
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Psychiatry, 06 February 2023
Sec. Psychopharmacology
Volume 14 - 2023 | https://doi.org/10.3389/fpsyt.2023.959590
This article is part of the Research TopicMDMA-Assisted Therapy for Treatment of PTSD and BeyondView all 10 articles
 Candace R. Lewis1,2*
Candace R. Lewis1,2* Joseph Tafur3Sophie Spencer1Joseph M. Green1
Joseph Tafur3Sophie Spencer1Joseph M. Green1 Charlotte Harrison4Benjamin Kelmendi5David M. Rabin6
Charlotte Harrison4Benjamin Kelmendi5David M. Rabin6 Rachel Yehuda7,8Berra Yazar-Klosinski4
Rachel Yehuda7,8Berra Yazar-Klosinski4 Baruch Rael Cahn9,10*
Baruch Rael Cahn9,10*Background: Previous research has demonstrated that epigenetic changes in specific hypothalamic-pituitary-adrenal (HPA) genes may predict successful psychotherapy in post-traumatic stress disorder (PTSD). A recent Phase 3 clinical trial reported high efficacy of 3,4-methylenedioxymethamphetamine (MDMA)-assisted therapy for treating patients with severe PTSD compared to a therapy with placebo group (NCT03537014). This raises important questions regarding potential mechanisms of MDMA-assisted therapy. In the present study, we examined epigenetic changes in three key HPA axis genes before and after MDMA and placebo with therapy. As a pilot sub-study to the parent clinical trial, we assessed potential HPA epigenetic predictors for treatment response with genomic DNA derived from saliva (MDMA, n = 16; placebo, n = 7). Methylation levels at all 259 CpG sites annotated to three HPA genes (CRHR1, FKBP5, and NR3C1) were assessed in relation to treatment response as measured by the Clinician-Administered PTSD Scale (CAPS-5; Total Severity Score). Second, group (MDMA vs. placebo) differences in methylation change were assessed for sites that predicted treatment response.
Results: Methylation change across groups significantly predicted symptom reduction on 37 of 259 CpG sites tested, with two sites surviving false discovery rate (FDR) correction. Further, the MDMA-treatment group showed more methylation change compared to placebo on one site of the NR3C1 gene.
Conclusion: The findings of this study suggest that therapy-related PTSD symptom improvements may be related to DNA methylation changes in HPA genes and such changes may be greater in those receiving MDMA-assisted therapy. These findings can be used to generate hypothesis driven analyses for future studies with larger cohorts.
Recent investigations demonstrate significant benefits of MDMA (3,4-methylenedioxymethamphetamine)- vs. placebo with therapy in the treatment of PTSD. A pooled analysis of six Phase 2 trials demonstrated that 54.2% of persons treated with MDMA-assisted therapy no longer met criteria for PTSD at study end compared to 22.6% in the control group, and gains continued to be sustained over time (1, 2). These results were recapitulated in a more recent randomized, double-blind, placebo-controlled Phase 3 study (3). The placebo with therapy also resulted in a rather high rate of PTSD loss of diagnosis, though far less substantial than that observed in the MDMA-assisted therapy arm; 67% of MDMA participants no longer met criteria for PTSD at study end compared to 32% in the placebo group. These findings raise important questions about the biological changes that occur in association with treatment response after MDMA-assisted therapy. To date, however, there have not been molecular studies examining predictors of successful MDMA-assisted therapy for PTSD.
Epigenetic mechanisms, such as DNA methylation, particularly of certain hypothalamic-pituitary-adrenal (HPA) axis-related genes, have been implicated in mediating adaptations to life conditions and may potentially serve as molecular markers of brain-body health (4–9). Importantly, alterations of the HPA axis were one of the earliest findings in PTSD (10) and have been repeatedly replicated. There is now ample support for the idea that epigenetic alterations underlie neuroendocrine abnormalities in PTSD and may be implicated in conferring risk for PTSD following trauma exposure (11–13). Epigenetic marks on HPA axis genes have been associated with the prediction and successful outcome of psychotherapy in PTSD (14–17). These and other genes were recently confirmed in a second study examining epigenome-wide correlates of successful psychotherapy in PTSD (18). As such, epigenetic alterations on HPA axis genes may be markers, or even predictors, of successful psychotherapy in PTSD. Taken together, HPA axis gene methylation appears to be a promising epigenetic mechanism for treatment response in PTSD, motivating the current study focus.
Using a sub-sample from the recent Phase 3 clinical trial, we conducted a pilot study investigating the methylation of three key PTSD-relevant HPA genes in association with MDMA-assisted therapy treatment response (3); NR3C1, FKBP5, and CRHR1. The gene NR3C1 encodes for a glucocorticoid receptor (GR) which plays a role in the HPA negative feedback loop. The FKBP5 gene encodes a molecular co-chaperone that interacts with cortisol-GR complexes to regulate its downstream transcription-factor activity. The CRHR1 gene encodes corticotropin-releasing hormone (CRH) receptor 1, which is one of two receptors in this gene family. These genes were chosen based on prior studies demonstrating a change in DNA methylation associating with treatment response in PTSD (14, 15, 17). We hypothesized that (1) symptomatic reduction of PTSD after placebo and MDMA-assisted therapy would be predicted by changes in DNA methylation on NR3C1, FKBP5, and CRHR1; and (2) MDMA-assisted therapy group would exhibit more methylation change compared to placebo, related to the additional efficacy conferred by MDMA relative to placebo conditions.
Participants represent a subsample of patients with severe PTSD from a Phase 3 clinical trial (NCT03537014; N = 90 treated) who consented to be in the epigenetic sub-study (N = 33; MDMA n = 16; placebo n = 7) (3). For the parent clinical trial, participants were recruited at 15 study sites through print and internet advertisements, referrals from treatment providers, and by word of mouth. Of the 11 sites located in the USA, seven participated in recruiting for the epigenetic study.
Following an initial phone screening for the clinical trial, participants provided written informed consent and underwent further screening assessments for eligibility in the clinical trial. Eligible participants were enrolled in the trial and began psychiatric medication taper if needed, lasting from 0 days (no taper needed) to 103 days. Only participants who enrolled in the study and met criteria for severe PTSD after the taper period and preparatory therapy were offered the opportunity to proceed with the clinical trial and enroll in the present epigenetics sub-study. A total of N = 33 participants in the parent clinical trial consented to participate in this epigenetics sub-study; however 10 of the 33 did not provide post-treatment salivary samples due to COVID-19 related interruptions to the study, leaving a total of N = 23 (MDMA, n = 16; Placebo, n = 7) participants with both pre- and post-salivary samples for our analysis.
This study was conducted in accordance with the principles of the Declaration of Helsinki–for full information on the larger study, see here (3). Ethics approval for this epigenetics sub-study was obtained from University of Southern California Institutional Review Board.
For full information on the intervention in the parent clinical trial, see Mitchell et al. (3).
Participants were randomized and allocated 1:1 to either the MDMA-assisted therapy group or the placebo with therapy group. Randomization was stratified by site and occurred following enrollment confirmation. The treatment period consisted of three 8-h experimental sessions of either MDMA-assisted therapy or therapy with inactive placebo control, spaced ∼4 weeks apart. In each experimental session the participants received a single divided dose of 80–180 mg MDMA or placebo. Each experimental session was followed by three 90-min integration sessions that were spaced ∼1 week apart to allow the participant to understand and incorporate their experience. The first integration session always occurred on the morning after the experimental session, and the remaining two integration sessions occurred over the following 3–4 weeks.
