Abstract
Introduction:
The Delta variant posed an increased risk to global public health and rapidly replaced the pre-existent variants worldwide. In this study, the genetic diversity and the spatio-temporal dynamics of 662 SARS-CoV2 genomes obtained during the Delta wave across Tunisia were investigated.
Methods:
Viral whole genome and partial S-segment sequencing was performed using Illumina and Sanger platforms, respectively and lineage assignemnt was assessed using Pangolin version 1.2.4 and scorpio version 3.4.X. Phylogenetic and phylogeographic analyses were achieved using IQ-Tree and Beast programs.
Results:
The age distribution of the infected cases showed a large peak between 25 to 50 years. Twelve Delta sub-lineages were detected nation-wide with AY.122 being the predominant variant representing 94.6% of sequences. AY.122 sequences were highly related and shared the amino-acid change ORF1a:A498V, the synonymous mutations 2746T>C, 3037C>T, 8986C>T, 11332A>G in ORF1a and 23683C>T in the S gene with respect to the Wuhan reference genome (NC_045512.2). Spatio-temporal analysis indicates that the larger cities of Nabeul, Tunis and Kairouan constituted epicenters for the AY.122 sub-lineage and subsequent dispersion to the rest of the country.
Discussion:
This study adds more knowledge about the Delta variant and sub-variants distribution worldwide by documenting genomic and epidemiological data from Tunisia, a North African region. Such results may be helpful to the understanding of future COVID-19 waves and variants.
Introduction
Since December 2019, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2) emerged and spread rapidly all around the world, causing a global health, social and economic crises (1–3). The World Health Organization (WHO), all international expert networks and national public health authority institutions have been mobilized to monitor the evolution of SARS-CoV-2 and track emergent variants and sub-variants. Variant surveillance is based on genetic characterization and rapid sharing of viral genomic sequences, in order to limit their spread and control the pandemic (4). SARS-CoV-2 variants are classified as Variants Of Concern (VOCs), Variants Of Interest (VOIs) and Variants Under Monitoring (VUMs) (5). VOCs are associated to increased transmissibility and/or virulence, and/or to reduced efficacy of diagnostic tests, vaccines or treatments. VOIs are associated with significant community transmission with multiple foci in multiple countries and/or increasing prevalence. VUMs present unclear phenotypic or epidemiological repercussions requiring enhanced surveillance.
Among VOCs, the Delta variant posed an increased risk to the global public health and rapidly displaced the then dominant Alpha variant globally and changed the epidemiological infection landscape of this virus during 2021 (6). Delta variant is the common name used to refer to the B.1.617.2 Pango-lineage (7). In late 2020/early 2021, the parental strain B.1.617 emerged and rapidly spread to at least 60 countries (8–10). The first detected sub-lineage was B.1.617.1, also named Kappa variant, followed by B.1.617.2, the Delta variant, and B.1.617.3, the Epsilon variant (7). The Delta variant is one of the most contagious variants of the SARS-CoV-2. First detected on March 01 2021, it spread rapidly to almost all countries of the world, replacing the Alpha variant which was predominating globally (7, 11). Delta harbors mutations along the genome; among those in the gene encoding the spike protein, the substitutions T19R and T478K as well as the deletion E156-F157 may enhance antibodies' evasion causing escape from the host immune response (11, 12) and L452R, P681R may help cell entry by enforcing attachment to the ACE2 receptor and membrane fusion (13–15). Several studies demonstrated increased replication fitness (6) and reduced sensitivity to neutralizing antibodies from past infection (14) which contributed to the deadly epidemic wave in India during the first quarter of 2021, as well as the epidemic waves in England, South Africa and then almost all countries in the world (16–20). Up to February 2022, Delta continued to circulate globally, even after the emergence and spread of Omicron variant, infecting especially unvaccinated populations or in-complete vaccinated populations, including children. Furthermore, an important variability of this variant was reported in different areas of the world. Up to 2022-11-03, Delta variant has been shown to circulate into 245 different sub-lineages: AY.1-AY.134 (8).
At the beginning of COVID-19 pandemic, Tunisia adopted a very stringent lockdown regime from 10th of March to 3rd of May 2020 which helped contain the virus spread. Since July 2020, the country experienced several waves of COVID-19. The first two ones, in 2020 and up to January 2021, were caused by several SARS-CoV2 lineages (21–23). The first detected VOC was the Alpha variant that induced an extensive wave from March to June 2021. In May 2021, Delta was detected for the first time and started replacing the Alpha variant, causing a substantial increase in the number of cases with high transmission rates within the community (23). It became rapidly the dominant variant in the country with great impact on public health and healthcare systems, increasing the number of hospitlaisation requirement for oxygen. By the end of December 2021, a slight decline of COVID-19 incidence was notified but, since January 2022, the number of cases increased again causing another wave mainly induced by the Omicron variant.
This study reported demographic and genetic data of 662 SARS-CoV2 Tunisian sequences belonging to the Delta variant, obtained from oropharyngeal swabs collected from May to December 2021, during the Delta wave. The genetic diversity and the spatio-temporal dynamics of virus transmission in the country are described.
Materials and methods
Ethical statement
This study was performed under ethical standards according to the 1964 Declaration of Helsinki and its later amendments. The samples were collected in the context of COVID-19 diagnostic activities. They were used in the present study after de-identification with respect to patient anonymity and after the approval of the Bio-Medical Ethics Committee of Pasteur Institute of Tunis, Tunisia (2020/14/I/LR16IPT/V1). Written informed consent to participate in this study was provided by the participants ‘legal guardian. No animal studies are presented in this manuscript. No potentially identifiable human images or data is presented in this study.
Tunisian sequences
A total of 662 SARS-CoV-2 sequence, obtained from positive nasopharyngeal samples and lately identified as belonging to the Delta variant, were included in the present work. They were obtained from May, 26 to December 1st, 2021, during the Delta wave in Tunisia (Figure 1). The samples were collected by the Tunisian Ministry of Health personnel (MoH's personnel). They included samples collected from home-quarantined and hospitalized individuals. The collected samples were transported, under refrigeration and within 24 h, to the Pasteur Institute of Tunis where they were immediately processed for SARS-CoV-2 RNA detection by specific real-time reverse transcription PCRs (RT-qPCR) according to WHO-approved protocols (24, 25). Samples were tested by Charité, Berlin protocol [singleplex envelop (E) and singleplex RNA-dependent RNA polymerase (RdRp)] (24), Hong Kong Universiy, China protocol (singleplex nucleoprotein (N) and singleplex Open reading frame Orf1b) (25), according to the reagents availability, in the pandemic context. SARS-CoV-2 variant identification was assessed using a combination of next-generation sequencing-based approaches (N = 484 samples) and by partial sequencing of the S gene (N = 178 samples).
Figure 1
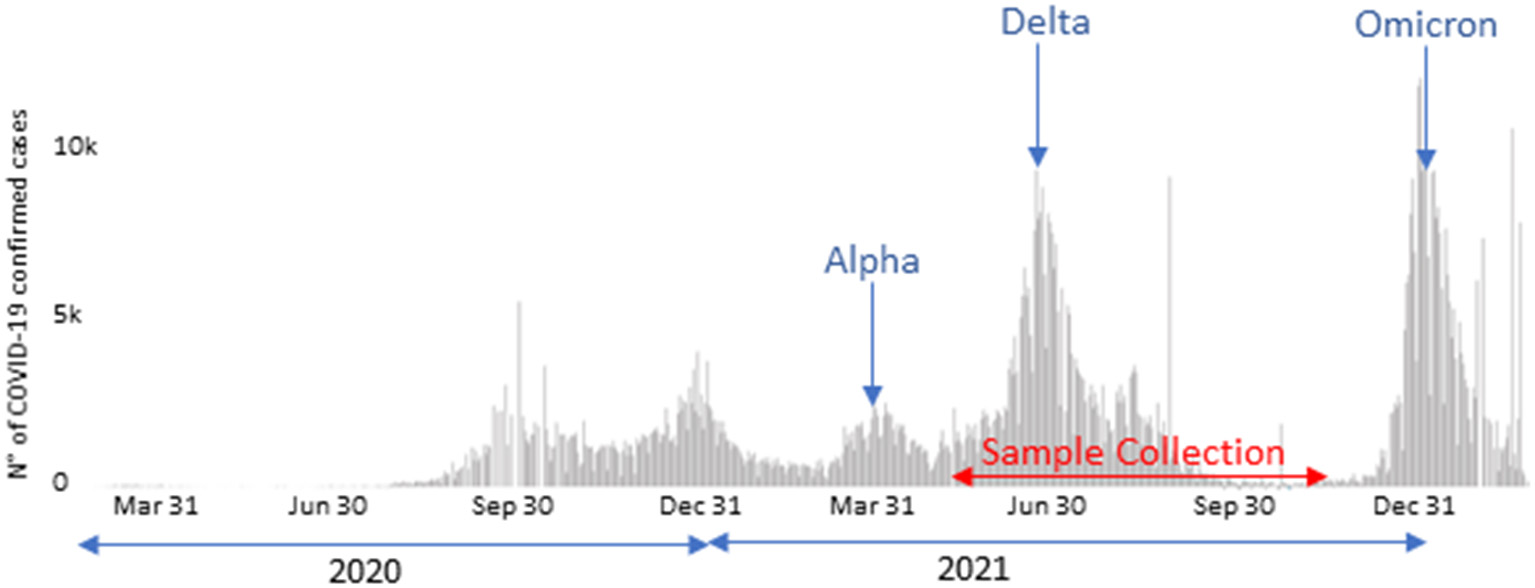
COVID-19 confirmed case counts in Tunisia starting from March 2020 (https://covid19.who.int/region/emro/country/tn, accessed by 18/03/2022) and sample collection period.
