 Albert Stuart Reece1,2*
Albert Stuart Reece1,2* Gary Kenneth Hulse1,2
Gary Kenneth Hulse1,2- 1Division of Psychiatry, University of Western Australia, Crawley, WA, Australia
- 2School of Medical and Health Sciences, Edith Cowan University, Joondalup, WA, Australia
Much recent attention has been directed toward the spatial organization of the cell nucleus and the manner in which three-dimensional topologically associated domains and transcription factories are epigenetically coordinated to precisely bring enhancers into close proximity with promoters to control gene expression. Twenty lines of evidence robustly implicate cannabinoid exposure with accelerated organismal and cellular aging. Aging has recently been shown to be caused by increased DNA breaks. These breaks rearrange and maldistribute the epigenomic machinery to weaken and reverse cellular differentiation, cause genome-wide DNA demethylation, reduce gene transcription, and lead to the inhibition of developmental pathways, which contribute to the progressive loss of function and chronic immune stimulation that characterize cellular aging. Both cell lineage-defining superenhancers and the superanchors that control them are weakened. Cannabis exposure phenocopies the elements of this process and reproduces DNA and chromatin breakages, reduces the DNA, RNA protein and histone synthesis, interferes with the epigenomic machinery controlling both DNA and histone modifications, induces general DNA hypomethylation, and epigenomically disrupts both the critical boundary elements and the cohesin motors that create chromatin loops. This pattern of widespread interference with developmental programs and relative cellular dedifferentiation (which is pro-oncogenic) is reinforced by cannabinoid impairment of intermediate metabolism (which locks in the stem cell-like hyper-replicative state) and cannabinoid immune stimulation (which perpetuates and increases aging and senescence programs, DNA damage, DNA hypomethylation, genomic instability, and oncogenesis), which together account for the diverse pattern of teratologic and carcinogenic outcomes reported in recent large epidemiologic studies in Europe, the USA, and elsewhere. It also accounts for the prominent aging phenotype observed clinically in long-term cannabis use disorder and the 20 characteristics of aging that it manifests. Increasing daily cannabis use, increasing use in pregnancy, and exponential dose-response effects heighten the epidemiologic and clinical urgency of these findings. Together, these findings indicate that cannabinoid genotoxicity and epigenotoxicity are prominent features of cannabis dependence and strongly indicate coordinated multiomics investigations of cannabinoid genome-epigenome-transcriptome-metabolome, chromatin conformation, and 3D nuclear architecture. Considering the well-established exponential dose-response relationships, the diversity of cannabinoids, and the multigenerational nature of the implications, great caution is warranted in community cannabinoid penetration.
1. Introduction
From recent investigations, four important overarching themes have emerged, which assist and direct an updated understanding of cannabinoid pathophysiology. In particular, the integrated multi-channel study of the genome, epigenome, transcriptome, proteome, metabolome, and numerous histone modifications has provided unprecedented insights into the control of gene transcription and cellular behavior both in normal growth and development and in numerous diseases (1).
First, by introducing DNA breaks (2–8) and inducing global DNA hypomethylation (9–13), cannabis directly drives cellular and organismal aging, including epigenomic DNA methylation age (14), increases cardiovascular–organismal age (15), and results in the increased incidence of acute and chronic physical and mental diseases, including protean psychiatric disorders (16).
Second, by disrupting the basic epigenomic machinery of DNA methylation (9–13, 17–20), as well as histone methylation and acetylation (21, 22), the coordination between the histone code and the methylome (11), and the machinery for nucleosome repositioning (11), cannabinoids change the way the cell nucleus processes information, including gene transcription. This includes perturbation of the chromatin looping structures from which genes are transcribed by altering the CTCF (CCCTC-binding factor) boundary elements that delimit and define the loops and by disrupting the cohesin motors that drive and form DNA loops (11). Together, these changes significantly alter the nuclear structure and enhancer–promoter interactions at an ultrafine resolution and can thereby prime cells for malignant transformation. Such actions on germ cells (eggs and sperm) lead to congenital anomalies and conceptus aging.