A blinded and centralized independent rater pool was used to conduct DSM-5 (The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition) diagnostic assessments at screening and to administer the primary outcome measure [i.e., Clinician-Administered PTSD Scale (CAPS-5) for DSM-5]. The independent rater measurements were conducted at baseline and ∼3 weeks after each of the first two experimental sessions via video interviews. The primary outcome assessment for the clinical trial was the final CAPS-5 conducted ∼8 weeks after the third experimental session (∼18 weeks after the baseline assessment). Saliva samples were collected from participants on days corresponding with the baseline and outcome CAPS-5 assessments to determine epigenetic changes associated with the clinical intervention and primary outcome of PTSD symptoms as measured by CAPS-5.
Saliva samples were collected with Oragene-DNA saliva kits (Ottawa, Ontario, Canada) from 33 participants during the baseline visit. Due to COVID-19 related interruptions to the study, 10 participants were unable to provide follow up samples at the final visit, providing a total of 23 participants with both baseline and final saliva samples (MDMA, n = 16; Placebo, n = 7). DNA was extracted with a DNA Genotek isolation kit (PT-L2P; Ottawa, Ontario, Canada). Sample yield and purity were assessed spectrophotometrically using a NanoDrop ND-1000 (Thermo Scientific, Wilmington, DE, United States). Approximately 500 ng of DNA was treated with sodium bisulfite using the EZ-96 DNA Methylation Kit (Zymo Research, Irvine, CA, United States). DNA methylation was quantified using the Infinium MethylationEPIC BeadChip (IlluminaEPICarray) run on an Illumina iScanSystem (Illumina, San Diego, CA, United States). Raw Intensity Data (IDAT) files were exported for preprocessing in R with the minfi package (19). A filter was applied to remove probes located on the sex chromosomes. Data was subjected to quality control analyses, which included quantile normalization, checking for sex mismatches, and excluding low-intensity samples (p < 0.01). All samples passed quality control. Using the minfi package, data were normalized and annotated with Illumina CpG site probe names. Using the R package EpiDISH (Epigenetic Dissection of Intra-Sample Heterogeneity, 3.8) Robust Partial Correlation (RPC) method, the proportion of estimated epithelial cells was used as a covariate in our statistical models. M-values were used for methylation analysis as has been recommended, especially for the homoscedasticity (20).
To ensure generalizability of the results, the epigenetics sub-sample and the parent clinical trial sample were compared on demographic variables and CAPS-5 baseline and outcome scores. Variables were tested for normality using the Shapiro–Wilk’s method. A Student’s t-test was used for normally distributed variables, a non-parametric Kruskal–Wallis rank sum test for non-normally distributed variables, and a χ2 test for categorical variables. The same procedures were used to compare the MDMA and placebo groups in the sample.
All sites annotated to the genes of interest on the Illumina Infinium MethylationEPIC BeadChip were analyzed. For all sites of interest, a Student’s t-test was used to compare baseline DNA methylation levels between placebo and MDMA groups. All sites significantly different in DNA methylation between groups pre-treatment were removed from further analyses. Multivariate linear regression modeling was conducted to assess if changes in DNA methylation predicted treatment response across groups (hypothesis 1). CpG change scores were used as an independent variable with CAPS-5 change score (final–baseline) as the dependent variable with sex, age, and proportion of estimated epithelial cells as covariates. Because prior associations between methylation of CpG sites on HPA genes with trauma have mixed directions (either hypermethylation, hypomethylation, mixed findings, or differential effects with no direction specified) [see Figure 4 in Watkeys et al. (21)], we allowed for either an increase or decrease in methylation to predict treatment response by calculating absolute value (absΔ) methylation changes scores to test in the same regression models. Because this is a small pilot study, we report both non-corrected and false-discovery rate (FDR < 0.1) corrected results (22–24).
Lastly, to determine if there was a group difference in methylation change, we tested if sites that significantly predicted treatment response across groups (FDR corrected) demonstrated significant group differences in methylation change (hypothesis 2). We compared MDMA and placebo groups on methylation change score with analysis of covariance (ANCOVA) models while controlling for sex, age, and proportion of estimated epithelial cells. Analyses were performed using various packages in R (25).
A note on nomenclature relevant to the following description of CpG genomic locations: CpG sites are often assessed in the CpG-rich islands in and around promotor regions of genes, and the methylation of these CpG sites are generally associated with gene silencing (26). If a CpG site occurs at a distance situated within 2 kb from an island, the location is referred to “north shore” or upstream and “south shore” or downstream from the island; if it occurs at a location within 2–4 kb then it is referred to as “north shelf” or “south shelf” and any CpG site located further than 4 kb from an island are referred to as “open sea” (27). This study assessed all sites annotated to the candidate genes on the Infinium MethylationEPIC BeadChip (IlluminaEPICarray). Beta- and M-values for all three analyzed genes can be found in Supplementary Data Sheets.
The sub-study sample did not differ from the clinical trial sample in age, sex, baseline CAPS-5, or outcome CAPS-5 scores (Table 1A; all p’s > 0.05). Within the sub-study sample, treatment groups (MDMA vs. placebo) did not significantly differ on age or baseline CAPS-5 score but did differ on sex (Table 1B). Sex is used as a covariate in later analyses to account for any bias due to the different group sex compositions. As expected, treatment groups significantly differed on final CAPS-5 score in favor of MDMA conferring more benefit than placebo (Figure 1). A comparison of DNA methylation levels measured from salivary DNA in this study and publicly available methylation level measured from brain can be found in Supplementary Tables 1–3.
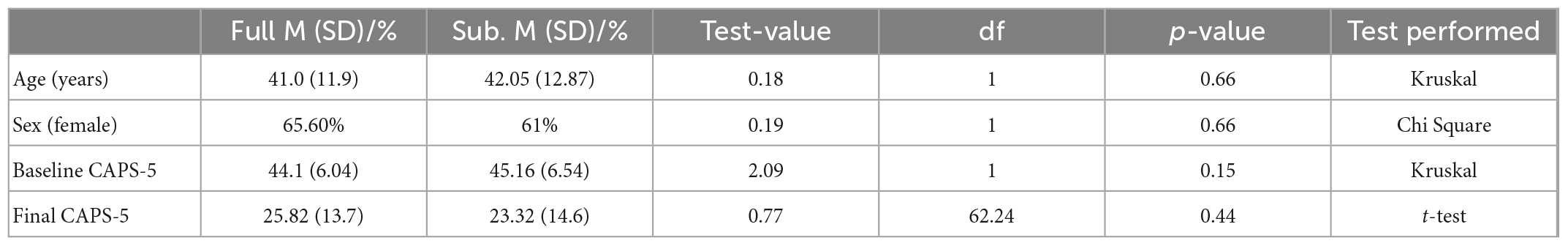
Table 1a. Full clinical study compared to sub-study.
