 Ronaldo da Silva Francisco Junior1
Ronaldo da Silva Francisco Junior1 Luiz G. P. de Almeida1
Luiz G. P. de Almeida1 Alessandra P. Lamarca1
Alessandra P. Lamarca1 Liliane Cavalcante1
Liliane Cavalcante1 Yasmmin Martins1
Yasmmin Martins1 Alexandra L. Gerber1
Alexandra L. Gerber1 Ana Paula de C. Guimarães1Ricardo Barbosa Salviano2Fernanda Leitão dos Santos2Thiago Henrique de Oliveira2
Ana Paula de C. Guimarães1Ricardo Barbosa Salviano2Fernanda Leitão dos Santos2Thiago Henrique de Oliveira2 Isabelle Vasconcellos de Souza3
Isabelle Vasconcellos de Souza3 Erika Martins de Carvalho3Mario Sergio Ribeiro4Silvia Carvalho4Flávio Dias da Silva5Marcio Henrique de Oliveira Garcia5Leandro Magalhães de Souza6Cristiane Gomes da Silva6Caio Luiz Pereira Ribeiro5Andréa Cony Cavalcanti6Claudia Maria Braga de Mello4
Erika Martins de Carvalho3Mario Sergio Ribeiro4Silvia Carvalho4Flávio Dias da Silva5Marcio Henrique de Oliveira Garcia5Leandro Magalhães de Souza6Cristiane Gomes da Silva6Caio Luiz Pereira Ribeiro5Andréa Cony Cavalcanti6Claudia Maria Braga de Mello4 Amilcar Tanuri2
Amilcar Tanuri2 Ana Tereza R. Vasconcelos1*
Ana Tereza R. Vasconcelos1*- 1Laboratório de Bioinformática, Laboratório Nacional de Computação Científica, Petrópolis, Brazil
- 2Departamento de Genética, Instituto de Biologia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 3Unidades de Apoio ao Diagnóstico da Covid-19, Rio de Janeiro, Brazil
- 4Secretaria Estadual de Saúde do Rio de Janeiro, Rio de Janeiro, Brazil
- 5Secretaria Municipal de Saúde Rio de Janeiro, Rio de Janeiro, Brazil
- 6Laboratório Central de Saúde Pública Noel Nutels, Rio de Janeiro, Brazil
In this study, we report the first case of intra-host SARS-CoV-2 recombination during a coinfection by the variants of concern (VOC) AY.33 (Delta) and P.1 (Gamma) supported by sequencing reads harboring a mosaic of lineage-defining mutations. By using next-generation sequencing reads intersecting regions that simultaneously overlap lineage-defining mutations from Gamma and Delta, we were able to identify a total of six recombinant regions across the SARS-CoV-2 genome within a sample. Four of them mapped in the spike gene and two in the nucleocapsid gene. We detected mosaic reads harboring a combination of lineage-defining mutations from each VOC. To our knowledge, this is the first report of intra-host RNA-RNA recombination between two lineages of SARS-CoV-2, which can represent a threat to public health management during the COVID-19 pandemic due to the possibility of the emergence of viruses with recombinant phenotypes.
Introduction
Since the first reports of patients coinfected by two genetically-distinct lineages of SARS-CoV-2 (1–7), the scientific community raised concerns about the recombination of intra-host viral RNA sequences as a possible mechanism underlying the emergence of novel variants. Indeed, this phenomenon occurs at a relatively high frequency among betacoronaviruses (8–10). In SARS-CoV-2, signals of ongoing recombination were observed in a few sequences deposited in GISAID and circulating in North America (11, 12). Both studies showed evidence of recombination between non-variants of concern. Given the emergence and widespread dispersion of variants of concern (VOC) harboring advantageous mutation able to alter the virus phenotype around the world, the possibility of recombination between them could represent a faster and more dangerous path to increase diversity facilitating the emergence of novel VOCs. The generation of recombinant genomes mainly requires (i) circulation of different lineages in the population, (ii) coinfection events, and (iii) intra-host recombination of the coinfected lineages. For this reason, we continuously monitored the presence of coinfected samples in the bam files of the samples originated from the state of Rio de Janeiro, Brazil. During the routine analysis across the 5,073 SARS-CoV-2 genomes generated by the Corona-ômica-RJ project (http://www.corona-omica.rj.lncc.br/#/), we identified the coinfection between Gamma and Delta reported next.
