 Szilvia Fiatal1,2*
Szilvia Fiatal1,2* Róza Ádány1,2,3
Róza Ádány1,2,3
- 1Department of Preventive Medicine, Faculty of Public Health, University of Debrecen, Debrecen, Hungary
- 2WHO Collaborating Centre on Vulnerability and Health, Department of Preventive Medicine, Faculty of Public Health, University of Debrecen, Debrecen, Hungary
- 3MTA-DE Public Health Research Group of the Hungarian Academy of Sciences, Faculty of Public Health, University of Debrecen, Debrecen, Hungary
Background: Although largely preventable, cardiovascular diseases (CVDs) are the biggest cause of death worldwide. Common complex cardiovascular disorders (e.g., coronary heart disease, hypertonia, or thrombophilia) result from a combination of genetic alterations and environmental factors. Recent advances in the genomics of CVDs have fostered huge expectations about future use of susceptibility variants for prevention, diagnosis, and treatment. Our aim was to summarize the latest developments in the field from a public health perspective focusing on the applicability of data on single-nucleotide polymorphisms (SNPs), through a systematic review of studies from the last decade on genetic risk estimating for common CVDs.
Methods: Several keywords were used for searching the PubMed, Embase, CINAHL, and Web of Science databases. Recent advances were summarized and structured according to the main public health domains (prevention, early detection, and treatment) using a framework suggested recently for translational research. This framework includes four recommended phases: “T1. From gene discovery to candidate health applications; T2. From health application to evidence-based practice guidelines; T3. From evidence-based practice guidelines to health practice; and T4. From practice to population health impacts.”
Results: The majority of translation research belongs to the T1 phase “translation of basic genetic/genomic research into health application”; there are only a few population-based impacts estimated. The studies suggest that an SNP is a poor estimator of individual risk, whereas an individual’s genetic profile combined with non-genetic risk factors may better predict CVD risk among certain patient subgroups. Further research is needed to validate whether these genomic profiles can prospectively identify individuals at risk to develop CVDs. Several research gaps were identified: little information is available on studies suggesting “Health application to evidence-based practice guidelines”; no study is available on “Guidelines to health practice.” It was not possible to identify studies that incorporate environmental or lifestyle factors in the risk estimation.
Conclusion: Currently, identifying populations having a larger risk of developing common CVDs may result in personalized prevention programs by reducing people’s risk of onset or disease progression. However, limited evidence is available on the application of genomic results in health and public health practice.
Introduction
Despite the fact that cardiovascular diseases (CVDs) are largely preventable they are the biggest cause of death worldwide, responsible for almost one-third of all global deaths (1). In 2012, CVDs were responsible for 46% of deaths caused by non-communicable diseases. Of these deaths, an estimated 7.4 million were due to ischemic heart disease and 6.7 million were due to stroke (2). This review focuses on common complex cardiovascular disorders with high public health importance caused by a combination of several genetic and environmental factors.
The role of social determinants (e.g., aging, employment, income, and education), behavior (e.g., unhealthy diet, tobacco use, physical inactivity, and harmful use of alcohol), and metabolic (e.g., obesity, diabetes, raised blood TG, and LDL-cholesterol level, low blood HDL-C level) risk factors have largely been well known for decades due to the large-scale longitudinal studies (e.g., Framingham study and Seven Countries study) (3–8). Nevertheless, the contribution of inherited (genetic) disposition is still the focus of intense research interest (2). Our knowledge of these non-genetic risk factors has been useful in disease prevention efforts, but hopefully, we may discover more effective ways of preventing and controlling CVDs if we understand the genetics underlying these diseases.
The large majority of the common CVDs are developed as a result of harmful interaction between heritable and environmental factors. Approaches to identifying the genetic causes of polygenic common CVDs (and also other polygenic diseases) became more prominent after completion of the Human Genome Project. Several genetic loci associated with cardiovascular traits have been identified by candidate gene and genome-wide association studies testing a set of genetic variants, mainly in case–control studies in populations of different ancestry and ethnicity. Evidence for the strong contribution of genetic factors in the development of common CVDs has consistently been reported in twin and family-based linkage studies. Heritability estimates from large twin studies suggest that genetic variations may account for about 30–50% of hypertension risk and about 50–60% of coronary artery disease or myocardial infarction risk (9–12). For these complex cardiovascular disorders, the main ambition of public health initiatives is to be able to prevent or predict diseases by identification of the subject at high risk (13). In addition, several variants for monogenic subtypes of, e.g., hypertension, congenital heart disease, or familial hypercholesterinaemia have been identified (14–16). For monogenic disorders, the major public health priorities are genetic screening and its effective use in health-care practice to arrange the best treatment and provide the best care for family members at high risk (13).
Rapid advances in the genomics of CVDs have fostered huge expectations about the future use of detecting susceptibility variants in prevention, diagnosis, and treatment. Although large-scale association studies promote the estimation and categorization of the predictive values related to genetic variants, and the large number of genetic loci associated with CVDs and cardiovascular risk factors have provided insights into the biologic pathways that underlie the cases of disease, the application of such findings to cardiovascular risk prediction, prevention, and treatment still needs to be elucidated (17). So far only a small number of findings in human genome research have resulted in evidence-based applications in the field of medicine and public health.
Genetic screenings, as Becker et al. indicate, aim at populations of asymptomatic individuals, or at subpopulations in which the risk is known to be increased, or in which the specific phase of life merits screening (pregnant women, newborns) (18). Screening for common complex CVDs would give us opportunities for preventive strategies related to lifestyle, medication, or intervention (18). In the last 10 years, there has been increased enquiry into the potential clinical and public health applications of genetic screening/genetic testing of CVD risk.
In this review, we will discuss the application of single-nucleotide polymorphisms (SNPs) related to risk estimates in identification of increased genetic susceptibility to common CVDs from a public health perspective and summarize the recent advances in translational research from the past decade using the comprehensive framework suggested by Khoury and his colleagues (19). This framework comprises four phases of evidence in translation research [“T1. From gene discovery to candidate health applications; T2. From health application to evidence-based practice guidelines; T3. From evidence-based practice guidelines to health practice; and T4. From practice to population health impact” (19)] and possible future applications of genetic screening/testing of CVDs in connection with these phases can be identified.
Methods
To identify relevant studies in the field of CVD genetics/genomics a structured literature search using compound terms (Table 1a–d) was performed using online database services of PubMed, Embase, CINAHL, and Web of Science. The timeframe of the search related to this review was from May 5, 2007 until May 5, 2017. The systematic search and selection process were conducted as proposed in the published PRISMA guideline (20) resulting in the final list of relevant publications (see adapted flowchart, Figure 1 in Section “Results”).
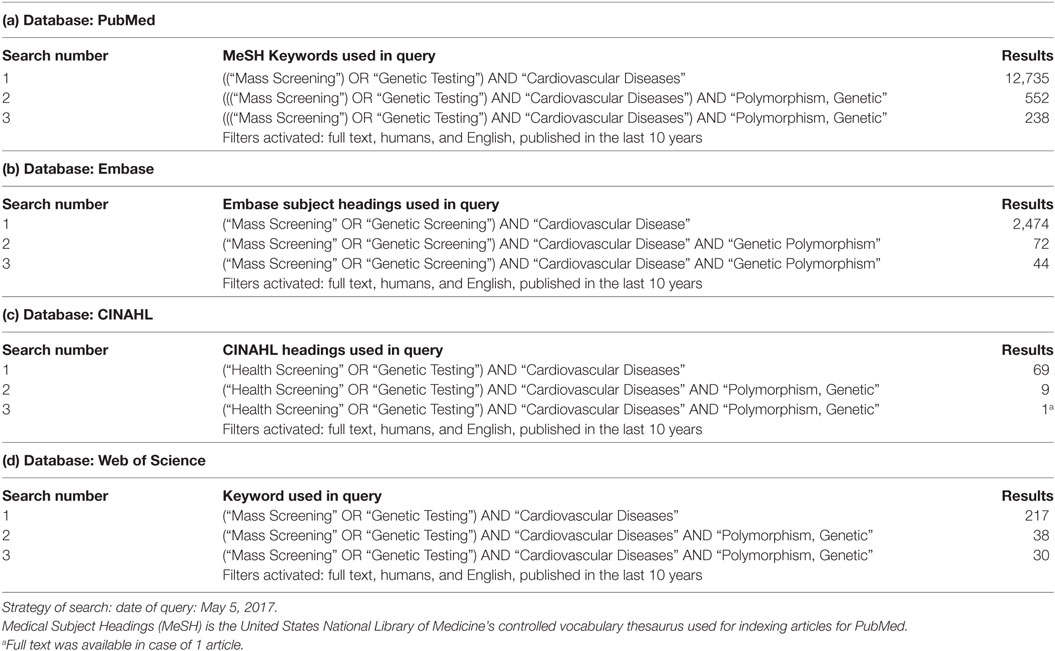
Table 1. Details on the systematic search.
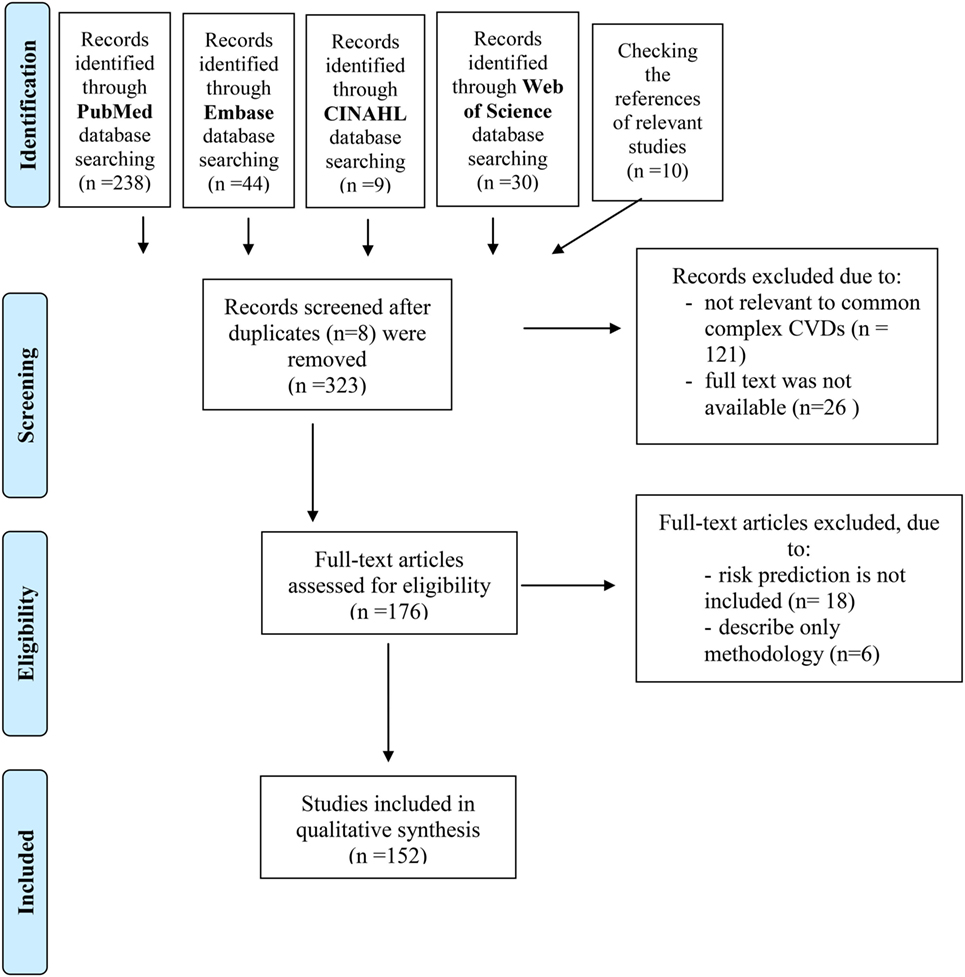
Figure 1. Flowchart shows study selection procedure. Adapted from Ref. (20).