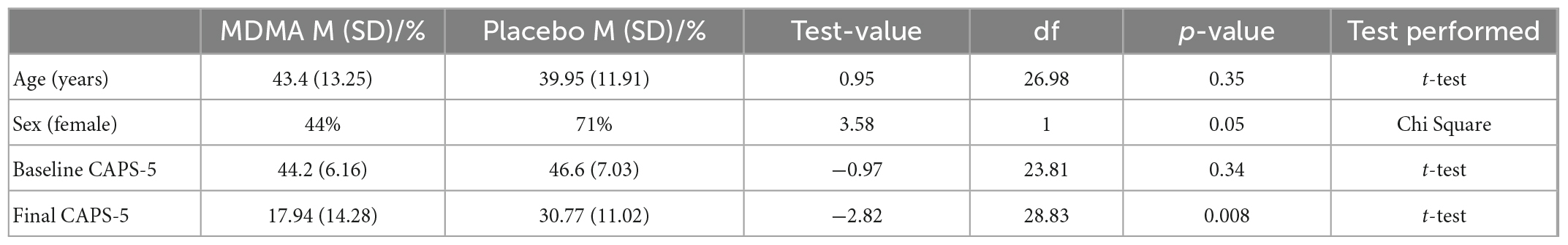
Table 1b. Sub-study sample 3,4-methylenedioxymethamphetamine (MDMA) compared to placebo.
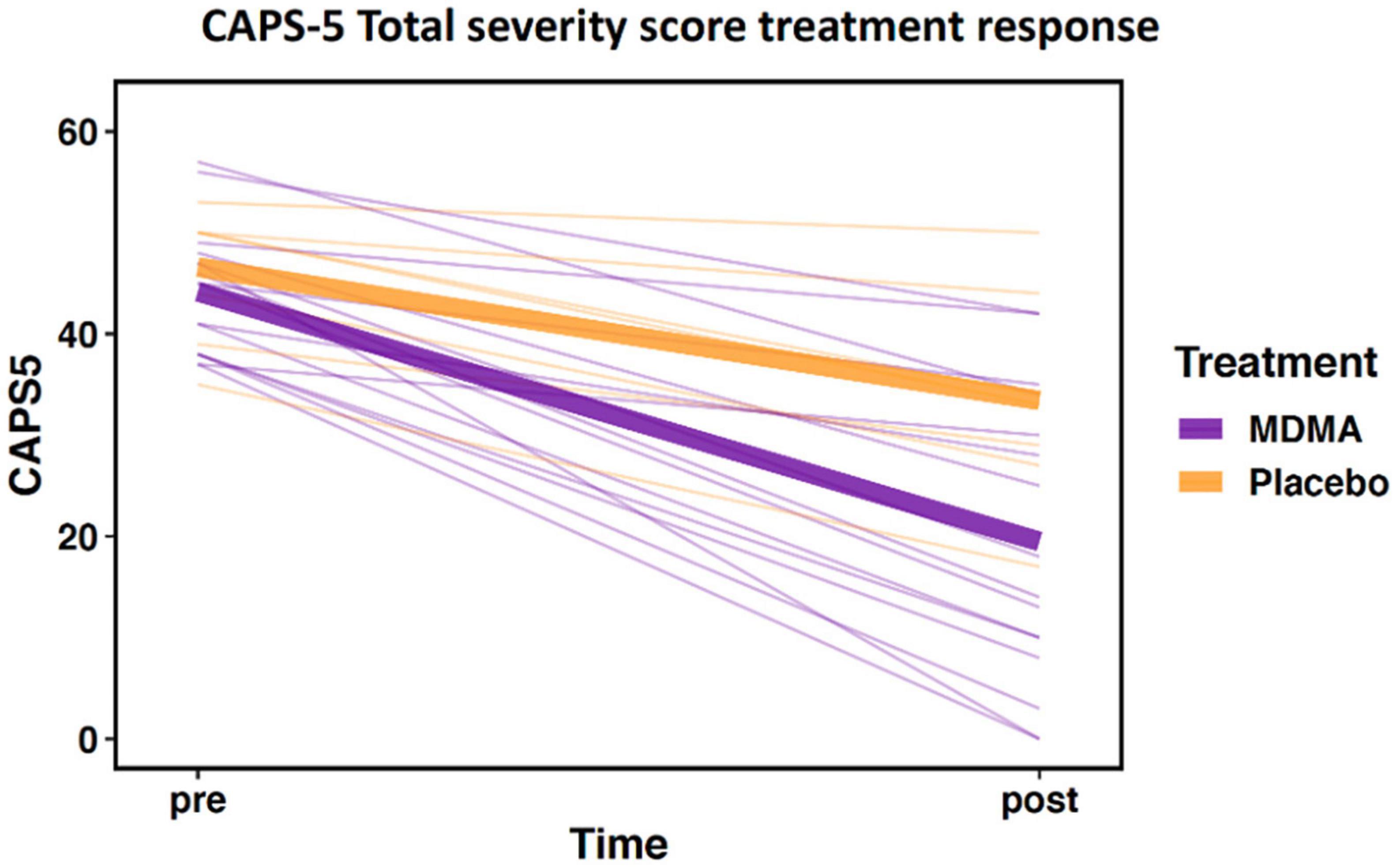
Figure 1. Change in Clinician-Administered PTSD Scale for DSM-5 (CAPS-5) total severity score from pre- to post-treatment. Data demonstrate differential treatment response between 3,4-methylenedioxymethamphetamine (MDMA)-assisted therapy compared to therapy alone (data represent the subset of the clinical trial included in this study; MDMA, n = 16; placebo, n = 7). Thick lines represent group means, thin lines represent individual scores.
Beta values (which represent percent methylation at each CpG site) can be visualized for all CpG sites that significantly predicted treatment response (Supplementary Figures 1–3). Correlation between beta values generated from saliva in this study and publicly available beta values generated from brain were compared. NR3C1 (r = 0.73, p = < 0.00001), CRHR1 (r = 0.88, p = < 0.00001), and FKBP5 (r = 0.83, p = < 0.00001) sites all show strong and significant correlations between saliva and brain samples (Supplementary Tables 1–3). Brain DNA methylation values were obtained from the Allen Brain Atlas BrainSpan data.1
Five CRHR1 sites, two FKBP5 sites, and three NR3C1 sites were significantly different between MDMA and placebo group at baseline thus were removed from further analyses (p’s < 0.05).
Methylation change on 20.9% (17 of 81 sites; 7 Δ; 10 absΔ) of sites annotated to CRHR1 predicted change in CAPS-5 score (p’s < 0.05; Table 2). Ten of these sites resided in open sea regions, two on south shore regions, two on north shore regions, two on an island, and one on a south shelf. The site with the largest Δ effect size survived FDR correction (open sea: cg08276280; B = 42.12, p = 0.005–pre-FDR correction) and is depicted in Figures 2A, B.
