 Ruiyun Li
Ruiyun Li Tao Zhang
Tao Zhang Yuqi Bai2
Yuqi Bai2 Yuhai Bi
Yuhai Bi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Public Health , 27 July 2018
Sec. Epidemiology
Volume 6 - 2018 | https://doi.org/10.3389/fpubh.2018.00210
The on-going reassortment, human-adapted mutations, and spillover events of novel A(H7N9) avian influenza viruses pose a significant challenge to public health in China and globally. However, our understanding of the factors that disseminate the viruses and drive their geographic distributions is limited. We applied phylogenic analysis to examine the inter-subtype interactions between H7N9 viruses and the closest H9N2 lineages in China during 2010–2014. We reconstructed and compared the inter-provincial live poultry trading and viral propagation network via phylogeographic approach and network similarity technique. The substitution rates of the isolated viruses in live poultry markets and the characteristics of localized viral evolution were also evaluated. We discovered that viral propagation was geographically-structured and followed the live poultry trading network in China, with distinct north-to-east paths of spread and circular transmission between eastern and southern regions. The epicenter of H7N9 has moved from the Shanghai–Zhejiang region to Guangdong Province was also identified. Besides, higher substitution rate was observed among isolates sampled from live poultry markets, especially for those H7N9 viruses. Live poultry trading in China may have driven the network-structured expansion of the novel H7N9 viruses. From this perspective, long-distance geographic expansion of H7N9 were dominated by live poultry movements, while at local scales, diffusion was facilitated by live poultry markets with highly-evolved viruses.
The detection of a novel H7N9 virus in March 2013 was the first time that a low-pathogenic avian influenza A(H7N9) virus was identified in humans (1). Since then, the disease has continued to evolve in avian and humans, causing 766 confirmed human infections in 19 provinces of mainland China in the following 3 years.1
Recent analyses demonstrated that the novel H7N9 virus was a reassortant, with surface and internal gene segments originating from wild birds and the H9N2 lineage in poultry, respectively. This indicates that wild birds were the most likely source of infection, introducing the virus into domestic ducks and chickens through sequential reassortment events, with a consequent spillover to humans by means of live poultry exposure (2–9). The resulting multiple H7N9 lineages and genotypes suggest that the evolution of H9N2 has facilitated the genesis of the internal segments of this novel reassortant (4, 10, 11). It has therefore exhibited greater genetic diversity compared with the surface genes (4, 12). As our preliminary researches have pointed out, this interaction at the wild birds–poultry–humans interface was common in the spread of infectious diseases on various scales (13–17).
During this process, live poultry trading network enclosing live poultry markets (LPMs) and live poultry transportations (LPTs), was vitally important to the further genetic evolution of avian influenza viruses (AIVs) and the poultry–humans sequential transmissions (Figure 1). LPMs were ideal environment for the co-circulation and co-infection of different subtypes and the spillover to humans (18–20). However, it will be efficient in the reduction of daily number and growth rate of new human cases through target control measures (e.g., market closure) (21, 22). Nevertheless, interventions mainly focused on LPMs was insufficient in restricting the geographic expansion of the viruses. The fact that the transmission of H7N9 virus into regionally-specific H9N2 gene pools and the consequent establishment of more diversified genotypes can be accelerated by the inter-provincial trading of live poultry (4, 8). In this case, preventions of viral spread along poultry movements should also be implemented practically. However, although restrictions in LPMs and LPTs have been put forward as one of the important prerequisites in limiting viral expansion, it was unclear about how live poultry trading shapes the geographic propagation of viruses.
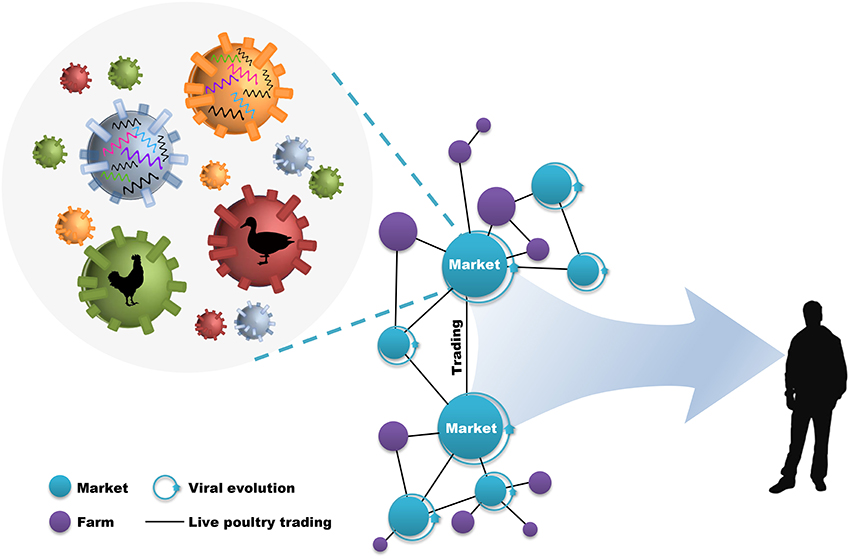
Figure 1. Live poultry trading network drives viral evolution and poultry–humans sequential transmissions. The live poultry trading network consist of live poultry markets, farms, and the trading among them. The genetic evolution of avian influenza viruses is facilitated by the co-circulation of different viral subtypes and hosts within markets, and further expanded by poultry trading. This poultry trading framework increases the potential for poultry–humans sequential transmissions.