Results
Studies Included in the Review by Implementing PRISMA Statement
The first step was the identification of the records. A primary search using PubMed, Embase, CINAHL, and Web of Science was performed to identify related publications using compound terms (Table 1a–d). Research communications published earlier than 2007 and not published in English were excluded. Only those studies that were conducted on human subjects and were available in full-text format were considered. This identification step resulted in 323 records. Moreover, by checking the references of the relevant studies an additional 10 studies were identified and included. The details of the search and the number of records identified in queries can be seen in Figure 1.
Next, the abstracts of the records that fit the abovementioned criteria were tested (screening of the records) for relevance to the topic, i.e., only those records were included that focused on common CVDs. Twenty-six studies were discarded because the full text of the research was not available, and 121 studies were excluded after reviewing the abstracts because it appeared that these papers clearly were not relevant to common complex CVDs.
Finally, eligibility and inclusion of the records were investigated: the full text of the remaining 176 publications was examined in more detail. A total of 152 studies were identified for inclusion in this review, after 24 studies were excluded because risk prediction was not included in those articles or only the methodology was described.
Translational Genomic Research in CVDs
During the last decade, voluminous research aimed at incorporating cardiovascular genetic/genomic discoveries into practice has been undertaken. Altogether, 152 studies were found to be relevant to this field. Khoury et al. suggested a framework for translational research that is required before applying genomic findings in clinical or public health practice. This framework includes the following four phases:
T1. From discovery to candidate health applications,
T2. From health application to evidence-based practice guidelines,
T3. From practice guidelines to health practice, and
T4. From practice to population health impact (19).
The major developments on CVDs’ genomics for each phase of the framework are summarized in Table 2. Consequently, this table also displays how close/far cardiovascular genetic applications are to/from clinical or public health practice.
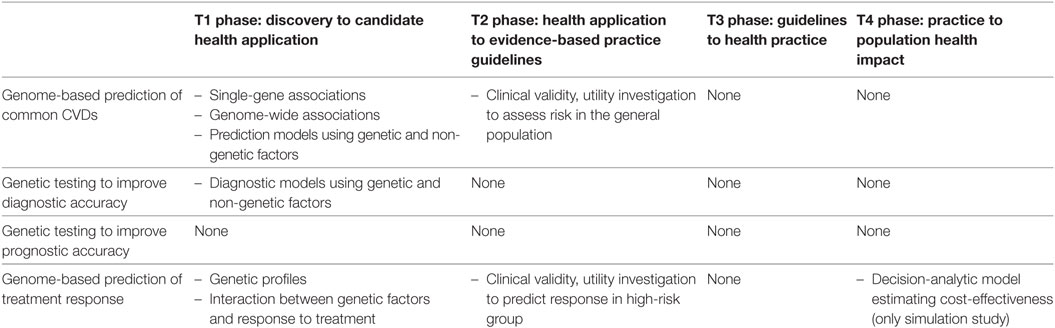
Table 2. Advances in genomic research on common cardiovascular diseases (CVDs) according to the translation research framework.
From Gene Discovery to Candidate Health Applications (T1)
Khoury et al. describe T1 research as follows: it begins after gene discovery, and its goal is the development of candidate health applications to be used in clinical and public health practice. Ideally, the outcome of this phase is the development of a single-gene test or construction of a genome profile that has high sensitivity, specificity, and predictive value. These applications can be used to foster clinical evaluation (predictive testing, screening, diagnostic, and prognostic testing) or selection of the most effective therapeutic option, usually by observational studies and clinical trials (phases I and II) (19).
Genome-Based Prediction of CVDs
Genetic Association Studies on SNPs. During the last 10 years, numerous association studies were performed in the field of cardiovascular genetics with a high representation of case–control design among them. These studies utilized a single candidate-gene approach almost without exception to estimate the potential cardiovascular risk. Few of them were genome-wide association studies examining SNPs across the genome, and only one linkage study and one segregation study were found (21–23).
Genetic alterations were associated with ischemic stroke (24–41), arrhythmias (42–51), coronary heart/artery disease (52–62), myocardial infarction (63–66), and carotid sclerosis (67–70) were intensely investigated, but less attention was paid to the following traits/phenotypes as an outcome: atrial fibrillation (71), abdominal aorta aneurysm (72), carotid intima-media thickness (73, 74), carotid plaque thickness (75), cardiovascular mortality/diseases in general (76–80), dilated cardiomyopathy (81, 82), venous thrombosis (83–85), familial hypercholesterinaemia (86), hypertrophic cardiomyopathy (87–89), plasma lipoproteins (90), hypertension (91–93), intracranial aneurysm (94, 95), intracerebral hemorrhage (96–98), ischemic heart disease (99), recanalization after ischemic stroke (100), subarachnoid hemorrhage (101–103), vasodilator reactivity (104), and lower extremity artery disease (105). During recent years, a number of review articles have appeared dealing with recent advances in genetics research (mostly reviews of association studies) of arrhythmia (106–108), coronary artery/heart disease (78, 109–114), sudden cardiac death (115, 116), sporadic heart failure (117, 118), cardiomyopathy (119), and thrombophilia (120, 121), that also discuss the recent and potential developments in the field (122–127).
Genetic association studies investigate a correlation between disease status and a genetic alteration(s) (e.g., SNPs, VNTRs, and CNVs) to identify risk or protective alleles that play a part in the development of a specific disease. An increased frequency of a risk allele or genotype in the individuals affected with a disease can result in the conclusion that the variant of interest increases the risk of a specific disease (128). According to our results, association studies still represent an important tool in identifying genes contributing susceptibility to several complex CVDs. Association studies (and meta-analyses) have reconfirmed that many different genetic variants affect disease risk, but each variant has only a relatively small effect. Single markers identified are unlikely to be considered for clinical use unless they yield a high effect size (characterized by odds ratio/beta coefficient).
Meta-analyses that combine the results of single-gene association studies provide an opportunity to obtain more robust effect sizes. In the last decade, meta-analyses were related to atrial fibrillation, MRI-defined brain infarct, ischemic stroke, and susceptibility to any type of atherosclerotic CVD, such as coronary artery disease, acute coronary syndrome, or ischemic heart diseases (129–135). It is worth mentioning that meta-analyses may be biased: publication bias, population stratification, control selection bias, and lack of genotype blinding exist, thus results should be interpreted with caution.
Genome-Wide Association Studies. Genome-wide association studies use high-throughput genotyping technologies to assay thousands of SNPs and correlate them to clinical conditions or measurable traits. GWA studies are very useful in discovering genetic variants related to different diseases but also have important limitations (summarized by Pearson and Manolio), “including false-positive and false-negative results and biases related to selection of study participants, and genotyping errors.” But most of the variants identified by GWA studies still have very modest effects on disease risk and explain only a small fraction of population risk or total estimated heritability (136).
It is important to point out that a variant with even small odds ratios can improve the indicative power of the predictive models, such as the 9p21 locus (114, 137–141). Despite several studies that show consistent associations of 9p21 locus with CVD traits, the biological role of the locus is still not well understood. In a study by Visel et al. (142), the results provide direct evidence that the coronary artery disease risk interval has a crucial role in regulation of cardiac Cdkn2a/b expression (a mouse ortholog of the 9p21 locus) and suggest that this region has an effect on the progression of coronary artery disease by modifying the dynamics of vascular cell proliferation. If it is confirmed this would represent a new mechanism for myocardial infarction that is unrelated to traditional risk factors (123).
Combining Candidate-Gene SNPs with or without Traditional Clinical Risk Factors—The Genetic Risk Score (GRS) Approach. Multiple markers with small effect sizes may be used in combination to generate high effect size. The simultaneous use of the most common and strongest risk markers (with or without other non-genetic traditional risk factors) may have the desired discriminatory accuracy (quantified by the C-statistics) to distinguish between diseased and healthy subjects. Information obtained from SNPs can be combined by first assigning a risk value of, e.g., 0 for a subject that is a non-carrier of risk allele, 1 if a “carrier,” or 2 if homozygous for that allele, and then calculating the overall score (GRS) for each individual in the study population (143).
Using this gene scoring approach, Aulchenko et al. (144) included a total of 17,797–22,562 persons, aged 18–104 years from the Nordic countries to Southern Europe. They investigated 22 loci known to be associated with serum lipid levels (total cholesterol, low-density lipoprotein, cholesterol, high-density lipoprotein cholesterol, and triglycerides). GRSs based on lipid loci explained 4.8, 3.4, and 3.0% of age-adjusted variances in HDL-C, LDL-C, and TG, respectively and were also associated with increased intima-media thickness (p = 0.001) and coronary heart disease incidence (p = 0.04). They tested for the association between the genetic risk profiles and intima-media thickness, and incident coronary heart disease. From the risk profiles, the total-cholesterol profile and the combined profile—including all associated SNPs of the four traits—were most strongly associated with the clinical outcomes. They concluded that the genetic profiles developed improve the identification of subjects at high risk of dyslipidemia but do not improve the prediction of atherosclerosis and CHD compared to classical risk factors.
In the study of Krarup et al. (145), the GRS of 45 risk variants was involved to estimate the effect on incidence and clinical predictability of myocardial infarction and coronary artery disease in 6,041 Danish individuals. Analyses using two different models (model 1: adjusted for age and sex; model 2: adjusted for age, sex, BMI, smoking status, and type 2 diabetes mellitus) detected allele-dependent association of GRS with myocardial infarction [hazard ratio (HR) (95% CI): model 1: 1.05 (1.01–1.10), p = 0.02 and model 2: 1.06 (1.02–1.11), p = 0.01]. No association with coronary artery disease was shown for either GRS model. They aimed to estimate the predictive capacity of GRS for only myocardial infarction, but no significant effect was identified on discriminative or reclassification ability by adding GRS to the European SCORE algorithm (age, sex, smoking status, systolic blood pressure, and total cholesterol).
In the recent study of Isaacs et al. (146), the cumulative effects of common genetic variants related to TC, LDL-C, HDL-C, and TG were associated with carotid plaque formation. As Isaacs et al. concluded, the relationship was the strongest for the LDL-C score, which increased plaque score by 0.102 per SD increase in GRS (p = 3.2 × 10−8). TC and LDL-C scores were significantly associated with incident myocardial infarction and coronary heart disease with HRs between 1.10 and 1.13 per SD increase in score. The Framingham risk score (FRS) discriminated myocardial infarction better than the GRSs (area under receiver-operating characteristic curve—AUC 0.65 vs. 0.62); after combining FRS and GRSs, the results improved slightly compared with the FRS AUC alone (AUC 0.66; p = 0.069). In cases of coronary heart disease, the results were similar. In conclusion, GRSs did not improve clinical AUCs.
Tikkanen et al. (147) genotyped 28 genetic variants in a Finnish cohort of 24,124 participants. A multilocus GRS was constructed, and its association with incident CVD events was evaluated. They reported that by adding genetic information to conventional risk factors the risk discrimination of coronary heart disease (C-index 0.856 vs. 0.851, p = 0.0002) and other end points (CVD: C-index 0.840 vs. 0.837, p = 0.0004; acute coronary sclerosis: C-index 0.859 vs. 0.855, p = 0.001) were improved. According to their model in a population of 100,000 individuals, additional genetic screening of subjects at intermediate risk for coronary heart disease would reclassify additional 2,144 subjects (12%) into the high-risk category.
According to the study by Weijmans et al. (148) in a group of patients (5,742 individuals) with symptomatic vascular disease, the GRS did not improve prediction of 10-year risk of cardiovascular events beyond clinical characteristics. The net reclassification index improved only in case of patients suffering from stable atherosclerosis (0.14, 95% CI: 0.03–0.25).
Ganna et al. (149) used data from 6 Swedish prospective cohort studies with 10,612 healthy participants. They investigated the clinical utility of GRS in primary prevention of CVDs. Several risk scores were developed: the overall GRS based on 395 SNPs was reported as being associated with cardiovascular traits: one coronary heart disease-specific GRS, including 46 SNPs, and 6 trait-specific GRS for each established CHD risk factor (body mass index, HDL-C, systolic blood pressure, TC, and smoking, type 2 diabetes mellitus). The overall and the coronary heart disease-specific GRS were significantly associated with CHD risk (HRs for fourth vs. first quartile, 1.54 and 1.52; p < 0.001) and improved risk classification beyond established risk factors (net reclassification improvement, 4.2 and 4.9%; p = 0.006 and 0.017). Discrimination improvement was modest (C-index improvement, 0.004).