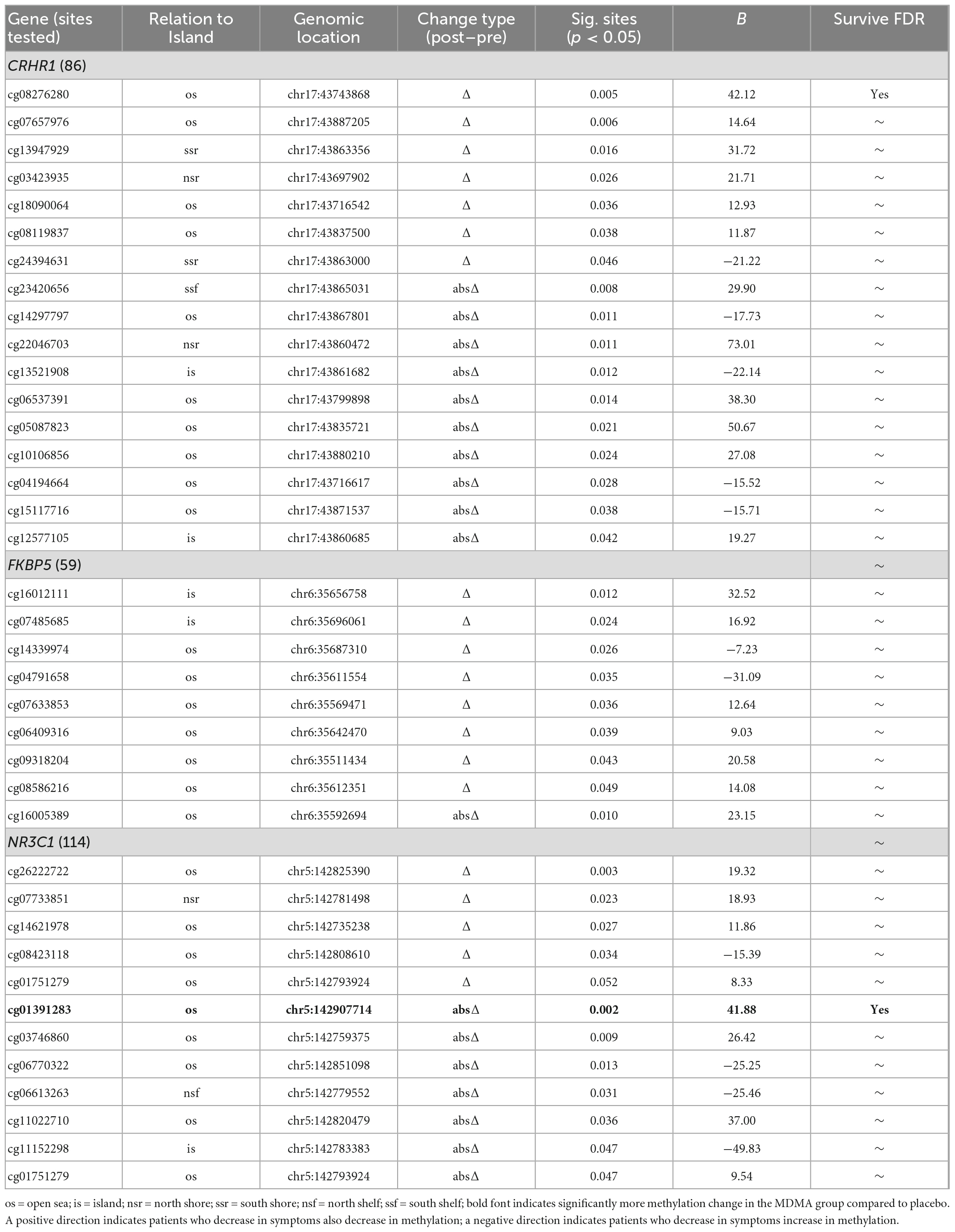
Table 2. Change in methylation predicting change in Clinician-Administered PTSD Scale for DSM-5 (CAPS-5).
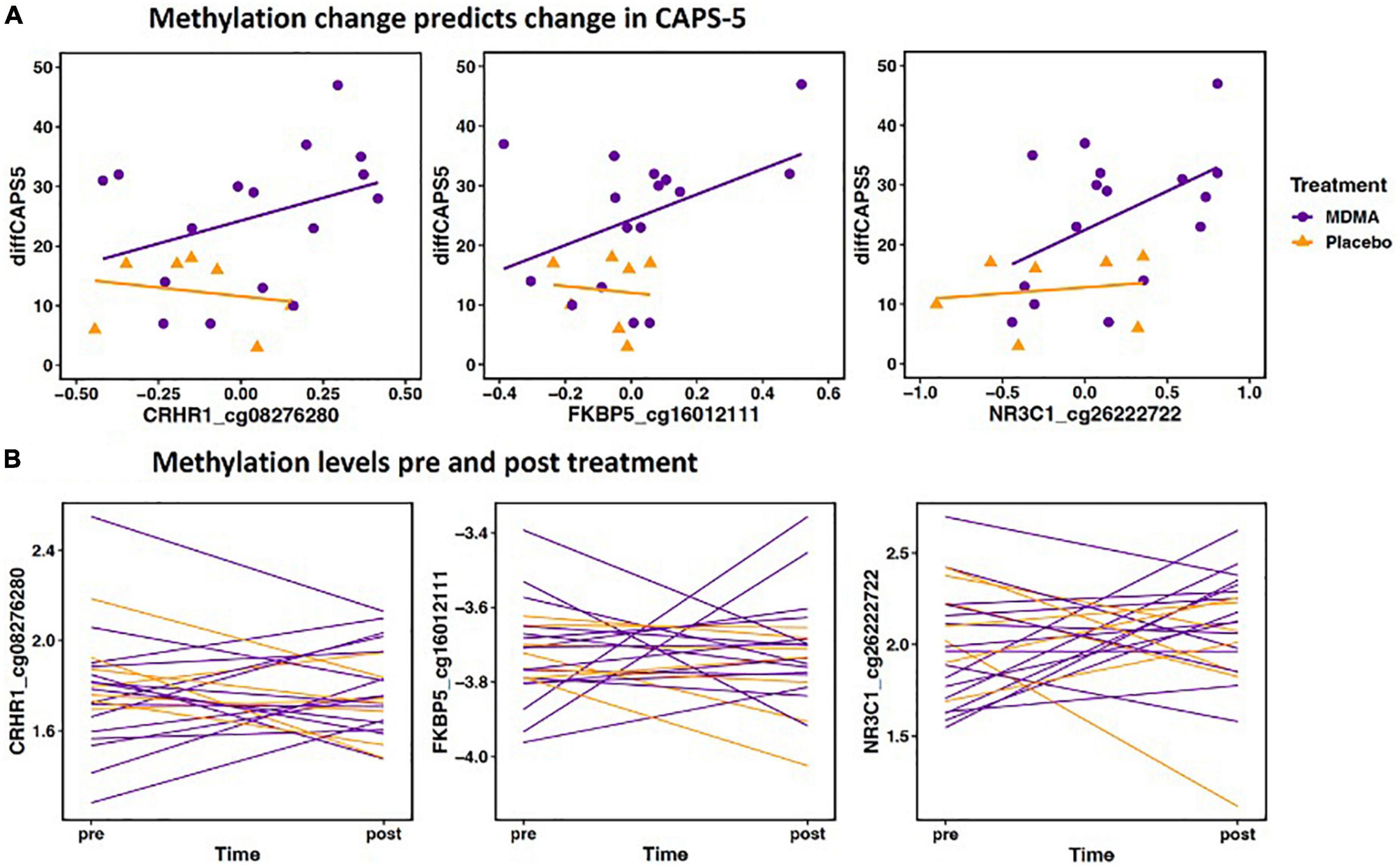
Figure 2. (A) Methylation change predicts change in Clinician-Administered PTSD Scale for DSM-5 (CAPS-5). The scatterplot depicts the three Δ change scores with the largest effect size from CRHR1, FKBP5, and NR3C1 (x axis) plotted with change scores in PTSD symptoms (CAPS-d). All three sites have a significant positive relationship, meaning decreased methylation is associated with decreased PTSD symptoms. The x axis change scores were calculated as post–treatment mVals–pre-treatment M-vals (positive values represent an increase in methylation). The y axis change scores were calculated as baseline–outcome (positive values represent a decrease in symptom severity). CRHR1 cg08276280 B = 42.12, p = 0.005; FKBP5 cg16012111 B = 32.52, p = 0.012; NR3C1 cg26222722 B = 19.32, p = 0.003. (B) Methylation levels from pre and post treatment (mVals). Figures highlight individual variation in methylation change across groups for the same three sites depicted above. M-values are plotted on the Y- axis; positive M-values represent more molecules are methylated than unmethylated, while negative M-values mean the opposite (20).
