 Zuhair Al-Hassnan1,2†Mazhor AlDosary3†Aljouhra AlHargan3†
Zuhair Al-Hassnan1,2†Mazhor AlDosary3†Aljouhra AlHargan3† Hanan AlQudairy3†Rawan Almass1†Khaled Omar Alahmadi4
Hanan AlQudairy3†Rawan Almass1†Khaled Omar Alahmadi4 Saif AlShahrani1
Saif AlShahrani1 Albandary AlBakheet3
Albandary AlBakheet3 Mohammad A. Almuhaizea2,5
Mohammad A. Almuhaizea2,5 Robert W. Taylor6,7‡
Robert W. Taylor6,7‡ Dilek Colak8‡
Dilek Colak8‡ Namik Kaya2,3*‡
Namik Kaya2,3*‡- 1Department of Medical Genomics, Center for Genomic Medicine, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
- 2College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
- 3Translational Genomics Department, Center for Genomic Medicine, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
- 4Department of Radiology, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
- 5Neuroscience Center, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
- 6Mitochondrial Research Group, Translational and Clinical Research Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne, United Kingdom
- 7National Health Service (NHS) Highly Specialised Mitochondrial Diagnostic Laboratory, Newcastle upon Tyne Hospitals National Health Service (NHS) Foundation Trust, Newcastle upon Tyne, United Kingdom
- 8Molecular Oncology Department, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
Background: Iron–sulfur cluster assembly 2 (ISCA2) deficiency is linked to an autosomal recessive disorder known as multiple mitochondrial dysfunctions syndrome 4 (MMDS4). This disorder is characterized by leukodystrophy and neuroregression. Currently, most of the reported patients are from Saudi Arabia. All these patients carry a homozygous founder variant (NM_194279.2:c.229G>A:p.Gly77Ser) in ISCA2.
Methods: We describe a patient who underwent full clinical evaluation, including metabolic, neurological, and radiological examinations. Standard genetic testing, including whole exome sequencing coupled with autozygome analysis, was undertaken, as were assessments of mitochondrial DNA (mtDNA) copy number and mtDNA sequencing on DNA extracted from blood and cultured fibroblasts. Functional workup consisted of splicing assessment of ISCA2 using RT-PCR, biochemical assessment of complex I status using dipstick assays, and mitochondrial respiration measurements using a Seahorse XFp analyzer.
Results: We present the clinical and functional characterization of a novel homozygous ISCA2 missense variant (NM_194279.3:c.70A>G:p.Arg24Gly), leading to aberrant splicing in a patient presenting with neuroregression, generalized spasticity with exaggerated deep tendon reflexes and head lag, and progressive loss of acquired milestones. The novel variant was fully segregated in a wider family and was absent in a large control cohort, ethnically matching in-house exomes, local databases such as CGMdb and Saudi Human Genome Program, and ClinVar.
Conclusions: Our analyses revealed that the variant is pathogenic, disrupting normal ISCA2 splicing and presumably leading to a truncated protein that disturbs metabolic pathways in patient-derived cells.
1 Introduction
Iron–sulfur clusters (ISC or Fe–S clusters) are conserved cofactors of iron and sulfur associated with the cysteine sulfurs of proteins. These clusters are common and present in a plethora of organisms, ranging from protists to eukaryotes (1). These cofactors in eukaryotic cells are involved in diverse functions and play critical roles in many fundamental molecular processes, such as electron transfer, structural stabilization, gene regulation, and enzymatic catalysis (1). Several proteins are involved in the assembly of Fe–S clusters. Some are located in the inner mitochondrial membrane and are related to electron transport chain function, including iron–sulfur cluster assembly 1 and 2 (ISCA1 and ISCA2), which are both encoded by nuclear genes (2). Depletion of both ISCA1 and ISCA2 has been linked to incorrect assembly of ISC and biosynthesis of mitochondrial ISC (3–6). ISCA2, an essential ISC assembly member, is crucial for multiple versatile cellular mechanisms such as DNA replication, RNA transcription, oxygen sensitization, and electron transport (7, 8). The utilization of ISC occurs in complexes I–III (9, 10). Recent findings suggest that in addition to being a vital component of normal mitochondrial functionality, ISCA2 is essential for erythroid differentiation and cell proliferation (11).
Previously, we reported a homozygous pathogenic variant (a founder mutation among Saudis) of ISCA2 in five unrelated consanguineous families comprising six infantile patients characterized by leukodystropy and neuroregression (8, 12). The syndrome is transmitted in an autosomal recessive mode of inheritance (8). Since then, the disease has been known as multiple mitochondrial dysfunctions syndrome 4 (MMDS4) (MIM Phenotype#616370; Gene/Locus MIM#615317) (13). Currently, more than 20 patients reported in the literature harbor ISCA2-related genetic defects, most of whom were from Saudi Arabia.
In this study, we report an additional case of a consanguineous Saudi family harboring the novel ISCA2 variant. We functionally characterized this variant and compared it to previously published Saudi cases with the founder variant, detailing the clinical and genetic features of MMDS4.
2 Materials and methods
2.1 Subjects
This study was conducted at the King Faisal Specialist Hospital and Research Center (KFSHRC) using an institutional review board-approved protocol (RAC#2120022/2180004). While visiting the medical genetics clinics, the patient (whose legal guardians) and family members were asked to sign an informed written consent before donating their samples. Subsequently, they were evaluated for the project and biological specimens were collected.
2.2 Sample collection and nucleic acid extraction
Venous blood samples (5–10 mL) were collected in EDTA tubes from each patient and their family members. DNA was extracted from the collected blood samples using a PureGene DNA Purification Kit (Gentra Systems, Inc., Minneapolis, MN, US). DNA quality and quantity were examined on 1%–2% agarose gels and then analyzed using NanoDrop® ND-1000 (NanoDrop Inc., Wilmington, DE, US).
2.3 Cell culture
Age-matched controls and index patient's (F1-II-2, Patient 1, Family 1) primary skin fibroblast cell lines were grown in high-glucose Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO, USA) containing 10% fetal calf serum, 1× non-essential amino acids, 50-U/mL penicillin, 50-μg/mL streptomycin, and 50-μg/mL uridine at 37°C and 5% CO2 in a humidified incubator. For harvesting, both the medium and trypsin were prewarmed at 37°C in an incubator prior to use. The cells were then washed with 1× PBS. Depending on the flask size, sufficient trypsin was added to cover the surface of the flask, which was then incubated at 37°C for 1–2 min to dislodge the cells from the flask. Complete growth medium was added to prevent further proteolysis by quenching trypsin and maintaining the cells in suspension. To harvest the cells, they were allowed to reach 90%–100% confluency, washed with 1× PBS, incubated in a sufficient amount of trypsin, and briefly incubated at 37°C. The cells were washed again with 1× PBS and lysed with complete lysis-M (EDTA-Free) buffer (Roche, Sigma Aldrich) according to the manufacturers’ protocol. DNA and RNA extraction was performed using fibroblasts and QIAGEN kits (Venlo, Netherlands). The nucleic acid quality and quantity were determined using a Bioanalyzer 1200 (Agilent Technologies, Santa Clara, CA, US).
