 Yuhan Gao
Yuhan Gao Wenqiao He
Wenqiao He Jiaqi Fu
Jiaqi Fu Yongzhi Li
Yongzhi Li Huan He
Huan He Qing Chen
Qing Chen- Guangdong Provincial Key Laboratory of Tropical Disease Research, Department of Epidemiology, School of Public Health, Southern Medical University, Guangzhou, China
Background: Murine Kobuvirus (MuKV) is a novel picornavirus of the genus Kobuvirus, and was first identified in the feces of murine rodents in the USA in 2011. There is limited information on the transmission route of MuKV. Thus, we conducted a study to investigate virus detection rates in fecal, serum, throat, and lung tissue samples from murine rodents.
Results: A total of 413 fecal samples, 385 lung samples, 269 throat swab samples, and 183 serum samples were collected from 413 murine rodents (Rattus norvegicus, Rattus tanezumi, and Rattus rattus) captured in urban Shenzhen. Kobuviruses were detected via RT-PCR. Only fecal samples were positive, with prevalence rates of 34.9% in Rattus norvegicus and 29.4% in Rattus tanezumi. Phylogenetic analysis based on partial 3D and complete VP1 sequence regions indicated that all of the MuKV sequences obtained belonged to Aichivirus A, and were genetically closely related to other MuKVs reported in China, Hungary, and the USA. Twenty-eight full-length MuKV sequences were acquired. Phylogenetic analysis of two sequences randomly selected from the two species (SZ59 and SZ171) indicated that they shared very high nucleotide and amino acid identity with one another (94.0 and 99.3%, respectively), and comparison with human Kobuvirus revealed amino acid identity values of ~80%. Additionally, a sewage-derived sequence shared high similarity with the rat-derived sequences identified in this study, with respective nucleotide and amino acid identity values from 86.5 and 90.7% to 87.2 and 91.1%.
Conclusion: The results of the current study provide evidence that murine Kobuvirus is transmitted via the fecal-oral route.
Introduction
Kobuviruses belong to the genus Kobuvirus, within the family Picornaviridae, and they are small (27–30 nm diameter), spherical, non-enveloped, positive-sense, single-stranded RNA viruses (1). They all share a common genome organization, with a length of 8.2–8.4 kb. The genome consists of a 5′ untranslated region (UTR), a single open reading frame (ORF) that encodes a large polyprotein of 2,400–2,500 amino acids which is divided into a leader (L) protein, three structural proteins (VP0, VP3, and VP1), seven non-structural proteins (2A−2C and 3A−3D), a 3′UTR, and a poly(A) tail (2). Of these, the VP1 protein is the most variable, whereas the 3D gene is highly conserved (3).
Kobuvirus was first isolated from a fecal sample from a patient with acute gastroenteritis in Japan in 1989 (4). Kobuviruses are widespread worldwide, and are associated with gastroenteritis, respiratory infections, and other clinical symptoms (5). They have been found in a wide range of hosts including humans (4), cattle (6), sheep (7), pigs (8), dogs (9), cats (10), foxes (11), ferrets (12), goats (13), rabbits (14), bats (15), mice (16), and rats (17). They can also be detected in the environment, such as in sewage (18). The International Committee on Taxonomy of Viruses has classified the genus Kobuvirus into six species; Aichivirus A (AiV A; formerly Aichi virus), Aichivirus B (AiV B; formerly bovine Kobuvirus), Aichivirus C (AiV C; formerly porcine Kobuvirus), Aichivirus D (AiV D; formerly Kagovirus 1), Aichivirus E (AiV E; formerly rabbit Kobuvirus), and Aichivirus F (AiV F; formerly bat Kobuvirus), and 20 genetic types (19). Human Kobuvirus, canine Kobuvirus, murine Kobuvirus (MuKV), feline Kobuvirus, and roller Kobuvirus belong to Aichivirus A (20).
Rodents are considered potential reservoirs of several novel pathogens including Kobuvirus. MuKV was first detected in Peromyscus crinitus and Peromyscus maniculatus feces in the USA in 2011 (16). MuKVs were subsequently found in Vietnam (17), Hungary (21, 22), and the USA (23, 24) in stool samples from various murine species including Rattus norvegicus, Rattus tanezumi, Rattus argentiventer, Rattus losea, Rattus exulans, Mus musculus, and Bandicota indica. We recently conducted studies using fecal samples to determine the circulation of MuKV from Rattus norvegicus in Guangdong, China (25), and investigated the prevalence and genetic characterization of MuKV in Rattus losea, Rattus tanezumi, and Rattus norvegicus in several regions in southern China (26). Related research was limited to fecal samples, however, and detailed data pertaining to Kobuvirus in different samples of several rodent species was lacking.
Multiple studies have shown that kobuviruses can cause gastroenteritis in humans and other animal species, but there is limited information on Kobuvirus transmission routes. In humans Kobuvirus has been detected in feces and serum samples (4, 27). Porcine Kobuvirus viraemia has been reported (28), and a seroprevalence study in dogs and cats suggests that high percentages of these animals contain IgG antibodies against Aichivirus A (29). MuKV has been found in lung, brain, heart, and liver samples from rodents (23, 24), and canine Kobuvirus was identified in the brain, lungs, tonsils, and liver of the same puppy (30), indicating that kobuviruses might have a broad tissue tropism. To date the transmission route of MuKV in murine rodents remains unknown. To expand knowledge of the transmission route of kobuviruses, the current study investigated detection rates of MuKV in fecal, serum, throat, and lung tissue samples from murine rodents.
Materials and Methods
Sample Collection
In total, 413 fecal samples, 385 lung samples, 269 throat samples, and 183 serum samples were collected from 413 murine rodents captured between October 2020 and October 2021 in Shenzhen, Guangdong Province, China (Figure 1). Urban rats were trapped once a month, using live capture traps (also known as cage traps) in residential areas, city parks, composite market, and bus station.
Figure 1. Location of the trapping site of the murine rodents in China. Map source: https://image.so.com/view?q=%E4%B8%AD%E5%9B%BD%E5%9C%B0%E5%9B%BE&src=srp&correct=%E4%B8%AD%E5%9B%BD%E5%9C%B0%E5%9B%BE&ancestor=list&cmsid=50712c46325496ee4c7fbfbded18f06e&cmras=0&cn=0&gn=0&kn=50&crn=0&bxn=20&fsn=130&cuben=0&pornn=0&manun=50&adstar=0&clw=233#id=812433445ba11b77cba553308beab1d9&currsn=0&ps=136&pc=136.
Sample collection was conducted in the sterile room. The rodents were anesthetized via 3% diethyl ether inhalation, and the dosage of diethyl ether was adjusted according to their heart rate, respiratory frequency, corneal reflection, and extremity muscle tension. Trained personnel wore filtering facepiece respirators, chemical safety goggles, anti-static uniforms, and chemical protective gloves to protect themselves from diethyl ether. Before animals were killed by cervical dislocation, blood was drawn via cardiac puncture and centrifuged to obtain serum samples. Individual fresh fecal and throat samples were immediately placed in RNase-free tubes with 1 mL phosphate-buffered saline (0.3% homogenate), and lung tissue samples were stored in RNAlater (Invitrogen, CA, USA). All specimens were stored at −80°C prior to further processing. The species of the trapped animals were determined by morphological identification and cytochrome B gene sequencing (31).