Previous studies have reported that interdisciplinary datasets and approaches provide a unique opportunity to infer the dynamic footprints and underlying mechanisms of AIVs (14, 17). In the present study, we employed sequence data and social-economic information to demonstrate the position of live poultry trading in dominating the local evolution and spatial diffusion structure of H7N9 viruses.
All animal work was approved by the Beijing Association for Science and Technology (approval SYXK [Beijing] 2007-0023). The laboratory animal research was performed in the microbiology laboratory of China Agricultural University, and in accordance with Beijing Laboratory Animal Welfare and Ethics guidelines issued by the Beijing Administration Committee of Laboratory Animals and China Agricultural University Institutional Animal Care and Use Committee guidelines (ID: SKLAB-B-2010-003).
Samples were collected from two different LPMs in Jiangxi Province during June 2014. These viruses were isolated from positive samples and sequenced following the high-throughput sequencing on an Illumina Hiseq2500 sequencer. The virus isolation rate was defined as the number of H9N2 samples divided by the total number of samples we collected.
Sequence data for all the internal gene segments of H7N9 isolates in March 2013–December 2014 and those of H9N2 isolates in January 2010–December 2014 were obtained from the GenBank database of the National Center for Biotechnology Information (23). The H9N2 isolates reported in previous studies were also included for the categorization and identification of the closest lineage to H7N9 isolates (11, 24). The integrated dataset including all these published H7N9 and H9N2 sequences and those we collected was used for further analysis. To address the issue of the unbalanced number of sequences among isolation locations, we reclassified locations by combining the geographically adjacent and social-economically similar provinces into an integral region. This location reassignment generated six regions/provinces for H9N2 isolates: Beijing-Tianjin-Hebei region (J-J-J region), Shanghai-Zhejiang region, and Shandong, Jiangsu, Jiangxi, and Guangdong Province. Since no H7N9 isolates were collected from J-J-J region and Shandong Province, the location of H7N9 isolates were assigned to one of the other four regions/provinces. In this study, both H7N9 and H9N2 isolates were assigned a specific information, containing the viral subtype (i.e., H9N2 or H7N9), spatial location (i.e., region or province) and sampling location (i.e., LPMs, farms, or others), and the additional epidemic waves for H7N9 isolates. In addition, isolates sampled from LPMs and farms were extracted to quantify and compare the substitution rates, or the rate at which mutations fix in a population, between subtypes and sampling locations.
The phylogenetic analysis of these sequence data was implemented to identify the evolutionary structure using Molecular Evolutionary Genetics Analysis (MEGA) software version 6.0 (25). In order to ensure the identity of phylogenetic topology, phylogenetic tree was constructed by both the maximum likelihood and neighbor-joining distance-based matrix algorithms. The H9N2 isolates during 2010–2014 was then grouped into each lineage, and those shared the highest similarity with H7N9 viruses were defined as the closest H9N2 lineage to H7N9 viruses (Supplementary Table 1).
The inference of the discrete phylogeography of internal segments of H7N9 and closest H9N2 was made in the Bayesian statistical inference framework using a Bayesian Markov chain Monte Carlo (MCMC) method in the BEAST package (version 1.8.0) (26, 27). In addition, substitution rate of isolates sampled from LPMs and farms were also estimated using strict molecular clock, Bayesian skyline coalescent prior, and HKY85+Gamma nucleotide substitution model. The convergence diagnosis of MCMC chain was inspected in Tracer (http://beast.community/tracer) by the trace plot of 20,000,000 iterations, with the effective sample size (ESS) greater than 200. The uncertainty analysis of alternative molecular evolutionary processes was also implemented by comparing the performance of different evolutionary models (i.e., the combination of molecular clock models and coalescent priors) using Bayes factor (Supplementary Table 2).
The set of risk factors in this study included national road density, the density of live poultry markets, and the relative production and consumption of poultry for the above regions/provinces. The national road data were extracted from the 1:1,000,000 geomatics dataset provided by the China State Information Center. The length of the pairwise national road between two locations refers to the total length of all the alternative national roads between them. In addition, on the basis of the big data for public health, the information of LPMs was derived from the search engine of Chinese Academy of Surveying and Mapping. This automated data mining process was executed with different terms, i.e., live poultry, agricultural market, fresh market, live bird market (Supplementary Table 3), and the replicated records were then removed. The collected pairwise road distance and number of LPMs were further transformed into national road density and LPMs density (i.e., the length of national road and the number of LPMs per square kilometers, respectively). The relative production and consumption of poultry in 2010–2013 were collected from the China Statistical Yearbook (28).
