 Veronica C. Jones
Veronica C. Jones Eric C. Dietze
Eric C. Dietze Tijana Jovanovic-Talisman
Tijana Jovanovic-Talisman Jeannine S. McCune
Jeannine S. McCune Victoria L. Seewaldt
Victoria L. Seewaldt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Public Health , 29 October 2020
Sec. Family Medicine and Primary Care
Volume 8 - 2020 | https://doi.org/10.3389/fpubh.2020.509714
This article is part of the Research Topic Perspectives in Primary Prevention Research for Breast Cancer: A Focus on Gene—Environment Interactions View all 15 articles
Currently, tamoxifen is the only drug approved for reduction of breast cancer risk in premenopausal women. The significant cardiovascular side effects of tamoxifen, coupled with lack of a survival benefit, potential for genotoxicity, and failure to provide a significant risk-reduction for estrogen receptor-negative breast cancer, all contribute to the low acceptance of tamoxifen chemoprevention in premenopausal women at high-risk for breast cancer. While other prevention options exist for postmenopausal women, there is a search for well-tolerated prevention agents that can simultaneously reduce risk of breast cancers, cardiovascular disease, and type-2 diabetes. Metformin is a well-tolerated oral biguanide hypoglycemic agent that is prescribed worldwide to over 120 million individuals with type-2 diabetes. Metformin is inexpensive, safe during pregnancy, and the combination of metformin, healthy lifestyle, and exercise has been shown to be effective in preventing diabetes. There is a growing awareness that prevention drugs and interventions should make the “whole woman healthy.” To this end, current efforts have focused on finding low toxicity alternatives, particularly repurposed drugs for chemoprevention of breast cancer, including metformin. Metformin's mechanisms of actions are complex but clearly involve secondary lowering of circulating insulin. Signaling pathways activated by insulin also drive biologically aggressive breast cancer and predict poor survival in women with breast cancer. The mechanistic rationale for metformin chemoprevention is well-supported by the scientific literature. Metformin is cheap, safe during pregnancy, and has the potential to provide heart-healthy breast cancer prevention. On-going primary and secondary prevention trials will provide evidence whether metformin is effective in preventing breast cancer.
Currently, tamoxifen is the only drug approved for reducing risk of breast cancer in premenopausal women. The approval of tamoxifen was based on the first National Surgical Adjuvant Breast and Bowel Project (NSABP) Breast Cancer Prevention Trial (P1) (1, 2). The P1 trial demonstrated that high-risk women who took tamoxifen had a “50% decrease in the incidence of estrogen receptor-positive breast cancer” (1). Results from the P1 trial underlined the decision of the US Food and Drug Administration (FDA) in October 1998 to approve tamoxifen as a chemoprevention agent for premenopausal high-risk women.
In 2013, the risk reduction benefit of tamoxifen was also shown in a meta-analysis of four randomized controlled trials (3): (1) Royal Marsden (4, 5), (2) International Breast Cancer Intervention Study (IBIS-1) (6, 7), (3) P1 (1, 2), and (4) Italian Randomized Tamoxifen Trial (8, 9). This analysis showed a 33% reduction (p < 0.0001) in all breast cancers (10, 11) in high-risk women who took tamoxifen chemoprevention vs. placebo controls (3). As in the P1 trial, the observed reduction was primarily due a decrease in the numbers ER-positive breast cancer (44% in invasive breast cancers (p < 0.0001) and DCIS (p = 0.009). Although tamoxifen-prevention was given for 5-years, follow-up evaluation of the high-risk subjects provide evidence that the long-term risk-reduction in subjects who took tamoxifen may persist up to 10 years (3).
The benefit of tamoxifen appears to be in risk-reduction of ER+ breast cancer; tamoxifen has failed to demonstrate in high-risk women (1) a significant risk reduction for ER- breast cancer and (2) a survival benefit. An extended analysis (median 16 years) of IBIS-I study participants, continues to shows in the tamoxifen vs. placebo arms “no difference in the number of breast cancer deaths (p = 0.8)” (12).
Despite initial recommendations by the FDA and American Society for Clinical Oncology, very few women take tamoxifen (11); it is estimated that only 5–12% of women offered tamoxifen chemoprevention elect to take tamoxifen (11).
Tamoxifen has been shown to increase risk for cardiovascular events, including venous thrombosis, pulmonary embolism, and stroke, and increases risk for endometrial cancer (12–14). Other side effects of tamoxifen include hot flashes, dyspareunia, depression, cataracts, weight gain, and bone loss in premenopausal women (12–15). Consistent with the increased risk of endometrial cancer in humans, a 2013 study in rats showed that 13-week tamoxifen treatment increased DNA point mutations in the liver (16). Lastly, a concern was raised that tamoxifen may be less active in the 5–10% of individuals who carried homozygous variant of the CYP2D2 gene; this gene variant has low activity to convert tamoxifen to its more active metabolite, 4-hydroxytamoxifen. Lacking in the analysis was a consideration of the concentration of 4-hydroxytamoxifen required to saturate ER; consequently, prospective clinical studies did not demonstrate a reduction in tamoxifen efficacy in individuals with the CYP2D2 variant (17).
While tamoxifen is the only agent approved for breast cancer prevention in premenopausal women, other agents have been approved for postmenopausal women. In the NSABP Study of Tamoxifen and Raloxifene (STAR) trial (raloxifene 60 mg vs. tamoxifen 20 mg), raloxifene was shown to reduce the incidence of breast cancer in postmenopausal women (18). Raloxifene does not increase the risk of endometrial cancer, however, the incidence of ischemic heart disease and stroke was equivalent to the risk associated with tamoxifen (18). IBIS-II tested anastrozole (1.0 mg) vs. placebo in postmenopausal women; the study found a significant decrease in breast cancer in women who took anastrozole; there was no increased incidence of fractures or cardiovascular disease (19). In the Mammary Prevention.3 trial (MAP.3) exemestane (25 mg) vs. placebo in postmenopausal women was associated with a decreased incidence of both ductal carcinoma in situ and invasive breast cancer; with a median follow-up of 3 years, side effects and impact on quality of life were minimal (20).
Women are not just at risk for breast cancer but also face the risk of developing heart disease, obesity, and type-2 diabetes. Furthermore, with the risk of currently available chemoprevention agents potentiating cardiovascular disease, there is a need to identify agents that can effectively target both conditions: breast cancer and cardiovascular disease. To this end, current efforts have focused on finding alternative prevention strategies that have the potential to reduce not just breast cancer but also reduce the risk for cardiometabolic diseases. Potential strategies have included exercise, aspirin, and metformin.
Metformin (1,1-dimethylbiguanide hydrochloride) is a well-tolerated oral agent that is prescribed for first-line treatment of type-2 diabetes (21, 22) and is approved for treatment of polycystic ovary and gestational diabetes (23). Metformin is well-tolerated by the majority of patients; common metformin side effects include lack of appetite, epigastric pain, nausea, and diarrhea (24). The most significant potential side effect is lactic acidosis; consequently, metformin is not prescribed in individuals with kidney and/or liver disease (23, 25). The mechanism of action of metformin remains a topic of current investigations. It is accepted that metformin inhibits hepatic gluconeogenesis and decreases intestinal absorption of glucose, secondarily decreasing circulating insulin (21, 26). Metformin is also thought to indirectly increase insulin sensitivity by increasing peripheral glucose utilization (21).
Until recently, most clinical care has focused on treatment of type-2 diabetes rather than its prevention. However, several well-controlled studies have shown that it is possible to prevent type-2 diabetes through a combination of diet, exercise, and metformin. The Diabetes Prevention Program/Diabetes Prevention Program Outcomes Study (DPP/DPPOS) is the largest and longest clinical trial of metformin for the prevention of type-2 diabetes (27, 28). Study participants in the DPP/DPPOS cohort have over 15 years prospective assessment of the impact of metformin and lifestyle modification on type-2 diabetes, cardiovascular events, safety, and fiscal outcomes (27). Metformin and intensive lifestyle modification resulted in a 50% type-2 diabetes risk-reduction in women with a history of type-2 diabetes (29). Based on findings from the DPP/DPPOS study, in 2014, the American Diabetes Association (ADA) published formal recommendations for prevention of type-2 diabetes (30). Recommendations included: (1) individuals with impaired glucose tolerance or a HgbA1c 5.7–6.4 should be referred to a life-style modification (7% weight loss target) and moderate physical activity (e.g., walking) for 150 min/week (30). These recommendations may also prove beneficial in modifying breast cancer risk; as outlined below, metformin is undergoing testing for primary and secondary breast cancer prevention.
