 Altaf Alabdali1
Altaf Alabdali1 Abir Ben Bacha1*
Abir Ben Bacha1* Mona Alonazi1
Mona Alonazi1 Laila Y. Al-Ayadhi2
Laila Y. Al-Ayadhi2 Abdullah S. J. Alanazi3
Abdullah S. J. Alanazi3 Afaf El‐Ansary4*
Afaf El‐Ansary4*- 1Biochemistry Department, Science College, King Saud University, Riyadh, Saudi Arabia
- 2Autism Research and Treatment Center, Department of Physiology, Faculty of Medicine, King Saud University, Riyadh, Saudi Arabia
- 3Biochemistry Laboratory, Qurayyat General Hospital, Qurayyat, Saudi Arabia
- 4Autism Center, Lotus Holistic Alternative Medical Center, Abu Dhabi, United Arab Emirates
Introduction: Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by social communication deficits and repetitive behaviors. An imbalance between the excitatory neurotransmitter glutamate and the inhibitory neurotransmitter gamma-aminobutyric acid (GABA) might play a crucial role in ASD. This study explores the biochemical markers associated with GABAergic and glutamatergic signaling in individuals with autism and healthy controls, aiming to identify potential diagnostic and therapeutic targets.
Methods: The study included 46 male individuals with autism and 26 age- and gender-matched healthy controls. The plasma levels of excitatory amino acid transporter 2 (EAAT2), potassium chloride co-transporter 2 (KCC2), Na–K–Cl co-transporter 1 (NKCC1), vitamin D3 (VD3), GABA, gamma aminobutyric acid type a receptor subunit alpha 5 (GABRA5), and glutamate were measured using ELISA. Statistical analyses, including correlation, multiple regression, and receiver operating characteristic (ROC) curve analysis, were performed to evaluate the diagnostic utility and interrelationships of these biomarkers.
Results: Significant biochemical differences were found between individuals with autism and healthy controls. Individuals with autism had notably lower levels of EAAT2, KCC2, NKCC1, VD3, GABA, and GABRA5, especially in the severe group. Altered KCC2/NKCC1 and GABA/glutamate ratios highlighted the imbalance in neurotransmission. The correlation and multiple regression analyses showed significant interconnections between biomarkers. The ROC analysis indicated that EAAT2, KCC2, GABA, and the ratios of KCC2/NKCC1 and GABA/glutamate have high diagnostic potential.
Conclusion: These findings support the hypothesis that GABA and glutamate imbalance is central to the pathophysiology of ASD. Significant disruptions in neurotransmitter signaling and chloride homeostasis, particularly in severe cases, provide insights into the neurobiological mechanisms of ASD. Restoring the GABA–glutamate balance could be an effective therapeutic strategy for ASD, warranting further research into these biochemical pathways for targeted treatments.
1 Introduction
Autism spectrum disorder (ASD) refers to a series of neurodevelopmental disorders characterized by social communication deficits and repetitive behaviors (1). Individuals with autism often have other conditions, such as intellectual disability, mood disorders, epilepsy, and gastrointestinal issues (2–4). The prevalence of autism has risen over time and is more common in male than in female individuals, with an estimated 1 in 100 children worldwide affected (5, 6). In Riyadh, Saudi Arabia, the prevalence is 2.51%, with a male-to-female ratio of 3:1 (7). Numerous studies have shown that genetic, epigenetic, and perinatal environmental factors, as well as their complex interactions, are involved in the neurophysiological mechanisms of autism (8–10). As the etiology of this disorder remains unknown, neither a specific treatment nor reliable diagnostic biomarkers are currently available. Due to the rapidly increasing frequency of ASD, there is an urgent need to discover diagnostic biomarkers (11).
Neurotransmitters are crucial for the development of both the central and peripheral nervous systems, and their dysfunction may be implicated in ASD. Gamma-aminobutyric acid (GABA) and glutamate homeostasis are vital for brain function and are affected in various neuropsychiatric disorders (12–14). Evidence suggests that an imbalance between excitatory (glutamate) and inhibitory (GABA) neurotransmission may be a key pathophysiological mechanism and a potential treatment target in ASD (15–18).
The excitatory/inhibitory (E/I) imbalance hypothesis, proposed by Rubinstein and Merzenich (15), suggests that autism symptoms result from an imbalance between the excitatory (glutamatergic) and inhibitory (GABAergic) brain mechanisms. Studies have identified causes such as overexcitation and overinhibition (15, 19–21). Understanding how E/I imbalance underlies autism symptoms is complex; however, evidence suggests that genetic and environmental risk factors disrupt the balance of excitatory and inhibitory neurotransmission. This disruption could help identify therapeutic targets for autism (19, 22, 23).
GABA is the primary inhibitory neurotransmitter in the brain, and its receptors play a crucial role in modulating neuronal excitability (23–25). Dysfunction in GABA receptors has been implicated in autism (15, 26–28). Studies have shown that individuals with autism often exhibit alterations in GABAergic signaling, which can contribute to the imbalance between excitation and inhibition in the brain (29–33). For instance, reduced activity of the GABA type A (GABAA) receptors has been observed in the brain of individuals with autism, which may lead to increased neuronal excitability and contribute to sensory hypersensitivity and other symptoms associated with autism (34, 35). During and after brain development, neuronal Cl− control plays a role in the dynamic regulation of GABAergic inhibition. This regulation depends mainly on two cation chloride co-transporters, such as sodium potassium chloride co-transporter 1 (NKCC1) and potassium chloride co-transporter 2 (KCC2), to regulate the intracellular chloride concentration, which in turn affects GABAergic signaling (36, 37). NKCC1 accumulates chloride inside neurons, making GABA an excitatory neurotransmitter during early development. In contrast, KCC2 reduces the intracellular chloride, making GABA inhibitory in mature neurons (38). Abnormalities in the expression or function of these transporters can disrupt GABAergic signaling (39). Studies have indicated that the developmental switch from NKCC1 to KCC2 might be delayed or dysfunctional in individuals with autism, resulting in improper inhibitory signaling and contributing to autism-related symptoms (40–43).
Glutamate is the primary excitatory neurotransmitter in the brain, and its transporters, such as excitatory amino acid transporter 1 (EAAT1) and transporter 2 (EAAT2), are responsible for the maintenance of the extracellular glutamate levels (44–46). Dysregulation of glutamate transporters can lead to excitotoxicity and neuronal damage (47–49). Research has shown that individuals with autism often have an altered glutamate transporter activity, which leads to increased extracellular glutamate levels and excitotoxicity. This E/I imbalance is thought to contribute to the neurological and behavioral symptoms observed in autism (22, 50).
Vitamin D plays a crucial role in the development and function of the brain (51). It regulates the expression of several genes involved in neurotransmission, neuroprotection, and neurodevelopment (52). There is growing evidence suggesting that vitamin D deficiency may be linked to the development and severity of autism (53–57). Some studies have indicated that adequate levels of vitamin D could help modulate GABAergic and glutamatergic signaling, potentially alleviating some of the symptoms of autism (58–60).
The purpose of this study was to assess factors related to glutamate excitotoxicity in plasma samples from individuals with autism in comparison to healthy controls. These factors include imbalanced GABA/glutamate levels resulting from dysfunction in the GABA receptors, chloride co-transporters, and glutamate transporters together with vitamin D deficiency. Through this study, it may be possible to ascertain how these biomarkers might influence the severity and the clinical presentation of ASD and how to best target them for treatment.
2 Materials and methods
2.1 Participants
The participants in this study were recruited from the Autism Research and Treatment Centre at King Khalid University Hospital Riyadh, King Saud University, Kingdom of Saudi Arabia. A total of 46 autistic male patients and 26 age- and gender-matched controls were recruited. All participants were screened and evaluated using the Diagnostic and Statistical Manual of Mental Disorders IV (DSM-IV). Scores were calculated to subclassify the participants into mild, moderate, or severe using the Childhood Autism Rating Scale (CARS) and the Social Responsiveness Scale (SRS).
2.2 Behavioral assessment
2.2.1 Childhood Autism Rating Scale
The CARS is a widely utilized screening tool for autism (61). It consists of 15 observational domains designed to distinguish ASD from other developmental conditions in children. The scale rates each domain on a continuum from 1 (indicative of typical behavior) to 4 (indicative of severe abnormality), with higher scores reflecting greater levels of impairment. The domains assessed include interpersonal relations, emotional responses, imitation abilities, use of body and objects, listening skills, fear or anxiety, verbal and nonverbal communication, activity level, intellectual response, adaptability to change, sensory responses (such as visual, taste, smell, and touch), and overall impressions. Total scores can range from 15 to 60, with scores below 30 suggesting a non-autistic range, scores between 30 and 36.5 indicating mild-to-moderate autism, and scores between 37 and 60 reflecting severe autism.
