 Xuan Wang
Xuan Wang Di Cao2†
Di Cao2†
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Psychiatry , 18 February 2025
Sec. Molecular Psychiatry
Volume 16 - 2025 | https://doi.org/10.3389/fpsyt.2025.1539596
Background: Major depressive disorder (MDD) is highly heterogeneous, which provides a significant challenge in the management of this disorder. However, the pathogenesis of major depressive disorder is not fully understood. Studies have shown that depression is highly correlated with gut flora. The objective of this study was to explore the potential of microbial biomarkers in the diagnosis of major depressive disorder.
Methods: In this study, we used a metagenomic approach to analyze the composition and differences of gut bacterial communities in 36 patients with major depressive disorder and 36 healthy individuals. We then applied a Support Vector Machine Recursive Feature Elimination (SVM-RFE) machine learning model to find potential microbial markers.
Results: Our results showed that the alpha diversity of the intestinal flora did not differ significantly in major depressive disorder compared to healthy populations. However, the beta diversity was significantly altered. Machine learning identified 8 MDD-specific bacterial biomarkers, with Alistipes, Dysosmobacter, Actinomyces, Ruthenibacterium, and Thomasclavelia being significantly enriched, while Faecalibacterium, Pseudobutyrivibrio, and Roseburia were significantly reduced, demonstrating superior diagnostic accuracy (area under the curve, AUC = 0.919). In addition, the gut bacteria performed satisfactorily in the validation cohort with an AUC of 0.800 (95% CI: 0.6334-0.9143).
Conclusion: This study reveals the complex relationship between gut microbiota and major depressive disorder and provides a scientific basis for the development of a microbiota-based diagnostic tool for depression.
Major depressive disorder (MDD) is a gradually debilitating global mental illness, more than 300 million people of all ages are suffering from depression, the prevalence of depression in China is 4.2%, and it is conservatively estimated that the number of people suffering from depression in China is more than 58 million (1, 2). Major depressive disorder is mainly driven by neuroendocrine, leading to neuroimmunity, metabolism or neurotransmitter imbalance, which is characterized by a low mood, slow thinking, reduced volitional activity, cognitive impairment and somatic symptoms (3, 4). In addition, depression has a serious suicidal tendency. The incidence of suicide among individuals with depression is approximately 10 times higher than in the general population, and approximately one-quarter of patients with depressive disorder have developed suicidal tendencies (5).
The mechanism of depression remains unclear. Studies have shown that the monoamine neurotransmitter hypothesis is the most widely accepted classical hypothesis, which suggests that decreased levels of dopamine (DA), serotonin (5-HT), and other monoamine neurotransmitters are an important mechanism in the onset and development of depression (6, 7). A decrease in gamma-aminobutyric acid (GABA) and an increase in glutamate also play an important role in the pathological development of depression (8, 9). In addition, there is a growing body of literature that supports and characterizes a gut-brain axis and elucidates a potential role for dysfunction of the gut microbiome in major depressive disorder. Animal studies have supported the possibility that dysbiosis (disruption of the microbiome) plays a causative role in depression-like behaviors. Broad-spectrum antibiotic administration in mice leads to dysbiosis, depression-like behaviors, and altered neuronal firing in the hippocampus (10, 11).
Machine learning is a subfield of artificial intelligence that develops models for prediction or decision-making through the analysis of data. The application of machine learning facilitates more effective processing and analysis of microbial datasets. In this study, we employed metagenomic techniques to analyze the intestinal bacterial communities of patients with MDD and healthy individuals and constructed a diagnostic model for MDD based on machine learning algorithms. We hope that machine learning modeling based on the analysis of gut bacterial communities will prove to be a valuable tool in the diagnosis of depression.
Metagenomic sequencing data from NCBI (SRA accession numbers: PRJNA762199 and PRJNA1083304) were used in this study. There were a total of 36 healthy individuals and 36 patients with MDD in the PRJNA762199 test dataset. There were a total of 20 healthy individuals and 16 patients with MDD in the PRJNA1083304 validation dataset. All patients met the diagnostic criteria for MDD, and the severity of anxiety and depression was assessed using the HAMD-17. And all of these patients were first-onset MDD who had not received medication and had no history of substance abuse.
To avoid the bias caused by different data processing methods, we chose sequence read archive (SRA) for raw sequencing metadata. The raw data in SRA format were converted to FASTQ format using the fastq-dump function of the SRA toolkit. The quality of the sequencing reads was assessed using FASTQC. Tool Kneaddata was first used to perform quality control on the metagenomic sequencing data (Specific parameters: –trimmomatic-options “SLIDINGWINDOW:4:20 MINLEN:50” –bowtie2-options “–very-sensitive –dovetail”). Sequences after quality control were then annotated using Kraken2 based on the bacterial database. Finally, Bracken was used to estimate the abundance of different microbial communities in each sample. All software invocations were performed on a Linux/Ubuntu system using bash commands.
All downstream data analyses of the metagenome were performed in R software (version 4.3.1). Sample data were normalized using the phyloseq package while filtering out some OTUs with lower abundance. Alpha diversity was analyzed using the vegan package and P < 0.05 was considered statistically significant. Ggplot2 and ggpubr packages were used to visualize alpha diversity. Beta diversity analysis was performed using permute, lattice, vegan, and ape packages, and principal coordinate analysis (PCoA) was plotted based on Bray-Curtis distance. Differences in bacterial communities between the MDD groups and healthy controls were analyzed using Statistical Analysis of Metagenomics Profiles (STAMP) (Welch t-test). Bacterial communities with high specificity and sensitivity were screened using the support vector machine recursive feature elimination (SVM-REF) to discriminate between patients with major depression and healthy individuals. The receiver operating characteristic (ROC) curves were generated with the MedCalc software.
