
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Public Health , 02 September 2020
Sec. Infectious Diseases: Epidemiology and Prevention
Volume 8 - 2020 | https://doi.org/10.3389/fpubh.2020.00451
 Chandler Roe1
Chandler Roe1 Charles H. D. Williamson1
Charles H. D. Williamson1 Adam J. Vazquez1
Adam J. Vazquez1 Kristen Kyger1Michael Valentine2
Kristen Kyger1Michael Valentine2 Jolene R. Bowers2
Jolene R. Bowers2 Paul D. Phillips1Veronica Harrison2Elizabeth Driebe2
Paul D. Phillips1Veronica Harrison2Elizabeth Driebe2 David M. Engelthaler2
David M. Engelthaler2 Jason W. Sahl1*
Jason W. Sahl1*Antimicrobial resistance (AMR) in the nosocomial pathogen, Acinetobacter baumannii, is becoming a serious public health threat. While some mechanisms of AMR have been reported, understanding novel mechanisms of resistance is critical for identifying emerging resistance. One of the first steps in identifying novel AMR mechanisms is performing genotype/phenotype association studies; however, performing these studies is complicated by the plastic nature of the A. baumannii pan-genome. In this study, we compared the antibiograms of 12 antimicrobials associated with multiple drug families for 84 A. baumannii isolates, many isolated in Arizona, USA. in silico screening of these genomes for known AMR mechanisms failed to identify clear correlations for most drugs. We then performed a bacterial genome wide association study (bGWAS) looking for associations between all possible 21-mers; this approach generally failed to identify mechanisms that explained the resistance phenotype. In order to decrease the genomic noise associated with population stratification, we compared four phylogenetically-related pairs of isolates with differing susceptibility profiles. RNA-Sequencing (RNA-Seq) was performed on paired isolates and differentially-expressed genes were identified. In these isolate pairs, five different potential mechanisms were identified, highlighting the difficulty of broad AMR surveillance in this species. To verify and validate differential expression, amplicon sequencing was performed. These results suggest that a diagnostic platform based on gene expression rather than genomics alone may be beneficial in certain surveillance efforts. The implementation of such advanced diagnostics coupled with increased AMR surveillance will potentially improve A. baumannii infection treatment and patient outcomes.
Antimicrobial resistance (AMR) has the potential to become a global health emergency and is expected to kill more people than cancer by the year 2050 (1). Multidrug resistance in Acinetobacter baumannii is now recognized as a major public health concern, resulting in the World Health Organization (WHO) declaring A. baumannii a priority 1 pathogen (2). A. baumannii is primarily a nosocomial pathogen (3) that affects immunocompromised patients, causing a variety of afflictions including pneumonia, septicemia, meningitis, and death (4, 5). Treatment of A. baumannii infections has become increasingly difficult due to the emergence of multidrug resistance; pan-resistant A. baumannii strains (6–8), including strains resistant to last-resort drugs such as colistin (9), have been identified in Asia and Europe.
Known mechanisms that confer AMR in A. baumannii include penicillin binding proteins (10), enzymes (11), porin defects (12), 16S rRNA methylation (13), and efflux (14). Efflux pumps that confer resistance in Acinetobacter are classified into four families: multidrug and toxic compound extrusion (MATE), resistance–nodulation–division (RND) family, major facilitator superfamily (MFS), and small multidrug resistance (SMR) (15). Additionally, mutations in promoter regions can lead to overexpression of some efflux systems, including AdeFGH (14), which has been shown to lead to resistance to multiple antimicrobial families.
Resistance mechanisms have also been reported for specific drug families used to treat A. baumannii infections. Perhaps the most studied family is beta-lactams, including carbapenems (e.g., meropenem and imipenem), which are used to treat recalcitrant nosocomial infections (16, 17). Carbapenem resistance has been associated with the action of carbapenem-hydrolyzing class D beta-lactamases (CHDLs), including blaOXA−23, blaOXA−24, and blaOXA−58 (18). The ampC cephalosporinase is a class C beta-lactamase that is broadly conserved across A. baumannii (19) and has been associated with resistance to narrow spectrum cephalosporins (20). Additionally, blaOXA−51−like genes are highly conserved across the A. baumannii species, as well as other Acinetobacter spp. (21); these genes confer resistance to carbapenems when in close proximity to the insertion element, ISAba1 (22).
Aminoglycoside resistance in A. baumannii has been associated with the actions of aminoglycoside modifying enzymes (AMEs) including aacC1, aphA6, aadA1, and aadB (23), the 16S rRNA methyltransferase armA (24), as well as through efflux action of AdeABC and AbeM, although the efflux effect is limited (23). Resistance to macrolides in A. baumannii has primarily been associated with target site alteration in Dfr (25) encoded by folA, the presence and activity of the tetM gene (26), and through the action of efflux pumps (27). Finally, quinolone resistance in A. baumannii has been linked to quinolone resistance-determining regions (QRDRs) (28), including mutations in parC, gyrA, and gyrB. Specifically, the gyrA S82L mutation has previously been shown to confer resistance to quinolones (29); two separate mutations, S83L and G80V, in gyrA have also been demonstrated to confer quinolone resistance in A. baumannii (30).
In recent years, multiple databases have been developed and maintained that include genomic regions associated with antimicrobial resistance. These databases include CARD (31), ResFinder (32), ARG-ANNOT (33), ARDB (34), and MEGARes (35). To identify potential resistance mechanisms, genomes are screened against these databases and if genes associated with resistance are identified, then resistance patterns are inferred (36). However, genomics doesn't capture expression profiles, including gene induction, which prevents accurate genotype to phenotype associations in some organisms (37).
Current treatment regimens for A. baumannii infections start with broad spectrum cephalosporins such as ceftazidime or cefepime, or a carbapenem (e.g., imipenem) (38). For drug resistant pathogens, polymyxins such as colistin are used, although emerging resistance has been reported (39) and the treatment can be toxic (40). Other drugs, including tigecycline (41) and minocycline (42) have been used to treat resistant strains, although resistance to these therapies has also been observed, prompting research into combination therapies (43) that overcome these limitations. However, pan-resistance in A. baumannii (44) has the potential to undermine all current treatment regimens and necessitates a better understanding of genotype/phenotype associations for improved surveillance efforts and targeted therapy.
Research into genotype/phenotype associations in A. baumannii is complicated by the highly plastic nature of the pan-genome (45). One example of this phenomenon is the biofilm associated protein (Bap) (46, 47), which, based on an in silico screen of more than 117 genomes annotated as A. baumannii, is found only in 2 genomes (unpublished). This demonstrates that mechanisms associated with a phenotype may not be broadly distributed across diverse isolates of this species. While true for virulence, similar patterns exist for AMR genes that are variably conserved within a highly plastic species (21).
In this study, we analyzed over 100 Acinetobacter isolates, largely isolated in Arizona, USA, in an effort to identify common as well as cryptic mechanisms of AMR. Implementing an iterative approach, we searched common AMR gene databases for known mechanisms, performed a bacterial genome wide association study (bGWAS) to identify potentially new mechanisms, and performed RNA-Sequencing (RNA-Seq) to compare gene expression profiles between isolates with variable AMR phenotypes. The results provide additional detail to understand AMR mechanisms in A. baumannii and identify targets for advanced diagnostics that will guide appropriate therapies for more effective patient treatment.
A total of 107, largely geographically confined isolates, were identified for sequencing based on collection from different body sites and clinical matrices. All isolates were classified as A. baumannii based on orthogonal, clinical laboratory techniques. A description of all sequenced isolates is shown in Table S1. Samples were streaked from glycerol stocks onto Mueller Hinton (MH) (Hardy Diagnostics, Santa Maria, CA) agar plates and incubated at 37°C for 24 h. Inoculated plates were checked for appropriate colony growth and morphology the following day prior to DNA extraction.
Genomic DNA was extracted from a single isolated colony for each sample using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA) following the recommended protocol for Gram-negative bacteria. Sample DNA was fragmented using the QSonica q800 ultrasonic liquid processor (QSonica, Newtown, CT, USA). Sonication parameters were optimized to produce fragment sizes of 600–700 base pairs (time: 3 min, pulse: 15 s (Pulse On), 15 s (Pulse Off), amplitude: 20%). Libraries were size selected using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA) in order to remove small and large fragments outside of the required size range. Genome libraries were prepared using the KAPA Hyper Library Preparation Kit with Standard PCR Library Amplification (Kapa Biosystems, Wilmington, MA) and sequenced on an Illumina MiSeq using V3 sequencing chemistry (Illumina Inc., San Diego, CA).
For MinION sequencing, DNA was extracted with the GenElute Bacterial Genomic DNA kit (Sigma-Aldrich Inc., St. Louis, MO), taking care to limit DNA shearing. Long read sequencing was performed using Oxford Nanopore technologies on a MK1B MinION device using a R9.4 flow cell. The DNA library was prepared using the SQK-LSK109 Ligation Sequencing kit in conjunction with the PCR-Free Native Barcode Expansion kit following manufacturer's protocol (downloaded from https://nanoporetech.com/resource-centre/protocols/ on March 20, 2019) without the optional shearing steps to select for long reads.
Illumina-derived whole genome sequence data was assembled with SPAdes v3.10 (48). Contigs that aligned against known contaminants or contained an anomalously low depth of coverage compared to the average depth of coverage on a per genome basis were manually removed. The MLST profiles were extracted from whole genome sequence (WGS) assemblies using BLAST-based methods (49) using both the Oxford (50) and Pasteur systems (51); novel alleles and sequence types were submitted to both databases. Annotation on all genomes was performed with Prokka v1.13 (52). Hybrid assemblies were generated with combined Illumina and MinION data with Unicycler v0.4.8-beta (53), which includes a polishing strep with Pilon v1.22 (54).
Acinetobacter genome assemblies were downloaded from GenBank on March 13th, 2018. All genome assemblies were aligned against the A. baumannii genome AB307-2094 (CP001172.1) with NUCmer v3.1 (55) in conjunction with NASP v1.1.2 (56). SNPs that fell within duplicated regions, based on a reference self-alignment with NUCmer, were filtered from downstream analyses. For rapid evaluation, an approximate maximum likelihood phylogeny was inferred on a concatenation of 1, 523, 968 single nucleotide polymorphisms (SNPs) with FastTree v2.1.8 (57); SNPs were retained if they were conserved in >90% of all genomes.
Antimicrobial resistance phenotypic profiles were identified for cefepime (FEP), cefuroxime (CXM), gentamicin (GEN), ceftazidime (CAZ), trimethoprim (TMP), azithromycin (AZM), ceftriaxone (CRO), aztreonam (ATM), erythromycin (ERY), piperacillin (PIP), levofloxacin (LVX), imipenem (IPM), and ciprofloxacin (CIP). A list of all drugs and resistance breakpoints used are shown in Table 1. Drugs were selected from published resistance patterns in the literature (58–60, 62–69). Samples were streaked from glycerol stocks onto Mueller Hinton (MH) (Hardy Diagnostics, Santa Maria, CA) agar plates and incubated at 37°C for 24 h. A single isolated colony was picked and inoculated into 10 mL of MH broth. Liquid cultures were incubated with shaking at 37°C overnight. The following morning, 100 μL of each overnight culture were transferred into 9.9 mL of fresh MH broth. Cultures were incubated with shaking at 37°C until optical density (OD600) measurements reached 0.5–0.8, indicating log phase growth. Fifty microliter of culture was inoculated onto new 15 × 150 mm MH agar plates and spread uniformly across the medium with a sterile cell spreader. Six different antimicrobial E-test strips (bioMérieux, France) were applied to the surface of the agar as directed by the manufacturer. Plates were incubated at 37°C for 16–18 h and minimum inhibitory concentrations (MIC) were determined by visual inspection following the recommended manufacturer guidelines. For paired isolates, MIC tests were performed on different days. If the MIC was above CLSI guidelines for resistance, the strain was given an “R” (resistant) for that drug. If the MIC was below guidelines it was given an “S” (susceptible); if the MIC was between thresholds, it was given an “I” (intermediate). If biological replicates differed by more than one MIC dilution, the test was repeated. If replicates differed by a single MIC dilution, the lower MIC value was reported.