PCR amplification and partial sequencing in the S gene
PCR amplification of S gene was performed as previously described (22) with minor modifications, using new designed primer's pair RBF_FW & IPT_REV, that allowed the amplification of a 1,112-nucleotides segment. Briefly, after viral RNA extraction using the QIAamp MinElute Virus Spin Kit (Qiagen, GmbH, Hilden, Germany), the amplification used the primer's pair RBF_FW: 5'ATTACAAACTTGTGCCCTTTT3' (positions 22535 to 22555 according to the SARS-CoV2 reference genome, NC_045512.2) and IPT_REV: 5′CTGCACCAAGTGACATAGTG3′ (positions 23666 to 23647) and the SuperScript III one-step RT-PCR system with the Platinum Taq DNA polymerase kit (Invitrogen). The S amplicons were purified using the ExoSAP-IT method (Invitrogen). Sequences were generated by mean of Big Dye terminator chemistry according to the manufacturer's recommendations (BigDye Terminator v3.1 kit, Applied Biosystems) and using the automated sequencer ABI 3130). The consensus sequences of approximately 1,071 nucleotides were deduced using Clustal X version 2.0 (http://www.clustal.org/) (26).
Whole genome sequencing
The sequencing of complete SARS-CoV2 viral genome was achieved using NGS technologies in 3 collaborating institutes: 396 at the Quadram Institute (Norwich, United Kingdom) on behalf of the UKHSA New Variant Assessment Platform, 50 at the Istituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise (Teramo, Italy) (IZS-Te) and 38 at Institut Pasteur de Dakar (Senegal). The three sequencing sites used almost equivalent sequencing protocols as previously described (22). RNA was extracted from a 140 μL nasopharyngeal sample with the Qiamp viral RNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The extracted RNAs were reverse transcribed to single-strand cDNA. The whole genome sequencing was assessed using the Illumina COVIDSeq Test, including reagents for cDNA amplification (CPP1 HT and the CCP2 HT primers), library preparation and Purification (Illumina Tune Bead) (Illumina Technology, USA). The quantification of amplification products was assessed by the Qubit 2.0 fluorometer (ThermoFisher Scientific, USA) and qualified on an Agilent Technologies 2100 Bioanalyser using a high-sensitivity DNA chip following the manufacturer's instructions. Generated libraries were pooled, denatured, diluted to 1.4 pM and sequenced using NextSeq 500/550 High output Kit version 2.5 (Illumina, Inc, USA) on a NextSeq500 or NextSeq550 instrument (Illumina, Inc, USA) providing 2 × 150 bp read length data.
The quality control of raw data was first checked using FastQC version 0.11.9 for (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Low-quality reads and adapters were filtered using Trimmomatic version 0.39 (27) with a Phred quality score of 30 as the threshold. Consensus sequences were generated by mapping to the SARS-CoV-2 reference genome (NC_045512) via Spades assembler version 3.15.0 (28), using thresholds of 80% for nucleotide sequence coverage and 90% for nucleotide similarity. The sequences were submitted to the GISAID database (https://www.gisaid.org) (29, 30).
Variant assignment and single nucleotide polymorphisms identification
The Fasta files of the whole sequences were uploaded into Pangolin online software version 3.1.17 (https://cov-lineages.org/pangolin.html; accessed on 01/16/2022) for variant and sub-lineage assignment (31). Sub-lineages within the Delta variant could be assigned for those having a coverage of more than 85% of the virus genome. The web-based software Nextclade v1.12.0 (https://clades.nextstrain.org/) was used for clade assignment, diversity and gene-wise amino acid mutations analysis (32). Maximum-likelihood trees were generated by the use of IQ-TREE multicore program version 1.6.12 (33).
For the 178 partial sequences and the NGS sequences with incomplete coverage of the whole genome, variant determination was based on the identification of key mutations specific to the Delta variant along the partial and complete S gene, respectively.
Mutational cascade analysis
Mutational cascade was analyzed for AY.122 variant of SARS-CoV-2 circulating in Tunisia using NUCmer pipeline (version 4.0.0) and the R script detailed by Bansal and Kumar (34). The SNP analysis was conducted by NUCmer v.4.0.0 using genome of first reported AY.122 variant (OV103802.1). This reference genome was retrieved from NCBI Genbank (https://www.ncbi.nlm.nih.gov/nuccore/OV103802.1/) and chosen for no N character in the nucleotide sequence as selection criteria. The SNP file generated by NUCmer v.4.0.0, the R script was launched to identify: most mutated samples, number of mutations per sample, mutation classes, and most frequent mutational events per type. In this analysis, a gff3 annotation file of the Wuhan reference genome (NC_045512.2) was used to extract coordinates of the proteins encoded by SARS-CoV-2 genome.
Phylogenetic analysis
Viral phylogeny was inferred on Tunisian generated sequences and others originating from different areas of the world, available at the NCBI GenBank (https://www.ncbi.nlm.nih.gov/labs/virus). Multiple alignment was first inferred by Mafft (V7.470) online platform (https://mafft.cbrc.jp/alignment/software/), using default parameters (35). A Maximum-likelihood tree was generated by IQ-TREE multicore program version 1.6.12, with a bootstrap replication of 1000 cycles (33, 36). The Wuhan reference genome (NC_045512.2) was used for rooting. The visualization of the obtained tree was assessed by the graphical viewer program, FigTree v1.4.4 (http://tree.bio.ed.ac.uk/) (37).
Phylogeographic analysis
To explore spatio-temporal distribution of investigated strains, the temporal signal was undertaken using TempEST v1.5.4 (38). Bayesian phylogeographic analysis used BEAST v1.8.4 (39), the discrete diffusion model approach was applied (40). The GTR (I+Γ4), strict clock and exponential growth models are applied as the best-fit models. To estimate the transition rates across regions, the Bayesian Stochastic Search Variable Selection (BSSVS) approach was inferred with application of the symmetric substitution model. A 200-million iteration was run, achieving a satisfying ESS value (ESS>200). By discarding 10% of burn-in, the time-scaled maximum clade credibility (MCC) tree was generated with Tree Annotator v1.8.4 (40) and visualized with FigTree v1.4.3 (37) programs. To evaluate the robustness of virus transitions between geographic locations, the Bayes factors (BF) were calculated by using the SpreaD3 program version 1.0.7 (41). A transition rate is considered as statistically valid when BF>3. The spatiotemporal dynamics of strains was studied by generating a KML file with the SpreaD3 program and then visualized via Google Earth Pro.
Data availability
Complete genome SARS-CoV-2 sequences generated in this study were submitted to the GISAID database (https://www.gisaid.org) (29, 30). Their accession numbers are provided in Supplementary Table S1.
Results
Epidemiological features of collected samples
The Delta sequences included in this study were obtained from individuals originating from 23 of the 24 Tunisian governorates. They were collected from symptomatic patients with a wide range of clinical presentations, as well as asymptomatic individuals sampled after contact with confirmed cases. The study population included 303 males and 359 females; their ages ranged from 1 month to 90 years. Figure 2 displays the distribution of SARS-CoV2 sequences according to the age. More than half of the sequences (52.6%) were obtained from individuals aged 25 to 50 years, with statistical significant difference (P = 0.0004 <0.001), obtained by student's t-test. An important proportion is also found among children and teenagers (20.0%) as well as seniors aged 50–65 (18.4%). Only 9.0% of sequences were obtained from individuals aged 65 years and more.
Figure 2
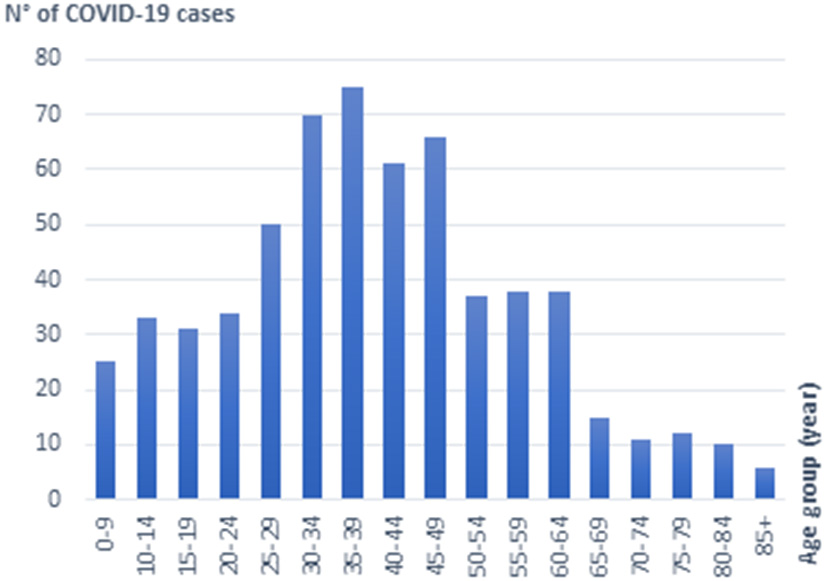
Distribution of Delta SARS-CoV2 sequences according to the age.