2.4 Genome-wide SNP genotyping and autozygome analysis
SNP genotyping was performed using Affymetrix Axiom Human Mapping assays on Axiom® 2.0. Samples from the families were registered on Gene Titan Array plates and processed on the Titan platform (Affymetrix, Santa Clara, CA, USA). All downstream applications, such as target labeling, hybridization, array scanning, image acquisition, and quality assessment of scanned images, were performed according to Affymetrix’s protocols and guidelines. Genome-wide SNP calls were then acquired, read, and analyzed using Affymetrix Genotyping Console. Genome-wide SNPs were transferred to AutoSNPa software, and homozygous blocks, also called runs of homozygosity (ROH), were determined (14).
2.5 ISCA2-related Sanger sequencing
Quality-assessed DNA samples were used for DNA amplification. Previously tested and published ISCA2 primers were used for the PCR (8). To assess the performance of the PCR products, amplified DNA samples were loaded and visualized on agarose gels. The amplified fragments were subjected to Sanger sequencing and analyzed using Lasergene from DNASTAR.
2.6 Mitochondrial DNA (mtDNA) sequencing
Genomic DNA was extracted from the fibroblasts and used as a template for mtDNA amplification. Complete genome amplification, sequencing, and analyses of sequencing results were performed as previously described (15, 16).
2.7 Multiple sequence alignment and protein modeling
Amino acid sequences from several species were extracted from Ensembl and aligned using the Clustal Omega software from the European Bioinformatics Institute. The sequences were aligned, and the variation site and surrounding region were targeted. Alignment and visualization were performed using the freely available Jalview and Protean tool (DNASTAR, Wisconsin, MD, USA).
2.8 Whole exome sequencing and variant detection
After quality assessment of the isolated DNA, sequence capture and library preparation were performed using SureSelect assays. The libraries were then sequenced on HiSeq 2000 (Illumina, San Diego, CA, USA) to obtain a minimum of 30-fold coverage. Sequences were aligned, and the resulting variant call format (VCF) file, which contains 55,091 variants, was filtered and analyzed using publicly and commercially available software and tools.
2.9 Assessment of mtDNA copy number using quantitative PCR (qPCR)
Depletion assays were carried out using NALCN (nuclear marker) and ND1 (mitochondrial gene) on DNA extracted from the index patient’s cultured fibroblasts, as previously described (8). The delta delta CT method was used for the copy number calculation.
2.10 Dipstick assay for complex I
Dipstick assay kit designed to measure complex I activity (MitoSciences/Abcam, Cambridge, UK) was utilized as done previously (8). Fibroblasts from index patients and controls were used in the experiments. The manufacturer’s protocols were strictly followed throughout the experiments.
2.11 RT-PCR analysis of the novel variant
High-quality and undegraded RNA was used for cDNA synthesis using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystem Corp., Waltham, MA, USA). For the primers used in RT-PCR, the Primer3 web tool was utilized for exon spanning primers to ensure successful and exclusive amplification of exonic regions without intrusion of genomic sequences. The expression of ISCA2 was compared to that of GAPDH (F-primer 5′-3′ CTGCCAACGTGTCAGTGGTG and R-primer 5′-3′ TCAGTGTAGCCCAGGATGCC) in the same cells.
2.12 Mitochondrial respiration measurements
We used Seahorse XFp analyzer (Agilent) to measure oxygen consumption rate (OCR) to determine key parameters of cellular metabolism using control and patients’ cells as described (17–19). One day prior to the assay, fibroblasts (20,000 cells/well) were seeded in a Seahorse 8-well mini-culture plate with growth medium (MEM supplemented with 10% FBS, 1% penicillin–streptomycin, and 1% L-glutamine), placed in a 37°C incubator with 5% CO2 and allowed to adhere overnight. The sensor cartridge was hydrated overnight in an XF calibrant at 37°C in a non-CO2 incubator. The next day, the cells were washed twice with warm assay medium, and 180 µL of assay medium was added per well. The cell culture mini-plate was then placed in a 37°C non-CO2 incubator for 45–60 min prior to the assay. Each sample was measured in duplicates (technical replicates). The results were calculated and produced using the Seahorse Wave software version 2.6.
2.13 Statistical analysis
All statistical analyses were performed using GraphPad Prism v6.0 (GraphPad Software, San Diego, CA, USA). Data are expressed as mean ± standard error of the mean (SEM). To compare the means between two groups, an unpaired Student’s t-test was performed, with Welch’s correction, if applicable. All statistical tests were two-sided, and a p-value <0.05 was considered statistically significant.
3 Results
3.1 Clinical findings
The details of the clinical examination results are presented in Table 1.
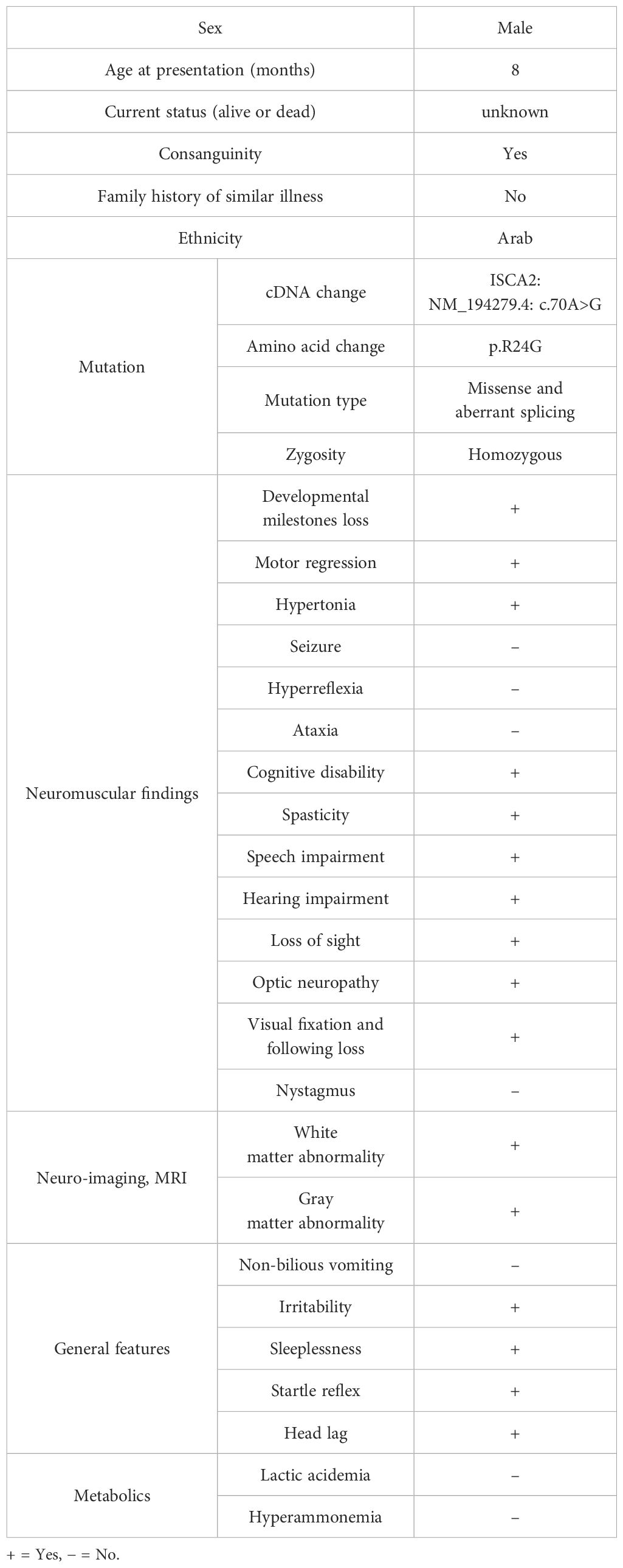
Table 1. Clinical details of the singleton carrying the novel ISCA2 variant.