Third, there is growing evidence that multiomics interactions between the metabolome, the microbiome, the immunome, the genome, and the epigenome are all interdependent and interrelated and cannot be properly understood without reference to one another. As cannabis is known to disrupt each level of this intercalated cascade, this must be borne in mind when considering its plethoric presentations. For example, it is known that the metabolome controls the epigenome in many ways (23, 24) and can reset the lineage determination set point of the cell away from full differentiation and toward dedifferentiation and premalignant preparedness for transformation (the Warburg effect) (25). Cannabis disrupts the post-translational tubulin code at several points (11, 26), inducing chromosomal missegregation, micronucleus formation (27–32), cell transformation, neurodevelopmental and congenital defects, and fetal loss (27, 33–37). The microbiome signals via the metabolome and the immunome (38–40). Similarly, the glycome bidirectionally interacts with the metabolome and the remaining cellular machinery and modulates the epigenome, the immunome, the microbiome, and aging (41–52).
Mitochondria are a major signaling hub within the cell (23, 24). The well-known inhibitory activities of cannabinoids on many mitochondrial functions (53–66) imply mitonuclear stress signaling to the nucleus (23, 24), endoplasmic stress response induction (67–72), and reduced supply of energy and metabolomic substrates to the epigenomic machinery (23, 24). Mitochondrial inhibition also increases cellular lactate (73, 74), which, in turn, increases the lactylation of major rate-limiting enzymes in glycolysis, oxidative phosphorylation, and related pathways (73, 74), alters the epigenomic structure and again dedifferentiates the cell, and primes it toward malignant pretransformation (73, 74). Increased shunting of glucose through the pentose phosphate pathway changes the synthesis of glycan groups for local and intercellular use (75) and can thus change the local tissue and tumor microenvironment.
Fourthly, the physiologic function of endocannabinoids under normal conditions is to signal the end of a synaptic trafficking event retrogradely from the post-synaptic membrane to the pre-synaptic membrane (76–78). It is well established that synapse formation and growth are activity-dependent and that much of the computation occurs based on the structure, size, strength, and chemical nature of the synapse (79–87), that is, the locus of many computations, including memory, is primarily synaptic (79–87). Flooding the synapse with xenophytocannabinoids, especially chronically, clearly grossly disrupts and perturbs this delicate process, thereby deranging the basic unit of brain computation (76, 88–90) and inducing downregulation of CB1Rs1 in the ventral striatum of the midbrain (91, 92). This cannabinoid synaptopathy is exacerbated by the usually pro-inflammatory actions of many cannabinoids on brain astrocytes and microglia (93–102), as well as the powerful negative effect of cannabinoids on oligodendroglial progenitor cells (OPCs). These cells create the myelin sheaths that nourish and preserve axons and white matter tracts. Additionally, the negative effects of cannabinoids on brain neuronogenesis (103–106) contribute to the exacerbation. These effects have been found to accumulate and cause a high degree of cortical white matter disconnection in chronic cannabis users (107).
1.1. Cannabinoid signaling
The complexity of the endocannabinoid system (ECS) in terms of its two main endogenous ligands and their synthesizing and metabolizing enzymes, as well as the many lipid molecules that interact with the ECS, has been described by many authors. However, for present purposes, it is important to appreciate that cannabinoid signals are neither simple nor binary, nor do they occur only at one locus. Endogenous cannabinoid receptors include but are not limited to the following: CB1R and CB2R2, vanilloid receptors TRPA1, TRPV1, TRPV4, and TRPV53 (108, 109), nuclear receptors PPARα and PPARγ4, and GPCR119, GPR18 and GPR555 receptors (110–112), the smoothened receptor in the sonic hedgehog pathway (11, 108, 111, 113–119), NMDAR6 (109), GABAARs,7 glycine receptors, 5HT3Rs8 (120), adenosine receptors (121), voltage-dependent anion channels (VDAC1–L-, N-, and P/Q-type calcium channels) (109, 122), and potentially others (112, 123). CB1Rs are located on the mitochondrial outer membrane and in the endoplasmic reticulum (66, 124–127). Between the inner and outer mitochondrial membranes and the intermembrane space, mitochondria possess all of the signaling machinery of the endocannabinoid system (66, 124–127). PPAR receptors are located in the cell nucleus (128–135). CB1R and CB2R activation leads to increased calcium fluxes into cells, increased potassium efflux from cells, and reduced cellular cAMP levels, which lead to the retrograde suppression of activity in the excitatory and inhibitory pre-synaptic nerve terminal (121).