Nucleic Acid Extraction and cDNA Synthesis
All samples were centrifuged at 8,000 rpm for 10 min at 4°C prior to collecting the supernatants. RNA and DNA were extracted from 200 μL of each supernatant using the MiniBEST Viral RNA/DNA Extraction Kit (TaKaRa, Kusatsu, Japan) in accordance with the manufacturer's instructions. The extracted RNA was reverse transcribed into cDNA using a Transcriptor First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland). The cDNA was used directly as a template for polymerase chain reaction (PCR) assays, or stored at −20°C prior to further analysis.
Molecular Detection of Murine Kobuvirus
Kobuviruses were detected via RT-PCR based on a previously described pair of consensus primers, UNIV-kobu-F/UNIV-kobu-R which amplifies a 216-nucleotide fragment of the conserved 3D region of all Kobuvirus species, encoding the RNA-dependent RNA polymerase (32). Among the 3D gene-positive samples, an 831-bp region of the VP1 gene was amplified via nested PCR using the Kobuvirus-VP1-os/oa and Kobuvirus-VP1-is/ia primer pair (17). The primer sequences are shown in Table 1. The PCR conditions were 94°C for 3 min, 40 cycles of 94°C for 30 s, 56°C for 1 min, and 72°C for 1 min, followed by a final extension at 72°C for 10 min. The amplified products were analyzed using 1.5% agarose gels, and visualized under ultraviolet light. Positive samples were sent to the Beijing Genomics Institute (Guangzhou, China) for sequencing.
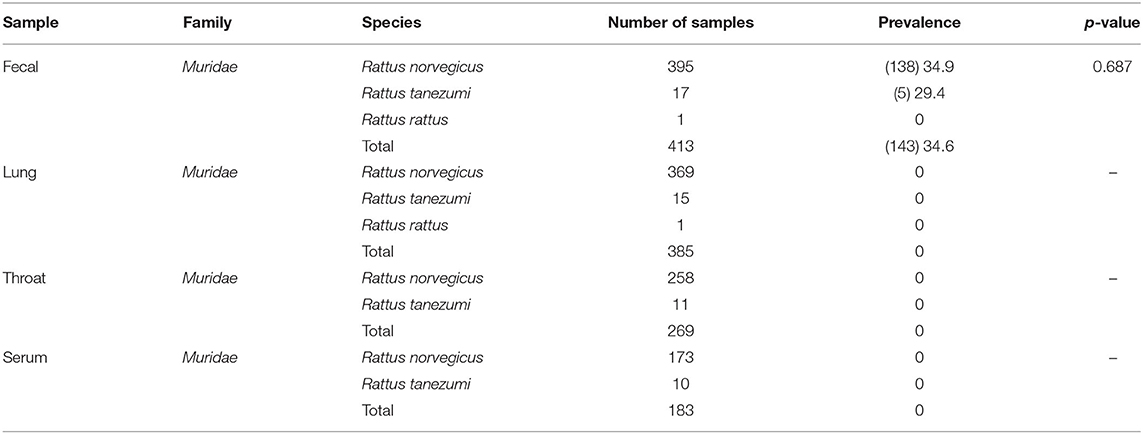
Table 1. Prevalence of Kobuvirus in fecal, lung, throat, and serum samples of rodents (n, %).
Genome Sequencing
Based on five murine Kobuvirus genome sequences from GenBank (accession numbers MF352432, MN116647, MN648600, MN648601, and MF175074), 13 pairs of primers designed to amplify the full-length viral genomes were generated (Supplementary Table 1). After sequencing, the nucleotide sequences were edited, validated, and assembled using Lasergene SeqMan software (DNASTAR, Inc. Madison, WI, USA).
Sequence and Phylogenetic Analyses
Sequences that were newly obtained in this study were aligned with Kobuvirus reference sequences from the GenBank database using the ClustalW multiple sequence alignment program in MEGA (version 7.0; Oxford Molecular Ltd., UK). To analyze evolutionary relationships, phylogenetic trees were generated based on the gene sequences obtained and representative gene sequences of murine Kobuvirus, rat Kobuvirus, mouse Kobuvirus, sewage Kobuvirus, canine Kobuvirus, feline Kobuvirus, human Kobuvirus, ovine Kobuvirus, bovine Kobuvirus, porcine Kobuvirus, bat Kobuvirus, and rabbit Kobuvirus, using the neighbor-joining (NJ) method in MEGA, with 1,000 bootstrap replicates. The single open reading frame (ORF) was identified using ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/), and potential proteolytic cleavage sites were predicted primarily based on the amino acid alignments of relevant sequences of kobuviruses. The GC content and nucleotide/amino acid identities among all sequences were calculated using the Sequence Identity Matrix program in BioEdit (version 7.2.5). Amino acid heterogeneity analysis was performed by aligning amino acids using DNAMAN (version 9.0). Similarity plot analysis of the almost full-length genome was conducted using SimPlot 3.5.1 software.
Statistical Analysis
Differences in Kobuvirus detection rates in the different species of animals, different seasons, and sexes were assessed using chi-square tests via Statistical Product and Service Solutions software (SPSS, version 25.0; IBM Corp., Armonk, NY, USA), with associations considered statistically significant at p < 0.05.
Ethics Statement
The study protocol was approved by the Animal Ethics and Welfare Committee of the School of Public Health, Southern Medical University, China, and adhered to the Rules for the Implementation of Laboratory Animal Medicine (1998) from the Ministry of Health, China. All surgical procedures were performed under anesthesia to minimize suffering. No endangered or protected species were involved in the study.
Results
Detection of Murine Kobuvirus
A total of 413 animals were trapped. Fecal, lung, throat swab, and serum samples were collected from 395 Rattus norvegicus, 17 Rattus tanezumi, and 1 Rattus rattus (Table 1). According to RT-PCR and nested PCR, 34.6% (143/413) of fecal specimens were positive for Kobuvirus RNA. Among them, the prevalence of Kobuvirus in different species of animals ranged from 0.0 to 34.9%, and there was no statistical difference in the positive rates of fecal samples from different species (χ2 = 0.751, p = 0.687) and sexes (χ2 = 0.004, p = 0.948; Tables 1, 2). However, the difference among seasons was significant (χ2 = 17.857, p < 0.001), and the prevalence in winter was significantly higher than other seasons (Table 2).