The patient was referred to our center at the age of 24 months for evaluation of neuroregression. He was a product of a full-term pregnancy with normal spontaneous vaginal delivery and an uneventful perinatal history. He was well until the age of 8 months, when he started to have irritability and poor eye contact. He progressively lost his acquired milestones and became spastic, with an inability to sit, hold objects, or babble. There was no history of seizures or abnormal movement. The parents were consanguineous. There were five other children who were alive and well. Physical examination revealed that both head circumference and height were at the 5th percentile, while weight was just below the 5th percentile. He was irritable and unable to fixate or follow. No dysmorphic features or skin stigmata were observed. He had generalized spasticity with exaggerated deep tendon reflexes and head lag. Organomegaly was not observed. Eye examination revealed a diffuse pallor of the optic disc.
3.1.1 Neuroradiological findings
Brain MRI showed confluent involvement of the white matter in the supratentorial brain parenchyma spanning from the periventricular to the subcortical area. It involves the corticospinal tracts in the posterior limbs of the internal capsules, tegmentum of the middle cerebral peduncles, and dorsal aspect of the brain stem. The white matter changes extended to the posterior structures, specifically the peridentate white matter of the cerebellar hemispheres. Diffuse atrophy of the corpus callosum is also observed. The brain MRI findings are shown in Figures 1A–D.
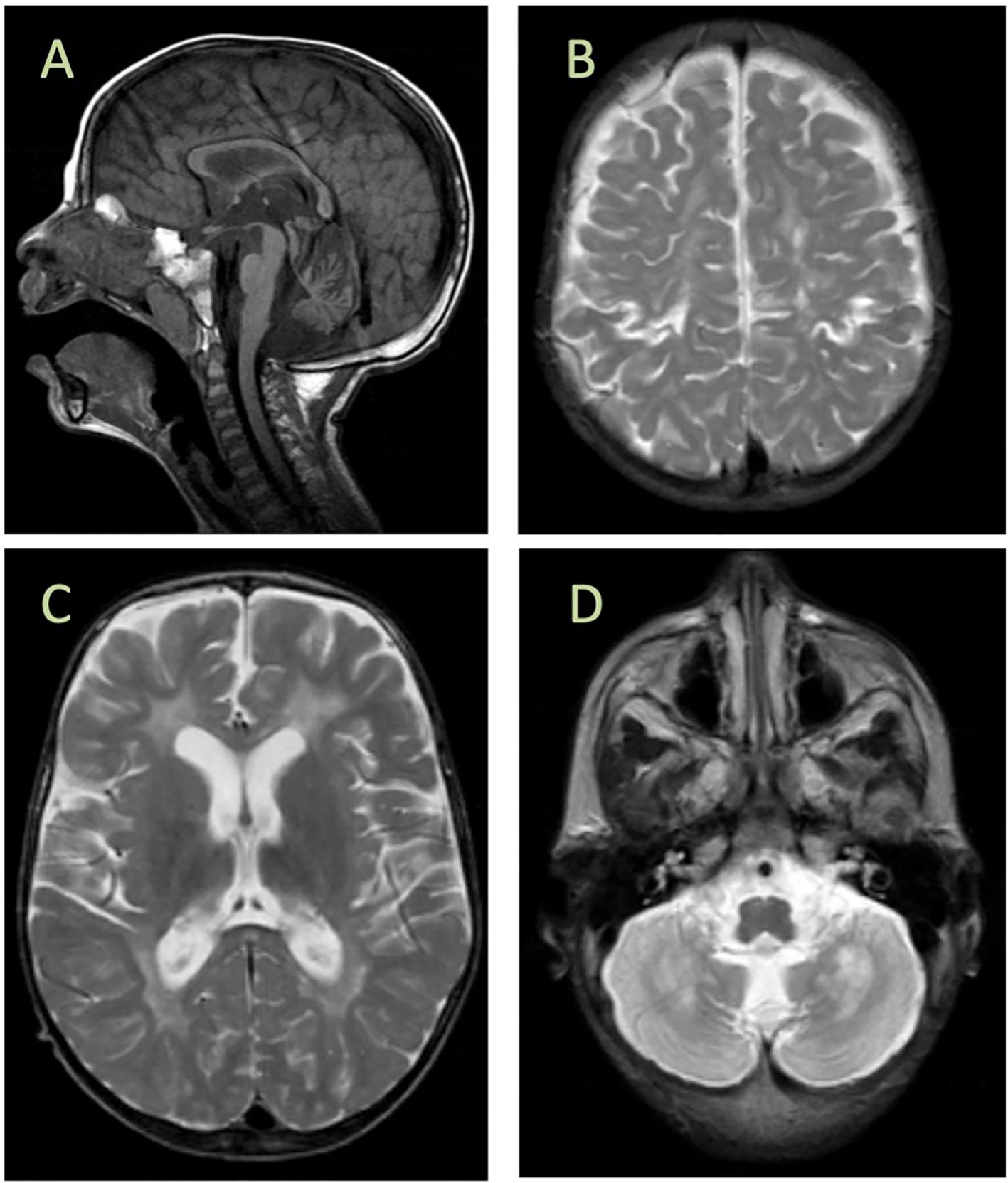
Figure 1. Brain MRI findings. (A) Cerebellar atrophy and decreased signal in the corpus callosum, diffuse increased signal, and cystic encephalomalacia in the white matter (B, C) with bilateral involvement of the gray matter around the central sulcus (D). MRI was performed at age of 2 years.
