbiol. (2003) 41:1240–4. doi: 10.1128/JCM.41.3.1240-1244.2003
78. Supply, P, Allix, C, Lesjean, S, Cardoso-Oelemann, M, Rüsch-Gerdes, S, Willery, E, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. (2006) 44:4498–510. doi: 10.1128/JCM.01392-06
79. Iakhiaeva, E, McNulty, S, Brown Elliott, BA, Falkinham, JO 3rd, Williams, MD, Vasireddy, R, et al. Mycobacterial interspersed repetitive-unit-variable-number tandem-repeat (MIRU-VNTR) genotyping of mycobacterium intracellulare for strain comparison with establishment of a PCR-based database. J Clin Microbiol. (2006) 51:409–16. doi: 10.1128/JCM.02443-12
80. Mougari, F, Raskine, L, Ferroni, A, Marcon, E, Sermet-Gaudelus, I, Veziris, N, et al. Clonal relationship and differentiation among Mycobacterium abscessus isolates as determined using the semiautomated repetitive extragenic palindromic sequence PCR-based DiversiLab system. J Clin Microbiol. (2014) 52:1969–77. doi: 10.1128/JCM.03600-13
81. Iakhiaeva, E, Howard, ST, Brown Elliott, BA, McNulty, S, Newman, KL, Falkinham, JO 3rd, et al. Variable-number tandem-repeat analysis of respiratory and household water biofilm isolates of "Mycobacterium avium subsp. hominissuis" with establishment of a PCR database. J Clin Microbiol. (2016) 54:891–901. doi: 10.1128/JCM.02409-15
82. Dou, HY, Lin, CH, Chen, YY, Yang, SJ, Chang, JR, Wu, KM, et al. Lineage-specific SNPs for genotyping of Mycobacterium tuberculosis clinical isolates. Sci Rep. (2017) 7:1425. doi: 10.1038/s41598-017-01580-z
83. Jolley, KA, and Maiden, MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinform. (2010) 11:595. doi: 10.1186/1471-2105-11-595
84. Matsumoto, Y, Kinjo, T, Motooka, D, Nabeya, D, Jung, N, Uechi, K, et al. Comprehensive subspecies identification of 175 nontuberculous mycobacteria species based on 7547 genomic profiles. Emerg Microbes Infect. (2019) 8:1043–53. doi: 10.1080/22221751.2019.1637702
85. Adékambi, T, and Drancourt, M. Dissection of phylogenetic relationships among 19 rapidly growing Mycobacterium species by 16S rRNA, hsp65, sodA, recA and rpoB gene sequencing. Int J Syst Evol Microbiol. (2004) 54:2095–105. doi: 10.1099/ijs.0.63094-0
86. Dalovisio, JR, Montenegro-James, S, Kemmerly, SA, Genre, CF, Chambers, R, Greer, D, et al. Comparison of the amplified Mycobacterium tuberculosis (MTB) direct test, Amplicor MTB PCR, and IS6110-PCR for detection of MTB in respiratory specimens. Clin Infect Dis. (1996) 23:1099–106; discussion 1107-8. doi: 10.1093/clinids/23.5.1099
87. Bloemberg, GV, Voit, A, Ritter, C, Deggim, V, and Böttger, EC. Evaluation of Cobas TaqMan MTB for direct detection of the Mycobacterium tuberculosis complex in comparison with Cobas Amplicor MTB. J Clin Microbiol. (2013) 51:2112–7. doi: 10.1128/JCM.00142-13
88. Rodríguez-Lázaro, D, Lloyd, J, Herrewegh, A, Ikonomopoulos, J, D'Agostino, M, Pla, M, et al. A molecular beacon-based real-time NASBA assay for detection of Mycobacterium avium subsp. paratuberculosis in water and milk. FEMS Microbiol Lett. (2004) 237:119–26. doi: 10.1016/j.femsle.2004.06.024
89. Barrett, A, Magee, JG, and Freeman, R. An evaluation of the BD ProbeTec ET system for the direct detection of Mycobacterium tuberculosis in respiratory samples. J Med Microbiol. (2002) 51:895–8. doi: 10.1099/0022-1317-51-10-895
90. Ichiyama, S, Iinuma, Y, Tawada, Y, Yamori, S, Hasegawa, Y, Shimokata, K, et al. Evaluation of gen-probe amplified Mycobacterium Tuberculosis direct test and Roche PCR-microwell plate hybridization method (AMPLICOR MYCOBACTERIUM) for direct detection of mycobacteria. J Clin Microbiol. (1996) 34:130–3. doi: 10.1128/jcm.34.1.130-133.1996
91. Kauppinen, J, Pelkonen, J, and Katila, ML. RFLP analysis of Mycobacterium malmoense strains using ribosomal RNA gene probes: an addition tool to examine intraspecies variation. J Microbiol Methods. (1994) 19:261–7. doi: 10.1016/0167-7012(94)90029-9
