 Zhiguo Liu
Zhiguo Liu Miao Wang3†
Miao Wang3† Xiaoping Dong
Xiaoping Dong Liping Gao
Liping Gao- 1National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
- 2Vocational and Technical College, Inner Mongolia Agricultural University, Baotou, China
- 3Comprehensive Office, Ulanqab Center for Disease Control and Prevention, Jining, Inner Mongolia, China
- 4National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
- 5Changzhi Medical College, Changzhi, Shanxi, China
Introduction: Brucellosis, a serious public health issue affecting animals and humans, is neglected in West Africa (WA).
Methods: In the present study, bio-typing, multi-locus sequence typing (MLST), multiple-locus variable-number tandem repeat analysis (MLVA), and whole genome sequencing single-nucleotide polymorphism (WGS-SNP) analysis were used to characterize the Brucella abortus (B. abortus) strains from WA.
Results: All of the 309 strains analyzed in this study were extracted and downloaded from the international MLVA bank and were from 10 hosts (cattle, humans, ovine, buffalo, dromedaries, horse, sheep, zebu, dog, and cat) distributed in 17 countries in WA. Based on the bio-typing, three biovars, dominated by B. abortus bv.3, were observed and reported across seven decades (1958–2019). With MLST, 129 B. abortus strains from the present study were sorted into 14 STs, with ST34 as the predicted founder. These 14 STs clustered into the global MLST data into three clone complexes (C I–C III) with the majority of strains clustering in C I, while C II forms an independent branch, and C III harbors three STs shared by different continents. These data revealed that most cases were caused by strains from native lineages. According to the MLVA-11 comparison, 309 strains were divided into 22 MLVA-11 genotypes, 15 of which were unique to WA and the remaining seven had a global distribution. MLVA-16 analysis showed that there were no epidemiological links among these strains. Based on the MLVA data, B. abortus strains from WA have high genetic diversity, and predominated genotypes were descended from a native lineage. While the MLVA-16 globally highlights that the dominant native and few introduced lineages (from Brazil, the USA, South Korea, Argentina, India, Italy, Portugal, the UK, Costa Rica, and China) co-driving the B. abortus ongoing prevalence in WA. The high-resolution SNP analysis implied the existence of introduced B. abortus lineages, which may be reasonably explained by the movement and trade of dominant hosts (cattle) and/or their products.
Discussion: Our results indicated that B. abortus strains in WA consist of native and introduced strains that necessitate control such as vaccination, testing, slaughtering, and movement control by the relevant country authorities to reduce brucellosis in livestock.
Introduction
Brucellosis, a widespread bacterial zoonotic disease that can cause considerable suffering in humans and massive economic losses in the animal industry, is caused by Gram-negative facultative intracellular bacteria of the genus Brucella (1–3). The genus includes 12 species, among which Brucella melitensis, B. abortus, and B. suis are the most important species and responsible for the majority of human and animal brucellosis cases (4). It has been reported that there are ~500,000 new cases of human infection annually, especially in low-income tropical countries (5). Cattle are natural hosts of the intracellular pathogen B. abortus, which can also be isolated in other hosts, such as sheep, camels, and horses, and imposes a significant burden on the health and reproduction of these important livestock (6, 7). Infected animals can have live offspring following the initial abortion, and they may continue to shed the bacteria (8). Transmission to human occurs through unprotected handling of tissues or body fluids from infected animals, consumption of unpasteurized milk and milk products, or inhalation of Brucella-contaminated aerosols (9, 10). Although B. abortus causes less severe infections than B. melitensis (11), back pain and arthralgia are common symptoms in patients infected with B. abortus in Turkey (12).
A previous study reported that the annual economic burden of bovine brucellosis in Latin America is ~$600 million (13). In 2015, in Kazakhstan, around $21 million was spent on compensation for slaughtered animals with brucellosis, and an additional $24 million was spent on testing and screening of animals (14). Therefore, brucellosis can not only lead to substantial economic losses in the animal industry but also pose an ongoing threat to public health.
Countries declared brucellosis-free are located in Europe and Oceania, whereas the prevalence is high in enzootic countries in central and South America, Africa, and parts of Asia (15). In these developing countries, especially in Africa, livestock husbandry development is continuously challenged by brucellosis (16, 17). Bovine brucellosis remains the most widespread disease in animals and the main concern in sub-Saharan countries (18). In African countries, B. abortus has been reported in Sudan, in both cattle and their handlers (19), as well as in Gambia, Mali, Niger, and more frequently in Nigeria, Senegal, and Ivory Coast (20). Therefore, a comprehensive analysis of the genetic diversity of B. abortus strains and the epidemiology of the disease in animals and humans in sub-Saharan countries is necessary.
Molecular characterization of the predominated circulating strains is critical to understanding B. abortus diversity and epidemiology in the country. Multi-locus sequence typing (MLST) is a reliable tool for the characterization of Brucella spp. populations and the determination of phylogenetic relationships (21, 22); however, this method yields less detailed typing results than MLVA and WGS-SNP due to the lower-resolution power (21, 23). Multiple locus variable-number tandem repeat analysis (MLVA) enables Brucella genotyping to infer genetic diversity and investigate the geographic clustering of the isolates (24–26). Moreover, whole genome sequencing single-nucleotide polymorphism (WGS-SNP) has higher discriminatory power to efficiently track the origin and spread of Brucella strains, leading to be targeted and effective control of disease spread (27, 28). At present, MLVA has been used to explore the diversity of B. abortus biovar 3 isolated in West Africa (29) and investigate the epidemiological links of the B. melitensis in Egypt (30). Moreover, studies that used MLVA and WGS-SNP (31, 32) indicated that WGS-SNP analysis allows a better differentiation than MLVA-16. WGS-SNP analysis has been used to type, discriminate and track outbreak strains (31–33). Therefore, the purpose of this study was to use a series of molecular typing assays, including MLST, MLVA, and WGS-SNP, to investigate the species population, genetic diversity, geographical origin, and molecular epidemiology of B. abortus strains from West Africa (WA), to provide insight into the comprehensive understanding of the B. abortus brucellosis epidemiology features in WA, and to facilitate surveillance and control strategy development.
Methods
Source of MLST and MLVA genotyping data
The MLST genotyping data of 129 B. abortus strains (Table 1; Supplementary Table S1) were downloaded from the PubMLST (https://pubmlst.org/organisms/brucella-spp), including the key (code) of isolates, sequence type (ST), allotype of nine loci (aroA/cobQ/dnaK/gap/glk/gyrB/int_hyp/omp25/typE), Brucella species/biovars, host, country, continent, and years isolated. Moreover, the MLVA-16 genotyping data of 309 B. abortus strains (Table 2; Supplementary Table S2) from 17 WA countries, including Senegal, Egypt, Togo, Nigeria, Rwanda, Kenya, Guinea-Bissau, Uganda, Niger, Zimbabwe, Cameroon, Sudan, Tanzania, Guinea, Ivory Coast, Chad, and Gambia, were abstracted from the MLVA bank database (https://microbesgenotyping.i2bc.paris-saclay.fr/databases). The key (code) of strains, number of tandem repeats at each of the MLVA-16 loci, MLVA-8 genotype, MLVA-11 genotype, hosts, species/biovars, isolated regions, years isolated, and lineage groups were obtained. The MLVA-8 genotype, MLVA-11 genotype, and lineage group data from 19 B. abortus MLVA-8 and 21 MLVA-11 genotypes were not previously identified genotypes in the international MLVA bank database.
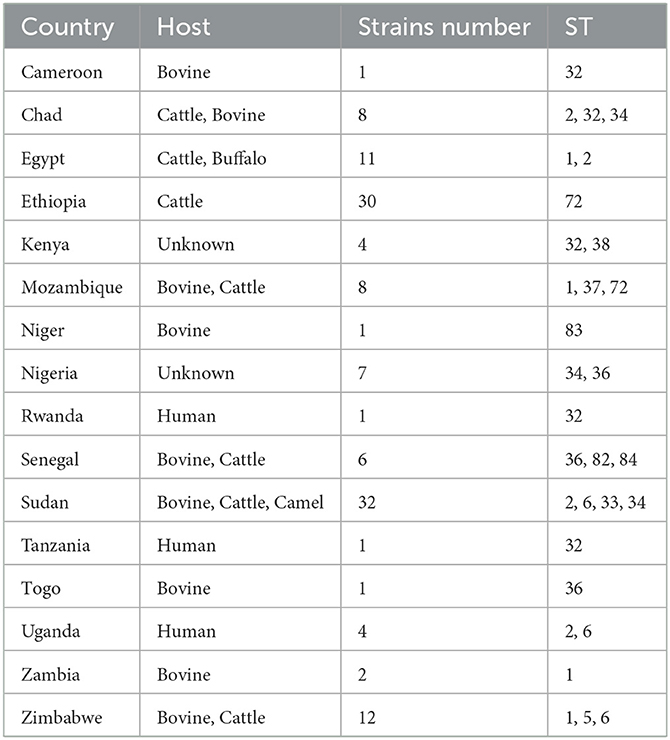
Table 1. Country, hosts, number of strains and sequencing type (ST) of 129 Brucella abortus strains from Africa used for multi-locus sequence typing (MLST) genotyping.
