
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Public Health , 16 May 2023
Sec. Infectious Diseases: Epidemiology and Prevention
Volume 11 - 2023 | https://doi.org/10.3389/fpubh.2023.1151805
This article is part of the Research Topic High-Throughput Genome Sequencing Technologies, Methods, and their Applications View all 9 articles
 Sareen Fatima1
Sareen Fatima1 Zaara Ishaq2†
Zaara Ishaq2† Muhammad Irfan3†
Muhammad Irfan3† Abdullah F. AlAsmari4Jahangir Khan Achakzai5Tahreem Zaheer6
Abdullah F. AlAsmari4Jahangir Khan Achakzai5Tahreem Zaheer6 Amjad Ali2
Amjad Ali2 Ali Akbar1*
Ali Akbar1*Introduction: Salmonella enterica serovar Typhi (S. Typhi) is a major cause of morbidity and mortality in developing countries, contributing significantly to the global disease burden.
Methods: In this study, S. Typhi strains were isolated from 100 patients exhibiting symptoms of typhoid fever at a tertiary care hospital in Pakistan. Antimicrobial testing of all isolates was performed to determine the sensitivity and resistance pattern. Three MDR strains, namely QS194, QS430, and QS468, were subjected to whole genome sequencing for genomic characterization.
Results and Discussion: MLST analysis showed that QS194, belonged to ST19, which is commonly associated with Salmonella enterica serovar typhimurium. In contrast, QS430 and QS468, belonged to ST1, a sequence type frequently associated with S. Typhi. PlasmidFinder identified the presence of IncFIB(S) and IncFII(S) plasmids in QS194, while IncQ1 was found in QS468. No plasmid was detected in QS430. CARD-based analysis showed that the strains were largely resistant to a variety of antibiotics and disinfecting agents/antiseptics, including fluoroquinolones, cephalosporins, monobactams, cephamycins, penams, phenicols, tetracyclines, rifamycins, aminoglycosides, etc. The S. Typhi strains possessed various virulence factors, such as Vi antigen, Agf/Csg, Bcf, Fim, Pef, etc. The sequencing data indicated that the strains had antibiotic resistance determinants and shared common virulence factors. Pangenome analysis of the selected S. Typhi strains identified 13,237 genes, with 3,611 being core genes, 2,093 shell genes, and 7,533 cloud genes. Genome-based typing and horizontal gene transfer analysis revealed that the strains had different evolutionary origins and may have adapted to distinct environments or host organisms. These findings provide important insights into the genetic characteristics of S. Typhi strains and their potential association with various ecological niches and host organisms.
Typhoid fever has been found as a major cause of mortality and illness in developing countries (1) This indicated its potential contribution in the social and economic cost of the diseases globally (2). Typhoid fever is caused by the Gram-negative Salmonella enterica Serovar Typhi. Around 14 million cases of typhoid and paratyphoid fever were observed in 2017, with over 130,000 fatalities. Among others, only South Asia accounted for approximately 70% among these deaths, indicating the importance of the pathogen, contributing for large percentage of the global disease burden (3, 4). Annually, around 0.155 M fatalities were observed to be caused from non-typhoidal Salmonella serovars as well, causing diarrhea and other symptoms (5, 6).
The Salmonella Reference Service (SRS) of Public Health England (PHE) received an estimated 8,000 isolates each year from local and regional hospital labs (2).
Typically, Salmonella Typhi was shown to be host-specific, monophyletic, seldom undergone recombination events and displayed differences due to genomic degradation Multiple virulence encoding genes, either chromosomally or on the virulence-associated plasmid, have been linked to its pathogenicity. Bacterial adhesion to the intestinal epithelium and cellular invasion is mediated, in part, by fimbriae virulence genes such those producing long polar fimbriae (lpf). The invA gene was also shown to have a role in host identification and invasion of intestinal mucosal epithelial cells (7). Similarly, msgA, pagC, and tolC, were shown to contribute to intracellular or macrophage survival. Evidence suggested that the spaN gene contributes to Salmonella’s invasiveness by allowing the pathogen to invade non-phagocytic cells and kill off macrophages. The cdtB gene, which encodes the toxin, was also discovered to have a role in host identification and invasion (8). According to reports, the spvC gene, which is found mostly on the virulence plasmid, helps Salmonella proliferate and survive within the host cell. Additionally, systemic Salmonella infections were linked to this phenomenon (9).
Acquisition of one or more specific pathways is commonly linked to the development of multidrug resistance (MDR) in Salmonella Typhi. The already high prevalence rates of typhoid fever have been exacerbated by the emergence of Salmonella Typhi isolates resistant to drugs prescribed for treatment. Antibiotic-resistant forms of Salmonella Typhi are a serious threat to public health, according to the Centers for Disease Control and Prevention (10). Typhoid was traditionally treated with the first-line antibiotics chloramphenicol, ampicillin, and trimethoprim-sulfamethoxazole. The emergence of multidrug-resistant Salmonella Typhi, which cannot be treated with standard antibiotics, dates back to the 1980s (11). MDR Salmonella Typhi infections might thereafter be treated with fluoroquinolones and third-generation cephalosporin. It is crucial to identify the potential negative effects of antibiotics on virulence mechanisms like motility because patients infected with antibiotic-resistant Salmonella isolates were more likely to experience morbidity than those infected with susceptible isolates (12).
Preceding the advent of polymerase chain reaction (PCR), Salmonella isolates were classified into serovars based on the reaction of rabbit antisera with lipopolysaccharide (O antigen encoded by rfb genes) and flagellar antigens (phases 1 and 2 of H antigen encoded by fliC and fljB). The organism was separated into almost 2,600 serovars using this strategy, which relied on their phenotypic variance. While initially promising, this approach was ultimately discredited due to its high cost, high expert labor requirements, and inability to reveal genetic differences among serovars (2). In contrast, numerous types of typing have been reported to be used routinely as a key component of detection and investigation, such as Multilocus variable number of tandem repeat analysis (MLVA) (13). Pulsed-field gel electrophoresis, ribotyping, Repetitive extra genic palindromic sequence-based PCR (rep-PCR), and a combined PCR and sequencing-based approach that directly targets O- and H-antigen. However, the development of Whole Genome Sequencing (WGS) technology has improved the notion by providing a low-cost, high-throughput alternative to conventional typing for public health monitoring and epidemic identification (14).The method also allowed for the resolution of bacterial strains down to the single nucleotide level, which aided in tracing the origin of an infection (10) and in classifying isolates into more refined taxonomic clones (such as those specified by serotyping) (2).