2.2.2 Social Responsiveness Scale
The SRS is a 65-item tool used to quantify the severity of autistic traits (62). It is a structured questionnaire completed by parents or teachers, which takes approximately 15–20 min, based on the child’s behavior observed over the previous 6 months. The SRS employs a standard four-point scale, where responses range from “0” (not true) to “3” (almost always true). The assessment is divided into five subscales: social awareness, social cognition, social communication, social motivation, and autistic mannerisms. An SRS score between 60 and 75 suggests mild-to-moderate social impairment, while a score of 76 or above indicates severe social difficulties.
2.3 Ethical approval
Institutional Review Board and Guidelines of Health Sciences Colleges Research on Human Subjects, King Saud University, College of Medicine, approved this study (no. 22/0122/IRB). An informed consent form was obtained from the parents or legal guardians of all participants prior to participation for approval for processing and publishing data. All experiments were performed in accordance with the relevant guidelines and regulations.
2.4 Blood samples
After an overnight fast, 10 ml blood samples were collected from both groups in test tubes containing sodium heparin as an anticoagulant. The tubes were centrifuged at 3,500 rpm at room temperature for 15 min and the resulting plasma frozen (at −80(C) until further analysis.
2.5 Biochemical analysis
2.5.1 Determination of excitatory amino acid transporter 2
The human EAAT2 ELISA kit (ELK 4324, Wuhan, China) was used in a quantitative sandwich enzyme immunoassay. The microtiter plate was pre-coated with an EAAT2-specific antibody. After addition of the standards or samples and a biotin-conjugated EAAT2 antibody, avidin–horseradish peroxidase (HRP) was added and incubated. The reaction was stopped with sulfuric acid, and the color change was measured at 450 nm. The detection range for EAAT2 was 0.32–20 ng/ml.
2.5.2 Determination of potassium chloride co-transporter 2
The levels of KCC2 were measured using an ELISA kit (ELK Biotechnology, ELK 0484, Wuhan, China). The assay used a sandwich format with a microtiter plate pre-coated with a monoclonal KCC2 antibody. The standards or samples were added to form antigen–antibody complexes. After washing, a biotin-conjugated KCC2 antibody and streptavidin–HRP were added. A substrate solution then produced a colorimetric reaction, with a detection range of 0.32–20 ng/ml.
2.5.3 Determination of sodium potassium chloride co-transporter 1
The human NKCC1 ELISA kit (ELK 0483, Wuhan, China) was used to quantitatively determine the NKCC1 concentrations in plasma using a sandwich enzyme immunoassay. The microtiter plate was pre-coated with an NKCC1-specific antibody. After addition of the standards or samples, a biotin-conjugated NKCC1 antibody and avidin–HRP were added. The reaction was stopped with sulfuric acid, and the color change was measured at 450 nm. The detection range was 0.32–20 ng/ml.
2.5.4 Determination of vitamin D3
The human VD3 ELISA kit (ELK 0811, Wuhan, China) was used in a competitive inhibition enzyme immunoassay. The microtiter plate was pre-coated with vitamin D3 (VD3) protein. After addition of the standards or samples and a biotin-conjugated VD3 antibody, avidin–HRP was added and incubated. The reaction was stopped with sulfuric acid, and the color change was measured at 450 nm. The detection range was 6.25–400 ng/ml.
2.5.5 Determination of gamma-aminobutyric acid
The human GABA ELISA kit (ELK 0753, Wuhan, China) was used in a competitive inhibition enzyme immunoassay. The microtiter plate was pre-coated with GABA protein. After addition of the standards or samples and a biotin-conjugated GABA antibody, avidin–HRP was added and incubated. The reaction was stopped with sulfuric acid, and the color change was measured at 450 nm. The detection range was 31.25–2,000 pg/ml.
2.5.6 Determination of glutamate
The plasma glutamate levels were measured using a double-sandwich ELISA kit (MyBioSource Ltd., San Diego, CA, USA). The pre-coated plate had a glutamate monoclonal antibody, and the detecting antibody was biotin-labeled. After addition of the samples and biotin-labeled antibodies, avidin–peroxidase conjugates were added. The reaction was measured at 450 nm, with a detection range of 0.312–20 nmol/ml.
2.5.7 Determination of gamma-aminobutyric acid receptor subunit alpha-5
The plasma levels of gamma aminobutyric acid type a receptor subunit alpha 5 (GABRA5) were measured using a quantitative sandwich ELISA kit (MyBioSource Ltd., San Diego, CA, USA). The microtiter plate was pre-coated with a GABRA5-specific antibody. The reaction was stopped with sulfuric acid and the color change measured at 450 nm. The detection range was 0.25–8 ng/ml.
2.6 Statistical analysis
Statistical analysis was conducted using SPSS software (SPSS Inc., Chicago, IL, USA). Data were expressed as the mean ± SD. One-way ANOVA and the Kruskal–Wallis test were used to assess group differences, followed by the least significant difference LSD) or the Mann–Whitney test for multiple comparisons. Receiver operating characteristic (ROC) curve analysis evaluated the diagnostic performance of the biochemical parameters, reporting the area under the curve (AUC), the sensitivity, and the specificity. Parameters with high AUC values were considered strong biomarker candidates. Stepwise multiple regression was used to identify significant predictors, and standardized regression coefficients were used to assess their importance. Spearman’s correlation analysis explored the relationships between the biochemical parameters, reporting the correlation coefficients (R) and p-values.
3 Results
Table 1 and Figure 1 show the primary data presented as the mean ± SD and the percent change for the measured variables. The presented data collectively highlighted significant differences between individuals with ASD and healthy controls, as well as among subgroups of ASD (i.e., mild-to-moderate and severe). The parameters EAAT2, KCC2, NKCC1, VD3, GABA, and GABRA5 exhibited markedly lower plasma levels in individuals with autism compared with controls, with the most substantial reductions observed in the severe group. The ratios of KCC2/NKCC1 and GABA/glutamate were also significantly altered in the autistic groups. While the glutamate levels were slightly elevated in individuals with autism, the increase was less pronounced than the changes observed in the other parameters.
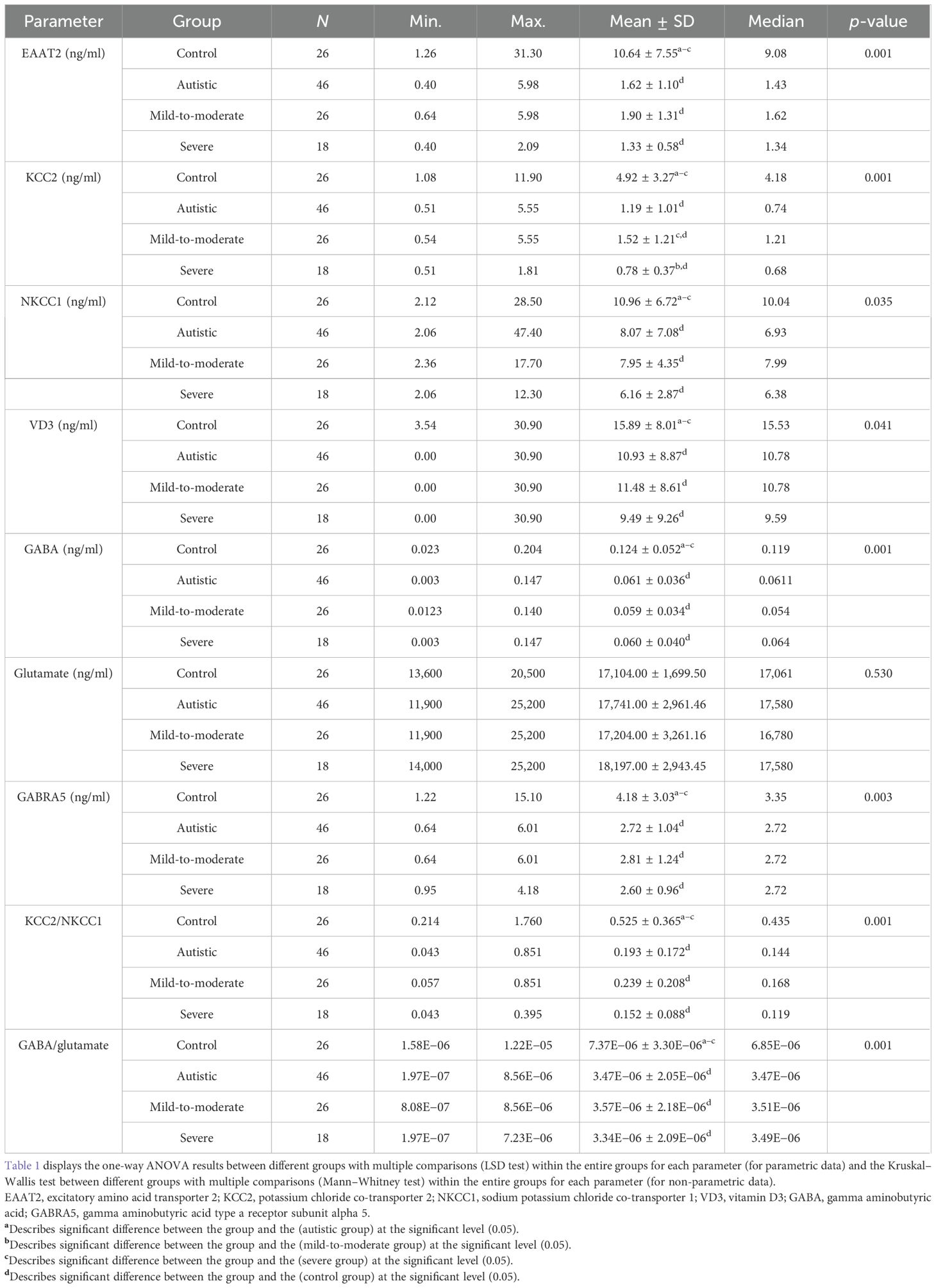
Table 1. Parameters assessed in the plasma of participants with autism in comparison to healthy controls.