Sequence analysis and variant assignement
Among the 484 whole genome sequences, variant and sub-variant could be assigned for 468 sequences displaying <15% ambiguous nucleotides.
Using Nextclade program, the 468 complete sequences belonged to clades 21J, 21I and 21A which correspond to the Delta variant. The majority of sequences constitute a group belonging to the 21J clade and other sporadic strains grouped within the 21A, 21I clades (Supplementary Figure S1). Similar group profile was obtained with the Maximum Likelihood (ML) tree obtained by Iqtree software (Supplementary Figure S2).
Using Pangolin program, the 468 analyzed sequences were classified into 12 different Delta sub-lineages: AY.122, B.1.617.2, AY.4, AY.43, AY.70, AY.127, AY.9.2, AY.52, AY.5.4, AY.36, AY.34, AY.119. The AY.122 sub-lineage was the most frequently detected (94.6%, n = 443) followed by the B1.617.2 (1.4%, n = 7) and the AY.4 (1.0%, n = 5) sub-lineages (Figure 3A). For the other minor sub-lineages, the number of sequences ranged from 1 to 3 each.
Figure 3
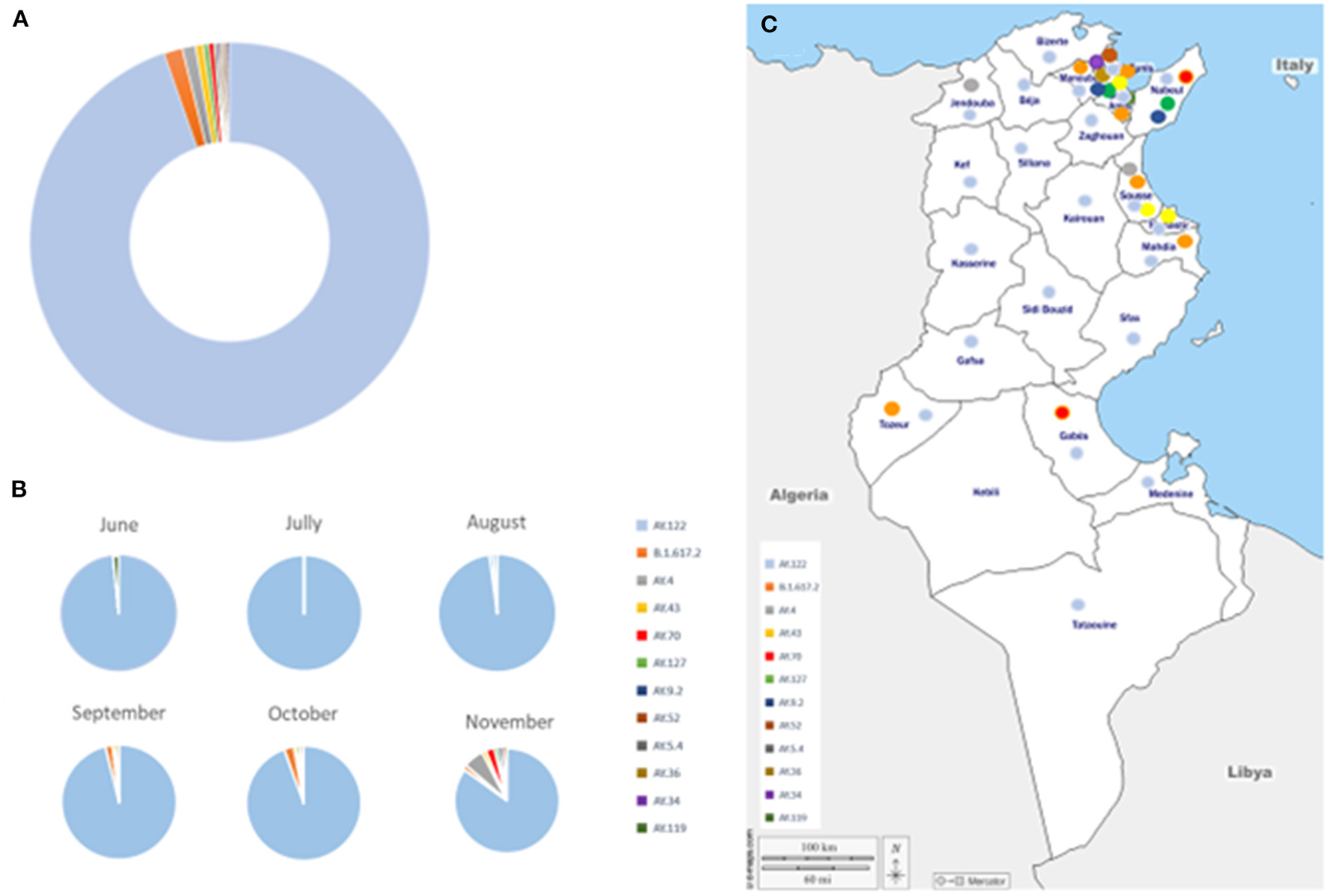
Distribution of Delta sub-lineages, during the study period. (A) Global distribution. (B) Temporal distribution. (C) Geographic distribution of Delta sub-lineages, during the study period. Delta sub-lineages are indicated by colored circles, as indicated in the legend.
The AY.122 sub-lineage was detected since May and circulated during the whole study period, up to December 2021 while the frequency of detection of the other sub-lineages increased since August 2021 and was the highest in November 2021 (Figure 3B). Four, five and eight different sub-lineages were detected in September, October and November 2021, respectively. AY.122 was detected in all regions of the country while the other sub-lineages were mainly detected in the North-east-coastal regions (Figure 3C).
Mutational cascade
For more investigation about evolution trait of emergent AY.122 variant (n = 165) in Tunisia, we constructed the mutational cascade based on comparison with the first reported AY.122 strain (Figure 4). According to the mutational analysis, the most mutant strains are: hCoV-19_TUN_Nab_S1343_27102021 and hCoV-19_TUN_Nab_S1349_02112021, where they accumulated more than 30 mutations in their genomes (Figure 4A). Figure 4B showed that most genomes accumulated 20 to 26 mutations during AY.122 variant emergence in Tunisia. According to the Figure 4C, the single nucleotide polymorphism (SNP) is the most prevalent mutation class among the AY.122 variant circulating in Tunisia. The mutational analysis also showed that C>T and A>G transitions are the most frequent mutation events (Figure 4D).
Figure 4
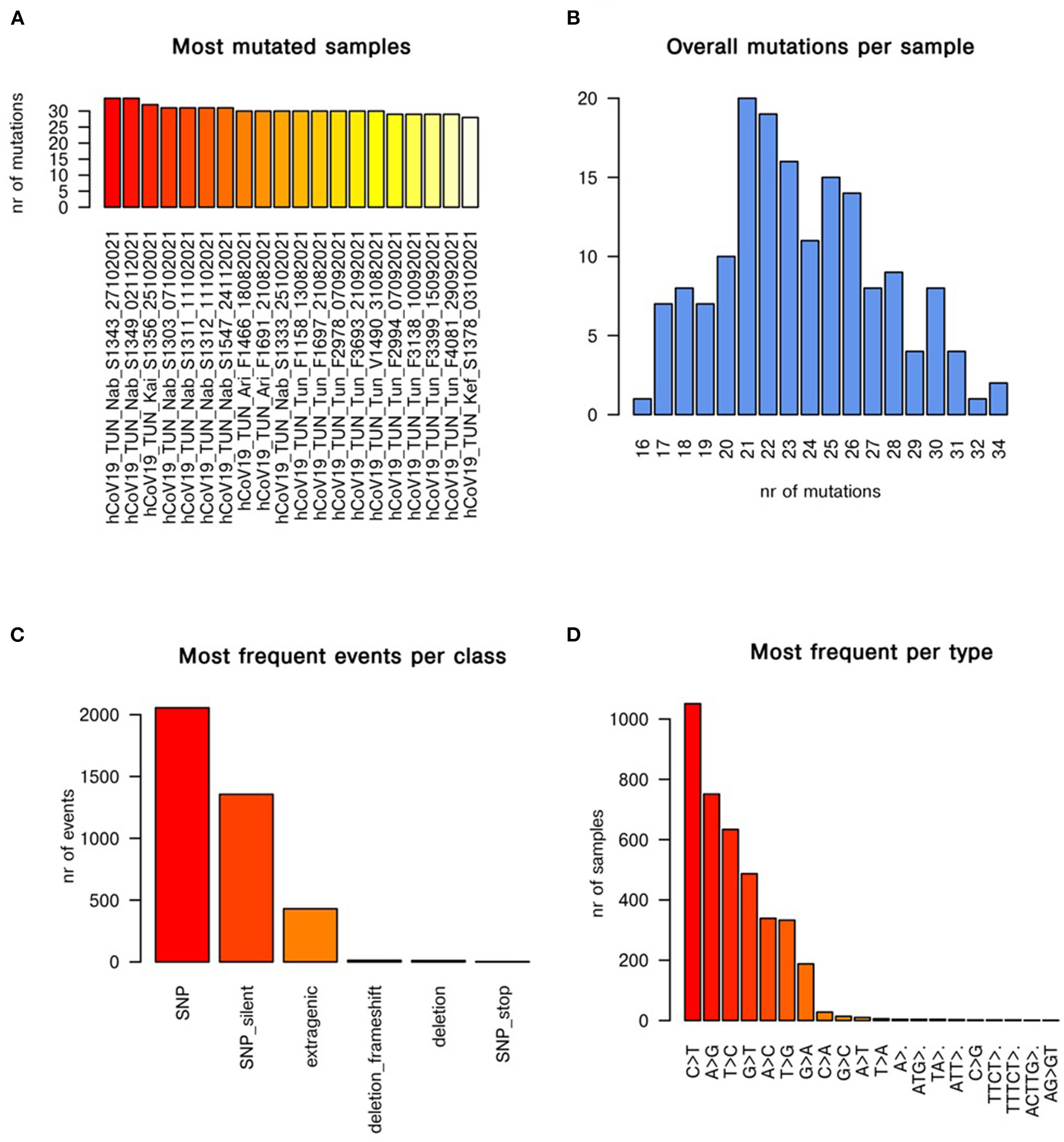
Mutational cascade of AY.122 variant emerged in Tunisia with respect to first reported AY.122 strain. The panels (A–D) show the most mutated samples, mutations per sample, most frequent events per class of mutation, most frequent mutational events per type, respectively.