Case Description
In late July, a 32-year-old male with mild flu-like symptoms of fever, headache, cough, fatigue, and sore throat was referred to the Santo Antônio de Pádua municipal health department in the northeastern region of the state of Rio de Janeiro, Brazil. After 2 days from the onset of symptoms, the diagnosis of COVID-19 was confirmed by the SARS-CoV-2 reverse transcriptase-polymerase chain reaction (RT-PCR) assay of a nasopharyngeal swab. Given the mild symptoms, no hospitalization was required and the patient was quarantined at home. No alterations in blood pressure or saturation were observed during the course of the disease. The patient did not report any comorbidities and had not been vaccinated by the time the sample was collected. The sample's relative quantification of the viral load (RQVL = 2−ΔCT) exhibited elevated values, with an RT-PCR cycle threshold (Ct) of 16.70 corresponding to the top 3% of samples with the highest viral load from Gamma variants in our database (n = 1,881). After 24 days from the symptoms onset, the patient presented the first negative RT-PCR test.
Genomic Evidence of Coinfection and Recombination
Whole-genome sequencing analysis detected 73 intra-host single nucleotide variants (iSNVs) with allele frequency >5% and depth >100 reads. Of these, 26 were lineage-defining mutations exclusively found in the variant of concern (VOC) Gamma (P.1) and 32 were from Delta (AY.33; Supplementary Table 1). By using a hypergeometric distribution approach (6), we estimate an overall haplotype frequency of approximately 16 and 82% for Delta and Gamma, respectively. Nevertheless, two iSNVs characteristics of VOC Delta (ORF1ab: I1091V—AF = 94%; and ORF7b: T40I—AF = 70%) were found with a high frequency in our analysis, suggesting that some genomic copies from Gamma haplotype could also harbor the same mutations (Supplementary Table 1).
We then sought to investigate the occurrence of within-host recombination events between the two lineages using next-generation sequencing. In theory, mutations characteristic of Gamma should be kept in different reads from those defining mutations of Delta. We divided the SARS-CoV-2 genome into windows with at least 100 nucleotides, which is smaller than the read length of the Illumina COVIDSeq Test (Illumina) sequencing kit used in this study. Next, we only selected windows that intersected at least two iSNVs, being one lineage-defining mutation from Gamma and the other from Delta. We aimed to detect unfragmented reads that covered the entire window harboring the iSNVs selected. A total of six recombinant candidate regions across the SARS-CoV-2 genome were found (see Supplementary Material), four mapped in the spike gene and two in the nucleocapsid gene (Table 1; Figure 1). We detected an average of two recombinant haplotypes supported by reads harboring a combination of the discriminating mutations from Gamma and Delta (Table 1; Supplementary Figures 1–6). In addition, the within-host recombinant sequences were formed by a single breakpoint event with a minimal and maximum interval of 15 and 75 nucleotides between two discriminate mutations where an event of recombination was observed, respectively.