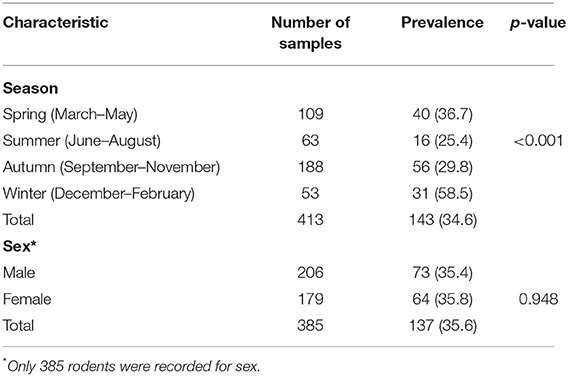
Table 2. Characteristics of prevalence kobuviruses in fecal samples of rodents (n, %).
Phylogenetic and Amino Acid Heterogeneity Analyses of 3D and VP1 Region Sequences
Neighbor-joining phylogenetic analysis based on partial 3D nucleotide sequences (634 nucleotides) was conducted using representative sequences detected in the study and other Kobuvirus reference sequences from GenBank (Figure 2). Sequences isolated from two different rodent species were highly similar, with respective nucleotide and amino acid identities of 92.6–100.0% and 99.0–100.0%. This demonstrates that the murine Kobuvirus strains from the same species were relatively clustered together, and the sequences identified in the study clustered with murine kobuviruses previously found in China and Hungary, within the Aichivirus A group, which also includes human Kobuvirus, canine Kobuvirus, feline Kobuvirus, and sewage Kobuvirus.
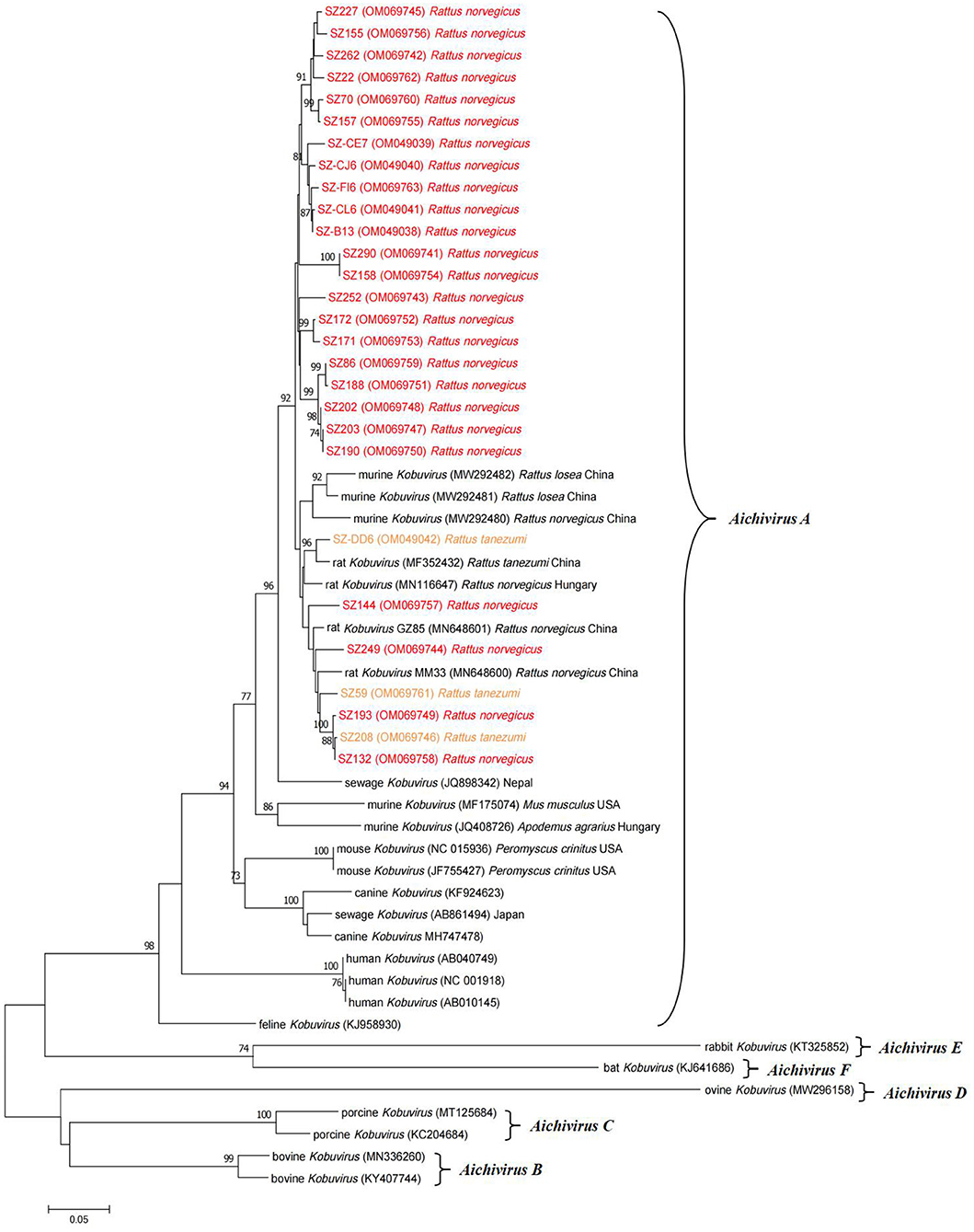
Figure 2. Phylogenetic tree of Kobuvirus based on the partial 3D nucleotide sequences (634 bp, 7,311–7,938 nt). The tree was generated by the neighbor-joining method with 1,000 bootstrap replicates, and the statistics values >70% are displayed above the tree branches. The sequences from samples collected in this study have been labeled with color. SZ, Shenzhen city in Guangdong province.
Complete VP1 sequences were obtained from samples positive for the partial 3D gene using nested PCR, and several representative sequences were selected to generate a phylogenetic tree (Figure 3) along with VP1 reference sequences from Kobuvirus strains in GenBank. The VP1 sequences of murine kobuviruses identified in the study shared respective nucleotide and amino acid identities of 89.8–98.9% and 97.8–100.0%. Phylogenetic analysis based on the VP1 nucleotide sequences indicated that the sequences identified in the study were more closely related to murine Kobuvirus strains and clustered within a major group, but origin-specific lineages of species were not apparent. Further analysis of amino acid heterogeneity and the DNAMAN output revealed that the sequences identified in the study were highly similar in 3D and VP1 regions. Compared with several reference sequences of other species, the VP1 region had more amino acid deletions and substitutions than the 3D region, with respective identities of 77.4 and 92.0% (Supplementary Figure 1).

Figure 3. Neighbor-joining tree for Kobuvirus nucleotide sequences based on the complete VP1 region nucleotide sequences (831 bp, 3,012–3,842 nt). Bootstrap support for branches (1,000 replications) is indicated, and the statistics values >70% are displayed above the tree branches. Murine Kobuvirus (MuKV) sequences obtained in this study are represented by color. SZ, Shenzhen city in Guangdong province.