Methylation change on 15.8% (9 of 57 sites; 8 Δ; 1 absΔ) of sites annotated to FKBP5 predicted change in CAPS-5 score [p’s < 0.05; Table 2]. Seven of these sites resided in open sea regions and two on an island. No sites from this gene survived FDR correction and the site with the largest Δ effect size for this gene (island site: cg16012111; B = 32.52, p = 0.012) is depicted in Figures 2A, B.
Methylation change on 10% (11 of 111 sites; 4 Δ; 6 absΔ; 1 Δ and absΔ) of sites annotated to NR3C1 predicted change in CAPS-5 score (p’s ≤ 0.05; Table 2). Nine of these sites resided in open sea regions, one on north shore regions, one on an island, and one on a north shelf. One site (absΔ; open sea; cg01391283; Figure 3) from this gene survived FDR correction. The site with the largest Δ effect size (open sea: cg6222722; B = 19.32, p = 0.003) is depicted in Figures 2A, B.
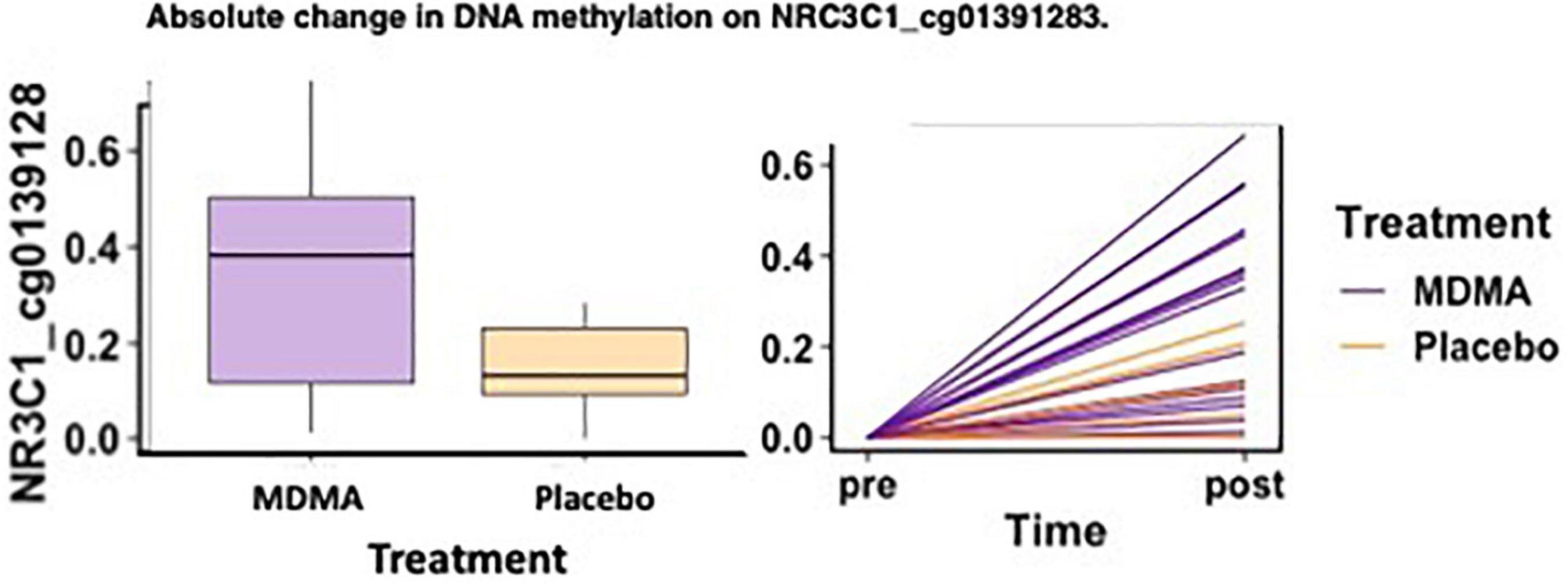
Figure 3. MDMA-assisted therapy group had more absolute change in methylation on NRC3C1_cg01391283 compared to the placebo group. Change in DNA methylation (absΔ) at NRC3C1_cg01391283 from pre-treatment to post-treatment [F(1, 18) = 4.78, p = 0.042].
Of the two sites that predicted treatment response across groups after FDR correction (CRHR1 Δ cg08276280 and NR3C1 absΔ cg01391283), one site also had a significant difference in methylation change between the MDMA and placebo groups [NR3C1; open sea; cg01391283 absΔ; F(1, 18) = 4.78, p = 0.042]. A figure depicting the absolute change score plotted from pre- to post-treatment can be found in Figure 3.
This is the first study to examine DNA methylation relationships with treatment response to MDMA-assisted therapy in patients with severe PTSD. Due to being a small pilot study, we report both non-corrected significant results and FDR-corrected results. Before FDR correction, seventeen CRHR1, nine FKBP5, and eleven NR3C1 CpG sites predicted treatment response across groups. After FDR correction, one CRHR1 (cg08276280) CpG site and one NR3C1 (cg01391283) CpG remained significant. The results reinforce previous research that symptom reduction after PTSD treatment correlates with DNA methylation changes on the CRHR1 and NR3C1 genes (14, 17). Of the two sites that predicted treatment response across groups after FDR correction, NR3C1 cg01391283 had a significantly larger change in methylation in the MDMA group compared to placebo.
Genes of the HPA axis have been extensively studied in the context of epigenetic response to trauma (8). The most commonly-studied epigenetic candidate gene in the HPA axis is the glucocorticoid receptor gene (nuclear receptor subfamily 3 group C member 1; NR3C1) (21). Glucocorticoid binding at this receptor regulates the stress response through a negative feedback loop (28). Much research to date demonstrates that childhood trauma and early stress alter the methylation status of this gene and its expression [for reviews see Watkeys et al. (21), Jiang et al. (29), and Palma-Gudiel et al. (30)]. While the majority of results find early life stress leads to an increase in methylation and a decrease of expression of NR3C1, opposing directional findings are reported (21). In addition to trauma-related methylation changes, two seminal papers found NR3C1 CpG methylation was associated with PTSD symptoms and many have replicated these findings (12, 31, 32). Taking into consideration the dynamic nature of epigenetic marks and their malleability in response to psycho-social experiences, recent research has focused on potential epigenetic changes in response to psychotherapy for stress-related disorders [for review see here (33)]. Our results add to this growing field, suggesting epigenetic change on the NR3C1 gene may relate to treatment response to MDMA-assisted therapy. Further, this pilot study was intended to inform hypothesis development, these results suggest specific sites to examine on three HPA genes in future studies with a larger sample size.