Genetic Testing to Improve Diagnostic Accuracy
Besides the several studies mentioned earlier on coronary heart disease, a study on risk models that predict a person’s risk for developing venous thrombosis was published by de Haan et al. (150). GRS based on 31 venous thrombosis-associated SNPs was developed for subjects of a large case–control study (2,712 patients and 4,634 controls). GRS computed from all the 31 SNPs or from the 5 most strongly associated SNPs performed very similarly (AUCs of 0.70 and 0.69, respectively). The AUC of a risk model based on known non-genetic risk factors was 0.77 (95% CI: 0.76–0.78). After combining the non-genetic and genetic risk models, the AUC improved to 0.82 (95% CI: 0.81–0.83), which indicates better diagnostic accuracy.
Genome-Based Prediction of Treatment Response
In addition to genetic testing that can improve the treatment by increasing drug efficacy and safety, a genetic test can be used to select patients for therapies that target-specific genes/gene products (151). An area where genome-based prediction of treatment response is important is the use of genetic testing for evaluating the antiplatelet effects of the antiplatelet drug clopidogrel. Several recent studies suggest that therapeutic responses to clopidogrel might depend on the genotype at the CYP2C19 gene; however, some findings are contradictory (152–156). It was shown that clopidogrel-treated patients who had an allele of CYP2C19 with reduced function [most commonly the CYP2C19*2 or CYP2C19*3 allele, roughly 30% of patients have loss-of-function (LOF) alleles] had less platelet inhibition, and consequently, a significantly higher risk of cardiovascular events than those who had a normally functioning CYP2C19 enzyme (157). Furthermore, it was shown that the CYP2C19*17 variant is the gain-of-function (GOF) allele (prevalence is between 3 and 21%) and is an independent factor in increased bleeding risk (157). Recently, Shen et al. (158) demonstrated the explicit clinical benefit of CYP2C19 genetic testing for guiding the antiplatelet therapy on a sample of 628 patients; clinical outcomes were analyzed at 1, 6, and 12 months after discharge. Individual antiplatelet therapy guided by CYP2C19 genetic testing significantly improved the prognosis of patients after percutaneous coronary intervention. The morbidity rates of “major adverse cardiovascular events” in the intervention group were decreased by 4.3, 4.6, and 5.2% compared with the routine group (conventionally treated with 75 mg daily of clopidogrel without CYP2C19 genetic testing) at 1, 6, and 12 months, respectively.
According to a review by Chan et al. (159), there is a good evidence of analytical validity for testing LOF polymorphisms in managing clopidogrel therapy. They highlighted that LOF polymorphisms are associated with reduced levels of the active clopidogrel metabolite and with reduced on-treatment inhibition of ADP-induced platelet activation. In percutaneous coronary intervention populations, there is consistent evidence for an association between LOF polymorphisms and adverse clinical outcomes (stent thrombosis and major adverse cardiovascular events). Evidence for clinical utility of CYP2C19 genotyping as a predictive biomarker is limited to subgroups with indecisive findings.
In a single-center study of 535 ischemic stroke patients who received clopidogrel, Yi et al. (160) found that for patients carrying the reduced function LOF polymorphisms the inhibition of platelet aggregation was significantly lower in patients treated with proton-pump inhibitors.
From Health Application to Evidence-Based Practice Guidelines (T2)
The second phase (T2) begins if there is convincing evidence on genetic test performance. In this phase, the so-called ACCE components (analytic and clinical validity, clinical utility and ethical, legal, and social issues) are investigated in the population settings for which the tests are intended. These evaluations depend on multidisciplinary research in the field of clinical medicine, laboratory sciences, economics, public health, ethics, behavioral, and social sciences. Results from this phase should result in evidence-based guidelines for both clinical and public health practice (19).
According to the Evaluation of Genomic Application in Practice and Prevention Working Group (EWG), testing for the 9p21 genetic variant or 57 other variants in 28 genes is not recommended to assess risk for CVD in the general population, specifically heart disease and stroke (161). The EWG highlighted that even if the 9p21 variants with heart disease had convincing evidence of per allele–odds ratio of between 1.2 and 1.3 (the highest among all variants they investigated) the magnitude of net health gain from use of any of these test (alone or in combination) is irrelevant. According to the guideline of the U.S. Preventive Services Task Force, genetic/genomic markers are not included among those non-traditional risk factors suggested in coronary heart disease risk assessment (carotid intima-media thickness and high sensitivity C-reactive protein) (162).
To date, several research studies have focused on the CYP2C19 gene (see relevant studies above) because its variants can reduce the formation of the active metabolite of clopidogrel and influence clopidogrel’s antiplatelet effects. While many studies showed that clopidogrel’s efficacy depends on CYP2C19 genetic polymorphisms, others did not find any association (163, 164). In fact, CYP2C19 LOF alleles account for only 12% of the variability in response to clopidogrel. This implies that most of the variabilities are caused by other factors not yet developed. To date, guidelines form the American College of Cardiology Foundation/American Heart Association recommended against routine genetic testing in patients with acute coronary syndrome (165). In addition, a IIb recommendation (evidence C) has been given to CYP2C19 genotyping by stating that: efficacy is less well established; only diverging expert opinion and case studies are available.
The MTHFR enzyme catalyzes the transition of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, the primary circulatory form of folate, and a cosubstrate for homocysteine remethylation to methionine. MTHFR polymorphism testing (for variant of c.665C → T and c.1286A → C) is frequently suggested by general practitioners as part of the clinical investigation for thrombophilia. The potential associations between MTHFR genotype status and several complications have been evaluated by case–control, cohort, Mendelian randomization, and meta-analysis because formerly it was suggested that reduced enzyme activity of MTHFR led to hyperhomocysteinemia, which amount to an increased risk for venous thromboembolism, coronary heart disease, and recurrent pregnancy loss (166). But a later meta-analysis has found that the association was not as strong as previously believed. Long and Goldblatt highlighted that “homozygosity for the 677C>T polymorphism is linked to a small increase in homocysteine levels; the increased risk of ischemic heart disease and stroke is more closely related to the serum levels of homocysteine rather than the presence of the MTHFR polymorphisms. Furthermore, there seems to be no increased risk of mortality from CVD to MTHFR 677C>T homozygotes (167).” Considering the fact that MTHFR polymorphism is only one out of many other factors contributing to the clinical picture, the utility of this testing is presently doubtful. There is growing evidence that MTHFR polymorphism testing has minimal clinical utility and therefore should not be prescribed as part of a routine evaluation for thrombophilia according to the American Congress of Obstetricians and Gynecologists, the American College of Medical Genetics and Genomics, and the British Society for Haematology (166–169).
Although the ethical, legal, and social issues related to genetic screening/testing of CVDs are rather important components of T2 research, they were not the focus of our interest.
From Evidence-Based Guidelines to Health Practice (T3)
The third phase addresses the spread and integration of knowledge yielded through the T2 phase research. The translation and dissemination of evidence-based guidelines into practice is challenging (19), and only one genomic application is ready for implementation in routine daily practice. The U.S. Preventive Services Task Force suggest BRCA mutation testing for predicting breast and ovarian cancers for women who have blood relatives with breast, ovarian, tubal, or peritoneal cancer (170), but no recommendations (published or in progress) are available regarding genetic testing of CVDs.
From Practice to Population Health Impact (T4)
This phase assesses how the adopted recommendations and guidelines make an impact. It focuses on clinical and public health outcomes of the guidelines obtained and includes measuring the incidence of the disease, quality of life indicators, clinical decision modeling, and cost-effectiveness analysis (19).
So far, only one study by Jiang and You (171) examined the clinical and economic outcomes of CYP2C19 LOF- and GOF-guided antiplatelet therapy in subjects with acute coronary syndrome undergoing percutaneous coronary intervention. They designed a lifelong decision-analytic model in a hypothetical cohort of 60-year-old patients to simulate outcomes of three strategies: clopidogrel, alternative P2Y12 inhibitors (prasugrel/ticagrelor), and LOF/GOF-guided therapy (LOF/GOF allele carriers received an alternative P2Y12 inhibitor and wild-type patients received clopidogrel). Direct costs, clinical event rates (including major cardiovascular events, stent thrombosis, and major bleeding), and quality-adjusted life-years gained were the model’s outcomes. They found that non-fatal myocardial infarction (5.62%) and stent thrombosis (1.2%) had the lowest rate in the alternative P2Y12 inhibitor arm, whereas non-fatal stroke (0.72%), cardiovascular death (2.42%), and major bleeding (2.73%) were the lowest in the LOF/GOF-guided group. The LOF/GOF-guided arm had the highest QALYs (7.5301 QALYs) at the lowest lifelong cost (USD 76,450). These finding suggest that personalized antiplatelet therapy driven by CYP2C19 LOF and GOF alleles appears to be the preferred antiplatelet strategy when compared to clopidogrel and alternative P2Y12 inhibitor therapy.
Quality Assessment of Studies Included
The full text of the articles (genome-wide association studies, genetic risk prediction studies, and meta-analyses) was screened, and data on recently published key reporting components (according to STREGA, GRIPS, and PRISMA) and methodological components (according to AMSTAR) of the studies were extracted (20, 172–174). The STREGA, GRIPS, PRISMA, and AMSTAR checklists have 22, 25, 27, and 11 items, respectively, that should be reported in the research articles. If each item is achieved, the maximum scores are identical to the item numbers. The mean STREGA score of all collected GWAs was 20.8 ± 1.64 (on average, 94.54% of the items properly reported). The per item STREGA analysis revealed that one item (item 19: discussing limitations of the study) was the least adhered to (40%) of the five GWAs included. The mean GRIPS score of all genetic risk prediction studies included was 21.43 ± 2.51 (85.72% of items were reported satisfactorily). The per item GRIPS analyses showed that one item had less than 57% adherence (item 14: report the number of individuals at each stage of the study) out of the seven risk prediction studies involved. Two items (item 10: specify the procedure and data used for validation of the risk model, and item 19: report any validation of the risk model) had 28.6% adherence. The overall mean PRISMA score of meta-analysis was 22 ± 4 (82.96% of the items adequately reported). The per item PRISMA analysis showed that one item (item 5: indicate if a review protocol exists and where it can be accessed) had 0% adherence out of the five meta-analyses included. Item 25 (discuss limitations at study and outcome level) has only 40% adherence. The mean AMSTAR score of meta-analyses was 6.2 ± 1.48 (56.36% of the items adequately reported). The per item AMSTAR analysis showed that three items (item 1: Was an a priori design provided? Item 4: Was the status of the publication—i.e., gray literature—used as an inclusion criterion? Item 5: Was the list of studies, included and excluded, provided?) had 0% adherence out of the five meta-analyses included. Based on our analyses the reporting qualities of recently published studies were good in general, although according to the per item analyses there is a need for improvement in the case of some items. Two meta-analyses (129, 133) were excluded from PRISMA and AMSTAR analyses owing to the fact that both were meta-analyses of genome-wide association studies developed by consortia and data were not obtained via systematic literature search.
Discussion
We have collected and reviewed the published literature according to an overarching framework for translational research recommended by Khoury et al. (19). The studies reviewed in this work represent the majority of current data available on genetic testing/screening for CVD risk. The relevant research papers were retrieved using a PubMed search and included original scientific papers, reviews, meta-analyses, and editorials. We have summarized the major findings of the research identified in each translation phase.
The overview of most significant outcomes in genetic research in common CVDs according to the framework for translational research is summarized in Table 3. The vast majority of the studies published relate to T1 research (24–127, 137–160), many fewer focus on T2 research (161–169); however, T3 research is missing, and only one simulation study (171) was identified as a part of the T4 research (Tables 2 and 3). These finding suggest that during the last 10 years very few cardiovascular genetic discoveries have led to evidence-based applications for medical or public health practice. Genetic prediction of the complex CVDs consists of multiple genes added to traditional risk factors (145, 146, 149). Several recent studies suggest that polymorphisms, mainly in candidate genes, may help to distinguish among several clusters/subgroups of patients. Several studies have identified certain risk profiles based on clusters of genes related to coronary heart disease or deep vein thrombosis but with low predictive values (147, 150). Further research is still needed to validate whether these genomic profiles can prospectively identify individuals at risk to develop CVDs.