Population-based studies provide evidence that cancer incidence and mortality decreased in individuals with cancer who took metformin (31–33). In a retrospective study of women with breast cancer who received neoadjuvant chemotherapy individuals who took metformin had a higher rate of pathologic complete remission vs. those did not [24 vs. 8%, p = 0.007; (34)]. In a 2014 meta-analysis, individuals who took metformin had a lower incidence of breast cancer (SRR = 0.94; 95% CI, 0.90–0.99) (35). These epidemiologic studies represent a starting point for recent prospective clinical trials testing the impact of metformin on primary and secondary breast cancer prevention.
Epidemiology studies investigating the impact of metformin on breast cancer incidence are limited by several factors. These factors include: (1) racial and ethnic differences in body mass index (BMI), (2) inability of BMI to precisely identify individuals who are metabolically unhealthy, and (3) the heterogeneity of breast cancer as a disease. A BMI ≥30 kg/m2 is the most frequently used measure of adiposity (36). BMI is an inexact measure of risk, particularly when comparing individuals of different race and ethnicity. Muscle tissue weighs significantly more per unit volume than adipose tissue; consequently fit, muscular individuals can be mistakenly identified as overweight (BMI 25–30 kg/m2) or obese.
BMI is not a precise measure of metabolic health. Over the past 20 years, the observation has been made that some individuals with a BMI > 30 kg/m2 are metabolically healthy, “metabolically healthy obese” (37). In contrast to individuals who are obese but metabolically healthy, there are also individuals with a normal BMI (BMI <25 kg/m2) who have abnormal metabolic profiles and are at increased risk for cardiovascular disease and type-2 diabetes. Current definition of metabolically unhealthy individuals with a normal BMI includes (1) BMI <25 kg/m2, (2) insulin-resistance, hypertriglyceridemia, (3) abdominal fat distribution, and (4) elevated blood pressure (37).
Type-2 diabetes is well-established to increase a woman's risk of developing breast cancer. The association between Type-2 diabetes and breast cancer subtypes, however, remains a work in progress, particularly since the majority of studies are underpowered. A case-control study of 916 postmenopausal women with breast cancer cases and 1,094 population-based controls conducted by Garcia-Esquinas et al. found that type-2 diabetes was associated with a 2.25-fold increased risk for triple negative breast cancer (TNBC) (38); this study was limited by a low number of TNBC and the study of only postmenopausal women. The Carolina Breast Cancer Study included 225 women with TNBC; no statistical association was found between type-2 diabetes and TNBC; unfortunately, this study did not test for the association between insulin-resistance and TNBC (39). A case-case study by Lara-Medina et al. of Latinas with breast cancer (469 women with TNBC) found no statistical association between type-2 diabetes and TNBC (40).
The most complete and well-designed epidemiologic study was a retrospective multi-center population-based case-case study of 4,557 women with breast cancer ages 20–69 years old performed by Chen et al.; 1,446 women had TNBC (41). The investigators identified that women with type-2 diabetes had a 38% (95% CI: 1.01–1.89) increased odds of having TNBC (vs. women without type-2 diabetes) (41).
Interestingly, Chen et al. also found that current and extended-time metformin use (13–24 months metformin) within 2 years of diagnosis, increased the odds of a woman having TNBC (OR = 1.54; 95% CI: 1.07–2.22 and OR = 1.80; 95% CI:1.13–2.85, respectively) (41). These latter results are puzzling, given the ability of insulin to activate signaling pathways that drive the aggressive biology of TNBC and the known ability of metformin to lower circulating insulin.
Epidemiologic studies are powerful tools for generating associations but do not test mechanisms. First off, as pointed out by Chen et al., it may be that the women who had the most poorly controlled diabetes (41), were the individuals who had the longest use of metformin; HgbA1c values for these individuals were not reported. While the number of women using metformin were carefully determined, it is not clear that the investigators incorporated insulin-use (insulin-dependent type-2 diabetes) in their risk models. Furthermore, these risk models do not account for individuals with insulin-resistance (Figure 1). Ultimately, the studies by Chen et al. are extremely important because they highlight how complex the associations between metformin-use, insulin-use, and TNBC are likely to be and underscore the importance of window-of-opportunity trials and ongoing prospective metformin prevention trials (such as MA-32, described below).
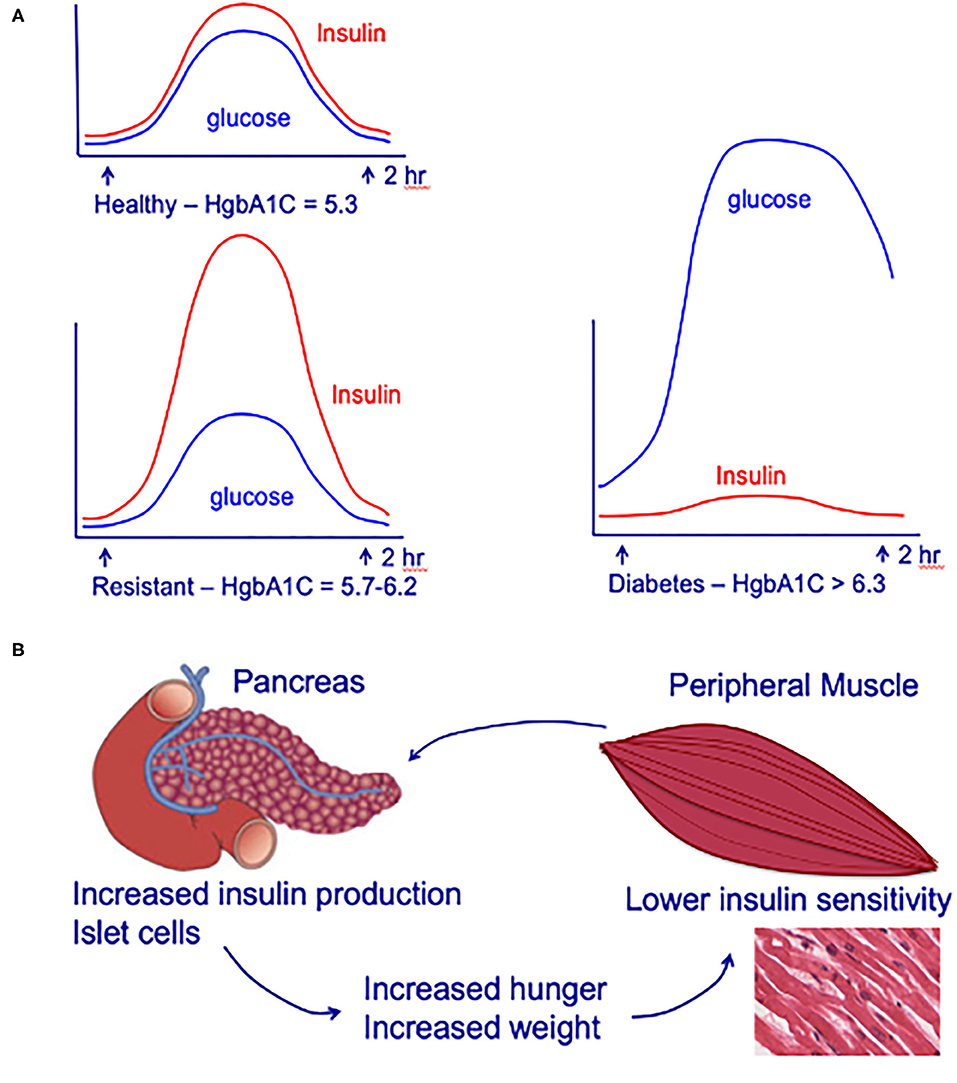
Figure 1. (A) Circulating insulin and glucose levels in healthy individuals (Healthy), insulin-resistant individuals (Resistant), and individuals with type-2 diabetes (Diabetes) at baseline and at 2 h after eating. (B) Impact of insulin-resistance on pancreatic islet cells, peripheral muscle, and individual. Insulin resistance in peripheral muscle tissue results in increased insulin demands from the pancreas. Increased circulating insulin drives hunger and increases weight, leading to a positive feedback loop that increases the chance of an individual developing type-2 diabetes. Adapted from (42).
After oral administration, the oral bioavailability is 55 ± 16% (mean ± standard deviation); metformin is predominantly absorbed in the small intestine (43). Metformin is excreted unchanged in the urine and has a half-life between 4 and 8 h (44). Metformin's absorption and renal clearance is primarily mediated by OCT2/MATE1/MATE2-K (organic cation transporter 2/multidrug and toxin extrusion 1/ multidrug and toxin extrusion 2-K) (45). There are frequent polymorphisms in OCT2, MATE1, and MATE2-K that impact clearance metformin [Table 1; (46, 62)]. Up to 9% of non-Hispanic Whites exhibit an “OCT1 null phenotype” (46). To date, there have been variable findings in pharmacogenomic studies in humans. However, there is evidence that cancer cell lines with high MATE2 expression may be resistant metformin's growth inhibitor effects (63).