Cannabinoid receptors have been observed to heterodimerize with opioid, dopamine, adrenergic, adenosine, serotonin, and angiotensin receptor type 2, as well as sonic hedgehog receptors (76, 77, 89–91, 108, 121, 136). CB1Rs also heterodimerize with tyrosine kinase receptors of the neurotrophin and epidermal growth factor receptor classes, among others (121). Indeed, heterodimerization between CB1R and CB2R has also been identified (121, 137). In most cases, the activities of these heterodimeric receptors are not well studied (121).
In this first paper, we aim to set out a narrative conceptual review of how and why gene expression is altered in cannabis use disorder and in the manner in which this disordered chromatin architecture is likely to underlie the findings of modern epidemiologic studies of cannabinoid teratogenesis and carcinogenesis in large nationwide and continental populations. Therefore, our perspective is both gene-centric and focused on a chromatin conformation-based analysis. Therefore, we first consider aging and then move on to epigenomics and the many ways in which these two major areas interact with each other.
2. Aging–epigenomic aging
2.1. Twenty stigmata of aging in cannabis dependence
Fifteen hallmarks of aging have been described in cannabis dependence, including (1) increased acute and chronic physical and mental illness (138), (2) acceleration of cardiovascular and organismal age (15), (3) endocrine disruption, particularly of the hypothalamo-pituitary-gonadal axis (139, 140), (4) mitochondrial inhibition (141–144), (5) DNA hypomethylation and advanced epigenetic age (14, 145–147), (6) neuroinflammation accompanying cannabis-associated mental illnesses (148–173), (7) cirrhosis (174–176), (8) degeneration of oocytes and sperm (177, 178), (9) increased carcinogenesis (28, 179–190), (10) heightened rates of many congenital anomalies and teratologic syndromes (27–29, 191–207), (11) telomerase inhibition (11, 208), (12) chromosomal damage (2, 4, 8, 178), (13) reduction in histones (5, 21, 26, 209–211), (14) immunostimulation (93, 94, 99–101, 212–217), and (15) elevated mortality rates in long-term users (218–229). These are elaborated in detail elsewhere (31, 185, 230).
To this list, an additional five features of aging that are also characteristic of cannabis dependence can be added. These include (16) a reduced respiratory exchange ratio (the amount of CO2 produced for oxygen taken up by tissues and organisms and clearly reduced due to the well-characterized inhibition of mitochondrial metabolism), (17) reduced ATP production by mitochondria, (18) increased extra-chromosomal DNA circles, (19) an increase in double-stranded DNA breaks, and (20) a reduction in lamin B (22). Double-stranded DNA breaks are a severe threat to cell survival, and the ability of cells to closely control their occurrence correlates well with organismal lifespan (22). Lamin B is a component of the internal nuclear envelope that functions to assist with gene silencing. Its reduction has been linked with increased senescence-associated β-galactosidase positive cell staining and an increase in the release of pro-inflammatory interleukin-6, CCL29, CCL2010, and LINE111 retrotransposons and inhibitors of apoptosis (IAP).
Therefore, these 20 features of aging together constitute strong and robust evidence for the acceleration of cellular aging by cannabis, similar to the evidence observed with tobacco use (22, 231). This implies that studies in aging have a direct relevance to understanding the effects of various cannabinoids on cell behavior.
2.2. Review of a key aging pathophysiology study
A team of 64 investigators from Harvard Medical School and their collaborators used an “Inducible Changes to the Epigenome” (ICE) protocol in mouse cells to show that the induction of only 20 double-stranded DNA breaks (DSBs) causes the epigenomic machinery on the genome to regenerate. This regeneration occurs in such a manner that the overall level of cell definition in the Waddington epigenetic landscape and the level of DNA methylation are reduced, while the epigenomic age is advanced. In the study, the rearrangement of the epigenomic apparatus was presumed to cause the redistribution of the DNA methylation machinery, which underlay the reduction in DNA methylation (22). The study showed that both gene activating (H3K27ac12 and H3K4me313) and gene repressive (H3K9me314 and H3K27me315) epigenetic marks were downregulated by DSBs so that the normal epigenomic definitions between cells were blurred.