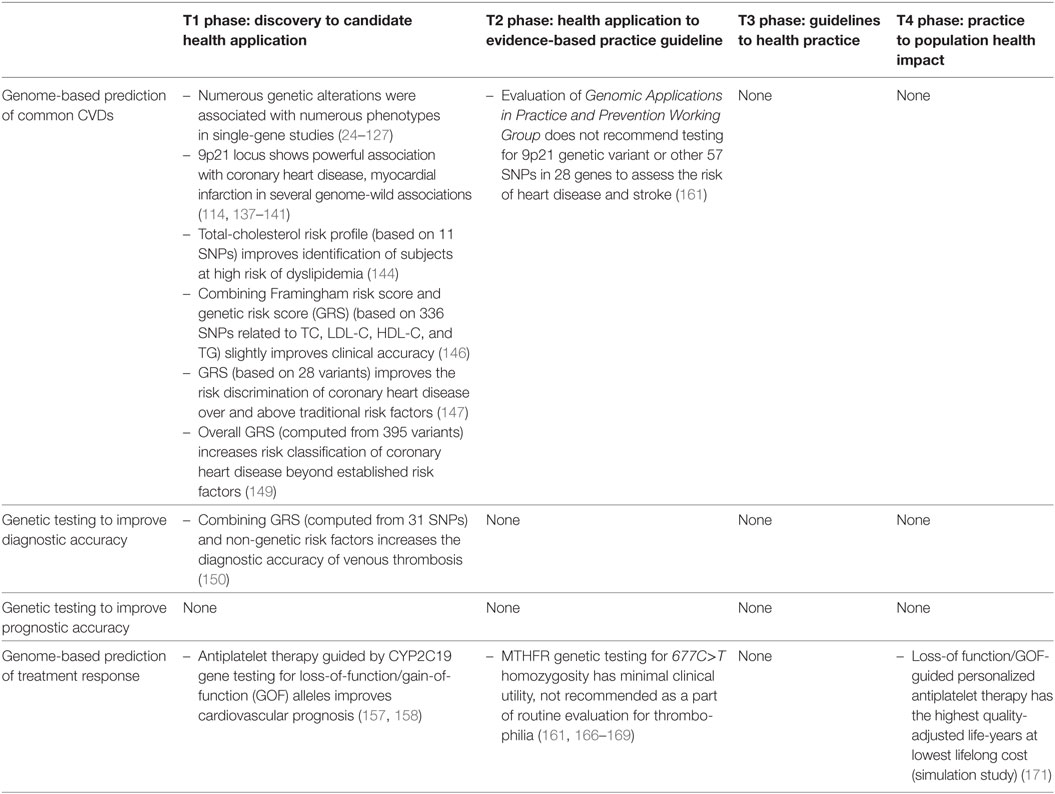
Table 3. Overview of most significant outcomes in genetic/genomic research in common cardiovascular diseases (CVDs) according to the framework for translational research.
The most important limitation of current cardiovascular T1 research is that only single-gene variants or several SNPs contributing to a small proportion of the genomic variation are investigated, but there are already prediction models available involving more complex system biology in large-scale and well-designed studies (144–150). We have also identified various research gaps including the following: little information is available on studies suggesting “Health application to evidence-based practice guidelines”; no study is available on “Guidelines to health practice.” Furthermore, it was not possible to identify studies that incorporated environmental or lifestyle factors into the risk estimation.
Single-gene association reports from the last decade clearly point out that the SNPs associated with increased or decreased cardiovascular risk have little impact in risk estimation. In a study reviewed, the results of more than 600 studies and 3,000 SNPs related to CVDs, the largest OR for the association between an SNP and cardiovascular trait was 1.6, and almost all OR were between 0.8 and 1.2 (175). These so-called “common variants with small effects” can explain only modest amount of heritability even if hundreds of genetic factors are used for risk prediction (176, 177). Beside these variants numerous “rare alleles” (population frequency is less than 0.5%) expected in the human genome. Rare variants have remarkable effect size and consequently might underlie the missing heritability of complex CVDs (178). To capture rare variants the GWAs approach or linkage study design are not powerful (178, 179); instead of genotyping a list of variants it is inevitable to sequence entire genome. The previous sequencing technology (known as “automated Sanger sequencing”) was expensive and time-consuming, but over the past decade, new high-throughput technologies, referred to as next-generation sequencing (NGS), were evolved. NGS technologies are cost-effective, able to explore the human genome in reasonable time and are suitable to discover full spectrum of sequence variations (180). Several recent studies suggest that the application of NGS technology in defining and characterizing inheritable components of CVDs is getting important (181–183). This strategy was successfully applied to find new genetic variants for Mendelian CVDs such as hypertension (KCNJ5, KLHL3 gene), dilated cardiomyopathy (BAG gene), or familial combined hypolipidemia (ANGPTL3 gene) (184–187). However, utilizing NGS technologies to discover novel variants contributing common CVDs is very challenging mainly because of stringent statistical requirements. Currently, this approach represents a field has not been widely explored (188, 189). A viewpoint paper from the European Society of Cardiology working group on myocardial and pericardial diseases and the members of the European Society of Human Genetics summarize their current opinion on the next-generation DNA sequencing. In routine care of patients whole exome/genome sequencing can be used as a diagnostic tool but only in case of recognized Mendelian disease genes (e.g., inherited cardiomyopathies, channelopathies, and familial dyslipidemias) (190–192). The collaboration between cardiologist, geneticist, molecular biologist, and bioinformatician are necessary in interpretation of sequencing results (190). All the challenges, advantages, and disadvantages of the NGS approach are beyond the scope of our wok.
Common CVDs are complex disorders in which gene–gene and gene–environment interactions play an important role. To date, the diagnosis of CVDs is mainly based on clinical signs and symptoms; however, the expectations surrounding genomic discoveries that improve presymptomatic testing, diagnosis, and treatment are huge. Genetic screening, a possible tool for disease prevention, is defined by the use of a set of diagnostic tests on a population to identify people who are carriers of specific genetic disorders, and who are consequently at higher risk of developing a certain disease. In the case of some common cardiovascular disorders, alteration in a single gene strongly affects the risk of development (these are referred to a “monogenic diseases,” e.g., familial hypercholesterinaemia, Mendelian forms of low and high blood pressure); however, in the case of the majority of common disorders, several genes, environmental factors, and also interactions between genes and between genes and environmental factors (considered “complex diseases,” e.g., coronary heart disease and venous thromboembolism) are required (188).
Smoking, harmful alcohol consumption, physical inactivity, and unhealthy eating habits are the most significant preventable behavioral risk factors of CVDs. As illustrated by several studies, gene–gene and gene–environment interactions contribute to the initiation and maintenance of these risk behaviors, which in turn increases risk for CVDs (193–196). Investigations of genetic and environmental attributes associated with ethnicity are an essential component of multidisciplinary research into the prevention of diseases, including those that differ in prevalence among ethnic groups such as CVDs (197, 198). The Roma population, which constitutes the largest ethnic minority in Europe, is the main subject of ethnicity-based studies because available data strongly suggest that Roma populations suffer from poorer health and lower life expectancy (199). Recently, our research group investigated whether genetic susceptibility contributes to the higher prevalence of smoking, harmful alcohol drinking habits and reduced HDL-C level in the Roma population compared to the general Hungarian population (200–202). Estimating the extent of genetic susceptibility might be important for designing and implementing targeted public health intervention programs among Roma. According to our results, harmful health behaviors (smoking and alcohol consumption) among Romani people have environmental/cultural underpinnings rather than inheritable attributes thus interventions aimed at smoking and alcohol consumption should preferentially target the cultural/environmental factors. However, in the case of reduced HDL-C levels, the contribution of genetic susceptibility was confirmed hence interventions aimed at this risk factor need to consider the increased genetic susceptibility of Roma.
Genetic testing offered for single-gene disorders known to be associated with CVDs (e.g., familial hypercholesterinaemia or Tangier disease) (18, 192). Presently, genetic screening is recommended for high-risk groups only in special cases (for example, cascade testing from known cases of Lynch syndrome and familial hypercholesterinaemia or testing for women at high risk of breast cancer because of their family history). In contrast with single-gene disorders, screening is limited for estimating susceptibility to multifactorial CVDs (203).
Caution is required before spreading out the use of such genetic screening tests to population level, because the positive predictive value of any variants found is low, and it would be hard to interpret the findings (13). Furthermore, several fundamental questions raised by Thanassoulis and Vasan concerning the genetic background of common CVDs still have not been resolved: “Can genetic markers really improve CVD risk prediction? How many SNPs are responsible for the genetic component of CVD? How many genetic markers will we need to discover to reliably improve risk prediction? What are the implications of the allelic architecture of CVD for risk prediction? What necessary steps are needed before bringing this information to patients?” (123).
Although genetic screening/testing for CVDs would ideally offer proper options concerning prevention, the discriminatory power of genetic screening to identify those who should or should not be the target of specific lifestyle advice and/or specific medication is still controversial, especially for variants having low individual relative risk and low predictive values (18).
In conclusion, we found that only a small proportion of the genetic/genomic research has advanced from discovery phase to an evidence-based health application. But recent findings and especially GWA studies and prediction studies offer a more advanced level of primary/secondary prevention interventions for those subjects who are at greater genetic risk, hopefully in the near future. Presumably, developments in public health practice will also inevitably facilitate effective implementation of genomic science.
Author Contributions
SF performed the literature search, interpreted the results, and wrote the manuscript. RÁ guided writing of the manuscript and was involved in finalizing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the GINOP-2.3.2-15-2016-00005 project. The project is cofinanced by the European Union under the European Social Fund and European Regional Development Fund, as well as by the Hungarian Academy of Sciences (TK2016-78). The work was funded by the PRECeDI project (Personalized PREvention of Chronic Diseases) funded by the European Commission in the H2020 Marie Skłodowska-Curie Research and Innovation Staff Exchange program (grant agreement number: 645740).