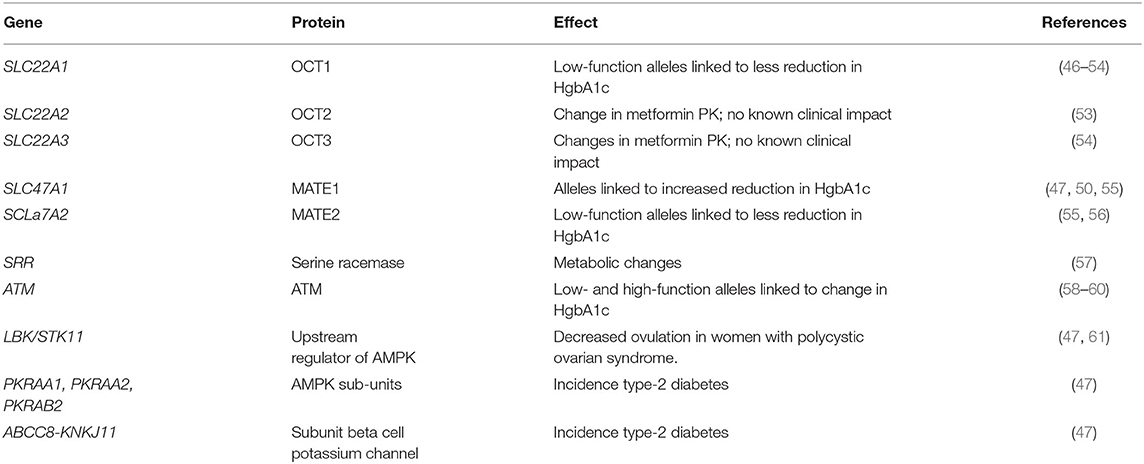
Table 1. Select list of clinically relevant known metformin pharmacokinetic and pharmacodynamic genes.
Despite metformin being one of our oldest medications, the precise molecular mechanism(s) underlying metformin's insulin-lowering effects, as well as its potential anti-neoplastic potential, are not completely understood. It is well-accepted that metformin inhibits hepatic gluconeogenesis and secondarily lowers circulating insulin. However, the precise mechanism(s) of metformin-action remains a work in progress. Two major pathways are thought to account for the main actions of metformin and metformin's proposed anti-cancer effects (Figure 2); both pathways converge on mammalian target of rapamycin (mTOR): (1) AMPK (adenosine monophosphate-activated protein kinase) independent, driven by metformin's ability to secondarily lower serum insulin and (2) AMPK-dependent, regulated by metformin-suppression inhibition of mitochondrial complex-I (complex-I).
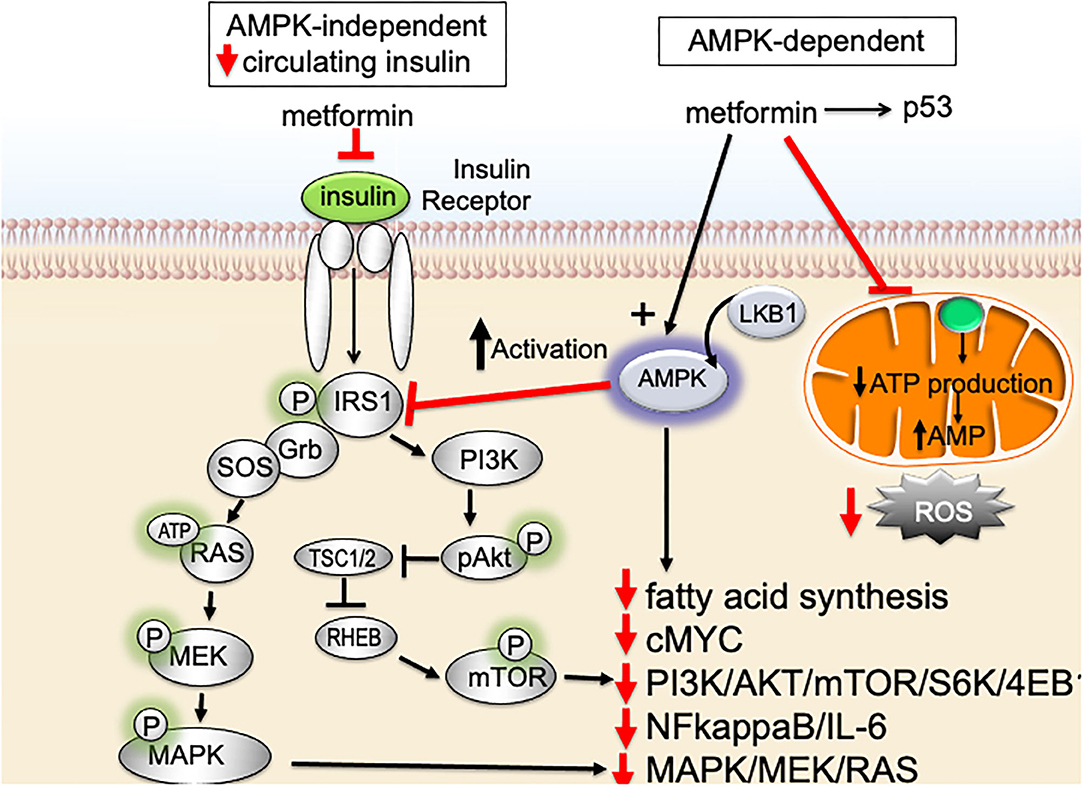
Figure 2. Impact of metformin on insulin-resistance, serum insulin, and signaling pathways important for breast cancer aggressive biology.
Metformin signals via an AMPK-independent pathway; in this pathway metformin secondarily lowers circulating insulin levels and inhibits insulin/insulin-like growth factor-1 (IGF-1)-signaling. Under nutrient-rich circumstances, IGF-1 binds to the IGF-1 receptor (IGF-1R) leading to activation of (1) PI3K (phosphatidylinositol-3-kinase)/AKT/mTOR-network signaling and (2) RAS/RAF/mitogen activated protein kinase (MAPK) [Figure 2; (64)]. Activation of PI3K/MAPK-pathways increase cell proliferation and activates signaling pathways associated with aggressive cancer biology in humans. By lowering circulating insulin, metformin inhibits IGF-1/IGF-1R signaling and inhibits PI3K- and MAPK-signaling pathways (Figure 2).
Metformin also signals through an AMPK-dependent pathway; in this pathway, metformin first inhibits the mitochondrial electron transport protein complex-I (65, 66). Inhibition of complex-I, in turn, blocks production of mitochondrial adenosine-5′-triphosphate (ATP), increases the AMP/ATP ratio, results in a reduction of AMP, and lowers hepatic energy state [Figures 2, 3; (65–69)]. This hepatic energy state restriction leads to AMP binding to AMPK and, thereby, increasing AMPK's affinity for serine-threonine liver kinase B1 (LKB1) (70, 71). AMPK-LKB1-activaiton inhibits AKT/mTOR-network signaling leading to downstream inhibition of S6-Kinase (S6K) and 4E binding protein-1 (4EB-1). Metformin's inhibition of mTOR suppress additional downstream cancer-promoting pathways including (1) Nuclear Factor kappa-light-chain-enhancer of activated B cells NFkB/interleukin-6 (IL6), (2) MAPK/Ras, and (3) cMyc [Figure 2; (64, 72, 73)]. NFkB, IL6, MAPK, Ras, and cMyc together play a role in tissue inflammation, metabolism, and immune cell signaling.
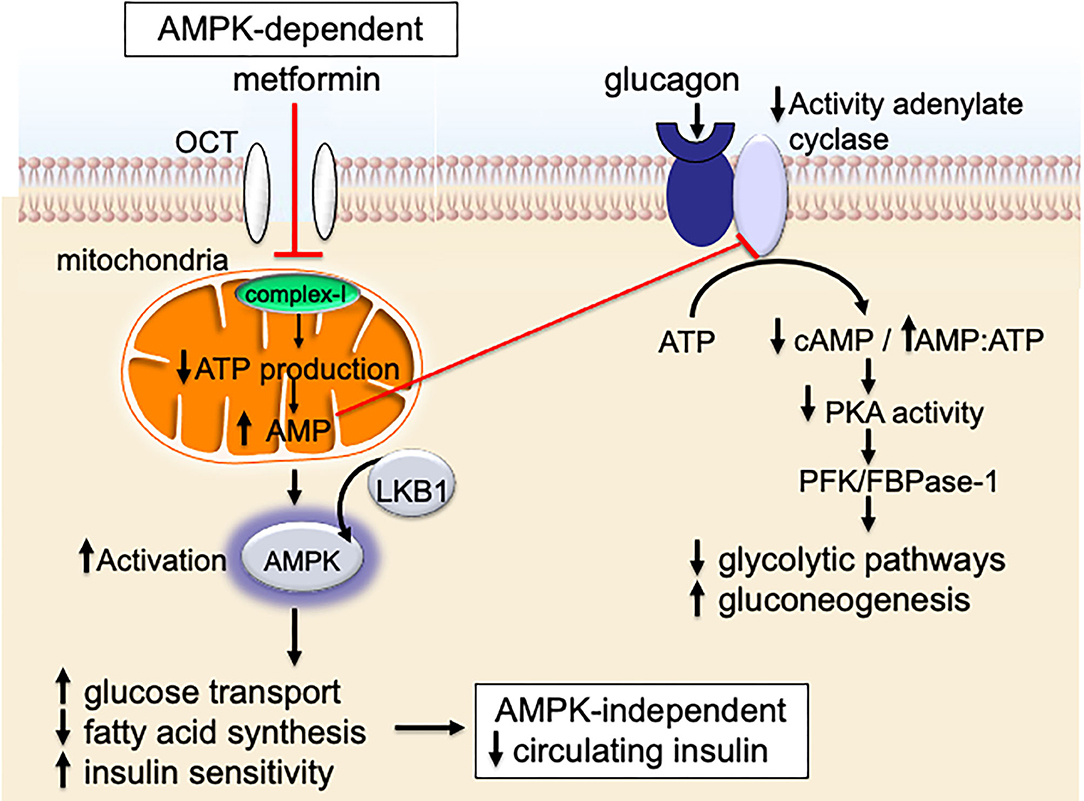
Figure 3. Metformin signaling in the liver.