Phylogenetic analysis
The phylogenetic analysis was performed on the AY.122 complete genome sequences, representing the most frequently detected Delta sub-lineage. The analysis involved a total of 391 AY.122 virus genomes including the Wuhan reference sequence as outgroup, 165 Tunisian sequences with complete genome coverage and 225 sequences from different geographic regions of the world. Indeed, to obtain reliable results, only complete sequences without ambiguous nucleotides (N) into the coding regions were considered. The 165 AY.122 complete genome Tunisian sequences originated from 17 different governorates and during the whole study period. Among the AY.122 complete genome sequences from other countries, available on Genbank, up to10 complete sequences per country and per month were first selected (n = 780) and then only those with without ambiguous nucleotides into the coding regions (n = 225) were retained. They included sequences for United Kingdom (n = 43), Germany (n = 38), United States of America (n = 29), India (n = 24), Egypt (n = 18), Bahrain (n = 14), Slovakia (n = 11), Italy (n = 10), Nigeria (n = 10), China (n = 8), Austria (n = 6), France (n = 6), Liechtenstein (n = 4), Mongolia (n = 2), Russia (n = 1).
Figure 5 represents the Maximum-Likelihood tree generated using the GTR+F+R2 model (Best fit model according to BIC) with a bootstrap replication of 1000 cycles. The majority of Tunisian sequences (163 out of 165) clustered together, independently from sequences of other countries. They were grouped into three major clusters. Cluster1 included 17 Tunisian sequences collected in limited areas of Tunisia (Tunis and Kairouan); they formed two groups according to their geographic origin. Cluster2 included a large number of Tunisian sequences (n = 105) from different governorates. Sequences from same region were highly related and likely to form different groups. This cluster included few sequences from UK. The Cluster 3 combines 43 sequences from three Tunisian regions (the region of Tunis, Nabeul and South Tunisia) with few sequences from Germany, Switzerland and UK.
Figure 5
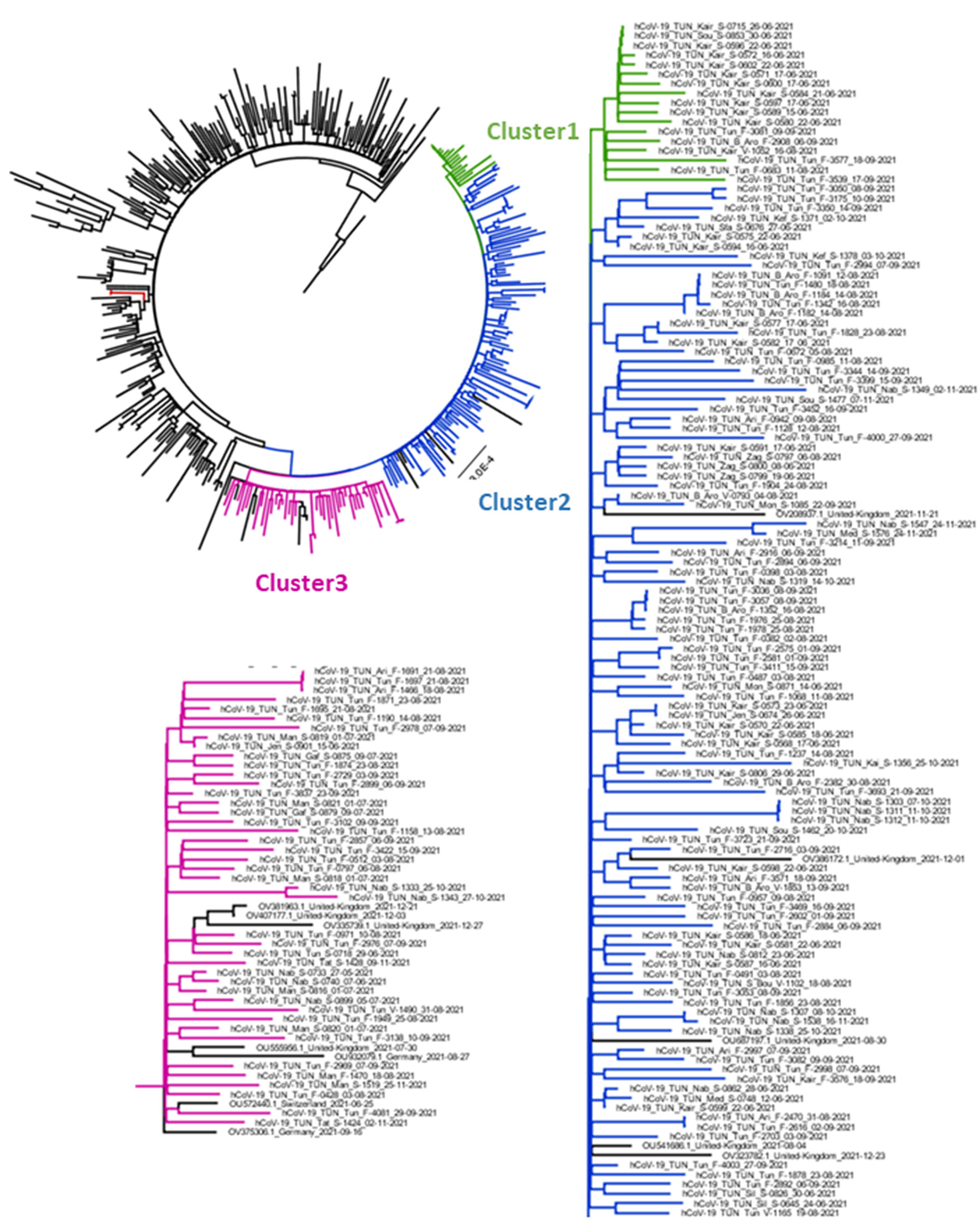
Phylogenetic analysis of AY.122 sub-lineage's Delta strains obtained in Tunisia between May and December. The Maximum Likelihood tree was obtained by comparison of 391 High quality AY.122 genomes obtained from Tunisia and other regions of the word. Tunisian sequences are indicated in Green (Cluster1), Blue (Cluster 2) and Pink (Cluster3) and other sequences different the world in Black. The tree was generated by IqTree software with 1000 boostrap cycles and visualized by FigTree software.
Figure 6 displays the mutational profile of the Tunisian sequences. The web-based software Nextclade v1.12.0 (https://clades.nextstrain.org/) was used for gene-wise amino acid mutations analysis (32). In addition to the amino-acid changes and mutations specific to the AY.122 sub-lineage, indicated in blue, all Tunisian sequences, from the three clusters, shared the amino-acid change A498V in ORF1a (indicated in dark purple), the synonymous mutations 2746T>C, 3037C>T, 8986C>T and 11332A>G in ORF1a and 23683C>T in the S gene (indicated in light purple). The same changes were also found with high frequency in all the 443 Tunisian AY.122 sequences: A498V was present in 441 sequences, 2746T>C, 3037C>T, 8986C>T, 11332A>G were found in 418, 441, 437 and 439 sequences, respectively and 23683C>T was found in 433AY.122 sequences.
Figure 6
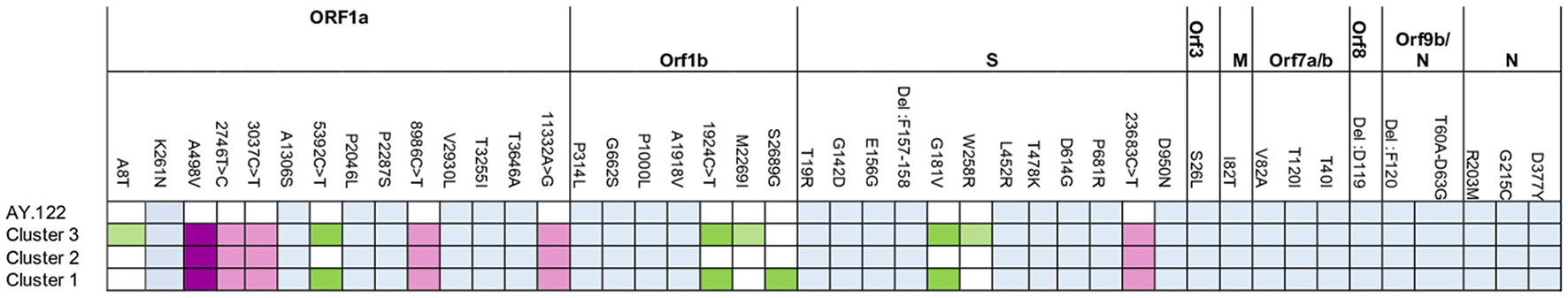
Mutational profile of Tunisian sequences from Cluster 1, 2 and 3 in comparison with the AY.122 sub-lineage.