Genomic and Phylogenetic Analyses of Complete Sequences
Twenty-eight full-length murine Kobuvirus genomes were successfully sequenced from Kobuvirus–positive samples, including 25 from Rattus norvegicus and 3 from Rattus tanezumi (Table 3). Multiple sequence alignments were performed with these sequences, and they shared 93.1–99.0% nucleotide and 93.3–99.1% amino acid identities with each other. Two sequences (SZ59 and SZ171) were then randomly selected from the two species to further analyze the genomic structure of murine Kobuvirus. The SZ59 Kobuvirus genome was 8,252 nucleotides in length and contained a 725-nucleotide 5′-UTR, a 237-nucleotide 3′-UTR, and an ORF (7,290 nucleotides) encoding a potential polyprotein of 2,429 amino acids, excluding the poly-(A) tail. The complete genome of SZ171 had 8,250 nucleotides, and contained a 724-nucleotide 5′-UTR and a 236-nucleotide 3′-UTR. We predicted the genome organization and potential cleavage sites for the complete polyprotein genes of SZ59 and SZ171, which were identical to other previously described murine Kobuvirus strains. The L protein was 492 nucleotides (164 amino acids) in length, whereas the complete P1, P2, and P3 regions were 2625 nucleotides (875 amino acids), 1,833 nucleotides (611 amino acids), and 2,340 nucleotides (779 amino acids) long, respectively. The two sequences yielded GC content of 55.9–56.3% (19.5–19.8% A, 21.1–21.3% G, 24.2–24.3% T, and 34.9–35.1% C), like other kobuviruses (52–59%), which is characteristic of the family Picornaviridae (11).
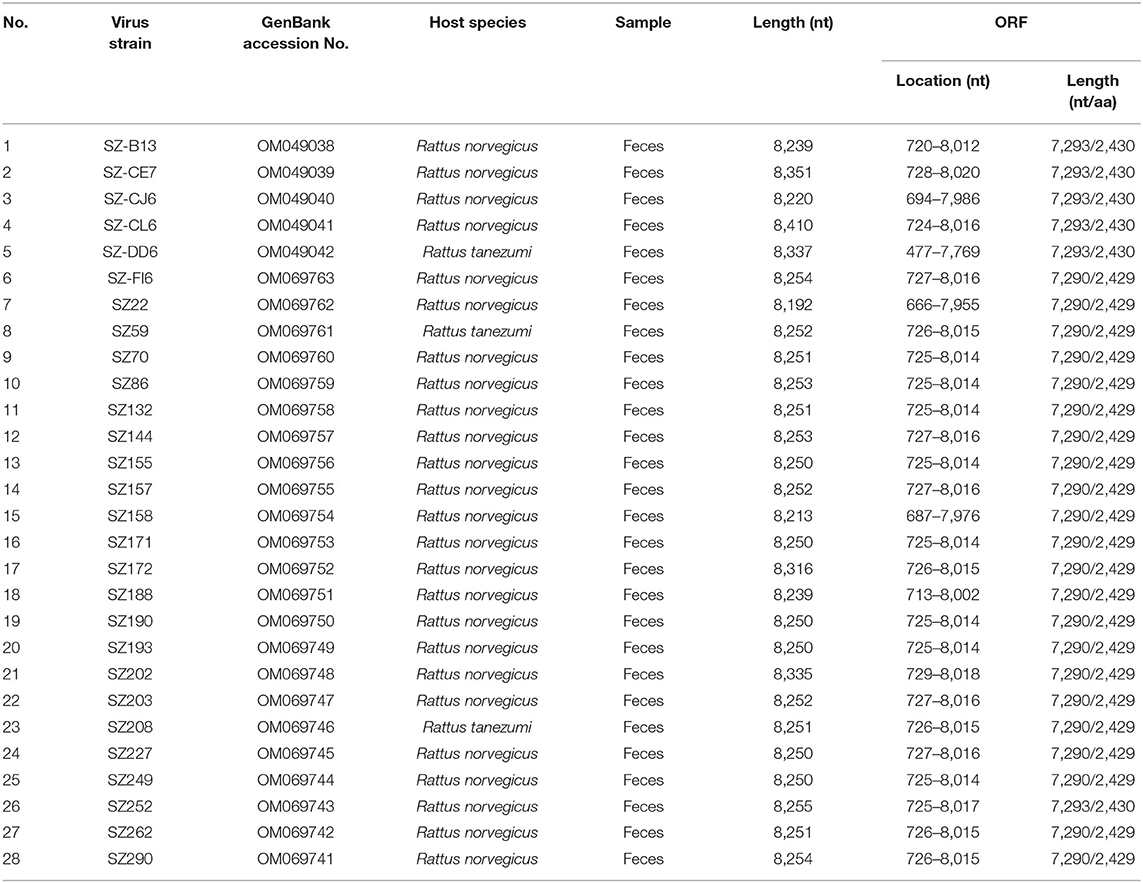
Table 3. Full-length murine Kobuvirus (MuKV) strains identified in this study.
The predicted protease-cleavage sites of the sequences identified in the current study and reference kobuviruses are shown in Supplementary Table 2, including Q/G, P/Q, Q/H, Q/S, Q/T, Q/A, Q/P, Y/V, Q/C, and A/T. The predicted protease cleavage sites between L and VP0 (Q/G), 2A and 2B (Q/G), 2B and 2C (Q/G), and 3C and 3D (Q/S) are conserved among kobuviruses isolated from different species. In comparison, the protease-cleavage site between 2C and 3A is Q/G for all the Kobuvirus sequences except for ovine Kobuvirus (A/T). The protease cleavage sites of the sequences identified in the current study are highly conserved, and are similar to those in other species.
Phylogenetic analysis based on the complete nucleotide sequences indicated that the sequences identified in the study shared higher nucleotide (93.7–95.6%) and amino acid (94.6–96.8%) identities with two other Chinese strains (MM33 and GZ85) (25) compared to other murine Kobuvirus strains. All murine Kobuvirus strains were clustered according to their geographical regions, albeit with limited sequence support (Figure 4). A sewage-derived Kobuvirus sequence (JQ898342) detected in Nepal (33), which was identified as a novel Kobuvirus in the Picornaviridae family and was named Kobuvirus sewage Kathmandu (KoV-SewKTM) after the location of its discovery, was clustered together with the rat-derived sequences identified in this study, with high respective nucleotide and amino acid identity values of 86.5 and 87.9% to 87.4 and 89.1%. A new Kobuvirus (34) closely related to canine Kobuvirus in sewage samples that was found in Aichi Prefecture, Japan, clustered within the Aichivirus A group. Comparisons between the full polyprotein and L/P1/P2/P3 fragments of the two representative sequences and reference Kobuvirus sequences from several other species and the environment are shown in Table 4. SZ59 shared 94.0% nucleotide identity and 99.3% amino acid identity with SZ171. Compared with other Kobuvirus reference strains, SZ59 and SZ171 had 75.9–76.0% nucleotide identities and 80.7% amino acid identities with the human Kobuvirus strain AB010145, and shared the highest sequence homologies with the murine Kobuvirus strain MN648601 at the nucleotide (93.3–93.6%) and amino acid (98.1–98.3%) levels. In the P1, P2, and P3 regions, SZ59 and SZ171 kobuviruses exhibited more than 74% nucleotide and 77% amino acid identity with human, rat, feline, canine, and untreated sewage-derived kobuviruses, which belong to Aichivirus A.