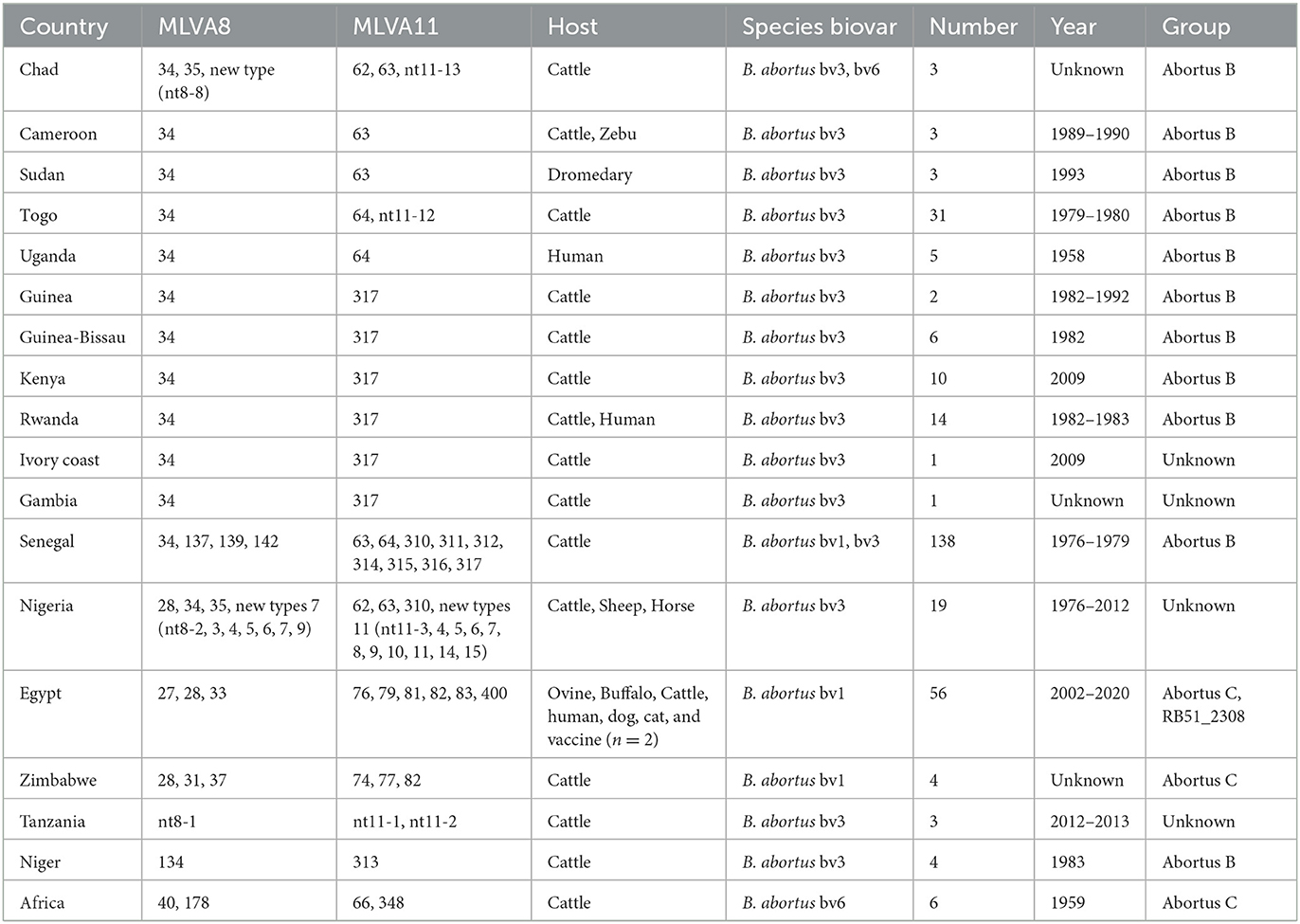
Table 2. Country, MLVA-8, MLVA-11, host, species/biovar, number, and year of 309 B. abortus strains used for MLVA genotyping.
Analysis and visualization of MLST and MLVA data
All data in this study were cleaned and processed using Excel 2016 software (Microsoft Corporation, Redmond, WA, USA). For MLST analysis, genetic similarities between the STs of 129 strains from WA and 631 strains from other continents (PUBMLST) (Supplementary Table S1) were investigated using eBURST software 2.0. Finally, the genetic relationships between the isolates and allelic profiles of MLST data were analyzed with the software PHYLOViZ version 2.0 using the goeBURST Full Minimum Spanning Tree algorithms (34). Subsequently, MLVA data were processed according to genotype information, species/biovars, hosts, location, and years isolated for all B. abortus strains. A minimum spanning tree (MST) of B. abortus strains was constructed using BioNumerics 8.0 software based on the 309 B. abortus strains' MLVA-11 data (Supplementary Table S2) to investigate the host lineage, genotype distribution, and the geographical origin of the B. abortus strains. At the global level, an MST was constructed using the MLVA-11 data of 1,746 strains (Supplementary Table S2) to explore the geographical origin feature of genotypes from this study at the global level. Moreover, the MST based on the MLVA-16 data of 1,746 B. abortus strains (Supplementary Table S2) was used to determinate the relationship among strains at a global scale. In this study, the MST trees were weighted according to MLVA panels, as described by previous reports (35).
Phylogenetic analysis based on the SNPs of 80 strains at the global level
WGS-SNP analysis was performed as previously described (36). Briefly, genomic alignment between the sample genome and reference genome (B. abortus 2308; Assembly ID: GCA_000054005.1) was performed using the MUMmer (37) and LASTZ (38) tools. The phylogenetic tree of 18 B. abortus strains from WA and 62 strains from outside WA (Supplementary Table S3) in GenBank was constructed using TreeBeST (39) based on the Maximum-Likelihood Phylogenies (PHYML) algorithm with 1,000 bootstrap replicates to investigate the genetic relationships of B. abortus strains.
Results
Species/biovars, host, and geographic distribution of 309 B. abortus strains
Of the identified biovars of 309 B. abortus African strains, 239 B. abortus bv 3, 9 B. abortus bv 1 and 6 B. abortus bv 6 were identified with 55 not typed to biovar level (Figure 1; Table 2). The B. abortus bv. 1 was distributed in Egypt (n = 6), Zimbabwe (n = 2), and Senegal (n = 1). B. abortus bv. 6 was found in Chad (n = 2), and the location of the remaining six is unknown. However, the B. abortus bv. 3 strains were distributed in 15 countries, i.e., all 17 countries except Egypt and Zimbabwe (Figure 1; Table 2). Based on the host, 280 strains were found in cattle, 8 in humans, 4 in ovine, 4 in buffalo, 3 in the dromedary, 2 in horse, 2 in sheep, 2 in zebu, 1 in a dog, and 1 in a cat, and the remaining two were vaccine strains (Table 2). A total of 309 B. abortus were collected from 17 countries, including 138 in Senegal, 56 in Egypt, 31 in Togo, 19 in Nigeria, 14 in Rwanda, and remaining countries contained 1–10 different numbers strains (Figure 1; Table 2). According to the MLVA data, strains were divided into the abortus B group (n = 209), the abortus C group (n = 55), and the abortus C_RB51_2308 group from Egypt (n = 11), and the remaining 24 strains were unknown. These strains were isolated between 1958 and 2020, including 5 in the 1950s, 147 in the 1970s, 43 in the 1980s, 7 in the 1990s, 13 in the 2000s, 46 in the 2010s, and 2 in 2020, and the year of isolation of the remaining isolates is unknown (Table 2; Supplementary Table S2).