This is a cross sectional study where three Salmonella isolates including QS194, QS430, and QS468 were collected between August 2019 and August 2020 from Sandeman Provisional Hospital, Quetta Balochistan, Pakistan. These isolates were selected for whole genome sequencing on the basis of drug resistance.
The collected blood sample was cultured on Salmonella–Shigella Agar and MacConkey agars to cultivate bacteria by incubating them at 37°C for 24 h. Later, non-lactose fermenting colonies were confirmed through biochemical testing and antibiotic sensitivity.
The disk diffusion technique was used to determine antimicrobial sensitivity and resistance after isolating colonies and growing them on Mueller-Hinton agar (Oxoid) at 37°C for 24 h. Following antibiotic disks (Oxoid) were used with the given concentrations: ampicillin (10 μg), chloramphenicol (30 μg), streptomycin (10 μg), sulfonamide (300 μg), trimethoprim (5 μg), ciprofloxacin (5 μg), tetracycline (30 μg), gentamicin (10 μg), nalidixic acid (30 μg), Cefotaxime (5 μg), Mecillinam (10 μg) and Imipenem (10 μg). The criteria published by the Clinical and Laboratory Standards Institute (CLSI) was used for the interpretation of the results.
Oligonucleotide primer sequence used in this study for the amplification of invA and fliC gene of Salmonella Typhi described earlier by Rahn et al. (15) and Song et al. (16) are shown in (Supplementary Table S1).
DNA library preparation using high-quality bacterial DNA (50 ng) was accomplished by following the manufacturer’s instructions for the Nextera XT kit (Illumina, San Diego, CA, US). After fragmenting genomic DNA, it was ligated to adapters with individual indexes and then amplified using polymerase chain reaction (PCR). The PCR products were purified by using Agencourt Ampure beads. The concentration of the purified DNA was evaluated by DNA high sensitivity qubit kit using Fluorometer 2.0 Qubit. Three DNA libraries were combined into one with a representational concentration of one. Size distribution of the pooled library was estimated by 3% agarose gel electrophoresis. The library concentration was adjusted from ng to 4 nM and was denatured using 0.2 N NaOH. The denatured library was diluted to final concentration of 16 pM by using hybridization buffer (HT1).
The library was subjected onto the MiSeq Illumina platform for paired-end high through put sequencing using 2 × 150 bp flowcell chemistry. With PE sequencing, both forward and reverse directions are sequenced on MiSeq Illumina. The total number of short sequenced reads 4,297,792 of QS194, 4,348,108 of QS430, and 4,179,946 of QS468 were generated.
FastQC package was used to assess the quality of the sequenced data and Trimmomatic v0.39 tool was employed to improve the quality of sequenced data by removing the adapter sequences. Adapter sequences, low quality bases, or such reads containing bases having size less than 36 were eliminated. After filtration, sequenced reads: 4270468 for QS194, 4.347290 for QS30 and 4,175,432 for QS468 were processed for de novo assembly using Unicycler (17) using SPAdes version 3.13.0 (18). The quality of the draft assembly was improved by pilon v1.23 tool (19). Filtered short sequenced reads of all the strains were aligned to the reference genome using BWA-MEM package to check the percentage of genome covered against the reference genome. The draft genome of all three Salmonella Typhi strains; QS194, QS430, and QS468 were assembled into contigs. Quast package was used to assess the quality of assembled draft genome of each strain. The protein-coding genes and RNA-coding genes of the draft were located using FASTA sequences obtained from the fully assembled genome. The genomes of all three strains were assembled, and then annotated using RAST and the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) v6.1.
In this study, we utilized the CGE services (1accessed on 28 March 2023) to conduct in silico epidemiological analysis of our isolates. We employed SeqSero 1.2 to predict the serotypes of Salmonella strains (20). While MLST 2.0 and PubMLST were utilized for multi locus sequence typing of the assembled genomes (21, 22). We used SPIFinder 2.0 to identify Salmonella Pathogenicity Islands, at default parameters (95% minimum identity threshold and 60% minimum coverage) (23). PlasmidFinder 2.1 was used to identify plasmids in the Salmonella isolates at default parameters (95% minimum identity threshold and 60% minimum coverage), the database selected was Enterobacteriales (24). Furthermore, we utilized ResFinder 4.1 to predict acquired antimicrobial resistance genes and chromosomal mutations with default parameters and the results were further confirmed by BLAST search at default parameters against Resistance Gene Identifier of Comprehensive Antimicrobial Resistance Database (CARD) (25, 26). CRISPRDetect 2.4 was employed at default parameters to predict and analyze CRISPR arrays in our isolates (27). We also identified and annotated virulence genes in our isolates using the Virulence Factor Database available at http://www.mgc.ac.cn/VFs/ accessed on 10 October 2022 (28).
One hundred fifty-six genome sequences of Salmonella Typhi and Typhimurium (95 complete genome sequences from different geographical locations and 61 WGS sequences of Pakistani strains) were downloaded from PATRIC (2accessed on 29 March 2023). Metadata regarding these genomes including strain name, BioSample, BioProject, accession numbers, GC% sequence type, isolate type and geographical location etc. is in Supplementary Table S2. For pangenome analysis, total 159 strains were used (95 complete genomes +61 Pakistani strains + Our 03 strains) which were then annotated using Prokka at default parameters (29). Then these annotated genomes were submitted to PanRV for pangenome estimation employing Roary, the core-genome SNPs based phylogenetic analysis was done and the tree was visualized and edited by iTOLv5 webserver (available at3 and accessed on 30 March 2023) (30–32).
The culture media showed non-lactose fermenting colorless colonies, suggestive of the presence of Salmonella. Following observation was done for the confirmation of Salmonella species through biochemical testing (Supplementary Table S3).
The antimicrobial susceptibility test for Salmonella Typhi has been performed on Mueller Hinton agar plate by the disk diffusion method. Result interpretation was conducted by following the Clinical and Laboratory Standards Institute (CLSI) guidelines (Supplementary Table S4).
The presence of Salmonella Typhi was confirmed by Agarose Gel Electrophoresis. 2% Agarose Gel was used, and for confirmation invA and fliC genes were used which were 284 bp and 495 bp in size, respectively. Positive and negative controls were use (M1/M2 DNA marker) which were 100 bp apart from each other (Supplementary Figure S1).