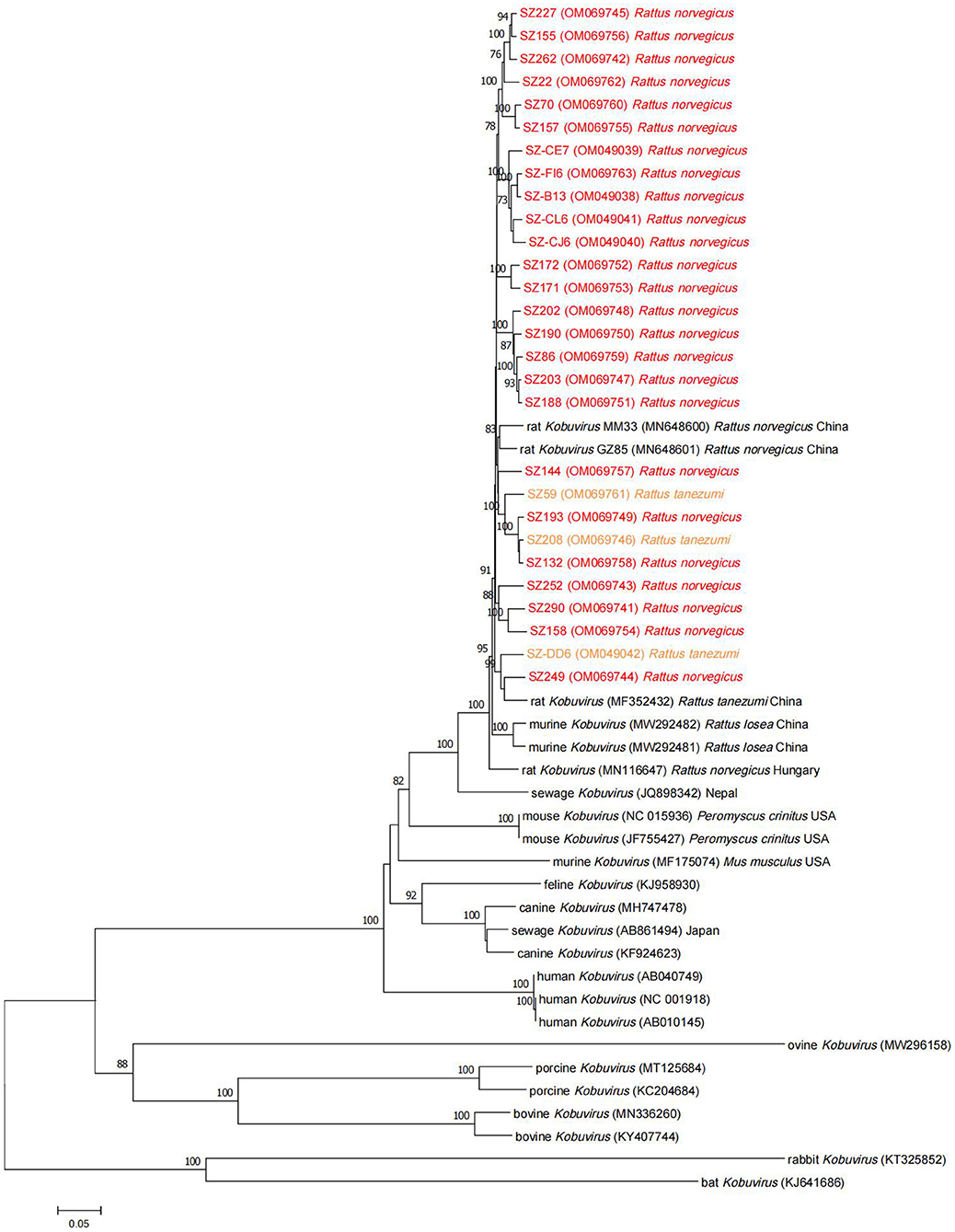
Figure 4. Phylogenetic tree of Kobuvirus based on the complete nucleotide sequences from different strains. The tree was generated by the neighbor-joining method with 1,000 bootstrap replicates, and the statistics values >70% are displayed above the tree branches. Murine Kobuvirus (MuKV) sequences identified in the present study are indicated by color. SZ, Shenzhen city in Guangdong province.
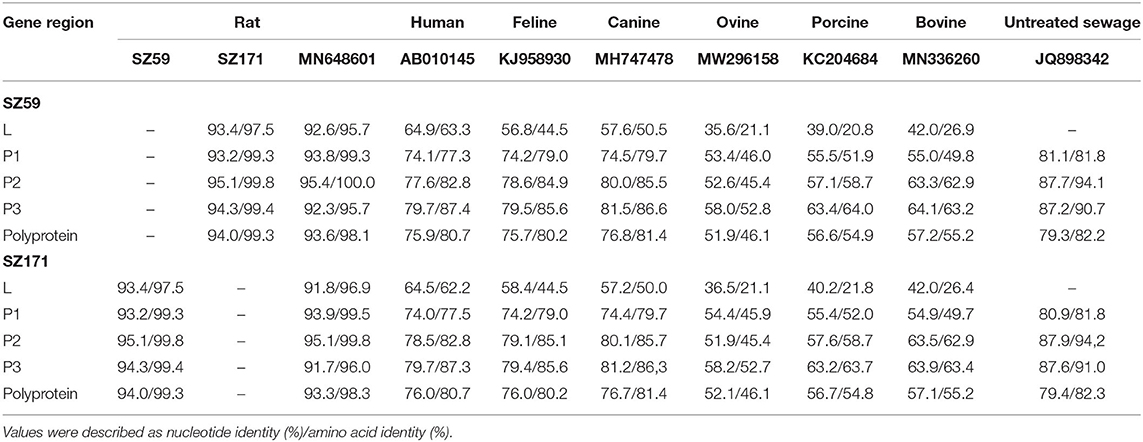
Table 4. Nucleotide and putative amino acid sequence identities of the murine kobuviruses (SZ59 and SZ171) with reference Kobuvirus strains from several species and the environment.
Similarity Plot Analysis
To further analyze the genetic characteristics of the complete polyprotein gene, similarity plot analysis was conducted using SimPlot 3.5.1 which compared polyprotein nucleotide sequences of SZ59, SZ171, and the human Kobuvirus sequence AB010145 (used as the out-group sequence) to murine Kobuvirus reference strain Wencheng-Rt386-2/MF352432 (used as the query sequence). SZ59 and SZ171 exhibited relatively high similarity to the query sequence in the 5′-UTR, VP3, 2A-2C, 3B-3D, and 3′-UTR regions, but less similarity in the L, VP0, VP1, 3A regions as well as a region near the start of the 3D gene (Supplementary Figure 2). A higher range of genetic variability was observed in the P1 coding region, and intergenotype recombination was not observed in the Kobuvirus coding regions.