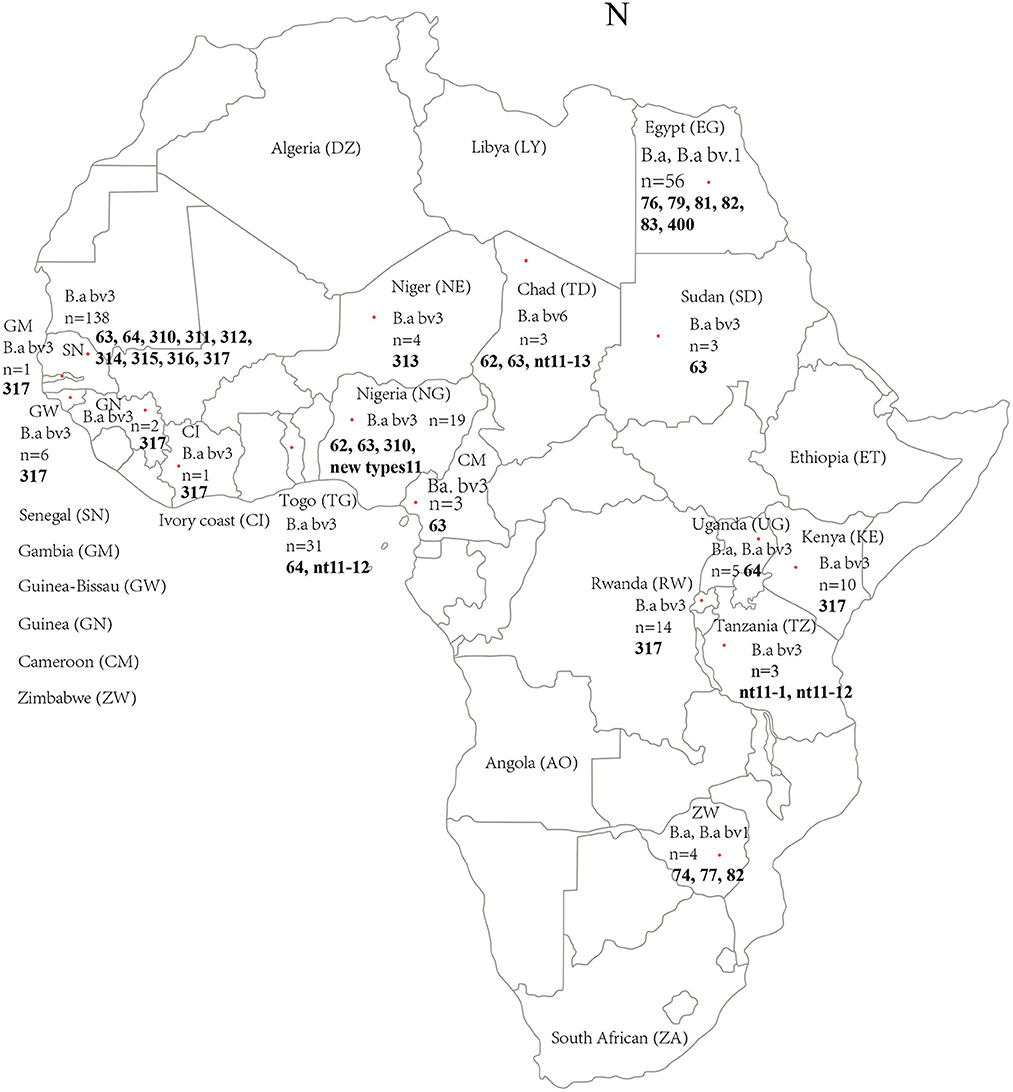
Figure 1. Geographic and genotype distribution features of 309 B. abortus in the present study. Red dot indicates the location (country) of the strains collected in this study. The letter in brackets is the abbreviation of the national name in the figure; n, Number of strains collected; figures marked with the bold refer into the MLVA-11 genotypes of strains from the location. The map of Africa used in this study was obtained from Google Maps (https://www.google.com/maps).
MLST genotyping characteristic of B. abortus strains in the present study
The results of eBURST analysis showed that the 129 B. abortus strains from the present study could be sorted into 14 STs, including ST1 (n = 16), ST2 (n = 8), ST5 (n = 3), ST6 (n = 9), ST32 (n = 8), ST33 (n = 4), ST34 (n = 31), ST36 (n = 8), ST37 (n = 5), ST38 (n = 3), ST72 (n = 31), ST82 (n = 1), ST83 (n = 1), and ST84 (n = 1) (Table 1). Of these, ST34 was the predicted founder and included two single locus variants, zero double locus variants, one triple locus variant, and 10 more distantly related (satellites) (Supplementary Figure S1). The ST34 is comprised of strains from three countries, including Sudan (n = 24), Nigeria (n = 4), and Chad (n = 3). The MST at the global scale indicated that among those strains (n = 760) from six continents, 14 STs in the present study were divided into three clone complexes (C I–C III) (Figure 2), of which C I and C II comprised strains from the present study and formed the independent complex. C I comprised eight STs (ST6, 32, 33, 34, 36, 82, 83, and 84; and strains from Cameroon, Chad, Kenya, Niger, Nigeria, Rwanda, Senegal, Sudan, Tanzania, Togo, Uganda and Zimbabwe) and C II contained three STs (ST37, 38, and 72; and strains from Ethiopia, Kenya, and Mozambique) (Figure 2). C III harbored three single STs (ST 1, 2 and 5; and strains from Chad, Egypt, Mozambique, Sudan, Uganda, Zambia, and Zimbabwe) that were shared between three and five continents: ST1 was shared by strains from Asia, Europe, North America, Oceania, and South America (Figure 2); ST2 was shared by strains from Asia, Europe, and South America; and ST5 was shared by strains from four continents, including Asia, Europe, North America, and South America (Figure 2).
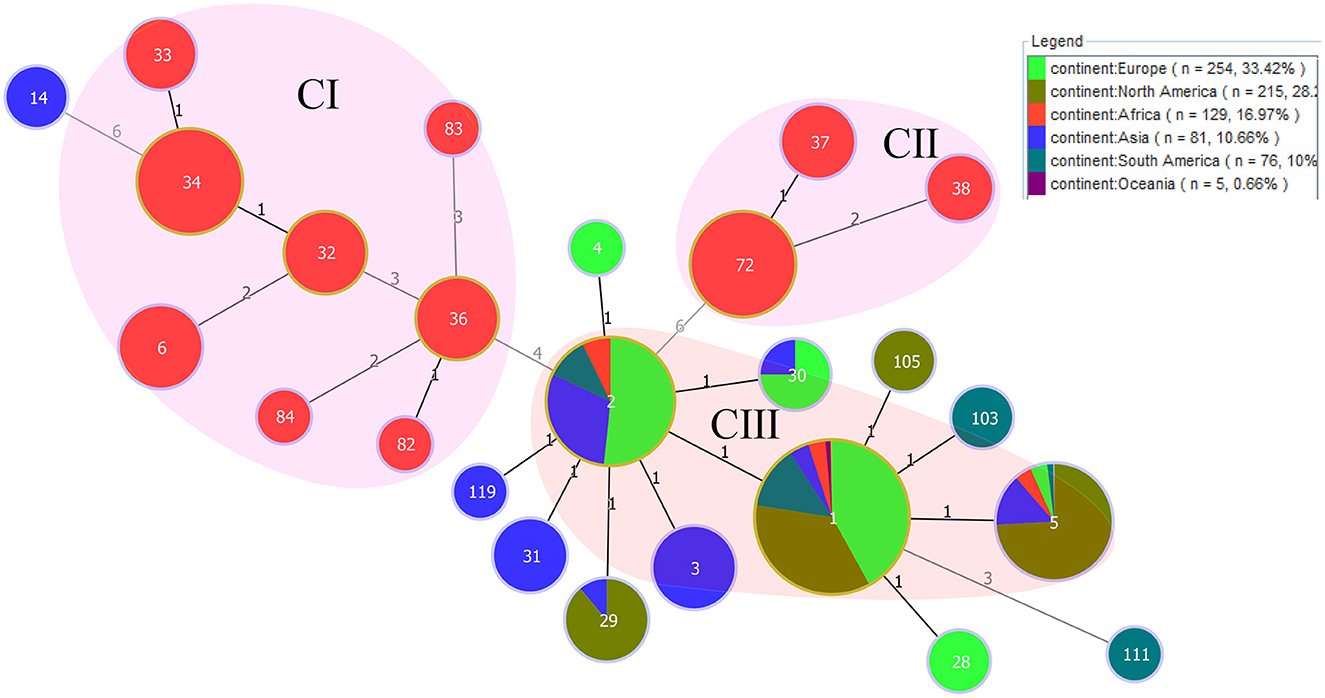
Figure 2. PHYLOViZ analysis showing the genetic relationships among STs of 760 B. abortus strains. Color coding according to continents. Circle size indicates the number of strains. Each circle represents a distinct ST, and the STs from the present study are shaded in pink (C I, C II) and light red (C III).
Diversity characteristics of MLVA-8 and MLVA-11 data of strains from this study
A total of 13 known MLVA-8 genotypes and nine new genotypes (nt8-1–nt8-9) were recorded among 309 strains, known 13 MLVA-8 genotypes including 27 (n = 11), 28 (n = 45), 31 (n = 1), 33 (n = 2), 34 (n = 206), 35 (n = 1), 37 (n = 1), 40 (n = 5), 134 (n = 3), 137 (n = 1), 139 (n = 1), 142 (n = 2), and 178 (n = 1) (Table 2). According to the MLVA-11 analysis showed that a total of 37 MLVA-11 genotypes identified among 309 strains, and 22 out of 37 MLVA-11 genotypes were previously identified including 62 (n = 1), 63 (n = 12), 64 (n = 80), 66 (n = 5), 74 (n = 1), 76 (n = 2), 77 (n = 1), 79 (n = 11), 81 (n = 6), 82 (n = 37), 83 (n = 1), 310 (n = 1), 311 (n = 1), 312 (n = 1), 313 (n = 3), 314 (n = 1), 315 (n = 5), 316 (n = 11), 317 (n = 98), 348 (n = 1), and 400 (n = 1) (Table 2); and the remaining 15 new MLVA genotypes were first found in this study, then named them as into nt11-1–nt11-15 (Table 2). MLVA-11 genotype 317 (n = 98) and 64 (n = 80) are the predominant circulating lineages (Figure 3). Moreover, MLVA-11 genotypes 317 and 64 were mainly distributed in Senegal, and MLVA-11 genotype 82 was mainly distributed in Egypt. MLVA-11 genotype 63 was observed in Senegal, Uganda, and Cameroon (Figure 3).