The market accessibility was defined as national road density to represent the transportation cost between pairwise markets. Specifically, the higher density of national road indicates the multiple alternative transportation approaches, and hence more ease and higher feasibility for live poultry trading.
The Fixation index (FST) was derived from the F-statistics proposed by Sewall Wright to study population structure (29). Here, we used FST to quantify the amount of genetic variance that can be explained by market accessibility. The value of FST varied from 0 and 1, and the two extreme estimates 0 and 1 indicated no and complete spatial structure of genetic variations, respectively. Specifically, a modified FST defined as was used (30), where Hw and Hb are the average genetic variations within and between provinces i and j, defined as
where δxy is the pairwise genetic distance between two nucleotide sequences (x and y); ni and nj are the numbers of sequences in i and j, respectively; and ix and jy are the xth and yth sequences in province i and j.
For each pair of locations with H7N9 isolates, we first calculated its FST and then proceeded to implement the linear regression and make an initial investigation of the relations between market accessibility and genetic divergence. Mantel test (31) was further employed to evaluate the strength of this genetic-geographic relations (Supplementary Figure 1).
Poultry transportation information was gathered from news reports on websites and statistical reports. The subtle intra-provincial movement routes were then combined or eliminated to give an illustrative inter-provincial transportation network. The corresponding undirected graph with only paths between regions/provinces was also generated for the subsequent correlation test with viral migration routes.
“Markov jump” counts (32) which provided an estimate of the expected number of spatial location transitions based on the reconstructed phylogeny, established the gene flow network among locations. With this quantitative assessment, we depicted a H9N2–H7N9 viral migration network for each internal gene segment to reflect the cross-subtype interactions and the H7N9 spatial pattern of spread. Statistically supported transmission paths were selected based on Bayes factor, with cutoff values of 3. These proposed networks were further merged into a single viral dynamic network where linkage type and strength indicated the persistence and the number of occurrence in all internal segments, respectively. This integrated network was then abstracted into an undirected viral propagation graph with only inter-provincial routes, to test the similarity with the structure of poultry transportations.
Considering the alternative viral migration process along unobserved networks, 1,000 random graphs were generated to ascertain the statistical significance of correlations between the topology of two graphs via quadratic assignment procedure (QAP) test (33).
Four hundred and ninety three samples were collected from two LPMs in Jiangxi Province during June 2014, among which 61 samples (12.37%) were H9N2-positive. Besides, the mixture sample of H9N2 and other subtypes were also identified, with the isolation rate to be 7.52% for the mixture of H9N2 and another subtype and 2.84% for those mixed with two or more subtypes.
Generally, both H7N9 and H9N2 viruses sampled during March 2013–December 2014 substituted at a higher rate compared with those in previous years (i.e., January 2010–December 2012), especially for viruses obtained in LPMs (Table 1). It is also noted that all the internal segments of H7N9 evolved even faster than the contemporary H9N2 viruses, particularly for the NS gene.
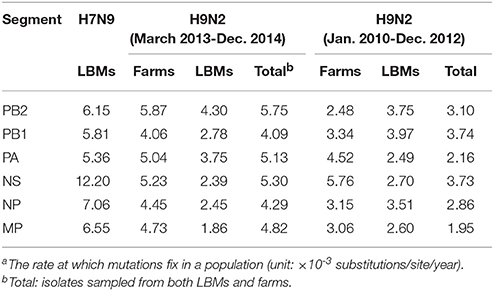
Table 1. Mean substitution ratesa of avian influenza samples isolated in different time periods and locations.
Statistically significant associations were observed between the genetic divergence and accessibility among markets, with the correlation coefficient values (R) of regression ranged from 0.54 to 0.76 (Supplementary Figure 1). There was a general decreasing trend of the regional genetic differentiation level with the increasing market accessibility. This result implied that market accessibility, or the feasibility of poultry trading, predominantly drove the genetic differentiation structure. Additionally, the higher road density benefited the live poultry trading by providing multiple alternative approaches and hence lowering the costs of transportations. However, the residual genetic variation not explained by the regression (around 24–46%) suggested that factors other than national road density may have also influenced the spatial differentiation pattern of internal genes.
The previous H9N2 viruses were closely related to the newly-emerged H7N9 viruses, forming a distinct north-to-east inter-provincial transmission route and a locally-evolved pattern in the southern region (Figure 2). Specifically, the H9N2 viruses that dispersed from northern area (i.e., J-J-J region and Shandong Province) may be the donor of the H7N9 internal genes in the Shanghai–Zhejiang region. However, we also noted that the H7N9 viruses in Jiangxi and Guangdong were more closely related to local than to external H9N2 viruses.