To characterize the gut bacteria of patients with MDD, we analyzed metagenomic sequencing data from different databases from different countries. One of the databases was used for modeling analyses, including 36 MDDs and 36 healthy individuals. Patients with MDD and healthy controls (HCs) were similar in age, sex, and body mass index (BMI) in training cohort (P > 0.05). MDD group had higher total Hamilton Rating Scale for Depression (HAMD-17) scores than the HC group (P < 0.05) (Table 1). After analyzing the metagenomic sequencing data, a total of 2,368,263,997 raw reads associated with bacteria were obtained from 72 libraries. An additional database of 16 MDDs and 20 healthy individuals was used for further validation analyses. Patients with MDD and HCs were similar in sex and body mass index (BMI) in validation cohort (P > 0.05). However, there was a difference in age between the two groups. MDD group had higher total Hamilton Rating Scale for Depression (HAMD-17) scores than the HC group in validation cohort (P < 0.05) (Table 2).
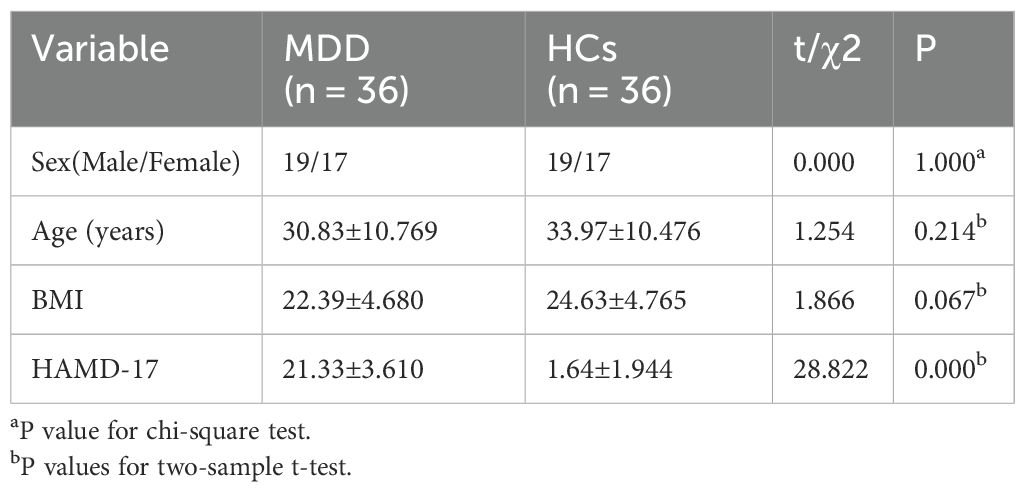
Table 1. Clinical characteristics in MDD and HCs (training cohort).
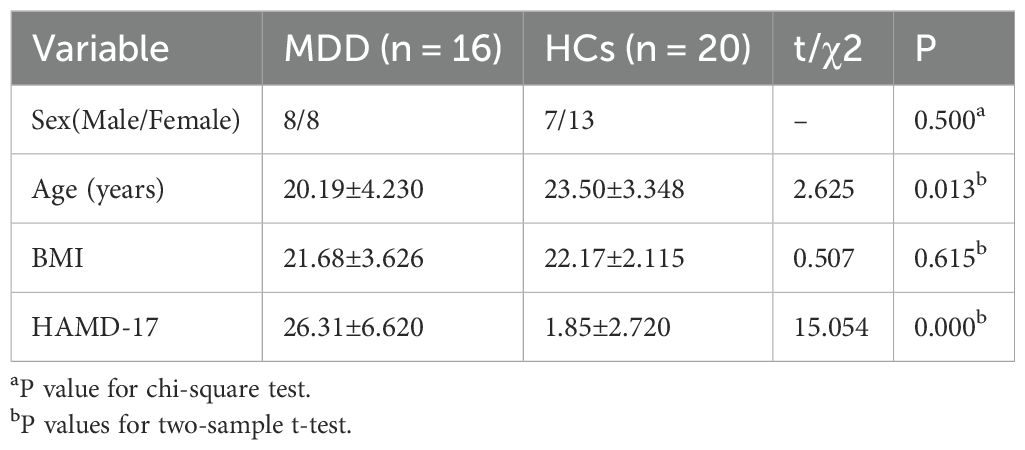
Table 2. Clinical characteristics in MDD and HCs (validation cohort).
First, species accumulation curve analysis was performed to verify that the sample size of the experiment was appropriate. The results showed that the species accumulation curve reached a plateau, indicating that the sample size was sufficient to reveal the characteristics of the gut bacteria (Figure 1A). We chose the Shannon–Wiener, Simpson, and Chao1 index to calculate the microbial alpha diversity. The results showed that there was no statistically significant difference in the Shannon, Simpson, and Chao1 index between MDD and healthy controls (P > 0.05, Wilcoxon test) (Figures 1B–D).