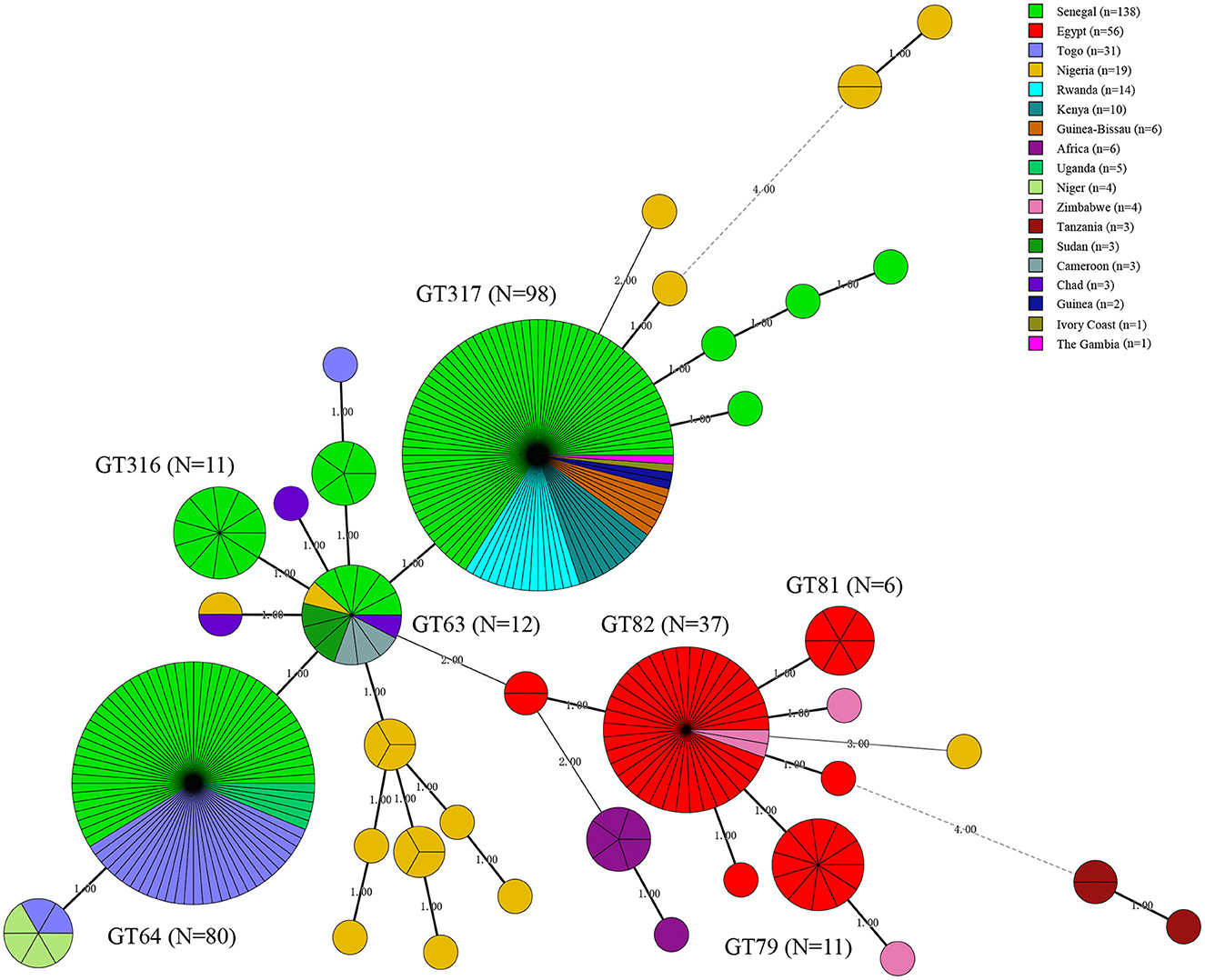
Figure 3. Minimal spanning tree (MST) of MLVA-11 genotype geographic distribution based on the multi-loci variable number of tandem repeats (VNTR) 11 data from West African Brucella abortus strains. Color coding according to countries distributed of MLVA-11 genotypes (the detail genotype profile is presented in Supplementary Figure S2), the predominated MLVA-11 genotypes in the present study were marked with the bold in figures. Circle size indicates the number of strains, GT indicates MLVA-11 Genotype. The number on the horizontal line is the number of site differences between genotypes.
Geographic origin and genetic relatedness of B. abortus strains on a global scale
Phylogenetic analysis of MLVA-11 data on B. abortus strains on a global scale showed that out of 22 MLVA-11 genotypes, 15 formed an independence lineage (Figure 4). Of these 15, except for three genotypes (MLVA-11 genotypes 74, 348, and 400) that were related to strains from other countries, the remaining 12 MLVA-11 genotypes formed a unique cluster (Figure 4). Moreover, strains from seven other MLVA-11 genotypes (MLVA-11 genotypes 66, 76, 77, 79, 81, 82, and 83) were shared with strains from 2 to 14 different countries (Figure 4). Of these, MLVA-11 genotype 66 was shared by strains from three countries, including Bangladesh, Brazil, and France; MLVA-11 genotype 76 was shared with strains from four countries, including Brazil, China, France, and Portugal; MLVA-11 genotype 76 was shared by strains from the present study and China; MLVA-11 genotype 79 was shared by strains from Costa Rica, India, Italia, Portugal, South Korea, Switzerland, and the USA (Figure 4); MLVA-11 genotype 81 was shared by strains from Italy and this study; MLVA-11 genotype 82 was shared by strains from Argentina, Belgium, Brazil, China, Costa Rica, France, Germany, India, Kazakhstan, Portugal, South Korea, Switzerland, the UK, and the USA (Figure 4); MLVA-11 genotype 83 was shared by strains from Brazil, China, Costa Rica, England, and the UK (Figure 4).
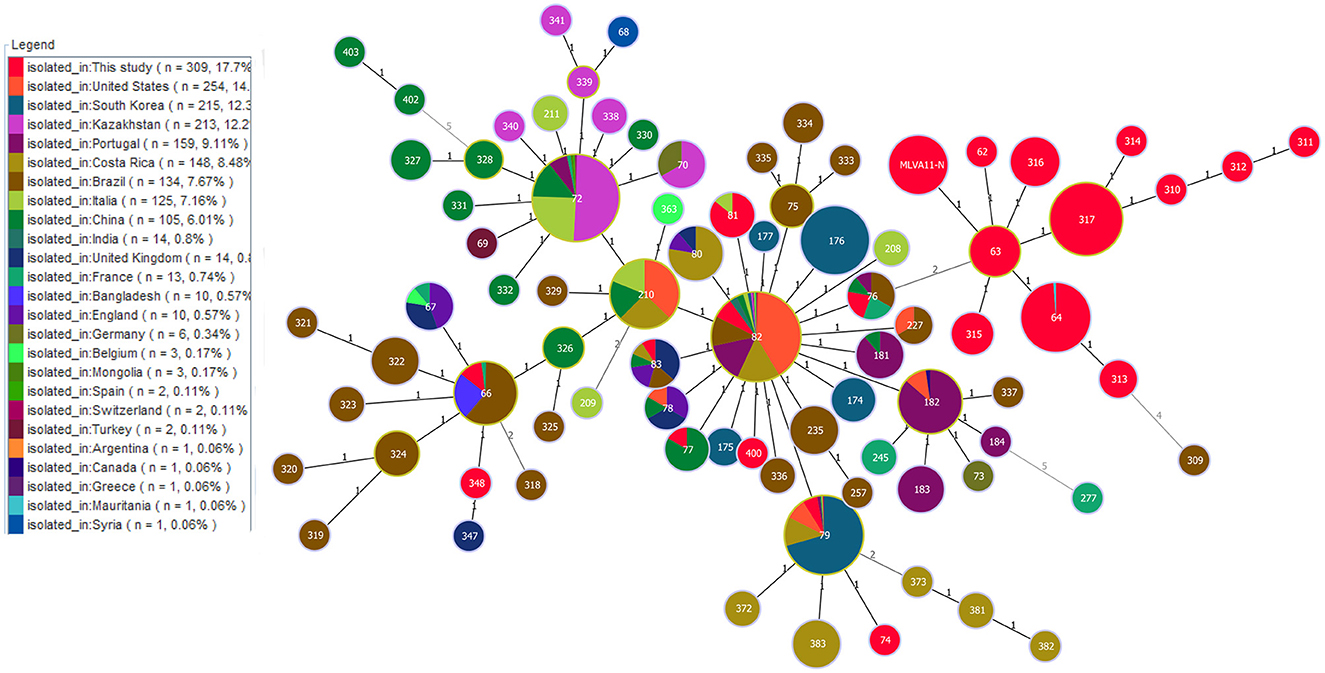
Figure 4. Geographic origin analysis of B. abortus strains in West Africa on a global scale based on the multi-loci variable number of tandem repeats (VNTR) 11 data worldwide. Color coding according to countries. Circle size indicates the number of strains.