Concomitantly, the boundary function that controls chromatin loop formation and gene expression and directs enhancer–promoter interactions was reduced so that the enhancer function became misdirected aberrantly toward anomalous promoters. In general, cells became less well differentiated. Several examples of anomalous cellular dedifferentiation were documented, including fibroblasts that expressed neuronal genes, muscle cells that expressed spleen and immune genes, including major histocompatibility class II genes, and muscle cells that increased epithelial–mesenchymal transition in renal glomerular parietal lining epithelial cells (22).
Genes expressed in development, such as HoxA and Wnt genes, were found to be specific targets of this epigenomic de-programming. This was believed to be because they were poised for activation to assist with tissue repair in the event of some local injury or insult. The proteins coded by these genes are part of the DSB repair machinery; therefore, they were recruited to the DNA break sites together with other complexes (22).
Many aspects of aging were accelerated in ICE mice, including reduced short- and long-term memory and reduced physical coordination when walking, reduced body weight, reduced mobility at night, reduced fat mass, reduced strength, reduced hearing, cataract formation, reduced glomerular size, reduced skeletal mass, shorter running time and distance, reduced muscle ATP, mitochondrial DNA, and muscle lactate, hair graying and thinner skin, and increased brain neuroinflammation, including 1.6x more activated astrocytes and 3.5 times more activated microglia. The epigenetic age of the blood and skeletal muscle was 50% advanced in ICE mice (22).
The expression of the classic senescence gene CDKN1A16 (encoding P21) was upregulated. The expression of the canonical epigenomic activators of gene expression H3K27ac and H3K56ac was downregulated. The expression of H3K27ac was inversely correlated with its baseline expression, implying that it was most reduced at promoters where it was previously highly expressed and vice versa. Since H3K27ac, the classic epigenomic signal for gene activation, is most enriched at the tissue- and cell lineage-defining superenhancers, these were the epigenomic loci most weakened by these rearrangements.
Significantly, 50% of the top 20 programs identified by a Gene Ontology search were involved in developmental and organ patterning processes (22). Organ systems that were inhibited by the ICE treatment included the adult and fetal brain, heart, lungs, gastrointestinal organs, and muscle cells. Gene Ontology terms that were suppressed included the following: regulation of blood coagulation, regulation of transmembrane receptor serine/threonine kinase pathways, negative regulation of endothelial cell proliferation, regulation of coagulation, skeletal system morphogenesis, single organism signaling, pattern specification processes, bone morphogenesis, tissue development, skeletal system development, organ development, transcription from RNA polymerase II promoter, cell communication, odontogenesis, negative regulation of cell adhesion, specification of organ identity, bone development, regulation of wound healing, regulation of smoothened signaling pathway (sonic hedgehog), and negative regulation of cell proliferation. It is clear from this extensive list that many key developmental processes were extensively suppressed.
Along with the weakening of superenhancers, superanchors were also weakened. This was demonstrated by showing that aberrant enhancer–promoter interactions occurred when the three-dimensional chromatin looping was assessed (22). Therefore, these findings together revealed that cellular identity was weakened and indeed disrupted.
Importantly, a highly broad and diverse spectrum of immune gene superenhancers exhibited an increase in H3K27ac in many cell types, while the transcriptional programs for other organ genes–such as heart, brain, livers, kidneys and muscle cells–were suppressed. Immune pathways that were increased by the application of the Gene Ontology analysis included cell activation, leukocyte activation, lymphocyte activation, T-cell activation, regulation of T-cell activation, regulation of lymphocyte activation, regulation of leukocyte activation, antigen processing, regulation of immune cell processes, lymphocyte differentiation, T-cell differentiation, peptide antigen processing via MHC, regulation of lymphocyte proliferation, positive regulation of lymphocyte activation, hemopoietic/lymphoid organ development, regulation of mononuclear cell proliferation, hemopoiesis, and leukocyte cell adhesion (22). The activity of the H3K27ac signal in immune superenhancers in the spleen was approximately double that of controls. This list demonstrates the profound extent of pro-inflammatory, pro-immune reprogramming created by the induced pro-aging genomic–epigenomic damage.