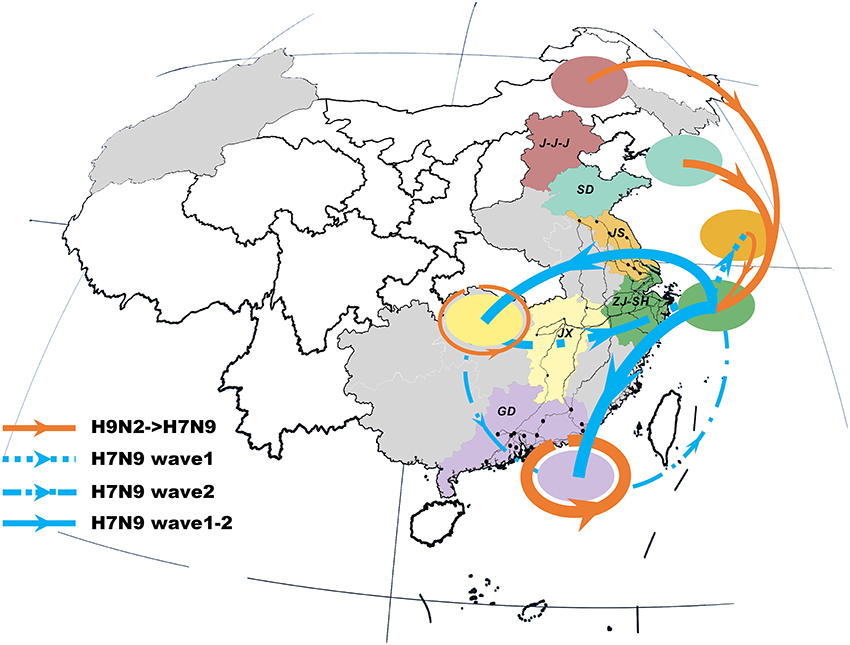
Figure 2. Viral propagation network for internal gene segments of H9N2-H7N9 viruses. Viral migration routes are distinguished by subtype, epidemic wave, and strength. Specifically, orange ones represent interaction between H9N2 and H7N9 viruses; while the blue dot lines, dash-dot lines and solid lines with arrow indicate the viral migration pattern of H7N9 in wave 1, wave 2 and persist during two epidemic waves, respectively. The linkage strength is represented by the number of occurrence in all internal segments. Locations with H7N9 human cases included in the study are colored and those not included are shaded gray. The primary national roads and cities with H7N9 human cases are represented by black lines and black dots, respectively. J-J-J, Beijing–Tianjin–Hebei region; SD, Shandong; JS, Jiangsu; SH-ZJ, Shanghai–Zhejiang region; JX, Jiangxi; GD, Guangdong.
The spatial dispersion patterns of the H7N9 viruses were diverse and highly-structured, with characteristics specific to each epidemic wave (Figure 2 and Table 2). In particular, the spatial spread of the H7N9 viruses was predominantly restricted to the vicinity of the Shanghai–Zhejiang region during the early period of wave 1, with only a small fraction reaching Jiangsu (the dot line with arrow). However, the long-distance spread from Shanghai–Zhejiang region to Jiangxi and Guangdong Provinces occurred at the end of wave1 and persisted as the major routes until the next wave (the solid lines with arrow). This wide-range migration was followed by the apparent backward transmission from Jiangxi and Guangdong Provinces to Shanghai–Zhejiang region and the established linkage between Jiangxi and Guangdong Province (the dash-dot lines with arrow). Thus, the viruses in the east and south region was linked through the bidirectional or circular transmission between them.
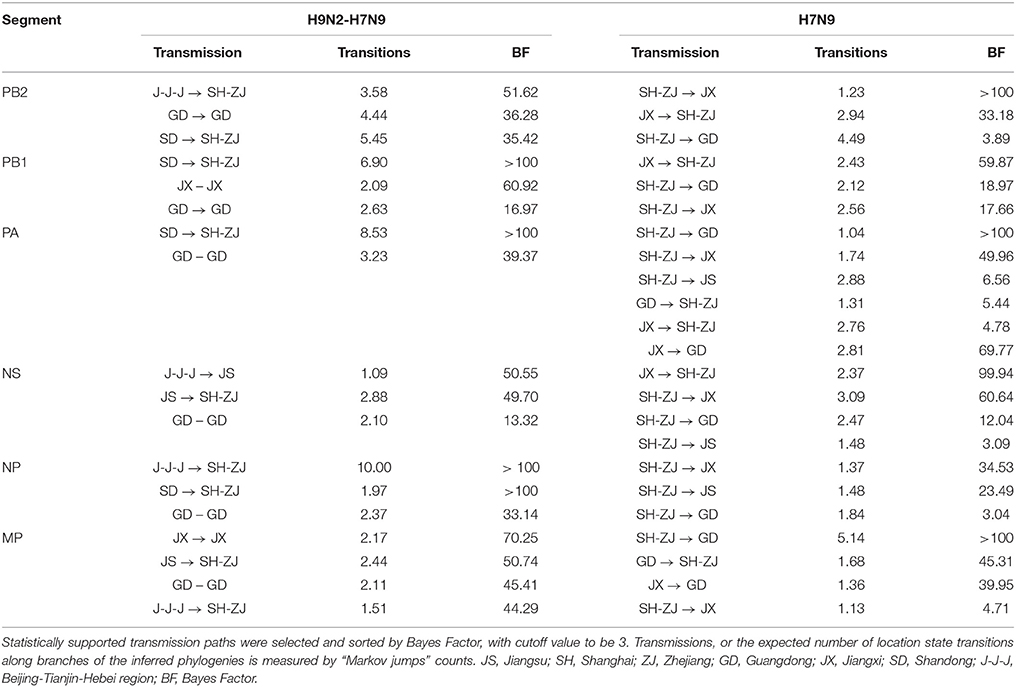
Table 2. Statistical performance of viral propagation paths.