Molecular epidemiological links of B. abortus in West Africa and on a global scale
Based on the MST of MLVA-16 data on 309 strains from 17 countries, the strains were divided into two branches (I and II). Branch I comprised strains from Egypt, Zimbabwe, and Uganda (Figure 5), and branch II contained strains from the other 14 countries (Figure 5). Remarkably, only one MVA-16 genotype was shared among two strains from two different countries (Senegal and Nigeria) (Figure 5). Based on the MST of 1,746 strains on a global scale, the majority of strains from this study formed independent clades, and only few strains have shared the same MLVA-16 genotype with strains from many countries, including Brazil, the USA, South Korea, Argentina, India, Italia, Portugal, the UK, Costa Rica, and China (Figure 6).
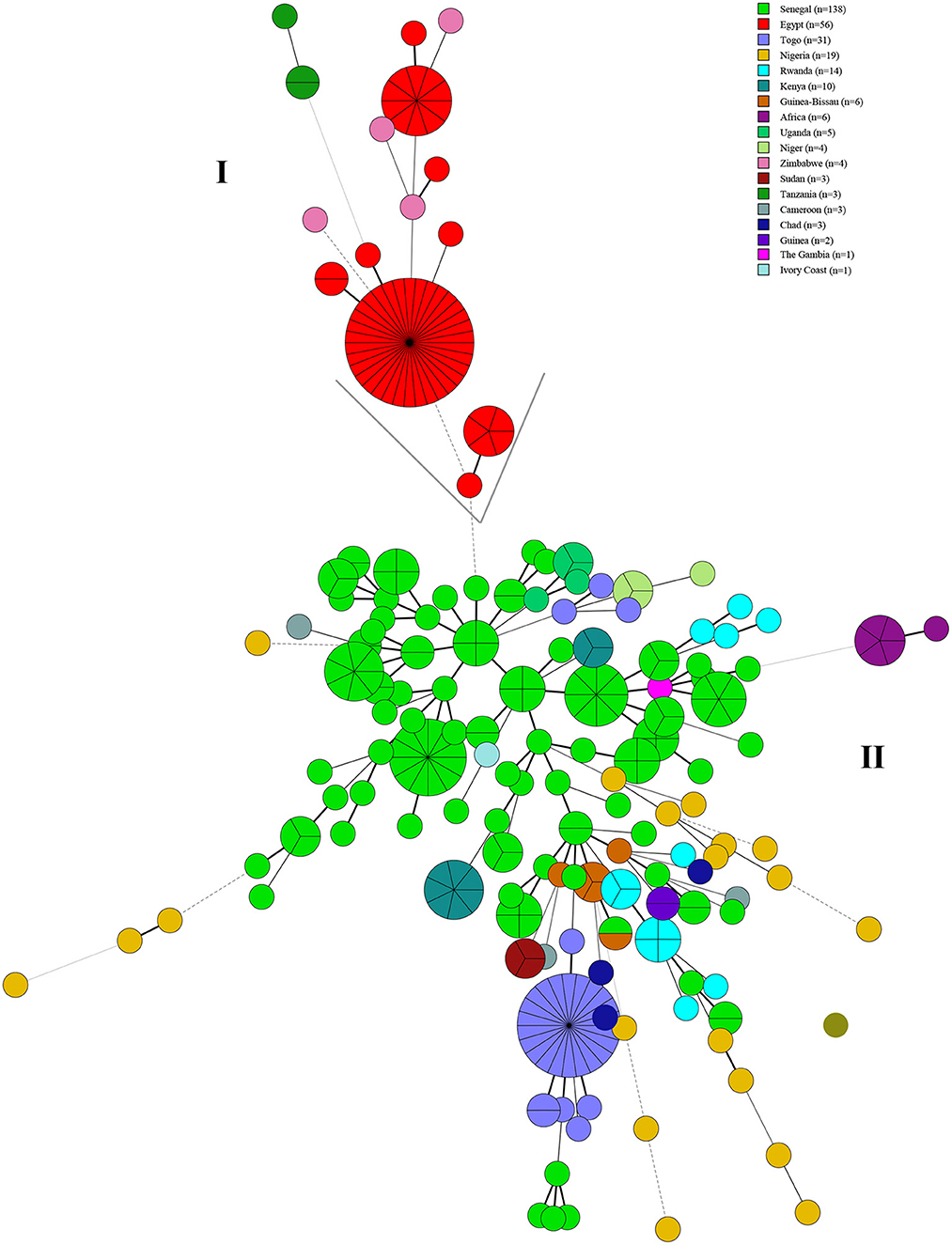
Figure 5. Molecular epidemiology relatedness of B. abortus strains from West Africa based on the MLVA-16 data of 309 B. abortus. Color coding according to the country in this study. Circle size indicates the number of strains. Branch (I) comprised strains from Egypt, Zimbabwe, and Uganda and branch (II) contained strains from the other 14 countries.
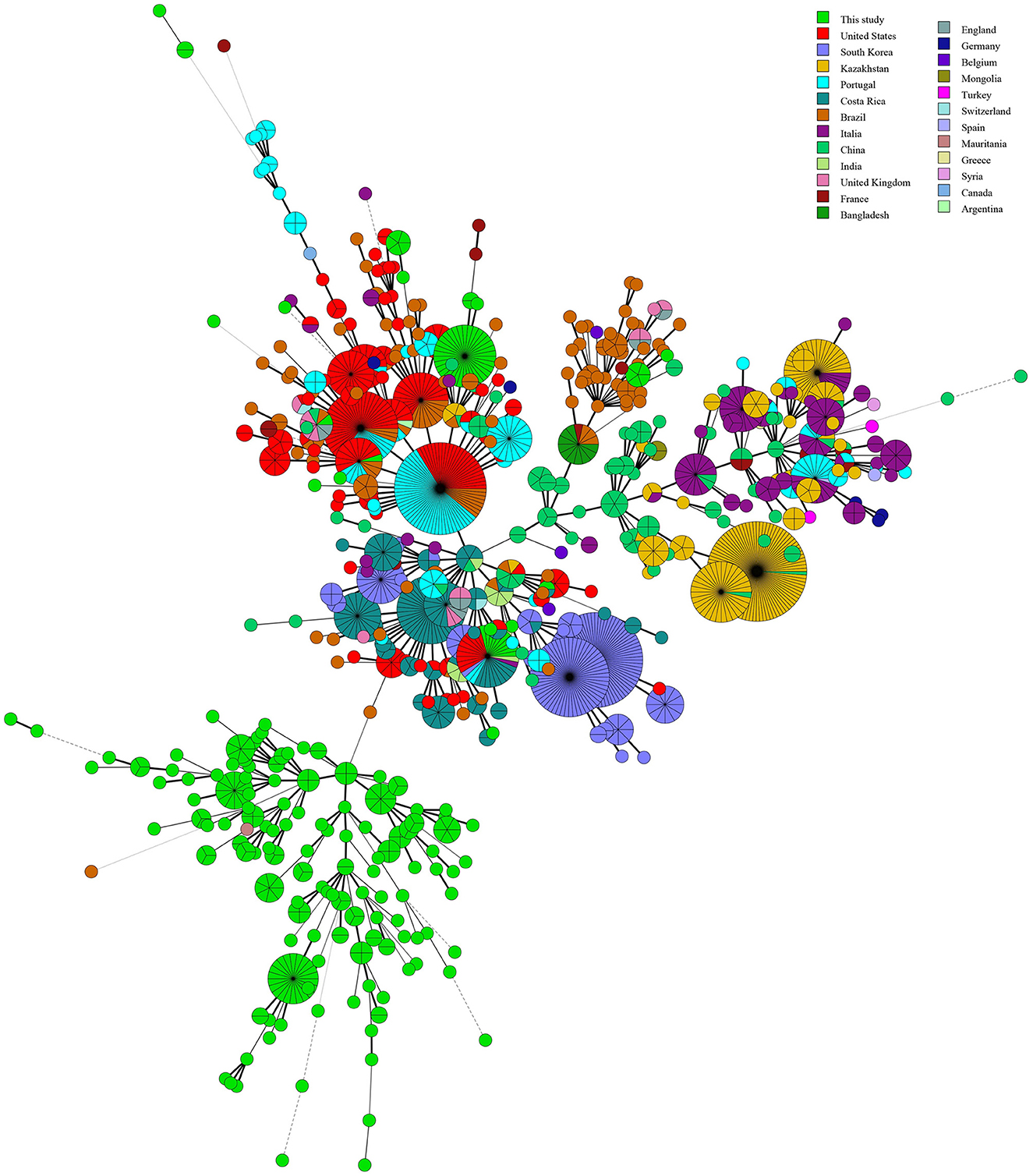
Figure 6. Molecular relationships between B. abortus strains from West Africa and worldwide strains based on the MLVA-16 data of 1,476 B. abortus strains globally. Color coding according to countries. Circle size indicates the number of strains.
Additionally, the MST based on our WGS-SNP matrix analysis showed that the 18 strains from the present study formed 17 SNP genotypes, and which one was shared by two strains from Mozambique and Zimbabwe (Figure 7). Furthermore, five out of 17 SNP genotypes contained only strains from this study (Figure 7), the remaining 12 were shared SNP genotypes that comprised strains from this study and strains from eight different countries, including the USA (Nigeria and Zimbabwe), the UK (Uganda and Senegal), Germany (Chad), France (Egypt), Poland (Sudan), Spain (Chad), New Zealand (Zimbabwe), Bolivia (Mozambique and Zimbabwe), and China (Sudan) (Figure 7).