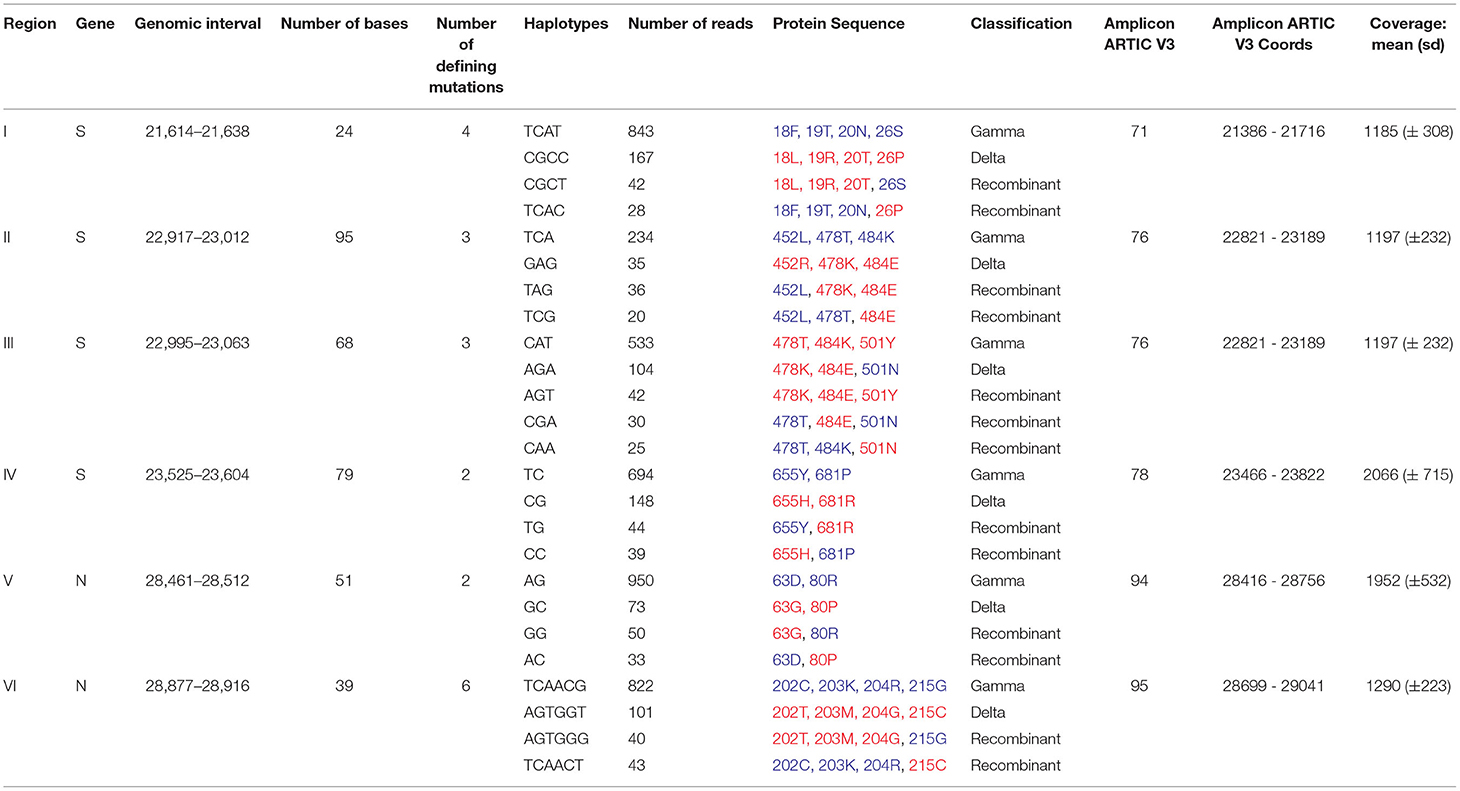
Table 1. Evidence of emergence of recombinant haplotypes within a sample coinfected by Gamma and Delta variants with the mutations characteristic of Gamma and Delta are highlighted in blue and red, respectively.