Increasing attention has been paid to identifying molecular mechanisms that promote chemotherapy-resistance. Kevin Struhl's group first showed in 2009 that 0.1 mM metformin in vitro blocked transformation and killed cancer-like stem cells (74). The combination of metformin and doxorubicin in a mouse xenograft model (metformin 100 μg/ml) exhibited synergy. These results provided a potentially novel mechanism of action for metformin and an experimental rationale for using the combination of metformin and chemotherapy. The metformin doses in this study, however, were supratherapeutic and this very interesting mechanism of metformin-action remains an area of active investigation.
There is also evidence that metformin acts on the tumor microenvironment. Metformin increases intracellular oxygen; this increase is thought to reduce tumor hypoxia (75). Metformin's decrease in hypoxia has been shown to inhibit hypoxia-inducible factor 1 (HIF1) and vascular endothelial growth factor A (VEGFA) driven angiogenesis; there is also evidence for a direct anti-tumor effect on endothelial cells (76, 77). Metformin's increase in tumor oxygenation and or activation of AMPK is thought to shift cancer associated macrophages from a M2 to an M1 phenotype (78). Metformin has been shown to reduce programmed death-ligand 1 (PD-L1) expression on cancer cells, increase lymphocyte anti-tumor cytotoxicity, and downregulate myeloid derived tumor cell activity (79–82). Taken together, these findings highlight a potential role for metformin to be used in concert with immune-therapy.
While the study of metformin's molecular mechanisms of actions remain an area of active research, there is a growing consensus of the key signaling targets of metformin. The following consensus statement for metformin's key mechanisms of actions is updated from Pernicova and Korbonits (83):
• Metformin alters cellular energy metabolism and promotes metabolic reprogramming.
• Metformin acts to lower glucose and increase insulin-sensitivity: (1) primarily by inhibiting hepatic gluconeogenesis and glucagon-signaling and (2) to a lesser degree, in the skeletal muscle by increasing glucose uptake.
• Metformin lowers circulating glucose by inhibiting hepatic gluconeogenesis and opposing glucagon-action.
• Mitochondria complex-1 is a key target of metformin-signaling.
• Antihyperglycemic effect of metformin remains an area of active investigation, more work is needed.
• Metformin impacts lipid metabolism primarily via activation of 5′-AMP-AMPK.
• Anti-cancer effects of metformin are hypothesized to be: (1) indirect—decrease in circulating insulin and (2) direct—energetic stress. However, additional studies are needed.
• Metformin induces energetic stress in cancer cells.
• AMPK-mediation inhibition of mTOR is important for much of metformin's anticancer activity.
• Impact of metformin on cancer stem-like cells needs validation in vivo and in human clinical trials.
• Metformin may have direct and indirect anti-tumor effects on the tumor microenvironment.
In breast cancer, particularly TNBC and basal-type breast cancer, activation of PI3K/AKT/mTOR-signaling pathway is associated with poor prognosis (84, 85). Activation of the PI3K/AKT/mTOR results in cell cycle progression, apoptosis-resistance, and invasion (86, 87). PI3K/AKT/mTOR is a regulator of glucose metabolism and aerobic glycolysis (Warburg effect) (88–90). The Warburg effect is directly linked to aggressive cancer biology due to its impact on glycolysis/glucose-uptake; increased glycolysis/glucose-uptake promotes increased growth, mitochondrial dysfunction, and apoptosis-resistance. Metformin targets the PI3K/AKT/mTOR pathway and promotes metabolic reprogramming. These actions support the use of metformin for prevention of biologically aggressive breast cancers (Figures 2–4).
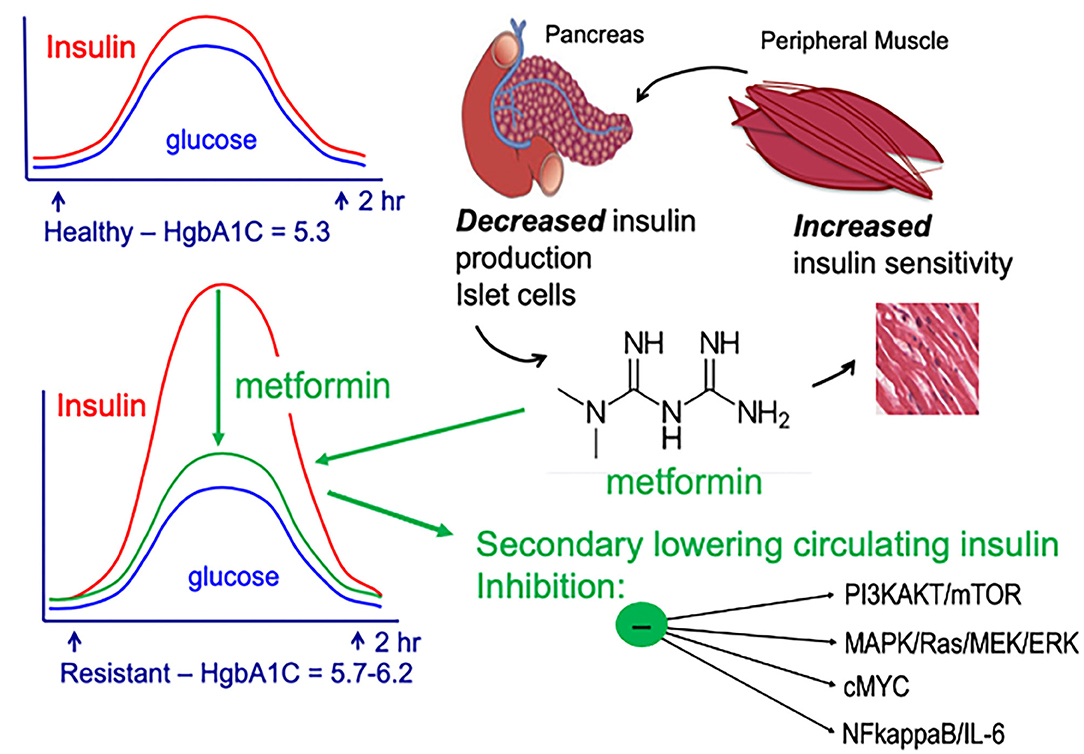
Figure 4. AMPK-dependent and AMPK-independent metformin signaling. Adapted from (42).
Prevention options for premenopausal women who carry a deleterious germline BRCA mutation are limited. There is strong scientific rationale for testing metformin in chemoprevention of breast cancer in BRCA mutation carriers: (1) metformin activates AMPK and (2) signaling networks regulated by both AMPK and BRCA1, include PTEN, p53, and acetyl coenzyme A carboxylase alpha (ACCA) (83, 91, 92). AMPK regulates the phosphorylation/dephosphorylation cycles of ACCA (93, 94). Given that AMPK and BRCA1 both inactivate ACCA, it is hypothesized that metformin might compensate for BRCA1-loss. Further rational for metformin prevention in BRCA1 mutation carriers has been provided by Cuyas et al. (95). Introduction of BRCA1 mutation185delAG in MCF10A cells resulted in metabolic reprograming including (1) mitochondrial activation, (2) increased glucose- and glutamine-dependent activation of the tricarboxylic acid cycle (TCA), and (3) increased production of acetyl-CoA and malonyl-CoA (95). Metformin was shown in vitro to inhibit (1) mitochondrial biosynthetic capacity, (2) the TCA cycle, and (3) generation of lipogeneic precursors. The authors hypothesize that the ability of metformin to block (“starve”) mitochondrial-generated biosynthesis, might provide further rationale for using metformin for cancer prevention in women with germline BRCA1-mutation (95). As described below, to date, the epidemiologic and clinical trials using metformin have yielded conflicting results. The ability of metformin to prevent biologically aggressive breast cancers, particularly TNBC, requires the completion of the on-going prospective trials, such as MA-32.