3.1.2 Molecular genetic findings
Previously, we reported six patients with MMDS4 and later expanded the phenotypic spectrum of the disease in our country (8, 20). Most reported cases were from Saudi Arabia, and the first reported mutation (NM_194279.2:c.229G>A:p.Gly77Ser) is an ancient founder that appears in the central and western regions of the country, and we expected to encounter more cases in this region. Hence, we continuously searched for additional patients with this disease. Interestingly, we encountered a singleton from a consanguineous Saudi family whose genomic DNA was comprehensively subjected to diagnostic and research-based genetic testing, including whole mitochondrial DNA (mtDNA) sequencing. After exhaustive filtering of whole-exome sequencing data, the analysis revealed a missense and homozygous variant (NM_194279.3:c.70A>G:p.Arg24Gly) in ISCA2. The filtering approach and criteria were performed using previously reported protocols (21–27). The variant was fully segregated in the family members (Figure 2A; Supplementary Figure 1) using Sanger sequencing. Moreover, our autozygome analysis revealed that ISCA2 sits in a long stretch of a homozygous run of homozygosity (ROH) block on chromosome14q24.3 specific to the affected individual (Figure 2B). We then searched our in-house exomes and local databases such as the Saudi Human Genome Database (SHGP) and King Abdullah International Medical Research Center (KAIMRC) Genomic Database (KGD), as well as ethnically matched control data sets (totals to more than 2,703) that did not have any positive match in the homoallelic state. This variant was not encountered in ClinVar and was classified as PP3 (Strong), PM2 (Supporting), or PB1 (Supporting) based on the American College of Medical Genetics and Genomics (ACMG) criteria. The variant was also not reported in GnomAD in the homozygous state (allele frequency: 0.000003554; allele count: 5; number of homozygotes: 0).
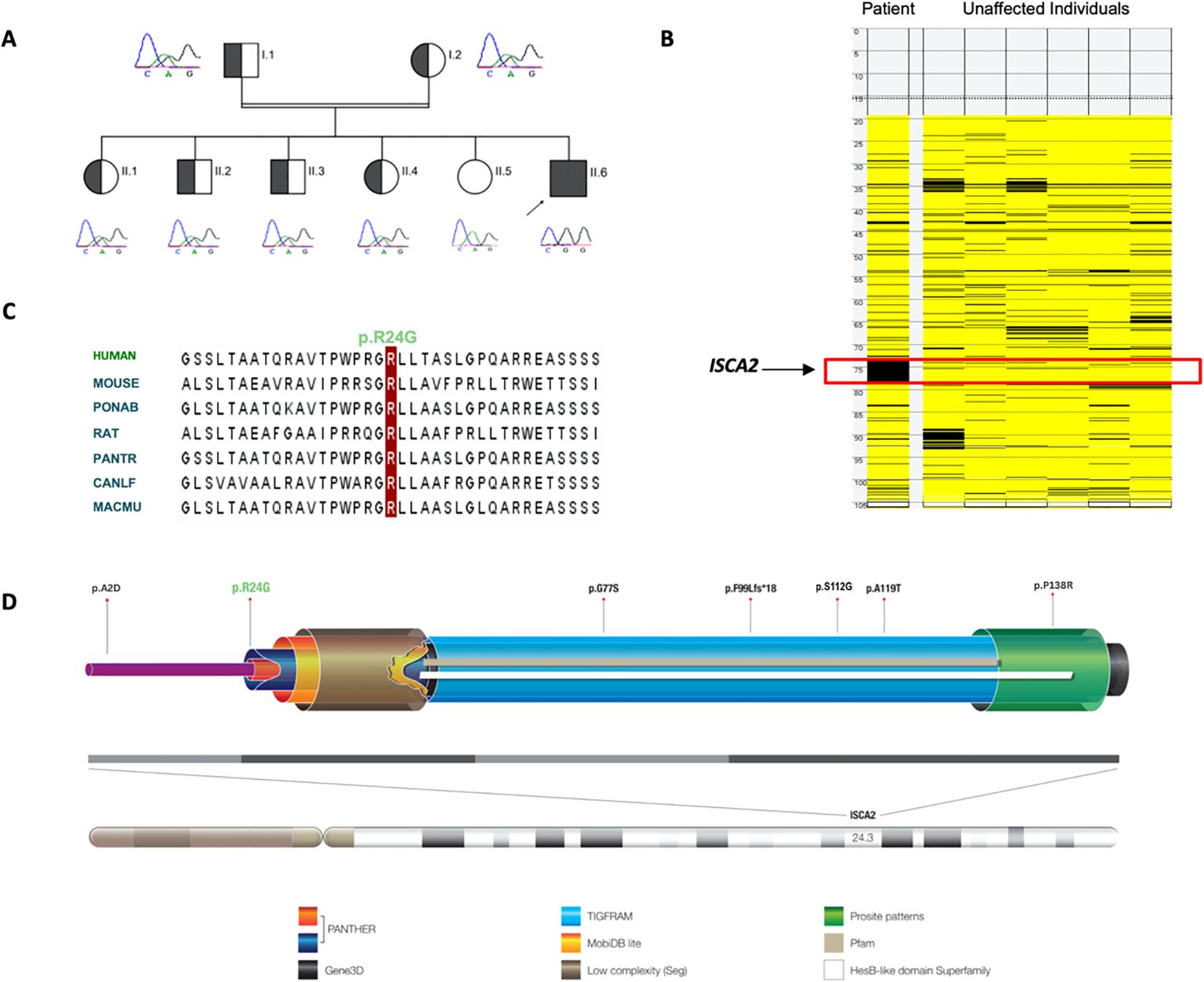
Figure 2. (A) Pedigree showing the affected individual and carriers of the novel and pathogenic ISCA2 variant c.70A>G; p.Arg24Gly and full segregation of the phenotype in the family, indicating a recessive inheritance pattern. Short Sanger sequencing images are presented below. (B) Autozygome analysis of the patient and unaffected individuals revealed a unique homozygosity block on the long arm of chromosome 14 in the affected individual. (C) Alignment of amino acid sequences at position 24 (as indicated by the red column) within ISCA2, showing high conservation in different species. (D) Structure and localization of the human ISCA2: The ISCA2 gene is located on chromosome 14q24.3 and consists of four exons shown as light and dark gray boxes (bottom). The ISCA2 protein has one essential functional domain for iron–sulfur (Fe–S) biogenesis, which overlaps with the predictions of PANTHER, Gene3D, TIGRFAM, MobiDB lite, Prosite pattern, Superfamily, and Pfam protein databases. Local complexity in amino acid sequences is also represented as ‘low complexity segments domain. The novel ISCA2 mutation site and previously reported mutations in ISCA2 are indicated (top panel).
The novel variant (c.70A>G) in ISCA2 replaces an A to G, leading to a change from arginine to glycine in the 24th amino acid position that sits in proximity to the splicing site on a highly conserved region and important domain (Figures 2C, D). After searching the frequency of the variant in a wide range of databases to ensure its novelty, the variant’s functionally deleterious potential was investigated using various experimental approaches. Initially, because the variant is close to the exon/intron junction, we suspected that it may cause a splicing error. To test this hypothesis, we used total RNA samples extracted from the index patient fibroblasts as well as those of the control samples (n = 3) and performed RT-PCR analysis. This experiment revealed multiple bands (n = 3) that were displayed on an agarose gel (2%) in comparison to those of the controls (Figure 3A). Sequencing of the bands revealed a normal product (~250 bp), an alternative splicing product (~300 bp), and a non-specific PCR band (~560 bp) (Figures 3A, B). Normal and aberrant transcript maps are shown in Figure 3C. All three bands consistently appeared in the patient’s sample, although several optimization attempts were made using different primer sets to remove the upper band. Sequencing of the RT-PCR product showed retention of the intronic sequence presumably leading to protein truncation (Supplementary Figure 2).