In addition, the Tunisian sequences from Clusters 1 and 3 differ from the cluster 2 by the presence of the amino-acid changes S: G181V and the synonymous mutations 5392C>T, 19245C>T (indicated in Green). Also, strains from Cluster 3differ from the other by the presence of ORF1b: S2689G amino acid change.
Along the genome of Tunisian sequences, other amino-acid changes were present at important levels in the S gene: G142D (121/165 sequences), S: W258R (33/43 sequences from the Cluster3) and ORF1b gene M2269I (9/43sequences from the Cluster 3) and also ORF7a gene A8T (5/43 sequences from Cluster 3).
Spatio-temporal dynamics of AY.122 Tunisian sequences
From the data set composed of the 165 AY.122 Tunisian sequences, with no ambiguous nucleotides, an ML tree was constructed and used to explore the temporal signal of the selected dataset. The root-to-tip regression analysis shows a positive correlation (R2 = 0.37) between genetic distances and sampling dates (Supplementary Figure S3).
Molecular clock analysis shows that the TMRCA (Time to the Most Recent Common Ancestor) for the root of AY.122 variant in Tunisia was estimated to be around October 03th, 2020 [July 19th, 2020–January 02th, 2021], which could correspond to the date of its first emergence in the world (6).
To gain insight into spatio-temporal dynamics of the AY.122 variant dispersal in Tunisia, we performed a Bayesian analysis based on phylogeography framework reconstruction. In the time-scaled phylogeographic MCC (Maximum Clade Credibility) tree, we observed that most nodes and branches have colors assigning the inferred location of Tunis (Supplementary Figure S4). Furthermore, the root state posterior probability (RSPP) analysis shows that Tunis has the highest value (RSPP = 0.95). This finding indicates that Tunis might be responsible for the spread of the AY.122 Delta SARS-CoV-2 variant in the country.
The spatial estimates of the MCC tree were than mapped on Google Earth (Figure 7, Supplementary material 1). The generated configuration of spatial dynamics shows that the AY.122 dispersal in Tunisia started from Tunis (the capital) to Nabeul (North-east). The virus was transmitted then from Nabeul to Kairouan, in the center of the country. Soon after, the role of Nabeul as primary epicenter of AY.122 variant became clearer; transitions were observed transitions with Manouba (North-east) and then with Gafsa (central-west). In this period, the AY.122 infection zones started to expand across the country from Nabeul, Tunis and Kairouan. Subsequently, Tunis and Kairouan became secondary epicenters of intensified AY.122 outbreaks which affected more governorates in the North, in the Center, and the South.
Figure 7
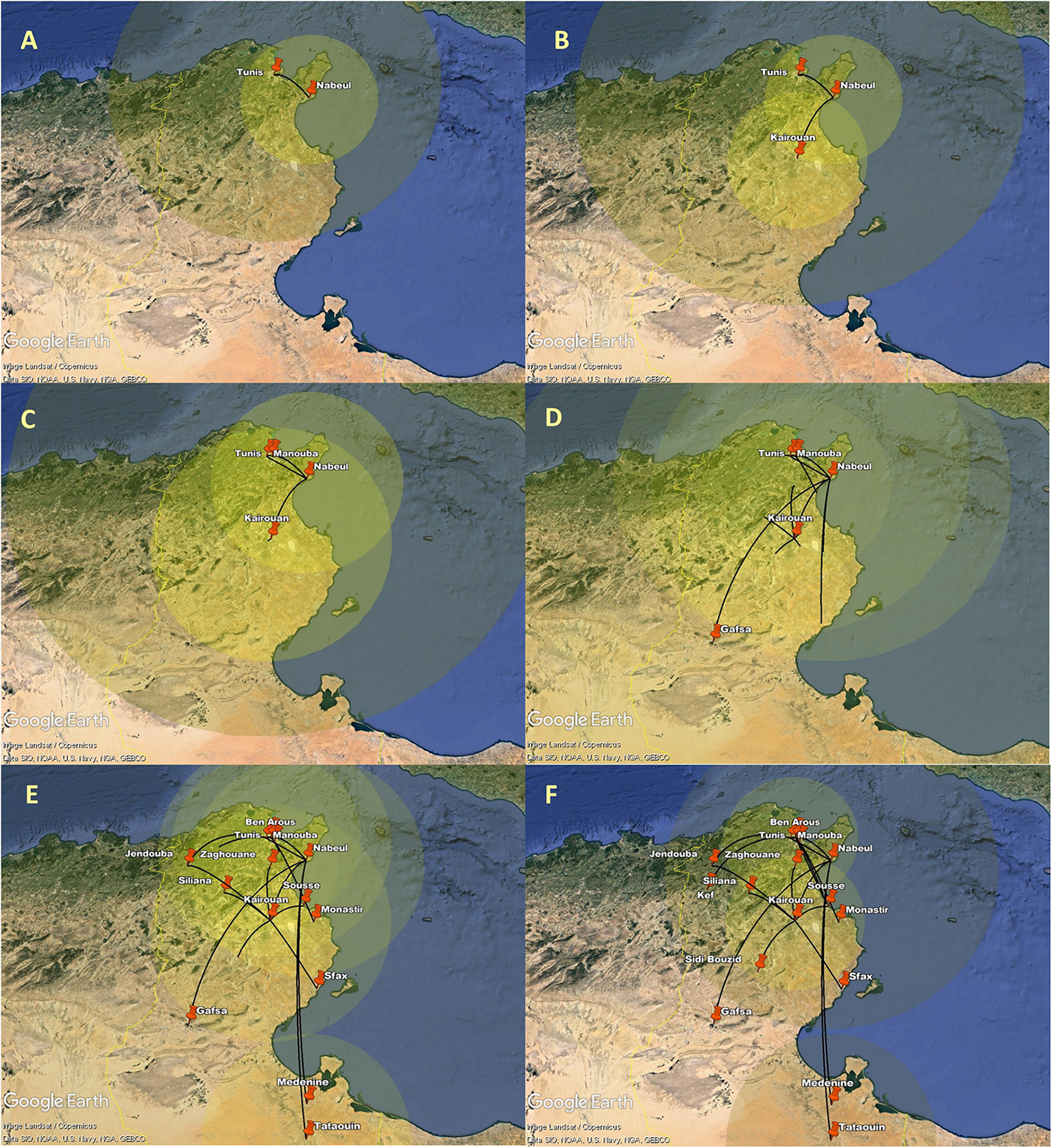
Spatial dynamics of AY.122 Delta variant of SARS-CoV-2 among different Tunisian governorates. The snapshots (A–F) show the different stages of AY.122 variant spread in Tunisian governorates. Transition lines linking different locations represent the branches in the MCC. Diameters of the circles are proportional to square root of the number of MCC branches maintaining a particular location state at each time point.
To provide statistical support for the phylodynamic reconstruction, we determined the Bayes Factor (BF) for the identified transitions between the different Tunisian governorates (Figure 8A, Supplementary Table S2). We found that all the transitions between the governorates are statistically supported (BF>3) except for the transition “Nabeul–Tataouine” (BF = 2.58). The highest Bayes Factor (BF>100) was recorded for the transitions linking: Tunis to Ariana (BF = 137653.83), Tunis to Nabeul (BF = 12132.57), Tunis to Ben Arous (BF = 1376535.83), Nabeul to Medenine (BF = 259.69), Kairouan to Siliana (BF = 201.58) and Kairouan to Jendouba (BF = 117.36).