The NR3C1 gene consists of nine non-coding first exons and seven of these are located within a CpG island spanning 3 kb along the proximal promoter region of the NR3C1 gene (30). The majority of studies assessing NR3C1 methylation in relation to stress, psychopathology, and treatment response have focused on differential methylation primarily in this CpG island. This work has shaped general discussions about the function of experience-dependent DNA methylation changes. There has been much discussion over the notion of epigenetic modifications turning gene regulation “up or down” or “on or off” since methylation in the immediate vicinity of the transcriptional start site inhibits the initiation of transcription. CpG sites with differential methylation are located all along the NR3C1 gene sequence and the position of methylation across the gene influences its functionality. For example, methylation in the gene body does not block and might even stimulate transcription elongation or have an impact on splicing (34, 35). While the functional implications of upstream methylation is less understood, some data suggests it may increase expression (36). Indeed, others have found methylation of the NR3C1 promoter region regulates transcript expression and levels (37–39). In this study, the two sites that predicted change in PTSD symptoms are both in open sea regions. To date, little is known about the functional consequences of DNA methylation in open sea positions. Therefore, it is difficult to speculate on the potential downstream effects of open sea DNA methylation changes on the HPA system.
While the majority of research to date has found increased NR3C1 promotor methylation associated with trauma, the results are not always consistent. A 2018 systematic review summarized results across studies and effectively visualized how CpG sites in the 1F region of NR3C1 have been associated with hypermethylation, hypomethylation, non-significant findings, mixed findings, or differential effects with no direction specified, in association with both depression and childhood trauma (21). This suggests that trauma may dysregulate the HPA axis through differing routes, i.e., increase or decrease in methylation at various CpG sites, and that both hyper- and hypo-methylation can be maladaptive (30). Therefore, it stands to reason that psychotherapeutic effects mediating treatment response may occur in opposite directions depending on the individual, gene, and CpG site. For this reason, we chose to not only assess raw difference scores as treatment predictors, but also absolute difference scores which allow for a change in either direction. This novel analytic approach highlighted interesting findings such that the CpG methylation absolute difference score was more sensitive at predicting treatment response than the raw difference scores for NR3C1 and CRHR1. Taken together, these results suggest that both trauma-induced dysregulation and psychotherapy-induced changes of the epigenome may entail either hyper- or hypo-methylation dependent on the individual.
MDMA induces a physiological response similar to an activated stress response, such as increased heart rate, blood pressure, oral temperature, and cortisol levels (40). MDMA and acute stress also increase levels of serotonin, dopamine, and norepinephrine (41–44). While the mechanisms behind acute stress induced changes in DNA methylation are not known, it is plausible that the increased monoamine and cortisol exposure induced by psychological stress may be involved in inducing a transient state of epigenetic-malleability (45, 46). Especially taken into consideration that the cortisol-bound glucocorticoid receptor directly up- and downregulates thousands of genes as a transcription factor and other mechanisms (47). Further, the glucocorticoid receptor may directly influence DNA methylation through decreasing activity of a transcription factor, p53, known to regulate DNA methyltransferase (DNMT) (48, 49). However, MDMA-induced psychological effects are quite opposite to an acute stressor or traumatic experience. MDMA produces unique subjective properties such as reduced anxiety, acute positive affect, increased insight, accelerated thinking and euphoria, and increased sense of trust and bonding (50–52). Taken together, MDMA may induce a transient epigenetic-malleable state similarly to that of stress but a psychological state highly conducive to successful psychotherapy. Therefore, MDMA-assisted therapy may serve as an “inverse trauma experience,” such that acute stress and MDMA produce similar physiological states and a highly salient psychological experience. However, trauma has the potential to alter epigenetics underlying symptom formation, whereas MDMA-assisted therapy has the potential to alter epigenetics underlying symptom reduction.
While this study highlights potential biological mechanisms underlying MDMA-assisted therapy for PTSD, there are limitations to address. This initial pilot study was small, intended to assess preliminary evidence to support a larger future investigation. As such, additional studies with more power should be conducted to validate these results, however, these results provide specific loci to be studied in the future which may negate the need for p-value correction. The small sample size also necessitated the use of a candidate gene approach; while this study focuses on HPA axis genes, there are other potentially informative genes not assessed here [e.g., immune/inflammatory genes such as IL-12 and IFN-γ, for example see Morrison et al. (53)]. However, our results corroborate a large body of literature associating HPA gene methylation and stress-related disorders. While we accounted for participant age, sex, and cell count in our analyses, we were not powered to control for other factors such as race, smoking, ancestry, or trauma histories. Of note, the Infinium EPIC array probes we utilized do not fully incorporate all CpG sites that have been investigated previously. For example, previous studies have focused on the promoter region in intron seven of the FKBP5 gene, demonstrating epigenetic changes correlating with trauma exposure and PTSD symptomatology (14, 15, 54). However, these sites were not included in the EPIC array so we cannot ascertain if methylation changes in this region were related to MDMA-assisted therapy efficacy. Finally, it is important to note the limitations of using peripheral samples, which cannot elucidate associations in relevant brain tissue.
More than ever, it has become clear that epigenetic changes in response to trauma or stress co-occur with behavioral and physiological symptoms associated with stress-related disorders. Here, we add to a growing number of studies demonstrating that psychotherapeutic experiences may also lead to alterations in the epigenetic landscape underlying symptom improvement. With the small pilot study sample size, it is difficult to determine if the greater efficacy of MDMA-assisted therapy compared to placebo therapy, is driven by greater changes in epigenetic regulation of HPA genes. However, these findings do suggest potential epigenetic mechanisms of MDMA-assisted therapy and their role in symptom reduction is worthy of continued investigation.
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the University of Southern California Institutional Review Board. The patients/participants provided their written informed consent to participate in this study.
BC, JT, DR, BK, CL, RY, and BY-K conceived and planned the experiments. BC organized the data acquisition and IRB approval in collaboration with CH, BY-K, and other MAPS Study Site personnel. BC and CH created the study protocol, instruction manual, trained sites on this sub-study. CL carried out DNA sample preparations, methylation microarrays, conducted all data analyses, and wrote the manuscript. CL and SS processed DNA methylation data. CL and JG created tables and figures. CL, BC, JT, RY, and BY-K contributed to the interpretation of the results. All authors contributed to manuscript revision, read and approved the submitted version.
Funding for this study was provided by Modern Spirit, a non-profit organization via a grant to Principal Investigator BC.
BY-K and CH received payment for full-time employment from the MAPS or the MAPS Public Benefit Corporation throughout their work on the parent trial. JT is the Executive Director of Modern Spirit, the non-profit organization that funded this study. BC is on the Board of Directors for Modern Spirit.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2023.959590/full#supplementary-material
HPA, hypothalamic-pituitary-adrenal; PTSD, post-traumatic stress disorder; MDMA, 3,4-methylenedioxymethamphetamine.