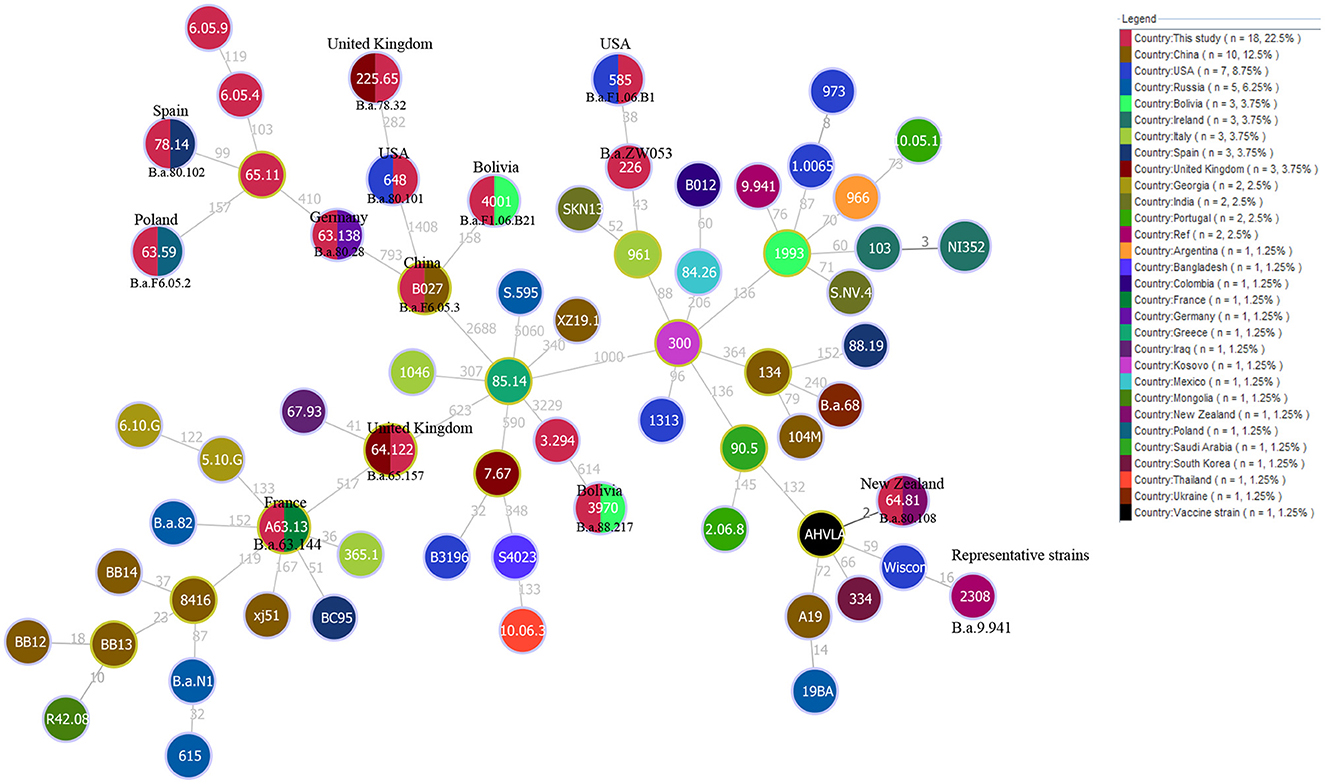
Figure 7. WGS-SNP phylogeny analysis of B. abortus strains from the present study and worldwide strains based on the SNP matrix of 80 B. abortus. Strains code are marked in black if they share the same SNP genotype with the strain in the circle. The countries marked in the figures indicate that strains from this country have identical SNP genotypes with strains from the present study.
Discussion
In this study, three molecular methods, including MLST, MLVA, and WGS-SNP, were applied to analyze the genetic diversity, population structure, and molecular relationship among 309 B. abortus strains from WA with global strains. Our analysis highlights that B. abortus strains have a high species/biovars, host diversity, as well as wide geographic distribution in WA. In the present study, more than 90% (280/309) of B. abortus strains were isolated from cattle, and the remaining strains were obtained from 10 other hosts, including wildlife. B. abortus predominantly infect cattle but also infect other host species and seems to be a spill-over from the dominant host species to other species due to farming and grazing (40), which is also evident from in the present study. A previous study showed that 273 Brucella strains from Africa were identified and typed since 1976, among which 272 strains were from cattle and ~95% (260/270) were from native animals showing hygromas (41). Similar when focusing on WA, B. abortus strains were historically prevalent mainly in cattle (42–44). Bio-typing identified three biovars in WA, and B. abortus bv.3 was the predominant biovar (45).
MLVA11 identified B. abortus bv 3 isolated from cattle as new major clusters from WA. In contrast to WA where B. abortus biovar 3 strains occur in cattle, B. abortus biovar 1 is the dominate biovar in South Africa (46). These data suggest that the predominated B. abortus biovars are unique in WA compared to southern Africa, but further investigation is needed to better understand the epidemiological trait of B. abortus in Africa.
Cattle production plays a crucial role in Africa including WA, as cattle are traded for status and serve as a “savings account” in nomadic systems in Nigeria (47). It has been reported that 20% of cattle are imported in Nigeria, mostly from Chad and Niger (48). Moreover, according to literature, brucellosis is one of the major transboundary animal diseases in North African countries, while the illegal animal movement was identified as one of the major constraining factors (49).
In cattle, B. abortus mainly invades the reproductive organs and causes symptoms such as abortion and a decline in milk productivity, resulting in substantial economic losses (50). Therefore, strengthening disease surveillance in cattle is necessary to map the epidemiology of B. abortus. However, strains were found in 10 wildlife species in WA, indicating that the B. abortus strains circulate among animal species that are not the preferential hosts. These reservoirs pose a crucial risk to domestic animals and humans. A previous study showed that exploring the wildlife reservoir of brucellosis may represent a new challenge to be faced by the medical and veterinarian community in the twenty-first century (51). Brucellosis in wildlife in this region cannot be neglected, detection and surveillance should be implemented to block the transmission chain.
Although MLVA has a higher resolution power for Brucella strains than MLST, both the MLST and MLVA are crucial to epidemiologic surveillance and investigation into the geographical distribution of B. abortus strains. MLST analysis highlighted that the predominant circulating B. abortus population of WA was native lineages, while three STs were shared by many continents (Figure 2). However, these shared populations were predominant in Europe (ST1 and 2) and America (ST1 and 5), implying that the introduced B. abortus lineage was from outside WA, but a further survey into the trade activities of cattle is needed.
The MLVA-11 highlights the high genetic diversity and multiple geographic original of strains from the present study as reflected by the 37 MLVA-11 genotypes were identified among 309 strains that include 22 known and 15 new genotypes. Two predominant MLVA-11 genotypes (317 and 64) were observed in WA (52, 53), while genotype 317 was distributed in Kenya, Senegal, Guinea, Guinea-Bissau, and Rwanda (52, 53), and 64 was also distributed in Senegal and Togo (53). Comparison analysis of global MLVA-11 data suggests that these genotypes originate from WA. Three MLVA-11 genotypes (66, 79, and 82) were shared with strains from Brazil (54), the USA (55), Italy, Portugal (56), France, and South Korea (26). The WA B. abortus strains from the present study existed multiple geographic original that suggest introduction from multiple regions outside WA similar to study by Wareth et al. (57).
The global analysis of B. abortus strains base on MLVA-16 highlights that the native dominant strains in WA are driving the ongoing prevalence of B. abortus in this region. This was similar to findings by Liu et al. (58) suggesting that B. abortus strains were transmitted within the national borders of a country despite common geographical origins of the strain in countries along the silk road. B. abortus bv 3 strains from Algeria grouped in two separate clusters using MLVA11 which most strains clustering with European isolates from France and Spain while a few strains cluster in African lineage (59). These studies thus indicate presence of introduced strains as well as native strains in WA. Therefore, further extensive bacteriological and molecular investigations are necessary for a comprehensive understanding of the epidemiology of cattle brucellosis in African countries.
WGS-SNP analysis identified B. abortus lineages circulating in WA was introduced through the import of animals and/or animal products from multiple countries as 12 of 17 SNP genotypes were from nine countries (regions). The MLST, MLVA, and SNP analyses demonstrated that native strains and introduced lineages are co-driving with the persistence of B. abortus circulating in WA. Similarly, WGS-SNP analysis demonstrated that brucellosis in South Africa spreads within the herd on some farms, whereas the introduction of infected animals is the mode of transmission on other farms (33). WGS phylodynamics analysis identified the main B. abortus lineage circulate in Costa Rica are widespread while new introductions seem to be more geographically restricted, which might be similar in various other middle- and low-income countries where brucellosis is endemic with similar farming practices and lack of control (60).
Therefore, a control program involving improved surveillance, animal movement restrictions, public health education is suggested. Moreover, there is a strong need for more sustainable molecular data on prevailing Brucella strains in WA, and on all susceptible species, including humans, to comprehensively analyze the relatedness between field strains of Brucella and the epidemiology of brucellosis within WA countries (29).