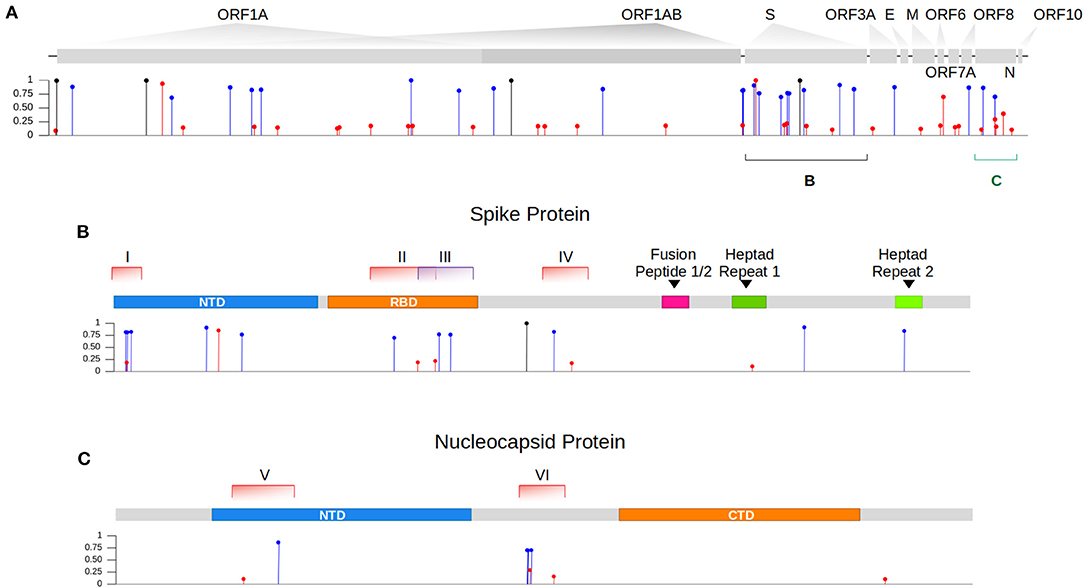
Figure 1. Intra-host single nucleotide variant frequencies evidence coinfection by two genetically distinct lineages. (A) Identification of iSNVs across the different genes of the SARS-CoV-2 genome within our sample. In blue and red, we showed the lineage-defining mutations from the variant Gamma and Delta, respectively. iSNVs represented in black are mutations commonly found in both lineages that descend from their ancestor B.1. (B) Zoom in gene S showing the location of each genomic window (I-IV) where recombination events were investigated. (C) Zoom in gene N showing the distribution of both selected regions (V and VI) across the gene.
From the six regions selected in our analysis, four overlapped the spike gene and two the nucleocapsid gene (Figure 1). All recombinant haplotypes were found at a low frequency compared to the reads supporting the sequences from each VOC (Table 1). Region I is located at the 5' region of the spike gene and includes three lineage-defining mutations from Gamma (S: L18F, T20N, and P26S) and one from Delta (S: T19R; Supplementary Figure 1). We identified two regions (II and III; Supplementary Figures 2, 3) overlapping the receptor-binding domain (RBD). Region II included two amino acid residue alterations from Delta (S: L452R, T478K) and one from Gamma (S: E484K; Supplementary Figure 2). The T478K and E484K mutations were also present in Region III (Supplementary Figure 3), which harbored a second alteration from Gamma (S: N501Y). This region was the only window where three different recombinant haplotypes were observed (Table 1; Supplementary Figure 3). The last region (IV) in the spike includes only one mutation from Gamma (S: H655Y) and one from Delta (S: 681R; Supplementary Figure 4). At the 5′ portion of the nucleocapsid gene, we selected a window (region V; Supplementary Figure 5) containing a single mutation from Delta (N: D63G) and from Gamma (N: P80R). Finally, the largest region selected (VI) was also mapped in N and included four mutations from Gamma (N: S202C, S202T, R203K, G204R) and two from Delta (R203M, G215C; Supplementary Figure 6).
The recombination in our sample was detected during the first reports of Delta in the city of Santo Antônio de Pádua and an ongoing replacement of the variant Gamma by the Delta in the state of Rio de Janeiro. Nevertheless, genomic surveillance data from the state of Rio de Janeiro indicated a frequency of 47% of sequences from Delta and 37% from Gamma (13) in July 2021. Thus, the epidemiological, geographic, and genomic data provide a conducive scenario to SARS-CoV-2 recombination.