Discussion
To the best of our knowledge, this is the first study to investigate the prevalence and genomes of kobuviruses in multiple samples (fecal, lung, throat swab, and serum) from different murine rodent species (Rattus norvegicus, Rattus tanezumi, and Rattus rattus). We investigated 413 fecal samples, of which 143 (34.6%) were positive for Kobuvirus, with detection rates of 34.9% in Rattus norvegicus, 29.4% in Rattus tanezumi, and 0% in Rattus rattus. In our previous study, detection rates were 23.7% in Rattus tanezumi, 10.2% in Rattus losea, and 28.1% in Rattus norvegicus in southern China (26), and the prevalence in Rattus norvegicus was 12.6% in Guangzhou (25). MuKVs were detected in 50% of Rattus norvegicus fecal samples in New York in 2014 (24), and were present in 55.5% of free-living and laboratory rats (Rattus norvegicus) in Hungary (22). In New York (Manhattan and Queens sites) in 2018, MuKV was detected in 17.5% of Mus musculus samples (23), and in Vietnam the prevalence rate of MuKV in rats was 17% (17). These findings reveal that kobuviruses are prevalent among different species of murine rodents. In this report, although the prevalence of kobuviruses in fecal samples did not differ significantly between the species, the difference among seasons was significant, with the highest prevalence in winter, suggesting that murine Kobuvirus is prevalent throughout the year, but more attention should be paid to disease prevention and control in winter. Furthermore, both female and male rats can carry Kobuvirus, and there was no statistical difference between the sexes.
Previous studies suggest that kobuviruses may play a role as causative agents in gastroenteritis diseases (5), and are often found in gastrointestinal tract samples from humans and other animals (5, 9–20), suggesting that Kobuvirus is transmitted via the fecal-oral route or consumption of contaminated food or water. The detection of kobuviruses in sewage samples in South Africa (18) and Tunisia (35), in surface waters in Venezuela (36), in sewage and surface waters in the Netherlands (37), and in sewage and river waters in Japan (38), and a new Kobuvirus closely related to canine Kobuvirus detected in sewage samples in Japan (34) supports this contention. In the current study, only MuKV was found in rodents feces, and in phylogenetic analyses all sequences obtained clustered together with the sewage-derived Kobuvirus found in Nepal (33), and shared more than 86% nucleotide and 87% amino acid identities, confirming that MuKV is mainly transmitted via the fecal-oral route through contaminated water and raw and treated sewage, which is concordant with previous Tunisian, Japanese, and Dutch studies (35, 37, 38). Furthermore, we recently conducted two initial epidemiological studies investigating MuKV in murine rodent stool samples (25, 26) which also provide evidence that is congruent with this finding.
Seroepidemiologic studies in Japan, Germany, France, Spain, and Tunisia (27, 39–42) indicate general circulation of kobuviruses in human populations, suggesting an important role of Kobuvirus infection in gastroenteritis. However, a report of porcine viremia suggests that Kobuvirus can escape the gastrointestinal tract into the circulatory system in immunocompetent virus-infected hosts (28), and its high prevalence in dog and cat serum samples indicates the importance of Kobuvirus detection in animal serum specimens (29). Further investigations are required to confirm whether the digestive tract is the main location of Kobuvirus replication. Notably, kobuviruses have also been identified in other sample types. MuKV was found in feces and liver samples from Mus musculus (23), and in fecal, lung, brain, and heart specimens from an infected Rattus norvegicus (24). Interestingly, canine Kobuvirus was detected in brain, lung, tonsil, and liver samples from a puppy, but feces and gastrointestinal tract samples from that same puppy were negative (30). These findings confirm widespread viral dissemination in multiple tissues of animals, but also expand the potential tissue tropisms and transmission routes of kobuviruses.
In neighbor-joining phylogenetic analysis based on partial 3D or VP1 regions, the MuKV sequences isolated from Rattus norvegicus and Rattus tanezumi in the current study clustered with other rat-derived Kobuvirus sequences identified in China, Hungary, and the USA. In phylogenetic tree analysis based on partial 3D regions, the sequences obtained clustered tightly together in a clade according to the species, confirming that the 3D gene is conserved. Notably, however, phylogenetic analysis based on the VP1 regions indicated that such origin-specific lineages of species were not clear. Although the above results suggest that MuKV has distinctive characteristics that differ from other Aichivirus A viruses, the possibility of cross-species transmission in Kobuvirus cannot be ignored. Historical transmissions between rabbits and bats, between rodents and bats, and between cows and pigs have been reported (17, 43, 44), raising the possibility of cross-species transmission in the future; which could result in a sustained threat to public health due to increased contact between humans and animals. More evidence is required to prove cross-species transmission of Kobuvirus, and assess the pathogenicity posed to human health.
Amino acid mutations cause changes in protein phenotype, and are considered most deleterious. Both the 3D and VP1 regions investigated in the current study are important in the stability of the Kobuvirus genome, and its replication. In amino acid heterogeneity analysis, the sequences identified in the study were highly similar, and the 3D region was relatively conserved whereas the VP1 region had more amino acid deletions and substitutions. These results are concordant with a previous review indicating that VP1 is the most variable structural protein and the 3D gene is highly conserved among kobuviruses (3). Estimation of these changes in genomic regions would be helpful with respect to the characteristics of kobuviruses.
In total, 28 complete gene sequences of MuKV were successfully acquired in this study. The polyproteins of the sequences were all cleaved into 11 viral proteins, including L, P1 (VP0, VP3, and VP1), P2 (2A−2C), and P3 (3A−3D), and the predicted cleavage sites were Q/G, P/Q, Q/H, Q/S, Q/T, Q/A, Q/P, Y/V, Q/C, and A/T, according to alignment with known Kobuvirus cleavage sites. Comparisons of the protease cleavage sites in Kobuvirus sequences isolated from humans and animals indicated that the protease cleavage site sequences are highly conserved, supporting a previous review suggesting that the predicted cleavage sites are similar to those in other picornaviruses (3). As representatives of two species, SZ59 shared relatively high nucleotide and amino acid identity with SZ171 (94.0 and 99.3%, respectively), and with other full reference sequences. SZ59 and SZ171 had the highest similarity to the MuKV sequences (93.3–93.6% nucleotide and 98.1–98.3% amino acid identities), with identity values <60% similar to ovine, porcine, and bovine sequences. In similarity plot analysis, SZ59 and SZ171 had lower similarities at the L, VP0, VP1, and 3A regions, and the region at the start of the 3D gene, indicating possible changes in these regions.