Figure 8
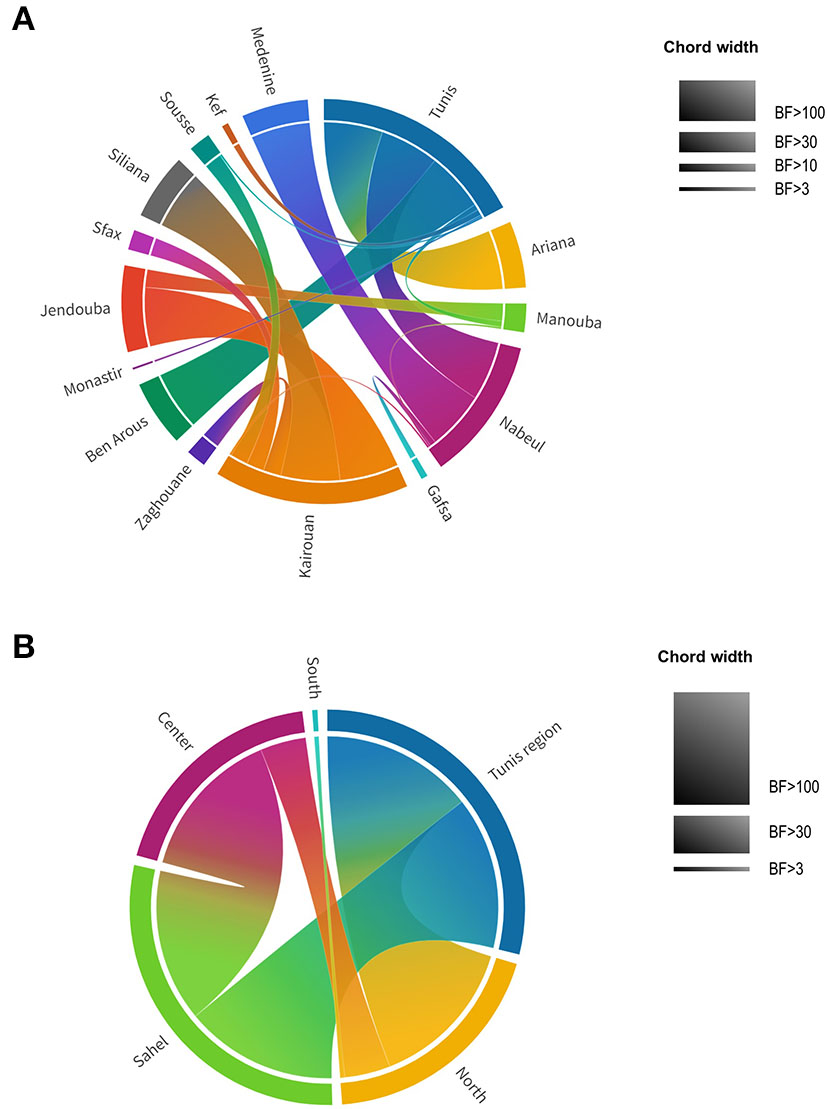
Chord diagram of theAY.122 SARS-CoV-2 variant representing Bayes Factor (BF) of transitions between different locations in Tunisia. (A) BF of transitions between Tunisian governorates. (B) BF of transitions between Tunisian regions. Tunisian governorates and regions are presented by different colors. Chord width is proportional to BF support levels: BF <3 no support, BF > 3 substantial support, BF > 10 strong support, BF > 30 very strong support, BF > 100 decisive support.
Higher decisive support of the Bayes Factor was found when governorates were assembled by regions (Figure 8B, Supplementary Table S3): Tunis region (Tunis, Manouba, Ariana, Ben Arous), North (Bizerte, Nabeul, Jendouba, Béjà, Zaghouan), Sahel (Sousse, Monastir, Mahdia, Sfax), Center (Siliana, Sidi Bouzid, Kasserine, Kairouan), South (Tataouin, Mednine, Kebili, Gafsa, Tozeur). For the region trait, all the Bayes Factors were superior to 3 indicating the statistical validation for the identified transitions. The transitions having a decisive support of Bayes Factor (BF > 100) are those of: “Tunis region–North” (BF = 294254.38), “Tunis region–Sahel” (BF = 104.44) and “Center–Sahel” (BF = 269.06).
Discussion
Since May 2021, the Delta variant dominated all the other SARS-CoV-2 variants and continued to largely shape the pandemic up to February 2022 (15). It overwhelmed health care systems, infecting mainly unvaccinated and non-fully vaccinated populations, especially in countries with limited access to vaccines (41, 42).
The Delta wave started in Tunisia, in May 2021. The present work shows that Delta infections occurred mainly in young adults. The age distribution of investigated cases showed a large peak between the ages of 25 to 50 years, but several cases were also found in children and teenagers, as well as seniors aged 50 to 65 years. Indeed, COVID-19 vaccination started in Tunisia only by March 13, 2021, with a slow cadence. Health worker and individuals aged 75years and over were first vaccinated, followed by individuals from age groups 65–74 and then 60–64 years (43). Low vaccination coverage was achieved by May 2021, when Delta was introduced, and slow progression of vaccination campaigns was reported. Vaccination of individuals from age groups 50–59, 40–49, 30–39 was initiated progressively from June 1, to August 9, 2021(44–46). Vaccination of the age group 18–29 started later, by October 10, 2021 (47). Thus, the stuttering vaccination campaign, coupled the advent of the delta variant may be the cause of the deterioration of the health situation in Tunisia and explain the age distribution of Delta cases that we report herein. Furthermore, when Delta was introduced in the country, most of the already vaccinated peoples received only one dose which has been reported to be insufficient to induce effective protection against the Delta variant. Indeed, clinical evidence indicated decline in vaccine's effectiveness in the case of Delta variant infection (48). In vitro tests also showed lower neutralization activity of vaccine-induced antibodies against the Delta variant (49), as compared with the previous variants. Only administration of two COVID-19 vaccine doses may provide effective protection against the Delta variant (42, 48, 50, 51).
Worldwide, The Delta variant was characterized by a huge genetic variability, including the largest number of sub-lineages among VOCs. As of 2022-03-26, 234 different sub-lineages were reported (AY.1 to AY.133), represented by 4 259 793 sequences shared via GISAID, up (7). AY.4 is the most predominant sub-lineage in the world (19.5% of available Delta sequences), followed by AY.43, AY.103, AY.44; AY.122 comes in the sixth position (7). In our series and among all the Delta sub-lineages reported globally, twelve were detected in Tunisia with a large predominance of the AY.122 sub-lineage, which represented more than 94% of detected sequences. This sub-lineage was present in the country, from the beginning of the delta wave in May 2021 till its end in December 2021. It also spread to almost all regions of the country, including rural and urban regions. This suggests that it has established an autochthone transmission. Although, the number of samples obtained from rural regions is limited, the localization of main Tunisian population into the coastal regions around the big tows and the high connection between population in rural and urban regions are online with our finding. In the other hand, the eleven other variants were detected during shorter periods of time and in more restricted geographical areas, especially in the North-East region where are found the biggest cities and the main airports. This suggests that they were introduced through multiple importation events, mainly since August 2021, and that these minor sub-lineages were not able to establish autochthone transmission in the presence of the already circulating AY.122 sub-lineage. The restricted number of circulating sub-lineages during the Delta wave in Tunisia may also reflect the efficiency of public health measures taken place at the national gateways and entry points connected to the Tunisian border. The observation of isolation measures by travelers after arrival at the national territory may have modulated dispersion of numerous sub-lineages. In other contexts, such as in India, multiple introductions at the same time resulted in a rapid community spread and a wide dispersion of sub-lineages among the population (52). In Tunisia, the main population is located at coastal regions with important exchanges between rural and urban populations.
The AY.122 sub-lineage was first detected in India, 2020-09-07 (8). It's mainly circulating in Eastern-European regions such as Belarus, Armania, Russia (>40% of sequenced samples) and is also frequently detected in other European countries such as Monaco (>40%) and in Asian regions such as Kazakhstan and China (41 and 27%, respectively) (7). AY.122 also represents 70% sequenced samples in Seychelles (7). Given the strategic localization of Tunisia in the middle of the Mediterranean Sea, the high trade exchange with Europe, the high number of tourists visiting Tunisia and also the localization of the Tunisian population mainly in coastal regions, AY.122 may have been imported from Europe, especially from Eastern-Europe region. Indeed, an increased number of tourists coming from this region during the last years was recorded. In the other hand, countries such as Monaco and Seychelles also constitute privileged destinations for Tunisian tourists (53). Spatio-temporal analyses conducted in the present work suggest the introduction of the AY.122 sub-lineage first in Tunis, the capital and then its propagation to two other big cities that constituted after-on, together with Tunis, epicenters for the propagation of the virus all over the country. The mutational cascade describing important evolution of virus obtained in November-December, from big Tunisian cities confirm also our finding. Similar widespread of the same sub-lineage in Russia was recently reported (54). In Tunisia, and especially in big cities, it is evident that the local commercial exchanges, regular market visits and families' celebrations, during the summer time may have accelerated community transmission. The low SARS-CoV2 vaccine coverage achieved during the Delta wave as well as the decline in prevention measures within the population may explain the establishment of autochthone transmission.
Noticeably, the AY.122 Tunisian investigated sequences showed were highly related to each other and constituted an individualized genetic group independently from most of the sequences from other countries included in the phylogenetic tree. The mutational profile reveals that most of the Tunisian sequences share the A498V amino acid change in the ORF1a and 5 other synonymous mutations: 2746T>C, 3037C>T, 8986C>T, 11332A>G in ORF1a and 23683C>T in the S gene. In addition, the amino acid changes S: G181V; W258R were found with important rates. To the best of our knowledge, this is the first report of such amino acid changes among the AY.122 sub-lineage. The substitution A498V in ORF1a was previously reported among Wuhan-like strains Malaysian sequences, obtained during the first and second SARS-CoV2 waves, January-February 2020 (55). The G181V was reported in the Brazilian P.1 variant, but its impact was not investigated (56). The W258R in the S gene was reported among COVID-19 family clusters, including severe cases (57). The spread of viral lineages may rely on different factors such as virus properties, host genetic and immunological background, demography and socio-cultural behavior of the population and, probably, the combination of different factors. The AY.122 Tunisian strains may owe their successful spread to the presence of particular amino-acid substitutions. Even no evidence of increased virulence or transmission related to these changes were reported, It will be relevant to investigate their impact on protein structure and function to better understand the AY.122 spread.
In conclusion, this study adds to the knowledge on the Delta variant and sub-variant distribution worldwide and documents genomic data from Tunisia, North Africa. The introduction of a restricted number of delta variants in Tunisia may reflect the efficiency of public health measures taken place at the national gateways and entry points connected to the Tunisian border. Nevertheless, the decline in prevention measures within the population may explain the establishment of autochthone transmission of AY.122 sublineage.