Discussion
The designation of VOC prioritizes genome sequences harboring genetic markers with demonstrated evidence for increasing virus transmissibility, virulence, or decreasing the effectiveness of control measures. In this context, the VOC Gamma and Delta were notable by causing an elevated number of Covid-19 cases and deaths in Brazil and worldwide, respectively. Both VOC are known to enhance SARS-CoV-2 transmissibility and induce an immune-escape response. Therefore, recombination between both sequences could represent the emergence of novel variants of concern with new phenotypic combinations. For example, we noticed the presence of the mutation N501Y, a lineage-defining mutation from Gamma, in a haplotype carrying predominantly mutations from Delta in Region III. This mutation is widely known to increase ACE2 binding (14). We also observed the emergence of recombinant haplotypes that lost advantageous alleles such as those from Gamma harboring 484E in region II, 501N in region III, and from Delta carrying 681P in region IV.
Homologous recombination in positive-sense RNA viruses is driven by a copy-choice replication mechanism with a switch from an RNA template by the RNA-polymerase complex during replication (15, 16). Thus, the recombinant haplotypes found across the different regions selected can represent multiple events of switches occurring within a cell or a few switches from RNA templates simultaneously occurring at multiple cells. These variations could be associated with a mechanism of escaping the host's immune response and should be further investigated for the development of antiviral therapy and vaccines. Although the number of reads intersecting the discriminating iSNVs in the recombinant haplotypes seems to be small, we also observed a huge number of reads covering each mutation separately (Supplementary Table 1; median read depth: 1947). Thus, the read coverage and minor allele frequency in each site suggest that these mutations were not caused by low mapping quality or miscalling variant issues.
The hypothesis that the recombination might be the product of laboratory contamination is highly discouraged due to the protocol used by us. All viral particles are inactivated before RNA extraction, which stops replication and, consequently, impairs recombination from happening due to sample contamination. In addition, the high efficiency of ARTIC pool of primers utilized in our library construction rule out the possibility of the recombinant fragment being made by the initial PCR amplification step. Moreover, the 100 nucleotide window used in our genome scan and the similarity of sequence breakpoints analyzed in different genome regions make it difficult to imagine that it can be produced by the PCR step used in our library construction. One alternative speculation about the presence of mosaic reads in our sample is that the sequences could be generated by reverse mutational processes that rescue the ancestral state of the Wuhan reference sequence in each haplotype. Nevertheless, to consider an elevated number of reverse mutations happening at the same time in a coinfected sample would exceed the rate of intra-host evolution previously observed (4, 17). Even if this high rate would be accepted as real, it is unlikely that these mutations would occur only in the “reverse direction” instead of generating a myriad of new substitutions.
Recent studies reported the detection of SARS-CoV-2 recombinant lineages circulating at a low frequency (11, 12, 18–21). Nevertheless, these studies were restricted to detecting the inter-host dissemination of genomes post-recombination events, which could be biased by spurious mutations generated due to library preparation, laboratory contamination, and unavailability of raw sequenced data. Our identification of a reliable recombination event between VOCs raises a red alert for the possible emergence of a new VOC in the future that combines advantageous mutations from independent lineages. Such an event could represent an evolutionary jump of SARS-CoV-2, immediately increasing the fitness of the VOC in a way that this hypothetical variant would fastly dominate current variants and generate a new wave worldwide (as currently seen with Omicron). While the recombination detected in this work involves mutations associated with fitness increase (L452R, E484K, N501Y), the absence of the recombinant sequences in SARS-CoV-2 samples was obtained posteriorly to this case suggests that it was not enough to supplant the circulating variants.