Dr. Pamela Goodwin has been a pioneer in the use of metformin for lowering insulin and breast cancer chemoprevention; she has developed some of the first trials testing metformin. In a trial of 32 women (4 dropout) with early stage breast cancer and fasting insulin of ≥45 pmol/L and glucose <7.0 mmol/L, administration of metformin 1500 mg per day for 6 months was associated with a 22.4% decrease in serum insulin [p = 0.024; (34)]. This study provided the rational for subsequent randomized clinical trials using metformin vs. placebo.
Window-of-opportunity trials provide important insight into metformin's mechanisms of action but have had conflicting results. In a Scottish trial, Hadad et al. tested the impact of metformin 500 mg ramp up and then 1,000 mg twice a day on Ki-67 and gene expression on 8 pilot women and a further 47 women with primary breast cancer; 7/32 women receiving metformin withdrew due to gastrointestinal upset (96). In women receiving metformin, Ki-67 fell significantly following metformin in both the pilot study (p = 0.041) and in the metformin arm (p = 0.027) but was unchanged in women who did not take metformin (96). Gene expression studies showed a decrease in mRNA expression in genes regulating AMPK; further analysis demonstrated that tumor necrosis factor receptor signaling, and mTOR- and AMPK-signaling were impacted by metformin (96).
The results by Hadad et al. contrast with a second window of opportunity trial. In a double-blind pre-surgical trial Bonanni et al. (2008-004912-10) randomized 200 non-diabetic women to metformin 850 mg/day vs. placebo for 4 weeks prior to surgery (97). Unlike findings by Hadad et al., Bonanni et al. observed no statistical difference in Ki-67 between arms (97). However, there was a differential impact on Ki-67 based on insulin-resistance (measured by homeostatic model assessment—HOMA). In women with HOMA >2.8 there was a 10.5% decrease in mean Ki-67 vs. an 11% increase in women with HOMA <2.8 (p-interaction = 0.045); women with Luminal B breast cancer had the greatest benefit [p = 0.005; (97)]. Further, biomarker analysis showed that this trial represented a significant accomplishment, given the difficulty of coordinating window-of-opportunity trials; importantly, this trial provided a key piece of evidence that non-diabetic metabolically unhealthy women may benefit from metformin chemoprevention (97). A third window-of-opportunity trial reported by Kalinsky et al. in women with early stage breast cancer and a BMI ≥30 reported that in women taking 1,500 mg metformin there were no significant differences in Ki-67 for either DCIS or invasive breast cancer (98). There has been significant discussion about the differences observed in these: trials; one potential difference is that women in the Scottish trial had larger breast cancers and therefore, had larger tumors for analysis [see Kalinsky and Hershman for a more in-depth analysis (99)]. Still, given the short duration of window-of-opportunity trials, longer duration trials with a cancer endpoint are required. See Table 2A for additional clinical and window-of-opportunity metformin trials in women with breast cancer.
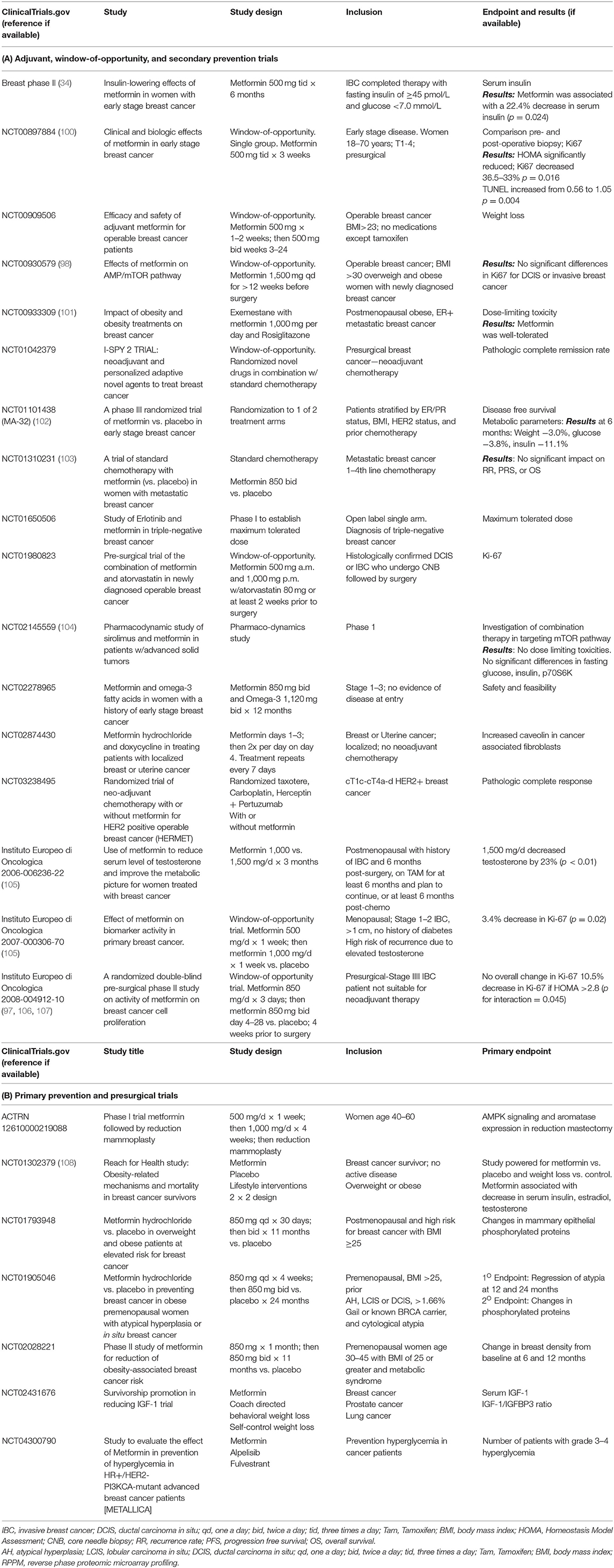
Table 2. Review of metformin in breast cancer treatment or prevention.
Currently many ongoing prospective clinical studies are testing the metformin for primary and secondary prevention of breast cancer (Tables 2A,B). Together, these clinical studies represent an important investment by the National Institute of Health, United States (NIH), European Cancer trials groups, and the National Cancer Institute, Canada (NCIC) (Table 2). The largest adjuvant (secondary prevention) trial is NCIC MA-32, comparing metformin 850 mg p.o. twice a day vs. placebo (NCT01101438) in women with breast cancer; the endpoint of this trial is breast cancer recurrence. After 2,382 women were enrolled, in 2012, the eligibility criteria were amended to mandate TNBC status for patients with T1cN0 disease and at least one adverse tumor characteristic for patients with T2N0 tumors. Interim analysis of the first 500 women taking metformin entered in MA-32, showed at 6 months there was a significant decrease in weight (−3.0%), serum glucose (−3.8%), and serum insulin (−11.1%) (102); further results from this trial are pending. ACTRN12610000219088 is currently testing the impact of metformin (1,000 mg) on LKB1 and AMPK signaling; NCT0430079 tests the impact of metformin in preventing grade 3–4 in (1) men and (2) post-menopausal women receiving treatment for ER/PR+, HER2-not amplified advanced breast cancer, with a PI3K-mutation [METALLICA trial]. Primary prevention studies include (1) NCT01793948: randomized testing the impact of metformin on postmenopausal women with high breast density, (2) NCT01905046: metformin vs. placebo in high-risk premenopausal women (including BRCA mutation carriers) with cytologic atypia, and (3) NCT01905046: randomized testing of whether metformin alters breast density, serum IGF-1/IGFBP-e ratios, IGF-2, and leptin/adiponectin ratios, body weight/body composition (109). See Table 2B for additional trials. Given the wealth of primary and secondary metformin chemoprevention trials, it is anticipated that over the next 5 years, these trials will provide important insights into whether metformin is a viable chemoprevention agent for breast cancer.
Metformin is cheap, safe during pregnancy, and has shown to prevent type-2 diabetes. There is a need for prevention drugs that target both ER+ and ER- breast cancer as well as providing prevention for cardiometabolic disease. Metformin clearly lowers insulin-signaling; signaling pathways activated by insulin are known to drive biologically aggressive breast cancer and predict poor survival in women with breast cancer. Despite the fact that metformin targets many key breast cancer pathways, there is much to be learned about whether metformin can prevent breast cancer and/or breast cancer recurrence. Window-of-opportunity trials provide important clues to metformin's impact on normal and malignant breast tissue, but results have not been entirely consistent. Currently, it is unclear which breast cancer subtypes may benefit the most from metformin. It is likely that MA-32 will provide answers to many of these questions. There is also much to be learned about metformin, insulin resistance, and BMI; specifically, whether metformin's impact is only in women who are metabolically unhealthy and/or have high BMI, or whether metformin can benefit all women. Biomarker studies that define key signaling pathways impacted by metformin will be critical to design and inform future clinical trials. Over the next 5 years on-going primary and secondary prevention trials will show (or not show) the ability of metformin to prevent breast cancer. Hopefully, these studies will not just provide a yes/no answer also provide the biomarkers to determine which women will maximally benefit from metformin. In the words of several of my patients “Please do not quote statistics at me; these statistics are about other women. If I take a prevention agent, I want to know if the prevention agent is working in my breasts.”