Paired end short sequenced data 1,055 MB, 1065 MB, and 1,028 MB of QS194, QS430, and QS468, respectively, was generated from the MiSeq Illumina. After removing the poor quality data using trimmomatic the obtained good quality sequenced reads, i.e., 4,270,468, 4,347,290, and 4,175,432 for QS194, QS30 and QS468, respectively, were de novo assembled by Unicycler followed by fine polishing using pilon package. The obtained high quality short sequenced reads data was mapped against the reference genome and found that 98.34% genome size of QS194 was covered against the reference genome followed by 89.4% QS430 and 87.48% for QS468. The entire genome was not assembled as a result of reference mapping. The sequenced data of all three strains QS194, QS430 and QS468 was assembled into draft genome of 4.59 MB, 4.48 MB and 4.38 MB, respectively, [Supplementary Figures S3a-c]. Annotation of these strains were performed by Prokaryotic Genome Annotation Pipeline (PGAP). The GC content of the sequenced strain was found on average of 52.39%. Table 1 summarizes the genome characteristics of three strains namely QS194, QS430 and QS468.
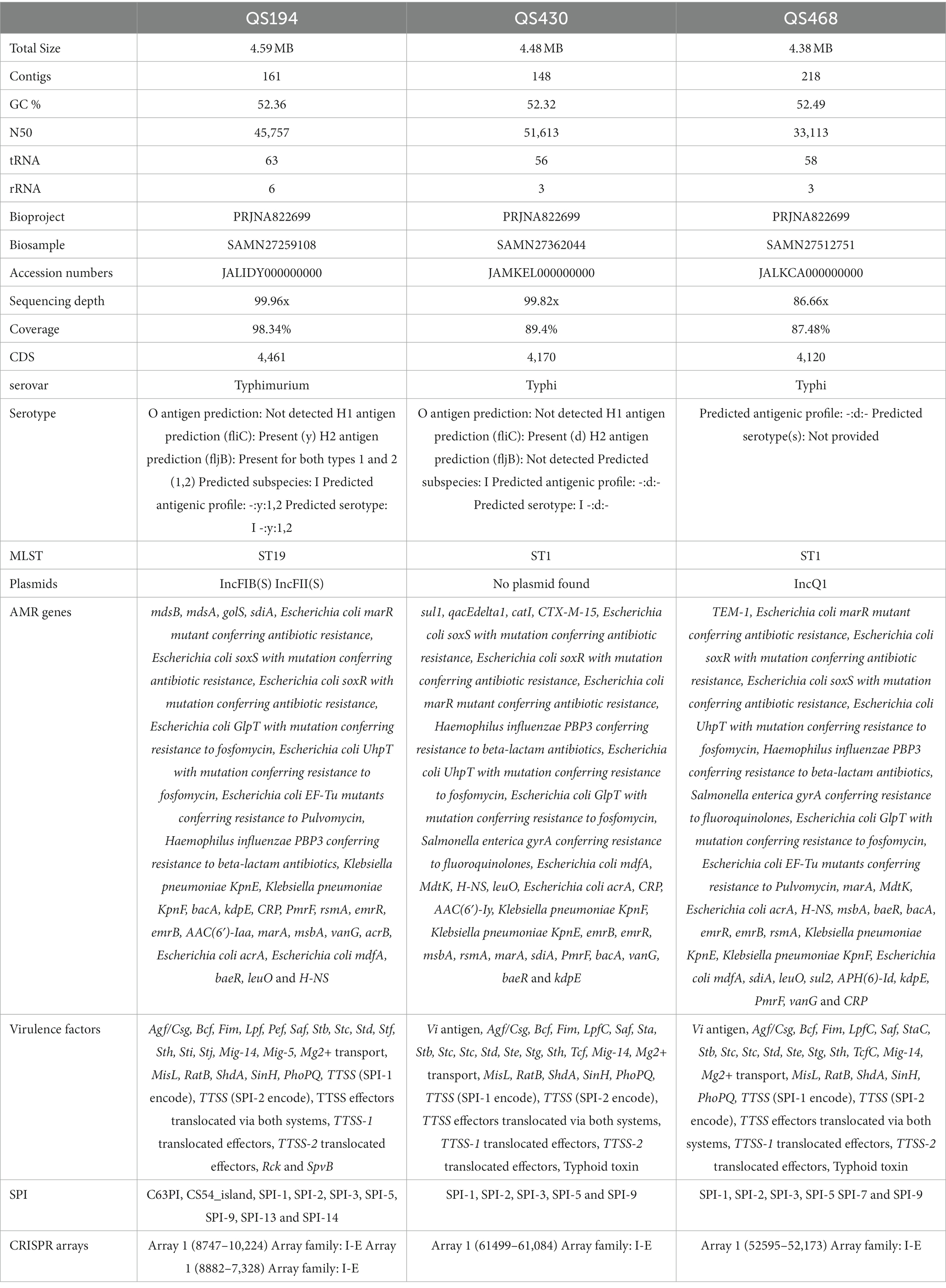
Table 1. Summary of genomic characteristics of three strains.
The strains have been found to share several antibiotic resistance determinants including sdiA, golS, mdsA, mdsB, H-NS, baeR, vanG, acrB, acrA from E. coli kdpE, mdfA from E. coli, msbA, KpnE and KpnF from K. pneumonia, emrR, emrB, rsmA and CRP. These determinants provide resistance by antibiotic efflux, target alteration, antibiotic inactivation and reduced membrane permeability. Furthermore, mutations in genes such as PmrF, bacA, PBP3 from H. influenzae, GlpT mutant from E. coli, EF-Tu mutants from E. coli, and UhpT from E. coli also confer resistance by target alteration and reduced membrane permeability. Mutations in AcrAB-TolC and soxR from E. coli result in resistance by target alteration and efflux, while mutations in soxS from E. coli provide resistance by target alteration, efflux, and reduced permeability. Additionally, QS430 has been found to possess these antibiotic determinants CTX-M-15, catI, qacEdelta1, sul1 and Salmonella enterica gyrA conferring antibiotic resistance by antibiotic efflux, target alteration and antibiotic inactivation. On the other hand, strain QS468 has found to possess TEM-1 conferring resistance to beta lactamases by antibiotic inactivation. All these results are summarized in Table 1 above and have been visualized in Supplementary Figures S2–S4, respectively. Although the strains exhibited some common virulence factors, there were notable differences among them (Table 1; Supplementary Figures S2–S4). Specifically, the typhoid toxin genes cdtB and pltA were present in QS430 and QS468 but absent in QS194, indicating that QS194 is distinct from the other strains in this regard.