Epigenomic factors known to be involved in DSB repair included SIRT117, SIRT6, HDAC118, and PARP119. It was shown that they relocalized from the genome to the sites of DSBs. This mobilization of epigenomic silencers, in turn, induced the mobilization of retrotransposons and mobile elements of the genome, both of which lead to genomic instability and potently stimulate innate immune pathways (22).
Importantly, they also showed that increased epigenetic age was linked to an increase in DSBs. Thus, DSBs were shown to drive epigenomic age, and epigenomic age was shown to drive DSBs, forming a positive feedback loop.
Importantly, all of these adverse changes could be reversed by using three of the Yamanaka stem cell factors Oct3/4, Sox2, and Klf4 (OSK), thereby demonstrating that aging could be modulated both forward and backward by manipulating the genome (through DSBs) and epigenome (22). When the OSK regenerative factors were administered by intravitreal injection into the eyeball, there was a marked regeneration of the retinal ganglion cells, which in older mice are normally highly degenerative. This phenotype was replicated in ICE mice. Gene Ontology pathways that were enriched in these optic nerves and retinae included nervous system development, system development, neurogenesis, generation of neurons, multicellular organism development, regulation of multicellular processes, development of anatomic structures, developmental processes, regulation of localization, regulation of biologic quality, regulation of transsynaptic signaling, modulation of chemical synaptic signaling, regulation of ion transport, neuronal differentiation, response to external stimuli, neuronal development, regulation of transport, multicellular organismal processes, synaptic signaling, and cellular development processes (22). Thus, many key neural regenerative pathways were strongly restored by OSK therapy.
Therefore, these workers could ascribe the aging process itself to a loss of epigenomic information, which was bidirectionally coordinated with related processes such as genomic breaks, immune stimulation, and stem cell impairment, as well as developmental and regenerative programs.
2.3. Relevance to cannabinoid pathophysiology
As indicated above, these epigenomic and functional studies of aging are directly relevant to patients exposed to cannabis for many reasons. As the authors state, there is no question that such findings apply to tobacco exposure (22), and since cannabis has currently been shown to be a more potent genotoxin than tobacco in multiple studies (189, 191, 202, 232), these observations apply even more so to cannabinoids.
It is important to note how closely cannabis phenocopies this described process. DSBs (2–8), DNA hypomethylation (9–13, 233), and weakened CTCF boundary elements (11), which are the core components of the above schema, are all well described following cannabis exposure (11).
The involvement of key developmental processes Wnt, HoxA, and sonic hedgehog in the above results explains for stroke the implication of cannabinoids in a wide variety of teratogenic, developmental, and neurodevelopmental congenital anomalies, as documented in Colorado, Hawaii, the USA, Canada, Australia, and Europe (27–29, 191–205, 207). This description fits well with the wide variety of congenital anomalies that have been linked with cannabis, including those of the cardiovascular, central nervous, gastrointestinal, chromosomal, limb, uronephrological, body wall, and orofacial systems, as well as in the general embryo (27–29, 191–205). Congenital anomalies that have been linked to cannabis exposure in the USA were anophthalmia/microphthalmia, anotia/microtia, aortic valve stenosis, atrial septal defect, biliary atresia, bladder extrophy, choanal atresia, cleft palate alone, cleft lip alone, cleft lip with cleft palate, cleft lip with or without cleft palate, cloacal extrophy, club foot, coarctation of the aorta, common truncus, congenital cataract, congenital dislocation of the hip, congenital posterior urethral valve, deletion of 22q11.2, diaphragmatic hernia, Ebstein's anomaly, encephalocele, epispadias, esophageal atresia with or without tracheesophageal atresia, Hirschsprung's disease, congenital megacolon, hydrocephalus without spina bifida, hypospadias, interrupted aortic arch, microcephalus, obstructive genitourinary defect, omphalocele, patent ductus arteriosus, pulmonary valve atresia, pulmonary valve atresia and stenosis, rectal and large intestinal atresia and stenosis, reduction deformity upper limbs, reduction deformity lower limbs, renal agenesis and hypoplasia, small intestinal atresia/stenosis, trisomy 13, trisomy 18, trisomy 21 (Down's syndrome), Turner's syndrome, and ventricular septal defect (192, 202, 205).