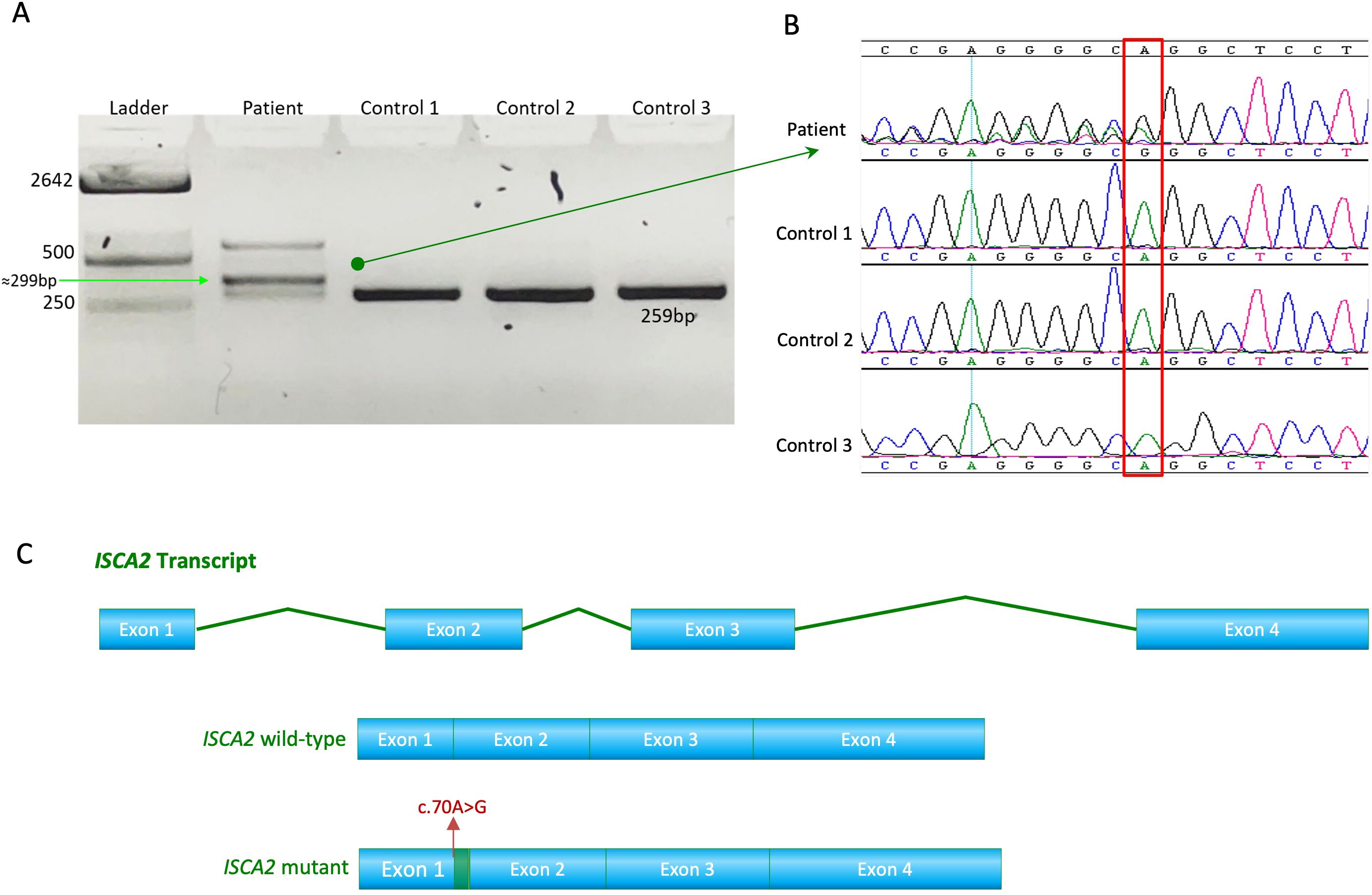
Figure 3. (A) RT-PCR analysis of RNA extracted from the patient’s fibroblasts showed faulty splicing due to the transition compared to the controls. (B) Sequence chromatogram showing the novel and pathogenic ISCA2 variant c.70A>G; p.Arg24Gly in the RNA extracted from the patient's fibroblasts. (C) Transcript maps of ISCA2 showing the normal transcript vs aberrant transcript. The novel variant, the aberrant transcript, causes faulty splicing, resulting in a retention of RNA transcript.
Next, we sought to determine whether the variant led to any depletion of the cultured fibroblasts in the index case. As previously described (8), we utilized a qPCR approach to investigate mitochondrial copy number changes. The results revealed increased mtDNA copy number in the patient’s fibroblasts (Figure 4A).
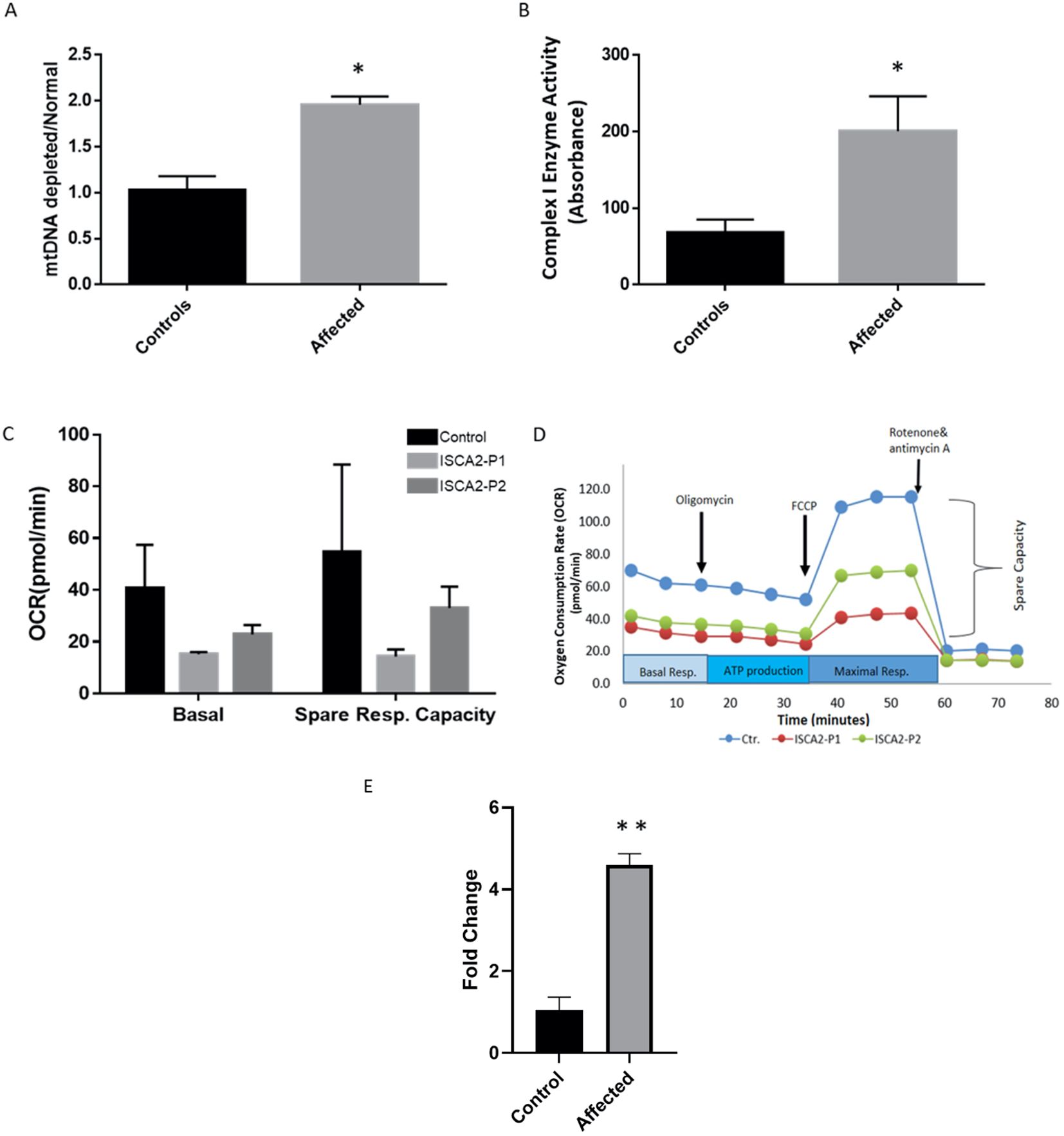
Figure 4. (A) Real-time PCR analysis of individual and control fibroblasts to assess mtDNA depletion. The assay indicated increased levels of mitochondria in the patient’s fibroblasts compared with the controls. The data represent the mean ± standard error (SE) of three controls and one patient (each sample was run in triplicate). The patient showed a significant increase compared to the controls. (B) Complex I enzyme activity dipstick assay. The results showed considerable evidence of mitochondrial respiratory chain abnormalities in fibroblasts from the index patient compared with controls. The data represent the mean ± standard error (SE) of four controls and one patient (each sample was run in triplicate). The patient showed a significant increase compared to the controls (p <0.05). (C, D) Seahorse XF cell mitochondrial stress test profile showing mitochondrial respiration parameters measured in fibroblasts from control subjects and two patients. Oxygen consumption rates measured in healthy subjects and individuals with ISCA2 deficiency. Spare respiratory capacity is the difference between basal and maximal respiration and measures the ability of cells to respond to increased energy demand. ISCA2-P1 was the index patient carrying the novel variant reported in this study, whereas ISCA2-P2 was another patient with a homoallelic missense founder mutation (NM_194279.2;c.229G>A;p.Gly77Ser). Two replicates were used for each experiment. Data are presented as the mean ± SEM. Both mitochondrial basal respiration and spare respiratory capacity were reduced in both patients compared to the control; however, the difference was not statistically significant. (E) Quantitative RT-PCR was performed on RNA extracted from the index patient fibroblasts and compared with that of the control fibroblasts. The results showed significantly increased expression levels of ISCA2 in cultured fibroblasts of the index case. The data represent the mean ± standard error (SE) of four controls and one patient (each sample was run in triplicate). The patient showed a significant increase compared with the control group (p <0.0001). In the figures, black indicates the control(s) and light gray indicates the affected individual(s). Two-tailed Student’s t-tests with Welch’s corrections were performed; *p <0.05 and **p <0.0001 indicate statistical significance.
3.1.3 Biochemical findings
Next, we used a complex I enzyme activity dipstick assay to investigate the functional consequences of this variant and the likely deficiency in fibroblasts from the patient. Dipstick tests revealed increased levels of complex I activity in fibroblasts from patients compared to those from the controls (Figure 4B).