In our case, the MLST analysis revealed that QS194 belongs to ST19, a sequence type that is commonly associated with Salmonella enterica serovar Typhimurium. This result suggests that QS194 is likely to be closely related to other strains of S. Typhimurium with the same sequence type. This could indicate that QS194 may have originated from the same geographical region or has a similar ecological niche as other S. Typhimurium strains that share this sequence type (Table 1).
On the other hand, QS430 and QS468 were found to have ST1, which is a different sequence type from QS194. This suggests that these strains are likely to have a different evolutionary origin and may have adapted to different environments or host organisms. It is also worth noting that ST1 is a common sequence type associated with Salmonella enterica serovar Typhi, which causes typhoid fever in humans. The presence of typhoid toxin genes cdtB and pltA in QS430 and QS468 further supports their potential association with S. Typhi (Table 1).
PlasmidFinder identified two plasmids in QS194 strain with 100% similarity with Salmonella enterica subsp. enterica serovar Typhimurium strain D23580 plasmid pSLT-BT and Salmonella enterica subsp. enterica serovar Paratyphi C strain RKS4594 plasmid pSPCV (Table 1). It identified one plasmid in QS468 strain with 100% identity with Escherichia coli plasmid RSF1010. No plasmid was detected in QS430 strain (Table 1).
SPIFinder identified that our isolates share 5 Salmonella pathogenicity islands, SPI-1, SPI-2, SPI-3, SPI-5 and SPI-9. SPI-1 and SPI-2 are the most well-studied SPIs and are critical for Salmonella’s ability to invade and replicate within host cells. SPI-3 contains genes that enable Salmonella to evade the host immune response, while SPI-5 is involved in the transport and utilization of nutrients within host cells. Additionally, SPI-13, SPI-14, C63PI and CS54_islands were detected in QS194. SPI-9, SPI-13, and SPI-14 are involved in the transport and metabolism of specific nutrients that are important for Salmonella survival within the host. C63PI (Chromosome 63 Pathogenicity Island) contains several genes that are associated with virulence, including genes encoding type III secretion system (T3SS) effectors and regulatory proteins. CS54_island (Chromosome segment 54 island) is a pathogenicity island found in some strains of Salmonella and contains several genes that are associated with virulence, antibiotic resistance, and fitness, including genes encoding efflux pumps, iron acquisition systems, and proteins involved in biofilm formation. Additionally, SPI-7 was detected in QS468 which is a relatively large genomic region that contains several genes that have been implicated in the virulence of Salmonella.
CRISPRDetect identified two CRISPR arrays in QS164. The first array, located at positions 8,747–10,224 in the genome, belongs to the I-E family of CRISPR arrays. This suggests that QS164 possesses the genetic machinery necessary for a type I CRISPR-Cas system, which can be used by the bacterium to defend against foreign DNA elements such as bacteriophages and plasmids.
The second array, located at positions 8,882–7,328 in the genome, also belongs to the I-E family of CRISPR arrays. The presence of two CRISPR arrays in the QS164 genome suggests that this strain has a robust defense system against foreign DNA elements, which may contribute to its virulence and survival in different environments. While in QS430 and QS468 one array per each genome had been detected (Table 1).
The analyzed Salmonella strains exhibit an open pangenome which consists of total 13,237 genes, of which 3,611 (27.28%) are part of core genes, 2093 (15.8%) are shell and 7,533 (56.9%) are cloud genes. The statistics confirm the diverse nature of these strains with a large proportion of cloud genes, indicating a high degree of genomic plasticity.
The core-genome SNP based phylogenetics analysis revealed that strain QS430 and QS468 belong to the prevalent ST1 sequence type (both globally and locally) by sharing same clade with other local strains 1,382, 10,109, 3,679, MDUST418, MDUST175, 3,484, MDUST374, MDUST269, MDUST372, MDUST266, Rwp1-Pk, CFSAN059648, CFSAN059647, MDUST257, MDUST201, MDUST156, 1,093, 19, 22, 7, 18 and 2,176 with same ST. While QS194 clustered with other ST19 strains reported from worldwide and was found to be most closely related to NCCP16345 strain from South Africa and PNCS014851 strain from Canada (Figure 1). ST19 was found to be second most prevalent sequence type globally after ST34. These results highlight the importance of studying the diversity and evolution of Salmonella strains to better understand their global epidemiology and pathogenic potential.
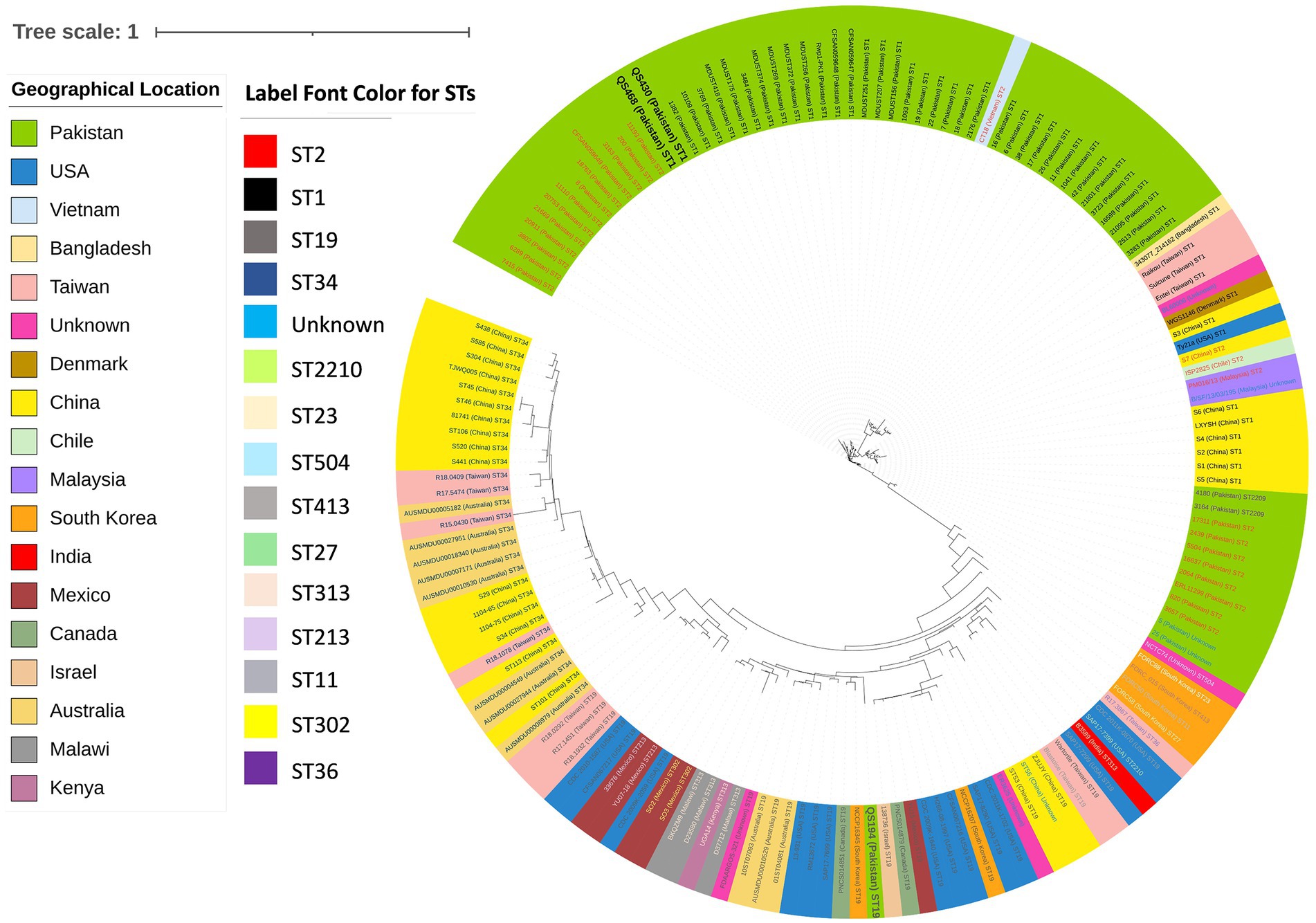
Figure 1. Core-genome SNPs based phylogenetic analysis of 159 (test strains n = 3, local strains n = 61, and global strains n = 95) publicly available Salmonella Typhi and Typhimurium strains showing diversity among local and global strains. Color key indicating geographical location and label font color for STs is provided along with.
Figure 1 indicates circularized core-genome SNP tree of 159 Salmonella strains showing same clade of QS430 and QS468 whereas QS194 belongs to the different clade. The most dominant sequence type was found to be ST1, including QS430 and QS468. QS164 belonged to ST19. Color key indicating STs and geographical location is provided alongside the circular tree.