92. Kusunoki, S, Ezaki, T, Tamesada, M, Hatanaka, Y, Asano, K, Hashimoto, Y, et al. Application of colorimetric microdilution plate hybridization for rapid genetic identification of 22 Mycobacterium species. J Clin Microbiol. (1991) 29:1596–603. doi: 10.1128/jcm.29.8.1596-1603.1991
93. Wang, HY, Kim, H, Kim, S, Bang, H, Kim, DK, and Lee, H. Evaluation of PCR-reverse blot hybridization assay for the differentiation and identification of Mycobacterium species in liquid cultures. J Appl Microbiol. (2015) 118:142–51. doi: 10.1111/jam.12670
94. Lee, MR, Cheng, A, Huang, YT, Liu, CY, Chung, KP, Wang, HC, et al. Performance assessment of the DR. TBDR/NTM IVD kit for direct detection of Mycobacterium tuberculosis isolates, including rifampin-resistant isolates, and nontuberculous mycobacteria. J Clin Microbiol. (2012) 50:3398–401. doi: 10.1128/JCM.01862-12
95. Yu, Q, Wang, Y, Gao, Z, Yang, H, Liu, S, Tan, J, et al. DNA microarray chip assay in new use: early diagnostic value in cutaneous mycobacterial infection. Front Cell Infect Microbiol. (2023) 13:1183078. doi: 10.3389/fcimb.2023.1183078
96. Huang, JJ, Li, YX, Zhao, Y, Yang, WH, Xiao, M, Kudinha, T, et al. Prevalence of nontuberculous mycobacteria in a tertiary hospital in Beijing, China, January 2013 to December 2018. BMC Microbiol. (2020) 20:158. doi: 10.1186/s12866-020-01840-5
97. Shukla, S, Shukla, SK, and Sharma, R. Species identification of aquatic mycobacterium isolates by sequencing and PCR-RFLP of the 16S–23S rDNA internal transcribed spacer (ITS) region. Gene Reports. (2018) 10:26–32. doi: 10.1016/j.genrep.2017.10.013
98. Bakuła, Z, Brzostek, A, Borówka, P, Żaczek, A, Szulc-Kiełbik, I, Podpora, A, et al. Molecular typing of Mycobacterium kansasii using pulsed-field gel electrophoresis and a newly designed variable-number tandem repeat analysis. Sci Rep. (2018) 8:4462. doi: 10.1038/s41598-018-21562-z
99. Sabat, AJ, Budimir, A, Nashev, D, Sá-Leão, R, van Dijl, JM, Laurent, F, et al. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill. (2013) 18:20380. doi: 10.2807/ese.18.04.20380-en
100. Zhang, Y, Rajagopalan, M, Brown, BA, and Wallace, RJ Jr. Randomly amplified polymorphic DNA PCR for comparison of Mycobacterium abscessus strains from nosocomial outbreaks. J Clin Microbiol. (1997) 35:3132–9. doi: 10.1128/jcm.35.12.3132-3139.1997
101. Risjani, Y, and Abidin, G. Genetic diversity and similarity between green and brown morphotypes of Kappaphycus alvarezii using RAPD. J Appl Phycol. (2020) 32:2253–60. doi: 10.1007/s10811-020-02223-z
102. Christianson, S, Wolfe, J, Soualhine, H, and Sharma, MK. Comparison of repetitive-sequence-based polymerase chain reaction with random amplified polymorphic DNA analysis for rapid genotyping of nontuberculosis mycobacteria. Can J Microbiol. (2012) 58:953–64. doi: 10.1139/w2012-068
103. Mougari, F, Guglielmetti, L, Raskine, L, Sermet-Gaudelus, I, Veziris, N, and Cambau, E. Infections caused by Mycobacterium abscessus: epidemiology, diagnostic tools and treatment. Expert Rev Anti-Infect Ther. (2016) 14:1139–54. doi: 10.1080/14787210.2016.1238304
104. Cooksey, RC, Jhung, MA, Yakrus, MA, Butler, WR, Adékambi, T, Morlock, GP, et al. Multiphasic approach reveals genetic diversity of environmental and patient isolates of Mycobacterium mucogenicum and Mycobacterium phocaicum associated with an outbreak of bacteremias at a Texas hospital. Appl Environ Microbiol. (2008) 74:2480–7. doi: 10.1128/AEM.02476-07
105. Vogiatzakis, E, Stefanou, S, Skroubelou, A, Anagnostou, S, Marinis, E, and Matsiota-Bernard, P. Molecular markers for the investigation of Mycobacterium gordonae epidemics. J Hosp Infect. (1998) 38:217–22. doi: 10.1016/s0195-6701(98)90277-8
106. van Ingen, J, Boeree, MJ, de Lange, WC, de Haas, PE, Dekhuijzen, PN, and van Soolingen, D. Clinical relevance of Mycobacterium szulgai in the Netherlands. Clin Infect Dis. (2008) 46:1200–5. doi: 10.1086/529443