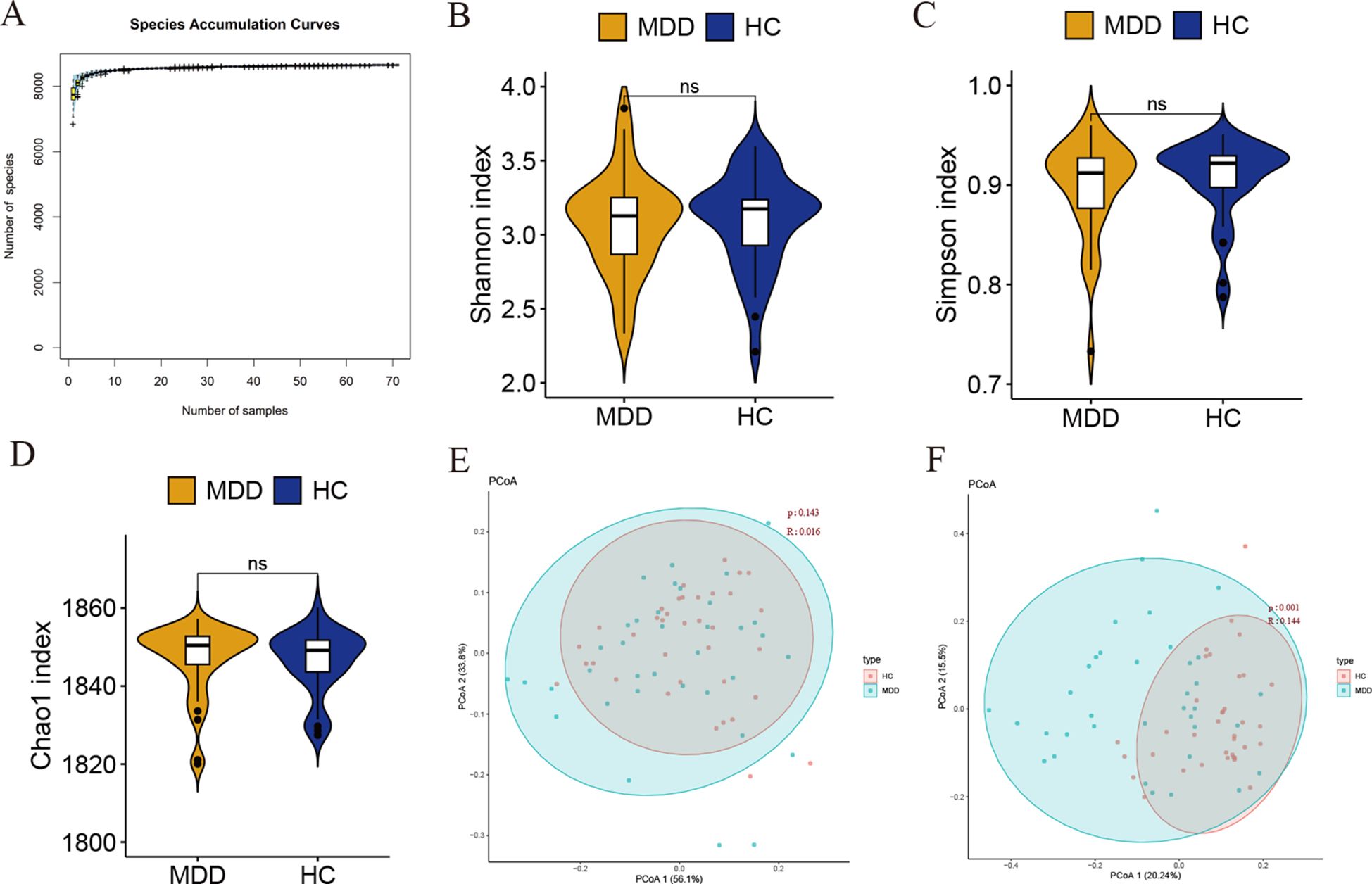
Figure 1. (A) Species accumulation curve. The abscissa represents samples and the ordinate represents the number of the cumulative number of species found. (B–D) Alpha diversity was estimated by the Shannon index, Simpson, and Chao1 index. Ns indicates not statistically significant difference. (E, F) Beta diversity analysis of MDD and healthy controls at the phylum and genus levels.
We then calculated the beta diversity between the groups based on the Bray-Curtis distance and performed a principal coordinate analysis (PCoA). Unweighted UniFrac analyses showed that PCoA could discriminate the healthy controls and MDD groups at the genus level (Figure 1F). Unfortunately, we did not observe significant differences between the two groups at the phylum level (Figure 1E). Taken together, these results suggest that gut bacterial communities may be different between MDD and control groups.
At the phylum level, Bacillota, Bacteroidota, and Actinomycetota were the three most abundant bacteria in all groups. At the genus level, Bacteroides, Faecalibacterium, and Blautia were the most abundant bacteria in the gut (Figures 2A, B). Statistical Analysis of Metagenomics Profiles (STAMP) was used to identify the gut bacterial communities that distinguish differences between patients with MDD and healthy controls. At the phylum level, Verrucomicrobiota, Pseudomonadota, and Bacillota were significantly different. At the genus level, Faecalibacterium, Roseburia, Pseudobutyrivibrio, Actinomyces, Vescimonas, Alistipes, Dysosmobacter, Akkermansia, Escherichia, Lacrimispora, Ruthenibacterium, Christensenella, Paraprevotella, Lachnoclostridium, Thomasclavelia, Anaerocolumna, Novisyntrophococcus, Intestinimonas, Avibacterium were significantly different. A total of 22 different bacterial communities were identified at the phylum and genus levels. Thirteen bacterial communities were significantly enriched in MDD groups compared to healthy controls, including Verrucomicrobiota, Pseudomonadota, Alistipes, Akkermansia, and Escherichia. Meanwhile, 9 bacterial communities were significantly reduced, including Bacillota, Faecalibacterium, Roseburia, Paraprevotella, Avibacterium, etc. (Figures 2C, D).