Conclusions
The present study revealed the constant circulation of B. abortus strains in cattle throughout WA and neighboring countries. These strains exhibited high species/biovars, host spectrum, and genetic diversity as well as multiple geographic origins. Moreover, most cases were caused by native strains, and few cases resulted from introduced lineages. The surveillance and control of B. abortus in WA should be made a priority which can be enhanced by improving molecular databases that can elucidate epidemiological relationships between strains. Regulations should be strictly implemented when introducing animals to prevent the spread of this species (48).
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
LG and ZLiu performed the data collection and data analysis and drafted the manuscript. MW and QS conducted the data check and critically reviewed the manuscript. ZLiu, ZLi, and XD participated in the design of the study and managed the project. All authors read and approved the final manuscript.
Funding
This work was supported by the Strategic Research and Consulting Project of the Chinese Academy of Engineering (No. 2022-XBZD-18, Project Name: Prevention and Control of Emerging Infectious Diseases, Institute No. 29163) and the Youth Science Foundation of the State Key Laboratory of Infectious Disease Prevention and Control, Chinese Center for Disease Control and Prevention (No. 2021SKLID503). The funders had no role in the study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
Acknowledgments
We are grateful to all of the authors who have previously published data on brucellosis in WA.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2023.1106361/full#supplementary-material
Supplementary Figure S1. Genetic relationships based on the STs of B. abortus strains (n = 129) in this study using eBURST. Re-sampling for bootstrapping = 1,000; minimum number of identical loci for group definition = 3; minimum number of SLVs for subgroup definition = 1. Strains in ST34 are from Sudan (n = 24), Nigeria (n = 4), and Chad (n = 3).
Supplementary Figure S2. Minimal spanning tree (MST) of MLVA-11 genotype diversity based on the multi-loci variable number of tandem repeats (VNTR) 11 data from West African Brucella abortus strains. Color coding according to MLVA-11 genotypes. Circle size indicates the number of strains.
Supplementary Table S1. MLST data sets of B. abortus strains used in the present study. 129 strains from the present study (Africa) and 631 strains from the outside of Africa.
Supplementary Table S2. The MLVA-16 data sets of B. abortus strains used for the present study. 309 marked with gray-used from the present study, MLVA-11 data from the 1,746 B. abortus strains used for geographical origin analysis globally and all data were used to determinate the relationship among strains at a global scale.
Supplementary Table S3. A total of 80 B. abortus strains genome data from GenBank (n = 62) and this study (n = 18) were used in the present study for WGS-SNP phylogeny analysis globally.
References
1. Carvalho Neta AV, Mol JP, Xavier MN, Paixao TA, Lage AP, Santos RL. Pathogenesis of bovine brucellosis. Vet J. (2010) 184:146–55. doi: 10.1016/j.tvjl.2009.04.010
2. Migisha R, Dan N, Boum Y, Page AL, Zúñiga-Ripa A, Conde-Álvarez R, et al. Prevalence and risk factors of brucellosis among febrile patients attending a community hospital in south western Uganda. Sci Rep. (2018) 8:15465. doi: 10.1038/s41598-018-33915-9
3. Mufinda FC, Boinas F, Nunes C. Prevalence and factors associated with human brucellosis in livestock professionals. Rev Saude Publ. (2017) 51:57. doi: 10.1590/s1518-8787.2017051006051
4. Alton GG, Jones LM, Angus R, Verger J. Techniques for the Brucellosis Laboratory. Paris: Institut National de la recherche Agronomique (INRA) (1988).
5. Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. The new global map of human brucellosis. Lancet. (2006) 6:91–9. doi: 10.1016/S1473-3099(06)70382-6
7. Fiebig A, Vrentas CE, Le T, Huebner M, Boggiatto PM, Olsen SC, Crosson S. Quantification of Brucella abortus population structure in a natural host. Proc Natl Acad Sci USA. (2021) 118:e2023500118. doi: 10.1073/pnas.2023500118
8. OIE. Brucellosis. (2018). Available online at: https://www.woah.org/en/disease/brucellosis/
9. Kaufmann AF, Fox MD, Boyce JM, Anderson DC, Potter ME, Martone WJ, et al. Airborne spread of brucellosis. Ann NY Acad Sci. (1980) 353:105–14. doi: 10.1111/j.1749-6632.1980.tb18912.x
10. Corbel MJ. Recent advances in brucellosis. J Med Microbiol. (1997) 46:101–3. doi: 10.1099/00222615-46-2-101
12. Dokuzoguz B, Ergönül O, Baykam N, Esener H, Kiliç S, Celikbaş A, et al. Characteristics of B. melitensis versus B. abortus bacteraemias. J Infect. (2005) 50:41–5. doi: 10.1016/j.jinf.2004.02.005
13. Seleem MN, Boyle SM, Sriranganathan N. Brucellosis: a re-emerging zoonosis. Vet Microbiol. (2010) 140:392–8. doi: 10.1016/j.vetmic.2009.06.021
14. Charypkhan D, Sultanov AA, Ivanov NP, Baramova SA, Taitubayev MK, Torgerson PR. Economic and health burden of brucellosis in Kazakhstan. Zoonoses Public Health. (2019) 66:487–94. doi: 10.1111/zph.12582
15. Cárdenas L, Awada L, Tizzani P, Cáceres P, Casal J. Characterization and evolution of countries affected by bovine brucellosis (1996–2014). Transbound Emerg Dis. (2019) 66:1280–90. doi: 10.1111/tbed.13144
16. Ekiri AB, Kilonzo C, Bird BH, VanWormer E, Wolking DJ, Smith WA, et al. Utility of the rose Bengal test as a point-of-care test for human brucellosis in endemic African settings: a systematic review. J Trop Med. (2020) 2020:6586182. doi: 10.1155/2020/6586182
17. Woodburn DB, Steyl J, Du Plessis EC, Last RD, Reininghaus B, Mitchell EP. Pathological findings in African buffaloes (Syncerus caffer) in South Africa. J S Afr Vet Assoc. (2021) 92:e1–e11. doi: 10.4102/jsava.v92i0.2117
18. Bronsvoort BM, Koterwas B, Land F, Handel IG, Tucker J, Morgan KL, et al. Comparison of a flow assay for brucellosis antibodies with the reference cELISA test in West African Bos indicus. PLoS ONE. (2009) 4:e5221. doi: 10.1371/journal.pone.0005221
19. Madut NA, Muwonge A, Nasinyama GW, Muma JB, Godfroid J, Jubara AS, et al. The sero-prevalence of brucellosis in cattle and their herders in Bahr el Ghazal region, South Sudan. PLoS Negl Trop Dis. (2018) 12:e0006456. doi: 10.1371/journal.pntd.0006456
20. Sanogo M, Abatih E, Thys E, Fretin D, Berkvens D, Saegerman C. Importance of identification and typing of Brucellae from West African cattle: a review. Vet Microbiol. (2013) 164:202–11. doi: 10.1016/j.vetmic.2013.02.009
21. Scholz HC, Vergnaud G. Molecular characterisation of Brucella species. Rev Sci Tech. (2013) 32:149–62. doi: 10.20506/rst.32.1.2189
22. Liu ZG, Wang M, Zhao HY, Piao DR, Jiang H, Li ZJ. Investigation of the molecular characteristics of Brucella isolates from Guangxi Province, China. BMC Microbiol. (2019) 19:292. doi: 10.1186/s12866-019-1665-6
23. Kydyshov K, Usenbaev N, Berdiev S, Dzhaparova A, Abidova A, Kebekbaeva N, et al. First record of the human infection of Brucella melitensis in Kyrgyzstan: evidence from whole-genome sequencing-based analysis. Infect Dis Poverty. (2022) 11:120. doi: 10.1186/s40249-022-01044-1
24. Zhu X, Zhao Z, Ma S, Guo Z, Wang M, Li Z, et al. Brucella melitensis, a latent “travel bacterium,” continual spread and expansion from Northern to Southern China and its relationship to worldwide lineages. Emerg Microbes Infect. (2020) 9:1618–27. doi: 10.1080/22221751.2020.1788995
25. Pelerito A, Nunes A, Grilo T, Isidro J, Silva C, Ferreira AC, et al. Genetic Characterization of Brucella spp.: whole genome sequencing-based approach for the determination of multiple locus variable number tandem repeat profiles. Front Microbiol. (2021) 12:740068. doi: 10.3389/fmicb.2021.740068
26. Le Flèche P, Jacques I, Grayon M, Al Dahouk S, Bouchon P, Denoeud F, et al. Evaluation and selection of tandem repeat loci for a Brucella MLVA typing assay. BMC Microbiol. (2006) 6:9. doi: 10.1186/1471-2180-6-9