Interestingly, there was a strong similarity between the pattern of viral propagation and LPTs. More precisely, the poultry movement path, which originated from primary poultry production area in the north to the main poultry consumption region in the east (i.e., Shanghai–Zhejiang region) (Figure 3), was consistent with the north-to-east spread pattern of AIVs. Furthermore, the circular poultry transportation paths connecting the Zhejiang–Shanghai region with Jiangxi and Guangdong Provinces were also similar to and may therefore have forced the H7N9 dissemination routes among them.
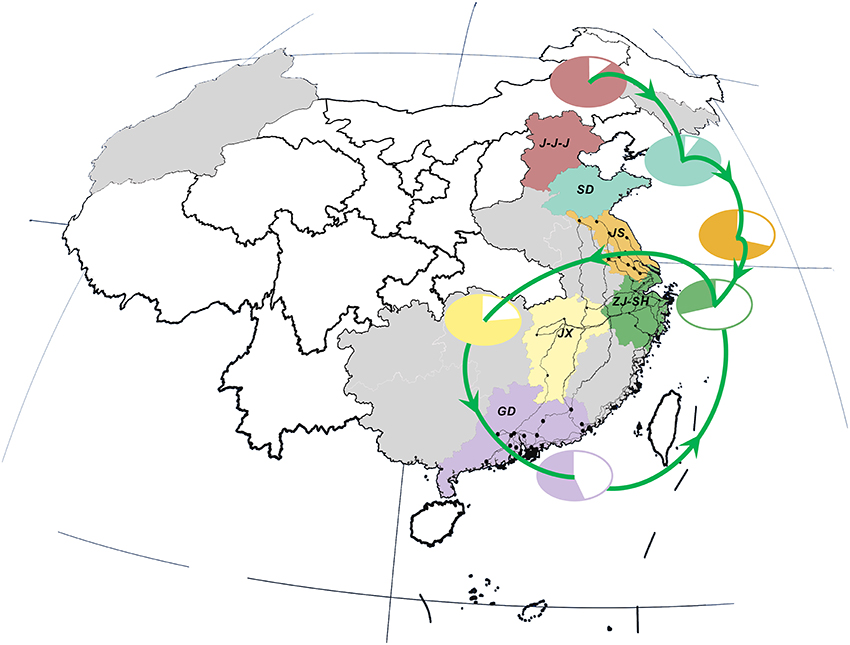
Figure 3. Live poultry transportation network and relative poultry production and consumption in study area. Locations, national roads and cities are colored in consistent with those in Figure 2. Poultry transmission paths are illustrated in green lines with arrows. The relative percentage of poultry production (color area) and consumption (white area) in each region/province is distinguished by the same color as the corresponding locations.
This similarity was further verified by the result of correlation analysis which rejected the hypotheses of random viral migration process (0.764, p-value: 0.037) (Supplementary Figure 2), and hence indicated a statistically significant association between the topology of viral diffusion and poultry movement graphs. In other words, the observed pattern of gene flow was highly-structured and can be best fitted by the migration process along the LPTs network.
Our results demonstrated that the network-structured diffusion of the internal gene segments of AIVs followed and can therefore be explained by the consistent inter-provincial paths of live poultry trading. However, the mode or mechanism of poultry trading in shaping the inter-subtype viral interactions differed between locations. More specifically, the genesis of H7N9 in eastern China may have been facilitated by the previous H9N2 viruses disseminated from the northern region along poultry movement paths (11). Contrast with this inter-regional viral interaction, the localized H9N2–H7N9 transmission contributed to the evolution of H7N9 in Guangdong Province. Thus, the H7N9 virus endemic in Guangdong may jointly be resulted from the continuous reassortment with local environmental and avian H9N2 viruses (22, 34, 35) and the H7N9 gene flow through circular poultry movements. The time-specific spatial propagations of the H7N9 viruses suggested that each location played distinct yet crucial role during different epidemic stages. In the first epidemic wave, Shanghai–Zhejiang may have acted as the epicenter, primarily diffusing virus to the adjacent provinces and introducing the viruses into Jiangxi and Guangdong Provinces. This dominant position was then taken over by Guangdong, with a higher emigration rate (12) and backward south-to-east dynamics during the second wave. These two established epicenters of novel H7N9 paralleled those of H9N2 virus (10) and therefore indicated the hotspots for viral evolution and surveillance in China. Further, we presented the H7N9 propagation network based on the phylogeny of all the internal genes, which provided a more comprehensive diffusion pattern compared with the former graph derived from only NA gene segment (4). This may reflect the fact that the internal gene segments affected by both reassorment and poultry movements displayed more diversity than the surface genes.