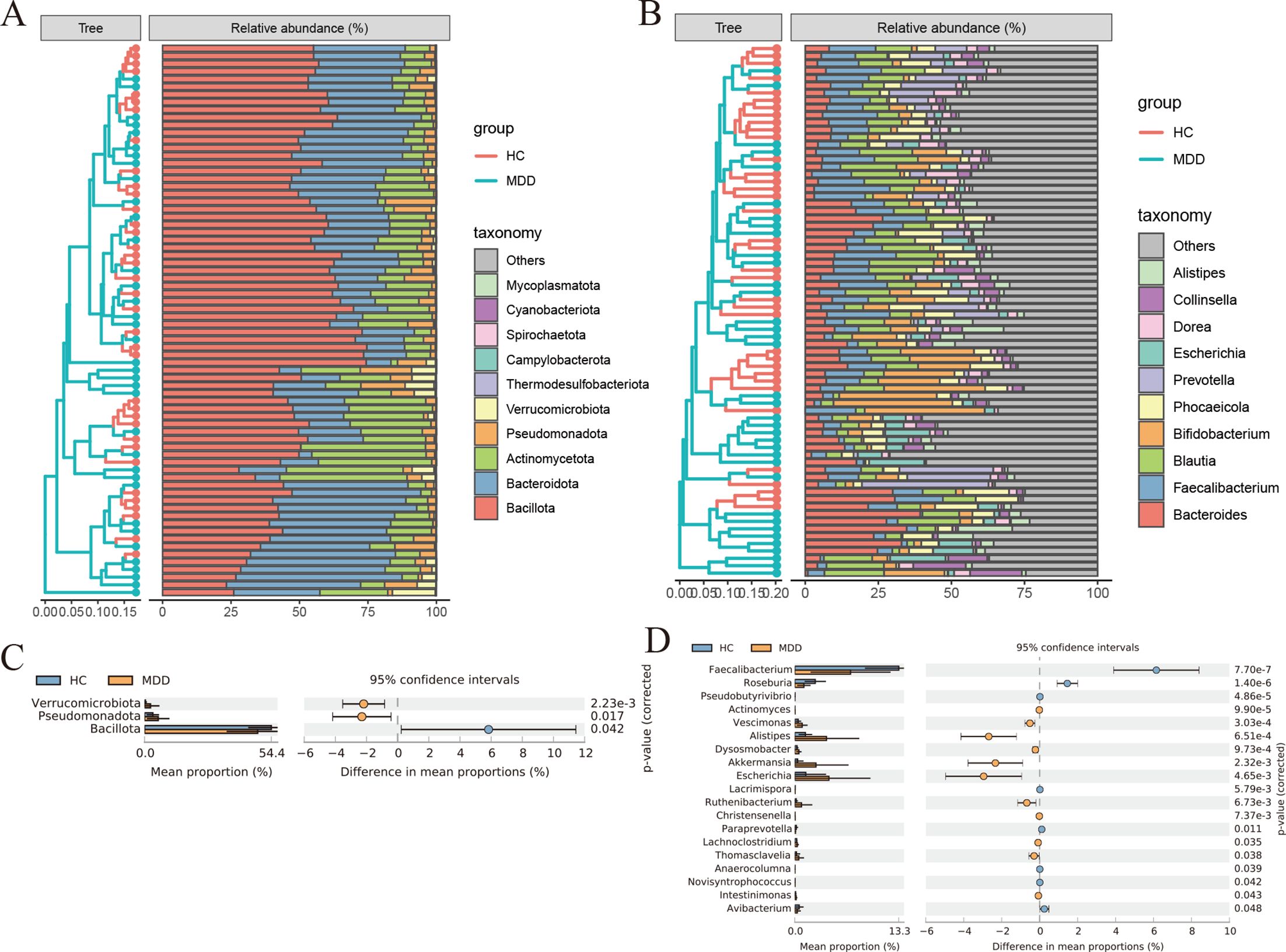
Figure 2. (A) Histogram of species abundance at phylum level. (B) Histogram of species abundance at genus level. (C, D) Analysis of differences in gut bacterial community composition between MDD and healthy controls using STAMP at the phylum and genus levels. Significance values shown were calculated using two-sided Welch t-tests.
The SVM-REF algorithm was used to screen potential microbial markers that could be used as diagnostic indicators from different bacterial communities of MDD. For the SVM-REF algorithm, the classifier error is minimized when the number of features is 8. Eight characteristic bacterial communities were finally identified (Figure 3A). The PCOA results showed that characteristic bacterial communities could distinguish healthy individuals with MDD (Figure 3B). In the heatmap we can observe the difference in the abundance of the 8 bacterial communities between MDD and the healthy groups (Figure 3C). Then, we compared the abundance of the characterized bacterial communities and showed that the abundance of the 8 bacterial communities was significantly different between MDD and healthy individuals (Figures 3D–K).