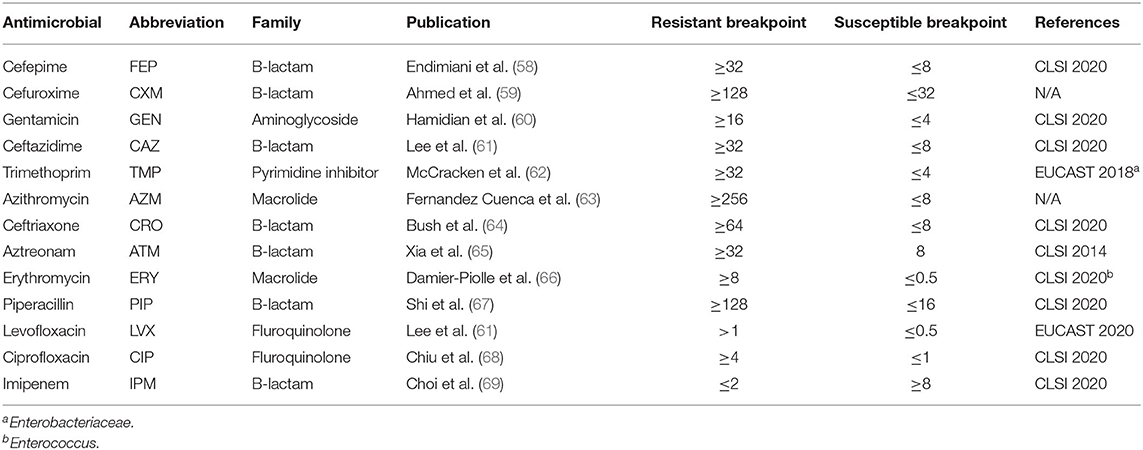
Table 1. Breakpoints of antimicrobials screened in this study.
Once the confirmed set of A. baumannii were identified, a phylogeny was generated for 84 genomes. Raw WGS data were aligned against AB307-0294 with BWA-MEM v0.7.7 (70) and single nucleotide polymorphisms (SNPs) were identified with the UnifiedGenotyper method in GATK v3.3.1 (71, 72). SNPs that fell into duplicate regions of the reference, based on a NUCmer self-alignment, were removed from downstream analyses. All SNP calling methods were wrapped by the NASP pipeline. A maximum likelihood phylogeny was inferred on a concatenation of 182,766 SNPs with IQ-TREE v1.6.1, using the TVMe + ASC + R5 model. Paired genomes were identified by low phylogenetic distance and variable MIC profiles (Table S2).
All A. baumannii genomes (n = 3,218) were downloaded from the Assembly database in GenBank (73) on September 19th, 2018. Genomes were filtered if they: (1) contained >200 ambiguous nucleotides (n = 860); (2) contained >400 contigs (n = 189); (3) had a genome assembly size <3, 684, 234 or >4, 297, 137 (n = 51), or; (4) had an average MASH (74) distance >0.0252 (~97.5% average nucleotide identity) (n = 20). Genomes passing through all filters (n = 2183) were aligned against A. baumannii AB307-2094 (6) with NASP in conjunction with NUCmer. A maximum likelihood phylogeny was inferred on a concatenation of 101,608 SNPs with IQ-TREE v1.6.1, using the TVM+F+ASC+R10 model, and rooted with an A. nosocomialis genome sequenced in this study (TG22170; RFEG00000000); the smaller number of SNPs is due to the reduced size of the core genome due to the inclusion of fragmented genome assemblies in GenBank.
To identify coding region differences between paired isolates, the large-scale blast score ratio (LS-BSR) (75) tool was run on paired genomes in conjunction with BLAT (76). The order of genes between isolates was visualized with genoPlotR (77). The conservation of genes was compared between resistant and susceptible groups using a Mann-Whitney test implemented in Scipy v1.4.1 (78) and significance was applied at an alpha of 0.01.
To identify genotype/phenotype associations, regions identified by LS-BSR were compared between resistant/susceptible phenotypes from each drug. Regions were first identified that had a blast score ratio (BSR) value (79) of >0.8 in one phenotype and a BSR value of <0.4 in the other phenotype. The BSR is calculated by dividing the query Blast bit score by the self-Blast bit score; a BSR value of 0.8 is equivalent to 80% identity over 100% of the alignment length. In addition to bulk differences in coding region sequences (CDSs), differences between individual SNPs and indels were identified through the analysis of Kmers. In this approach, the reverse complement was taken for all genome assemblies so that both strands were included in the analysis. All 21-mers were then identified with Ray-surveyor (80) and placed into a presence absence matrix; the choice of 21-mers was to ensure a short enough length to hopefully identify single mutations. The frequency of Kmers in each phenotype was then calculated with a custom Python script (https://gist.github.com/jasonsahl/e9516b2d940ad2474ba6e97f5b856440) and significance was assessed with a Mann-Whitney test implemented in Scipy with a Bonferroni p-value correction. In addition to these analyses, SNPs associated with resistance phenotypes were identified with default parameters in treeWAS v1.0 (81).
From the raw sequence data, all possible 54-mers at a minimum frequency of 4 per genome were also processed with pyseer (82). A similarity matrix was generated with pyseer and included in the analysis to account for population structure. Pyseer was run using default settings, a similarity matrix, and a “maf” value of 0.40. Only Kmers with good chi-square values were included and were grouped by locus tag.
Three machine learning models were tested within the scikit-learn python module (83): gradient boosting classification (GBC), random forest (RF), and penalized logistic regression (LR). Parameters were tuned for each model over 1,000 50–50% train-test splits. For each antimicrobial studied, all three models were analyzed on the same 500 replicates of 75–25% train-test split cross validation. Each iteration began with a random seed to ensure different train-test splits.
To identify the conservation of previously characterized antimicrobial resistance mechanisms, we screened all genomes against proteins from the Comprehensive Antimicrobial Resistance Database (CARD) (31) with LS-BSR in conjunction with Diamond (84).
Samples identified as paired isolates based on phylogenetic relatedness and differing AMR susceptibility profiles were streaked for isolation from glycerol stocks onto MH agar plates and incubated overnight at 37°C. For each sample a single colony was picked and inoculated into 10 mL of MH broth and incubated with shaking at 37°C overnight. The following morning 100 μL of each culture was inoculated into 9.9 mL of fresh media and OD600nm was monitored until cultures reached log phase growth OD600nm of ~0.5–0.8. Five-hundred microliter of each sample, as well as the susceptible control strain, Staphylococcus aureus subsp. aureus (ATCC 29213), were aliquoted into 2 mL microcentrifuge tubes. Each sample was treated with sub-MIC concentrations of the designated antimicrobial, at one half of the previously recorded MIC value. Cultures were then incubated for 30 min with shaking at 37°C. Two volumes of RNAprotect Bacteria Reagent (Qiagen, Valencia, CA, USA) were added to all samples and incubated at room temperature for 5 min, followed by centrifugation for 10 min at room temperature, at a speed of 5,000 × g. The supernatant was decanted and the treated cell pellets were stored at −80°C. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) following recommended protocol #4 beginning at step 7 and continuing to protocol #7. A DNase I treatment was included for step 2 in protocol #7. Extracted RNA was immediately stored at −80°C. All mRNA extractions were performed in biological triplicate with cultures grown on separate days.
RNA quality and quantity were checked by Agilent 2100 Bioanalyzer with the RNA 6000 Nano Kit (Agilent Technologies, Santa Clara, CA, USA). mRNA was isolated from total RNA using the MICROBExpress kit (Thermo Fisher Scientific, Waltham, MA) following the manufacturer's protocol. Isolated mRNA was quantified and checked for rRNA depletion on the bioanalyzer with an additional RNA Nano chip prior to sequencing.
Previously isolated mRNA was prepared for transcriptome sequencing using the TruSeq Stranded mRNA, HT kit (Illumina, San Diego, CA) following the High Sample (HS) protocol. Prepared samples were quantified and checked for quality, then pooled in equimolar concentrations. Library pools were loaded into an Illumina High Output NextSeq 2 × 150bp kit, according to manufacturer recommendations for sequencing on the Illumina NextSeq 550 platform. The transcriptomes were assembled with metaSPAdes (85) using default settings. For targeted amplicon studies, complementary DNA (cDNA) was generated with the SuperScript IV VILO RT-PCR Master Mix with ezDNase enzyme (Invitrogen, Carlsbad, California), following manufacturer's recommendations.
For each isolate pair, coding and intergenic regions identified with LS-BSR and prodigal were combined for complete genomes, then dereplicated with USEARCH v10 at an ID of 0.98. RNA-Seq reads were aligned against these regions with BWA-MEM and read counts were called on the resulting BAM file with Salmon v0.13.1 (86). Differential expression (DE) analysis was performed with DESeq2 (87). The p-values were corrected using the Benjamini-Hochberg (88) correction. For each pair, the resistant strain expression, including those samples grown in sub-MIC concentrations of antimicrobials, were individually compared to the expression of the susceptible strain.
Polymerase chain reaction (PCR) primers were designed for differentially expressed regions identified in the RNA-Seq analysis (Table S3); a constitutively expressed target (locus tag: IX87_18340), based on analysis of RNA-Seq data, was included for normalization. cDNA was amplified with the following protocol: 1X Promega PCR Master Mix (Promega, Fitchburg, WI), 2.5 μL cDNA template, and multiplexed primer concentrations listed in Table S3. Gene specific PCR parameters were as follows: initial denaturation at 95°C for 2 m, 30 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 45 s, with a final extension at 72°C for 5 m. Included on each primer was a universal tail (89), which facilitated Illumina index ligation. Samples were indexed with the following final concentrations: 1X HiFi HotStart Readymix (Kapa Biosystems Inc., Wilmington, MA), 0.4 μM of each indexing primer, and 2 μL of gene specific PCR product. The indexing PCR parameters were as follows: initial denaturation at 98°C for 2 m, 6 cycles of denaturation at 98°C for 30 s, annealing at 60°C for 20 s, and extension at 72°C for 30 s, with a final extension at 72°C for 5 m. Following each PCR, a 1X Agencourt AMPure bead (Beckman Coulter, Brea, CA) clean-up was performed according to manufacturer's instructions. All amplicons were normalized with SequalPrep (Thermo Fisher Scientific, Applied Biosystems), pooled, and sequenced on the Illumina MiSeq platform (Illumina Inc., San Diego, CA).
Raw AmpSeq data were aligned against predicted amplicons with Kallisto v0.45.0 (90); predicted amplicons were identified by the in silico extraction of sequence between matching primer pairs. Counts were normalized based on the median read counts between all samples. The difference, or delta, between the raw read counts of the target and a constitutively expressed reference housekeeping gene was identified for each sample. The average delta was then identified for each set of resistant and intermediate genomes and the delta Ct was calculated. The average deltas were compared between resistant and intermediate samples and a p-value was calculated with a Mann-Whitney U test.
All data were deposited to appropriate databases and linked under BioProject PRJNA497581. Accession information for specific samples is shown in Table S1.
In this study, we sequenced DNA from 107 isolates identified by laboratory methods to be A. baumannii. These isolates were retrospectively identified from our collection and sequenced to reflect a range of years and isolation sources (Table S1). Of the 107 genomes sequenced in this study, only 84 were confirmed A. baumannii (Table S1) isolates based on a global WGS phylogenetic analysis (Figure 1). In order to define and add context to the phylogenetic diversity of genomes sequenced in this study, more than 3,000 publicly available A. baumannii genomes were included in the analysis (Figure S1).