References
3. Dawber TR, Kannel WB, Gordon T. Coffee and cardiovascular disease. Observations from the Framingham study. N Engl J Med (1974) 291:871–4. doi:10.1056/NEJM197410242911703
4. Jacobs DR Jr, Adachi H, Mulder I, Kromhout D, Menotti A, Nissinen A, et al. Cigarette smoking and mortality risk: twenty-five-year follow-up of the Seven Countries Study. Arch Intern Med (1999) 159:733–40. doi:10.1001/archinte.159.7.733
5. Kannel WB, Brand N, Skinner JJ Jr, Dawber TR, McNamara PM. The relation of adiposity to blood pressure and development of hypertension. The Framingham study. Ann Intern Med (1967) 67:48–59. doi:10.7326/0003-4819-67-1-48
6. Kannel WB, Gordon T, Schwartz MJ. Systolic versus diastolic blood pressure and risk of coronary heart disease. The Framingham study. Am J Cardiol (1971) 27:335–46. doi:10.1016/0002-9149(71)90428-0
7. Kannel WB, LeBauer EJ, Dawber TR, McNamara PM. Relation of body weight to development of coronary heart disease. The Framingham study. Circulation (1967) 35:734–44. doi:10.1161/01.CIR.35.4.734
8. Verschuren WM, Jacobs DR, Bloemberg BP, Kromhout D, Menotti A, Aravanis C, et al. Serum total cholesterol and long-term coronary heart disease mortality in different cultures. Twenty-five-year follow-up of the seven countries study. JAMA (1995) 274:131–6. doi:10.1001/jama.274.2.131
10. Dai X, Wiernek S, Evans JP, Runge MS. Genetics of coronary artery disease and myocardial infarction. World J Cardiol (2016) 8:1–23. doi:10.4330/wjc.v8.i1.1
11. Luft FC. What have we learned from the genetics of hypertension? Med Clin North Am (2017) 101:195–206. doi:10.1016/j.mcna.2016.08.015
12. Samani NJ, Burton P, Mangino M, Ball SG, Balmforth AJ, Barrett J, et al. A genomewide linkage study of 1,933 families affected by premature coronary artery disease: The British Heart Foundation (BHF) Family Heart Study. Am J Hum Genet (2005) 77:1011–20. doi:10.1086/498653
13. Burton H, Jackson C, Abubakar I. The impact of genomics on public health practice. Br Med Bull (2014) 112:37–46. doi:10.1093/bmb/ldu032
14. Farhan SM, Hegele RA. Genetics 101 for cardiologists: rare genetic variants and monogenic cardiovascular disease. Can J Cardiol (2013) 29:18–22. doi:10.1016/j.cjca.2012.10.010
15. Simonetti GD, Mohaupt MG, Bianchetti MG. Monogenic forms of hypertension. Eur J Pediatr (2012) 171:1433–9. doi:10.1007/s00431-011-1440-7
16. Zaidi S, Brueckner M. Genetics and genomics of congenital heart disease. Circ Res (2017) 120:923–40. doi:10.1161/CIRCRESAHA.116.309140
17. Cappola TP, Margulies KB. Functional genomics applied to cardiovascular medicine. Circulation (2011) 124:87–94. doi:10.1161/CIRCULATIONAHA.111.027300
18. Becker F, van El CG, Ibarreta D, Zika E, Hogarth S, Borry P, et al. Genetic testing and common disorders in a public health framework: how to assess relevance and possibilities. Background Document to the ESHG recommendations on genetic testing and common disorders. Eur J Hum Genet (2011) 19(Suppl 1):S6–44. doi:10.1038/ejhg.2010.249
19. Khoury MJ, Gwinn M, Yoon PW, Dowling N, Moore CA, Bradley L. The continuum of translation research in genomic medicine: how can we accelerate the appropriate integration of human genome discoveries into health care and disease prevention? Genet Med (2007) 9:665–74. doi:10.1097/GIM.0b013e31815699d0
20. Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med (2009) 6:e1000097. doi:10.1371/journal.pmed.1000097
21. Foroud T, Sauerbeck L, Brown R, Anderson C, Woo D, Kleindorfer D, et al. Genome screen to detect linkage to intracranial aneurysm susceptibility genes: the Familial Intracranial Aneurysm (FIA) study. Stroke (2008) 39:1434–40. doi:10.1161/STROKEAHA.107.502930
22. Hiura Y, Shen CS, Kokubo Y, Okamura T, Morisaki T, Tomoike H, et al. Identification of genetic markers associated with high-density lipoprotein-cholesterol by genome-wide screening in a Japanese population: the Suita study. Circ J (2009) 73:1119–26. doi:10.1253/circj.CJ-08-1101
23. Harakalova M, van der Smagt J, de Kovel CG, Van’t Slot R, Poot M, Nijman IJ, et al. Incomplete segregation of MYH11 variants with thoracic aortic aneurysms and dissections and patent ductus arteriosus. Eur J Hum Genet (2013) 21:487–93. doi:10.1038/ejhg.2012.206
24. Akhter MS, Biswas A, Rashid H, Devi L, Behari M, Saxena R. Screening of the GPX3 gene identifies the “T” allele of the SNP -861A/T as a risk for ischemic stroke in young Asian Indians. J Stroke Cerebrovasc Dis (2014) 23:2060–8. doi:10.1016/j.jstrokecerebrovasdis.2014.03.010
25. Cheng J, Liu J, Li X, Peng J, Han S, Zhang R, et al. Insulin-like growth factor-1 receptor polymorphism and ischemic stroke: a case–control study in Chinese population. Acta Neurol Scand (2008) 118:333–8. doi:10.1111/j.1600-0404.2008.01040.x
26. Liu Y, Li S, Chen X, Zheng L, Yang Y, Jin L, et al. Association of natriuretic peptide receptor-C gene with ischemic stroke and hypertension in Chinese Han population. Clin Exp Hypertens (2012) 34:504–9. doi:10.3109/10641963.2012.681719
27. Man BL, Baum L, Fu YP, Chan YY, Lam W, Hui CF, et al. Genetic polymorphisms of Chinese patients with ischemic stroke and concurrent stenoses of extracranial and intracranial vessels. J Clin Neurosci (2010) 17:1244–7. doi:10.1016/j.jocn.2010.01.050
28. Oh SH, Min KT, Jeon YJ, Kim MH, Kim OJ, Shin BS, et al. Association between common genetic variants of alpha2A-, alpha2B-, and alpha2C-adrenergic receptors and ischemic stroke. Clin Neurol Neurosurg (2013) 115:26–31. doi:10.1016/j.clineuro.2012.04.002
29. Szolnoki Z, Serly J, Kondacs A, Mandi Y, Somogyvari F. Evaluation of the genetic variants of kinesin motor protein in ischemic stroke. J Stroke Cerebrovasc Dis (2009) 18:360–2. doi:10.1016/j.jstrokecerebrovasdis.2009.01.004
30. Gschwendtner A, Ripke S, Freilinger T, Lichtner P, Muller-Myhsok B, Wichmann HE, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) is associated with an increased risk of ischemic stroke in white Europeans. Stroke (2008) 39:1593–6. doi:10.1161/STROKEAHA.107.502179
31. Hong SH, Park HM, Ahn JY, Kim OJ, Hwang TS, Oh D, et al. ACE I/D polymorphism in Korean patients with ischemic stroke and silent brain infarction. Acta Neurol Scand (2008) 117:244–9. doi:10.1111/j.1600-0404.2007.00932.x
32. Matarin M, Brown WM, Singleton A, Hardy JA, Meschia JF, ISGS Investigators. Whole genome analyses suggest ischemic stroke and heart disease share an association with polymorphisms on chromosome 9p21. Stroke (2008) 39:1586–9. doi:10.1161/STROKEAHA.107.502963
33. Meschia JF, Nalls M, Matarin M, Brott TG, Brown RD Jr, Hardy J, et al. Siblings with ischemic stroke study: results of a genome-wide scan for stroke loci. Stroke (2011) 42:2726–32. doi:10.1161/STROKEAHA.111.620484
34. Rexrode KM, Ridker PM, Hegener HH, Buring JE, Manson JE, Zee RY. Genetic variation of the androgen receptor and risk of myocardial infarction and ischemic stroke in women. Stroke (2008) 39:1590–2. doi:10.1161/STROKEAHA.107.508218
35. Biswas A, Ranjan R, Meena A, Akhter MS, Yadav BK, Munisamy M, et al. Homocystine levels, polymorphisms and the risk of ischemic stroke in young Asian Indians. J Stroke Cerebrovasc Dis (2009) 18:103–10. doi:10.1016/j.jstrokecerebrovasdis.2008.09.014
36. Gromadzka G, Baranska-Gieruszczak M, Sarzynska-Dlugosz I, Ciesielska A, Czlonkowska A. The APOE polymorphism and 1-year outcome in ischemic stroke: genotype-gender interaction. Acta Neurol Scand (2007) 116:392–8. doi:10.1111/j.1600-0404.2007.00880.x
37. Lovkvist H, Olsson S, Hoglund P, Melander O, Jern C, Sjogren M, et al. A large-sample assessment of possible association between ischaemic stroke and rs12188950 in the PDE4D gene. Eur J Hum Genet (2012) 20:783–9. doi:10.1038/ejhg.2012.4
38. MacClellan LR, Howard TD, Cole JW, Stine OC, Giles WH, O’Connell JR, et al. Relation of candidate genes that encode for endothelial function to migraine and stroke: the Stroke Prevention in Young Women study. Stroke (2009) 40:e550–7. doi:10.1161/STROKEAHA.109.557462
39. Shin BS, Oh SY, Kim YS, Kim KW. The paraoxonase gene polymorphism in stroke patients and lipid profile. Acta Neurol Scand (2008) 117:237–43. doi:10.1111/j.1600-0404.2007.00929.x
40. Wang N, Xue XH, Lin Y, Fang L, Murong S, Wu ZY. The R219K polymorphism in the ATP-binding cassette transporter 1 gene has a protective effect on atherothrombotic cerebral infarction in Chinese Han ethnic population. Neurobiol Aging (2010) 31:647–53. doi:10.1016/j.neurobiolaging.2008.05.032
41. Della-Morte D, Guadagni F, Palmirotta R, Testa G, Caso V, Paciaroni M, et al. Genetics of ischemic stroke, stroke-related risk factors, stroke precursors and treatments. Pharmacogenomics (2012) 13:595–613. doi:10.2217/pgs.12.14
42. Campuzano O, Alcalde M, Berne P, Castro V, Guzzo G, Iglesias A, et al. Genetic testing of candidate genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Med Genet (2012) 55:225–34. doi:10.1016/j.ejmg.2012.02.007
43. Christophersen IE, Holmegard HN, Jabbari J, Sajadieh A, Haunso S, Tveit A, et al. Rare variants in GJA5 are associated with early-onset lone atrial fibrillation. Can J Cardiol (2013) 29:111–6. doi:10.1016/j.cjca.2012.08.002
44. Fukuyama M, Wang Q, Kato K, Ohno S, Ding WG, Toyoda F, et al. Long QT syndrome type 8: novel CACNA1C mutations causing QT prolongation and variant phenotypes. Europace (2014) 16:1828–37. doi:10.1093/europace/euu063
45. Lahtinen AM, Marjamaa A, Swan H, Kontula K. KCNE1 D85N polymorphism – a sex-specific modifier in type 1 long QT syndrome? BMC Med Genet (2011) 12:11. doi:10.1186/1471-2350-12-11
46. Lopez-Ayala JM, Gomez-Milanes I, Sanchez Munoz JJ, Ruiz-Espejo F, Ortiz M, Gonzalez-Carrillo J, et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: characterizing a phenotype. Europace (2014) 16:1838–46. doi:10.1093/europace/euu128
47. Risgaard B, Jabbari R, Refsgaard L, Holst AG, Haunso S, Sadjadieh A, et al. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin Genet (2013) 84:489–95. doi:10.1111/cge.12126
48. Selga E, Campuzano O, Pinsach-Abuin ML, Perez-Serra A, Mademont-Soler I, Riuro H, et al. Comprehensive genetic characterization of a Spanish Brugada syndrome cohort. PLoS One (2015) 10:e0132888. doi:10.1371/journal.pone.0132888
49. Subbiah RN, Gollob MH, Gula LJ, Davies RW, Leong-Sit P, Skanes AC, et al. Torsades de pointes during complete atrioventricular block: genetic factors and electrocardiogram correlates. Can J Cardiol (2010) 26:208–12. doi:10.1016/S0828-282X(10)70369-X
50. Uzieblo-Zyczkowska B, Gielerak G, Siedlecki P, Pajak B. Genetic diversity of SCN5A gene and its possible association with the concealed form of Brugada syndrome development in Polish group of patients. Biomed Res Int (2014) 2014:462609. doi:10.1155/2014/462609
51. Weeke P, Parvez B, Blair M, Short L, Ingram C, Kucera G, et al. Candidate gene approach to identifying rare genetic variants associated with lone atrial fibrillation. Heart Rhythm (2014) 11:46–52. doi:10.1016/j.hrthm.2013.10.025
52. Brinkley TE, Kume N, Mitsuoka H, Brown MD, Phares DA, Ferrell RE, et al. Variation in the human lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) gene is associated with plasma soluble LOX-1 levels. Exp Physiol (2008) 93:1085–90. doi:10.1113/expphysiol.2008.042267
53. Galluccio E, Cassina L, Russo I, Gelmini F, Setola E, Rampoldi L, et al. A novel truncated form of eNOS associates with altered vascular function. Cardiovasc Res (2014) 101:492–502. doi:10.1093/cvr/cvt267
54. Gupta N, Singh S, Maturu VN, Sharma YP, Gill KD. Paraoxonase 1 (PON1) polymorphisms, haplotypes and activity in predicting cad risk in North-West Indian Punjabis. PLoS One (2011) 6:e17805. doi:10.1371/journal.pone.0017805
55. Hinohara K, Nakajima T, Yasunami M, Houda S, Sasaoka T, Yamamoto K, et al. Megakaryoblastic leukemia factor-1 gene in the susceptibility to coronary artery disease. Hum Genet (2009) 126:539–47. doi:10.1007/s00439-009-0698-6
56. Kim MP, Wahl LM, Yanek LR, Becker DM, Becker LC. A monocyte chemoattractant protein-1 gene polymorphism is associated with occult ischemia in a high-risk asymptomatic population. Atherosclerosis (2007) 193:366–72. doi:10.1016/j.atherosclerosis.2006.06.029
57. Vasku A, Meluzin J, Blahak J, Kincl V, Goldbergova MP, Sitar J, et al. Matrix metalloproteinase 13 genotype in rs640198 polymorphism is associated with severe coronary artery disease. Dis Markers (2012) 33:43–9. doi:10.3233/DMA-2012-0902
58. Matoo S, Fallah MS, Daneshpour MS, Mousavi R, Sedaghati Khayat B, Hasanzad M, et al. Increased risk of CHD in the presence of rs7865618 (A allele): Tehran lipid and glucose study. Arch Iran Med (2017) 20:153–7.