107. Kauppinen, J, Mäntyjärvi, R, and Katila, ML. Random amplified polymorphic DNA genotyping of Mycobacterium malmoense. J Clin Microbiol. (1994) 32:1827–9. doi: 10.1128/jcm.32.7.1827-1829.1994
108. Hoheisel, JD. Microarray technology: beyond transcript profiling and genotype analysis. Nat Rev Genet. (2006) 7:200–10. doi: 10.1038/nrg1809
109. Stevenson, K. Genetic diversity of Mycobacterium avium subspecies paratuberculosis and the influence of strain type on infection and pathogenesis: a review. Vet Res. (2015) 46:64. doi: 10.1186/s13567-015-0203-2
110. Stinear, TP, Seemann, T, Pidot, S, Frigui, W, Reysset, G, Garnier, T, et al. Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Genome Res. (2007) 17:192–200. doi: 10.1101/gr.5942807
111. Cho, YJ, Yi, H, Chun, J, Cho, SN, Daley, CL, Koh, WJ, et al. The genome sequence of 'Mycobacterium massiliense' strain CIP 108297 suggests the independent taxonomic status of the Mycobacterium abscessus complex at the subspecies level. PLoS One. (2013) 8:e81560. doi: 10.1371/journal.pone.0081560
112. Roetzer, A, Diel, R, Kohl, TA, Rückert, C, Nübel, U, Blom, J, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. (2013) 10:e1001387. doi: 10.1371/journal.pmed.1001387
113. Walker, TM, Ip, CL, Harrell, RH, Evans, JT, Kapatai, G, Dedicoat, MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. (2013) 13:137–46. doi: 10.1016/S1473-3099(12)70277-3
114. Kato-Maeda, M, Ho, C, Passarelli, B, Banaei, N, Grinsdale, J, Flores, L, et al. Use of whole genome sequencing to determine the microevolution of Mycobacterium tuberculosis during an outbreak. PLoS One. (2013) 8:e58235. doi: 10.1371/journal.pone.0058235
115. Niemann, S, Köser, CU, Gagneux, S, Plinke, C, Homolka, S, Bignell, H, et al. Genomic diversity among drug sensitive and multidrug resistant isolates of Mycobacterium tuberculosis with identical DNA fingerprints. PLoS One. (2009) 4:e7407. doi: 10.1371/journal.pone.0007407
116. Gardy, JL, Johnston, JC, Ho Sui, SJ, Cook, VJ, Shah, L, Brodkin, E, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. (2011) 364:730–9. doi: 10.1056/NEJMoa1003176
117. Wlodarska, M, Johnston, JC, Gardy, JL, and Tang, P. A microbiological revolution meets an ancient disease: improving the management of tuberculosis with genomics. Clin Microbiol Rev. (2015) 28:523–39. doi: 10.1128/CMR.00124-14
118. Beviere, M, Reissier, S, Penven, M, Dejoies, L, Guerin, F, Cattoir, V, et al. The role of next-generation sequencing (NGS) in the Management of Tuberculosis: practical review for implementation in routine. Pathogens. (2023) 12:978. doi: 10.3390/pathogens12080978
119. Yadav, D, Patil-Takbhate, B, Khandagale, A, Bhawalkar, J, Tripathy, S, and Khopkar-Kale, P. Next-generation sequencing transforming clinical practice and precision medicine. Clin Chim Acta. (2023) 551:117568. doi: 10.1016/j.cca.2023.117568
120. Sarkar, MMH, Rahman, MS, Islam, MR, Rahman, A, Islam, MS, Banu, TA, et al. Comparative phylogenetic analysis and transcriptomic profiling of dengue (DENV-3 genotype I) outbreak in 2021 in Bangladesh. Virol J. (2023) 20:127. doi: 10.1186/s12985-023-02030-1
121. Wei, Y, Cai, Y, Han, X, Han, Z, Zhang, Y, Xu, Y, et al. Genetic diversity and molecular evolution of Seoul virus in Hebei province. China Infect Genet Evol. (2023) 114:105503. doi: 10.1016/j.meegid.2023.105503
122. Satam, H, Joshi, K, Mangrolia, U, Waghoo, S, Zaidi, G, Rawool, S, et al. Next-generation sequencing technology: current trends and advancements. Biology. (2023) 12:997. doi: 10.3390/biology12070997
123. Dougherty, CE, and Graf, E. Next generation sequencing for outbreak investigation in the clinical microbiology laboratory. Amer Soci Clin Lab Sci. (2019):ascls.119.001750. doi: 10.29074/ascls.119.001750