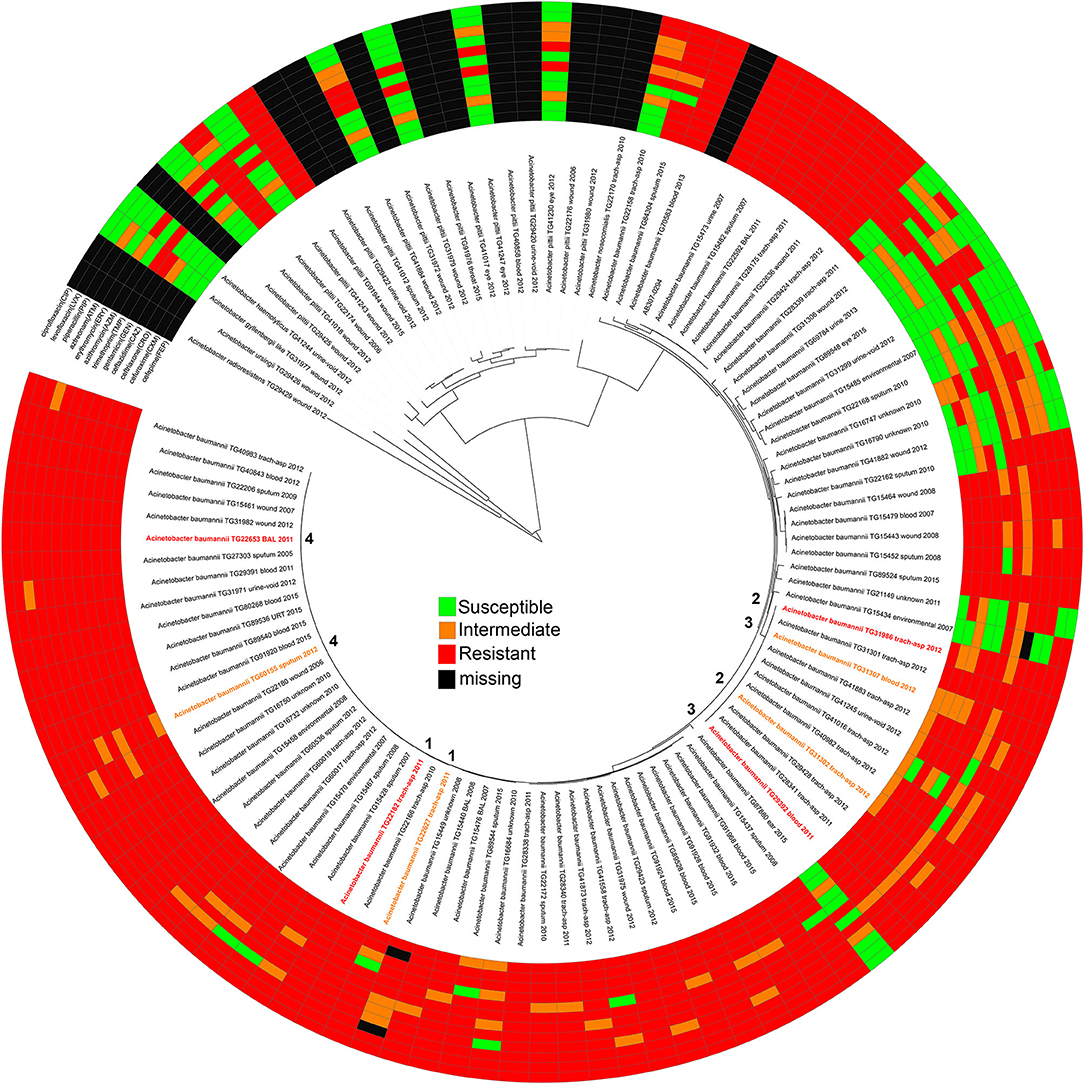
Figure 1. A maximum-likelihood phylogeny of Acinetobacter genomes sequenced in this study based on a concatenation of 50, 869 core genome SNPs. Each genome is annotated with its antimicrobial susceptibility profile across 12 drugs. The annotations were visualized with the Interactive tree of life (91). The pair information (1–4) is shown in the middle of the phylogeny.
AMR profiles were identified for 95 of the isolates across 12 drugs, including all A. baumannii (Figure 1, Table S2). Twelve isolates were excluded due to either difficult to interpret MIC results or inconsistent results across replicates. Some test strips were discontinued during the course of this experiment and were therefore marked as missing in the AMR profile. Although not analyzed further, the AMR profiles for non-baumannii genomes may be useful for researchers studying emerging resistance in other hospital-associated Acinetobacter species.
Antibiograms were obtained for the majority of isolates across tested drugs (Table S2) using E tests; selected drugs were chosen based on treatment suggestions in previous publications (58–68). The MIC values were mapped against a phylogeny of A. baumannii genomes (Figure 1) inferred from a concatenation of 182,916 SNPs. Resistant, susceptible, or intermediate calls were determined based on identified breakpoints (Table 1). Four of the drugs used in this study do not have an identified breakpoint for Acinetobacter. Breakpoints were applied for two of these drugs based on other organisms. For two additional drugs, breakpoints were applied that only includes the highest and lowest values of the E test range. This conservative approach is potentially useful for grouping isolates into categories to identify mechanisms associated with the largest differences in MIC values, but may not be clinically relevant.
From the A. baumannii phylogeny, isolates were identified that were closely related based on phylogenetic distance, but differed in their antibiograms. These isolate pairs (Figure 1, Table 2) were the subject of additional investigation in order to identify cryptic resistance mechanisms based on a common genomic background.
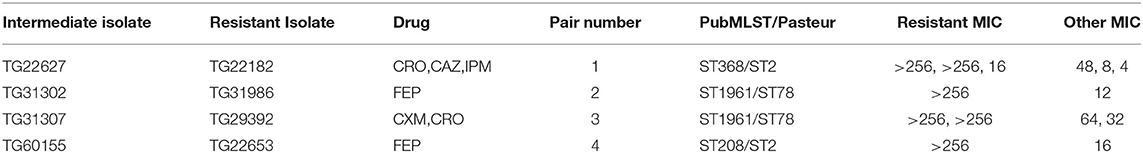
Table 2. Paired isolate antimicrobial susceptibility.
The 84 confirmed A. baumannii genomes were screened for the presence of AMR-associated genes from the CARD database with LS-BSR (Table S4). For 2 isolate pairs, no obvious differences were observed in resistance genes between variably resistant pairs (Figure S2, Table S5). For TG22182 (R) and TG22627 (I), one CARD gene was differentially present (aminoglycoside phosphotransferase (CAE51638)], although no differences were observed in resistance to the tested aminoglycoside, gentamicin; these genes potentially confer resistance to different aminoglycosides that were not screened in this study. Multiple differences were observed between the distribution of CARD genes between TG22653 (R) and TG60155 (I) (Figure S2), although the antibiograms only differed in the resistance to two antimicrobials and the genomes differed by only 27 core genome SNPs.
All proteins from the CARD database (n = 2,420) were aligned against 84 sequenced A. baumannii genomes. Proteins that were highly conserved in at least 2 genomes demonstrate variable conservation of AMR-associated proteins (Figure 2). Some proteins had a clear phylogenetic distribution, where they were either found across almost all Acinetobacter [e.g., blaOXA−64 (OXA-51 family)], conserved across phylogenetic groups in A. baumannii [e.g., aminoglycoside resistance genes (APH(6)-Id): AAC23556.1], or were variably present [e.g., aminoglycoside adenyltransferase (ANT(2”)-Ia): AAC64365.1], suggesting horizontal gene transfer.
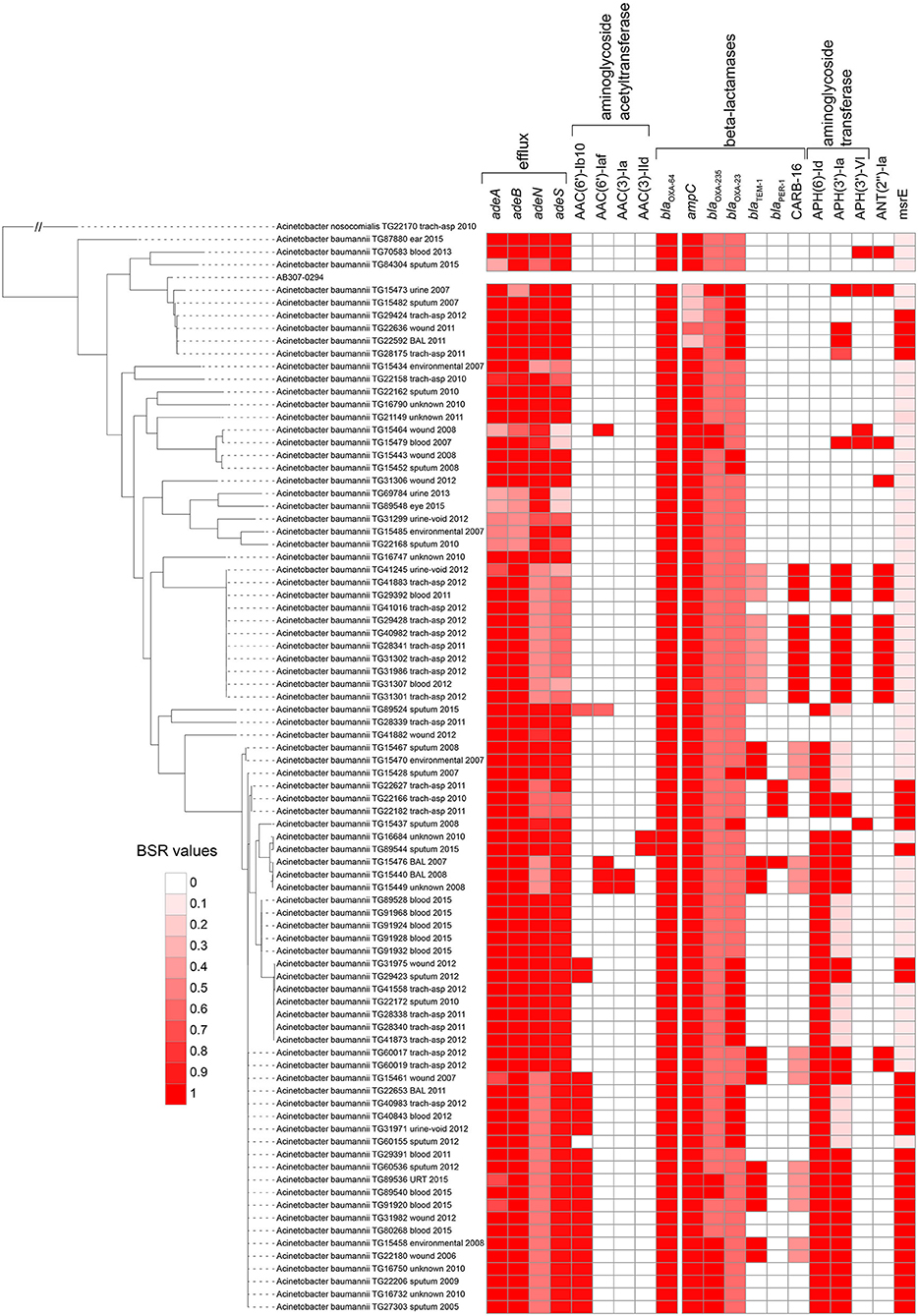
Figure 2. Screen of selected CARD proteins (31) across all Acinetobacter baumannii genomes sequenced in this study. The heatmap is associated with the blast score ratio (BSR) (79) values of each gene across each genome. The BSR values were visualized with the Interactive tree of life (91).
In addition to blaOXA−64, the beta-lactamase genes blaOXA−23 (92) and blaOXA−235 (93) were identified across a subset of sequenced A. baumannii genomes (Figure 2, Table S4). The extended spectrum beta lactamase blaTEM−1 (94) was present in 15 of 84 of sequenced genomes. Finally, 10 genomes contained a CARB-family (CARB-16), class A beta-lactamase (95). Imipenem was only screened across paired isolates in this study and 3 isolates were resistant based on established breakpoints (Table 1). Two of these resistant genomes (TG60155, TG22653) contained blaOXA−23, while 3 susceptible isolate genomes contained CARB-16. One resistant isolate genome (TG22627) was negative for all screened carbapenemase genes with the exception of blaOXA−64.
In addition to differentially-present CARD genes (Table S4), genomes were also screened for mechanisms associated with resistance to the following specific drugs:
The gyrA S82L mutation has previously been shown to confer resistance to quinolones in Acinetobacter (29). Of the resistant A. baumannii strains (n = 73), 72 (~99%) contained the leucine (L) residue at position 82; the one exception was TG22162, which had the serine (S) residue. All susceptible strains had the serine residue at this position, suggesting that this mutation is the primary mechanism conferring quinolone resistance in analyzed strains.
All tested A. baumannii isolates were resistant to trimethoprim. An in silico screen of folA, which has been associated with target site alteration and trimethoprim resistance (25), demonstrated that all genomes contained this gene, although there was some variation in the peptide identities (Table S4). As susceptible strains were not identified through screening, the genotype/phenotype relationship for this compound cannot be tested, although based on published results, folA appears to be the associated mechanism.