In summary, we report in this work the first case of intra-host SARS-CoV-2 recombination during a coinfection by the VOC Delta (AY.33) and Gamma (P.1) supported by sequencing reads harboring a mosaic of lineage-defining mutations as well as putative sequences breakpoints. We did not detect the circulation of the recombinant sequences in the state, so far. Nevertheless, such identification deeply depends on the access to raw read data from the state of Rio de Janeiro and other locations across the world. The faster detection and surveillance of recombinant SARS-CoV-2 genomes require an increase in the number of sequencing, specially in countries with an elevated number of cases, and rapid deposition of both assembled genomes and raw reads in public databases.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA774631.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee (30161620.0.1001.5257 and 34025020.0.0000.5257). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
ATRV: conceptualization. LC, ALG, APCG, RBS, FLS, and THO: DNA extraction and sequencing. IVS, EMC, MSR, SC, FDS, MHOG, LMS, CGS, CLPR, ACC, and CMBM: project administration. RSFJ, LGPA, APL, and YM: formal analysis. RSFJ and LGPA: data curation. ATRV: funding. RSFJ, APL, LGPA, and ATRV: writing – original draft. AT and ATRV: writing – review and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was developed in the frameworks of Corona-ômica-RSFJ (FAPERJ = E-26/210.179/2020 and E-26/211.107/2021). ATRV is supported by CNPq (303170/2017-4) and FAPERJ (E-26/202.903/20). AT is supported by FAPERJ E-26/010.002434/2019 and E-26/210.178/2020. RSFJ is a recipient of a graduate fellowship from CNPq. APL is granted a post-doctoral scholarship (DTI-A) from CNPq. We acknowledge the support from the Rede Corona-ômica BR MCTI/FINEP affiliated to RedeVírus/MCTI (FINEP 01.20.0029.000462/20, CNPq 404096/2020-4).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the patient for taking part in this study. We also acknowledge the authors and administrators of the GISAID database, which allowed this study of genomic epidemiology to be conducted properly.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2022.849978/full#supplementary-material
References
1. Samoilov AE, Kaptelova VV, Bukharina AY, Shipulina OY, Korneenko EV, Saenko SS, et al. Case report: change of dominant strain during dual SARS-CoV-2 infection. BMC Infect Dis. (2021) 21:959. doi: 10.1186/s12879-021-06664-w
2. Hashim HO, Mohammed MK, Mousa MJ, Abdulameer HH, Alhassnawi ATS, Hassan SA, et al. Unexpected co-infection with different strains of SARS-CoV-2 in patients with COVID-19. Preprints. (2020) 1:1–17. doi: 10.20944/preprints202009.0375.v1
3. Tonkin-Hill G, Martincorena I, Amato R, Lawson AR, Gerstung M, Johnston I, et al. Patterns of within-host genetic diversity in SARS-CoV-2. Elife. (2021) 10:e66857. doi: 10.7554/eLife.66857
4. Lythgoe KA, Hall M, Ferretti L, de Cesare M, MacIntyre-Cockett G, Trebes A, et al. SARS-CoV-2 within-host diversity and transmission. Science. (2021) 372: 0821. doi: 10.1126/science.abg0821
5. Dezordi FZ, Resende PC, Naveca FG, do Nascimento VA, de Souza VC, Paixão ACD, et al. Unusual SARS-CoV-2 intra-host diversity reveals lineages superinfection. MedRxiv [Preprint]. (2021). doi: 10.1101/2021.09.18.21263755
6. Zhou H-Y, Cheng Y-X, Xu L, Li J-Y, Tao C-Y, Ji C-Y, et al. Genomic evidence for divergent co-infections of SARS-CoV-2 lineages. bioRxiv [Preprint]. (2021). doi: 10.1101/2021.09.03.458951
7. Francisco R da S Jr, Benites LF, Lamarca AP, de Almeida LGP, Hansen AW, Gularte JS, et al. Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. (2021) 296:198345. doi: 10.1016/j.virusres.2021.198345