124. Buckwalter, SP, Olson, SL, Fida, M, Epperson, LE, Hasan, NA, Khare, R, et al. Mycobacterium abscessus subspecies identification using the Deeplex Myc-TB targeted NGS assay. J Clin Microbiol. (2023) 61:e0048923. doi: 10.1128/jcm.00489-23
125. He, Y, Gong, Z, Zhao, X, Zhang, D, and Zhang, Z. Comprehensive determination of Mycobacterium tuberculosis and nontuberculous mycobacteria from targeted capture sequencing. Front Cell Infect Microbiol. (2020) 10:449. doi: 10.3389/fcimb.2020.00449
126. Deng, Z, Li, C, Wang, Y, Wu, F, Liang, C, Deng, W, et al. Targeted next-generation sequencing for pulmonary infection diagnosis in patients unsuitable for bronchoalveolar lavage. Front Med. (2023) 10:1321515. doi: 10.3389/fmed.2023.1321515
127. Gaston, DC, Miller, HB, Fissel, JA, Jacobs, E, Gough, E, Wu, J, et al. Evaluation of metagenomic and targeted next-generation sequencing workflows for detection of respiratory pathogens from Bronchoalveolar lavage fluid specimens. J Clin Microbiol. (2022) 60:e0052622. doi: 10.1128/jcm.00526-22
128. Redgrave, LS, Sutton, SB, Webber, MA, and Piddock, LJ. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. (2014) 22:438–45. doi: 10.1016/j.tim.2014.04.007
129. Larsson, DGJ, and Flach, CF. Antibiotic resistance in the environment. Nat Rev Microbiol. (2022) 20:257–69. doi: 10.1038/s41579-021-00649-x
130. Brown-Elliott, BA, and Woods, GL. Antimycobacterial susceptibility testing of nontuberculous mycobacteria. J Clin Microbiol. (2019) 57:e00834–19. doi: 10.1128/JCM.00834-19
131. Cowman, S, Burns, K, Benson, S, Wilson, R, and Loebinger, MR. The antimicrobial susceptibility of NTM. J Infect. (2016) 72:324–31. doi: 10.1016/j.jinf.2015.12.007
132. Sharma, M, Malhotra, B, and Khandelwal, S. Drug susceptibiity testing of nontuberculous mycobacteria by broth microdilution method. Indian J Med Microbiol. (2021) 39:306–10. doi: 10.1016/j.ijmmb.2021.03.015
133. Alffenaar, JW, Märtson, AG, Heysell, SK, Cho, JG, Patanwala, A, Burch, G, et al. Therapeutic drug monitoring in non-tuberculosis mycobacteria infections. Clin Pharmacokinet. (2021) 60:711–25. doi: 10.1007/s40262-021-01000-6
134. Nguyen, TNA, Anton-Le Berre, V, Bañuls, AL, and Nguyen, TVA. Molecular diagnosis of drug-resistant tuberculosis. Lit Rev Front Microbiol. (2019) 10:794. doi: 10.3389/fmicb.2019.00794
135. Koch, A, Cox, H, and Mizrahi, V. Drug-resistant tuberculosis: challenges and opportunities for diagnosis and treatment. Curr Opin Pharmacol. (2018) 42:7–15. doi: 10.1016/j.coph.2018.05.013
136. Meier, A, Kirschner, P, Springer, B, Steingrube, VA, Brown, BA, Wallace, RJ Jr, et al. Identification of mutations in 23S rRNA gene of clarithromycin-resistant Mycobacterium intracellulare. Antimicrob Agents Chemother. (1994) 38:381–4. doi: 10.1128/AAC.38.2.381
137. Nash, KA, and Inderlied, CB. Genetic basis of macrolide resistance in Mycobacterium avium isolated from patients with disseminated disease. Antimicrob Agents Chemother. (1995) 39:2625–30. doi: 10.1128/AAC.39.12.2625
138. Thapa, J, Chizimu, JY, Kitamura, S, Akapelwa, ML, Suwanthada, P, Miura, N, et al. Characterization of DNA gyrase activity and elucidation of the impact of amino acid substitution in GyrA on fluoroquinolone resistance in Mycobacterium avium. Microbiol Spectr. (2023) 11:e0508822. doi: 10.1128/spectrum.05088-22
139. Burman, WJ, Stone, BL, Brown, BA, Wallace, RJ Jr, and Böttger, EC. AIDS-related Mycobacterium kansasii infection with initial resistance to clarithromycin. Diagn Microbiol Infect Dis. (1998) 31:369–71. doi: 10.1016/s0732-8893(98)00013-3
140. Calvet-Seral, J, Crespo-Yuste, E, Mathys, V, Rodriguez-Villalobos, H, Ceyssens, PJ, Martin, A, et al. Targeted chromosomal barcoding establishes direct genotype-phenotype associations for antibiotic resistance in Mycobacterium abscessus. Microbiol Spectr. (2023) 11:e0534422. doi: 10.1128/spectrum.05344-22