The ampC gene in A. baumannii is a class C beta-lactamase (96). A screen of the ampC peptide sequence against A. baumannii isolates sequenced in this study indicates that almost all genomes have a highly conserved ampC gene (EA665_005650) (Figure 2, Table S4), but have widely different antibiograms (Table S2). A comparison of the ampC gene identified that a significant genotype/phenotype relationship was only observed for cefepime (FEP) (p = 0.006). This demonstrates that the presence/absence of this gene alone is not broadly associated with resistance to beta-lactams screened in this study.
The insertion element ISAba1, in conjunction with blaOXA−51−like genes, has been shown to confer resistance to beta-lactams (22). There was a significant difference in the conservation of ISAba1, based on a Mann-WhitneyU test of BSR values for resistant and susceptible genomes, for 5 beta-lactams (cefepime, cefuroxime, ceftazidime, piperacillin, ceftriaxone) (p < 0.01). Many of the ISAba1 transposases were split across multiple contigs in Illumina assemblies, likely due to a repeat region that could not be resolved during assembly; this limited our ability to determine the proximity of ISAba1 to the blaOXA−51−like genes. Furthermore, copies of ISAba1 were likely collapsed during the short read assembly. For example, for the 8 isolates for which draft genomes and complete genomes were generated in this study, only a single copy of ISAba1 was observed in each draft genome, while 9–26 copies were observed in complete genomes. Additionally, all of the paired isolates in this study contained ISAba1 and a blaOXA−51−like gene but showed variable antibiograms to at least one beta-lactam, suggesting that the conservation of this region alone did not explain the resistance phenotype in the absence of proximity information.
Genes associated with efflux (adeA, adeB) were missing from six genomes (Figure 2, Table S4) that showed susceptibility to a number of antimicrobials including cefepime, cefuroxime, ceftazidime, and gentamicin (Table S2). Other genomes contained these genes but were also susceptible to beta-lactams, suggesting multiple genotypes result in the same resistance or susceptibility phenotype.
Although ermB has been associated with macrolide resistance in A. baumannii, the gene was not detected in any genome sequenced in this study, based on a LS-BSR analysis (Table S4). The mefA gene was also screened, as it has been demonstrated to provide macrolide resistance, but the gene (Table S4) was present in all 8 azithromycin susceptible strains (Table S2). Resistance to macrolides has also been associated with efflux, although differences in efflux cannot be investigated with genomics alone.
CARD genes associated with aminoglycoside resistance were screened against genomes with LS-BSR. Of the 4 regions [AAC(3)-Ia, APH(3′)-VIa, aadA1, aadB] previously associated with aminoglycoside resistance, AAC(3)-Ia and aadA1 both showed a significant association (p < 0.01) with gentamicin resistance in genomes screened in this study.
To identify genotype/phenotype associations, a bGWAS analysis was performed by splitting up isolates into resistant/susceptible groups for each drug based on defined thresholds (Table 1); for this analysis, isolates with an intermediate phenotype were ignored in order to isolate the mechanism. Using gene conservation, we failed to identify a clear genotype/phenotype relationship across all Acinetobacter across all drugs. This suggests that genomic analyses alone can fail to comprehensively identify AMR mechanisms in Acinetobacter. When the analysis was repeated for only A. baumannii genomes using genes, SNPs, and Kmers, significant associations were identified (Table 3, Table S6). The method testing genes and Kmers did not consider population structure, suggesting that the significance of these hits cannot be separated from population differences. TreeWas does incorporate population structure data and identified four SNPs associated with two different antimicrobials (gentamicin, levofloxacin). There was only one SNP that was associated with a non-synonymous mutation in a gene annotated as a rRNA methyltransferase. Ribosomal methylation has been demonstrated to confer resistance to aminoglycosides in multiple bacterial genera, including Acinetobacter (13).
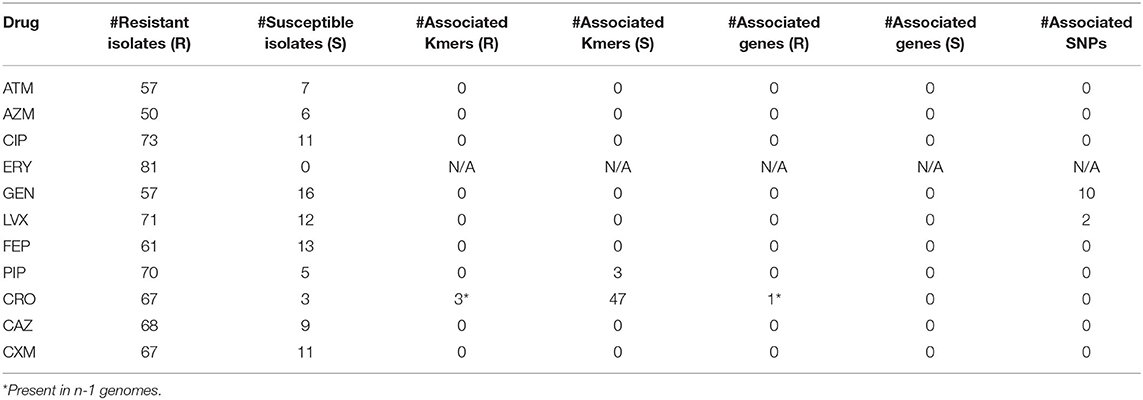
Table 3. Differences in Kmers, genes, and SNPs between resistant (R) and susceptible (S) isolates.
For ceftriaxone (CRO), all susceptible strains (n = 3) were missing ISAba1 (ABLAC_32600), while 66 of 67 resistant strains contained this region; the small number of genomes analyzed limits the power of this analysis. In spite of this correlation, the lack of broadly conserved genomic regions associated with resistance directed a paired genome analysis into the identification of novel or cryptic genotype/phenotype associations.
An analysis with pyseer was also performed and found associated locus tags for 6 antimicrobials (Table S6). The locus ABBFA_01412, coding for a MFS transporter, was identified as significant by both TreeWas and pyseer. Pyseer also found a significant association of parC (ABBFA_03297) with resistance to levofloxacin and gentamicin; the association of levofloxacin resistance with mutations in parC have been observed previously in Acinetobacter (97) and may synergize with observed mutations in gyrA that were not identified by pyseer. For cefuroxime resistance, pyseer identified 3 Kmers that were significantly associated with ampC (ABBFA_001076). Although pyseer failed to identify a significant association with ISAba1, the ampC results suggests that the co-location of these regions is likely associated with the resistance phenotype.
Machine learning approaches were applied to a gene presence/absence matrix generated with LS-BSR. The results demonstrate that associations were identified for 7 of the tested antimicrobials (Table 4); the different machine learning algorithms found different regions associated with the resistance phenotype, designated by “N/A” in terms of rank of importance. While none of these genes can uniformly discriminate between resistant and susceptible genotypes, they may add to the resistance phenotype and could be the subject of additional investigations.
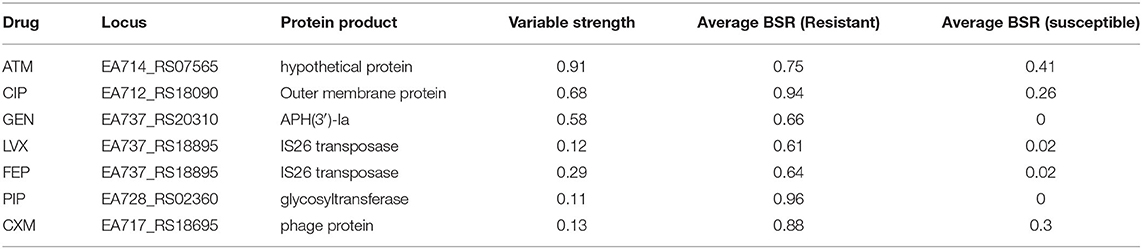
Table 4. Machine learning results.
For isolate pairs where a clear genotype/phenotype relationship was not identified, RNA was extracted and cDNA was sequenced. Despite implementing methods to enrich mRNA, ~20% rRNA + tRNA presence was observed in all samples. For each pair, all coding and intergenic sequences were combined into a single file and de-replicated. Reads were mapped against these regions, normalized, and differential expression was identified using DESeq2. Results were then identified for the following isolate pairs:
TG22627 (I) and TG22182 (R) showed variable susceptibility to ceftriaxone (CRO), imipenem (IPM), and ceftazidime (CAZ). Sixty-eight differentially expressed genes were identified that were upregulated in TG22182 (Table S7) using a Wald stat threshold of 10; this threshold was used to prioritize targets for further investigation, but may not capture all biologically-relevant differential expression. One of these regions (Table 5) was a PER-1 beta-lactamase gene (blaPER−1) (EA714_008075) that is not broadly conserved across sequenced genomes (Table S4) and appears to be present on a transposon. A screen of this gene against ST368 genomes isolated from diverse geographic locations suggested that there was an acquisition of this region in a single sequence type and a clear phylogenetic effect (Figure S3). Indeed, a genomic island was identified in both the resistant and intermediate genomes between two transposases (Figure 3A) that includes a Glutathione S-transferase gene (EA674_08405) that was also upregulated in the resistant strain (11.2x up-regulation); this region has previously been associated with beta-lactam resistance (98). The operon structure was similar between resistant and intermediate strains, with the exception of an IS91 transposon situated between an IS26 transposon and blaPER−1. The operon structure for the resistant strain that contained an IS91 transposon was determined to be highly similar with an ISCR1 (Insertion sequence Common Region) element. Within the ISCR1 element is an oriIS (origin of replication) region that allows for rolling-circle replication and transposition of the ISCR1 element. Within the oriIS are two outward-oriented promoters (POUT) that have been shown to affect downstream gene expression (99). The resistant strain, TG22182, has both POUT promoters associated with increased gene expression directly upstream of the blaPER−1 gene (Figure 3A). The more susceptible strain, TG22627, has neither of the POUT promoters upstream of its blaPER−1 gene. Additionally, the composition of the blaPER−1 gene between isolates was different (Table 5), with a different coding length as well as composition in the first 12 amino acids of the peptide.
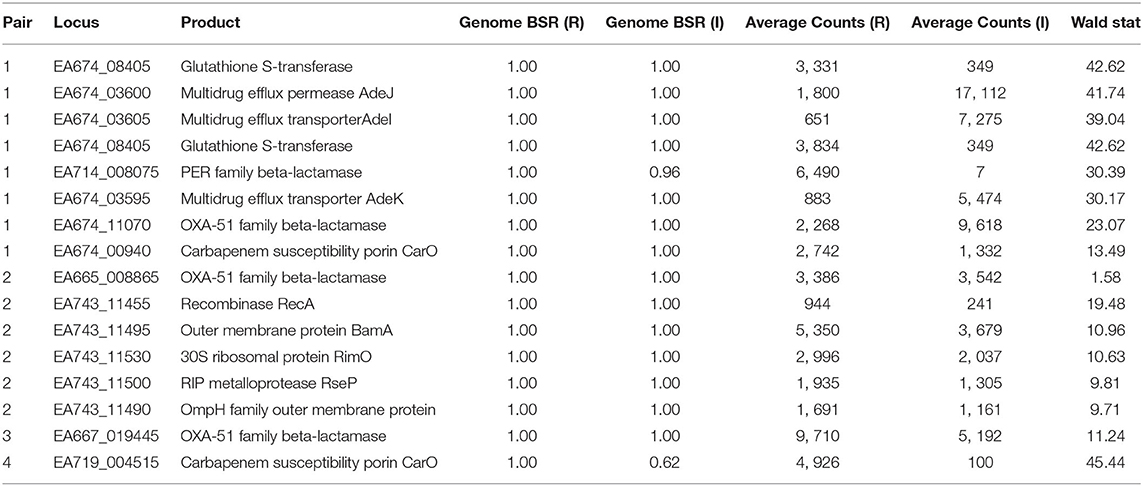
Table 5. Conservation and expression of differentially-expressed loci between resistant (R) and intermediate (I) strains.