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by National Institutes of Health/National Cancer Institute (NIH/NCI) grants R01CA170851, P20CA24619, R01CA192914, and U01CA189283 (all to VS) and P30CA033572-Biomarkers Core. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and bowel project P-1 study. J Natl Cancer Inst. (1998) 90:1371–88. doi: 10.1093/jnci/90.18.1371
2. Fisher B, Costantino JP, Wickerham DL, Cecchini RS, Cronin WM, Robidoux A, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and bowel project P-1 study. J Natl Cancer Inst. (2005) 97:1652–62. doi: 10.1093/jnci/dji372
3. Cuzick J, Sestak I, Bonanni B, Costantino JP, Cummings S, DeCensi A, et al. Selective oestrogen receptor modulators in prevention of breast cancer: an updated meta-analysis of individual participant data. Lancet. (2013) 381:1827–34. doi: 10.1016/S0140-6736(13)60140-3
4. Powles T, Eeles R, Ashley S, Easton D, Chang J, Dowsett M, et al. Interim analysis of the incidence of breast cancer in the Royal Marsden Hospital tamoxifen randomised chemoprevention trial. Lancet. (1998) 352:98–101. doi: 10.1016/S0140-6736(98)85012-5
5. Powles TJ, Ashley S, Tidy A, Smith IE, Dowsett M. Twenty-year follow-up of the Royal Marsden randomized, double-blinded tamoxifen breast cancer prevention trial. J Natl Cancer Inst. (2007) 99:283–90. doi: 10.1093/jnci/djk050
6. Cuzick J, Forbes J, Edwards R, Baum M, Cawthorn S, Coates A, et al. First results from the International Breast Cancer Intervention Study (IBIS-I): a randomised prevention trial. Lancet. (2002) 360:817–24. doi: 10.1016/S0140-6736(02)09962-2
7. Cuzick J, Forbes JF, Sestak I, Cawthorn S, Hamed H, Holli K, et al. Long-term results of tamoxifen prophylaxis for breast cancer−96-month follow-up of the randomized IBIS-I trial. J Natl Cancer Inst. (2007) 99:272–82. doi: 10.1093/jnci/djk049
8. Veronesi U, Maisonneuve P, Costa A, Sacchini V, Maltoni C, Robertson C, et al. Prevention of breast cancer with tamoxifen: preliminary findings from the Italian randomised trial among hysterectomised women. Italian Tamoxifen Prevention study. Lancet. (1998) 352:93–7. doi: 10.1016/S0140-6736(98)04394-3
9. Veronesi U, Maisonneuve P, Rotmensz N, Bonanni B, Boyle P, Viale G, et al. Tamoxifen for the prevention of breast cancer: late results of the Italian Randomized Tamoxifen prevention trial among women with hysterectomy. J Natl Cancer Inst. (2007) 99:727–37. doi: 10.1093/jnci/djk154
10. Taylor R, Taguchi K. Tamoxifen for breast cancer chemoprevention: low uptake by high-risk women after evaluation of a breast lump. Ann Fam Med. (2005) 3:242–7. doi: 10.1370/afm.284
11. Donnelly LS, Evans DG, Wiseman J, Fox J, Greenhalgh R, Affen J, et al. Uptake of tamoxifen in consecutive premenopausal women under surveillance in a high-risk breast cancer clinic. Br J Cancer. (2014) 110:1681–7. doi: 10.1038/bjc.2014.109
12. Cuzick J, Sestak I, Cawthorn S, Hamed H, Holli K, Howell A, et al. Tamoxifen for prevention of breast cancer: extended long-term follow-up of the IBIS-I breast cancer prevention trial. Lancet Oncol. (2015) 16:67–75. doi: 10.1016/S1470-2045(14)71171-4
13. Francis PA, Regan MM, Fleming GF. Adjuvant ovarian suppression in premenopausal breast cancer. N Engl J Med. (2015) 372:1673. doi: 10.1056/NEJMc1502618
14. Prasad V, Diener-West M. Primary chemoprevention of breast cancer: are the adverse effects too burdensome? CMAJ. (2015) 187:E276–8. doi: 10.1503/cmaj.141627
15. Sestak I, Harvie M, Howell A, Forbes JF, Dowsett M, Cuzick J. Weight change associated with anastrozole and tamoxifen treatment in postmenopausal women with or at high risk of developing breast cancer. Breast Cancer Res Treat. (2012) 134:727–34. doi: 10.1007/s10549-012-2085-6
16. Kawamura Y, Hayashi H, Kurata Y, Hiratsuka K, Masumura K, Nohmi T. Evaluation of the genotoxicity of tamoxifen in the liver and kidney of F344 gpt delta transgenic rat in 3-week and 13-week repeated dose studies. Toxicology. (2013) 312:56–62. doi: 10.1016/j.tox.2013.07.014
17. Regan MM, Leyland-Jones B, Bouzyk M, Pagani O, Tang W, Kammler R, et al. CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: the breast international group 1-98 trial. J Natl Cancer Inst. (2012) 104:441–51. doi: 10.1093/jnci/djs125
18. Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP study of tamoxifen and raloxifene (STAR) P-2 trial. JAMA. (2006) 295:2727–41. doi: 10.1001/jama.295.23.joc60074
19. Cuzick J, Sestak I, Forbes JF, Dowsett M, Cawthorn S, Mansel RE, et al. Use of anastrozole for breast cancer prevention (IBIS-II): long-term results of a randomised controlled trial. Lancet. (2020) 395:117–22. doi: 10.1016/S0140-6736(19)32955-1
20. Goss PE, Ingle JN, Ales-Martinez JE, Cheung AM, Chlebowski RT, Wactawski-Wende J, et al. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med. (2011) 364:2381–91. doi: 10.1056/NEJMoa1103507
21. Maruthur NM, Tseng E, Hutfless S, Wilson LM, Suarez-Cuervo C, Berger Z, et al. Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med. (2016) 164:740–51. doi: 10.7326/M15-2650
23. Lautatzis ME, Goulis DG, Vrontakis M. Efficacy and safety of metformin during pregnancy in women with gestational diabetes mellitus or polycystic ovary syndrome: a systematic review. Metabolism. (2013) 62:1522–34. doi: 10.1016/j.metabol.2013.06.006
24. Triggle CR, Ding H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol. (2017) 219:138–51. doi: 10.1111/apha.12644
25. Lipska KJ, Bailey CJ, Inzucchi SE. Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care. (2011) 34:1431–7. doi: 10.2337/dc10-2361
26. Horakova O, Kroupova P, Bardova K, Buresova J, Janovska P, Kopecky J, et al. Metformin acutely lowers blood glucose levels by inhibition of intestinal glucose transport. Sci Rep. (2019) 9:6156. doi: 10.1038/s41598-019-42531-0
27. Aroda VR, Knowler WC, Crandall JP, Perreault L, Edelstein SL, Jeffries SL, et al. Metformin for diabetes prevention: insights gained from the diabetes prevention program/diabetes prevention program outcomes study. Diabetologia. (2017) 60:1601–11. doi: 10.1007/s00125-017-4361-9
28. Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. (2002) 346:393–403. doi: 10.1056/NEJMoa012512
29. Ratner RE, Christophi CA, Metzger BE, Dabelea D, Bennett PH, Pi-Sunyer X, et al. Prevention of diabetes in women with a history of gestational diabetes: effects of metformin and lifestyle interventions. J Clin Endocrinol Metab. (2008) 93:4774–9. doi: 10.1210/jc.2008-0772
30. American Diabetes A. 3. Prevention or delay of type 2 Diabetes: standards of medical care in Diabetes-2020. Diabetes Care. (2020) 43(Suppl. 1):S32–6. doi: 10.2337/dc20-S003
31. Zhou YY, Zhu GQ, Liu T, Zheng JN, Cheng Z, Zou TT, et al. Systematic review with network meta-analysis: antidiabetic medication and risk of hepatocellular carcinoma. Sci Rep. (2016) 6:33743. doi: 10.1038/srep33743
32. Yu H, Zhong X, Gao P, Shi J, Wu Z, Guo Z, et al. The potential effect of metformin on cancer: an umbrella review. Front Endocrinol. (2019) 10:617. doi: 10.3389/fendo.2019.00617
33. Rennert G, Rennert HS, Gronich N, Pinchev M, Gruber SB. Use of metformin and risk of breast and colorectal cancer. Diabetes Res Clin Pract. (2020) 165:108232. doi: 10.1016/j.diabres.2020.108232
34. Goodwin PJ, Pritchard KI, Ennis M, Clemons M, Graham M, Fantus IG. Insulin-lowering effects of metformin in women with early breast cancer. Clin Breast Cancer. (2008) 8:501–5. doi: 10.3816/CBC.2008.n.060