141. Nessar, R, Reyrat, JM, Murray, A, and Gicquel, B. Genetic analysis of new 16S rRNA mutations conferring aminoglycoside resistance in Mycobacterium abscessus. J Antimicrob Chemother. (2011) 66:1719–24. doi: 10.1093/jac/dkr209
142. Prammananan, T, Sander, P, Brown, BA, Frischkorn, K, Onyi, GO, Zhang, Y, et al. A single 16S ribosomal RNA substitution is responsible for resistance to amikacin and other 2-deoxystreptamine aminoglycosides in Mycobacterium abscessus and Mycobacterium chelonae. J Infect Dis. (1998) 177:1573–81. doi: 10.1086/515328
143. Wallace, RJ Jr, Meier, A, Brown, BA, Zhang, Y, Sander, P, Onyi, GO, et al. Genetic basis for clarithromycin resistance among isolates of Mycobacterium chelonae and Mycobacterium abscessus. Antimicrob Agents Chemother. (1996) 40:1676–81. doi: 10.1128/AAC.40.7.1676
144. Mougari, F, Bouziane, F, Crockett, F, Nessar, R, Chau, F, Veziris, N, et al. Selection of resistance to clarithromycin in Mycobacterium abscessus subspecies. Antimicrob Agents Chemother. (2016) 61:e00943–16. doi: 10.1128/AAC.00943-16
145. Vester, B, and Douthwaite, S. Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob Agents Chemother. (2001) 45:1–12. doi: 10.1128/AAC.45.1.1-12.2001
146. Sander, P, Prammananan, T, Meier, A, Frischkorn, K, and Böttger, EC. The role of ribosomal RNAs in macrolide resistance. Mol Microbiol. (1997) 26:469–80. doi: 10.1046/j.1365-2958.1997.5811946.x
147. Pfister, P, Risch, M, Brodersen, DE, and Böttger, EC. Role of 16S rRNA Helix 44 in ribosomal resistance to Hygromycin B. Antimicrob Agents Chemother. (2003) 47:1496–502. doi: 10.1128/AAC.47.5.1496-1502.2003
148. Vilchèze, C, Weisbrod, TR, Chen, B, Kremer, L, Hazbón, MH, Wang, F, et al. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob Agents Chemother. (2005) 49:708–20. doi: 10.1128/AAC.49.2.708-720.2005
149. Springer, B, Kidan, YG, Prammananan, T, Ellrott, K, Böttger, EC, and Sander, P. Mechanisms of streptomycin resistance: selection of mutations in the 16S rRNA gene conferring resistance. Antimicrob Agents Chemother. (2001) 45:2877–84. doi: 10.1128/AAC.45.10.2877-2884.2001
150. Taniguchi, H, Chang, B, Abe, C, Nikaido, Y, Mizuguchi, Y, and Yoshida, SI. Molecular analysis of kanamycin and viomycin resistance in Mycobacterium smegmatis by use of the conjugation system. J Bacteriol. (1997) 179:4795–801. doi: 10.1128/jb.179.15.4795-4801.1997
151. Svetlov, MS, Syroegin, EA, Aleksandrova, EV, Atkinson, GC, Gregory, ST, Mankin, AS, et al. Structure of Erm-modified 70S ribosome reveals the mechanism of macrolide resistance. Nat Chem Biol. (2021) 17:412–20. doi: 10.1038/s41589-020-00715-0
152. Richard, M, Gutiérrez, AV, and Kremer, L. Dissecting erm(41)-mediated macrolide-inducible resistance in Mycobacterium abscessus. Antimicrob Agents Chemother. (2020) 64:e01879–19. doi: 10.1128/AAC.01879-19
153. Moon, SM, Park, HY, Kim, SY, Jhun, BW, Lee, H, Jeon, K, et al. Clinical characteristics, treatment outcomes, and resistance mutations associated with macrolide-resistant Mycobacterium avium complex lung disease. Antimicrob Agents Chemother. (2016) 60:6758–65. doi: 10.1128/AAC.01240-16
154. Zhu, JH, Wang, BW, Pan, M, Zeng, YN, Rego, H, and Javid, B. Rifampicin can induce antibiotic tolerance in mycobacteria via paradoxical changes in rpoB transcription. Nat Commun. (2018) 9:4218. doi: 10.1038/s41467-018-06667-3
155. Bakuła, Z, Modrzejewska, M, Pennings, L, Proboszcz, M, Safianowska, A, Bielecki, J, et al. Drug susceptibility profiling and genetic determinants of drug resistance in Mycobacterium kansasii. Antimicrob Agents Chemother. (2018) 62:e01788–17. doi: 10.1128/AAC.01788-17
156. Kim, HJ, Lee, JS, Kwak, N, Cho, J, Lee, CH, Han, SK, et al. Role of ethambutol and rifampicin in the treatment of Mycobacterium avium complex pulmonary disease. BMC Pulm Med. (2019) 19:212. doi: 10.1186/s12890-019-0982-8