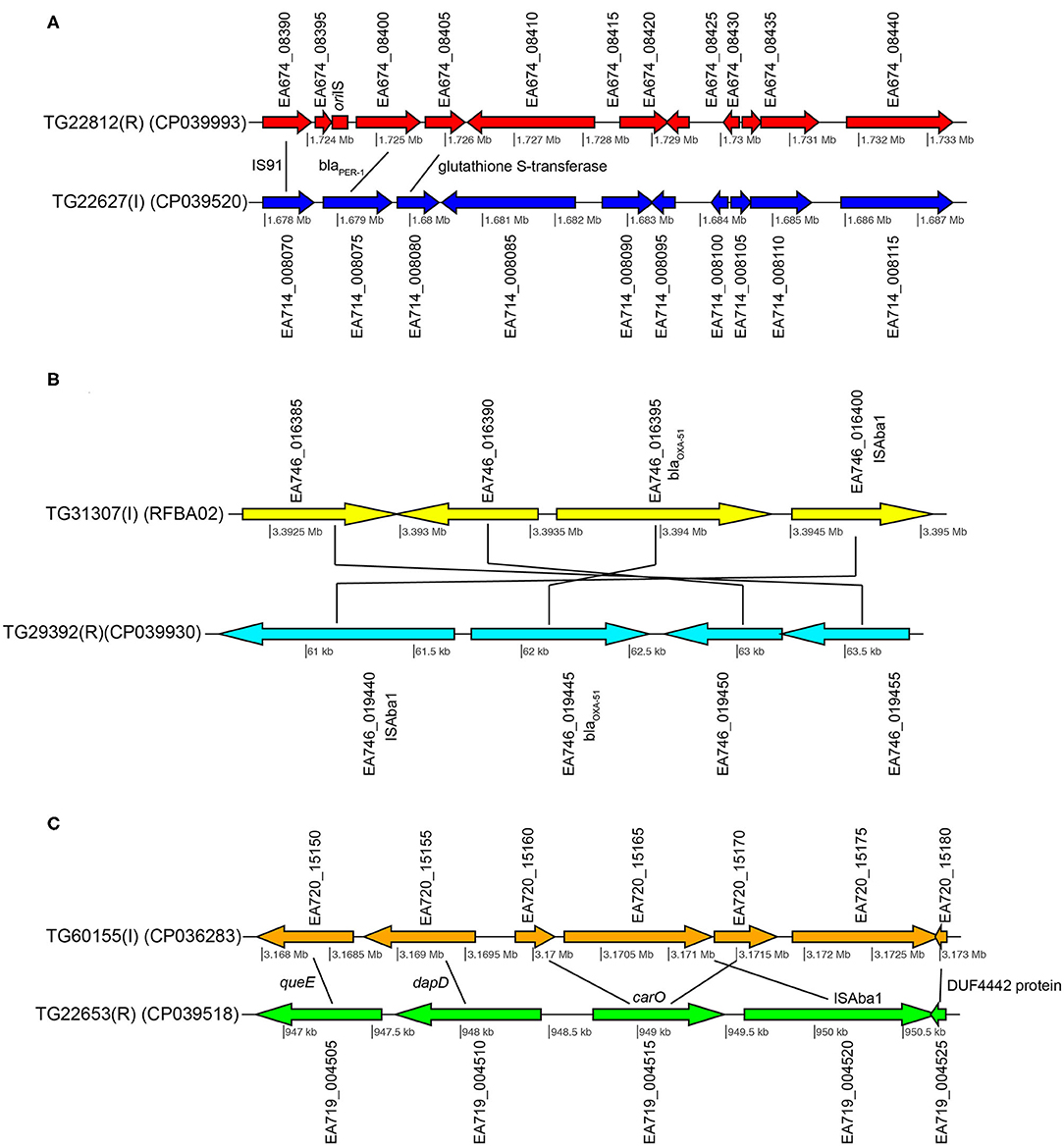
Figure 3. Gene content comparisons between paired isolates in pair 1 (A), pair 3 (B), and pair 4 (C). All figure panels were generated with genoPlotR (77).
Additionally, a glutathione S-transferase family gene (EA674_008405) and the carbapenem susceptibility porin carO (EA674_000940) gene were highly expressed in TG22182 (R) in comparison to TG22627 (I). Both of these genes have been shown to confer resistance to beta-lactams and specifically carbapenems (100). Both isolates in Pair 1 also contain an OXA-51 beta-lactamase gene (blaOXA−51) (EA674_011070), although the gene is slightly up-regulated (4.5x) in TG22627 (I). TG22627 also showed higher expression of other genes associated with AMR including adeI (EA674_003605) (11x), adeB (EA674_009735) (9.5x), adeA (EA674_009730) (9.6x), and adeJ (EA674_003600) (9.6x); however, the up-regulation of genes in the AdeABC pump have previously been demonstrated to not confer aminoglycoside resistance (23).
These results suggest that the action of the PER-1 beta-lactamase has substrate specificity to ceftriaxone (CRO) and ceftazidime (CAZ). The PER-1 beta-lactamase protein has been associated with virulence in A. baumannii (101) and has also been associated with resistance to beta-lactams (102). However, the increased resistance of TG22627 to imipenem is likely due to the up-regulation of efflux-associated genes, as has been demonstrated previously (103).
The two pair 2 strains (TG3103, TG31986) showed variable resistance to cefepime (FEP), cefuroxime (CXM), and ceftriaxone (CRO). Twenty-four differentially-expressed regions were observed between Pair 2 isolates at a Wald stat value of 10 (Table S8). None of these regions were associated with known mechanisms of AMR in A. baumannii. The two isolates contained a class-D beta-lactamase (blaOXA−51) and a class-C beta-lactamase gene (ampC). There was no significant difference in gene expression of these regions between isolate pairs.
The composition of each beta-lactamase was determined at the nucleotide and peptide level. A protein alignment of ampC between TG31302 (I) and TG31986 (R) revealed a single amino acid difference (R to G) at position 172 in the PAZ domain. This non-synonymous mutation falls within the second of three characteristic conserved motifs, RxY150xN, for all class C serine beta-lactamase sequences (104). Although this mutation has not been previously associated with increased activity or misfolding of the protein, other mutations in ampC gene have (105), suggesting that this mutation may confer increased hydrolyzing activity against beta-lactams, although additional validation work is required to test this hypothesis.
A continuous stretch of 17 genes was upregulated in TG31986 (R) (EA743_011445 - EA743_011530) (Table S8). Nine genes within this region were in the top 30 differentially expressed genes in this analysis, including: recombinase recA (EA743_011455), outer membrane protein assembly factor bamA (EA743_011495), 30S ribosomal protein S12 methylthiotransferase rimO (EA743_011530), UMP kinase gene pyrH (EA665_011490), RIP metalloprotease rseP (EA743_011500), a gene coding for an OmpH family outer membrane protein (EA743_011490), a phosphatidate cytidylyltransferase gene (EA665_011475), a 1-deoxy-D-xylulose-5phosphate reductoisomerase gene (EA665_011470), and a di-trans,poly-cis-decaprenylcistransferase gene (EA665_011480). The outer membrane protein (OMP) H, a homolog of the Skp protein in E. coli (106), has been classified as a chaperone protein involved in the folding of BamA. Previous research has shown a correlation between upregulation of molecular chaperones when exposed to antimicrobials and the bacterium's improved ability to tolerate antimicrobial stress (107). Researchers have demonstrated that the skp gene in E. coli is an important stress-associated gene (108) that may be associated with AMR (109).
Pair 3 genomes (TG31307, TG29392) demonstrated variable resistance to cefepime (FEP), cefuroxime (CXM), ceftriaxone (CRO), and ceftazidime (CAZ). Of 94 genomic regions that were significantly differentially expressed (Walt stat of 10) (Table S9) between this isolate pair, one of the significant differences was between a blaOXA−51 family beta-lactamase gene (EA667_019445), which showed 1.9x up-regulation in the resistant strain (Table 5). Interestingly, 79 bases separated the end of insertion element ISAba1 and the start codon of blaOXA−51 in sample TG29392 (R); based on MinION sequencing, this version of blaOXA−51 is present on a plasmid in TG29392 (CP039931.1) and on the chromosome in TG31307. Previous research has demonstrated that this blaOXA−51−like gene is ubiquitous across A. baumannii lineages, but only genomes containing the ISAba1 directly upstream of blaOXA−51 show resistance to carbapenems. It is likely that ISAba1 is acting as the promoter for blaOXA−51 in TG29392, conferring resistance to carbapenems (22). The intermediate resistance strain, TG31307, has both ISAba1 and blaOXA−51; however, ISAba1 is downstream of blaOXA−51 and therefore not functioning as a strong promoter for the beta-lactamase gene (Figure 3B). An analysis of the coding region of the blaOXA−51 gene in both isolates revealed no differences, suggesting that differences in resistance are likely due to gene expression.
Interestingly, several of the genes that were differentially expressed in the resistant strain in Pair 2 were up-regulated in the intermediate Pair 3 strain (TG31307). For example, recA was the most differentially expressed gene in Pair 3 genomes, but was up-regulated in TG31307 (Table S9). This suggests that the mechanisms of resistance have complex interactions that need to be investigated through the identification of epistatic interactions.
Susceptibility differences were observed between Pair 4 genomes for cefepime (FEP), gentamicin (GEN), and azithromycin (AZM). Of the 195 differences in gene expression (Wald stat of 10) between the resistant (TG22653) and intermediate strain (TG60155), perhaps the most striking is in the expression of carO (Table 5) (EA719_004515), an outer membrane porin (Table S10). Previous analyses have demonstrated that insertion sequences that disrupt carO are associated with decreased activity against beta-lactams (110). The genome assembly of the intermediate strain, TG60155, shows that carO is interrupted by the insertion sequence, ISAba1 (EA720_015165) (Figure 3C). An analysis of the transcriptome of TG60155 also failed to identify an intact carO transcript, which was present in TG22653.
In an effort to observe induced antimicrobial resistance in the four paired isolates, resistant strains were grown in sub-inhibitory concentrations of select antimicrobials. Differential expression of each sub-inhibitory isolate was compared to the isolate grown under inhibitory concentrations using the Wald statistic produced from DESeq2. Additionally, the resistant isolate TG22653 was grown under two different sub-inhibitory concentrations of cefepime (16 and 258 μg/mL) and differential expression was compared. No significant differential expression was observed in the four analyses based on an FDR-adjusted p-value of 0.05. Likewise, using the Wald statistic from these analyses also failed to identify significant differential expression between the differing antimicrobial concentrations using the chosen threshold. This suggests that differential expression is due to constitutively expressed mechanisms that are not inducible.
Amplicon sequencing was performed on cDNA to not only confirm the RNA-Seq results, but also to provide a proof of concept as an advanced AMR diagnostic. Comparative expression was identified through comparison of ratios of the number of read counts of each targeted gene compared to a housekeeping gene (IX87_18340); the housekeeping gene, an ABC transporter permease, was identified as a gene with consistent, and relatively high, expression in the RNA-Seq data. For pair 1, the blaPER−1 and aphA1 genes were significantly upregulated in the resistant strain compared to the intermediate strain (Table 6). For pair 2, the expression of the ampC gene was not significantly different, suggesting that differential expression of this region doesn't fully explain the resistance phenotype and is consistent with the RNA-Seq data. For pair 3, the blaOXA−51 gene was confirmed to be significantly (p = 0.0003) up-regulated in the resistant strain compared to the intermediate strain. For pair 4, the carO gene was significantly (p < 0.0001) up-regulated in the resistant strain, which is consistent with the RNA-Seq results and is likely the primary mechanism of resistance. A gene associated with a chaperone-usher pili (CsuA/B) was highly up-regulated in the resistant strain. While not directly associated with AMR, this validation demonstrates consistency between the AmpSeq and RNA-Seq data.
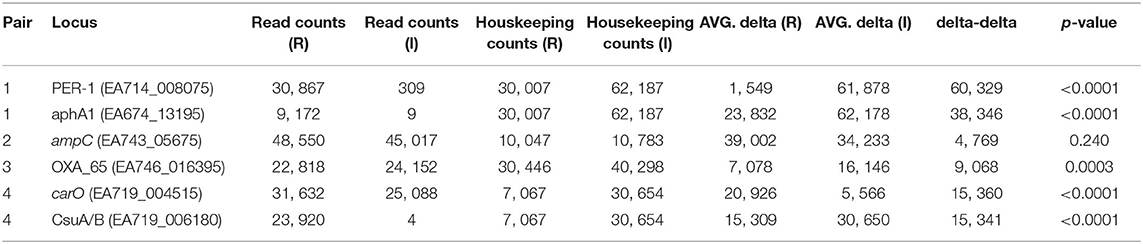
Table 6. Differences in AmpSeq count data between resistant (R) and intermediate (I) isolate pairs.