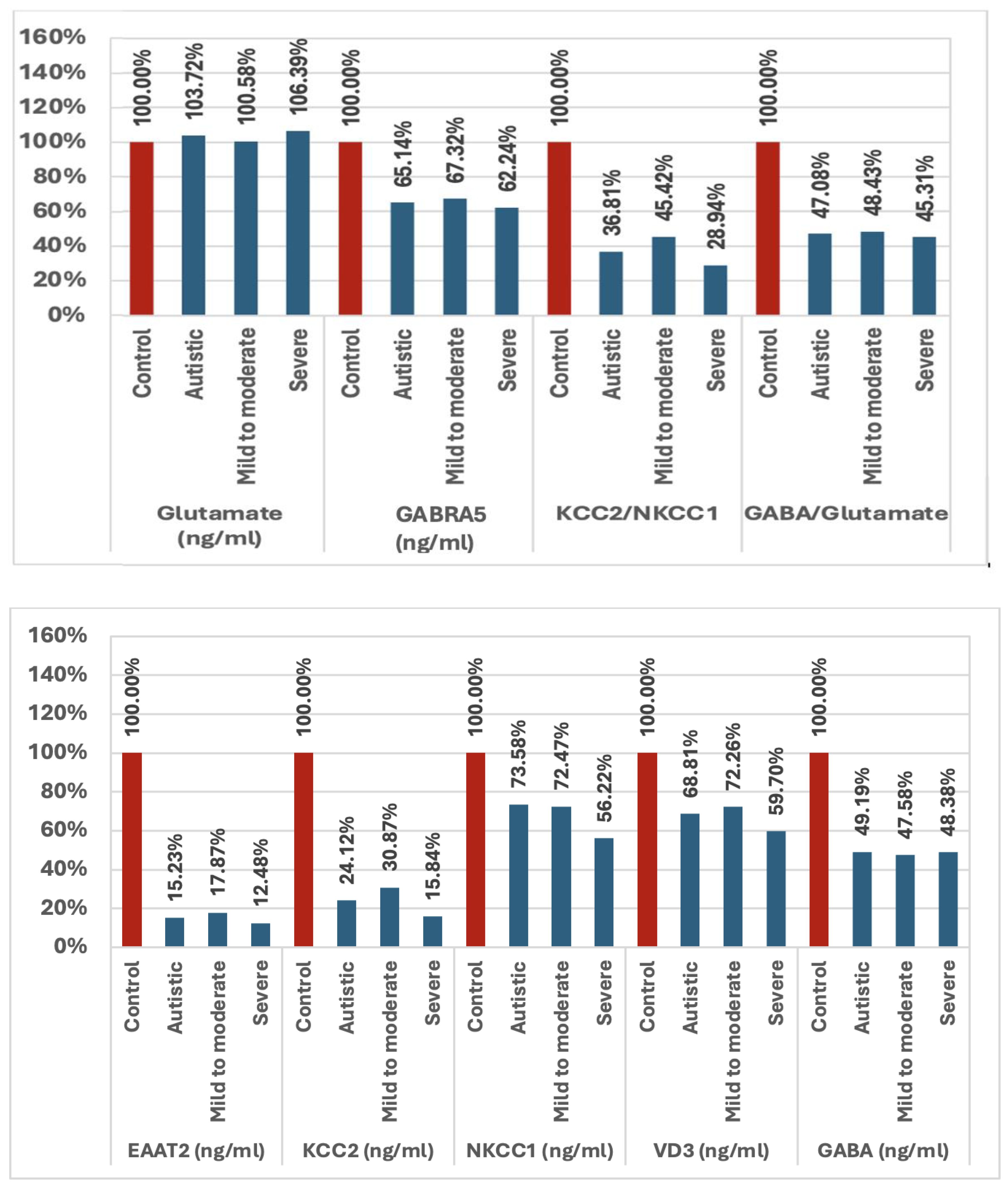
Figure 1. Percentage change in all variables examined in mild-to-moderate and severe participants with autism versus healthy controls. EAAT2, excitatory amino acid transporter 2; KCC2, potassium chloride co-transporter 2; NKCC1, sodium potassium chloride co-transporter 1; VD3, vitamin D3; GABA, gamma aminobutyric acid; GABRA5, gamma aminobutyric acid type A receptor subunit alpha 5.
Table 2 presents the correlations between the different biochemical parameters using Spearman’s correlation, showing correlation coefficients (R) and p-values. EAAT2 showed strong positive correlations with KCC2, NKCC1, GABA, KCC2/NKCC1, and GABA/glutamate. KCC2 was also correlated positively with NKCC1, GABA, GABRA5, KCC2/NKCC1, and GABA/glutamate. NKCC1 showed negative correlations with glutamate, but a positive correlation with GABRA5. VD3 was positively correlated with GABA, GABRA5, KCC2/NKCC1, and GABA/glutamate. GABA had a very strong positive correlation with GABA/glutamate.
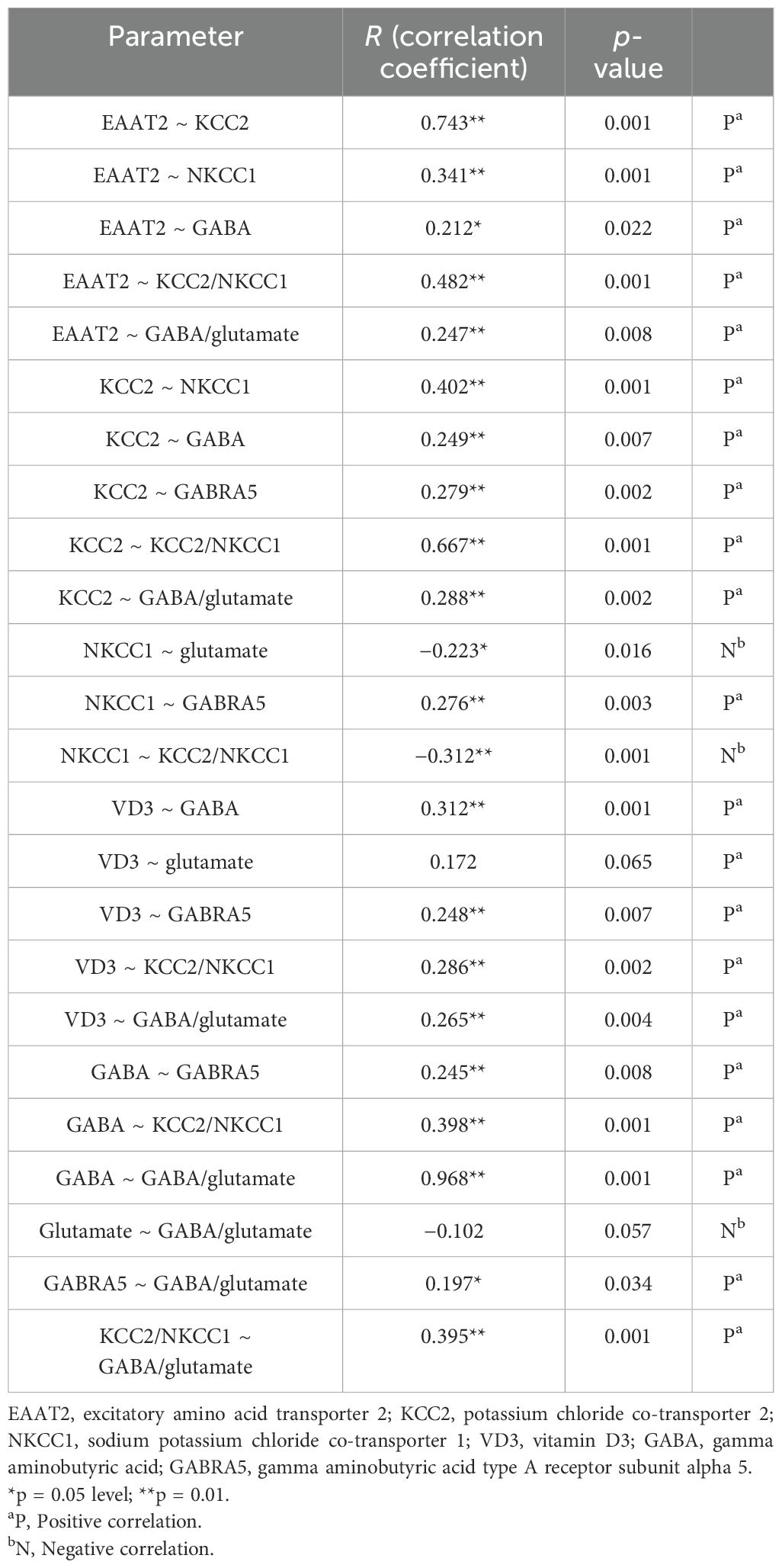
Table 2. Spearman’s correlations among all the measured variables.