157. Johansen, MD, Herrmann, JL, and Kremer, L. NTM and the rise of Mycobacterium abscessus. Nat Rev Microbiol. (2020) 18:392–407. doi: 10.1038/s41579-020-0331-1
158. Rominski, A, Roditscheff, A, Selchow, P, Böttger, EC, and Sander, P. Intrinsic rifamycin resistance of Mycobacterium abscessus is mediated by ADP-ribosyltransferase MAB_0591. J Antimicrob Chemother. (2017) 72:376–84. doi: 10.1093/jac/dkw466
159. Ruth, MM, Sangen, JJN, Remmers, K, Pennings, LJ, Svensson, E, Aarnoutse, RE, et al. A bedaquiline/clofazimine combination regimen might add activity to the treatment of clinically relevant NTM. J Antimicrob Chemother. (2019) 74:935–43. doi: 10.1093/jac/dky526
160. Chen, Y, Chen, J, Zhang, S, Shi, W, Zhang, W, Zhu, M, et al. Novel mutations associated with Clofazimine resistance in Mycobacterium abscessus. Antimicrob Agents Chemother. (2018) 62:e00544–18. doi: 10.1128/AAC.00544-18
161. Griffith, DE, Aksamit, T, Brown-Elliott, BA, Catanzaro, A, Daley, C, Gordin, F, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. (2007) 175:367–416. doi: 10.1164/rccm.200604-571ST
162. Nash, KA, Brown-Elliott, BA, and Wallace, RJ Jr. A novel gene, erm(41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob Agents Chemother. (2009) 53:1367–76. doi: 10.1128/AAC.01275-08
163. Kim, HY, Kim, BJ, Kook, Y, Yun, YJ, Shin, JH, Kim, BJ, et al. Mycobacterium massiliense is differentiated from Mycobacterium abscessus and Mycobacterium bolletii by erythromycin ribosome methyltransferase gene (erm) and clarithromycin susceptibility patterns. Microbiol Immunol. (2010) 54:347–53. doi: 10.1111/j.1348-0421.2010.00221.x
164. Bouzinbi, N, Marcy, O, Bertolotti, T, Chiron, R, Bemer, P, Pestel-Caron, M, et al. Evaluation of the GenoType NTM-DR assay performance for the identification and molecular detection of antibiotic resistance in Mycobacterium abscessus complex. PLoS One. (2020) 15:e0239146. doi: 10.1371/journal.pone.0239146
165. Ruedas-López, A, Tato, M, Broncano-Lavado, A, Esteban, J, Ruiz-Serrano, MJ, Sánchez-Cueto, M, et al. Subspecies distribution and antimicrobial susceptibility testing of Mycobacterium abscessus clinical isolates in Madrid, Spain: a retrospective multicenter Study. Microbiol Spectr. (2023) 11:e0504122. doi: 10.1128/spectrum.05041-22
166. Wetzstein, N, Diricks, M, Kohl, TA, Wichelhaus, TA, Andres, S, Paulowski, L, et al. Molecular epidemiology of Mycobacterium abscessus isolates recovered from German cystic fibrosis patients. Microbiol Spectr. (2022) 10:e0171422. doi: 10.1128/spectrum.01714-22
167. Lipworth, S, Hough, N, Leach, L, Morgan, M, Jeffery, K, Andersson, M, et al. Whole-genome sequencing for predicting clarithromycin resistance in Mycobacterium abscessus. Antimicrob Agents Chemother. (2018) 63:e01204–18. doi: 10.1128/AAC.01204-18
168. Yoshida, M, Sano, S, Chien, JY, Fukano, H, Suzuki, M, Asakura, T, et al. A novel DNA chromatography method to discriminate Mycobacterium abscessus subspecies and macrolide susceptibility. EBioMedicine. (2021) 64:103187. doi: 10.1016/j.ebiom.2020.103187
169. Realegeno, S, Mirasol, R, Garner, OB, and Yang, S. Clinical whole genome sequencing for clarithromycin and amikacin resistance prediction and subspecies identification of Mycobacterium abscessus. J Mol Diagn. (2021) 23:1460–7. doi: 10.1016/j.jmoldx.2021.07.023
170. Chae, H, and Shin, SJ. Importance of differential identification of Mycobacterium tuberculosis strains for understanding differences in their prevalence, treatment efficacy, and vaccine development. J Microbiol. (2018) 56:300–11. doi: 10.1007/s12275-018-8041-3
171. Lane, DJ, Pace, B, Olsen, GJ, Stahl, DA, Sogin, ML, and Pace, NR. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci USA. (1985) 82:6955–9. doi: 10.1073/pnas.82.20.6955
172. Hugenholtz, P, Pitulle, C, Hershberger, KL, and Pace, NR. Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol. (1998) 180:366–76. doi: 10.1128/JB.180.2.366-376.1998