27. Armstrong GL, MacCannell DR, Taylor J, Carleton HA, Neuhaus EB, Bradbury RS, et al. Pathogen genomics in public health. N Engl J Med. (2019) 381:2569–80. doi: 10.1056/NEJMsr1813907
28. Ferdinand AS, Kelaher M, Lane CR, da Silva AG, Sherry NL, Ballard SA, et al. An implementation science approach to evaluating pathogen whole genome sequencing in public health. Genome Med. (2021) 13:121. doi: 10.1186/s13073-021-00934-7
29. Sanogo M, Fretin D, Thys E, Saegerman C. Exploring the diversity of field strains of Brucella abortus biovar 3 isolated in West Africa. Front Microbiol. (2017) 8:1232. doi: 10.3389/fmicb.2017.01232
30. Sayour AE, Elbauomy E, Abdel-Hamid NH, Mahrous A, Carychao D, Cooley MB, et al. Fingerprinting of Brucella melitensis circulating among livestock and cases of sporadic human illness in Egypt. Transbound Emerg Dis. (2020) 67:2435–45. doi: 10.1111/tbed.13581
31. Ledwaba MB, Gomo C, Lekota KE, Le Flèche P, Hassim A, Vergnaud G, et al. Molecular characterization of Brucella species from Zimbabwe. PLoS Negl Trop Dis. (2019) 13:e0007311. doi: 10.1371/journal.pntd.0007311
32. Holzer K, Wareth G, El-Diasty M, Abdel-Hamid NH, Hamdy MER, Moustafa SA, et al. Tracking the distribution, genetic diversity and lineage of Brucella melitensis recovered from humans and animals in Egypt based on core-genome SNP analysis and in silico MLVA-16. Transbound Emerg Dis. (2022) 69:3952–63. doi: 10.1111/tbed.14768
33. Ledwaba MB, Glover BA, Matle I, Profiti G, Martelli PL, Casadio R, et al. Whole genome sequence analysis of Brucella abortus isolates from various regions of South Africa. Microorganisms. (2021) 9:e570. doi: 10.3390/microorganisms9030570
34. Nascimento M, Sousa A, Ramirez M, Francisco AP, Carriço JA, Vaz C, et al. 2.0: providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics. (2016) 33:128–9. doi: 10.1093/bioinformatics/btw582
35. Al Dahouk S, Flèche PL, Nöckler K, Jacques I, Grayon M, Scholz HC, et al. Evaluation of Brucella MLVA typing for human brucellosis. J Microbiol Methods. (2007) 69:137–45. doi: 10.1016/j.mimet.2006.12.015
36. Li Z, Wang XM, Zhu X, Wang M, Cheng H, Li D, et al. Molecular characteristics of Brucella isolates collected from humans in Hainan Province, China. Front Microbiol. (2020) 11:452. doi: 10.3389/fmicb.2020.00452
37. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. (2004) 5:R12. doi: 10.1186/gb-2004-5-2-r12
38. Chiaromonte F, Yap VB, Miller W. Scoring pairwise genomic sequence alignments. Pac Symp Biocomput. (2002) 2002:115–26. doi: 10.1142/9789812799623_0012
39. Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. (2009) 19:327–35. doi: 10.1101/gr.073585.107
40. Simpson G, Thompson PN, Saegerman C, Marcotty T, Letesson JJ, de Bolle X, et al. Brucellosis in wildlife in Africa: a systematic review and meta-analysis. Sci Rep. (2021) 11:5960. doi: 10.1038/s41598-021-85441-w
41. Verger JM, Grayon M. Characteristics of 273 strains of Brucella abortus of African origin. Dev Biol Stand. (1984) 56:63–71.
42. Akoko JM, Pelle R, Lukambagire AS, Machuka EM, Nthiwa D, Mathew C, et al. Molecular epidemiology of Brucella species in mixed livestock-human ecosystems in Kenya. Sci Rep. (2021) 11:8881. doi: 10.1038/s41598-021-88327-z
43. D'Anastasio R, Zipfel B, Moggi-Cecchi J, Stanyon R, Capasso L. Possible brucellosis in an early hominin skeleton from sterkfontein, South Africa. PLoS ONE. (2009) 4:e6439. doi: 10.1371/journal.pone.0006439
44. Mathew C, Stokstad M, Johansen TB, Klevar S, Mdegela RH, Mwamengele G, et al. First isolation, identification, phenotypic and genotypic characterization of Brucella abortus biovar 3 from dairy cattle in Tanzania. BMC Vet Res. (2015) 11:156. doi: 10.1186/s12917-015-0476-8
45. Bertu WJ, Ducrotoy MJ, Muñoz PM, Mick V, Zúñiga-Ripa A, Bryssinckx W, et al. Phenotypic and genotypic characterization of Brucella strains isolated from autochthonous livestock reveals the dominance of B. abortus biovar 3a in Nigeria. Vet Microbiol. (2015) 180:103–8. doi: 10.1016/j.vetmic.2015.08.014
46. Matle I, Ledwaba B, Madiba K, Makhado L, Jambwa K, Ntushelo N. Characterisation of Brucella species and biovars in South Africa between 2008 and 2018 using laboratory diagnostic data. Vet Med Sci. (2020) 7:1245–53. doi: 10.1002/vms3.483
47. Alausa OK. The investigation and control of a large-scale community outbreak of brucellosis in Nigeria. Public Health. (1979) 93:185–93. doi: 10.1016/S0033-3506(79)80124-9
49. Kardjadj M. Epidemiological situation of transboundary animal diseases in North African countries-proposition of a regional control strategy. Trop Anim Health Prod. (2018) 50:459–67. doi: 10.1007/s11250-017-1453-y
50. Godfroid J, Cloeckaert A, Liautard JP, Kohler S, Fretin D, Walravens K, et al. From the discovery of the Malta fever's agent to the discovery of a marine mammal reservoir, brucellosis has continuously been a re-emerging zoonosis. Vet Res. (2005) 36:313–26. doi: 10.1051/vetres:2005003
51. Maurin M. Brucellosis at the dawn of the 21st century. Med Mal Infect. (2005) 35:6–16. doi: 10.1016/j.medmal.2004.08.003
52. Muendo EN, Mbatha PM, Macharia J, Abdoel TH, Janszen PV, Pastoor R, et al. Infection of cattle in Kenya with Brucella abortus biovar 3 and Brucella melitensis biovar 1 genotypes. Trop Anim Health Prod. (2012) 44:17–20. doi: 10.1007/s11250-011-9899-9
53. Vergnaud G, Hauck Y, Christiany D, Daoud B, Pourcel C, Jacques I, et al. Genotypic expansion within the population structure of classical brucella species revealed by MLVA16 typing of 1404 brucella isolates from different animal and geographic origins, 1974–2006. Front Microbiol. (2018) 9:1545. doi: 10.3389/fmicb.2018.01545
54. Minharro S, Silva Mol JP, Dorneles EM, Pauletti RB, Neubauer H, Melzer F, et al. Biotyping and genotyping (MLVA16) of Brucella abortus isolated from cattle in Brazil, 1977 to 2008. PLoS ONE. (2013) 8:e81152. doi: 10.1371/journal.pone.0081152
55. Her M, Kang SI, Cho DH, Cho YS, Hwang IY, Heo YR, et al. Application and evaluation of the MLVA typing assay for the Brucella abortus strains isolated in Korea. BMC Microbiol. (2009) 9:230. doi: 10.1186/1471-2180-9-230
56. Ferreira AC, Chambel L, Tenreiro T, Cardoso R, Flor L, Dias IT, et al. MLVA16 typing of Portuguese human and animal Brucella melitensis and Brucella abortus isolates. PLoS ONE. (2012) 7:e42514. doi: 10.1371/journal.pone.0042514
57. Wareth G, El-Diasty M, Melzer F, Schmoock G, Moustafa SA, El-Beskawy M, et al. MLVA-16 genotyping of Brucella abortus and Brucella melitensis isolates from different animal species in Egypt: geographical relatedness and the Mediterranean Lineage. Pathogens. (2020) 9:498. doi: 10.3390/pathogens9060498
58. Liu Z, Wang C, Wei K, Zhao Z, Wang M, Li D, et al. Investigation of genetic relatedness of brucella strains in countries along the silk road. Front Vet Sci. (2020) 7:539444. doi: 10.3389/fvets.2020.539444
59. Khames M, Mick V, de Miguel MJ, Girault G, Conde-Álvarez R, Khelef D, et al. The characterization of Brucella strains isolated from cattle in Algeria reveals the existence of a B. abortus lineage distinct from European and Sub-Saharan Africa strains. Vet Microbiol. (2017) 211:124–8. doi: 10.1016/j.vetmic.2017.10.008
Keywords: Brucella abortus, species/biovars, genetic diversity, MLST, MLVA, WGS-SNP
Citation: Liu Z, Wang M, Shi Q, Dong X, Gao L and Li Z (2023) Original and introduced lineages co-driving the persistence of Brucella abortus circulating in West Africa. Front. Public Health 11:1106361. doi: 10.3389/fpubh.2023.1106361
Received: 23 November 2022; Accepted: 20 February 2023;
Published: 15 March 2023.
Edited by:
Shuchao Wang, Changchun Veterinary Research Institute (CAAS), ChinaReviewed by:
Henriette Van Heerden, University of Pretoria, South AfricaMaryam Dadar, Razi Vaccine and Serum Research Institute, Iran
Copyright © 2023 Liu, Wang, Shi, Dong, Gao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liping Gao, Z2FvbHBAaXZkYy5jaGluYWNkYy5jbg==; Zhenjun Li, emhlbmp1bmxpQGljZGMuY24=
†These authors have contributed equally to this work