59. Rotger M, Glass TR, Junier T, Lundgren J, Neaton JD, Poloni ES, et al. Contribution of genetic background, traditional risk factors, and HIV-related factors to coronary artery disease events in HIV-positive persons. Clin Infect Dis (2013) 57:112–21. doi:10.1093/cid/cit196
60. Roberts R, Stewart AF. 9p21 and the genetic revolution for coronary artery disease. Clin Chem (2012) 58:104–12. doi:10.1373/clinchem.2011.172759
61. Kanth VV, Golla JP, Sastry BK, Naik S, Kabra N, Sujatha M. Genetic interactions between MTHFR (C677T), methionine synthase (A2756G, C2758G) variants with vitamin B12 and folic acid determine susceptibility to premature coronary artery disease in Indian population. J Cardiovasc Dis Res (2011) 2:156–63. doi:10.4103/0975-3583.85262
62. Benn M, Schwartz M, Nordestgaard BG, Tybjaerg-Hansen A. Mitochondrial haplogroups: ischemic cardiovascular disease, other diseases, mortality, and longevity in the general population. Circulation (2008) 117:2492–501. doi:10.1161/CIRCULATIONAHA.107.756809
63. Buysschaert I, Carruthers KF, Dunbar DR, Peuteman G, Rietzschel E, Belmans A, et al. A variant at chromosome 9p21 is associated with recurrent myocardial infarction and cardiac death after acute coronary syndrome: the GRACE Genetics Study. Eur Heart J (2010) 31:1132–41. doi:10.1093/eurheartj/ehq053
64. Corsetti JP, Ryan D, Moss AJ, Rainwater DL, Zareba W, Sparks CE. Plasminogen activator inhibitor-1 polymorphism (4G/5G) predicts recurrence in nonhyperlipidemic postinfarction patients. Arterioscler Thromb Vasc Biol (2008) 28:548–54. doi:10.1161/ATVBAHA.107.155556
65. Cui Y, Wang H, Chen H, Pang S, Wang L, Liu D, et al. Genetic analysis of the SIRT1 gene promoter in myocardial infarction. Biochem Biophys Res Commun (2012) 426:232–6. doi:10.1016/j.bbrc.2012.08.071
66. Nasibullin TR, Timasheva YR, Sadikova RI, Tuktarova IA, Erdman VV, Nikolaeva IE, et al. Genotype/allelic combinations as potential predictors of myocardial infarction. Mol Biol Rep (2016) 43:11–6. doi:10.1007/s11033-015-3933-3
67. Belfer I, Wu T, Hipp H, Walter J, Scully M, Nyquist PA, et al. Linkage of large-vessel carotid atherosclerotic stroke to inflammatory genes via a systematic screen. Int J Stroke (2010) 5:145–51. doi:10.1111/j.1747-4949.2010.00422.x
68. Lan MY, Chang YY, Chen WH, Tseng YL, Lin HS, Lai SL, et al. Association between MIF gene polymorphisms and carotid artery atherosclerosis. Biochem Biophys Res Commun (2013) 435:319–22. doi:10.1016/j.bbrc.2013.02.129
69. Zhang Y, Wang L, Zhang Z, Zhang Z, Zhou S, Cao L, et al. Shared and discrepant susceptibility for carotid artery and aortic arch calcification: a genetic association study. Atherosclerosis (2015) 241:371–5. doi:10.1016/j.atherosclerosis.2015.05.030
70. Zhao R, Wang Y, Shen R, Sun Y. Relationship between CX3CR1 genetic polymorphism and carotid atherosclerosis. Artif Cells Blood Substit Immobil Biotechnol (2010) 38:19–23. doi:10.3109/10731190903495728
71. Jabbari J, Olesen MS, Yuan L, Nielsen JB, Liang B, Macri V, et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ Cardiovasc Genet (2015) 8:64–73. doi:10.1161/HCG.0000000000000022
72. Smallwood L, Warrington N, Allcock R, van Bockxmeer F, Palmer LJ, Iacopetta B, et al. Matrix metalloproteinase-2 gene variants and abdominal aortic aneurysm. Eur J Vasc Endovasc Surg (2009) 38:169–71. doi:10.1016/j.ejvs.2009.04.019
73. Armstrong C, Abilleira S, Sitzer M, Markus HS, Bevan S. Polymorphisms in MMP family and TIMP genes and carotid artery intima-media thickness. Stroke (2007) 38:2895–9. doi:10.1161/STROKEAHA.107.491696
74. Lanktree MB, Hegele RA, Yusuf S, Anand SS. Multi-ethnic genetic association study of carotid intima-media thickness using a targeted cardiovascular SNP microarray. Stroke (2009) 40:3173–9. doi:10.1161/STROKEAHA.109.556563
75. Gardener H, Beecham A, Cabral D, Yanuck D, Slifer S, Wang L, et al. Carotid plaque and candidate genes related to inflammation and endothelial function in Hispanics from northern Manhattan. Stroke (2011) 42:889–96. doi:10.1161/STROKEAHA.110.591065
76. Aijala M, Ronkainen J, Huusko T, Malo E, Savolainen ER, Savolainen MJ, et al. The fat mass and obesity-associated (FTO) gene variant rs9939609 predicts long-term incidence of cardiovascular disease and related death independent of the traditional risk factors. Ann Med (2015) 47:655–63. doi:10.3109/07853890.2015.1091088
77. Holzhauser L, Zolty R. Endothelin receptor polymorphisms in the cardiovascular system: potential implications for therapy and screening. Heart Fail Rev (2014) 19:743–58. doi:10.1007/s10741-014-9426-y
78. Casazza K, Page GP, Fernandez JR. The association between the rs2234693 and rs9340799 estrogen receptor alpha gene polymorphisms and risk factors for cardiovascular disease: a review. Biol Res Nurs (2010) 12:84–97. doi:10.1177/1099800410371118
79. Kepp K, Org E, Sober S, Kelgo P, Viigimaa M, Veldre G, et al. Hypervariable intronic region in NCX1 is enriched in short insertion-deletion polymorphisms and showed association with cardiovascular traits. BMC Med Genet (2010) 11:15. doi:10.1186/1471-2350-11-15
80. Schurks M, Zee RY, Buring JE, Kurth T. Interrelationships among the MTHFR 677C>T polymorphism, migraine, and cardiovascular disease. Neurology (2008) 71:505–13. doi:10.1212/01.wnl.0000316198.34558.e5
81. Biagi DG, Mill JG, Mansur AJ, Krieger JE, Pereira AC. A negative screen for mutations in calstabin 1 and 2 genes in patients with dilated cardiomyopathy. J Negat Results Biomed (2012) 11:4. doi:10.1186/1477-5751-11-4
82. Elliott P, O’Mahony C, Syrris P, Evans A, Rivera Sorensen C, Sheppard MN, et al. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet (2010) 3:314–22. doi:10.1161/CIRCGENETICS.110.937805
83. Patrignani P, Di Febbo C, Tacconelli S, Douville K, Guglielmi MD, Horvath RJ, et al. Differential association between human prostacyclin receptor polymorphisms and the development of venous thrombosis and intimal hyperplasia: a clinical biomarker study. Pharmacogenet Genomics (2008) 18:611–20. doi:10.1097/FPC.0b013e328301a774
84. Sode BF, Allin KH, Dahl M, Gyntelberg F, Nordestgaard BG. Risk of venous thromboembolism and myocardial infarction associated with factor V Leiden and prothrombin mutations and blood type. CMAJ (2013) 185:E229–37. doi:10.1503/cmaj.121636
85. Yapijakis C, Antoniadi T, Salavoura K, Voumvourakis C, Vairaktaris E. Potential prevention of thromboembolism by genetic counseling and testing for two common thrombophilia mutations. In Vivo (2012) 26:165–72.
86. Braenne I, Kleinecke M, Reiz B, Graf E, Strom T, Wieland T, et al. Systematic analysis of variants related to familial hypercholesterolemia in families with premature myocardial infarction. Eur J Hum Genet (2016) 24:191–7. doi:10.1038/ejhg.2015.100
87. Tanjore RR, Rangaraju A, Kerkar PG, Calambur N, Nallari P. MYBPC3 gene variations in hypertrophic cardiomyopathy patients in India. Can J Cardiol (2008) 24:127–30. doi:10.1016/S0828-282X(08)70568-3
88. Mouton JM, van der Merwe L, Goosen A, Revera M, Brink PA, Moolman-Smook JC, et al. MYBPH acts as modifier of cardiac hypertrophy in hypertrophic cardiomyopathy (HCM) patients. Hum Genet (2016) 135:477–83. doi:10.1007/s00439-016-1649-7
89. Das KJ, Ingles J, Bagnall RD, Semsarian C. Determining pathogenicity of genetic variants in hypertrophic cardiomyopathy: importance of periodic reassessment. Genet Med (2014) 16:286–93. doi:10.1038/gim.2013.138
90. Ferreira CN, Carvalho MG, Fernandes AP, Lima LM, Loures-Valle AA, Dantas J, et al. Comparative study of apolipoprotein-E polymorphism and plasma lipid levels in dyslipidemic and asymptomatic subjects, and their implication in cardio/cerebro-vascular disorders. Neurochem Int (2010) 56:177–82. doi:10.1016/j.neuint.2009.09.016
91. Oniki K, Hori M, Takata K, Yokoyama T, Mihara S, Marubayashi T, et al. Association between glutathione S-transferase A1, M1 and T1 polymorphisms and hypertension. Pharmacogenet Genomics (2008) 18:275–7. doi:10.1097/FPC.0b013e3282f56176
92. Tabara Y, Kohara K, Miki T; Millennium Genome Project for Hypertension. Hunting for genes for hypertension: the Millennium Genome Project for Hypertension. Hypertens Res (2012) 35:567–73. doi:10.1038/hr.2012.41
93. Liu Y, Li Y, Wang X, Ma Q, Zhu C, Li Z, et al. Mitochondrial tRNA mutations in Chinese hypertensive individuals. Mitochondrion (2016) 28:1–7. doi:10.1016/j.mito.2016.02.007
94. Joo SP, Kim TS, Lee IK, Lee JK, Seo BR, Kim JH, et al. The role of collagen type I alpha2 polymorphisms: intracranial aneurysms in Koreans. Surg Neurol (2009) 72:48–53. doi:10.1016/j.surneu.2009.02.009
95. Ruigrok YM, Tan S, Medic J, Rinkel GJ, Wijmenga C. Genes involved in the transforming growth factor beta signalling pathway and the risk of intracranial aneurysms. J Neurol Neurosurg Psychiatry (2008) 79:722–4. doi:10.1136/jnnp.2007.128041
96. James ML, Blessing R, Bennett E, Laskowitz DT. Apolipoprotein E modifies neurological outcome by affecting cerebral edema but not hematoma size after intracerebral hemorrhage in humans. J Stroke Cerebrovasc Dis (2009) 18:144–9. doi:10.1016/j.jstrokecerebrovasdis.2008.09.012
97. Somarajan BI, Kalita J, Misra UK, Mittal B. A study of alpha1 antichymotrypsin gene polymorphism in Indian stroke patients. J Neurol Sci (2010) 290:57–9. doi:10.1016/j.jns.2009.11.009
98. Yang J, Wu B, Lin S, Zhou J, Li Y, Dong W, et al. Genetic variations of MMP9 gene and intracerebral hemorrhage susceptibility: a case-control study in Chinese Han population. J Neurol Sci (2014) 341:55–7. doi:10.1016/j.jns.2014.03.049
99. Stengard JH, Frikke-Schmidt R, Tybjaerg-Hansen A, Nordestgaard BG, Sing CF. Variation in 5’ promoter region of the APOE gene contributes to predicting ischemic heart disease (IHD) in the population at large: the Copenhagen City Heart Study. Ann Hum Genet (2007) 71:762–71. doi:10.1111/j.1469-1809.2007.00370.x
100. Fernandez-Cadenas I, Del Rio-Espinola A, Giralt D, Domingues-Montanari S, Quiroga A, Mendioroz M, et al. IL1B and VWF variants are associated with fibrinolytic early recanalization in patients with ischemic stroke. Stroke (2012) 43:2659–65. doi:10.1161/STROKEAHA.112.657007
101. Fontanella M, Gallone S, Panciani PP, Garbossa D, Stefini R, Latronico N, et al. Vascular endothelial growth factor gene polymorphisms and intracranial aneurysms. Acta Neurochir (Wien) (2013) 155:1511–5. doi:10.1007/s00701-013-1779-9
102. Rueffert H, Gumplinger A, Renner C, Dengl M, Reske A, Kaisers UX, et al. Search for genetic variants in the ryanodine receptor 1 gene in patients with symptomatic cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Neurocrit Care (2011) 15:410–5. doi:10.1007/s12028-011-9542-7
103. Ruigrok YM, Slooter AJ, Rinkel GJ, Wijmenga C, Rosendaal FR. Genes influencing coagulation and the risk of aneurysmal subarachnoid hemorrhage, and subsequent complications of secondary cerebral ischemia and rebleeding. Acta Neurochir (Wien) (2010) 152:257–62. doi:10.1007/s00701-009-0505-0
104. Aschauer S, Brunner M, Wolzt M, Haslacher H, Ristl R, Muller M. Forearm vasodilator reactivity in healthy male carriers of the 3q22.3 rs9818870 polymorphism. Microvasc Res (2015) 102:33–7. doi:10.1016/j.mvr.2015.08.005
105. Popa S, Aursulesi V, Gorduza EC II. Preliminary evaluation of proptein convertase subtilisin/kexin type 5 mutations in lower extremity artery disease. Biomed Res India (2017) 28:4676–9.