Statements
Data availability statement
Complete genome SARS-CoV-2 sequences generated in this study were submitted to the GISAID database (https://www.gisaid.org) (29, 30). Their accession numbers are provided in Supplementary Table S1.
Ethics statement
The studies involving human participants were reviewed and approved by Bio-Medical Ethics Committee of Pasteur Institute of Tunis, Tunisia (2020/14/I/LR16IPT/V1). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
SH-B, OS, ABK, and HTr: conceptualization. SH-B, ABK, MA, OS, AC, WF, MG, ALa, and HTr: data curation. SH-B, MA, AC, WF, OS, KE, SS, CC, ALa, MD, AD, MG, HTo, and ALo: investigation. MM, HK, SG, OB, AT, OK, NH, YC, HS, KM, SBB, SF, MZ, MS, NA, IB, SF, and HTr: resources. HTr: funding acquisition. SH-B, OS, ABK, MG, and HTr: supervision. SH-B, ABK, OS, and HTr: validation. SH-B, MA, ABK, OS, and HTr: visualization and original draft preparation. SH-B, MA, and OS: writing. SH-B and HTr: writing—review and editing. All authors reviewed the manuscript and agreed to its submission to this journal.
Funding
This study was co-funded by the French Ministry for Europe and Foreign Affairs via the project REPAIR COVID-19-Africa coordinated by the Pasteur International Network, the Tunisian Ministry of Health and the Tunisian Ministry of Higher Education and Research (LR20IPT02, Projects: TriOMICS-CoV, PRFD5P1-Lutte COVID-19, and PRF4-CIC2016IPT02).
Acknowledgments
The authors gratefully acknowledge the New Variant Assessment Program (NVAP) and Quadram Institute Bioscience UK on behalf of the International COVID-19 response working to strengthen genomic surveillance of SARS-CoV-2 and data sharing, the Instituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise G. Caporale (IZSAM), Teramo, Italy, for their contribution in the whole-genome sequencing (WGS), as well as the Tunisian WHO Africa CDC and the WHO office in Tunisia for covering sample shipment fees to IPD and IZSAM, respectively. IZSAM was funded by the Italian Ministry of Health (IZSAM 05/20Ricerca Corrente 2020 PanCO: epidemiologia e patogenesi dei coronavirus umani ed animali. The authors also acknowledge the dedication of the technical staff of the Laboratory of Clinical Virology at the Pasteur Institute of Tunis for their efforts in real-time PCR COVID-19 diagnosis and biobank building, the staff of the health departments in the 24 governorates of Tunisia and the National Observatory of New and Emerging Diseases (ONMNE) for coordinating sample and data collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2022.990832/full#supplementary-material
References
1.
Hu B Guo H Zhou P . Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. (2021) 19:141–54. 10.1038/s41579-020-00459-7
2.
Motayo BO Oluwasemowo OO Olusola BA Akinduti PA Arege OT Obafemi YD et al . Evolution and genetic diversity of SARS-CoV-2 in Africa using whole genome sequences. Int J Infect Dis. (2021) 103:282–7. 10.1016/j.ijid.2020.11.190
3.
Rockett RJ Arnott A Lam C Sadsad R Timms V Gray KA et al . Revealing COVID-19 transmission in Australia by SARS-CoV-2 genome sequencing and agent-based modeling. Nat Med. (2020) 26:1398–404. 10.1038/s41591-020-1000-7
4.
Koyama T Platt D Parida L . Variant analysis of SARS-CoV-2 genomes. Bull World Health Organ. (2020) 98:495–504. 10.2471/BLT.20.253591
5.
World Health Organization. Tracking SARS-CoV2 variants. (2022). Available online at: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed February 28, 2022).
6.
Mlcochova P Kemp SA Dhar MS Papa G Meng B Ferreira IATM et al . SARS-CoV-2 B16172 Delta variant Replication and immune Evasion. Nature. (2021) 599:114–9. 10.1038/s41586-021-03944-y
7.
Tsueng G Mullen JL Alkuzweny M Cano M Rush B Haag E et al . Outbreak.info Research Library: A standardized, searchable platform to discover and explore COVID-19 resources. bioRxiv [Preprint]. (2022). 10.1101/2022.01.20.477133
8.
Scher E O'Toole A Andrew Rambaut A . SARS-CoV-2 Lineages. Available online at: https://cov-lineages.org/lineage_list.html (accessed November 03, 2022).
9.
Kumar S Bansal K . Cross-sectional genomic perspective of epidemic waves of SARS-CoV-2: a pan India study. Virus Res. (2022) 308:198642. 10.1016/j.virusres.2021.198642
10.
Ahammad I Hossain MU Rahman A Chowdhury ZM Bhattacharjee A et al . Wave-wise comparative genomic study for revealing the complete scenario and dynamic nature of COVID-19 pandemic in Bangladesh. PLoS ONE. (2021) 16:e0258019. 10.1371/journal.pone.0258019
11.
Tao K Tzou PL Nouhin J Gupta RK de Oliveira T Kosakovsky Pond SL et al . The biological and clinical significance of emerging SARS-CoV-2 variants. Nature Rev Genetics. (2021) 22:757–3. 10.1038/s41576-021-00408-x
12.
Zhang J Xiao T Cai Y Lavine CL Peng H Zhu H et al . Membrane fusion and immune Evasion by the Spike Protein of SARS-CoV-2 delta variant. Science. (2021) 373:1353–60. 10.1126/science.abl9463
13.
Gobeil SM Janowska K McDowell S Mansouri K Parks R Manne K et al . D614G mutation alters SARS-CoV-2 spike conformation and enhances protease cleavage at the S1/S2 junction. Cell Rep. (2021) 34:108630. 10.1016/j.celrep.2020.108630
14.
Motozono C Toyoda M Zahradnik J Saito A Nasser H Tan TS et al . SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe. (2021) 29:1124–36.e11. 10.1016/j.chom.2021.06.006
15.
Takeda M . Proteolytic activation of SARS-CoV-2 spike protein. Microbiol Immunol. (2022) 66:15–23. 10.1111/1348-0421.12945
16.
Malani A Shah D Kang G Lobo GN Shastri J Mohanan M et al . Seroprevalence of SARS-CoV-2 in slums versus non-slums in Mumbai, India. Lancet Glob Health. (2021) 9:e110–1. 10.1016/S2214-109X(20)30467-8
17.
Mishra S Mindermann S Sharma M Whittaker C Mellan TA Wilton T et al . Changing composition of SARS-CoV-2 lineages and rise of Delta variant in England. E Clinical Medicine. (2021) 39:101064. 10.1016/j.eclinm.2021.101064
18.
Rob P,. Britain thinks it can out-vaccinate the Delta variant. The world isn't so sure. CNN. (2021). Available online at: https://edition.cnn.com/2021/06/30/health/uk-delta-covid-variant-lessons-cmd-gbr-intl/index.html (accessed July 02, 2021).
19.
Ellyatt H . New COVID Wave Could be Imminent as Delta Variant Sweeps Europe, WHO says. CNBC. (2021). Available online at: https://techilive.in/new-covid-wave-could-be-imminent-as-delta-variant-sweeps-europe-who-says/ (accessed July 02, 2021).
20.
Callaway E . Delta coronavirus variant: scientists brace for impact. Nature. (2021) 595:17–8. 10.1038/d41586-021-01696-3
21.
Fares W Chouikha A Ghedira K Gdoura M Rezig D Haddad-Boubaker S et al . Whole genome sequencing and phylogenetic analysis of six SARS-CoV-2 strains isolated during COVID-19 pandemic in Tunisia, North Africa. BMC Genomics. (2021) 22:540. 10.1186/s12864-021-07870-1
22.
Fares W Ghedira K Gdoura M Chouikha A Haddad-Boubaker S Khedhiri M et al . Sequencing using a two-step strategy reveals high genetic diversity in the S Gene of SARS-CoV-2 after a high-transmission period in Tunis, Tunisia. Microbiol Spectr. (2021) 9:e0063921. 10.1128/Spectrum.00639-21
23.
Chouikha A Fares W Laamari A Haddad-Boubaker S Belaiba Z Ghedira K et al . Molecular epidemiology of SARS-CoV-2 in Tunisia (North Africa) through several successive waves of COVID-19. Viruses. (2022) 14:624. 10.3390/v14030624
24.
Corman VM Landt O Kaiser M Molenkamp R Meijer A Chu DK et al . Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. (2020) 25:2000045. 10.2807/1560-7917.ES.2020.25.3.2000045
25.
Chu DKW Pan Y Cheng SMS Hui KPY Krishnan P Liu Y et al . Molecular diagnosis of a novel coronavirus (2019-nCoV) causing an outbreak of pneumonia. Clin Chem. (2020) 66:549–55. 10.1093/clinchem/hvaa029
26.
Larkin MA Blackshields G Brown NP Chenna R McGettigan PA McWilliam H et al . Clustal X version 2.0. Bioinformatics. (2007) 23:2947–48. 10.1093/bioinformatics/btm404
27.
Bolger AM Lohse M Usadel B . Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. 10.1093/bioinformatics/btu170
28.