Typhoid fever has been recognized as a great health concern throughout the world, affecting the major economy of many developing and developed countries. Greater disease burden from typhoid and paratyphoid fever has been recorded particularly in low-income areas of Asia, Southeast Asia, and Sub-Saharan Africa in 2015 (33). This accounted for more than 15 million cases globally, only in 2015. However, the recent research stated that this number has been raised to more than 20 million cases annually around the globe. As Salmonella species have been recognized as the potential contributor in the emergence of the disease, there drug-resistant strains could increase its prevalence in high-risk countries (11, 34).
Antibiotic resistance is a significant public health threat, and Salmonella is a leading cause of antibiotic-resistant bacterial infections in humans. Several previous studies have reported the presence of antibiotic resistance determinants in Salmonella strains isolated from human hosts. A study by Randall et al. identified the presence of genes conferring resistance to multiple antibiotics, including tetracycline, sulfonamides, and beta-lactams, in Salmonella strains isolated from human patients in humans and animals (35). Similarly, a study by Xia et al. found that Salmonella strains isolated from human patients in China exhibited resistance to multiple antibiotics, including tetracycline, ampicillin, and ciprofloxacin, through the acquisition of antibiotic resistance genes (36). These studies highlight the urgent need for effective antibiotic stewardship programs and the development of new treatment strategies to combat antibiotic-resistant Salmonella infections in human hosts.
The findings of our study are consistent with previous studies that have characterized the genomic diversity of Salmonella isolates. A study by Zhou et al. analyzed the genomes of 1,500 Salmonella isolates from around the world and found that there is a high degree of genomic diversity among different serotypes and sequence types. In particular, they observed that certain sequence types, such as ST19 and ST34, are associated with multiple serotypes and are prevalent globally (37). This is consistent with our finding that QS194 belongs to ST19 and is closely related to other S. Typhimurium strains with the same sequence type.
Our study identified several antibiotic resistance determinants in the three Salmonella strains, including genes conferring resistance to beta-lactams, aminoglycosides, quinolones, and tetracyclines. Interestingly, QS430 and QS468 were found to possess the typhoid toxin genes cdtB and pltA, which are commonly associated with S. Typhi, the causative agent of typhoid fever. The presence of these genes in Salmonella strains has been previously reported and is a cause for concern as it suggests a potential transfer of these genes between different Salmonella serovars (38, 39).
Our study also identified mutations in several genes that confer antibiotic resistance by target alteration and reduced membrane permeability. This is in line with previous studies that have reported the role of mutations in conferring antibiotic resistance in Salmonella strains (40, 41).
Our observation that QS430 and QS468 have ST1, a sequence type commonly associated with S. Typhi, is also consistent with previous studies that have identified ST1 as a prevalent sequence type among S. Typhi isolates (42). Moreover, the presence of typhoid toxin genes in QS430 and QS468 further supports the potential association of these strains with S. Typhi.
Our analysis of plasmids and pathogenicity islands in the Salmonella isolates is also consistent with previous studies that have characterized the genomic features of Salmonella strains. A study by Ahmer et al. analyzed the plasmid content of 92 Salmonella isolates and identified a wide variety of plasmids with different sizes and functions (43). Similarly, a study by Langridge et al. (44) characterized the diversity of pathogenicity islands in Salmonella and identified a total of 26 different pathogenicity islands in the genomes of 90 Salmonella isolates (44).
The findings of the comparative pangenomics and phylogenetic inference analysis of the Salmonella strains are consistent with previous studies that have reported the high degree of genomic plasticity and diverse nature of these bacteria. A study by Leekitcharoenphon et al. found that the pangenome of Salmonella enterica consists of approximately 18,000 genes, with a large proportion of accessory genes that contribute to the variability and adaptability of the species (45). Similarly, a study by Ashton et al. identified a high degree of genomic diversity among Salmonella strains, with a large proportion of variable genes and a core genome that varies in size depending on the clade or serotype being analyzed (46).
The identification of ST1 and ST19 as prevalent sequence types in the Salmonella strains analyzed in the present study is consistent with global trends reported in previous studies. A study by Díaz-Torres et al. reported that ST1 and ST19 are among the most prevalent sequence types of Salmonella enterica worldwide (47). Similarly, a study by Wong et al. found that ST1, ST19, and ST34 are the most common sequence types of Salmonella enterica in Southeast Asia (48).