106. Szeliga MA, Hedley PL, Green CP, Moller DV, Christiansen M. Long QT syndrome – a genetic cardiac channelopathy. Kardiol Pol (2010) 68:575–83.
107. Vatta M. Intronic variants and splicing errors in cardiovascular diseases. Heart Rhythm (2009) 6:219–20. doi:10.1016/j.hrthm.2008.12.006
108. Mahida S, Ellinor PT. New advances in the genetic basis of atrial fibrillation. J Cardiovasc Electrophysiol (2012) 23:1400–6. doi:10.1111/j.1540-8167.2012.02445.x
109. Kalea AZ, Harrison SC, Stephens JW, Talmud PJ. Genetic susceptibility for coronary heart disease and type 2 diabetes complications. Clin Chem (2012) 58:818–20. doi:10.1373/clinchem.2012.182725
110. Lanktree M, Oh J, Hegele RA. Genetic testing for atherosclerosis risk: inevitability or pipe dream? Can J Cardiol (2008) 24:851–4. doi:10.1016/S0828-282X(08)70194-6
111. McPherson R, Tybjaerg-Hansen A. Genetics of coronary artery disease. Circ Res (2016) 118:564–78. doi:10.1161/CIRCRESAHA.115.306566
112. Pranavchand R, Reddy BM. Current status of understanding of the genetic etiology of coronary heart disease. J Postgrad Med (2013) 59:30–41. doi:10.4103/0022-3859.109492
113. Roberts R. A customized genetic approach to the number one killer: coronary artery disease. Curr Opin Cardiol (2008) 23:629–33. doi:10.1097/HCO.0b013e32830e6b4e
114. Superko HR, Roberts R, Agatston A, Frohwein S, Reingold JS, White TJ, et al. Genetic testing for early detection of individuals at risk of coronary heart disease and monitoring response to therapy: challenges and promises. Curr Atheroscler Rep (2011) 13:396–404. doi:10.1007/s11883-011-0198-8
115. Prutkin JM, Sotoodehnia N. Genetics of sudden cardiac arrest. Prog Cardiovasc Dis (2008) 50:390–403. doi:10.1016/j.pcad.2007.10.006
116. Priori SG. Genetic testing to predict sudden cardiac death: current perspectives and future goals. Indian Heart J (2014) 66(Suppl 1):S58–60. doi:10.1016/j.ihj.2013.11.004
117. Dorn GW II. The genomic architecture of sporadic heart failure. Circ Res (2011) 108:1270–83. doi:10.1161/CIRCRESAHA.110.229260
118. Yang J, Xu WW, Hu SJ. Heart failure: advanced development in genetics and epigenetics. Biomed Res Int (2015) 2015:352734. doi:10.1155/2015/352734
119. Wilde AA, Behr ER. Genetic testing for inherited cardiac disease. Nat Rev Cardiol (2013) 10:571–83. doi:10.1038/nrcardio.2013.108
120. Reitsma PH. Genetics in thrombophilia. An update. Hamostaseologie (2015) 35:47–51. doi:10.5482/HAMO-14-11-0062
121. Daraban A, Trifa A, Popp R, Botezatu D, Şerban M, Uscatescu V, et al. Thrombophilia genetic testing in Romanian young women with acute thrombotic events: role of Factor V Leiden, Prothrombin G20210A, MTHFR C677T and A1298C polymorphisms. Revista Romana de Medicina de Laborator (2016) 24:291–305. doi:10.1515/rrlm-2016-0032
122. Thanassoulis G, Peloso GM, O’Donnell CJ. Genomic medicine for improved prediction and primordial prevention of cardiovascular disease. Arterioscler Thromb Vasc Biol (2013) 33:2049–50. doi:10.1161/ATVBAHA.113.301814
123. Thanassoulis G, Vasan RS. Genetic cardiovascular risk prediction: will we get there? Circulation (2010) 122:2323–34. doi:10.1161/CIRCULATIONAHA.109.909309
124. Paynter NP, Ridker PM, Chasman DI. Are genetic tests for atherosclerosis ready for routine clinical use? Circ Res (2016) 118:607–19. doi:10.1161/CIRCRESAHA.115.306360
125. Prudente S, Dallapiccola B, Pellegrini F, Doria A, Trischitta V. Genetic prediction of common diseases. Still no help for the clinical diabetologist! Nutr Metab Cardiovasc Dis (2012) 22:929–36. doi:10.1016/j.numecd.2012.04.010
126. Roberts R, Stewart AF. Genes and coronary artery disease: where are we? J Am Coll Cardiol (2012) 60:1715–21. doi:10.1016/j.jacc.2011.12.062
127. Herrington DM. Cardiovascular genomics: outcomes and implications. Can J Cardiol (2010) 26(Suppl A):60A–3A. doi:10.1016/S0828-282X(10)71065-5
128. Lewis CM, Knight J. Introduction to genetic association studies. Cold Spring Harb Protoc (2012) 2012:297–306. doi:10.1101/pdb.top068163
129. Traylor M, Zhang CR, Adib-Samii P, Devan WJ, Parsons OE, Lanfranconi S, et al. Genome-wide meta-analysis of cerebral white matter hyperintensities in patients with stroke. Neurology (2016) 86:146–53. doi:10.1212/WNL.0000000000002263
130. Song DH, Zhou PZ, Xiu XL, Zhou GH, Sun YX, Song C. Relationships of OPG genetic polymorphisms with susceptibility to cardiovascular disease: a meta-analysis. Med Sci Monit (2016) 22:1223–31. doi:10.12659/MSM.895434
131. Smith JG, Almgren P, Engstrom G, Hedblad B, Platonov PG, Newton-Cheh C, et al. Genetic polymorphisms for estimating risk of atrial fibrillation: a literature-based meta-analysis. J Intern Med (2012) 272:573–82. doi:10.1111/j.1365-2796.2012.02563.x
132. Maguire JM, Thakkinstian A, Sturm J, Levi C, Lincz L, Parsons M, et al. Polymorphisms in platelet glycoprotein 1balpha and factor VII and risk of ischemic stroke: a meta-analysis. Stroke (2008) 39:1710–6. doi:10.1161/STROKEAHA.107.507228
133. Debette S, Bis JC, Fornage M, Schmidt H, Ikram MA, Sigurdsson S, et al. Genome-wide association studies of MRI-defined brain infarcts: meta-analysis from the CHARGE Consortium. Stroke (2010) 41:210–7. doi:10.1161/STROKEAHA.109.569194
134. Kitsios G, Zintzaras E. ACE (I/D) polymorphism and response to treatment in coronary artery disease: a comprehensive database and meta-analysis involving study quality evaluation. BMC Med Genet (2009) 10:50. doi:10.1186/1471-2350-10-50
135. Jiang N, Yang G, Peng CL. ESR1 gene polymorphisms PvuII (rs2234693T>C) and XbaI (rs9340799A>G) may not be directly correlated with cardiovascular disease risk. Genet Mol Res (2015) 14:13932–44. doi:10.4238/2015.October.29.14
136. Pearson TA, Manolio TA. How to interpret a genome-wide association study. JAMA (2008) 299:1335–44. doi:10.1001/jama.299.11.1335
137. Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature (2007) 447:661–78. doi:10.1038/nature05911
138. Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, et al. Genomewide association analysis of coronary artery disease. N Engl J Med (2007) 357:443–53. doi:10.1056/NEJMoa072366
139. Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet (2009) 41:334–41. doi:10.1038/ng.327
140. McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science (2007) 316:1488–91. doi:10.1126/science.1142447
141. Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science (2007) 316:1491–3. doi:10.1126/science.1142842
142. Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature (2010) 464:409–12. doi:10.1038/nature08801
143. Talmud PJ, Hingorani AD, Cooper JA, Marmot MG, Brunner EJ, Kumari M, et al. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. BMJ (2010) 340:b4838. doi:10.1136/bmj.b4838
144. Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet (2009) 41:47–55. doi:10.1038/ng.269
145. Krarup NT, Borglykke A, Allin KH, Sandholt CH, Justesen JM, Andersson EA, et al. A genetic risk score of 45 coronary artery disease risk variants associates with increased risk of myocardial infarction in 6041 Danish individuals. Atherosclerosis (2015) 240:305–10. doi:10.1016/j.atherosclerosis.2015.03.022
146. Isaacs A, Willems SM, Bos D, Dehghan A, Hofman A, Ikram MA, et al. Risk scores of common genetic variants for lipid levels influence atherosclerosis and incident coronary heart disease. Arterioscler Thromb Vasc Biol (2013) 33:2233–9. doi:10.1161/ATVBAHA.113.301236
147. Tikkanen E, Havulinna AS, Palotie A, Salomaa V, Ripatti S. Genetic risk prediction and a 2-stage risk screening strategy for coronary heart disease. Arterioscler Thromb Vasc Biol (2013) 33:2261–6. doi:10.1161/ATVBAHA.112.301120
148. Weijmans M, de Bakker PI, van der Graaf Y, Asselbergs FW, Algra A, Jan de Borst G, et al. Incremental value of a genetic risk score for the prediction of new vascular events in patients with clinically manifest vascular disease. Atherosclerosis (2015) 239:451–8. doi:10.1016/j.atherosclerosis.2015.02.008
149. Ganna A, Magnusson PK, Pedersen NL, de Faire U, Reilly M, Arnlov J, et al. Multilocus genetic risk scores for coronary heart disease prediction. Arterioscler Thromb Vasc Biol (2013) 33:2267–72. doi:10.1161/ATVBAHA.113.301218
150. de Haan HG, Bezemer ID, Doggen CJ, Le Cessie S, Reitsma PH, Arellano AR, et al. Multiple SNP testing improves risk prediction of first venous thrombosis. Blood (2012) 120:656–63. doi:10.1182/blood-2011-12-397752
151. Weeke P, Roden DM. Pharmacogenomics and cardiovascular disease. Curr Cardiol Rep (2013) 15:376. doi:10.1007/s11886-013-0376-0
152. Cannon CP. Clopidogrel: to test or not to test? That is the question – still. Clin Chem (2011) 57:659–61. doi:10.1373/clinchem.2010.158709
153. Jia DM, Chen ZB, Zhang MJ, Yang WJ, Jin JL, Xia YQ, et al. CYP2C19 polymorphisms and antiplatelet effects of clopidogrel in acute ischemic stroke in China. Stroke (2013) 44:1717–9. doi:10.1161/STROKEAHA.113.000823
154. Yang E. Current antiplatelet agents: place in therapy and role of genetic testing. J Thromb Thrombolysis (2015) 39:328–36. doi:10.1007/s11239-015-1195-5
155. Ford NF, Taubert D. Clopidogrel, CYP2C19, and a Black Box. J Clin Pharmacol (2013) 53:241–8. doi:10.1002/jcph.17
156. Fuster V, Sweeny JM. Clopidogrel and the reduced-function CYP2C19 genetic variant: a limited piece of the overall therapeutic puzzle. JAMA (2010) 304:1839–40. doi:10.1001/jama.2010.1566
157. Campo G, Miccoli M, Tebaldi M, Marchesini J, Fileti L, Monti M, et al. Genetic determinants of on-clopidogrel high platelet reactivity. Platelets (2011) 22:399–407. doi:10.3109/09537104.2011.579648