1. Mithoefer M, Feduccia A, Jerome L, Mithoefer A, Wagner M, Walsh Z, et al. MDMA-assisted psychotherapy for treatment of PTSD: study design and rationale for phase 3 trials based on pooled analysis of six phase 2 randomized controlled trials. Psychopharmacology. (2019) 236:2735–45. doi: 10.1007/s00213-019-05249-5
2. Jerome L, Feduccia A, Wang J, Hamilton S, Yazar-Klosinski B, Emerson A, et al. Long-term follow-up outcomes of MDMA-assisted psychotherapy for treatment of PTSD: a longitudinal pooled analysis of six phase 2 trials. Psychopharmacology. (2020) 237:2485–97. doi: 10.1007/s00213-020-05548-2
3. Mitchell J, Bogenschutz M, Lilienstein A, Harrison C, Kleiman S, Parker-Guilbert K, et al. MDMA-assisted therapy for severe PTSD: a randomized, double-blind, placebo-controlled phase 3 study. Nat Med. (2021) 27:1025–33. doi: 10.1038/s41591-021-01336-3
4. McEwen B. Epigenetic interactions and the brain-body communication. Psychother Psychosom. (2017) 86:1–4. doi: 10.1159/000449150
5. Vaiserman A. Epigenetic programming by early-life stress: evidence from human populations. Dev Dyn. (2015) 244:254–65. doi: 10.1002/dvdy.24211
6. Matosin N, Cruceanu C, Binder EB. Preclinical and clinical evidence of DNA methylation changes in response to trauma and chronic stress. Chronic Stress. (2017) 1:247054701771076. doi: 10.1177/2470547017710764
7. Lewis CR, Olive MF. Early-life stress interactions with the epigenome: potential mechanisms driving vulnerability toward psychiatric illness. Behav Pharmacol. (2014) 25:341–51. doi: 10.1097/FBP.0000000000000057
8. Murgatroyd C, Spengler D. Epigenetic programming of the HPA axis: early life decides. Stress. (2011) 14:581–9. doi: 10.3109/10253890.2011.602146
9. Lewis C, Breitenstein R, Henderson A, Sowards H, Piras I, Huentelman M, et al. Harsh parenting predicts novel HPA receptor gene methylation and NR3C1 methylation predicts cortisol daily slope in middle childhood. Cell Mol Neurobiol. (2021) 41:783–93. doi: 10.1007/s10571-020-00885-4
10. Yehuda R, Giller EL, Southwick SM, Lowy MT, Mason JW. Hypothalamic-pituitary-adrenal dysfunction in posttraumatic stress disorder. Biol Psychiatry. (1991) 30:1031–48. doi: 10.1016/0006-3223(91)90123-4
11. Zannas AS, Provençal N, Binder EB. Epigenetics of posttraumatic stress disorder: current evidence, challenges, and future directions. Biol Psychiatry. (2015) 78:327–35. doi: 10.1016/j.biopsych.2015.04.003
12. Mehta D, Miller O, Bruenig D, David G, Shakespeare-Finch J. A systematic review of DNA methylation and gene expression studies in posttraumatic stress disorder, posttraumatic growth, and resilience. J Trauma Stress. (2020) 33:171–80. doi: 10.1002/jts.22472
13. Fischer S, Schumacher T, Knaevelsrud C, Ehlert U, Schumacher S. Genes and hormones of the hypothalamic-pituitary-adrenal axis in post-traumatic stress disorder. What is their role in symptom expression and treatment response? J Neural Transm. (2021) 128:1279–86. doi: 10.1007/s00702-021-02330-2
14. Yehuda R, Daskalakis N, Desarnaud F, Makotkine I, Lehrner A, Koch E, et al. Epigenetic biomarkers as predictors and correlates of symptom improvement following psychotherapy in combat veterans with PTSD. Front Psychiatry. (2013) 4:118. doi: 10.3389/fpsyt.2013.00118
15. Bishop J, Lee A, Mills L, Thuras P, Eum S, Clancy D, et al. Methylation of FKBP5 and SLC6A4 in relation to treatment response to mindfulness based stress reduction for posttraumatic stress disorder. Front Psychiatry. (2018) 9:418. doi: 10.3389/fpsyt.2018.00418
16. Vinkers C, Geuze E, van Rooij S, Kennis M, Schür R, Nispeling D, et al. Successful treatment of post-traumatic stress disorder reverses DNA methylation marks. Mol Psychiatry. (2021) 26:1264–71. doi: 10.1038/s41380-019-0549-3
17. Pape J, Carrillo-Roa T, Rothbaum B, Nemeroff C, Czamara D, Zannas A, et al. DNA methylation levels are associated with CRF1 receptor antagonist treatment outcome in women with post-traumatic stress disorder. Clin Epigenetics. (2018) 10:136. doi: 10.1186/s13148-018-0569-x
18. Yang R, Xu C, Bierer L, Flory J, Gautam A, Bader H, et al. Longitudinal genome-wide methylation study of PTSD treatment using prolonged exposure and hydrocortisone. Transl Psychiatry. (2021) 11:398. doi: 10.1038/s41398-021-01513-5
19. Aryee M, Jaffe A, Corrada-Bravo H, Ladd-Acosta C, Feinberg A, Hansen K, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics. (2014) 30:1363–9. doi: 10.1093/bioinformatics/btu049
20. Du P, Zhang X, Huang C, Jafari N, Kibbe W, Hou L, et al. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. (2010) 11:587. doi: 10.1186/1471-2105-11-587
21. Watkeys OJ, Kremerskothen K, Quidé Y, Fullerton JM, Green MJ. Glucocorticoid receptor gene (NR3C1) DNA methylation in association with trauma, psychopathology, transcript expression, or genotypic variation: a systematic review. Neurosci Biobehav Rev. (2018) 95:85–122. doi: 10.1016/j.neubiorev.2018.08.017
22. Guma E, Devenyi G, Malla A, Shah J, Chakravarty M, Pruessner M. Neuroanatomical and symptomatic sex differences in individuals at clinical high risk for psychosis. Front Psychiatry. (2017) 8:291. doi: 10.3389/fpsyt.2017.00291
23. Son S, Park B, Choi J, Roh H, Kim N, Sin J, et al. Psychological resilience enhances the orbitofrontal network in the elderly with mild cognitive impairment. Front Psychiatry. (2019) 10:615. doi: 10.3389/fpsyt.2019.00615
24. Yuferov V, Zhang Y, Liang Y, Zhao C, Randesi M, Kreek M. Oxycodone self-administration induces alterations in expression of integrin, semaphorin and ephrin genes in the mouse striatum. Front Psychiatry. (2018) 9:257. doi: 10.3389/fpsyt.2018.00257
25. R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing (2020).
26. Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. (2008) 9:465–76. doi: 10.1038/nrg2341
27. Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana M, Bibikova M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. (2011) 6:692–702. doi: 10.4161/epi.6.6.16196
28. Herman J, McKlveen J, Ghosal S, Kopp B, Wulsin A, Makinson R, et al. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr Physiol. (2016) 6:603–21. doi: 10.1002/cphy.c150015
29. Jiang S, Postovit L, Cattaneo A, Binder EB, Aitchison KJ. Epigenetic modifications in stress response genes associated with childhood trauma. Front Psychiatry. (2019) 10:808. doi: 10.3389/fpsyt.2019.00808
30. Palma-Gudiel H, Córdova-Palomera A, Leza J, Fañanás L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: a critical review. Neurosci Biobehav Rev. (2015) 55:520–35. doi: 10.1016/j.neubiorev.2015.05.016
31. Labonté B, Azoulay N, Yerko V, Turecki G, Brunet A. Epigenetic modulation of glucocorticoid receptors in posttraumatic stress disorder. Transl Psychiatry. (2014) 4:e368.