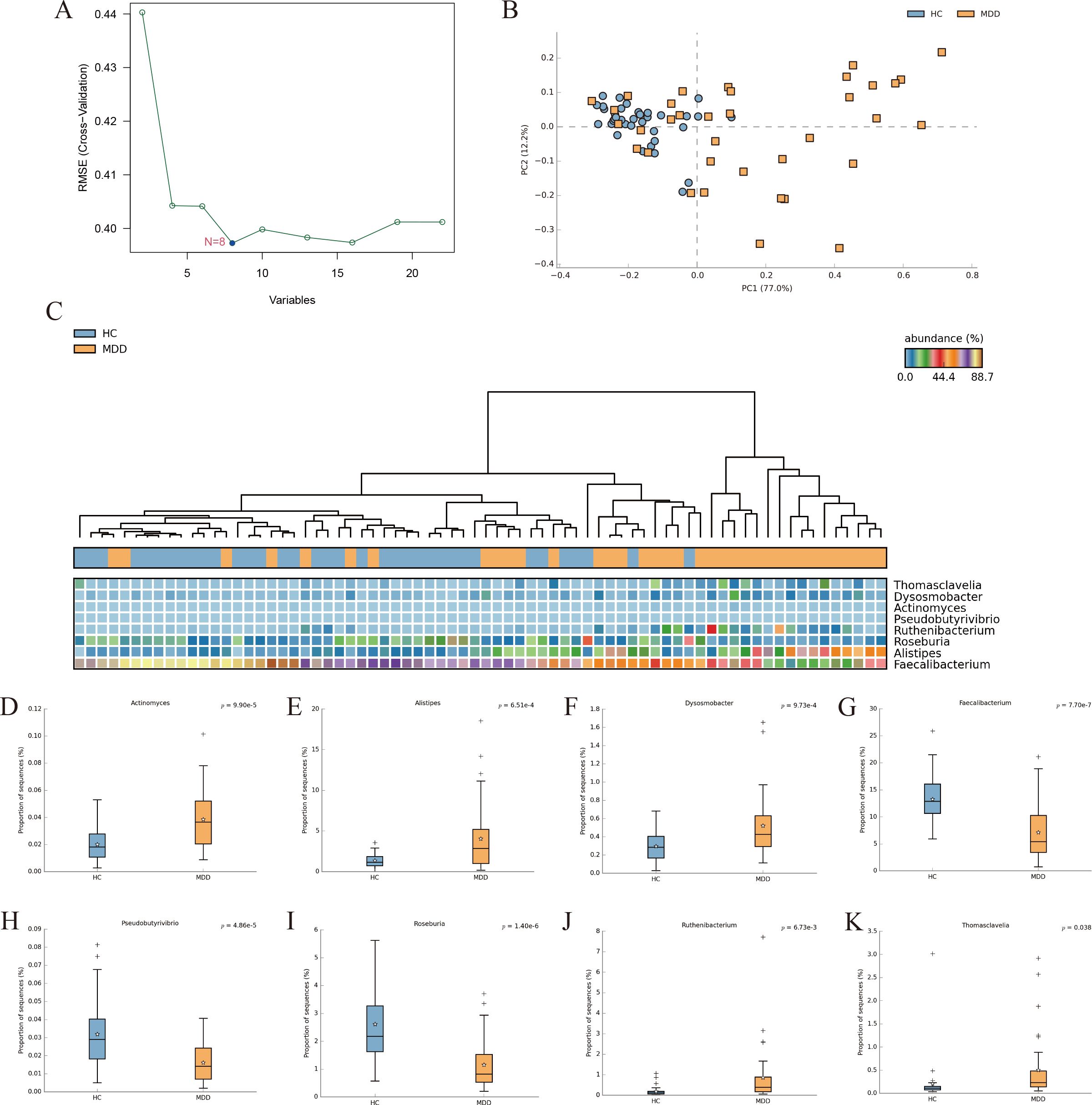
Figure 3. (A) SVM-REF model for screening bacterial biomarkers that distinguish MDD from healthy individuals. (B) PCoA of the 8 differential bacterial communities. (C) Heat map of the 8 differential bacterial communities. (D–K) Differential analysis of the abundance of characteristic gut bacterial communities was performed using STAMP (Welcht’s test).
Finally, we evaluated the predictive performance of the bacterial communities for the diagnosis of MDD using ROC curves. The results showed that the model constructed by the SVM-REF algorithm had a good predictive performance with an AUC value of 0.919 (95% CI: 0.8303-0.9702), indicating that these characteristic bacterial communities can be used as diagnostic indicators for MDD (Figure 4A). To further confirm the diagnostic potential of the bacterial communities in other samples, an independent test was conducted using an external validation cohort from Shanxi Province to confirm the reliability of the model. To further confirm the diagnostic potential of the gut bacteria, we validated the reliability of the model using an external validation cohort from Shanxi Province. The results showed that the validation cohort had an AUC of 0.800 (95% CI: 0.6334-0.9143), which was a good prediction (Figure 4B). Therefore, we can conclude that the characteristic bacterial communities have good predictive performance for the diagnosis of MDD.