In the current study, all the lung, throat swab, and serum samples were negative for Kobuvirus. This may be related to an insufficient sample size, lack of representation in rat species, and geographic distribution. We also screened Kobuvirus-positive samples for Rosavirus, Bocavirus, and Hunnivirus, and 52, 43, and 6 samples, respectively, were co-infected with these viruses. These results are consistent with previous studies demonstrating that kobuviruses are frequently present in mixed infections with other pathogens (45, 46).
Conclusion
The present study provides evidence of fecal-oral transmission of MuKV, and increases the knowledge of molecular characteristics and evolution of MuKV. Further epidemiological and laboratory studies are needed to verify fecal-oral transmission of MuKV.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by Animal Ethics and Welfare Committee of the School of Public Health, Southern Medical University.
Author Contributions
YG and QC conceived of the project. QC obtained the funding. YG contributed to the writing of the article and analyzed the data. YG, WH, and JF performed the experiment. YG, WH, JF, YL, and HH collected the samples. All authors have read and approved the manuscript for publication. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81973107). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Minyi Zhang, Fei Wu, Qiushuang Li, Juxian Xian, Liting Zheng, and Minyi Liang for their participation in the collection of samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2022.865605/full#supplementary-material
References
1. Zell R. Picornaviridae-the ever-growing virus family. Arch Virol. (2018) 163:299–317. doi: 10.1007/s00705-017-3614-8
2. Akagami M, Ito M, Niira K, Kuroda M, Masuda T, Haga K, et al. Complete genome analysis of porcine kobuviruses from the feces of pigs in Japan. Virus Genes. (2017) 53:593–602. doi: 10.1007/s11262-017-1464-9
3. Reuter G, Boros A, Pankovics P. Kobuviruses - a comprehensive review. Rev Med Virol. (2011) 21:32–41. doi: 10.1002/rmv.677
4. Yamashita T, Kobayashi S, Sakae K, Nakata S, Chiba S, Ishihara Y, et al. Isolation of cytopathic small round viruses with BS-C-1 cells from patients with gastroenteritis. J Infect Dis. (1991) 164:954–7. doi: 10.1093/infdis/164.5.954
5. Khamrin P, Maneekarn N, Okitsu S, Ushijima H. Epidemiology of human and animal kobuviruses. Virusdisease. (2014) 25:195–200. doi: 10.1007/s13337-014-0200-5
6. Yamashita T, Ito M, Kabashima Y, Tsuzuki H, Fujiura A, Sakae K. Isolation and characterization of a new species of Kobuvirus associated with cattle. J Gen Virol. (2003) 84(Pt 11):3069–77. doi: 10.1099/vir.0.19266-0
7. Abi KM, Zhang Q, Jing ZZ, Tang C. First detection and molecular characteristics of caprine Kobuvirus in goats in China. Infect Genet Evol. (2020) 85:104566. doi: 10.1016/j.meegid.2020.104566
8. Reuter G, Boldizsár A, Kiss I, Pankovics P. Candidate new species of Kobuvirus in porcine hosts. Emerg Infect Dis. (2008) 14:1968–70. doi: 10.3201/eid1412.080797
9. Li L, Pesavento PA, Shan T, Leutenegger CM, Wang C, Delwart E. Viruses in diarrhoeic dogs include novel kobuviruses and sapoviruses. J Gen Virol. (2011) 92(Pt 11):2534–41. doi: 10.1099/vir.0.034611-0
10. Chung JY, Kim SH, Kim YH, Lee MH, Lee KK, Oem JK. Detection and genetic characterization of feline kobuviruses. Virus Genes. (2013) 47:559–62. doi: 10.1007/s11262-013-0953-8
11. Di Martino B, Di Profio F, Melegari I, Robetto S, Di Felice E, Orusa R, et al. Molecular evidence of kobuviruses in free-ranging red foxes (Vulpes vulpes). Arch Virol. (2014) 159:1803–6. doi: 10.1007/s00705-014-1975-9
12. Smits SL, Raj VS, Oduber MD, Schapendonk CM, Bodewes R, Provacia L, et al. Metagenomic analysis of the ferret fecal viral flora. PLoS ONE. (2013) 8:e71595. doi: 10.1371/journal.pone.0071595
13. Lee MH, Jeoung HY, Lim JA, Song JY, Song DS, An DJ. Kobuvirus in South Korean black goats. Virus Genes. (2012) 45:186–9. doi: 10.1007/s11262-012-0745-6
14. Pankovics P, Boros Á, Bíró H, Horváth KB, Phan TG, Delwart E, et al. Novel picornavirus in domestic rabbits (Oryctolagus cuniculus var. domestica). Infect Genet Evol. (2016) 37:117–22. doi: 10.1016/j.meegid.2015.11.012
15. Li L, Victoria JG, Wang C, Jones M, Fellers GM, Kunz TH, et al. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol. (2010) 84:6955–65. doi: 10.1128/JVI.00501-10
16. Phan TG, Kapusinszky B, Wang C, Rose RK, Lipton HL, Delwart EL. The fecal viral flora of wild rodents. PLoS Pathog. (2011) 7:e1002218. doi: 10.1371/journal.ppat.1002218
17. Lu L, Van Dung N, Ivens A, Bogaardt C, O'Toole A, Bryant JE, et al. Genetic diversity and cross-species transmission of kobuviruses in Vietnam. Virus Evol. (2018) 4:vey002. doi: 10.1093/ve/vey002
18. Onosi O, Upfold NS, Jukes MD, Luke GA, Knox C. The first molecular detection of Aichi virus 1 in raw sewage and mussels collected in South Africa. Food Environ Virol. (2019) 11:96–100. doi: 10.1007/s12560-018-9362-4
19. Adams MJ, King AM, Carstens EB. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses 2013. Arch Virol. (2013) 158:2023–30. doi: 10.1007/s00705-013-1688-5
20. Zell R, Delwart E, Gorbalenya AE, Hovi T, King AMQ, Knowles NJ, et al. ICTV virus taxonomy profile: Picornaviridae. J Gen Virol. (2017) 98:2421–2. doi: 10.1099/jgv.0.000911
21. Farkas T, Fey B, Keller G, Martella V, Egyed L. Molecular detection of novel astroviruses in wild and laboratory mice. Virus Genes. (2012) 45:518–25. doi: 10.1007/s11262-012-0803-0
22. Boros Á, Orlovácz K, Pankovics P, Szekeres S, Földvári G, Fahsbender E, et al. Diverse picornaviruses are prevalent among free-living and laboratory rats (Rattus norvegicus) in Hungary and can cause disseminated infections. Infect Genet Evol. (2019) 75:103988. doi: 10.1016/j.meegid.2019.103988