A LS-BSR analysis of genes in the CARD database between the genome and transcriptome demonstrated that some genomic regions, such as the adeF gene (EA677_11720), were highly conserved in the genome, but were largely absent from the transcriptome (Table S11). Other genes associated with efflux, including adeA (EA679_RS16030), adeH (EA677_20370), adeG (EA686_07885), and an MFS transporter permease (EA708_RS14915) were also not expressed in laboratory growth conditions. These findings demonstrate the importance of incorporating gene expression when trying to understand phenotypic differences.
Antimicrobial resistance (AMR) is a significant, emerging threat, with A. baumannii being recently classified as a priority 1 pathogen (2). Some mechanisms associated with AMR in A. baumannii are understood, especially with regards to documented beta lactamases (111–114) and efflux pumps (15, 115, 116). However, the highly plastic pan-genome of A. baumannii (45) suggests that the identification of single, universal AMR mechanisms may be unlikely for some drugs. This same trend has been observed in other species with highly plastic genomes, such as Pseudomonas aeruginosa (117), and complicates surveillance and targeted therapy efforts. As such, A. baumannii is not only an emerging threat, but represents a critical challenge to the development of both novel drugs and molecular diagnostics.
In this study, we sequenced 107 genomes reported to be A. baumannii based on testing in the clinical laboratory. Typing based on WGS analyses identified 23 of the genomes were misclassified and belonged to other Acinetobacter species (Table S1). These incorrect clinical laboratory typing results highlight the need for improved clinical diagnostics of A. baumannii. An additional 35 genomes in the GenBank assembly database were incorrectly annotated as A. baumannii and belonged to other species (not shown), which further demonstrates the difficulty in typing as well as and the impact of mis-annotated genomes on population structure analysis in Acinetobacter. Typing strains using WGS should be a first step in any large comparative genomics study to limit the analysis to a targeted group, clade, or species.
We generated antibiograms for 12 drugs, representing multiple drug families, across the 84 isolates confirmed to belong to A. baumannii by WGS analysis. Some of the drugs screened in this study aren't typically used in current treatment regimens for A. baumannii infections. However, with growing resistance emerging to next generation drugs, clinicians are exploring older drugs (e.g., chloramphenicol) to treat emerging threats (42, 118). In this study, we sought to identify genomic differences that could explain the variable resistance phenotypes using established antimicrobial resistance gene databases as a method to predict AMR from genomics data (31, 32, 35, 68). A screen of regions from the Comprehensive Antimicrobial Resistance Database (CARD) against genomes sequenced in this study failed to identify characterized resistance mechanisms that largely explain the resistance phenotype. These results demonstrate the limitations to this approach in highly plastic species and suggest that alternative approaches, including RNA-Seq, may be required for a comprehensive understanding of AMR mechanisms in A. baumannii.
We then employed reference independent, bacterial genome wide association study (bGWAS) methods to identify genomic differences between susceptible/intermediate/resistant phenotypes. These types of associations have been used in other pathogens to identify genotype/phenotype associations (119). In general, we failed to identify a clear association between the genotype (21 bp Kmers, coding regions, SNPs) and the resistance phenotype when comparing either all Acinetobacter or just A. baumannii genomes (Table 3) without accounting for population structure. Methods that do incorporate population structure using SNPs (treeWAS) or Kmers (pyseer) identified associations for some drugs and could be the focus of additional work. One limiting factor in bGWAS studies is the number of samples in each group. While our study also suffered from small sample sizes, this work provides a framework for future bGWAS studies in A. baumannii.
Recent research has demonstrated the difficulty in identifying complex mechanisms, or under-or-over-represented phenotypes, using a GWAS approach (120). As a way to focus on sparsely distributed AMR mechanisms, a paired isolate approach was utilized in order to reduce noise in the genomic background. In this analysis, four isolate pairs were individually compared across four antimicrobials (Table 2). RNA-Sequencing (RNA-Seq) of these four paired resistant/intermediate isolates that shared a common genetic background was employed. This paired approach identified several novel and previously identified mechanisms, but also highlights the difficult in identifying mechanisms through standard comparative genomics approaches. While known resistance mechanisms were identified in the resistant strains, some of those regions were also identified in intermediate strains; previous studies of A. baumannii transcriptomes have also observed up-regulation of resistance and efflux genes in susceptible strains (5). RNASeq data allowed for the identification of antimicrobial resistance mechanisms that while present in both the resistant and susceptible genomes, were differentially expressed due to an upstream insertion element. These results also highlight the possibility that expression of a single AMR gene does not always confer resistance and it is likely that combinations of genes are responsible for observed resistance. Differential expression differences were confirmed using a cDNA-AmpSeq approach and largely confirmed the differential expression of targeted regions. Previous research has demonstrated a bias of differentially expressed regions when applying a multiplexed PCR approach (121). We addressed this issue by optimizing primer concentrations using genomic DNA and including a single copy number gene for normalization.
The results of this study demonstrate that, due to the plastic nature of A. baumannii's pan-genome, comprehensive AMR surveillance cannot solely be achieved through genomics methods alone, especially with current AMR databases and commonly used analytical methods. This study demonstrates that AMR genes are not conserved across A. baumannii lineages with similar AMR profiles and that solely relying on genomics methods for AMR surveillance and discovery, such as gene presence/absence, will fail to detect novel or recently acquired AMR mechanisms. For instance, identifying only the position of insertion sequence (IS) elements throughout a genome using informatics tools provides little resolution in the identification of AMR genes upregulated by the presence of upstream IS elements. However, by utilizing transcriptome data, AMR genes upregulated by these elements as well as novel AMR mechanisms were identified; these targets were used to design cDNA amplicon sequencing targets for these mechanisms to improve surveillance and diagnostic efforts.
The datasets generated for this study can be found in the PRJNA497581.
Ethical approval for this study was not required in accordance with local legislation and national guidelines. All human-derived samples were collected under ethics protocols from clinical laboratories. Patients included in this study were anonymized and no written informed consent was acquired because of the retrospective nature of the study.
CR performed laboratory experiments, analyzed data, and helped to write the manuscript. CW and KK helped to write the manuscript. AV performed laboratory experiments. MV and VH performed laboratory analyses. JB and ED was involved with experimental design. PP was involved in data analysis. DE was involved in experimental design. JS was involved with experimental design, analyzed data, and helped to write the manuscript. All authors contributed to the article and approved the submitted version.
Funding for this project was provided by an R21 grant awarded to JS (1R21AI121738-01). DE was supported in part by CDC contract 200-2016-92313.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
An earlier version of this manuscript has been released as a pre-print (122).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2020.00451/full#supplementary-material
Figure S1. A maximum-likelihood phylogeny of global A. baumannii genomes inferred from an alignment of 11,687 concatenated SNPs. Red dashes indicate genomes sequenced in this study.
Figure S2. A maximum-likelihood phylogeny of paired isolates. The conservation of selected proteins from the CARD database, based on blast score ratio (BSR) values, is shown as a heatmap. The BSR values were visualized with the Interactive tree of life (91).
Figure S3. A maximum likelihood phylogeny of select A. baumannii genomes. The distribution of the blaPER−1 beta-lactamase gene in ST368 genomes, based on blast score ratio (BSR) values, was visualized as a heatmap with the Interactive tree of life (91).
Table S1. Metadata for genomes sequenced in this study.
Table S2. MIC values for screened antimicrobials across all genomes.
Table S3. PCR primers for targeted amplicon sequencing.
Table S4. BSR values for differentially-present CARD proteins.
Table S5. The distribution of CARD genes against paired isolate genomes.
Table S6. bGWAS associations between resistant and susceptible phenotypes. Differences in Kmer frequences were identified with a Mann-Whitney test with Bonferroni correction.
Table S7. DESeq2 results of Pair 1 RNA-Seq datasets. Biological replicates denoted with (a,b,c). Isolates grown in sub-inhibitory concentrations are labeled as “sub” with the corresponding antimicrobial.
Table S8. DESeq2 results of Pair 2 RNA-Seq datasets. Biological replicates denoted with (a,b,c). Isolates grown in sub-inhibitory concentrations are labeled as “sub” with the corresponding antimicrobial.
Table S9. DESeq2 results of Pair 3 RNA-Seq datasets. Biological replicates denoted with (a,b,c). Isolates grown in sub-inhibitory concentrations are labeled as “sub” with the corresponding antimicrobial.
Table S10. DESeq2 results of Pair 4 RNA-Seq datasets. Biological replicates denoted with (a,b,c). Isolates grown in sub-inhibitory concentrations are labeled as “sub” with the corresponding antimicrobial. Two different concentrations of cefepime (FEP) were used to test induction (16, 128).
Table S11. A BSR matrix of CARD genes in the genome and transcriptome of each paired isolate.
1. O'Neill J. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. Rev Antimicrob Resist. (2014) 20:1–16.
2. WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics (2018).
3. Dijkshoorn L, Nemec A, Seifert H. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat Rev Microbiol. (2007) 5:939–51. doi: 10.1038/nrmicro1789
4. Cisneros JM, Reyes MJ, Pachon J, Becerril B, Caballero FJ, Garcia-Garmendia JL, et al. Bacteremia due to Acinetobacter baumannii: epidemiology, clinical findings, and prognostic features. Clin Infect Dis. (1996) 22:1026–32. doi: 10.1093/clinids/22.6.1026
5. Qin H, Lo NW, Loo JF, Lin X, Yim AK, Tsui SK, et al. Comparative transcriptomics of multidrug-resistant Acinetobacter baumannii in response to antibiotic treatments. Sci Rep. (2018) 8:3515. doi: 10.1038/s41598-018-21841-9
6. Adams MD, Goglin K, Molyneaux N, Hujer KM, Lavender H, Jamison JJ, et al. Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii. J Bacteriol. (2008) 190:8053–64. doi: 10.1128/JB.00834-08
7. Huang H, Yang ZL, Wu XM, Wang Y, Liu YJ, Luo H, et al. Complete genome sequence of Acinetobacter baumannii MDR-TJ and insights into its mechanism of antibiotic resistance. J Antimicrob Chemother. (2012) 67:2825–32. doi: 10.1093/jac/dks327
8. Zhu L, Yan Z, Zhang Z, Zhou Q, Zhou J, Wakeland EK, et al. Complete genome analysis of three Acinetobacter baumannii clinical isolates in China for insight into the diversification of drug resistance elements. PLoS ONE. (2013) 8:e66584. doi: 10.1371/journal.pone.0066584
9. Li J, Rayner CR, Nation RL, Owen RJ, Spelman D, Tan KE, et al. Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother. (2006) 50:2946–50. doi: 10.1128/AAC.00103-06
10. Gehrlein M, Leying H, Cullmann W, Wendt S, Opferkuch W. Imipenem resistance in Acinetobacter baumanii is due to altered penicillin-binding proteins. Chemotherapy. (1991) 37:405–12. doi: 10.1159/000238887
11. Manchanda V, Sanchaita S, Singh N. Multidrug resistant acinetobacter. J Glob Infect Dis. (2010) 2:291–304. doi: 10.4103/0974-777X.68538
12. Smani Y, Fabrega A, Roca I, Sanchez-Encinales V, Vila J, Pachon J. Role of OmpA in the multidrug resistance phenotype of Acinetobacter baumannii. Antimicrob Agents Chemother. (2014) 58:1806–8. doi: 10.1128/AAC.02101-13
13. Doi Y, Arakawa Y. 16S ribosomal RNA methylation: emerging resistance mechanism against aminoglycosides. Clin Infect Dis. (2007) 45:88–94. doi: 10.1086/518605
14. Coyne S, Rosenfeld N, Lambert T, Courvalin P, Perichon B. Overexpression of resistance-nodulation-cell division pump AdeFGH confers multidrug resistance in Acinetobacter baumannii. Antimicrob Agents Chemother. (2010) 54:4389–93. doi: 10.1128/AAC.00155-10
15. Coyne S, Courvalin P, Perichon B. Efflux-mediated antibiotic resistance in Acinetobacter spp. Antimicrob Agents Chemother. (2011) 55:947–53. doi: 10.1128/AAC.01388-10
16. Martinez Lacasa J, Garau J. [The role of carbapenems in the treatment of nosocomial infection]. Enferm Infecc Microbiol Clin. (1997) 115 (Suppl.):78–85.