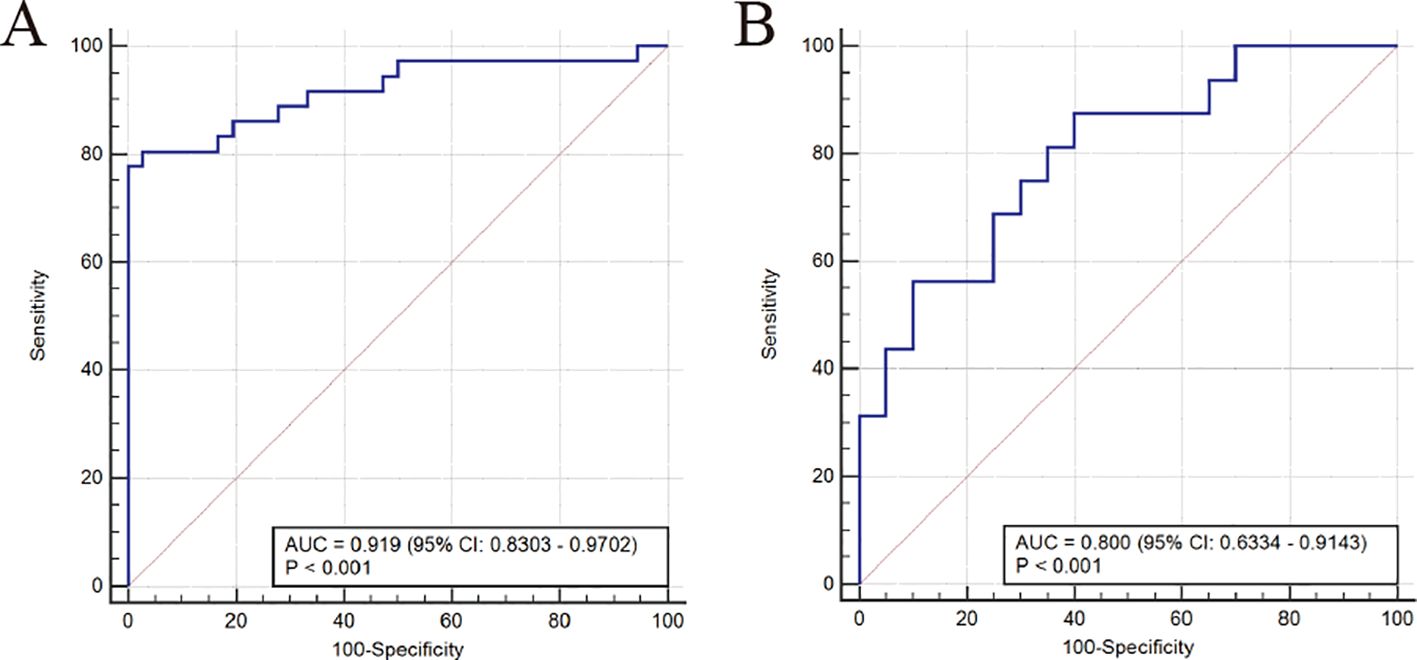
Figure 4. Area under the ROC curve (AUC) was used to evaluate the diagnostic performance. ROC curve of the models in the training group (A) and validation group (B).
Depression, as one of the most prevalent psychiatric disorders globally, exhibits a sharp annual increase in incidence rates and is the leading cause of global disability burden (12, 13). Clinical manifestations of major depressive disorder (MDD) are diverse and complex, with core characteristics including mood dysregulation, cognitive decline (such as memory impairment), motor function impairment (presented as reduced physical capacity), diminished energy (with increased fatigue), and a lowered sense of self-worth. Moreover, patients with MDD face a high risk of severe disability, which further exacerbates the negative impact on individual quality of life and socio-economic outcomes (14).
The gut microbiota plays a pivotal role in maintaining physiological functions in the gastrointestinal tract, including but not limited to regulating intestinal secretion, facilitating digestive processes, enhancing nutrient absorption, and synthesizing various vitamins and short-chain fatty acids such as acetate, butyrate, propionate, and lactate (15). Furthermore, the gut microbiota is involved in the biosynthesis of neurotransmitters and their precursors, including dopamine, norepinephrine, γ-aminobutyric acid (GABA), and acetylcholine. They also secrete and upregulate a range of essential proteins and metabolites that play key roles in the release of neuropeptides and gut hormones, thereby impacting neuroendocrine signaling. Additionally, the gut microbiota, through its metabolic activities and immune-modulatory functions, can finely tune the host’s immune responses, including the promotion of anti-inflammatory and immune tolerance mechanisms, as well as the regulation of inflammatory signaling pathways. These functions are crucial for maintaining the homeostasis of the gut-brain axis and overall health (16). Numerous previous studies have clearly indicated that the imbalance of the structure and function of the gut microbiota, known as dysbiosis, along with the associated dysfunction of the microbiota-gut-brain axis, may be a direct pathological mechanism in the occurrence and development of depression. These studies reveal the complex interplay between the gut microbiota and the central nervous system, as well as their key roles in mood regulation and behavioral expression (15–19).
In this study, we employed Statistical Analysis of Metagenomic Profiles (STAMP) to identify and differentiate key bacterial populations within the gut microbiota between patients with major depressive disorder (MDD) and healthy individuals. Additionally, we utilized a Support Vector Machine Recursive Feature Elimination (SVM-REF) model, which, while retaining the advantages of the Support Vector Machine (SVM), optimizes variable selection in the predictive model by reducing the number of feature vectors (20). Leveraging the superior predictive capacity of the SVM-REF model, we identified eight bacterial communities with statistically significant differences at the phylum and genus levels, which are considered potential microbial biomarkers for the diagnosis of MDD. Specifically, the relative abundance of Alistipes, Dysosmobacter, Actinomyces, Ruthenibacterium, and Thomasclavelia was significantly higher in MDD patients compared to healthy controls, while the abundance of Faecalibacterium, Pseudobutyrivibrio, and Roseburia was significantly reduced in MDD patients.
The relative abundance of five bacterial communities is elevated in patients with major depressive disorder, as detailed below: Alistipes, an indole-positive organism, can reduce the bioavailability of serotonin and metabolizes glutamate to γ-aminobutyric acid (GABA) through the expression of glutamate decarboxylase, and an increase in its abundance may disrupt the function of the gut-brain axis (21). Although literature on Dysosmobacter is scarce, studies suggest that it may modulate immune responses through interactions with the host’s immune system (22). Actinomyces has been reported to be more abundant in patients with major depressive disorder (23). Bacteria of the genus Ruthenibacterium are implicated in COVID-19 pathogenesis, with affected patients exhibiting reduced immune cell levels and refractory hypoxemia (24). Thomasclavelia may be associated with chronic inflammatory diseases of the gut (25).