158. Shen DL, Wang B, Bai J, Han Q, Liu C, Huang XH, et al. Clinical value of CYP2C19 genetic testing for guiding the antiplatelet therapy in a Chinese population. J Cardiovasc Pharmacol (2016) 67:232–6. doi:10.1097/FJC.0000000000000337
159. Chan NC, Eikelboom JW, Ginsberg JS, Lauw MN, Vanassche T, Weitz JI, et al. Role of phenotypic and genetic testing in managing clopidogrel therapy. Blood (2014) 124:689–99. doi:10.1182/blood-2014-01-512723
160. Yi X, Han Z, Zhou Q, Cheng W, Lin J, Wang C. Concomitant use of proton-pump inhibitors and clopidogrel increases the risk of adverse outcomes in patients with ischemic stroke carrying reduced-function CYP2C19*2. Clin Appl Thromb Hemost (2018) 24(1):55–62. doi:10.1177/1076029616669787
161. Evaluation of Genomic Applications in P, Prevention Working G. Recommendations from the EGAPP Working Group: genomic profiling to assess cardiovascular risk to improve cardiovascular health. Genet Med (2010) 12:839–43. doi:10.1097/GIM.0b013e3181f872c0
162. Force USPST. Using nontraditional risk factors in coronary heart disease risk assessment: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med (2009) 151:474–82. doi:10.7326/0003-4819-151-7-200910060-00008
163. Goswami S, Cheng-Lai A, Nawarskas J. Clopidogrel and genetic testing: is it necessary for everyone? Cardiol Rev (2012) 20:96–100. doi:10.1097/CRD.0b013e3182455744
164. Hulot JS, Hajjar R, Montalescot G. Clopidogrel and CYP2C19 testing: ready for clinical prime time? Clin Chem (2012) 58:154–7. doi:10.1373/clinchem.2011.165829
165. Wright RS, Anderson JL, Adams CD, Bridges CR, Casey DE Jr, Ettinger SM, et al. 2011 ACCF/AHA focused update of the guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation (2011) 123:2022–60. doi:10.1161/CIR.0b013e31820f2f3e
166. Hickey SE, Curry CJ, Toriello HV. ACMG practice guideline: lack of evidence for MTHFR polymorphism testing. Genet Med (2013) 15:153–6. doi:10.1038/gim.2012.165
167. Long S, Goldblatt J. MTHFR genetic testing: controversy and clinical implications. Aust Fam Physician (2016) 45:237–40.
168. Varga EA. Genetic counseling for inherited thrombophilias. J Thromb Thrombolysis (2008) 25:6–9. doi:10.1007/s11239-007-0056-2
169. Ray JG. Hyperhomocysteinemia: no longer a consideration in the management of venous thromboembolism. Curr Opin Pulm Med (2008) 14:369–73. doi:10.1097/MCP.0b013e328307ee38
170. Moyer VA; U.S. Preventive Services Task Force. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med (2014) 160:271–81. doi:10.7326/M13-2747
171. Jiang M, You JH. CYP2C19 LOF and GOF-guided antiplatelet therapy in patients with acute coronary syndrome: a cost-effectiveness analysis. Cardiovasc Drugs Ther (2017) 31:39–49. doi:10.1007/s10557-016-6705-y
172. Little J, Higgins JP, Ioannidis JP, Moher D, Gagnon F, von Elm E, et al. STrengthening the REporting of Genetic Association studies (STREGA) – an extension of the STROBE statement. Eur J Clin Invest (2009) 39:247–66. doi:10.1111/j.1365-2362.2009.02125.x
173. Janssens AC, Ioannidis JP, van Duijn CM, Little J, Khoury MJ, Group G. Strengthening the reporting of genetic risk prediction studies: the GRIPS statement. Eur J Epidemiol (2011) 26:255–9. doi:10.1007/s10654-011-9552-y
174. Shea BJ, Hamel C, Wells GA, Bouter LM, Kristjansson E, Grimshaw J, et al. AMSTAR is a reliable and valid measurement tool to assess the methodological quality of systematic reviews. J Clin Epidemiol (2009) 62:1013–20. doi:10.1016/j.jclinepi.2008.10.009
175. Palomaki GE, Melillo S, Neveux L, Douglas MP, Dotson WD, Janssens AC, et al. Use of genomic profiling to assess risk for cardiovascular disease and identify individualized prevention strategies – a targeted evidence-based review. Genet Med (2010) 12:772–84. doi:10.1097/GIM.0b013e3181f8728d
176. Pepe MS, Gu JW, Morris DE. The potential of genes and other markers to inform about risk. Cancer Epidemiol Biomarkers Prev (2010) 19:655–65. doi:10.1158/1055-9965.EPI-09-0510
177. Simmonds M. Genetic profiling tests in screening for cardiovascular disease. J Med Screen (2011) 18:56–7. doi:10.1258/jms.2011.011003
178. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature (2009) 461:747–53. doi:10.1038/nature08494
179. McCarthy MI, Hirschhorn JN. Genome-wide association studies: potential next steps on a genetic journey. Hum Mol Genet (2008) 17:R156–65. doi:10.1093/hmg/ddn289
180. Metzker ML. Sequencing technologies – the next generation. Nat Rev Genet (2010) 11:31–46. doi:10.1038/nrg2626
181. Xu F, Wang Q, Zhang F, Zhu Y, Gu Q, Wu L, et al. Impact of next-generation sequencing (NGS) technology on cardiovascular disease research. Cardiovasc Diagn Ther (2012) 2:138–46. doi:10.3978/j.issn.2223-3652.2012.06.01
182. Roberts R, Marian AJ, Dandona S, Stewart AF. Genomics in cardiovascular disease. J Am Coll Cardiol (2013) 61:2029–37. doi:10.1016/j.jacc.2012.12.054
183. Celestino-Soper PB, Gao H, Lynnes TC, Lin H, Liu Y, Spoonamore KG, et al. Validation and utilization of a clinical next-generation sequencing panel for selected cardiovascular disorders. Front Cardiovasc Med (2017) 4:11. doi:10.3389/fcvm.2017.00011
184. Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science (2011) 331:768–72. doi:10.1126/science.1198785
185. Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature (2012) 482:98–102. doi:10.1038/nature10814
186. Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med (2010) 363:2220–7. doi:10.1056/NEJMoa1002926
187. Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet (2011) 88:273–82. doi:10.1016/j.ajhg.2011.01.016
188. Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell (2012) 148:1242–57. doi:10.1016/j.cell.2012.03.001
189. Faita F, Vecoli C, Foffa I, Andreassi MG. Next generation sequencing in cardiovascular diseases. World J Cardiol (2012) 4:288–95. doi:10.4330/wjc.v4.i10.288
190. Mogensen J, van Tintelen JP, Fokstuen S, Elliott P, van Langen IM, Meder B, et al. The current role of next-generation DNA sequencing in routine care of patients with hereditary cardiovascular conditions: a viewpoint paper of the European Society of Cardiology working group on myocardial and pericardial diseases and members of the European Society of Human Genetics. Eur Heart J (2015) 36:1367–70. doi:10.1093/eurheartj/ehv122
191. Parikh VN, Ashley EA. Next-generation sequencing in cardiovascular disease: present clinical applications and the horizon of precision medicine. Circulation (2017) 135:406–9. doi:10.1161/CIRCULATIONAHA.116.024258
192. Giudicessi JR, Kullo IJ, Ackerman MJ. Precision cardiovascular medicine: state of genetic testing. Mayo Clin Proc (2017) 92:642–62. doi:10.1016/j.mayocp.2017.01.015
193. Breitling LP. Current genetics and epigenetics of smoking/tobacco-related cardiovascular disease. Arterioscler Thromb Vasc Biol (2013) 33:1468–72. doi:10.1161/ATVBAHA.112.300157
194. Buscemi L, Turchi C. An overview of the genetic susceptibility to alcoholism. Med Sci Law (2011) 51(Suppl 1):S2–6. doi:10.1258/msl.2010.010054
195. de Vilhena e Santos DM, Katzmarzyk PT, Seabra AF, Maia JA. Genetics of physical activity and physical inactivity in humans. Behav Genet (2012) 42:559–78. doi:10.1007/s10519-012-9534-1
196. Grimm ER, Steinle NI. Genetics of eating behavior: established and emerging concepts. Nutr Rev (2011) 69:52–60. doi:10.1111/j.1753-4887.2010.00361.x
197. Race, Ethnicity, and Genetics Working Group. The use of racial, ethnic, and ancestral categories in human genetics research. Am J Hum Genet (2005) 77:519–32. doi:10.1086/491747
198. Dobranici M, Buzea A, Popescu R. The cardiovascular risk factors of the Roma (gypsies) people in Central-Eastern Europe: a review of the published literature. J Med Life (2012) 5:382–9.
199. Foldes ME, Covaci A. Research on Roma health and access to healthcare: state of the art and future challenges. Int J Public Health (2012) 57:37–9. doi:10.1007/s00038-011-0312-2
200. Dioszegi J, Fiatal S, Toth R, Moravcsik-Kornyicki A, Kosa Z, Sandor J, et al. Distribution characteristics and combined effect of polymorphisms affecting alcohol consumption behaviour in the Hungarian general and Roma populations. Alcohol Alcohol (2017) 52:104–11. doi:10.1093/alcalc/agw052
201. Fiatal S, Toth R, Moravcsik-Kornyicki A, Kosa Z, Sandor J, McKee M, et al. High prevalence of smoking in the Roma population seems to have no genetic background. Nicotine Tob Res (2016) 18:2260–7. doi:10.1093/ntr/ntw161
202. Piko P, Fiatal S, Kosa Z, Sandor J, Adany R. Genetic factors exist behind the high prevalence of reduced high-density lipoprotein cholesterol levels in the Roma population. Atherosclerosis (2017) 263:119–26. doi:10.1016/j.atherosclerosis.2017.05.028
Keywords: genetic screening, genetic susceptibility, single-nucleotide polymorphism, translational research, cardiovascular diseases, literature search
Citation: Fiatal S and Ádány R (2018) Application of Single-Nucleotide Polymorphism-Related Risk Estimates in Identification of Increased Genetic Susceptibility to Cardiovascular Diseases: A Literature Review. Front. Public Health 5:358. doi: 10.3389/fpubh.2017.00358
Received: 22 June 2017; Accepted: 15 December 2017;
Published: 31 January 2018
Edited by:
Rumen Stefanov, Plovdiv Medical University, BulgariaReviewed by:
Iveta Nagyova, University of Pavol Jozef Šafárik, SlovakiaGuvenc Kockaya, Beykent University, Turkey
Copyright: © 2018 Fiatal and Ádány. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Szilvia Fiatal, ZmlhdGFsLnN6aWx2aWFAc3BoLnVuaWRlYi5odQ==