The results of the multiple regression analyses highlighted significant predictors for various dependent variables, demonstrating the complex biochemical interactions in ASD. For EAAT2 as the dependent variable (Table 3), KCC2 was identified as a significant predictor, explaining 49.6% of the EAAT2 variance with a coefficient of 1.578. In contrast, KCC2 as the dependent variable (Table 4) was influenced by EAAT2, KCC2/NKCC1, NKCC1, and GABRA5 as independent variables, collectively accounting for up to 79% of its variance. NKCC1 (Table 5) was strongly predicted by KCC2 and the KCC2/NKCC1 ratio, indicating their crucial roles in the regulation of NKCC1. The VD3 levels (Table 6) were significantly associated with GABA, albeit explaining a modest 8.8% of its variance. The GABA/glutamate ratio (Table 7) emerged as a primary positive predictor for the GABA levels, accounting for 98.3% of its variance, while GABRA5 also played a notable role. Similarly, KCC2 (Table 8) was a significant predictor of GABRA5, explaining 6.5% of its variance. The KCC2/NKCC1 ratio (Table 9) was positively influenced by KCC2 and negatively by NKCC1, explaining up to 68.7% of its variance. Finally, the GABA/glutamate ratio and GABRA5 (Table 10) were significant predictors for GABA/glutamate, with the former explaining up to 98.3% of the variance and the latter showing a smaller but a significant negative effect.

Table 3. Multiple regression using the stepwise method for EAAT2 as a dependent variable.
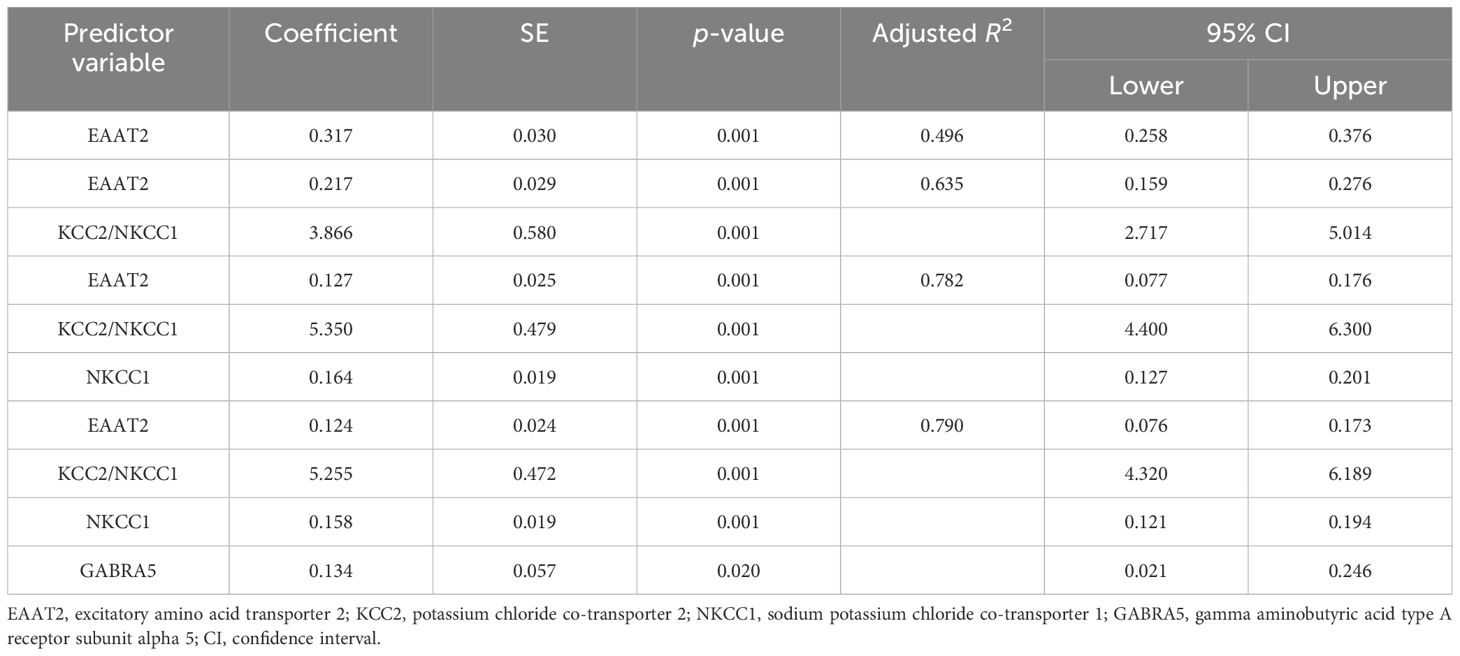
Table 4. Multiple regression using the stepwise method for KCC2 as a dependent variable.
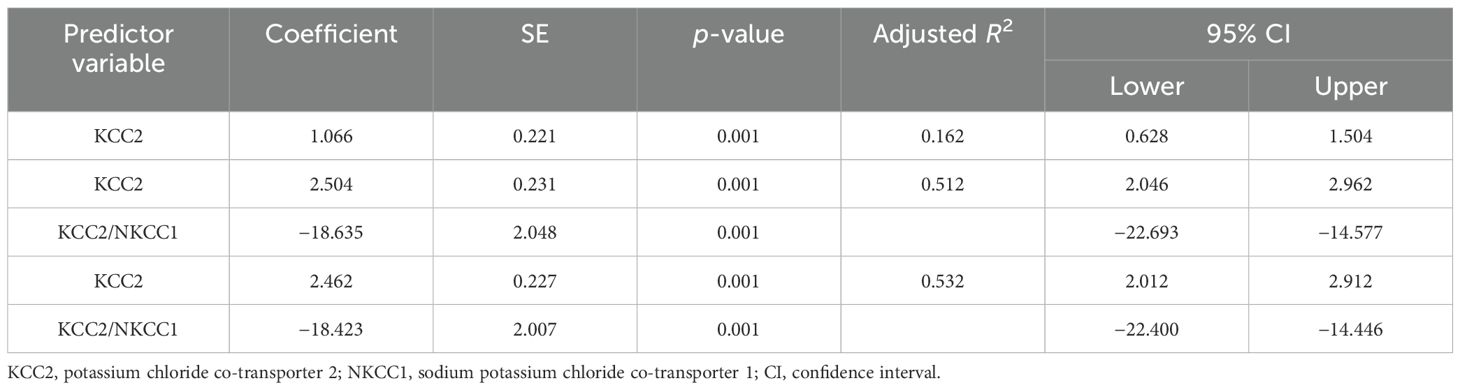
Table 5. Multiple regression using the stepwise method for NKCC1 as a dependent variable.