The abundance of the following three bacterial genera is significantly lower in patients with major depressive disorder: bacteria of the genus Faecalibacterium are associated with anti-inflammatory activity, particularly in inflammatory bowel disease, where a reduction in their numbers correlates with inflammatory conditions. Furthermore, Faecalibacterium species can produce short-chain fatty acids such as acetate, propionate, and butyrate. This genus can also modulate the host’s immune system, including upregulating the expression of IL-10 and enhancing T-cell proliferation, playing a significant role in the immune regulation of the gut-brain axis (26). In non-psychiatric conditions such as spinal cord injury and drug-induced liver injury, the metabolic pathways of gut bacteria are impaired, and the abundance of Pseudobutyrivibrio is consistently lower (27, 28). Roseburia, capable of producing short-chain fatty acids, significantly increases the levels of 5-hydroxytryptamine (5-HT) in the brain and colon, inhibits the expression of rate-limiting enzymes, and can reverse the stress-induced conversion of tryptophan to kynurenine in the brain and colon, demonstrating antidepressant functions. Additionally, Roseburia has been confirmed to efficiently predict major depressive disorder in adolescents (29).
When assessing the risk of developing major depressive disorder, the predictive power of individual bacterial populations is limited and does not provide accurate and reliable results. In contrast, the eight bacterial populations selected in this study, due to their significant differences in composition, exhibit higher specificity and sensitivity, thereby being more effective in predicting major depressive disorder. Furthermore, through external validation analyses, the predictive model constructed using these eight bacterial populations was able to accurately identify patients with major depressive disorder, confirming the model’s high predictive accuracy and practical utility.
This study successfully identified eight bacterial populations significantly associated with major depressive disorder at the phylum and genus levels using Statistical Analysis of Metagenomic Profiles (STAMP) and Support Vector Machine Recursive Feature Elimination (SVM-REF) models. These bacterial populations, as potential microbial biomarkers, demonstrate significant application value in the diagnosis of major depressive disorder. The study not only unravels the complex relationship between the gut microbiota and major depressive disorder but also provides a scientific basis for the development of microbiome-based diagnostic tools for depression, highlighting the importance of multi-population analysis in enhancing predictive accuracy and practicality. However, this study also has certain limitations, such as: limited sample size and cross-sectional design cannot determine causality. Future studies should focus on addressing these limitations, by increasing the sample size, adopting a longitudinal study design, and considering more potential confounding factors, to further verify these findings and explore their potential for application in clinical practice.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. The manuscript presents research on animals that do not require ethical approval for their study.
XW: Conceptualization, Writing – original draft, Writing – review & editing. DC: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. HZ: Visualization, Writing – original draft, Writing – review & editing. WC: Visualization, Writing – original draft, Writing – review & editing. JS: Writing – original draft, Writing – review & editing. HH: Conceptualization, Methodology, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pitsillou E, Bresnehan SM, Kagarakis EA, Wijoyo SJ, Liang J, Hung A, et al. The cellular and molecular basis of major depressive disorder: towards a unified model for understanding clinical depression. Mol Biol Rep. (2020) 47:753–70. doi: 10.1007/s11033-019-05129-3
2. Kang S-G, Cho S-E. Neuroimaging biomarkers for predicting treatment response and recurrence of major depressive disorder. Int J Mol Sci. (2020) 21(6):2148. doi: 10.3390/ijms21062148
3. Trivedi MH. Major depressive disorder in primary care: strategies for identification. J Clin Psychiatry. (2020) 81(2):UT17042BR1C. doi: 10.4088/JCP.UT17042BR1C
4. Su YA, Ye C, Xin Q, Si T. Major depressive disorder with suicidal ideation or behavior in Chinese population: A scoping review of current evidence on disease assessment, burden, treatment and risk factors. J Affect Disord. (2023) 340:732–42. doi: 10.1016/j.jad.2023.08.106