173. Janda, JM, and Abbott, SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol. (2007) 45:2761–4. doi: 10.1128/JCM.01228-07
174. Schulz, F, Eloe-Fadrosh, EA, Bowers, RM, Jarett, J, Nielsen, T, Ivanova, NN, et al. Towards a balanced view of the bacterial tree of life. Microbiome. (2017) 5:140. doi: 10.1186/s40168-017-0360-9
175. Konstantinidis, KT, and Tiedje, JM. Towards a genome-based taxonomy for prokaryotes. J Bacteriol. (2005) 187:6258–64. doi: 10.1128/JB.187.18.6258-6264.2005
176. Parks, DH, Chuvochina, M, Waite, DW, Rinke, C, Skarshewski, A, Chaumeil, PA, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. (2018) 36:996–1004. doi: 10.1038/nbt.4229
177. Wu, D, Jospin, G, and Eisen, JA. Systematic identification of gene families for use as "markers" for phylogenetic and phylogeny-driven ecological studies of bacteria and archaea and their major subgroups. PLoS One. (2013) 8:e77033. doi: 10.1371/journal.pone.0077033
178. Fedrizzi, T, Meehan, CJ, Grottola, A, Giacobazzi, E, Fregni Serpini, G, Tagliazucchi, S, et al. Genomic characterization of nontuberculous mycobacteria. Sci Rep. (2017) 7:45258. doi: 10.1038/srep45258
179. Tortoli, E, Fedrizzi, T, Meehan, CJ, Trovato, A, Grottola, A, Giacobazzi, E, et al. The new phylogeny of the genus Mycobacterium: the old and the news. Infect Genet Evol. (2017) 56:19–25. doi: 10.1016/j.meegid.2017
180. Gupta, RS, Lo, B, and Son, J. Phylogenomics and comparative genomic studies robustly support division of the genus Mycobacterium into an emended genus Mycobacterium and four novel genera. Front Microbiol. (2018) 9:67. doi: 10.3389/fmicb.2018.00067
181. Chew, KL, Octavia, S, Jureen, R, Ng, OT, Marimuthu, K, Lin, RTP, et al. Molecular epidemiology and phylogenomic analysis of Mycobacterium abscessus clinical isolates in an Asian population. Microb Genom. (2021) 7:000708. doi: 10.1099/mgen.0.000708
182. Das, S, Pettersson, BMF, Behra, PRK, Mallick, A, Cheramie, M, Ramesh, M, et al. Extensive genomic diversity among Mycobacterium marinum strains revealed by whole genome sequencing. Sci Rep. (2018) 8:12040. doi: 10.1038/s41598-018-30152-y
183. Uchiya, KI, Tomida, S, Nakagawa, T, Asahi, S, Nikai, T, and Ogawa, K. Comparative genome analyses of Mycobacterium avium reveal genomic features of its subspecies and strains that cause progression of pulmonary disease. Sci Rep. (2017) 7:39750. doi: 10.1038/srep39750
184. Stinear, TP, Seemann, T, Harrison, PF, Jenkin, GA, Davies, JK, Johnson, PD, et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. (2008) 18:729–41. doi: 10.1101/gr.075069.107
185. Qi, W, Käser, M, Röltgen, K, Yeboah-Manu, D, and Pluschke, G. Genomic diversity and evolution of Mycobacterium ulcerans revealed by next-generation sequencing. PLoS Pathog. (2009) 5:e1000580. doi: 10.1371/journal.ppat.1000580
186. Liang, Q, Shang, Y, Huo, F, Xue, Y, Li, Y, Dong, L, et al. Assessment of current diagnostic algorithm for detection of mixed infection with Mycobacterium tuberculosis and nontuberculous mycobacteria. J Infect Public Health. (2020) 13:1967–71. doi: 10.1016/j.jiph.2020.03.017
187. Zhang, C, Xiu, L, Xiao, Y, Xie, Z, Ren, L, and Peng, J. Simultaneous detection of key bacterial pathogens related to pneumonia and meningitis using multiplex PCR coupled with mass spectrometry. Front Cell Infect Microbiol. (2018) 8:107. doi: 10.3389/fcimb.2018.00107
188. Wu, X, Tan, G, Yang, J, Guo, Y, Huang, C, Sha, W, et al. Prediction of Mycobacterium tuberculosis drug resistance by nucleotide MALDI-TOF-MS. Int J Infect Dis. (2022) 121:47–54. doi: 10.1016/j.ijid.2022.04.061
189. Kriegsmann, M, Arens, N, Endris, V, Weichert, W, and Kriegsmann, J. Detection of KRAS, NRAS and BRAF by mass spectrometry - a sensitive, reliable, fast and cost-effective technique. Diagn Pathol. (2015) 10:132. doi: 10.1186/s13000-015-0364-3