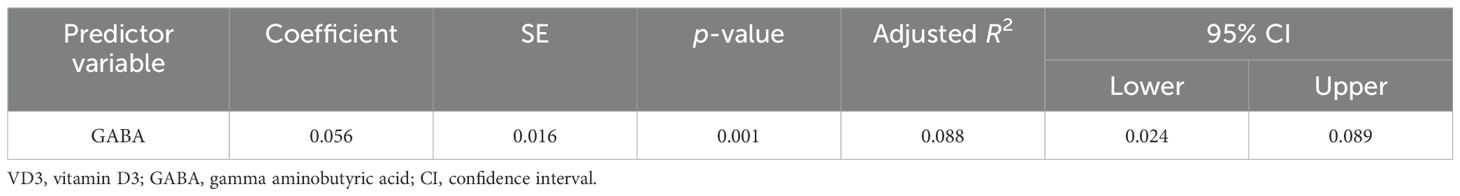
Table 6. Multiple regression using the stepwise method for VD3 as a dependent variable.
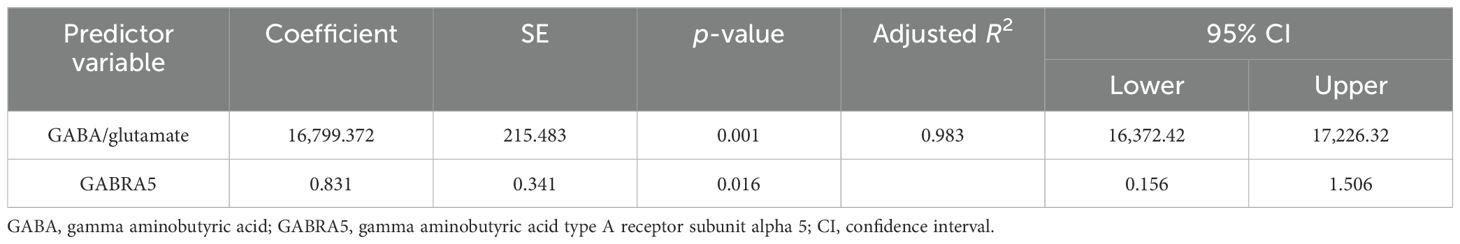
Table 7. Multiple regression using the stepwise method for GABA as a dependent variable.

Table 8. Multiple regression using the stepwise method for GABRA5 as a dependent variable.
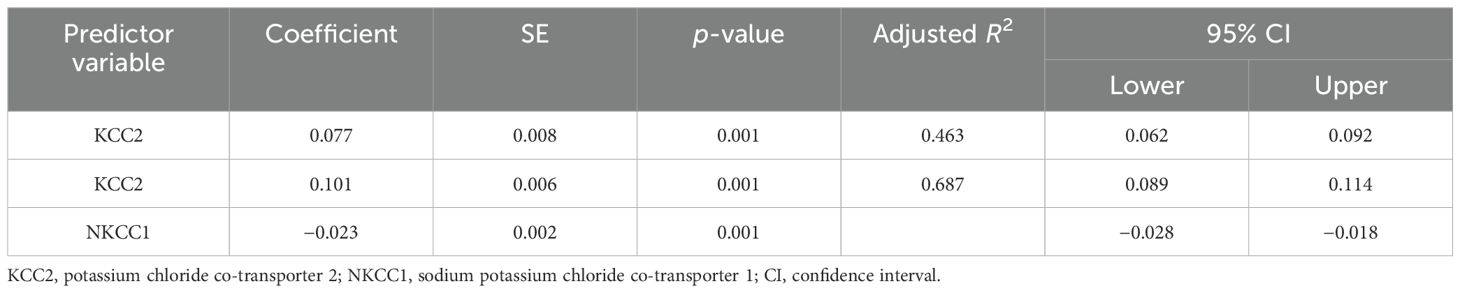
Table 9. Multiple regression using the stepwise method for KCC2/NKCC1 as a dependent variable.
Table 10. Multiple regression using the stepwise method for GABA/glutamate as a dependent variable.
The ROC analysis evaluated the diagnostic accuracy of specific biomarkers in distinguishing individuals with autism from controls and among different autism severity levels. For the autistic group compared with the controls, Table 11 shows that EAAT2 (AUC = 0.952) had a sensitivity of 93.5% and a specificity of 84.6% (p = 0.001), while KCC2 (AUC = 0.931) had a sensitivity of 89.1% and a specificity of 88.5% (p = 0.001), indicating their strong diagnostic potential. The KCC2/NKCC1 ratio (AUC = 0.883) had a sensitivity of 73.9% and a specificity of 100.0% (p = 0.001), while the GABA/glutamate ratio (AUC = 0.836) showed a sensitivity of 84.8% and a specificity of 69.2% (p = 0.001). Another notable biomarker was GABA (AUC = 0.827, sensitivity = 84.8%, specificity = 73.1%, p = 0.001).
Table 11. ROC results for the autistic group according to the control group.
4 Discussion
This study showed significant biochemical differences between individuals with autism and healthy controls, with specific emphasis on the subgroups of autism (mild-to-moderate and severe). These findings help to enhance our understanding of the neurobiological mechanisms of autism, aligning with previous research while offering new insights. Individuals with autism, particularly those in the severe group, exhibited reduced levels of EAAT2, KCC2, NKCC1, VD3, GABA, and GABRA5. These disruptions in neurotransmitter signaling and chloride homeostasis likely contribute to the E/I imbalance observed in ASD. The modulation of glutamate homeostasis is most recently being investigated and has been highlighted as a potential critical target for the treatment of neurodevelopmental disorders, among which is ASD (63).
This study showed significantly lower levels of EAAT2 in individuals with autism, especially in the severe group. Although the glutamate levels were slightly elevated, the change was less pronounced. This aligns with research indicating that EAAT2 dysregulation leads to extracellular glutamate accumulation and excitotoxicity. Rothstein et al. (64) found that beta-lactam antibiotics upregulated EAAT2, which reduced the toxicity of glutamate, suggesting their potential therapeutic benefits for ASD. Moreover, EAAT2 modulation, as a therapeutic approach, alleviated the symptoms of glutamate dysregulation in mouse models of neurodegeneration (65). These findings underscore the crucial function of EAAT2 in autism and suggest that the regulation of its expression could aid in managing the neurobiological problems in ASD by resolving the E/I imbalance.
This study found significantly lower levels of KCC2 and NKCC1 in individuals with autism, particularly in the severe subgroup. KCC2 maintains low intracellular chloride for GABAergic inhibition, while NKCC1 accumulates chloride, making GABA excitatory. The dysregulation of these transporters likely contributes to the E/I imbalance in ASD. Duarte et al. (66) showed that altered KCC2 and NKCC1 expression affected neuronal Cl− gradients, impacting GABAergic inhibition. Adjustment of these transporters can reverse GABA polarity, exacerbating the symptoms of ASD. Clinical trials using the NKCC1 inhibitor bumetanide improved autistic behaviors, supporting the therapeutic potential of targeting these transporters (67). These findings suggest that the modulation of KCC2 and NKCC1 could offer a new therapeutic approach for ASD.
The levels of VD3 were significantly lower in individuals with autism, supporting previous research linking vitamin D deficiency to an increased risk of ASD (68, 69). This study highlighted the role of VD3 in the neurodevelopment and neurotransmitter systems, suggesting that supplementation could benefit the management of autism. Guo et al. (53) found that VD3 deficiency worsened the neuroinflammatory responses in autism. Two studies demonstrated the neuroprotective effects of VD3, showing behavioral improvements with supplementation (59, 70). El-Ansary (22) reported that vitamin D deficiency decreased the glutamic acid decarboxylase levels, disrupting the glutamate–glutamine–GABA cycle associated with autism, potentially reducing glutamate excitotoxicity. These findings suggest a promising approach for therapeutic interventions targeting VD3 deficiency in ASD.
This study found significant reductions in GABA and its receptor GABRA5 in individuals with autism, especially in the severe subgroup, indicating that disruptions in GABAergic signaling are critical for maintaining neuronal balance. This aligns with previous studies reporting decreased GABAergic activity in autism (29, 71). Fatemi et al. (34) found similar reductions in GABAergic markers, highlighting consistent GABAergic dysfunction in autism. Lower GABRA5 levels suggest a compromised neurotransmission, contributing to the E/I imbalance in ASD. Previous studies (72, 73) showed that the GABAA receptor subtypes, particularly the α5 subunit, are associated with autistic behaviors, supporting our findings on disrupted GABA signaling.