Given the higher substitution rate of both H7N9 and H9N2 isolates during 2013–2014, LPMs had broad potential to affect viral mergence and evolution. Specifically, the co-circulation and co-infection of different subtypes can provide some cross-immunity to infections from other subtypes, and lower the infection rate, the number of susceptibles, and the consequent probability of pandemic emergence. However, it may also result in the viral evasion from host immunity response through continuous and rapid mutations to better adapt to and efficiently transmit in new environment. Still, the highly-substituted internal genes of H7N9 viruses indicated that this novel AIVs was in the expansion period during 2013–2014. This inference was rational and supported by the modeling work where viral lineage from epidemic regions was found to have higher substitution rate than those from endemic regions (36). Therefore, compared with the inter-regional viral dynamics facilitated by LPTs, LPMs served as the local reservoir for the emergence and evolution of novel viruses.
In the perspective of live poultry trading network in the spatially-structured spread of AIVs presented here, additional interventions should be jointly implemented to restrict viral expansion along LPTs paths and targeted at live poultry workers. Despite the effectiveness in reducing the daily number and growth rate of new human cases (21, 22) and the amount and detection rate of viable viruses (37) the mandatory closure of LBMs alone was unlikely to eliminate the zoonotic threat (38) accounting for the LPTs as the pathway with the highest likelihood of viral spread2. It is also reported that humans who engaged in the transportation work of live chickens and ducks was particularly susceptible to infections from AIVs (39). These facts ascertained the role of live poultry trading in the spillover to humans at avian–human interface and the occurrence of H7N9 human cases. Therefore, a multi-sector, cost-effective approach and even international collaboration will be essential for the substantial reduction in the risk of disease spread and the build of a safer trade in animals3.
These findings were based on the assumptions themed around our research interests which should be taken into account when making interpretations and generalizations. Firstly, since we focused on the inter-provincial or macroscopic live poultry trading patterns and genetic differentiation of H7N9 viruses, we assume that the inter-provincial/regional LPTs occurred only along the national roads connecting pairwise locations. Similarly, market accessibility was also defined by the density of national road. However, this dominant role of national road was scale-dependent and cannot be directly generated to other spatial scales or the contribution of other factors. More precisely, previous studies have shown that detailed and localized LPTs along provincial roads (40), illegal trades (38), and even cross-border transports4 have also made crucial contributions to shaping the viral diffusions. Additionally, although our inferences were implemented using the integrated molecular dataset in Bayesian statistical framework, sampling bias may still have potential effect on the reconstructed phylogeny and the inferred transmission networks. Whereas, the inference system was partially evaluated, our study demonstrated the role of the live poultry trading in dominating the spatially-structured evolution and expansion of H7N9 viruses and the need for active surveillance and interventions on poultry trading.
The genetic sequence data analyzed for this study can be found in the GenBank database of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/pubmed/). The poultry production and consumption data are available from the China Statistical Yearbook (http://www.stats.gov.cn/english/Statisticaldata/AnnualData/).
The H9N2 sequence data collected in Jiangxi Province, poultry transportation and the national road data supporting the conclusions of this manuscript are available on request.
RL and BX deigned the study. TZ and JC performed the field and laboratory experiments. HL and YW provided the geographic information data. RL, TZ, YBai, YBi, JC, and BX analyzed and interpreted the data. RL and BX wrote the paper.