190. Body, BA, Beard, MA, Slechta, ES, Hanson, KE, Barker, AP, Babady, NE, et al. Evaluation of the Vitek MS v3.0 matrix-assisted laser desorption ionization-time of flight mass spectrometry system for identification of Mycobacterium and Nocardia species. J Clin Microbiol. (2018) 56:e00237–18. doi: 10.1128/JCM.00237-18
191. Alcaide, F, Amlerová, J, Bou, G, Ceyssens, PJ, Coll, P, Corcoran, D, et al. How to: identify non-tuberculous Mycobacterium species using MALDI-TOF mass spectrometry. Clin Microbiol Infect. (2018) 24:599–603. doi: 10.1016/j.cmi.2017.11.012
192. Rodriguez-Temporal, D, Rodríguez-Sánchez, B, and Alcaide, F. Evaluation of MALDI Biotyper interpretation criteria for accurate identification of nontuberculous mycobacteria. J Clin Microbiol. (2020) 58:e01103–20. doi: 10.1128/JCM.01103-20
193. Panagea, T, Pincus, DH, Grogono, D, Jones, M, Bryant, J, Parkhill, J, et al. Mycobacterium abscessus complex identification with matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol. (2015) 53:2355–8. doi: 10.1128/JCM.00494-15
194. Luo, L, Liu, W, Li, B, Li, M, Huang, D, Jing, L, et al. Evaluation of matrix-assisted laser desorption ionization-time of flight mass spectrometry for identification of Mycobacterium abscessus subspecies according to whole-genome sequencing. J Clin Microbiol. (2016) 54:2982–9. doi: 10.1128/JCM.01151-16
195. Kehrmann, J, Wessel, S, Murali, R, Hampel, A, Bange, FC, Buer, J, et al. Principal component analysis of MALDI TOF MS mass spectra separates M. abscessus (sensu stricto) from M. massiliense isolates. BMC Microbiol. (2016) 16:24. doi: 10.1186/s12866-016-0636-4
196. Weis, C, Cuénod, A, Rieck, B, Dubuis, O, Graf, S, Lang, C, et al. Direct antimicrobial resistance prediction from clinical MALDI-TOF mass spectra using machine learning. Nat Med. (2022) 28:164–74. doi: 10.1038/s41591-021-01619-9
197. Ceyssens, PJ, Soetaert, K, Timke, M, Van den Bossche, A, Sparbier, K, De Cremer, K, et al. Matrix-assisted laser desorption ionization-time of flight mass spectrometry for combined species identification and drug sensitivity testing in mycobacteria. J Clin Microbiol. (2017) 55:624–34. doi: 10.1128/JCM.02089-16
198. Shinnick, TM, and Good, RC. Diagnostic mycobacteriology laboratory practices. Clin Infect Dis. (1995) 21:291–9. doi: 10.1093/clinids/21.2.291
199. Butler, WR, and Guthertz, LS. Mycolic acid analysis by high-performance liquid chromatography for identification of Mycobacterium species. Clin Microbiol Rev. (2001) 14:704–26. doi: 10.1128/CMR.14.4.704-726.2001
200. Forbes, BA, Hall, GS, Miller, MB, Novak, SM, Rowlinson, MC, Salfinger, M, et al. Practical guidance for clinical microbiology laboratories: mycobacteria. Clin Microbiol Rev. (2018) 31:e00038–17. doi: 10.1128/CMR.00038-17
201. Tortoli, E. Microbiological features and clinical relevance of new species of the genus Mycobacterium. Clin Microbiol Rev. (2014) 27:727–52. doi: 10.1128/CMR.00035-14
202. Ashour, ME, Byrum, AK, Meroni, A, Xia, J, Singh, S, Galletto, R, et al. Rapid profiling of DNA replication dynamics using mass spectrometry-based analysis of nascent DNA. J Cell Biol. (2023) 222:e202207121. doi: 10.1083/jcb.202207121
Keywords: non-tuberculous mycobacterium, molecular diagnostics, whole genome sequencing, next-generation sequencing, drug resistance gene
Citation: Zhang H, Tang M, Li D, Xu M, Ao Y and Lin L (2024) Applications and advances in molecular diagnostics: revolutionizing non-tuberculous mycobacteria species and subspecies identification. Front. Public Health. 12:1410672. doi: 10.3389/fpubh.2024.1410672
Edited by:
Giovanni Ghielmetti, University of Zurich, SwitzerlandReviewed by:
Seyed Ehtesham Hasnain, Sharda University, IndiaCopyright © 2024 Zhang, Tang, Li, Xu, Ao and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liangkang Lin, bGlhbmdrYW5nbGluMTk5N0AxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.