The ratios of KCC2/NKCC1 and GABA/glutamate were significantly altered in the autistic groups, especially in the severe subgroup, supporting the E/I imbalance hypothesis by Rubenstein and Merzenich (15). Several studies further support these findings, linking abnormalities in the GABA and glutamate levels to the sensory and behavioral issues in ASD (19, 32, 74). Altered KCC2/NKCC1 ratios indicate a disrupted chloride homeostasis, impairing GABAergic neurotransmission and contributing to ASD symptoms. Duarte et al. (66) suggest that targeting this imbalance could offer new therapeutic perspectives.
The results of the correlation analysis (Table 2) revealed significant interconnections between the biochemical parameters in individuals with autism. A strong positive correlation between EAAT2 and the KCC2/NKCC1 ratio suggests efficient glutamate clearance as EAAT2 is linked to neuronal E/I balance, mediated by KCC2 and NKCC1. This supports the findings by Duarte et al. (66) indicating that alterations in the glutamate transporter activity are linked to shifts in the E/I balance in the brain. The positive correlation between KCC2 and the GABA/glutamate ratio indicates that higher levels of KCC2 enhance GABAergic inhibition, aligning with the research by Sernagor et al. (75) suggesting that boosting the function of KCC2 can correct the E/I imbalance seen in neurodevelopmental disorders. GABA and GABRA5 also showed a positive correlation, emphasizing the regulatory mechanism between neurotransmitters and receptors, supported by Robertson et al. (32). This relationship is pivotal in stabilizing neural circuits and is often disrupted in ASD, which leads to behavioral and cognitive impairments. The significant correlations between VD3 and both GABA and GABRA5 suggest the role of VD3 in modulating GABAergic signaling, as proposed by Cannell (68). These correlations highlight potential therapeutic targets, suggesting that enhancing the function of EAAT2 or KCC2 and optimizing the levels of VD3 could mitigate the core ASD symptoms.
The ROC analysis results highlighted the diagnostic potential of the biochemical markers for ASD. EAAT2 and KCC2 showed excellent diagnostic accuracy. Green et al. (76) emphasized the role of EAAT2 in modulating synaptic glutamate and preventing excitotoxicity, which are relevant to ASD. Moreover, KCC2 is important in the maintenance of GABAergic inhibition, suggesting its potential as a therapeutic target (67). NKCC1 and VD3 had moderate AUC values, indicating their lower diagnostic reliability, but still supported as markers for ASD (36, 77). These findings suggest that EAAT2 and KCC2 are effective diagnostic markers for ASD and that targeting these markers could help treat the underlying neurochemical imbalances.
The results of the multiple regression analyses highlighted the significant relationships between these markers. KCC2 strongly predicted the EAAT2 levels, suggesting its crucial role in modulating EAAT2 expression. Two studies support the importance of KCC2 in chloride transport and synaptic inhibition, underscoring its significance in neurological disorders (78, 79). Similarly, the GABA/glutamate ratio and GABRA5 significantly predicted the GABA levels, corroborating the findings by previous studies that highlighted the complex regulatory interactions in ASD (15, 50, 73).
In the current study, it is worth noting that severe individuals had considerably more observed alterations in the variables that were measured than mild-to-moderate participants with autism (Table 1, Figure 1), showing that an altered GABA inhibition is a significant contributing factor to the severity of ASD. Several studies have revealed that hypo-inhibition and/or hyper-excitation, which boosts the E/I ratio, are the primary reasons for the social processing deficiencies, sensory sensitivity, and elevated anxiety reactivity in social contexts in autism, which are important characteristics of the disorder (80, 81). GABAergic inhibition is essential for social interactions as it filters information, regulates inhibition, and processes rewards. Moreover, pharmacologically, different parts of the brain and circuits that govern repetitive behaviors as another clinical presentation of ASD react differently to local and systemic glutamatergic inhibitors and GABAergic medications (82).
5 Conclusions
This study highlights the importance of biochemical parameters such as EAAT2, KCC2, NKCC1, VD3, GABA, and GABRA5 in understanding ASD. Significant disruptions in neurotransmitter signaling and chloride homeostasis, especially in severe ASD, provide insights into its neurobiological mechanisms. These findings support previous research and reveal novel aspects for future studies and therapies. The results of the multiple regression analyses underscore the potential of these parameters as therapeutic targets and biomarkers for better ASD diagnosis and intervention. Further research is needed to explore these biochemical pathways for more effective ASD treatments.
5.1 Limitations and future research
The small sample size, together with the absence of female participants, could be considered as selection bias and a limitation of the current study. Future research could expand the sample size and include more diverse people to confirm the generalizability of the findings. We will endeavor to implement the reviewers’ helpful recommendation of converting data from one format to another and of incorporating autism severity scores into our future investigations. Conducting longitudinal research to determine how these markers change with age or therapy regimens in people with autism is highly recommended. Further research will be important to shed light on the implications of age- and sex-related GABAergic alterations in ASD phenotypic conditions in order to design improved and more effective personalized treatments that enhance the inhibitory effects of GABA.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Institutional Review Board and Guidelines of Health Sciences Colleges Research on Human Subjects, King Saud University, College of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
AA: Formal analysis, Investigation, Methodology, Writing – original draft. AB: Conceptualization, Supervision, Validation, Visualization, Writing – review & editing. MA: Data curation, Investigation, Project administration, Resources, Writing – review & editing. LA-A: Conceptualization, Data curation, Funding acquisition, Writing – review & editing. ASJA: Investigation, Methodology, Writing – review & editing. AE-A: Conceptualization, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article.
Acknowledgments
This project was funded by the National Plan for Science Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology; Kingdom of Saudi Arabia (award number: 08-MED 510-02).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Happé F. Autism: Cognitive deficit or cognitive style? Trends Cognit Sci. (1999) 3:216–22. doi: 10.1016/s1364-6613(99)01318-2
2. Richard AE, Scheffer IE, Wilson SJ. Features of the broader autism phenotype in people with epilepsy support shared mechanisms between epilepsy and autism spectrum disorder. Neurosci Biobehav Rev. (2017) 75:203–33. doi: 10.1016/j.neubiorev.2016.12.036
3. Tye C, Runicles AK, Whitehouse AJO, Alvares GA. Characterizing the interplay between autism spectrum disorder and comorbid medical conditions: An integrative review. Front Psychiatry. (2019) 9:751. doi: 10.3389/fpsyt.2018.00751
4. Al-Beltagi M. Autism medical comorbidities. World J Clin Pediatr. (2021) 10:15–28. doi: 10.5409/wjcp.v10.i3.15
5. Zeidan J, Fombonne E, Scorah J, Ibrahim A, Durkin MS, Saxena S, et al. Global prevalence of autism: A systematic review update. Autism Res. (2022) 15:778–90. doi: 10.1002/aur.2696
6. Kriegel G, Paul S, Leonard KH, Sandor P. Prevalence of autism spectrum disorder (ASD) in inpatient adolescent psychiatric population. J Autism Dev Disord. (2025) 55(3):1138–45. doi: 10.1007/s10803-023-05923-w
7. AlBatti TH, Alsaghan LB, Alsharif MF, Alharbi JS, BinOmair AI, Alghurair HA, et al. Prevalence of autism spectrum disorder among Saudi children between 2 and 4 years old in Riyadh. Asian J Psychiatr. (2022) 71:103054. doi: 10.1016/j.ajp.2022.103054
8. Belmonte MK, Cook EH Jr, Anderson GM, Rubenstein JL, Greenough WT, Beckel-Mitchener A, et al. Autism as a disorder of neural information processing: Directions for research and targets for therapy. Mol Psychiatry. (2004) 9:646–63. doi: 10.1038/sj.mp.4001499
9. Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. (2011) 68:1095–102. doi: 10.1001/archgenpsychiatry.2011.76
10. Cattane N, Richetto J, Cattaneo A. Prenatal exposure to environmental insults and enhanced risk of developing schizophrenia and autism spectrum disorder: Focus on biological pathways and epigenetic mechanisms. Neurosci Biobehav Rev. (2020) 117:253–78. doi: 10.1016/j.neubiorev.2018.07.001
11. Shen L, Liu X, Zhang H, Lin J, Feng C, Iqbal J. Biomarkers in autism spectrum disorders: Current progress. Clin Chim Acta. (2020) 502:41–54. doi: 10.1016/j.cca.2019.12.009