This research was supported by the National Key Research and Development Program of China (2016YFA0600104).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2018.00210/full#supplementary-material
1. ^FAO. H7N9 situation updates. (2016) Available online at: http://www.fao.org/ag/againfo/programmes/en/empres/H7N9/situation_update.html
2. ^FAO. Qualitative risk assessment update. Addressing avian influenza A(H7N9). Emergency Prevention System (EMPRES) Program. Rome. (2014) Available online at: http://www.fao.org/3/a-i3813e.pdf
3. ^FAO. Viet Nam – China aim for safer trade of animals and animal products. (2016) Available online at: http://www.fao.org/vietnam/news/detail-events/en/c/381699/
4. ^FAO. Approaches to controlling, preventing and eliminating H5N1 highly pathogenic avian influenza in endemic countries. (2011) Available from: http://www.fao.org/docrep/014/i2150e/i2150e.pdf
1. Parry J. H7N9 avian flu infects humans for the first time. BMJ (2013) 346:f2151. doi: 10.1136/bmj.f2151
2. Liu D, Shi W, Shi Y, Wang D, Xiao H, Li W, et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: phylogenetic, structural, and coalescent analyses. Lancet (2013) 381:1926–32. doi: 10.1016/S0140-6736(13)60938-1
3. Wu A, Su C, Wang D, Peng Y, Liu M, Hua S, et al. Sequential reassortments underlie diverse influenza H7N9 genotypes in China. Cell Host Microbe (2013) 14:446–52. doi: 10.1016/j.chom.2013.09.001
4. Cui L, Liu D, Shi W, Pan J, Qi X, Li X, et al. Dynamic reassortments and genetic heterogeneity of the human-infecting influenza A (H7N9) virus. Nat Commun. (2014) 5:3142. doi: 10.1038/ncomms4142
5. Qi W, Shi W, Li W, Huang L, Li H, Wu Y, et al. Continuous reassortments with local chicken H9N2 virus underlie the human-infecting influenza A(H7N9) virus in the new influenza season, Guangdong, China. Protein Cell (2014) 5:878–82. doi: 10.1007/s13238-014-0084-6
6. Cowling BJ, Jin L, Lau EH, Liao Q, Wu P, Jiang H, et al. Comparative epidemiology of human infections with avian influenza A H7N9 and H5N1 viruses in China: a population-based study of laboratory-confirmed cases. Lancet (2013) 382:129–37. doi: 10.1016/S0140-6736(13)61171-X
7. Lam TT, Wang J, Shen Y, Zhou B, Duan L, Cheung CL, et al. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature (2013) 502:241–4. doi: 10.1038/nature12515
8. Lam TT, Zhou B, Wang J, Chai Y, Shen Y, Chen X, et al. Dissemination, divergence and establishment of H7N9 influenza viruses in China. Nature (2015) 522:102–5. doi: 10.1038/nature14348
9. Li Q, Zhou L, Zhou M, Chen Z, Li F, Wu H, et al. Epidemiology of human infections with avian influenza A(H7N9) virus in China. N Engl J Med. (2014) 370:520–32. doi: 10.1056/NEJMoa1304617
10. Jin Y, Yu D, Ren H, Yin Z, Huang Z, Hu M, et al. Phylogeography of avian influenza A H9N2 in China. BMC Genomics (2014) 15:1110. doi: 10.1186/1471-2164-15-1110
11. Pu J, Wang S, Yin Y, Zhang G, Carter RA, Wang J, et al. Evolution of the H9N2 influenza genotype that facilitated the genesis of the novel H7N9 virus. Proc Natl Acad Sci USA. (2015) 112:548–53. doi: 10.1073/pnas.1422456112
12. Wang D, Yang L, Zhu W, Zhang Y, Zou S, Bo H, et al. Two outbreak sources of influenza A(H7N9) viruses have been established in China. J Virol. (2016) 90:5561–73. doi: 10.1128/JVI.03173-15
13. Tian H, Cui Y, Dong L, Zhou S, Li X, Huang S, et al. Spatial, temporal and genetic dynamics of highly pathogenic avian influenza A (H5N1) virus in China. BMC Infect Dis. (2015) 15:54. doi: 10.1186/s12879-015-0770-x
14. Tian H, Zhou S, Dong L, Boeckel TPV, Cui Y, Newman SH, et al. Avian influenza H5N1 viral and bird migration networks in Asia. Proc Natl Acad Sci USA. (2015) 112:172–7. doi: 10.1073/pnas.1405216112
15. Wang Y, Jiang Z, Jin Z, Tan H, Xu B. Risk factors for infectious diseases in backyard poultry farms in the Poyang Lake area, China. PLoS ONE (2013) 8:e67366. doi: 10.1371/journal.pone.0067366
16. Wang G, Zhang T, Li X, Jiang Z, Jiang Q, Chen Q, et al. Serological evidence of H7, H5 and H9 avian influenza virus co-infection among herons in a city park in Jiangxi, China. Sci Rep. (2014) 4:6345. doi: 10.1038/srep06345
17. Li R, Jiang Z, Xu B. Global spatiotemporal and genetic footprint of the H5N1 avian influenza virus. Int J Health Geogr. (2014) 13:14. doi: 10.1186/1476-072X-13-14
18. Bi Y, Chen Q, Wang Q, Chen J, Jin T, Wong G, et al. Genesis, evolution and prevalence of H5N6 avian influenza viruses in China. Cell Host Microbe (2016) 20:1–12. doi: 10.1016/j.chom.2016.10.022
19. Shi J, Deng G, Liu P, Zhou J, Guan L, Li W, et al. Isolation and characterization of H7N9 viruses from live poultry markets—implication of the source of current H7N9 infection in humans. Sci Bull. (2013) 58:1857–63. doi: 10.1007/s11434-013-5873-4
20. Zhang T, Bi Y, Tian H, Li X, Liu D, Wu Y, et al. Human infection with influenza virus A(H10N8) from live poultry markets, China. Emerg Infect Dis. (2014) 20:2076–9. doi: 10.3201/eid2012.140911
21. Chowell G, Simonsen L, Towers S, Miler MA, Viboud C. Transmission potential of influenza A/H7N9, February to May 2013, China. BMC Med. (2013) 11:214. doi: 10.1186/1741-7015-11-214
22. Yu H, Wu JT, Cowling BJ, Liao Q, Gang VJ, Zhou S, et al. Effect of closure of live poultry markets on poultry-to-person transmission of avian influenza A H7N9 virus: an ecological study. Lancet (2014) 383:541–8. doi: 10.1016/S0140-6736(13)61904-2
23. Genbank. National Center for Biotechnology Information. (2011). Available online at: Available online at: http://www.ncbi.nlm.nih.gov/pubmed/(Accessed on March 8, 2015).