23. Williams SH, Che X, Garcia JA, Klena JD, Lee B, Muller D, et al. Viral diversity of house mice in New York City. mBio. (2018) 9:e01354-17. doi: 10.1128/mBio.01354-17
24. Firth C, Bhat M, Firth MA, Williams SH, Frye MJ, Simmonds P, et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. mBio. (2014) 5:e01933-14. doi: 10.1128/mBio.01933-14
25. You FF, Zhang MY, He H, He WQ, Li YZ, Chen Q. Kobuviruses carried by Rattus norvegicus in Guangdong, China. BMC Microbiol. (2020) 20:94. doi: 10.1186/s12866-020-01767-x
26. Zhang M, You F, Wu F, He H, Li Q, Chen Q. Epidemiology and genetic characteristics of murine Kobuvirus from faecal samples of Rattus losea, Rattus tanezumi and Rattus norvegicus in southern China. J Gen Virol. (2021) 102:001646. doi: 10.1099/jgv.0.001646
27. Yamashita T, Sakae K, Ishihara Y, Isomura S, Utagawa E. Prevalence of newly isolated, cytopathic small round virus (Aichi strain) in Japan. J Clin Microbiol. (1993) 31:2938–43. doi: 10.1128/jcm.31.11.2938-2943.1993
28. Reuter G, Kecskémeti S, Pankovics P. Evolution of porcine Kobuvirus infection, Hungary. Emerg Infect Dis. (2010) 16:696–8. doi: 10.3201/eid1604.090937
29. Carmona-Vicente N, Buesa J, Brown PA, Merga JY, Darby AC, Stavisky J, et al. Phylogeny and prevalence of kobuviruses in dogs and cats in the UK. Vet Microbiol. (2013) 164:246–52. doi: 10.1016/j.vetmic.2013.02.014
30. Ribeiro J, Headley SA, Diniz JA, Pereira AH, Lorenzetti E, Alfieri AA, et al. Extra-intestinal detection of canine Kobuvirus in a puppy from Southern Brazil. Arch Virol. (2017) 162:867–72. doi: 10.1007/s00705-016-3164-5
31. Kocher TD, Thomas WK, Meyer A, Edwards SV, Pääbo S, Villablanca FX, et al. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci USA. (1989) 86:6196–200. doi: 10.1073/pnas.86.16.6196
32. Reuter G, Boldizsár A, Pankovics P. Complete nucleotide and amino acid sequences and genetic organization of porcine Kobuvirus, a member of a new species in the genus Kobuvirus, family Picornaviridae. Arch Virol. (2009) 154:101–8. doi: 10.1007/s00705-008-0288-2
33. Ng TF, Marine R, Wang C, Simmonds P, Kapusinszky B, Bodhidatta L, et al. High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage. J Virol. (2012) 86:12161–75. doi: 10.1128/JVI.00869-12
34. Yamashita T, Adachi H, Hirose E, Nakamura N, Ito M, Yasui Y, et al. Molecular detection and nucleotide sequence analysis of a new Aichi virus closely related to canine Kobuvirus in sewage samples. J Med Microbiol. (2014) 63(Pt 5):715–20. doi: 10.1099/jmm.0.070987-0
35. Sdiri-Loulizi K, Hassine M, Aouni Z, Gharbi-Khelifi H, Sakly N, Chouchane S, et al. First molecular detection of Aichi virus in sewage and shellfish samples in the Monastir region of Tunisia. Arch Virol. (2010) 155:1509–13. doi: 10.1007/s00705-010-0744-7
36. Alcalá A, Vizzi E, Rodríguez-Díaz J, Zambrano JL, Betancourt W, Liprandi F. Molecular detection and characterization of Aichi viruses in sewage-polluted waters of Venezuela. Appl Environ Microbiol. (2010) 76:4113–5. doi: 10.1128/AEM.00501-10
37. Lodder WJ, Rutjes SA, Takumi K, de Roda Husman AM. Aichi virus in sewage and surface water, the Netherlands. Emerg Infect Dis. (2013) 19:1222–30. doi: 10.3201/eid1908.130312
38. Kitajima M, Haramoto E, Phanuwan C, Katayama H. Prevalence and genetic diversity of Aichi viruses in wastewater and river water in Japan. Appl Environ Microbiol. (2011) 77:2184–7. doi: 10.1128/AEM.02328-10
39. Goyer M, Aho LS, Bour JB, Ambert-Balay K, Pothier P. Seroprevalence distribution of Aichi virus among a French population in 2006-2007. Arch Virol. (2008) 153:1171–4. doi: 10.1007/s00705-008-0091-0
40. Oh DY, Silva PA, Hauroeder B, Diedrich S, Cardoso DD, Schreier E. Molecular characterization of the first Aichi viruses isolated in Europe and in South America. Arch Virol. (2006) 151:1199–206. doi: 10.1007/s00705-005-0706-7
41. Ribes JM, Montava R, Téllez-Castillo CJ, Fernández-Jiménez M, Buesa J. Seroprevalence of Aichi virus in a Spanish population from 2007 to 2008. Clin Vaccine Immunol. (2010) 17:545–9. doi: 10.1128/CVI.00382-09
42. Sdiri-Loulizi K, Ambert-Balay K, Gharbi-Khelifi H, Sakly N, Hassine M, Chouchane S, et al. Molecular epidemiology of norovirus gastroenteritis investigated using samples collected from children in Tunisia during a four-year period: detection of the norovirus variant GGII.4 Hunter as early as January 2003. J Clin Microbiol. (2009) 47:421–9. doi: 10.1128/JCM.01852-08
43. Allocati N, Petrucci AG, Di Giovanni P, Masulli M, Di Ilio C, De Laurenzi V. Bat-man disease transmission: zoonotic pathogens from wildlife reservoirs to human populations. Cell Death Discov. (2016) 2:16048. doi: 10.1038/cddiscovery.2016.48
44. Khamrin P, Maneekarn N, Hidaka S, Kishikawa S, Ushijima K, Okitsu S, et al. Molecular detection of Kobuvirus sequences in stool samples collected from healthy pigs in Japan. Infect Genet Evol. (2010) 10:950–4. doi: 10.1016/j.meegid.2010.06.001
45. Yang Z, Jin W, Zhao Z, Lin W, Zhang D, Yu E, et al. Genetic characterization of porcine Kobuvirus and detection of coinfecting pathogens in diarrheic pigs in Jiangsu Province, China. Arch Virol. (2014) 159:3407–12. doi: 10.1007/s00705-014-2204-2
Keywords: genetic characteristic, Kobuvirus, murine rodent, phylogenetic analysis, prevalence, transmission route
Citation: Gao Y, He W, Fu J, Li Y, He H and Chen Q (2022) Epidemiological Evidence for Fecal-Oral Transmission of Murine Kobuvirus. Front. Public Health 10:865605. doi: 10.3389/fpubh.2022.865605
Received: 02 February 2022; Accepted: 28 March 2022;
Published: 19 April 2022.
Edited by:
Nur A. Hasan, EzBiome Inc., United StatesReviewed by:
Christian Timothy Chimimba, University of Pretoria, South AfricaMd Niaz Rahim, Manitoba Government, Canada
Copyright © 2022 Gao, He, Fu, Li, He and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Chen, MTgwMDIyNzAzMDgmI3gwMDA0MDsxNjMuY29t