The unequivocal demonstration that cellular dedifferentiation occurs due to DNA demethylation, weakening of superenhancers and superanchors, aberrant promoter–enhancer communication, and retrotransposon activation clearly explains why many diverse tissues are primed by cannabis for malignant transformation, which addresses the issue of why so many cancers have been epidemiologically linked with cannabis (25, 28, 32, 179–188, 220, 234–247). Cancers that were linked with cannabis exposure in Europe were all cancers, excluding non-melanoma skin cancer, bladder, brain, breast, colorectal, Hodgkin's, kidney, larynx, liver, lung, melanoma, multiple myeloma, myeloid and lymphoid leukemias, non-Hodgkin's lymphoma, and esophagus, oropharynx, ovary, pancreas, prostate, stomach, testis, thyroid, and uterine cervix cancers (189).
For many of these tumors, positive dose-response effects have been described (220, 238, 240, 241). There are also many examples of inheritable tumors due to the intergenerational transmission of major genotoxic lesions (248, 249), including acute lymphoid and myeloid leukemias, rhabdomyosarcoma, and neuroblastoma (28, 186, 188, 250–252).
Importantly, cannabis has been shown to be a driver of rising rates of breast, testicular, liver and pancreatic cancers in adults (28, 183, 184, 187, 190, 234, 253–255) and of total pediatric cancer (188) and acute lymphoid leukemia (188) in children. Most of the studies referred to in this paragraph were conducted in space–time contexts and in causal inferential paradigms to allow for the formal quantitative investigation of epidemiologically causal pathways to be investigated.
Indeed, a question has been formally posed (190, 234) regarding whether cannabis might be a major factor underlying the modern resurgence of several types of cancer developing in patients younger than 50 years (235).
The close, reciprocal, and mutually reinforcing relationship between the DSB-inducing actions of cannabinoids and epigenomic dysregulation is also clarified. Moreover, the manner in which the classically described DSB induction and chromosomal clastogenicity are linked to the newly defined epigenomic dysregulation is also explicated.
Multiple cannabinoids are known to impede mitochondrial and intermediate metabolism (55, 56, 65, 66, 122, 256–263). This necessarily reduces the availability of methyl and acetyl groups for methylation and acetylation reactions, which, by definition, reduces both the epigenomic instructions written to the DNA and gene availability and, thereby, “flattens” the epigenomic landscape [related Waddington's epigenomic valleys (264)].
Furthermore, DSB induction and various levels of epigenomic dysregulation also clarify not only the occurrence of cannabinoid-induced aging but also some of its likely cellular mechanisms.
With this argument established on theoretical grounds, all of these features require verification in the cellular models of cannabinoid cytotoxicity, genotoxicity, epigenotoxicity, and aging.
3. Epigenomics
3.1. Enhancer–promoter interactions
The human genome has approximately 25,000 genes and 1,000,000 enhancers (265). There is significant enthusiasm within the scientific community due to the development of low input chromosome conformation capture techniques for interrogating three-dimensional genome architecture within the nucleus, which allows for a detailed description of the manner in which genes are transcribed from chromatin loops that are formed when cohesin motors extrude DNA loops through their lumen (266). The cohesin complex is known to form loops around chromatin during chromosomal pairing, which occurs at the mitotic metaphase and also during gene transcription (267). These looping structures are constrained by boundary elements, which is most often CTCF20 (266–270) being the most common element. These boundary elements divide the chromatin into topologically defined domains for transcription (269). The minichromosome maintenance (MCM) complex has also been shown to block cohesin loop extrusion and act as a boundary element (271). These domains are carefully constrained to usually contain both the gene promoter and the enhancers acting in cis (on the same chromosome), albeit some enhancers act at large distances over one megabase or on different chromosomes (in trans). Importantly, DNA methylation prevents the binding of CTCF to chromatin (272). These topologically defined domains are organized and clustered together inside the three-dimensional space of the nucleus into transcription factories. At present, this looping model has been demonstrated in many different tissues in both physiologic and pathologic states, including during embryonic development (273–276), during chondrogenesis (277), in normal tissues (278, 279), in the heart (280–282), in the brain (283–290), in T-cell differentiation (269, 291), for stem cells (292) during cellular reprogramming and dedifferentiation (22, 293, 294), and within many cancers (269, 291, 295–302). Thus, these looping structures bring together both the promoter and enhancers, usually within 300 nm, to control gene transcription. Indeed, it has been reported that 90% of the risk genes identified in genome-wide studies are located within non-coding genomic regions, especially in enhancers (265). Experimental and biostatistical studies have shown that clusters of enhancers work together synergistically and combinatorially (265, 270, 278).