To gain further functional insight into the likely consequences of the c.70A>G novel mutation, we performed a mitochondrial respiration test on fibroblasts from the control and two patients, our index patient and another patient who carried the homozygous missense founder mutation (NM_194279.2:c.229G>A:p.Gly77Ser). Both patients’ cells showed a decrease in mitochondrial basal respiration and spare-respiratory capacity as compared to those of the control (Figures 4C, D). Moreover, quantitative RT-PCR results showed a significant increase in the expression levels of ISCA2 in the cultured fibroblasts of the index case compared to those of the controls (p <0.0001) (Figure 4E).
4 Discussion
The first ISCA2 human mutation (NM_194279.2 c.229G>A; p.Gly77Ser), a homozygous missense founder, was identified in Saudi patients belonging to unrelated consanguineous families. Additional disease-causing variants have been discovered over the past few years. Among these, the most commonly encountered mutation is the Saudi founder variant. The total number of MMDS4 patients reached 44, mostly with the Saudi founder mutation (n = 37) except for six non-Saudis. Subsequent cases have been reported in non-Saudi Arab patients with different types of mutations. For example, an Italian patient was identified to carry a different mutation: a paternal frameshift (c.295delT:p.Phe99Leufs*18), resulting in a premature stop codon and a maternal missense variant (c.334A>G:p.Ser112Gly) with compound heterozygosity (30). Moreover, two unrelated patients of French origin were identified as carrying homozygous missense variants in ISCA2 (c.154C>T, p.Leu52Phe and c.313A>G, p.Arg105Gly) that resulted in reduced activity of complex I (28). More recently, an Iranian patient was found to have an ISCA2 variant (c.355G>A, p.Ala119Thr) (29).
Here, we present the clinical and functional characterization of a novel homozygous ISCA2 missense variant (NM_194279.3:c.70A>G; p.Arg24Gly) leading to aberrant splicing. This patient presented with neuroregression, generalized spasticity with exaggerated deep tendon reflexes, and head lag with progressive loss of acquired milestones that started with irritability and poor eye contact. The clinical presentation of the patient was consistent with the ISCA2-related mitochondrial disease phenotype (8, 13, 21). This study identified the second Saudi ISCA2 variant among Saudi Arabs, expanding the genotypic spectrum of MMDS4 even in a highly consanguineous population, indicating that more cases remain to be identified.
The novel transition, as predicted by SpliceAI, causes a splicing error due to its location. We investigated this using RT-PCR on the total RNA extracted from patient fibroblasts. RT-PCR revealed faulty splicing due to this transition. The splicing error caused retention of the part of the intronic sequence, resulting in a larger transcript compared to control transcripts (Figures 3A–C).
In addition, this novel mutation caused increased mtDNA copy number and may also lead to increased complex I activity in the patient’s fibroblasts. However, we were unable to confirm this increase in activity using another method. Moreover, we could not compare citrate synthase activity to complex I activity; therefore, our results in this regard are not conclusive. Since ISCs are known to play a major role in oxidation–reduction reactions for mitochondrial electron transport, mainly complex I and complex II of oxidative phosphorylation (1, 30), this novel mutation is expected to cause such aberrant activity, as other studies have shown (6, 28). Using the Seahorse approach, our assessment of mitochondrial respiration revealed a decrease in the mitochondrial respiration parameters in the patients’ cells compared to those of the control. Similar findings were reported in a previous study of two patients with a founder homozygous missense mutation (NM_194279.2 c.229G>A; p.Gly77Ser) in ISCA2, which showed an increase in the activity of complex I and decreased mitochondrial respiration (31). The likely mitochondrial respiration deficiency observed in our patient may indicate the involvement of this novel mutation in pathogenicity by affecting electron transport through the mitochondrial respiratory chain complexes.