The identification of the relatedness between QS194 and ST19 strains reported from worldwide, such as NCCP16345 strain from South Africa and PNCS014851 strain from Canada, highlights the global distribution of Salmonella strains and their potential for causing outbreaks on a global scale. This finding is consistent with previous studies that have reported the global distribution of Salmonella strains and the importance of monitoring their diversity and evolution to better understand their epidemiology and pathogenic potential (45, 47, 48).
The correct surveillance and monitoring of control programs rely on the availability of rapid, simple, cost-effective, and improved diagnostics for pathogens identification. Finding biomarkers in typhoid carrier strains that need in-depth experimental examination is a difficult task, but whole genome sequencing has been a game-changer in this regard. There is currently no animal model that accurately represents human illness, and the enormous expenses of conducting experiments make this method difficult. Research into the typhoid carrier-state mechanism has allegedly started, but this area remains mostly unexplored. Consequently, there is still a need to incorporate significant efforts to solve this puzzling topic.
The study concluded that Salmonella Typhi has very diverse nature of strains among which certain genes were primarily involved in disease and antibiotic resistance, whereas others were involved in metabolic processes. Due to the ability to generate invasive infections and spread antimicrobial resistance (AMR) genes, Salmonella Typhi remains a formidable therapeutic challenge. There are no newer antityphoid medications in development, thus it is critical to examine full genomes to learn about their traits and look for new diagnostic targets.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, JALKCA000000000; https://www.ncbi.nlm.nih.gov/, JAMKEL000000000; https://www.ncbi.nlm.nih.gov/, JALIDY000000000.
The study was approved by the Departmental Committee and Advanced Studies and Research Board with reference No. GSO/74, 01/2020. The patients/participants provided written informed consent to participate in this study.
AAk and SF: conceptualization, methodology, experimentation, data analysis, manuscript writing, and review and editing. ZI, MI, and AA helped in experimental data analysis and revision. JKA, TZ and AFA helped in resources provision and review of draft. AAk: final manuscript proof reading. All authors read and approved the final manuscript.
The authors are thankful to the researchers supporting project number (RSP2023R335), King Saud University, Riyadh, Saudi Arabia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2023.1151805/full#supplementary-material
1. Buckle, GC, Walker, CLF, and Black, RE. Typhoid fever and paratyphoid fever: systematic review to estimate global morbidity and mortality for 2010. J Glob Health. (2012) 2. doi: 10.7189/jogh.01.010401
2. Ashton, PM, Nair, S, Peters, TM, Bale, JA, Powell, DG, Painset, A, et al. Identification of Salmonella for public health surveillance using whole genome sequencing. PeerJ. (2016) 4:e1752. doi: 10.7717/peerj.1752
3. Akbar, A, Sadiq, MB, Ali, I, Muhammad, N, Rehman, Z, Khan, MN, et al. Synthesis and antimicrobial activity of zinc oxide nanoparticles against foodborne pathogens Salmonella Typhimurium and Staphylococcus aureus. Biocatal Agric Biotechnol. (2019) 17:36–42. doi: 10.1016/j.bcab.2018.11.005
4. Knudsen, A, and Collaborators, GN. Disease burden in the Nordic region. Results from the global burden of disease study 2017 (GBD 2017). Eur J Pub Health. (2019) 29:161. doi: 10.1093/eurpub/ckz185.161
5. Akbar, A, and Anal, AK. Isolation of Salmonella from ready-to-eat poultry meat and evaluation of its survival at low temperature, microwaving and simulated gastric fluids. J Food Sci Technol. (2015) 52:3051–7. doi: 10.1007/s13197-014-1354-2
6. Majowicz, SE, Musto, J, Scallan, E, Angulo, FJ, Kirk, M, O'Brien, SJ, et al. The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis. (2010) 50:882–9. doi: 10.1086/650733
7. Bahramianfard, H, Derakhshandeh, A, Naziri, Z, and Khaltabadi Farahani, R. Prevalence, virulence factor and antimicrobial resistance analysis of Salmonella enteritidis from poultry and egg samples in Iran. BMC Vet Res. (2021) 17:1–8. doi: 10.1186/s12917-021-02900-2
8. Webber, D, and Parker, S. Estimating unaccounted fishing mortality in the Ross Sea region and Amundsen Sea (CCAMLR subareas 88.1 and 88.2) bottom longline fisheries targeting Antarctic toothfish. CCAMLR Sci. (2012) 19:17–30.
9. Gritli, A, Daboussi, T, Moussa, MB, and Abassi, M. Prevalence and characterizaton of Salmonella in chicken consumed in military cantines. J. New Sci. (2015) 12:908–14.