32. Yehuda R, Flory J, Bierer L, Henn-Haase C, Lehrner A, Desarnaud F, et al. Lower methylation of glucocorticoid receptor gene promoter 1F in peripheral blood of veterans with posttraumatic stress disorder. Biol Psychiatry. (2015) 77:356–64. doi: 10.1016/j.biopsych.2014.02.006
33. Schiele MA, Gottschalk MG, Domschke K. The applied implications of epigenetics in anxiety, affective and stress-related disorders - A review and synthesis on psychosocial stress, psychotherapy and prevention. Clin Psychol Rev. (2020) 77:101830. doi: 10.1016/j.cpr.2020.101830
34. Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. (2013) 23:1256–69. doi: 10.1038/cr.2013.110
35. Jones P. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. (2012) 13:484–92. doi: 10.1038/nrg3230
36. Rauscher G, Kresovich J, Poulin M, Yan L, Macias V, Mahmoud A, et al. Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC Cancer. (2015) 15:816. doi: 10.1186/s12885-015-1777-9
37. McGowan P, Sasaki A, D’Alessio A, Dymov S, Labonté B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. (2009) 12:342–8. doi: 10.1038/nn.2270
38. Labonte B, Yerko V, Gross J, Mechawar N, Meaney M, Szyf M, et al. Differential glucocorticoid receptor exon 1(B), 1(C), and 1(H) expression and methylation in suicide completers with a history of childhood abuse. Biol Psychiatry. (2012) 72:41–8. doi: 10.1016/j.biopsych.2012.01.034
39. Leenen FAD, Muller CP, Turner JD. DNA methylation: conducting the orchestra from exposure to phenotype? Clin Epigenetics. (2016) 8:92. doi: 10.1186/s13148-016-0256-8
40. Tancer M, Johanson CE. Reinforcing, subjective, and physiological effects of MDMA in humans: a comparison with d-amphetamine and mCPP. Drug Alcohol Depend. (2003) 72:33–44. doi: 10.1016/s0376-8716(03)00172-8
41. Schenk S, Highgate Q. Methylenedioxymethamphetamine (MDMA): serotonergic and dopaminergic mechanisms related to its use and misuse. J Neurochem. (2021) 157:1714–24. doi: 10.1111/jnc.15348
42. Morilak D, Barrera G, Echevarria D, Garcia A, Hernandez A, Ma S, et al. Role of brain norepinephrine in the behavioral response to stress. Prog Neuropsychopharmacol Biol Psychiatry. (2005) 29:1214–24. doi: 10.1016/j.pnpbp.2005.08.007
43. Vaessen T, Hernaus D, Myin-Germeys I, van Amelsvoort T. The dopaminergic response to acute stress in health and psychopathology: a systematic review. Neurosci Biobehav Rev. (2015) 56:241–51. doi: 10.1016/j.neubiorev.2015.07.008
44. Chaouloff F, Berton O, Mormède P. Serotonin and stress. Neuropsychopharmacology. (1999) 21:28S–32.
45. Glad C, Andersson-Assarsson J, Berglund P, Bergthorsdottir R, Ragnarsson O, Johannsson G. Reduced DNA methylation and psychopathology following endogenous hypercortisolism - a genome-wide study. Sci Rep. (2017) 7:44445. doi: 10.1038/srep44445
46. Müller D, Grevet E, da Silva B, Charão M, Rovaris D, Bau C. The neuroendocrine modulation of global DNA methylation in neuropsychiatric disorders. Mol Psychiatry. (2021) 26:66–9. doi: 10.1038/s41380-020-00924-y
47. Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR. Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol. (2017) 18:159–74. doi: 10.1038/nrm.2016.152
48. Sengupta S, Vonesch JL, Waltzinger C, Zheng H, Wasylyk B. Negative cross-talk between p53 and the glucocorticoid receptor and its role in neuroblastoma cells. EMBO J. (2000) 19:6051–64. doi: 10.1093/emboj/19.22.6051
49. Lara H, Wang Y, Beltran A, Juárez-Moreno K, Yuan X, Kato S, et al. Targeting serous epithelial ovarian cancer with designer zinc finger transcription factors. J Biol Chem. (2012) 287:29873–86. doi: 10.1074/jbc.M112.360768
50. Dumont G, Sweep F, van der Steen R, Hermsen R, Donders A, Touw D, et al. Increased oxytocin concentrations and prosocial feelings in humans after ecstasy (3,4-methylenedioxymethamphetamine) administration. Soc Neurosci. (2009) 4:359–66. doi: 10.1080/17470910802649470
51. Kirkpatrick M, Delton AW, Robertson TE, De Wit H. Prosocial effects of MDMA: a measure of generosity. J Psychopharmacol. (2015) 29:661–8. doi: 10.1177/0269881115573806
52. Liechti ME, Vollenweider FX. Which neuroreceptors mediate the subjective effects of MDMA in humans? A summary of mechanistic studies. Hum Psychopharmacol. (2001) 16:589–98. doi: 10.1002/hup.348
53. Morrison FG, Miller MW, Logue MW, Assef M, Wolf EJ. DNA methylation correlates of PTSD: recent findings and technical challenges. Prog Neuropsychopharmacol Biol Psychiatry. (2019) 90:223–34. doi: 10.1016/j.pnpbp.2018.11.011
Keywords: PTSD, MDMA, MDMA-assisted therapy, HPA, NR3C1, glucocorticoid receptor, epigenetics, DNA methylation
Citation: Lewis CR, Tafur J, Spencer S, Green JM, Harrison C, Kelmendi B, Rabin DM, Yehuda R, Yazar-Klosinski B and Cahn BR (2023) Pilot study suggests DNA methylation of the glucocorticoid receptor gene (NR3C1) is associated with MDMA-assisted therapy treatment response for severe PTSD. Front. Psychiatry 14:959590. doi: 10.3389/fpsyt.2023.959590
Received: 01 June 2022; Accepted: 17 January 2023;
Published: 06 February 2023.
Edited by:
Peter Schuyler Hendricks, University of Alabama at Birmingham, United StatesReviewed by:
Gilles Maussion, Montreal Neurological Institute, CanadaCopyright © 2023 Lewis, Tafur, Spencer, Green, Harrison, Kelmendi, Rabin, Yehuda, Yazar-Klosinski and Cahn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Candace R. Lewis,  Y2FuZGFjZS5sZXdpc0Bhc3UuZWR1; Baruch Rael Cahn, cmFlbC5jYWhuQHVzYy5lZHU=
Y2FuZGFjZS5sZXdpc0Bhc3UuZWR1; Baruch Rael Cahn, cmFlbC5jYWhuQHVzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.