Notably, this novel variant increased both the mtDNA copy number and mRNA expression in our index patient, as opposed to the founder homozygous missense mutation (NM_194279.2 c.229G>A:p.Gly77Ser), which showed decreased levels (8, 31). This increase as well as the possible increase in complex I activity in our patient, may be attributed to a compensatory response triggered by a defect in ISCA2 function because of this variant. Moreover, mitochondrial mass was found to be increased in fibroblasts due to a defect in the respiratory chain (32), which could also explain the increased mtDNA copy number in patient fibroblasts.
Generally, the affected individuals with MMDS4 are characterized by neurodegeneration, developmental regression, failure to thrive, quadriplegia, truncal hypotonia, optic atrophy, and leukoencephalopathy (8, 13). The c.229G>A (p.Gly77Ser) variant caused decreased oxygen consumption and overall respiration, ATP production, and mitochondrial membrane potential, and significantly decreased complex II and minimal reduction in complex IV (31). Interestingly, an increase was also observed for complex I subunit NDUFB8. Further investigations also pointed out the negative impact of the variant on the functioning of proteins, specifically for 4Fe–4S clusters, but not 2Fe–2S proteins (29). However, a patient with bi-allelic variants p.Ala2Asp and p.Pro138Arg was found to have a milder phenotype with a longer life span, better psychomotor function, and cavitating leukodystrophy (30).
Taken together, these data indicate that this novel variant causes pathogenicity in our patient; however, it is important to note that technical issues related to the maintenance of patient-derived cells prevented us from performing confirmatory functional studies.
5 Conclusion
Here, we report the first case of a variant other than the founder mutation in Saudi Arabia. Our study reports the second Saudi ISCA2 variant among Saudi Arabs. This study expands the genotypic spectrum of MMDS4, even in a highly consanguineous population in which a founder variant is frequently encountered in medical genetics clinics, considering that the disease is a rare disorder. MMDS type 4 is still more common in Saudi Arabia than in any other population studied to date.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board (or Ethics Committee) of King Faisal Specialist Hospital and Research Centre (KFSHRC, RAC#2120022; 23 August 2021, and RAC#2180004 03 March 2022). Informed consents were obtained from all subjects participating in the study.
Author contributions
ZA-H: Conceptualization, Investigation, Writing – original draft, Writing – review & editing, Formal analysis. MA: Formal analysis, Investigation, Writing – original draft, Writing – review & editing, Methodology, Software, Visualization. AljA: Formal analysis, Investigation, Visualization, Writing – review & editing. HA: Formal analysis, Investigation, Visualization, Writing – review & editing. RA: Formal analysis, Investigation, Writing – review & editing. KA: Formal analysis, Investigation, Writing – review & editing, Visualization. SA: Investigation, Writing – review & editing. AlbA: Writing – review & editing, Formal analysis, Supervision. MAA: Writing – review & editing, Investigation. RT: Writing – review & editing. DC: Writing – review & editing, Formal analysis, Investigation, Software, Visualization. NK: Investigation, Writing – review & editing, Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the King Salman Center for Disability Research (grant no. KSRG-2022-105).
Acknowledgments
We thank the patients and their families for their participation in this study. The authors extend their appreciation to the King Salman Center for Disability Research for funding this work through Research Group no. KSRG-2022-105.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2024.1428175/full#supplementary-material
Supplementary Figure 1 | Sanger sequencing results of the PCR with novel ISCA2 variant.
Supplementary Figure 2 | Aberrant transcript presumably leads to a protein truncation.
References
1. Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, et al. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim Biophys Acta. (2012) 1823:1491–508. doi: 10.1016/j.bbamcr.2012.05.009
2. Lill R, Muhlenhoff U. Iron-sulfur-protein biogenesis in eukaryotes. Trends Biochem Sci. (2005) 30:133–41. doi: 10.1016/j.tibs.2005.01.006
3. Jensen LT, Culotta VC. Role of saccharomyces cerevisiae isa1 and isa2 in iron homeostasis. Mol Cell Biol. (2000) 20:3918–27. doi: 10.1128/MCB.20.11.3918-3927.2000
4. Kaut A, Lange H, Diekert K, Kispal G, Lill R. Isa1p is a component of the mitochondrial machinery for maturation of cellular iron-sulfur proteins and requires conserved cysteine residues for function. J Biol Chem. (2000) 275:15955–61. doi: 10.1074/jbc.M909502199
5. Pelzer W, Muhlenhoff U, Diekert K, Siegmund K, Kispal G, Lill R. Mitochondrial isa2p plays a crucial role in the maturation of cellular iron-sulfur proteins. FEBS Lett. (2000) 476:134–9. doi: 10.1016/s0014-5793(00)01711-7
6. Sheftel AD, Wilbrecht C, Stehling O, Niggemeyer B, Elsasser HP, Muhlenhoff U, et al. The human mitochondrial isca1, isca2, and iba57 proteins are required for [4fe-4s] protein maturation. Mol Biol Cell. (2012) 23:1157–66. doi: 10.1091/mbc.E11-09-0772
7. Ajit Bolar N, Vanlander AV, Wilbrecht C, van der Aa N, Smet J, De Paepe B, et al. Mutation of the iron-sulfur cluster assembly gene iba57 causes severe myopathy and encephalopathy. Hum Mol Genet. (2013) 22:2590–602. doi: 10.1093/hmg/ddt107
8. Al-Hassnan ZN, Al-Dosary M, Alfadhel M, Faqeih EA, Alsagob M, Kenana R, et al. Isca2 mutation causes infantile neurodegenerative mitochondrial disorder. J Med Genet. (2015) 52:186–94. doi: 10.1136/jmedgenet-2014-102592
9. Shi R, Hou W, Wang Z-Q, Xu X. Biogenesis of iron–sulfur clusters and their role in DNA metabolism. Front Cell Dev Biol. (2021) 9:735678. doi: 10.3389/fcell.2021.735678
10. Read A, Bentley R, Archer S, Dunham-Snary K. Mitochondrial iron-sulfur clusters: structure, function, and an emerging role in vascular biology. Redox Biol. (2021) 47:102164. doi: 10.1016/j.redox.2021.102164
11. Wang J, Jiang M, Yue G, Zhu L, Wang X, Liang M, et al. Isca2 deficiency leads to heme synthesis defects and impaired erythroid differentiation in K562 cells by indirect ros-mediated irp1 activation. Biochim Biophys Acta Mol Cell Res. (2022) 1869:119307. doi: 10.1016/j.bbamcr.2022.119307
12. Al-Hassnan ZN, Kaya N. Isca2-related Mitochondrial Disorder. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al, editors. Genereviews((R)). Seattle (WA) (1993).