10. Dallman, TJ, Byrne, L, Ashton, PM, Cowley, LA, Perry, NT, Adak, G, et al. Whole-genome sequencing for national surveillance of Shiga toxin–producing Escherichia coli O157. Clin Infect Dis. (2015) 61:305–12. doi: 10.1093/cid/civ318
11. Crump, JA, and Mintz, ED. Global trends in typhoid and paratyphoid fever. Clin Infect Dis. (2010) 50:241–6. doi: 10.1086/649541
12. Irazoki, O, Aranda, J, Zimmermann, T, Campoy, S, and Barbé, J. Molecular interaction and cellular location of RecA and CheW proteins in Salmonella enterica during SOS response and their implication in swarming. Front Microbiol. (2016) 7:1560. doi: 10.3389/fmicb.2016.01560
13. Campioni, F, Davis, M, Medeiros, MIC, Falcão, JP, and Shah, DH. MLVA typing reveals higher genetic homogeneity among S. enteritidis strains isolated from food, humans and chickens in Brazil in comparison to the north American strains. Int J Food Microbiol. (2013) 162:174–81. doi: 10.1016/j.ijfoodmicro.2013.01.008
14. Köser, CU, Ellington, MJ, Cartwright, EJ, Gillespie, SH, Brown, NM, Farrington, M, et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. (2012) 8:e1002824. doi: 10.1371/journal.ppat.1002824
15. Rahn, K, De Grandis, S, Clarke, R, McEwen, S, Galan, J, Ginocchio, C, et al. Amplification of an invA gene sequence of Salmonella Typhimurium by polymerase chain reaction as a specific method of detection of Salmonella. Mol Cell Probes. (1992) 6:271–9. doi: 10.1016/0890-8508(92)90002-F
16. Song, J-H, Cho, H, Park, MY, Na, DS, Moon, HB, and Pai, CH. Detection of Salmonella Typhi in the blood of patients with typhoid fever by polymerase chain reaction. J Clin Microbiol. (1993) 31:1439–43. doi: 10.1128/jcm.31.6.1439-1443.1993
17. Wick, RR, Judd, LM, Gorrie, CL, and Holt, KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. (2017) 13:e1005595. doi: 10.1371/journal.pcbi.1005595
18. Bankevich, A, Nurk, S, Antipov, D, Gurevich, AA, Dvorkin, M, Kulikov, AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
19. Walker, BJ, Abeel, T, Shea, T, Priest, M, Abouelliel, A, Sakthikumar, S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. (2014) 9:e112963. doi: 10.1371/journal.pone.0112963
20. Zhang, S, Yin, Y, Jones, MB, Zhang, Z, Deatherage Kaiser, BL, Dinsmore, BA, et al. Salmonella serotype determination utilizing high-throughput genome sequencing data. J Clin Microbiol. (2015) 53:1685–92. doi: 10.1128/jcm.00323-15
21. Jolley, KA, Bray, JE, and Maiden, MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. (2018) 3:124. doi: 10.12688/wellcomeopenres.14826.1
22. Thomsen, MC, Ahrenfeldt, J, Cisneros, JL, Jurtz, V, Larsen, MV, Hasman, H, et al. A bacterial analysis platform: an integrated system for analysing bacterial whole genome sequencing data for clinical diagnostics and surveillance. PLoS One. (2016) 11:e0157718. doi: 10.1371/journal.pone.0157718
23. Roer, L, Hendriksen Rene, S, Leekitcharoenphon, P, Lukjancenko, O, Kaas Rolf, S, Hasman, H, et al. Is the evolution of Salmonella enterica subsp. enterica linked to restriction-modification systems? mSystems. (2016) 1:e00009–16. doi: 10.1128/mSystems.00009-16
24. Carattoli, A, Zankari, E, García-Fernández, A, Voldby Larsen, M, Lund, O, Villa, L, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. (2014) 58:3895–903. doi: 10.1128/aac.02412-14
25. Florensa, AF, Kaas, RS, Clausen, P, Aytan-Aktug, D, and Aarestrup, FM. ResFinder–an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genom. (2022) 8:748. doi: 10.1099/mgen.0.000748
26. McArthur, AG, Waglechner, N, Nizam, F, Yan, A, Azad, MA, Baylay, AJ, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. (2013) 57:3348–57. doi: 10.1128/AAC.00419-13
27. Biswas, A, Staals, RHJ, Morales, SE, Fineran, PC, and Brown, CM. CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genomics. (2016) 17:356. doi: 10.1186/s12864-016-2627-0
28. Liu, B, Zheng, D, Jin, Q, Chen, L, and Yang, J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. (2019) 47:D687–92. doi: 10.1093/nar/gky1080
29. Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
30. Letunic, I, and Bork, P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. (2021) 49:W293–6. doi: 10.1093/nar/gkab301
31. Naz, K, Naz, A, Ashraf, ST, Rizwan, M, Ahmad, J, Baumbach, J, et al. PanRV: Pangenome-reverse vaccinology approach for identifications of potential vaccine candidates in microbial pangenome. BMC Bioinformatics. (2019) 20:123. doi: 10.1186/s12859-019-2713-9
32. Page, AJ, Cummins, CA, Hunt, M, Wong, VK, Reuter, S, Holden, MT, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. (2015) 31:3691–3. doi: 10.1093/bioinformatics/btv421
33. Klemm, EJ, Shakoor, S, Page, AJ, Qamar, FN, Judge, K, Saeed, DK, et al. Emergence of an extensively drug-resistant Salmonella enterica serovar Typhi clone harboring a promiscuous plasmid encoding resistance to fluoroquinolones and third-generation cephalosporins. MBio. (2018) 9:e00105–18. doi: 10.1128/mBio.00105-18
34. Procaccianti, M, Motta, A, Giordani, S, Riscassi, S, Guidi, B, Ruffini, M, et al. First case of typhoid fever due to extensively drug-resistant Salmonella enterica serovar Typhi in Italy. Pathogens. (2020) 9:151. doi: 10.3390/pathogens9020151
35. Randall, L, Cooles, S, Osborn, M, Piddock, L, and Woodward, MJ. Antibiotic resistance genes, integrons and multiple antibiotic resistance in thirty-five serotypes of Salmonella enterica isolated from humans and animals in the UK. J Antimicrob Chemother. (2004) 53:208–16. doi: 10.1093/jac/dkh070
36. Xia, S, Hendriksen, RS, Xie, Z, Huang, L, Zhang, J, Guo, W, et al. Molecular characterization and antimicrobial susceptibility of Salmonella isolates from infections in humans in Henan Province, China. J Clin Microbiol. (2009) 47:401–9. doi: 10.1128/JCM.01099-08
37. Zhou, Z, Lundstrøm, I, Tran-Dien, A, Duchêne, S, Alikhan, NF, Sergeant, MJ, et al. Pan-genome analysis of ancient and modern Salmonella enterica demonstrates genomic stability of the invasive Para C lineage for millennia. Curr Biol. (2018) 28:2420–2428.e10. doi: 10.1016/j.cub.2018.05.058