24. Dong G, Luo J, Zhang H, Wang C, Duan M, Deliberto TJ, et al. Phylogenetic diversity and genotypical complexity of H9N2 influenza A viruses revealed by genomic sequence analysis. PLoS ONE (2011) 6:e17212. doi: 10.1371/journal.pone.0017212
25. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
26. Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Comput Biol. (2009) 5:e1000520. doi: 10.1371/journal.pcbi.1000520
27. Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. (2007) 7:214. doi: 10.1186/1471-2148-7-214
28. National Bureau of Statistics of China. China Statistical Yearbook (2010-2013). Available online at: http://www.stats.gov.cn/english/Statisticaldata/AnnualData/
29. Wright S. Statistical genetics and evolution. Bull Am Math Soc. (1942) 48:223–46. doi: 10.1090/S0002-9904-1942-07641-5
30. Hudson RR, Slatkin M, Maddison WP. Estimation of levels of gene flow from DNA sequence data. Genetics (1992) 132:583–9.
31. Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. (1967) 27:209–20.
32. Minin VN, Suchard MA. Counting labeled transitions in continuous-time Markov models of evolution. J Math Biol. (2008) 56:391–412. doi: 10.1007/s00285-007-0120-8
33. Krackarct D. QAP partialling as a test of spuriousness. Soc Netw. (1987) 9:171–86. doi: 10.1016/0378-8733(87)90012-8
34. Ke C, Lu J, Wu J, Guan D, Zou L, Song T, et al. Circulation of reassortant influenza A(H7N9) viruses in poultry and humans, Guangdong Province, China, 2013. Emerg Infect Dis. (2014) 20:2034–40. doi: 10.3201/eid2012.140765
35. Lu J, Wu J, Zeng X, Guan D, Zou L, Yi L, et al. Continuing reassortment leads to the genetic diversity of influenza virus H7N9 in Guangdong, China. J Virol. (2014) 88:8297–306. doi: 10.1128/JVI.00630-14
36. Scholle SO, Ypma RJF, Lloyd AL, Koelle K. Viral substitution rate variation can arise from the interplay between within-host and epidemiological dynamics. Am Nat. (2013) 182:494–513. doi: 10.1086/672000
37. Yuan J, Lau EH, Li K, Leung YHC, Yang Z, Xie C, et al. Effect of live poultry market closure on avian influenza A(H7N9) virus activity in Guangzhou, China, 2014. Emerg Infect Dis. (2015) 21:1784–93. doi: 10.3201/eid2110.150623
38. Fournié G, Pfeiffer DU. Can closure of live poultry markets halt the spread of H7N9? Lancet (2014) 383:496–7. doi: 10.1016/S0140-6736(13)62109-1
39. Zhou X, Wang Y, Liu H, Guo F, Doi SA, Smith C, et al. Effectiveness of market-level biosecurity at reducing exposure of poultry and humans to avian influenza: a systematic review and meta-analysis. J Infect Dis. (2018). doi: 10.1093/infdis/jiy400
Keywords: H7N9, live poultry trade, network, evolution, propagation
Citation: Li R, Zhang T, Bai Y, Li H, Wang Y, Bi Y, Chang J and Xu B (2018) Live Poultry Trading Drives China's H7N9 Viral Evolution and Geographical Network Propagation. Front. Public Health 6:210. doi: 10.3389/fpubh.2018.00210
Received: 09 May 2018; Accepted: 09 July 2018;
Published: 27 July 2018.
Edited by:
Jimmy Thomas Efird, University of Newcastle, AustraliaReviewed by:
Lin Wang, University of Hong Kong, Hong KongCopyright © 2018 Li, Zhang, Bai, Li, Wang, Bi, Chang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianyu Chang, Y2hhbmdqaWFueXVAY2F1LmVkdS5jbg==
Bing Xu, YmluZ3h1QHRzaW5naHVhLmVkdS5jbg==
†Present Address: Ruiyun Li, MRC Center for Outbreak Analysis and Modeling, Department of Infectious Disease Epidemiology, Imperial College London, London, United Kingdom
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.