12. Schür RR, Draisma LW, Wijnen JP, Boks MP, Koevoets MG, Joëls M, et al. Brain GABA levels across psychiatric disorders: A systematic literature review and meta-analysis of (1)H-MRS studies. Hum Brain Mapp. (2016) 37:3337–52. doi: 10.1002/hbm.23244
13. Chen YP, Wang C, Xu JP. Chronic unpredictable mild stress induced depression-like behaviours and glutamate-glutamine cycling dysfunctions in both blood and brain of mice. Pharm Biol. (2019) 57:280–6. doi: 10.1080/13880209.2019.1598445
14. Wenneberg C, Glenthøj BY, Hjorthøj C, Buchardt Zingenberg FJ, Glenthøj LB, et al. Cerebral glutamate and GABA levels in high-risk of psychosis states: A focused review and meta-analysis of 1H-MRS studies. Schizophr Res. (2020) 215:38–48. doi: 10.1016/j.schres.2019.10.050
15. Rubenstein JL, Merzenich MM. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. (2003) 2:255–67. doi: 10.1034/j.1601-183x.2003.00037.x
16. Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea D J, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. (2011) 477:171–8. doi: 10.1038/nature10360
17. Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. GABA system dysfunction in autism and related disorders: From synapse to symptoms. Neurosci Biobehav Rev. (2012) 36:2044–55. doi: 10.1016/j.neubiorev.2012.07.005
18. Johnson AJ, Shankland E, Richards T, Relationships between GABA. glutamate, and GABA/glutamate and social and olfactory processing in children with autism spectrum disorder. Psychiatry Res Neuroimaging. (2023) 336:111745. doi: 10.1016/j.pscychresns.2023.111745
19. Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron. (2015) 87:684–98. doi: 10.1016/j.neuron.2015.07.033
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
20. Foss-Feig JH, Adkinson BD, Ji JL, Yang G, Srihari VH, McPartland JC, et al. Searching for cross-diagnostic convergence: Neural mechanisms governing excitation and inhibition balance in schizophrenia and autism spectrum disorders. Biol Psychiatry. (2017) 81:848–61. doi: 10.1016/j.biopsych.2017.03.005
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
21. Horder J, Petrinovic MM, Mendez MA, Bruns A, Takumi T, Spooren W, et al. Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models. Transl Psychiatry. (2018) 8:106. doi: 10.1038/s41398-018-0155-1
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
22. El-Ansary-A. GABA. and glutamate imbalance in autism and their reversal as novel hypothesis for effective treatment strategy. Autism Dev Disord. (2020) 18:46–63. doi: 10.17759/autdd.2020180306
23. McCormick DA. GABA as an inhibitory neurotransmitter in human cerebral cortex. J Neurophysiol. (1989) 62:1018–27. doi: 10.1152/jn.1989.62.5.1018
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
24. Möhler H. GABA(A) receptor diversity and pharmacology. Cell Tissue Res. (2006) 326:505–16. doi: 10.1007/s00441-006-0284-3
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
25. Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: A pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. (2007) 87:1215–84. doi: 10.1152/physrev.00017.2006
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
26. Blatt GJ, Fatemi SH. Alterations in GABAergic biomarkers in the autism brain: Research findings and clinical implications. Anat Rec. (2011) 294:1646–52. doi: 10.1002/ar.21252
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
27. Zikopoulos B, Barbas H. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front Hum Neurosci. (2013) 7:609. doi: 10.3389/fnhum.2013.00609
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
28. Zhao H, Mao X, Zhu C, Zou X, Peng F, Yang W, et al. GABAergic system dysfunction in autism spectrum disorders. Front Cell Dev Biol. (2022) 9:781327. doi: 10.3389/fcell.2021.781327
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
29. Pizzarelli R, Cherubini E. Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. (2011) 2011:297153. doi: 10.1155/2011/297153
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
30. Harada M, Taki MM, Nose A, Kubo H, Mori K, Nishitani H, et al. Non-invasive evaluation of the GABAergic/glutamatergic system in autistic patients observed by MEGA-editing proton MR spectroscopy using a clinical 3 tesla instrument. J Autism Dev Disord. (2011) 41:447–54. doi: 10.1007/s10803-010-1065-0
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
31. Cellot G, Cherubini E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front Pediatr. (2014) 2:70. doi: 10.3389/fped.2014.00070
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
32. Robertson CE, Ratai EM, Kanwisher N. Reduced GABAergic action in the autistic brain. Curr Biol. (2016) 26:80–5. doi: 10.1016/j.cub.2015.11.019
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
33. Hollestein V, Poelmans G, Forde NJ, Beckmann CF, Ecker C, Mann C, et al. Excitatory/inhibitory imbalance in autism: The role of glutamate and GABA gene-sets in symptoms and cortical brain structure. Transl Psychiatry. (2023) 13:18. doi: 10.1038/s41398-023-02317-5
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
34. Fatemi SH, Reutiman TJ, Folsom TD, Thuras PD. GABA(A) receptor downregulation in brains of subjects with autism. J Autism Dev Disord. (2009) 39:223–30. doi: 10.1007/s10803-008-0646-7
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
35. Oblak AL, Gibbs TT, Blatt GJ. Reduced GABAA receptors and benzodiazepine binding sites in the posterior cingulate cortex and fusiform gyrus in autism. Brain Res. (2011) 1380:218–28. doi: 10.1016/j.brainres.2010.09.021
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
36. Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, et al. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol. (2008) 4:490–503. doi: 10.1038/ncpneuro0883
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
37. Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. (2009) 61:820–38. doi: 10.1016/j.neuron.2009.03.003
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
38. Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci. (2014) 15:637–54. doi: 10.1038/nrn3819
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
39. Lam P, Newland J, Faull RLM, Kwakowsky A. Cation-chloride cotransporters KCC2 and NKCC1 as therapeutic targets in neurological and neuropsychiatric disorders. Molecules. (2023) 28:1344. doi: 10.3390/molecules28031344
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
40. Lemonnier E, Ben-Ari Y. The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr. (2010) 99:1885–8. doi: 10.1111/j.1651-2227.2010.01933.x
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
41. Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. (2014) 343:675–9. doi: 10.1126/science.1247190
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
42. Amin H, Marinaro F, De Pietri Tonelli D, Berdondini L. Developmental excitatory-to-inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: A potential target for clinical therapeutics. Sci Rep. (2017) 7:15752. doi: 10.1038/s41598-017-15793-9
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
43. Abruzzo PM, Panisi C, Marini M. The alteration of chloride homeostasis/GABAergic signaling in brain disorders: Could oxidative stress play a role? Antioxidants (Basel). (2021) 10:1316. doi: 10.3390/antiox10081316
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
44. Anderson CM, Swanson RA. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia. (2000) 32:1–14. doi: 10.1002/(SICI)1098-1136(200010)32:1<1:AID-GLIA10>3.0.CO;2-C
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
45. Danbolt NC. Glutamate uptake. Prog Neurobiol. (2001) 65:1–105. doi: 10.1016/s0301-0082(00)00067-8
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
46. Fontana AC. Current approaches to enhance glutamate transporter function and expression. J Neurochem. (2015) 134:982–1007. doi: 10.1111/jnc.13200
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
47. Foran E, Trotti D. Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid Redox Signal. (2009) 11:1587–602. doi: 10.1089/ars.2009.2444
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
48. Zhou X, Liang J, Wang J, Fei Z, Qin G, Zhang D, et al. Up-regulation of astrocyte excitatory amino acid transporter 2 alleviates central sensitization in a rat model of chronic migraine. J Neurochem. (2020) 155:370–89. doi: 10.1111/jnc.14944
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
49. Reeb LK, Gill SK, Temmermand R, Fontana CK. Glutamate transporters in health and disease. IntechOpen. (2024). doi: 10.5772/intechopen.1005544
50. Montanari M, Martella G, Bonsi P, Meringolo M. Autism spectrum disorder: Focus on glutamatergic neurotransmission. Int J Mol Sci. (2022) 23:3861. doi: 10.3390/ijms23073861
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
51. Cui X, Gooch H, Groves NJ, Sah P, Burne TH, et al. Vitamin D and the brain: Key questions for future research. J Steroid Biochem Mol Biol. (2015) 148:305–9. doi: 10.1016/j.jsbmb.2014.11.004