13. Amberger JS, Bocchini CA, Scott AF, Hamosh A. Omim.Org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. (2019) 47:D1038–D43. doi: 10.1093/nar/gky1151
14. Carr IM, Flintoff KJ, Taylor GR, Markham AF, Bonthron DT. Interactive visual analysis of snp data for rapid autozygosity mapping in consanguineous families. Hum Mutat. (2006) 27:1041–6. doi: 10.1002/humu.20383
15. Aldosary M, Al-Bakheet A, Al-Dhalaan H, Almass R, Alsagob M, Al-Younes B, et al. Rett syndrome, a neurodevelopmental disorder, whole-transcriptome, and mitochondrial genome multiomics analyses identify novel variations and disease pathways. OMICS. (2020) 24:160–71. doi: 10.1089/omi.2019.0192
16. Kaya N, Imtiaz F, Colak D, Al-Sayed M, Al-Odaib A, Al-Zahrani F, et al. Genome-wide gene expression profiling and mutation analysis of saudi patients with canavan disease. Genet Med. (2008) 10:675–84. doi: 10.1097/gim.0b013e31818337a8
17. Calton MA, Beaulieu MO, Benchorin G, Vollrath D. Method for measuring extracellular flux from intact polarized epithelial monolayers. Mol Vis. (2018) 24:425–33.
18. Leung DT, Chu S. Measurement of oxidative stress: mitochondrial function using the seahorse system. Preeclampsia. Springer. (2018) . p:285–93.
19. Aldosary M, Baselm S, Abdulrahim M, Almass R, Alsagob M, AlMasseri Z, et al. Slc25a42-associated mitochondrial encephalomyopathy: report of additional founder cases and functional characterization of a novel deletion. JIMD Rep. (2021) 60:75–87. doi: 10.1002/jmd2.12218
20. Alfadhel M, Nashabat M, Alrifai MT, Alshaalan H, Al Mutairi F, Al-Shahrani SA, et al. Further delineation of the phenotypic spectrum of isca2 defect: A report of ten new cases. Eur J Paediatr Neurol. (2018) 22:46–55. doi: 10.1016/j.ejpn.2017.10.003
21. AlMuhaizea M, AlMass R, AlHargan A, AlBader A, Medico Salsench E, Howaidi J, et al. Truncating mutations in yif1b cause a progressive encephalopathy with various degrees of mixed movement disorder, microcephaly, and epilepsy. Acta Neuropathol. (2020) 139:791–4. doi: 10.1007/s00401-020-02128-8
22. Al-Sayed MD, Al-Zaidan H, Albakheet A, Hakami H, Kenana R, Al-Yafee Y, et al. Mutations in nalcn cause an autosomal-recessive syndrome with severe hypotonia, speech impairment, and cognitive delay. Am J Hum Genet. (2013) 93:721–6. doi: 10.1016/j.ajhg.2013.08.001
23. Seidahmed MZ, Hamad MH, AlBakheet A, Elmalik SA, AlDrees A, Al-Sufayan J, et al. Ancient founder mutation in rubcn: A second unrelated family confirms salih ataxia (Scar15). BMC Neurol. (2020) 20:207. doi: 10.1186/s12883-020-01761-w
24. Al-Muhaizea MA, AlQuait L, AlRasheed A, AlHarbi S, Albader AA, AlMass R, et al. Pyrostigmine therapy in a patient with vamp1-related congenital myasthenic syndrome. Neuromuscul Disord. (2020) 30:611–5. doi: 10.1016/j.nmd.2020.04.007
25. Kaya N, Alsagob M, D'Adamo MC, Al-Bakheet A, Hasan S, Muccioli M, et al. Kcna4 deficiency leads to a syndrome of abnormal striatum, congenital cataract and intellectual disability. J Med Genet. (2016) 53:786–92. doi: 10.1136/jmedgenet-2015-103637
26. Chelban V, Alsagob M, Kloth K, Chirita-Emandi A, Vandrovcova J, Maroofian R, et al. Genetic and phenotypic characterization of nkx6-2-related spastic ataxia and hypomyelination. Eur J Neurol. (2020) 27:334–42. doi: 10.1111/ene.14082
27. Perenthaler E, Nikoncuk A, Yousefi S, Berdowski WM, Alsagob M, Capo I, et al. Loss of ugp2 in brain leads to a severe epileptic encephalopathy, emphasizing that bi-allelic isoform-specific start-loss mutations of essential genes can cause genetic diseases. Acta Neuropathol. (2020) 139:415–42. doi: 10.1007/s00401-019-02109-6
28. Lebigot E, Gaignard P, Dorboz I, Slama A, Rio M, de Lonlay P, et al. Impact of mutations within the [Fe-S] cluster or the lipoic acid biosynthesis pathways on mitochondrial protein expression profiles in fibroblasts from patients. Mol Genet Metab. (2017) 122:85–94. doi: 10.1016/j.ymgme.2017.08.001
29. Eidi M, Garshasbi M. A novel isca2 variant responsible for an early-onset neurodegenerative mitochondrial disorder: A case report of multiple mitochondrial dysfunctions syndrome. BMC Neurol. (2019) 19:1–7. doi: 10.1186/s12883-019-1387-2
30. Rouault TA. Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis Model Mech. (2012) 5:155–64. doi: 10.1242/dmm.009019
31. Alaimo JT, Besse A, Alston CL, Pang K, Appadurai V, Samanta M, et al. Loss-of-function mutations in isca2 disrupt 4fe-4s cluster machinery and cause a fatal leukodystrophy with hyperglycinemia and mtdna depletion. Hum Mutat. (2018) 39:537–49. doi: 10.1002/humu.23396
Keywords: ISCA2 founder variant, novel splicing variant, mtDNA, depletion, leukodystrophy, neuroregression
Citation: Al-Hassnan Z, AlDosary M, AlHargan A, AlQudairy H, Almass R, Alahmadi KO, AlShahrani S, AlBakheet A, Almuhaizea MA, Taylor RW, Colak D and Kaya N (2024) A novel missense mutation in ISCA2 causes aberrant splicing and leads to multiple mitochondrial dysfunctions syndrome 4. Front. Psychiatry 15:1428175. doi: 10.3389/fpsyt.2024.1428175
Received: 05 May 2024; Accepted: 26 August 2024;
Published: 18 October 2024.
Edited by:
Memoona Ramzan, University of the Punjab, PakistanReviewed by:
Mariana Santos, Universidade do Porto, PortugalValerija Dobricic, University of Lübeck, Germany
Copyright © 2024 Al-Hassnan, AlDosary, AlHargan, AlQudairy, Almass, Alahmadi, AlShahrani, AlBakheet, Almuhaizea, Taylor, Colak and Kaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Namik Kaya, bmtheWFAa2ZzaHJjLmVkdS5zYQ==
†These authors have contributed equally to this work
‡ORCID: Robert W. Taylor, orcid.org/0000-0002-7768-8873
Dilek Colak, orcid.org/0000-0001-6485-8768
Namik Kaya, orcid.org/0000-0001-8912-7507