35. Gandini S, Puntoni M, Heckman-Stoddard BM, Dunn BK, Ford L, DeCensi A, et al. Metformin and cancer risk and mortality: a systematic review and meta-analysis taking into account biases and confounders. Cancer Prev Res. (2014) 7:867–85. doi: 10.1158/1940-6207.CAPR-13-0424
36. Heymsfield SB, Peterson CM, Thomas DM, Heo M, Schuna JM Jr. Why are there race/ethnic differences in adult body mass index-adiposity relationships? A quantitative critical review. Obes Rev. (2016) 17:262–75. doi: 10.1111/obr.12358
37. Brandao I, Martins MJ, Monteiro R. Metabolically healthy obesity-heterogeneity in definitions and unconventional factors. Metabolites. (2020) 10:48. doi: 10.3390/metabo10020048
38. Garcia-Esquinas E, Guino E, Castano-Vinyals G, Perez-Gomez B, Llorca J, Altzibar JM, et al. Association of diabetes and diabetes treatment with incidence of breast cancer. Acta Diabetol. (2016) 53:99–107. doi: 10.1007/s00592-015-0756-6
39. Millikan RC, Newman B, Tse CK, Moorman PG, Conway K, Dressler LG, et al. Epidemiology of basal-like breast cancer. Breast Cancer Res Treat. (2008) 109:123–39. doi: 10.1007/s10549-007-9632-6
40. Lara-Medina F, Perez-Sanchez V, Saavedra-Perez D, Blake-Cerda M, Arce C, Motola-Kuba D, et al. Triple-negative breast cancer in Hispanic patients: high prevalence, poor prognosis, and association with menopausal status, body mass index, and parity. Cancer. (2011) 117:3658–69. doi: 10.1002/cncr.25961
41. Chen H, Cook LS, Tang MC, Hill DA, Wiggins CL, Li CI. Relationship between diabetes and diabetes medications and risk of different molecular subtypes of breast cancer. Cancer Epidemiol Biomarkers Prev. (2019) 28:1802–8. doi: 10.1158/1055-9965.EPI-19-0291
42. Yee LD, Mortimer JE, Natarajan R, Dietze EC, Seewaldt VL. Metabolic health, insulin, and breast cancer: why oncologists should care about insulin. Front Endocinol. (2020) 20:58. doi: 10.3389/fendo.2020.00058
43. Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. (2011) 50:81–98. doi: 10.2165/11534750-000000000-00000
44. Pawlyk AC, Giacomini KM, McKeon C, Shuldiner AR, Florez JC. Metformin pharmacogenomics: current status and future directions. Diabetes. (2014) 63:2590–9. doi: 10.2337/db13-1367
45. Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. (2012) 22:820–7. doi: 10.1097/FPC.0b013e3283559b22
46. Sundelin E, Gormsen LC, Jensen JB, Vendelbo MH, Jakobsen S, Munk OL, et al. Genetic polymorphisms in organic cation transporter 1 attenuates hepatic metformin exposure in humans. Clin Pharmacol Ther. (2017) 102:841–8. doi: 10.1002/cpt.701
47. Jablonski KA, McAteer JB, de Bakker PI, Franks PW, Pollin TI, Hanson RL, et al. Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes. (2010) 59:2672–81. doi: 10.2337/db10-0543
48. Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. (2007) 117:1422–31. doi: 10.1172/JCI30558
49. Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. (2008) 83:273–80. doi: 10.1038/sj.clpt.6100275
50. Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH. Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. Pharmacogenomics J. (2009) 9:242–7. doi: 10.1038/tpj.2009.15
51. Christensen MM, Brasch-Andersen C, Green H, Nielsen F, Damkier P, Beck-Nielsen H, et al. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet Genomics. (2011) 21:837–50. doi: 10.1097/FPC.0b013e32834c0010
52. Gambineri A, Tomassoni F, Gasparini DI, Di Rocco A, Mantovani V, Pagotto U, et al. Organic cation transporter 1 polymorphisms predict the metabolic response to metformin in women with the polycystic ovary syndrome. J Clin Endocrinol Metab. (2010) 95:E204–8. doi: 10.1210/jc.2010-0145
53. Shikata E, Yamamoto R, Takane H, Shigemasa C, Ikeda T, Otsubo K, et al. Human organic cation transporter (OCT1 and OCT2) gene polymorphisms and therapeutic effects of metformin. J Hum Genet. (2007) 52:117–22. doi: 10.1007/s10038-006-0087-0
54. Tzvetkov MV, Vormfelde SV, Balen D, Meineke I, Schmidt T, Sehrt D, et al. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin Pharmacol Ther. (2009) 86:299–306. doi: 10.1038/clpt.2009.92
55. Stocker SL, Morrissey KM, Yee SW, Castro RA, Xu L, Dahlin A, et al. The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther. (2013) 93:186–94. doi: 10.1038/clpt.2012.210
56. Choi JH, Yee SW, Ramirez AH, Morrissey KM, Jang GH, Joski PJ, et al. A common 5′-UTR variant in MATE2-K is associated with poor response to metformin. Clin Pharmacol Ther. (2011) 90:674–84. doi: 10.1038/clpt.2011.165
57. Dong M, Gong ZC, Dai XP, Lei GH, Lu HB, Fan L, et al. Serine racemase rs391300 G/A polymorphism influences the therapeutic efficacy of metformin in Chinese patients with diabetes mellitus type 2. Clin Exp Pharmacol Physiol. (2011) 38:824–9. doi: 10.1111/j.1440-1681.2011.05610.x
58. van Leeuwen N, Nijpels G, Becker ML, Deshmukh H, Zhou K, Stricker BH, et al. A gene variant near ATM is significantly associated with metformin treatment response in type 2 diabetes: a replication and meta-analysis of five cohorts. Diabetologia. (2012) 55:1971–7. doi: 10.1007/s00125-012-2537-x
59. GoDarts, Group UDPS, Wellcome Trust Case Control C, Zhou K, Bellenguez C, Spencer CC, et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. (2011) 43:117–20. doi: 10.1038/ng.735
60. Tkac I. Replication of the association of gene variant near ATM and response to metformin. Pharmacogenomics. (2012) 13:1331–2. doi: 10.2217/pgs.12.115
61. Legro RS. Impact of metformin, oral contraceptives, and lifestyle modification on polycystic ovary syndrome in obese adolescent women: do we need a new drug? J Clin Endocrinol Metab. (2008) 93:4218–20. doi: 10.1210/jc.2008-1994
62. Mannino GC, Andreozzi F, Sesti G. Pharmacogenetics of type 2 diabetes mellitus, the route toward tailored medicine. Diabetes Metab Res Rev. (2019) 35:e3109. doi: 10.1002/dmrr.3109
63. Chowdhury S, Yung E, Pintilie M, Muaddi H, Chaib S, Yeung M, et al. MATE2 expression is associated with cancer cell response to metformin. PLoS ONE. (2016) 11:e0165214. doi: 10.1371/journal.pone.0165214
64. Heckman-Stoddard BM, Gandini S, Puntoni M, Dunn BK, DeCensi A, Szabo E. Repurposing old drugs to chemoprevention: the case of metformin. Semin Oncol. (2016) 43:123–33. doi: 10.1053/j.seminoncol.2015.09.009
65. Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. (2014) 510:542–6. doi: 10.1038/nature13270
66. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. (2005) 310:1642–6. doi: 10.1126/science.1120781
67. Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. (2006) 3:403–16. doi: 10.1016/j.cmet.2006.05.005
68. Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. (2006) 116:817–24. doi: 10.1172/JCI27300
69. Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. (2013) 19:1649–54. doi: 10.1038/nm.3372
70. Hardie DG. Neither LKB1 nor AMPK are the direct targets of metformin. Gastroenterology. (2006) 131:973. doi: 10.1053/j.gastro.2006.07.032
71. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. (2014) 20:953–66. doi: 10.1016/j.cmet.2014.09.018
72. Mishra AK, Dingli D. Metformin inhibits IL-6 signaling by decreasing IL-6R expression on multiple myeloma cells. Leukemia. (2019) 33:2695–709. doi: 10.1038/s41375-019-0470-4
73. Sekino N, Kano M, Matsumoto Y, Sakata H, Akutsu Y, Hanari N, et al. Antitumor effects of metformin are a result of inhibiting nuclear factor kappa B nuclear translocation in esophageal squamous cell carcinoma. Cancer Sci. (2018) 109:1066–74. doi: 10.1111/cas.13523
74. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. (2009) 69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994