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
52. Moretti R, Morelli ME, Caruso P. Vitamin D in neurological diseases: A rationale for a pathogenic impact. Int J Mol Sci. (2018) 19:2245. doi: 10.3390/ijms19082245
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
53. Guo M, Zhu J, Yang T, Lai X, Lei Y, Chen J, et al. Vitamin A and vitamin D deficiencies exacerbate symptoms in children with autism spectrum disorders. Nutr Neurosci. (2019) 22:637–47. doi: 10.1080/1028415X.2017.1423268
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
54. Ali A, Vasileva S, Langguth M, Alexander S, Cui X, Whitehouse A, et al. Developmental vitamin D deficiency produces behavioral phenotypes of relevance to autism in an animal model. Nutrients. (2019) 11:1187. doi: 10.3390/nu11051187
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
55. Petruzzelli MG, Marzulli L, Margari F, De Giacomo A, Gabellone A, Giannico OV, et al. Vitamin D deficiency in autism spectrum disorder: A cross-sectional study. Dis Markers. (2020) 2020:9292560. doi: 10.1155/2020/9292560
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
56. García-Serna AM, Morales E. Neurodevelopmental effects of prenatal vitamin D in humans: Systematic review and meta-analysis. Mol Psychiatry. (2020) 25:2468–81. doi: 10.1038/s41380-019-0357-9
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
57. Principi N, Esposito S. Vitamin D deficiency during pregnancy and autism spectrum disorders development. Front Psychiatry. (2020) 10:987. doi: 10.3389/fpsyt.2019.00987
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
58. Patrick RP, Ames BN. Vitamin D hormone regulates serotonin synthesis. Part 1: relevance for autism. FASEB J. (2014) 28:2398–413. doi: 10.1096/fj.13-246546
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
59. Song L, Luo X, Jiang Q, Chen Z, Zhou L, Wang D, et al. Vitamin D supplementation is beneficial for children with autism spectrum disorder: A meta-analysis. Clin Psychopharmacol Neurosci. (2020) 18:203–13. doi: 10.9758/cpn.2020.18.2.203
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
60. Kasatkina LA, Tarasenko AS, Krupko OO, Kuchmerovska TM, Lisakovska OO, Trikash IO. Vitamin D deficiency induces the excitation/inhibition brain imbalance and the proinflammatory shift. Int J Biochem Cell Biol. (2020) 119:105665. doi: 10.1016/j.biocel.2019.105665
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
61. Moulton E, Bradbury K, Barton M, Fein D. Factor analysis of the Childhood Autism Rating Scale in a sample of two-year-olds with an autism spectrum disorder. J Autism Dev Disord. (2016) 49:2733–46. doi: 10.1007/s10803-016-2810-6
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
62. Schanding GT, Nowell KP, Goin-Kochel RP. Utility of the Social Communication Questionnaire-Current and Social Responsiveness Scale as teacher-report screening tools for autism spectrum disorders. J Autism Dev Disord. (2011) 42:1705–16. doi: 10.1007/s10803-011-1409-7
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
63. Nicosia N, Giovenzana M, Misztak P, Mingardi J, Musazzi L. Glutamate-mediated excitotoxicity in the pathogenesis and treatment of neurodevelopmental and adult mental disorders. Int J Mol Sci. (2024) 25:6521. doi: 10.3390/ijms25126521
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
64. Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. (2005) 433:73–7. doi: 10.1038/nature03180
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
65. Aida T, Yoshida J, Nomura M, Tanimura A, Iino Y, Soma M, et al. Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsychopharmacology. (2015) 40:1569–79. doi: 10.1038/npp.2015.26
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
66. Duarte ST, Armstrong J, Roche A, Ortez C, Pérez A, O'Callaghan delM M, et al. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PloS One. (2013) 8(7):e68851. doi: 10.1371/journal.pone.0068851
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
67. Lemonnier E, Degrez C, Phelep M, Tyzio R, Josse F, Grandgeorge M, et al. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl Psychiatry. (2012) 2(12):e202. doi: 10.1038/tp.2012.124
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
68. Cannell JJ. Autism and vitamin D. Med Hypotheses. (2008) 70:750–9. doi: 10.1016/j.mehy.2007.08.016
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
69. Fernell E, Bejerot S, Westerlund J, Miniscalco C, Simila H, Eyles D, et al. autism spectrum disorder and low vitamin D at birth: A sibling control study. Mol Autism. (2015) 6:3. doi: 10.1186/2040-2392-6-3
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
70. Saad K, Abdel-Rahman AA, Elserogy YM, Al-Atram AA, Cannell JJ, Bjørklund G, et al. Vitamin D status in autism spectrum disorders and the efficacy of vitamin D supplementation in autistic children. Nutr Neurosci. (2016) 19:346–51. doi: 10.1179/1476830515Y.0000000019
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
71. Fatemi SH, Halt AR, Stary JM, Kanodia R, Schulz SC, Realmuto GR. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry. (2002) 52:805–10. doi: 10.1016/s0006-3223(02)01430-0
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
72. Mendez MA, Horder J, Myers J, Coghlan S, Stokes P, Erritzoe D, et al. The brain GABA-benzodiazepine receptor alpha-5 subtype in autism spectrum disorder: A pilot [(11)C] Ro15-4513 positron emission tomography study. Neuropharmacology. (2013) 68:195–201. doi: 10.1016/j.neuropharm.2012.04.008
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
73. Zurek AA, Kemp SW, Aga Z, Walker S, Milenkovic M, Ramsey AJ, et al. [amp]]alpha;5GABAA receptor deficiency causes autism-like behaviors. Ann Clin Transl Neurol. (2016) 3:392–8. doi: 10.1002/acn3.303
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
74. El-Ansary A, Al-Ayadhi L. GABAergic/glutamatergic imbalance relative to excessive neuroinflammation in autism spectrum disorders. J Neuroinflammation. (2014) 11:189. doi: 10.1186/s12974-014-0189-0
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
75. Sernagor E, Young C, Eglen SJ. Developmental modulation of retinal wave dynamics: Shedding light on the GABA saga. J Neurosci. (2003) 23:7621–9. doi: 10.1523/JNEUROSCI.23-20-07621.2003
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
76. Green JL, Dos Santos WF, Fontana ACK. Role of glutamate excitotoxicity and glutamate transporter EAAT2 in epilepsy: Opportunities for novel therapeutics development. Biochem Pharmacol. (2021) 193:114786. doi: 10.1016/j.bcp.2021.114786
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
77. Azhari SA. 25-hydroxyvitamin D level in autism spectrum disorders and the efficacy of vitamin D supplementation in Saudi autistic children. J Pharm Res Int. (2023) 35:40–7. doi: 10.9734/jpri/2023/v35i107353
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
78. Takahashi K, Foster JB, Lin CL. Glutamate transporter EAAT2: Regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol Life Sci. (2015) 72:3489–506. doi: 10.1007/s00018-015-1937-8
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
79. Bilchak JN, Yeakle K, Caron G, Malloy D, Côté MP. Enhancing KCC2 activity decreases hyperreflexia and spasticity after chronic spinal cord injury. Exp Neurol. (2021) 338:113605. doi: 10.1016/j.expneurol.2021.113605
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
80. Courchesne E, Gazestani VH, Lewis NE. Prenatal origins of ASD: the when, what, and how of ASD development. Trends Neurosci. (2020) 43 :326–42. doi: 10.1016/j.tins.2020.03.005
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
81. Parrella N-F, Hill AT, Dipnall LM, Loke YJ, Enticott PG, Ford TC. Inhibitory dysfunction and social processing difficulties in autism: A comprehensive narrative review. J Psychiatr Res. (2024) 169:113–25. doi: 10.1016/j.jpsychires.2023.11.014
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
82. Gandhi T, Lee CC. Neural mechanisms underlying repetitive behaviors in rodent models of autism spectrum disorders. Front Cell Neurosci. (2021) 14:592710. doi: 10.3389/fncel.2020.592710
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
Keywords: autism spectrum disorder (ASD), gamma-aminobutyric acid (GABA), glutamate, KCC2, NKCC1, EAAT2, vitamin D3 (VD3), GABRA5
Citation: Alabdali A, Ben Bacha A, Alonazi M, Al-Ayadhi LY, Alanazi ASJ and El‐Ansary A (2025) Comparative evaluation of certain biomarkers emphasizing abnormal GABA inhibitory effect and glutamate excitotoxicity in autism spectrum disorders. Front. Psychiatry 16:1562631. doi: 10.3389/fpsyt.2025.1562631
Received: 20 January 2025; Accepted: 12 March 2025;
Published: 22 April 2025.
Edited by:
Wen-Xiong Chen, Guangzhou Medical University, ChinaReviewed by:
Dejan Budimirovic, Johns Hopkins University, United StatesXian Liu, Guangzhou Medical University, China
Copyright © 2025 Alabdali, Ben Bacha, Alonazi, Al-Ayadhi, Alanazi and El‐Ansary. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Afaf El‐Ansary, YWZhZmtlbGFuc2FyeUBnbWFpbC5jb20=; Abir Ben Bacha, YWFsZ2hhbm91Y2hpQGtzdS5lZHUuc2E=