38. Gaballa, A, Cheng, R, Harrand, A, Cohn, A, and Wiedmann, M. The majority of typhoid toxin-positive Salmonella Serovars encode ArtB, an alternate binding subunit. Msphere. (2021) 6:e01255–20. doi: 10.1128/mSphere.01255-20
39. Galán, JE. Typhoid toxin provides a window into typhoid fever and the biology of Salmonella Typhi. Proc Natl Acad Sci. (2016) 113:6338–44. doi: 10.1073/pnas.1606335113
40. Al-Ansari, MM, Aljubali, MM, Somily, AM, Albarrag, AM, and Masood, A. Isolation and molecular characterization of multidrug-resistant Salmonella enterica serovars. J Infect Public Health. (2021) 14:1767–76. doi: 10.1016/j.jiph.2021.10.011
41. Kuijpers, LMF, Le Hello, S, Fawal, N, Fabre, L, Tourdjman, M, Dufour, M, et al. Genomic analysis of Salmonella enterica serotype Paratyphi a during an outbreak in Cambodia, 2013-2015. Microb. Genom. (2016) 2:e000092. doi: 10.1099/mgen.0.000092
42. Stanaway, JD, Reiner, RC, Blacker, BF, Goldberg, EM, Khalil, IA, Troeger, CE, et al. The global burden of typhoid and paratyphoid fevers: a systematic analysis for the global burden of disease study 2017. Lancet Infect Dis. (2019) 19:369–81. doi: 10.1016/S1473-3099(18)30685-6
43. Ahmer, BM, Tran, M, and Heffron, F. The virulence plasmid of Salmonella Typhimurium is self-transmissible. J Bacteriol. (1999) 181:1364–8. doi: 10.1128/JB.181.4.1364-1368.1999
44. Langridge, GC, Fookes, M, Connor, TR, Feltwell, T, Feasey, N, Parsons, BN, et al. Patterns of genome evolution that have accompanied host adaptation in Salmonella. Proc Natl Acad Sci U S A. (2015) 112:863–8. doi: 10.1073/pnas.1416707112
45. Leekitcharoenphon, P, Hendriksen, RS, Le Hello, S, Weill, FX, Baggesen, DL, Jun, SR, et al. Global genomic epidemiology of Salmonella enterica Serovar Typhimurium DT104. Appl Environ Microbiol. (2016) 82:2516–26. doi: 10.1128/AEM.03821-15
46. Ashton, PM, Nair, S, Dallman, T, Rubino, S, Rabsch, W, Mwaigwisya, S, et al. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat Biotechnol. (2015) 33:296–300. doi: 10.1038/nbt.3103
47. Díaz-Torres, O, Lugo-Melchor, OY, De Anda, J, Gradilla-Hernández, MS, Amézquita-López, BA, and Meza-Rodríguez, D. Prevalence, distribution, and diversity of Salmonella strains isolated from a subtropical Lake. Front Microbiol. (2020) 11. doi: 10.3389/fmicb.2020.521146
48. Wong, VK, Baker, S, Pickard, DJ, Parkhill, J, Page, AJ, Feasey, NA, et al. Phylogeographical analysis of the dominant multidrug-resistant H58 clade of Salmonella Typhi identifies inter- and intracontinental transmission events. Nat Genet. (2015) 47:632–9. doi: 10.1038/ng.3281
49. Eng, S-K, Pusparajah, P, Ab Mutalib, N-S, Ser, H-L, Chan, K-G, and Lee, L-H. Salmonella: a review on pathogenesis, epidemiology and antibiotic resistance. Front. Life Sci. (2015) 8:284–93. doi: 10.1080/21553769.2015.1051243
50. Goay, YX, Chin, KL, Tan, CLL, Yeoh, CY, Jaafar, JAN, Zaidah, AR, et al. Identification of five novel Salmonella Typhi-specific genes as markers for diagnosis of typhoid fever using single-gene target PCR assays. Biomed Res Int. (2016) 2016:8905675. doi: 10.1155/2016/8905675
51. Jacob, JJ, Solaimalai, D, Muthuirulandi Sethuvel, DP, Rachel, T, Jeslin, P, Anandan, S, et al. A nineteen-year report of serotype and antimicrobial susceptibility of enteric non-typhoidal Salmonella from humans in southern India: changing facades of taxonomy and resistance trend. Gut Pathog. (2020) 12:1–9. doi: 10.1186/s13099-020-00388-z
52. Kariuki, S, Gilks, C, Revathi, G, and Hart, CA. Genotypic analysis of multidrug-resistant Salmonella enterica Serovar typhi, Kenya. Emerg. Infect. Dis. (2000) 6:649–51. doi: 10.3201/eid0606.000616
53. Katiyar, A, Sharma, P, Dahiya, S, Singh, H, Kapil, A, and Kaur, P. Genomic profiling of antimicrobial resistance genes in clinical isolates of Salmonella Typhi from patients infected with typhoid fever in India. Sci Rep. (2020) 10:1–15. doi: 10.1038/s41598-020-64934-0
54. Mengo, DM, Kariuki, S, Muigai, A, and Revathi, G. Trends in Salmonella enteric serovar Typhi in Nairobi, Kenya from 2004 to 2006. J. Infect. Dev. Countr. (2010) 4:393–6. doi: 10.3855/jidc.503
55. Oludairo, OO, Kwaga, JK, Kabir, J, Abdu, PA, Gitanjali, A, Perrets, A, et al. A review on Salmonella characteristics, taxonomy, nomenclature with special reference to non-Typhoidal and Typhoidal salmonellosis. Zagazig Vet. J. (2022) 50:160–71. doi: 10.21608/zvjz.2022.137946.1179
56. Saier, MH Jr, Reddy, VS, Moreno-Hagelsieb, G, Hendargo, KJ, Zhang, Y, Iddamsetty, V, et al. The transporter classification database (TCDB): 2021 update. Nucleic Acids Res. (2021) 49:D461–7. doi: 10.1093/nar/gkaa1004
57. Shetty, AK, Shetty, IN, Furtado, ZV, Antony, B, and Boloor, R. Antibiogram of Salmonella isolates from blood with an emphasis on nalidixic acid and chloramphenicol susceptibility in a tertiary care hospital in coastal Karnataka: a prospective study. J. Lab. Physicians. (2012) 4:074–7. doi: 10.4103/0974-2727.105585
58. Yap, K-P, Ho, WS, Gan, HM, Chai, LC, and Thong, KL. Global MLST of Salmonella Typhi revisited in post-genomic era: genetic conservation, population structure, and comparative genomics of rare sequence types. Front Microbiol. (2016) 7:270. doi: 10.3389/fmicb.2016.00270
59. Zhu, F, Han, B, Kumar, P, Liu, X, Ma, X, Wei, X, et al. Update of TTD: therapeutic target database. Nucleic Acids Res. (2009) 38:D787–91. doi: 10.1093/nar/gkp1014
Keywords: pathogenic bacteria, diseases, enteric infections, antibiotics resistance, superbugs
Citation: Fatima S, Ishaq Z, Irfan M, AlAsmari AF, Achakzai JK, Zaheer T, Ali A and Akbar A (2023) Whole-genome sequencing of multidrug resistance Salmonella Typhi clinical strains isolated from Balochistan, Pakistan. Front. Public Health. 11:1151805. doi: 10.3389/fpubh.2023.1151805
Edited by:
Li Tai Fang, Freenome Inc., United StatesReviewed by:
Mushal Allam, United Arab Emirates University, United Arab EmiratesCopyright © 2023 Fatima, Ishaq, Irfan, AlAsmari, Achakzai, Zaheer, Ali and Akbar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali Akbar, YWxpYWtiYXIudW9iQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.