75. Kurelac I, Umesh Ganesh N, Iorio M, Porcelli AM, Gasparre G. The multifaceted effects of metformin on tumor microenvironment. Semin Cell Dev Biol. (2020) 98:90–7. doi: 10.1016/j.semcdb.2019.05.010
76. Ye J, Chen K, Qi L, Li R, Tang H, Zhou C, et al. Metformin suppresses hypoxiainduced migration via the HIF1alpha/VEGF pathway in gallbladder cancer in vitro and in vivo. Oncol Rep. (2018) 40:3501–10. doi: 10.3892/or.2018.6751
77. Wang J, Li G, Wang Y, Tang S, Sun X, Feng X, et al. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF-1alpha/VEGF secretion axis. Oncotarget. (2015) 6:44579–92. doi: 10.18632/oncotarget.6373
78. Liu Q, Tong D, Liu G, Gao J, Wang LA, Xu J, et al. Metformin inhibits prostate cancer progression by targeting tumor-associated inflammatory infiltration. Clin Cancer Res. (2018) 24:5622–34. doi: 10.1158/1078-0432.CCR-18-0420
79. Incio J, Tam J, Rahbari NN, Suboj P, McManus DT, Chin SM, et al. PlGF/VEGFR-1 signaling promotes macrophage polarization and accelerated tumor progression in obesity. Clin Cancer Res. (2016) 22:2993–3004. doi: 10.1158/1078-0432.CCR-15-1839
80. Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res. (2017) 5:9–16. doi: 10.1158/2326-6066.CIR-16-0103
81. Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. (2018) 78:1779–91. doi: 10.1158/0008-5472.CAN-17-2460
82. Kim SH, Li M, Trousil S, Zhang Y, Pasca di Magliano M, Swanson KD, et al. phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J Invest Dermatol. (2017) 137:1740–8. doi: 10.1016/j.jid.2017.03.033
83. Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. (2014) 10:143–56. doi: 10.1038/nrendo.2013.256
84. Yang SX, Polley E, Lipkowitz S. New insights on PI3K/AKT pathway alterations and clinical outcomes in breast cancer. Cancer Treat Rev. (2016) 45:87–96. doi: 10.1016/j.ctrv.2016.03.004
85. Sobhani N, Roviello G, Corona SP, Scaltriti M, Ianza A, Bortul M, et al. The prognostic value of PI3K mutational status in breast cancer: a meta-analysis. J Cell Biochem. (2018) 119:4287–92. doi: 10.1002/jcb.26687
86. Xue G, Hemmings BA. PKB/Akt-dependent regulation of cell motility. J Natl Cancer Inst. (2013) 105:393–404. doi: 10.1093/jnci/djs648
87. Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. (2020) 20:74–88. doi: 10.1038/s41568-019-0216-7
88. Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. (2009) 19:25–31. doi: 10.1016/j.semcancer.2008.11.010
89. Wu Z, Wu J, Zhao Q, Fu S, Jin J. Emerging roles of aerobic glycolysis in breast cancer. Clin Transl Oncol. (2020) 22:631–46. doi: 10.1007/s12094-019-02187-8
90. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in Cancer. Recent Results Cancer Res. (2016) 207:39–72. doi: 10.1007/978-3-319-42118-6_3
91. Fabian CJ, Kimler BF. Chemoprevention for high-risk women: tamoxifen and beyond. Breast J. (2001) 7:311–20. doi: 10.1046/j.1524-4741.2001.21570.x
92. Kumar NB, Vadaparampil ST, Mahajan N, Lilienfeld HS, Lee JH, Laronga C, et al. Metformin- a promising agent for chemoprevention in BRCA1 carriers. Hereditary Genet. (2012) 1:104. doi: 10.4172/2161-1041.1000104
93. Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. (2003) 144:5179–83. doi: 10.1210/en.2003-0982
94. Dasgupta B, Chhipa RR. Evolving lessons on the complex role of AMPK in normal physiology and cancer. Trends Pharmacol Sci. (2016) 37:192–206. doi: 10.1016/j.tips.2015.11.007
95. Cuyas E, Fernandez-Arroyo S, Alarcon T, Lupu R, Joven J, Menendez JA. Germline BRCA1 mutation reprograms breast epithelial cell metabolism towards mitochondrial-dependent biosynthesis: evidence for metformin-based “starvation” strategies in BRCA1 carriers. Oncotarget. (2016) 7:52974–92. doi: 10.18632/oncotarget.9732
96. Hadad S, Iwamoto T, Jordan L, Purdie C, Bray S, Baker L, et al. Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res Treat. (2011) 128:783–94. doi: 10.1007/s10549-011-1612-1
97. Bonanni B, Puntoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gonzaga A, et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J Clin Oncol. (2012) 30:2593–600. doi: 10.1200/JCO.2011.39.3769
98. Kalinsky K, Crew KD, Refice S, Xiao T, Wang A, Feldman SM, et al. Presurgical trial of metformin in overweight and obese patients with newly diagnosed breast cancer. Cancer Invest. (2014) 32:150–7. doi: 10.3109/07357907.2014.889706
99. Kalinsky K, Hershman DL. Cracking open window of opportunity trials. J Clin Oncol. (2012) 30:2573–5. doi: 10.1200/JCO.2012.42.3293
100. Niraula S, Dowling RJ, Ennis M, Chang MC, Done SJ, Hood N, et al. Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res Treat. (2012) 135:821–30. doi: 10.1007/s10549-012-2223-1
101. Esteva FJ, Moulder SL, Gonzalez-Angulo AM, Ensor J, Murray JL, Green MC, et al. Phase I trial of exemestane in combination with metformin and rosiglitazone in nondiabetic obese postmenopausal women with hormone receptor-positive metastatic breast cancer. Cancer Chemother Pharmacol. (2013) 71:63–72. doi: 10.1007/s00280-012-1977-9
102. Goodwin PJ, Parulekar WR, Gelmon KA, Shepherd LE, Ligibel JA, Hershman DL, et al. Effect of metformin vs placebo on and metabolic factors in NCIC CTG MA.32. J Natl Cancer Inst. (2015) 107:djv006. doi: 10.1093/jnci/djv006
103. Pimentel I, Lohmann AE, Ennis M, Dowling RJO, Cescon D, Elser C, et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast. (2019) 48:17–23. doi: 10.1016/j.breast.2019.08.003
104. Sehdev A, Zha Y, Karrison TG, Janisch LA, Cohen EEW, Maitland ML, et al. A pharmacodynamic study of sirolimus and metformin in patients with advanced solid tumors. J Clin Oncol. (2017) 35(Suppl. 15):TPS11628. doi: 10.1200/JCO.2017.35.15_suppl.TPS11628
105. Campagnoli C, Pasanisi P, Abba C, Ambroggio S, Biglia N, Brucato T, et al. Effect of different doses of metformin on serum testosterone and insulin in non-diabetic women with breast cancer: a randomized study. Clin Breast Cancer. (2012) 12:175–82. doi: 10.1016/j.clbc.2012.03.004
106. Macis D, Gandini S, Guerrieri-Gonzaga A, Johansson H, Magni P, Ruscica M, et al. Prognostic effect of circulating adiponectin in a randomized 2 x 2 trial of low-dose tamoxifen and fenretinide in premenopausal women at risk for breast cancer. J Clin Oncol. (2012) 30:151–7. doi: 10.1200/JCO.2011.35.2237
107. Cazzaniga M, DeCensi A, Pruneri G, Puntoni M, Bottiglieri L, Varricchio C, et al. The effect of metformin on apoptosis in a breast cancer presurgical trial. Br J Cancer. (2013) 109:2792–7. doi: 10.1038/bjc.2013.657
108. Patterson RE, Marinac CR, Sears DD, Kerr J, Hartman SJ, Cadmus-Bertram L, et al. The effects of metformin and weight loss on biomarkers associated with breast cancer outcomes. J Natl Cancer Inst. (2018) 110:1239–47. doi: 10.1093/jnci/djy040
Keywords: breast cancer, prevention, metformin, chemoprevention, diabetes, heart disease
Citation: Jones VC, Dietze EC, Jovanovic-Talisman T, McCune JS and Seewaldt VL (2020) Metformin and Chemoprevention: Potential for Heart-Healthy Targeting of Biologically Aggressive Breast Cancer. Front. Public Health 8:509714. doi: 10.3389/fpubh.2020.509714
Received: 20 November 2019; Accepted: 24 September 2020;
Published: 29 October 2020.
Edited by:
Dorothy Teegarden, Purdue University, United StatesReviewed by:
Davide Serrano, European Institute of Oncology (IEO), ItalyCopyright © 2020 Jones, Dietze, Jovanovic-Talisman, McCune and Seewaldt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victoria L. Seewaldt, dnNlZXdhbGR0QGNvaC5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.