5. Rihmer Z, Rihmer A. Depression and suicide - the role of underlying bipolarity. Psychiatr Hung. (2019) 34:359–68.
6. Boku S, Nakagawa S, Toda H, Hishimoto A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Psychiatry Clin Neurosci. (2018) 72(1):3–12. doi: 10.1111/pcn.2018.72.issue-1
7. Heninger GR, Delgado PL, Charney DS. The revised monoamine theory of depression: a modulatory role for monoamines, based on new findings from monoamine depletion experiments in humans. Pharmacopsychiatry. (1996) 29(1):2–11. doi: 10.1055/s-2007-979535
8. Lener MS, Niciu MJ, Ballard ED, Park M, Park LT, Nugent AC, et al. Glutamate and gamma-aminobutyric acid systems in the pathophysiology of major depression and antidepressant response to ketamine. Biol Psychiatry. (2017) 81:886–97. doi: 10.1016/j.biopsych.2016.05.005
9. Yu H, Chen Z-Y. The role of BDNF in depression on the basis of its location in the neural circuitry. Acta Pharmacol Sin. (2011) 32(1):3–11. doi: 10.1038/aps.2010.184
10. Yano JM, Yu K, Donaldson GP, Shastri GG, Ann P, Ma L, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. (2015) 161:264–76. doi: 10.1016/j.cell.2015.02.047
11. Cheung SG, Goldenthal AR, Uhlemann AC, Mann JJ, Miller JM, Sublette ME. Systematic review of gut microbiota and major depression. Front Psychiatry. (2019) 10:34. doi: 10.3389/fpsyt.2019.00034
12. Stringaris A. Editorial: what is depression? J Child Psychol Psychiatry. (2017) 58:1287–9. doi: 10.1111/jcpp.12844
13. Dean J, Keshavan M. The neurobiology of depression: An integrated view. Asian J Psychiatry. (2017) 27:101–11. doi: 10.1016/j.ajp.2017.01.025
14. Song J, Kim YK. Animal models for the study of depressive disorder. CNS Neurosci Ther. (2021) 27:633–42. doi: 10.1111/cns.13622
15. Góralczyk-Bińkowska A, Szmajda-Krygier D, Kozłowska E. The microbiota–gut–brain axis in psychiatric disorders. Int J Mol Sci. (2022) 23(19):11245. doi: 10.3390/ijms231911245
16. Simpson CA, Diaz-Arteche C, Eliby D, Schwartz OS, Simmons JG, Cowan CSM. The gut microbiota in anxiety and depression – A systematic review. Clin Psychol Rev. (2021) 83:101943. doi: 10.1016/j.cpr.2020.101943
17. Radford-Smith DE, Anthony DC. Prebiotic and probiotic modulation of the microbiota–gut–brain axis in depression. Nutrients. (2023) 15(8):1880. doi: 10.3390/nu15081880
18. Liu L, Wang H, Chen X, Zhang Y, Zhang H, Xie P. Gut microbiota and its metabolites in depression: from pathogenesis to treatment. eBioMedicine. (2023) 90:104527. doi: 10.1016/j.ebiom.2023.104527
19. Bear TLK, Dalziel JE, Coad J, Roy NC, Butts CA, Gopal PK. The role of the gut microbiota in dietary interventions for depression and anxiety. Adv Nutr. (2020) 11:890–907. doi: 10.1093/advances/nmaa016
20. Dong B, Liu X, Yu S. Utilizing machine learning algorithms to identify biomarkers associated with diabetic nephropathy: A review. Medicine. (2024) 103(8):e37235. doi: 10.1097/MD.0000000000037235
21. Parker BJ, Wearsch PA, Veloo ACM, Rodriguez-Palacios A. The genus alistipes: gut bacteria with emerging implications to inflammation, cancer, and mental health. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.00906
22. Rong XQ, Shu QL. Modulating butyric acid-producing bacterial community abundance and structure in the intestine of immunocompromised mice with neutral polysaccharides extracted from Codonopsis pilosula. Int J Biol Macromol. (2024) 278:134959. doi: 10.1016/j.ijbiomac.2024.134959
23. Barandouzi ZA, Starkweather AR, Henderson WA, Gyamfi A, Cong XS. Altered composition of gut microbiota in depression: A systematic review. Front Psychiatry. (2020) 11. doi: 10.3389/fpsyt.2020.00541
24. Kovtun AS, Averina OV, Angelova IY, Yunes RA, Zorkina YA, Morozova AY, et al. Kostyuk GP et al: Alterations of the Composition and Neurometabolic Profile of Human Gut Microbiota in Major Depressive Disorder. Biomedicines. (2022) 10(9):2162. doi: 10.3390/biomedicines10092162
25. Wu X, Li Y, Shang Y. Application of two-dimensional polymerase chain reaction to detect four types of microorganisms in feces for assisted diagnosis of IBD. Clin Chim Acta. (2024) 555:117802. doi: 10.1016/j.cca.2024.117802
26. Martín R, Rios-Covian D, Huillet E, Auger S, Khazaal S, Bermúdez-Humarán LG, et al. Faecalibacterium: a bacterial genus with promising human health applications. FEMS Microbiol Rev. (2023) 47(4):fuad039. doi: 10.1093/femsre/fuad039
27. Sun J, Gungor B, Adiguzel E, Gursel I, Yilmaz B, Gursel M. Intestinal microbiota in patients with spinal cord injury. PloS One. (2016) 11(1):e0145878. doi: 10.1371/journal.pone.0145878
28. Rodriguez-Diaz-Cristina TB. García-García Alberto: Microbiota diversity in nonalcoholic fatty liver disease and in drug-induced liver injury. Pharmacol Res. (2022) 182:106348. doi: 10.1016/j.phrs.2022.106348
29. Zhou M, Fan Y, Xu L, Yu Z, Wang S, Xu H, et al. Wu L et al: Microbiome and tryptophan metabolomics analysis in adolescent depression: roles of the gut microbiota in the regulation of tryptophan-derived neurotransmitters and behaviors in human and mice. Microbiome. (2023) 11(1):145. doi: 10.1186/s40168-023-01589-9
Keywords: major depressive disorder, gut bacteria, metagenome, machine learning, diagnosing disease
Citation: Wang X, Cao D, Zhang H, Chen W, Sun J and Hu H (2025) Utilizing metagenomic profiling and machine learning model to identify bacterial biomarkers for major depressive disorder. Front. Psychiatry 16:1539596. doi: 10.3389/fpsyt.2025.1539596
Received: 04 December 2024; Accepted: 31 January 2025;
Published: 18 February 2025.
Edited by:
Melanie Föcking, Royal College of Surgeons in Ireland, IrelandCopyright © 2025 Wang, Cao, Zhang, Chen, Sun and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huimin Hu, MTI3MTI5MjgwMEBxcS5jb20=
†These authors have contributed equally to this work
‡ORCID: Huimin Hu, orcid.org/0000-0001-9476-7621
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.