8. Keck JG, Matsushima GK, Makino S, Fleming JO, Vannier DM, Stohlman SA, et al. In vivo RNA-RNA recombination of coronavirus in mouse brain. J Virol. (1988) 62:1810–3. doi: 10.1128/jvi.62.5.1810-1813.1988
9. Dudas G, Rambaut A. MERS-CoV recombination: implications about the reservoir and potential for adaptation. Virus Evol. (2016) 2:vev023. doi: 10.1093/ve/vev023
10. Hon C-C, Lam T-Y, Shi Z-L, Drummond AJ, Yip C-W, Zeng F, et al. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J Virol. (2008) 82:1819–26. doi: 10.1128/JVI.01926-07
11. VanInsberghe D, Neish AS, Lowen AC, Koelle K. Recombinant SARS-CoV-2 genomes circulated at low levels over the first year of the pandemic. Virus Evol. (2021) 7:1–12 doi: 10.1093/ve/veab059
12. Gutierrez B, Castelán Sánchez HG, da Silva Candido D, Jackson B, Fleishon S, Ruis C, et al. Emergence and widespread circulation of a recombinant SARS-CoV-2 lineage in North America. MedRxiv [Preprint]. (2021). doi: 10.1101/2021.11.19.21266601
13. Francisco Junior R da S, Lamarca AP, de Almeida LGP, Cavalcante L, Machado DT, Martins Y, et al. Turnover of SARS-CoV-2 lineages shaped the pandemic and enabled the emergence of new variants in the State of Rio de Janeiro, Brazil. Viruses. (2021) 13:2013. doi: 10.3390/v13102013
14. Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. (2020) 182:1295–310.e20. doi: 10.1016/j.cell.2020.08.012
15. Cooper PD, Steiner-Pryor A, Scotti PD, Delong D. On the nature of poliovirus genetic recombinants. J General Virol. (1974) 23:41–9. doi: 10.1099/0022-1317-23-1-41
16. Worobey M, Holmes EC. Evolutionary aspects of recombination in RNA viruses. J Gen Virol. (1999) 80:2535–43. doi: 10.1099/0022-1317-80-10-2535
17. Voloch CM, da Silva Francisco Jr R, de Almeida LGP, Brustolini OJ, Cardoso CC, Gerber AL, et al. Intra-host evolution during SARS-CoV-2 prolonged infection. Virus Evol. (2021) 7:veab078. doi: 10.1093/ve/veab078
18. Jackson B, Boni MF, Bull MJ, Colleran A, Colquhoun RM, Darby AC, et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell. (2021) 184:5179–88.e8. doi: 10.1016/j.cell.2021.08.014
19. Varabyou A, Pockrandt C, Salzberg SL, Pertea M. Rapid detection of inter-clade recombination in SARS-CoV-2 with Bolotie. Genetics. (2021) 218:1–8. doi: 10.1093/genetics/iyab074
20. Yi H. 2019 novel coronavirus is undergoing active recombination. Clin Infect Dis. (2020) 71:884–7. doi: 10.1093/cid/ciaa219
Keywords: recombination, SARS-CoV-2, COVID-19, iSNVs, coinfection
Citation: Francisco Junior RS, Almeida LGPd, Lamarca AP, Cavalcante L, Martins Y, Gerber AL, Guimarães APdC, Salviano RB, dos Santos FL, de Oliveira TH, de Souza IV, de Carvalho EM, Ribeiro MS, Carvalho S, da Silva FD, Garcia MHdO, de Souza LM, da Silva CG, Ribeiro CLP, Cavalcanti AC, de Mello CMB, Tanuri A and Vasconcelos ATR (2022) Emergence of Within-Host SARS-CoV-2 Recombinant Genome After Coinfection by Gamma and Delta Variants: A Case Report. Front. Public Health 10:849978. doi: 10.3389/fpubh.2022.849978
Received: 06 January 2022; Accepted: 24 January 2022;
Published: 22 February 2022.
Edited by:
Jessica L. Jones, United States Food and Drug Administration, United StatesReviewed by:
Safdar Ali, Aliah University, IndiaTofazzal Islam, Bangabandhu Sheikh Mujibur Rahman Agricultural University, Bangladesh
Maharshi Pandya, Snake Research Institute, India
Setya Haksama, Airlangga University, Indonesia
Copyright © 2022 Francisco Junior, Almeida, Lamarca, Cavalcante, Martins, Gerber, Guimarães, Salviano, dos Santos, de Oliveira, de Souza, de Carvalho, Ribeiro, Carvalho, da Silva, Garcia, de Souza, da Silva, Ribeiro, Cavalcanti, de Mello, Tanuri and Vasconcelos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Tereza R. Vasconcelos, YXRydiYjeDAwMDQwO2xuY2MuYnI=