17. Bush K. Past and present perspectives on beta-lactamases. Antimicrob Agents Chemother. (2018) 62:e01076–18. doi: 10.1128/AAC.01076-18
18. Poirel L, Nordmann P. Carbapenem resistance in Acinetobacter baumannii: mechanisms and epidemiology. Clin Microbiol Infect. (2006) 12:826–36. doi: 10.1111/j.1469-0691.2006.01456.x
19. Liu Y, Liu X. Detection of AmpC beta-lactamases in Acinetobacter baumannii in the Xuzhou region and analysis of drug resistance. Exp Ther Med. (2015) 10:933–6. doi: 10.3892/etm.2015.2612
20. Tian GB, Adams-Haduch JM, Taracila M, Bonomo RA, Wang HN, Doi Y. Extended-spectrum AmpC cephalosporinase in Acinetobacter baumannii: ADC-56 confers resistance to cefepime. Antimicrob Agents Chemother. (2011) 55:4922–5. doi: 10.1128/AAC.00704-11
21. Sahl JW, Gillece JD, Schupp JM, Waddell VG, Driebe EM, Engelthaler DM, et al. Evolution of a pathogen: a comparative genomics analysis identifies a genetic pathway to pathogenesis in Acinetobacter. PLoS ONE. (2013) 8:e54287. doi: 10.1371/journal.pone.0054287
22. Turton JF, Ward ME, Woodford N, Kaufmann ME, Pike R, Livermore DM, et al. The role of ISAba1 in expression of OXA carbapenemase genes in Acinetobacter baumannii. FEMS Microbiol Lett. (2006) 258:72–7. doi: 10.1111/j.1574-6968.2006.00195.x
23. Sheikhalizadeh V, Hasani A, Ahangarzadeh Rezaee M, Rahmati-Yamchi M, Hasani A, Ghotaslou R, et al. Comprehensive study to investigate the role of various aminoglycoside resistance mechanisms in clinical isolates of Acinetobacter baumannii. J Infect Chemother. (2017) 23:74–9. doi: 10.1016/j.jiac.2016.09.012
24. Kim JW, Heo ST, Jin JS, Choi CH, Lee YC, Jeong YG, et al. Characterization of Acinetobacter baumannii carrying bla(OXA-23), bla(PER-1) and armA in a Korean hospital. Clin Microbiol Infect. (2008) 14:716–8. doi: 10.1111/j.1469-0691.2008.02022.x
25. Mak JK, Kim MJ, Pham J, Tapsall J, White PA. Antibiotic resistance determinants in nosocomial strains of multidrug-resistant Acinetobacter baumannii. J Antimicrob Chemother. (2009) 63:47–54. doi: 10.1093/jac/dkn454
26. Ribera A, Ruiz J, Vila J. Presence of the Tet M determinant in a clinical isolate of Acinetobacter baumannii. Antimicrob Agents Chemother. (2003) 47:2310–2. doi: 10.1128/AAC.47.7.2310-2312.2003
27. Zhang T, Wang M, Xie Y, Li X, Dong Z, Liu Y, et al. Active efflux pump adeB is involved in multidrug resistance of Acinetobacter baumannii induced by antibacterial agents. Exp Ther Med. (2017) 13:1538–46. doi: 10.3892/etm.2017.4141
28. Park S, Lee KM, Yoo YS, Yoo JS, Yoo JI, Kim HS, et al. Alterations of gyrA, gyrB, and parC and activity of efflux pump in fluoroquinolone-resistant Acinetobacter baumannii. Osong Public Health Res Perspect. (2011) 2:164–70. doi: 10.1016/j.phrp.2011.11.040
29. Hujer KM, Hujer AM, Endimiani A, Thomson JM, Adams MD, Goglin K, et al. Rapid determination of quinolone resistance in Acinetobacter spp. J Clin Microbiol. (2009) 47:1436–42. doi: 10.1128/JCM.02380-08
30. Vila J, Ruiz J, Goni P, Marcos A, Jimenez de Anta T. Mutation in the gyrA gene of quinolone-resistant clinical isolates of Acinetobacter baumannii. Antimicrob Agents Chemother. (1995) 39:1201–3. doi: 10.1128/AAC.39.5.1201
31. Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. (2017) 45:D566–73. doi: 10.1093/nar/gkw1004
32. Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. (2012) 67:2640–4. doi: 10.1093/jac/dks261
33. Gupta SK, Padmanabhan BR, Diene SM, Lopez-Rojas R, Kempf M, Landraud L, et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. (2014) 58:212–20. doi: 10.1128/AAC.01310-13
34. Liu B, Pop M. ARDB–antibiotic resistance genes database. Nucleic Acids Res. (2009) 37:D443–7. doi: 10.1093/nar/gkn656
35. Lakin SM, Dean C, Noyes NR, Dettenwanger A, Ross AS, Doster E, et al. MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. (2017) 45:D574–80. doi: 10.1093/nar/gkw1009
36. Holt KE, Wertheim H, Zadoks RN, Baker S, Whitehouse CA, Dance D, et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci USA. (2015) 112:E3574–81. doi: 10.1073/pnas.1501049112
37. Ruppe E, Cherkaoui A, Lazarevic V, Emonet S, Schrenzel J. Establishing genotype-to-phenotype relationships in bacteria causing hospital-acquired pneumonia: a prelude to the application of clinical metagenomics. Antibiotics. (2017) 6:30. doi: 10.3390/antibiotics6040030
38. Karageorgopoulos DE, Falagas ME. Current control and treatment of multidrug-resistant Acinetobacter baumannii infections. Lancet Infect Dis. (2008) 8:751–62. doi: 10.1016/S1473-3099(08)70279-2
39. Adams MD, Nickel GC, Bajaksouzian S, Lavender H, Murthy AR, Jacobs MR, et al. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob Agents Chemother. (2009) 53:3628–34. doi: 10.1128/AAC.00284-09
40. Ordooei Javan A, Shokouhi S, Sahraei Z. A review on colistin nephrotoxicity. Eur J Clin Pharmacol. (2015) 71:801–10. doi: 10.1007/s00228-015-1865-4
41. Metan G, Alp E, Yildiz O, Percin D, Aygen B, Sumerkan B. Clinical experience with tigecycline in the treatment of carbapenem-resistant Acinetobacter infections. J Chemother. (2010) 22:110–4. doi: 10.1179/joc.2010.22.2.110
42. Goff DA, Bauer KA, Mangino JE. Bad bugs need old drugs: a stewardship program's evaluation of minocycline for multidrug-resistant Acinetobacter baumannii infections. Clin Infect Dis. (2014) 6 59 (Suppl.):S381–7. doi: 10.1093/cid/ciu593
43. Aydemir H, Akduman D, Piskin N, Comert F, Horuz E, Terzi A, et al. Colistin vs. the combination of colistin and rifampicin for the treatment of carbapenem-resistant Acinetobacter baumannii ventilator-associated pneumonia. Epidemiol Infect. (2013) 141:1214–22. doi: 10.1017/S095026881200194X
44. Valencia R, Arroyo LA, Conde M, Aldana JM, Torres MJ, Fernandez-Cuenca F, et al. Nosocomial outbreak of infection with pan-drug-resistant Acinetobacter baumannii in a tertiary care university hospital. Infect Control Hosp Epidemiol. (2009) 30:257–63. doi: 10.1086/595977
45. Sahl JW, Johnson JK, Harris AD, Phillippy AM, Hsiao WW, Thom KA, et al. Genomic comparison of multi-drug resistant invasive and colonizing Acinetobacter baumannii isolated from diverse human body sites reveals genomic plasticity. BMC Genomics. (2011) 12:291. doi: 10.1186/1471-2164-12-291
46. Erntell M, Sjobring U, Myhre EB, Kronvall G. Non-immune Fab- and Fc- mediated interactions of avian Ig with S. aureus and group C and G streptococci. APMIS. (1988) 96:239–49. doi: 10.1111/j.1699-0463.1988.tb05297.x
47. Kumar A, Pattabiraman TN. Elimination of factors interfering in the estimation of serum glycated albumin. Indian J Biochem Biophys. (1988) 25:703–7.
48. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
49. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. (1997) 25:3389–402. doi: 10.1093/nar/25.17.3389
50. Bartual SG, Seifert H, Hippler C, Luzon MA, Wisplinghoff H, Rodriguez-Valera F. Development of a multilocus sequence typing scheme for characterization of clinical isolates of Acinetobacter baumannii. J Clin Microbiol. (2005) 43:4382–90. doi: 10.1128/JCM.43.9.4382-4390.2005
51. Nemec A, Krizova L, Maixnerova M, Diancourt L, van der Reijden TJ, Brisse S, et al. Emergence of carbapenem resistance in Acinetobacter baumannii in the Czech Republic is associated with the spread of multidrug-resistant strains of European clone II. J Antimicrob Chemother. (2008) 62:484–9. doi: 10.1093/jac/dkn205
52. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
53. Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. (2017) 13:e1005595. doi: 10.1371/journal.pcbi.1005595
54. Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. (2014) 9:e112963. doi: 10.1371/journal.pone.0112963
55. Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. (2002) 30:2478–83. doi: 10.1093/nar/30.11.2478
56. Sahl JW, Lemmer D, Travis J, Schupp JM, Gillece JD, Aziz M, et al. NASP: an accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microbial Genomics. (2016) 2: e000074. doi: 10.1099/mgen.0.000074
57. Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. (2010) 5:e9490. doi: 10.1371/journal.pone.0009490
58. Endimiani A, Perez F, Bonomo RA. Cefepime: a reappraisal in an era of increasing antimicrobial resistance. Expert Rev Anti Infect Ther. (2008) 6:805–24. doi: 10.1586/14787210.6.6.805
59. Ahmed NH, Baba K, Clay C, Lekalakala R, Hoosen AA. In vitro activity of tigecycline against clinical isolates of carbapenem resistant Acinetobacter baumannii complex in Pretoria, South Africa. BMC Res Notes. (2012) 5:215. doi: 10.1186/1756-0500-5-215
60. Hamidian M, Nigro SJ, Hall RM. Variants of the gentamicin and tobramycin resistance plasmid pRAY are widely distributed in Acinetobacter. J Antimicrob Chemother. (2012) 67:2833–6. doi: 10.1093/jac/dks318
61. Lee H, Yong D, Yum JH, Roh KH, Lee K, Yamane K, et al. Dissemination of 16S rRNA methylase-mediated highly amikacin-resistant isolates of Klebsiella pneumoniae and Acinetobacter baumannii in Korea. Diagn Microbiol Infect Dis. (2006) 56:305–12. doi: 10.1016/j.diagmicrobio.2006.05.002
62. McCracken M, DeCorby M, Fuller J, Loo V, Hoban DJ, Zhanel GG, et al. Identification of multidrug- and carbapenem-resistant Acinetobacter baumannii in Canada: results from CANWARD 2007. J Antimicrob Chemother. (2009) 64:552–5. doi: 10.1093/jac/dkp225
63. Fernandez Cuenca F, Pascual A, Martinez Martinez L, Perea EJ. [In vitro activity of azithromycin against clinical isolates of Acinetobacter baumannii]. Rev Esp Quimioter. (2003) 16:204–8.
64. Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. (1995) 39:1211–33. doi: 10.1128/AAC.39.6.1211
65. Xia J, Zhang D, Xu Y, Gong M, Zhou Y, Fang X. A retrospective analysis of carbapenem-resistant Acinetobacter baumannii-mediated nosocomial pneumonia and the in vitro therapeutic benefit of cefoperazone/sulbactam. Int J Infect Dis. (2014) 23:90–3. doi: 10.1016/j.ijid.2014.01.017
66. Damier-Piolle L, Magnet S, Bremont S, Lambert T, Courvalin P. AdeIJK, a resistance-nodulation-cell division pump effluxing multiple antibiotics in Acinetobacter baumannii. Antimicrob Agents Chemother. (2008) 52:557–62. doi: 10.1128/AAC.00732-07
67. Shi ZY, Liu PY, Lau Y, Lin Y, Hu BS, Shir JM. Antimicrobial susceptibility of clinical isolates of Acinetobacter baumannii. Diagn Microbiol Infect Dis. (1996) 24:81–5. doi: 10.1016/0732-8893(96)00017-X
68. Chiu CH, Lee HY, Tseng LY, Chen CL, Chia JH, Su LH, et al. Mechanisms of resistance to ciprofloxacin, ampicillin/sulbactam and imipenem in Acinetobacter baumannii clinical isolates in Taiwan. Int J Antimicrob Agents. (2010) 35:382–6. doi: 10.1016/j.ijantimicag.2009.12.009
69. Choi JY, Kim CO, Park YS, Yoon HJ, Shin SY, Kim YK, et al. Comparison of efficacy of cefoperazone/sulbactam and imipenem/cilastatin for treatment of Acinetobacter bacteremia. Yonsei Med J. (2006) 47:63–9. doi: 10.3349/ymj.2006.47.1.63
70. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXivorg. (2013). Available online at: https://arxiv.org/abs/1303.3997v2
71. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. (2011) 43:491–8. doi: 10.1038/ng.806
72. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
73. Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. (2012) 40:D48–53. doi: 10.1093/nar/gkr1202
74. Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. (2016) 17:132. doi: 10.1186/s13059-016-0997-x
75. Sahl JW, Caporaso JG, Rasko DA, Keim P. The large-scale blast score ratio (LS-BSR) pipeline: a method to rapidly compare genetic content between bacterial genomes. PeerJ. (2014) 2:e332. doi: 10.7717/peerj.332
76. Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. (2002) 12:656–64. doi: 10.1101/gr.229202
77. Guy L, Kultima JR, Andersson SG. genoPlotR: comparative gene and genome visualization in R. Bioinformatics. (2010) 26:2334–5. doi: 10.1093/bioinformatics/btq413
78. Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. (2020) 17:261–72. doi: 10.1038/s41592-019-0686-2
79. Rasko DA, Myers GS, Ravel J. Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinformatics. (2005) 6:2. doi: 10.1186/1471-2105-6-2
80. Boisvert S, Laviolette F, Corbeil J. Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol. (2010) 17:1519–33. doi: 10.1089/cmb.2009.0238
81. Collins C, Didelot X. A phylogenetic method to perform genome-wide association studies in microbes that accounts for population structure and recombination. PLoS Comput Biol. (2018) 14:e1005958. doi: 10.1371/journal.pcbi.1005958
82. Lees JA, Galardini M, Bentley SD, Weiser JN, Corander J. pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics. (2018) 34:4310–2. doi: 10.1093/bioinformatics/bty539
83. Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in python. J Mach Learn Res. (2011) 12:2825–30.
84. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. (2015) 12:59–60. doi: 10.1038/nmeth.3176
85. Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. (2017) 27:824–34. doi: 10.1101/gr.213959.116
86. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. (2017) 14:417–9. doi: 10.1038/nmeth.4197
87. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
88. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. (1995) 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
89. Colman RE, Schupp JM, Hicks ND, Smith DE, Buchhagen JL, Valafar F, et al. Detection of low-level mixed-population drug resistance in Mycobacterium tuberculosis using high fidelity amplicon sequencing. PLoS ONE. (2015) 10:e0126626. doi: 10.1371/journal.pone.0126626
90. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. (2016) 34:525–7. doi: 10.1038/nbt.3519
91. Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. (2016) 44:W242–5. doi: 10.1093/nar/gkw290
92. Yang Y, Xu Q, Li T, Fu Y, Shi Y, Lan P, et al. OXA-23 is a prevalent mechanism contributing to sulbactam resistance in diverse Acinetobacter baumannii clinical strains. Antimicrob Agents Chemother. (2019) 63:e01676–18. doi: 10.1128/AAC.01676-18
93. Higgins PG, Perez-Llarena FJ, Zander E, Fernandez A, Bou G, Seifert H. OXA-235, a novel class D beta-lactamase involved in resistance to carbapenems in Acinetobacter baumannii. Antimicrob Agents Chemother. (2013) 57:2121–6. doi: 10.1128/AAC.02413-12
94. Krizova L, Poirel L, Nordmann P, Nemec A. TEM-1 beta-lactamase as a source of resistance to sulbactam in clinical strains of Acinetobacter baumannii. J Antimicrob Chemother. (2013) 68:2786–91. doi: 10.1093/jac/dkt275
95. Choury D, Szajnert MF, Joly-Guillou ML, Azibi K, Delpech M, Paul G. Nucleotide sequence of the bla(RTG-2) (CARB-5) gene and phylogeny of a new group of carbenicillinases. Antimicrob Agents Chemother. (2000) 44:1070–4. doi: 10.1128/AAC.44.4.1070-1074.2000
96. Jacoby GA. AmpC beta-lactamases. Clin Microbiol Rev. (2009) 22:161–82. doi: 10.1128/CMR.00036-08
97. Vakili B, Khorvash F, Fazeli H, Khaleghi M. Detection of quinolone-resistance mutations of parC gene in clinical isolates of Acinetobacter baumannii in Iran. J Res Med Sci. (2014) 19:567–70.
98. Rossjohn J, Polekhina G, Feil SC, Allocati N, Masulli M, Di Illio C, et al. A mixed disulfide bond in bacterial glutathione transferase: functional and evolutionary implications. Structure. (1998) 6:721–34. doi: 10.1016/S0969-2126(98)00074-4
99. Lallement C, Pasternak C, Ploy MC, Jove T. The role of ISCR1-borne POUT promoters in the expression of antibiotic resistance genes. Front Microbiol. (2018) 9:2579. doi: 10.3389/fmicb.2018.02579
100. Vuilleumier S. Bacterial glutathione S-transferases: what are they good for? J Bacteriol. (1997) 179:1431–41. doi: 10.1128/JB.179.5.1431-1441.1997
101. Lee CR, Lee JH, Park M, Park KS, Bae IK, Kim YB, et al. Biology of Acinetobacter baumannii: pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front Cell Infect Microbiol. (2017) 7:55. doi: 10.3389/fcimb.2017.00055
102. Yong D, Shin JH, Kim S, Lim Y, Yum JH, Lee K, et al. High prevalence of PER-1 extended-spectrum beta-lactamase-producing Acinetobacter spp. in Korea. Antimicrob Agents Chemother. (2003) 47:1749–51. doi: 10.1128/AAC.47.5.1749-1751.2003
103. Hou PF, Chen XY, Yan GF, Wang YP, Ying CM. Study of the correlation of imipenem resistance with efflux pumps AdeABC, AdeIJK, AdeDE and AbeM in clinical isolates of Acinetobacter baumannii. Chemotherapy. (2012) 58:152–8. doi: 10.1159/000335599
104. Brandt C, Braun SD, Stein C, Slickers P, Ehricht R, Pletz MW, et al. In silico serine beta-lactamases analysis reveals a huge potential resistome in environmental and pathogenic species. Sci Rep. (2017) 7:43232. doi: 10.1038/srep43232
105. Lai JH, Yang JT, Chern J, Chen TL, Wu WL, Liao JH, et al. Comparative phosphoproteomics reveals the role of AmpC beta-lactamase phosphorylation in the clinical imipenem-resistant strain Acinetobacter baumannii SK17. Mol Cell Proteomics. (2016) 15:12–25. doi: 10.1074/mcp.M115.051052
106. Schafer U, Beck K, Muller M. Skp, a molecular chaperone of gram-negative bacteria, is required for the formation of soluble periplasmic intermediates of outer membrane proteins. J Biol Chem. (1999) 274:24567–74. doi: 10.1074/jbc.274.35.24567
107. Eguale T, Marshall J, Molla B, Bhatiya A, Gebreyes WA, Engidawork E, et al. Association of multicellular behaviour and drug resistance in Salmonella enterica serovars isolated from animals and humans in Ethiopia. J Appl Microbiol. (2014) 117:961–71. doi: 10.1111/jam.12579
108. Sklar JG, Wu T, Kahne D, Silhavy TJ. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. (2007) 21:2473–84. doi: 10.1101/gad.1581007
109. Poole K. Bacterial stress responses as determinants of antimicrobial resistance. J Antimicrob Chemother. (2012) 67:2069–89. doi: 10.1093/jac/dks196
110. Zahn M, D'Agostino T, Eren E, Basle A, Ceccarelli M, van den Berg B. Small-molecule transport by CarO, an abundant eight-stranded beta-barrel outer membrane protein from Acinetobacter baumannii. J Mol Biol. (2015) 427:2329–39. doi: 10.1016/j.jmb.2015.03.016
111. Afzal-Shah M, Woodford N, Livermore DM. Characterization of OXA-25, OXA-26, and OXA-27, molecular class D beta-lactamases associated with carbapenem resistance in clinical isolates of Acinetobacter baumannii. Antimicrob Agents Chemother. (2001) 45:583–8. doi: 10.1128/AAC.45.2.583-588.2001
112. Danes C, Navia MM, Ruiz J, Marco F, Jurado A, Jimenez de Anta MT, et al. Distribution of beta-lactamases in Acinetobacter baumannii clinical isolates and the effect of Syn 2190 (AmpC inhibitor) on the MICs of different beta-lactam antibiotics. J Antimicrob Chemother. (2002) 50:261–4. doi: 10.1093/jac/dkf092
113. Kwon NY, Kim JD, Pai HJ. The resistance mechanisms of b-lactam antimicrobials in clinical isolates of Acinetobacter baumannii. Korean J Intern Med. (2002) 17:94–9. doi: 10.3904/kjim.2002.17.2.94
114. Raible KM, Sen B, Law N, Bias TE, Emery CL, Ehrlich GD, et al. Molecular characterization of beta-lactamase genes in clinical isolates of carbapenem-resistant Acinetobacter baumannii. Ann Clin Microbiol Antimicrob. (2017) 16:75. doi: 10.1186/s12941-017-0248-3
115. Marchand I, Damier-Piolle L, Courvalin P, Lambert T. Expression of the RND-type efflux pump AdeABC in Acinetobacter baumannii is regulated by the AdeRS two-component system. Antimicrob Agents Chemother. (2004) 48:3298–304. doi: 10.1128/AAC.48.9.3298-3304.2004
116. Su XZ, Chen J, Mizushima T, Kuroda T, Tsuchiya T. AbeM, an H+-coupled Acinetobacter baumannii multidrug efflux pump belonging to the MATE family of transporters. Antimicrob Agents Chemother. (2005) 49:4362–4. doi: 10.1128/AAC.49.10.4362-4364.2005
117. Lister PD, Wolter DJ, Hanson ND. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev. (2009) 22:582–610. doi: 10.1128/CMR.00040-09
118. Sood S. Chloramphenicol - a potent armament against multi-drug resistant (MDR) gram negative bacilli? J Clin Diagn Res. (2016) 10:DC01–3. doi: 10.7860/JCDR/2016/14989.7167
119. Chewapreecha C, Marttinen P, Croucher NJ, Salter SJ, Harris SR, Mather AE, et al. Comprehensive identification of single nucleotide polymorphisms associated with beta-lactam resistance within pneumococcal mosaic genes. PLoS Genet. (2014) 10:e1004547. doi: 10.1371/journal.pgen.1004547
120. Wheeler NE, Reuter S, Chewapreecha C, Lees JA, Blane B, Horner C, et al. Contrasting approaches to genome-wide association studies impact the detection of resistance mechanisms in Staphylococcus aureus. bioRxiv. (2019) doi: 10.1101/758144
121. Mamedov IZ, Britanova OV, Zvyagin IV, Turchaninova MA, Bolotin DA, Putintseva EV, et al. Preparing unbiased T-cell receptor and antibody cDNA libraries for the deep next generation sequencing profiling. Front Immunol. (2013) 4:456. doi: 10.3389/fimmu.2013.00456
Keywords: genomics, transcriptomics, bioinformatics, acinetobacter, AMR
Citation: Roe C, Williamson CHD, Vazquez AJ, Kyger K, Valentine M, Bowers JR, Phillips PD, Harrison V, Driebe E, Engelthaler DM and Sahl JW (2020) Bacterial Genome Wide Association Studies (bGWAS) and Transcriptomics Identifies Cryptic Antimicrobial Resistance Mechanisms in Acinetobacter baumannii. Front. Public Health 8:451. doi: 10.3389/fpubh.2020.00451
Received: 05 March 2020; Accepted: 21 July 2020;
Published: 02 September 2020.
Edited by:
Fumito Maruyama, Hiroshima University, JapanReviewed by:
Hirokazu Yano, Tohoku University, JapanCopyright © 2020 Roe, Williamson, Vazquez, Kyger, Valentine, Bowers, Phillips, Harrison, Driebe, Engelthaler and Sahl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason W. Sahl, amFzb24uc2FobEBuYXUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.