Bankevich A Nurk S Antipov D Gurevich AA Dvorkin M Kulikov AS et al . SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. 10.1089/cmb.2012.0021
29.
Elbe S Buckland-Merrett G . Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall. (2017)1:33–46. 10.1002/gch2.1018
30.
Shu Y McCauley J . GISAID global initiative on sharing all influenza data—from vision to reality. Euro Surveill. (2017) 22:30494. 10.2807/1560-7917.ES.2017.22.13.30494
31.
Rambaut A Holmes EC O'Toole À Hill V McCrone JT Ruis C et al . A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. (2020) 5:1403–7. 10.1038/s41564-020-0770-5
32.
Hadfield J Megill C Bell SM Huddleston J Potter B Callender C et al . Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. (2018) 34:4121–3. 10.1093/bioinformatics/bty407
33.
Minh BQ Schmidt HA Chernomor O Schrempf D Woodhams MD von Haeseler A et al . IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. (2020) 37:1530–4. 10.1093/molbev/msaa015
34.
Bansal K Kumar S . Mutational cascade of SARS-CoV-2 leading to evolution and emergence of omicron variant. Virus Res. (2022) 315:198765. 10.1016/j.virusres.2022.198765
35.
Katoh K Rozewicki J Yamada KD . MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. (2019) 20:1160–6. 10.1093/bib/bbx108
36.
Sagulenko P Puller V Neher RA . TreeTime: maximum-likelihood phylodynamic analysis. Virus Evol. (2018) 4:vex042. 10.1093/ve/vex042
37.
Rambaut A . FigTree v1.3.1. Edinburgh: Institute of Evolutionary Biology, University of Edinburgh (2010).
38.
Rambaut A Lam TT Max Carvalho L Pybus OG . Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. (2016) 2:vew007. 10.1093/ve/vew007
39.
Drummond AJ Rambaut A BEAST . Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. (2007) 7:214. 10.1186/1471-2148-7-214
40.
Dellicour S Durkin K Hong SL Vanmechelen B Martí-Carreras J Gill MS et al . Phylodynamic workflow to rapidly gain insights into the dispersal history and dynamics of SARS-CoV-2 LINEAGES. Mol Biol Evol. (2021) 38:1608–13. 10.1093/molbev/msaa284
41.
Bielejec F Rambaut A Suchard MA Lemey P SPREAD SPREAD spatial phylogenetic reconstruction of evolutionary dynamics . Bioinformatics. (2011) 27:2910–2. 10.1093/bioinformatics/btr481
42.
Thangaraj JWV Yadav P Kumar CG Shete A Nyayanit DA Rani DS et al . Predominance of delta variant among the COVID-19 vaccinated and unvaccinated individuals, India, May 2021. J Infect. (2022) 84:94–118. 10.1016/j.jinf.2021.08.006
43.
Mahumud RA Ali MA Kundu S Rahman MA Kamara JK . Renzaho AMN. Effectiveness of COVID-19 vaccines against Delta Variant (B16172): a meta-analysis. Vaccines (Basel). (2022) 10:277. 10.3390/vaccines10020277
44.
Ministère de la Santé,. Stratégie Nationale de la Vaccination contre la COVID-19 en Tunisie. (2022). Available online at: http://www.santetunisie.rns.tn/fr/34-infos-pratiques/126-programme-de-vaccination (accessed March 04, 2022).
45.
Jomni M,. Vaccination anti-COVID en Tunisie: convocation de la tranche d'âge 40-49 ans. Monde. (2022). Available online at: https://www.aa.com.tr/fr/afrique/vaccination-anti-covid-en-tunisie-convocation-de-la-tranche-d%C3%A2ge-50-60-ans/2260589 (accessed March 04, 2022).
46.
Jlassi W,. Vaccination anti-COVID en Tunisie: convocation de la tranche d'âge 40-49 ans. Monde. (2022). Available online at: https://www.aa.com.tr/fr/monde/vaccination-anti-covid-en-tunisie-convocation-de-la-tranche-d%C3%A2ge-40-49-ans/2318242 (accessed March 04, 2022).
47.
Business news,. Début de la vaccination des trente ans et plus. (2022). Available online at: https://www.businessnews.com.tn/mustapha-ferjani–debut-de-la-vaccination-des-30-ans-et-plus,520,111053,3 (accessed March 04, 2022).
48.
Jomni M,. Tunisie/Covid-19: Vaccination ouverte pour les 18ans et plus. Monde. (2022). Available online at: https://www.aa.com.tr/fr/afrique/tunisie-covid-19-vaccination-ouverte-pour-les-18-ans-et-plus-/2387698 (accessed March 04, 2022).
49.
Imai N Hogan AB Williams L Cori A Mangal TD Winskill P et al . Interpreting estimates of coronavirus disease 2019 (COVID-19) vaccine efficacy and effectiveness to inform simulation studies of vaccine impact: a systematic review. Wellcome Open Res. (2021) 6:185. 10.12688/wellcomeopenres.16992.1
50.
Davis C Logan N Tyson G Orton R Harvey WT Perkins JS et al . Reduced neutralisation of the Delta (B.1.617.2) SARS-CoV-2 variant of concern following vaccination. PLoSPathog. (2021) 17:e1010022. 10.1371/journal.ppat.1010022
51.
Noori M Nejadghaderi SA Arshi S Carson-Chahhoud K Ansarin K Kolahi AA et al . Potency of BNT162b2 and mRNA-1273 vaccine-induced neutralizing antibodies against severe acute respiratory syndrome-CoV-2 variants of concern: A systematic review of in vitro studies. Rev Med Virol. (2022) 32:e2277. 10.1002/rmv.2277
52.
Gupta N Kaur H Yadav PD Mukhopadhyay L Sahay RR Kumar A et al . Clinical characterization and genomic analysis of samples from COVID-19 breakthrough infections during the second wave among the various states of India. Viruses. (2021) 13:1782. 10.3390/v13091782
53.
Tunisian National Tourist Office. Le Tourisme Tunisien en chiffre. (2020). Available online at: https://www.ontt.tn/sites/default/files/inline-files/Extrait%20tourisme%20en%20chiffres%202020.pdf (accessed March 04, 2022).
54.
Klink GV Safina K Nabieva E Shvyrev N Garushyants S Alekseeva E et al . The rise and spread of the SARS-CoV-2 AY.122 lineage in Russia. MedRxiv preprint. (2021). 10.1101/2021.12.02.21267168
55.
Zawawi ZM Suppiah J Kalyanasundram J Azizan MA Mat-Sharani S Hisham HA et al . Whole genome sequence analysis of SARS-CoV-2 strains circulating in malaysia during first wave and early second wave of infections. Res Squ. [Preprint].10.21203/rs.3.rs-81152/v1
56.
Imai M Halfmann PJ Yamayoshi S Iwatsuki-Horimoto K Chiba S Watanabe T et al . Characterization of a new SARS-CoV-2 variant that emerged in Brazil. Proc Natl Acad Sci U S A. (2021) 118:e2106535118. 10.1073/pnas.2106535118
57.
Gunadi Wibawa H Hakim MS Marcellus Trisnawati I Khair RE et al . Molecular epidemiology of SARS-CoV-2 isolated from COVID-19 family clusters. BMC Med Genomics. (2021) 14:144. 10.1186/s12920-021-00990-3
Summary
Keywords
SARS-CoV2, Delta variant, AY.122, phylogeny, spatio-temporal dynamic, next-generation sequencing
Citation
Haddad-Boubaker S, Arbi M, Souiai O, Chouikha A, Fares W, Edington K, Sims S, Camma C, Lorusso A, Diagne MM, Diallo A, Boubaker IBB, Ferjani S, Mastouri M, Mhalla S, Karray H, Gargouri S, Bahri O, Trabelsi A, Kallala O, Hannachi N, Chaabouni Y, Smaoui H, Meftah K, Bouhalila SB, Foughali S, Zribi M, Lamari A, Touzi H, Safer M, Alaya NB, Kahla AB, Gdoura M and Triki H (2023) The Delta variant wave in Tunisia: Genetic diversity, spatio-temporal distribution and evidence of the spread of a divergent AY.122 sub-lineage. Front. Public Health 10:990832. doi: 10.3389/fpubh.2022.990832
Received
10 July 2022
Accepted
09 December 2022
Published
04 January 2023
Volume
10 - 2022
Edited by
Sanjay Kumar, Armed Forces Medical College, Pune, India
Reviewed by
Sanjeet Kumar, Max Planck Institute for the Science of Human History (MPI-SHH), Germany; Kanika Bansal, Institute of Microbial Technology (CSIR), India; Nicola Decaro, University of Bari Aldo Moro, Italy
Updates
Copyright
© 2023 Haddad-Boubaker, Arbi, Souiai, Chouikha, Fares, Edington, Sims, Camma, Lorusso, Diagne, Diallo, Boubaker, Ferjani, Mastouri, Mhalla, Karray, Gargouri, Bahri, Trabelsi, Kallala, Hannachi, Chaabouni, Smaoui, Meftah, Bouhalila, Foughali, Zribi, Lamari, Touzi, Safer, Alaya, Kahla, Gdoura and Triki.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sondes Haddad-Boubaker ✉ Sondeshaddadboubaker@gmail.com
†These authors have contributed equally to this work
This article was submitted to Infectious Diseases - Surveillance, Prevention and Treatment, a section of the journal Frontiers in Public Health
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.