Superenhancers are large groups of enhancers that are clustered on the genome and control the state of differentiation and cell lineage determination (267, 303–305). In other words, they are believed to determine whether a heart cell is a heart cell as opposed to a neuron or blood cell, for example. Superenhancers are extremely powerful and perform activities that are several orders of magnitude above ordinary enhancers; they may act either from the same chromosome or from another chromosome. The limits of superenhancers are protected by “superanchors,” which normally control their activity and reach (269). Clearly, their significant power confers great risk if their ability to stimulate transcription is misdirected, as indeed occurs in many cancers (267, 269, 300, 303, 304). These phenomena are referred to as “enhancer hijacking” and “silencer hijacking” (267). DNA hypomethylation caused the loss of CTCF boundary elements, resulting in the formation of neoloops even between adjacent chromosomes and leukaemogenesis through a gain of function related to this enhancer hijacking (267, 269, 306). Contrarily, the superenhancer dependence of many tumors becomes a particular vulnerability for therapeutic exploitation, and this is presently being intensively explored (307).
A crucial detailed longitudinal study of the changes in human and rat sperm induced by cannabis exposure and resolving after a period of cannabis abstinence has been published (11). Cannabis-dependent human volunteers and rats were exposed to cannabis and then underwent 11 weeks of documented abstinence from cannabis. Eleven weeks is the period one sperm cycle takes in humans. Epigenomic changes were then documented from a control state and longitudinally against earlier time points.
Since the control of enhancer–promoter looping interactions by boundary elements has currently become the basic model for controlling gene transcription, the observation in the Schrott dataset (11) that cannabis withdrawal disrupts the expression of CTCF carries profound implications, since CTCF is the basis of structure and order in the whole architecture of enhancer–promoter interactions. In the absence of proper CTCF boundary function, enhancers and promoters will inevitably be brought into inappropriate contact with severe sequelae, including disordered neurodevelopmental outcomes (269, 279, 283, 284, 287, 289, 308, 309) and many cancers (269, 295–302). Cancer can occur when a promoter region is inappropriately exposed to an enhancer region, thereby providing an inappropriate stimulus to gene transcription. Indeed, one powerful scenario is when a tissue defining superenhancer is brought adjacent to a strong oncogene, such as Myc or Notch, which can cause run away growth stimulation, which is a not uncommon scenario both in many leukaemias and solid tumours (265, 267, 269, 291, 296, 298–300, 310–312).
The main proteins comprising the cohesin ring may be listed as SMC121, SMC3, RAD5122, and STAG23 proteins. Cohesin is involved in post-replicative DNA repair and transcriptional regulation, and it also plays an important role in pairing chromosomes (313). Therefore, the finding that there were 96 DMRs in the Schrott dataset for the structural maintenance of chromosomes (SMC) genes, 9 DMRs for RAD51, and 152 DMRs for the STAG proteins, comprising 257 hits, is crucial (11). Indeed, the significance of RAD51 epigenomic inhibition is amplified by its primary role as a key enzyme in the high-fidelity DNA repair pathway known as homologous recombination. When RAD51 expression is disabled, alternative lower fidelity error-prone DNA repair processes, such as mismatch repair (in stem cells) or theta end joining (in oocytes and in many cells) (272), are employed, and these lower fidelity pathways are inherently mutagenic. Importantly, sperm were shown to be particularly susceptible to DNA damage owing to their largely unmethylated DNA state, their DNA compaction in protamine barrels that are six times more tightly compressed than normal, and the complete absence of DNA repair machinery (272). For this reason, 80% of congenital disorders diagnosed postnatally have been ascribed to paternal contribution (272).
For example, in acute lymphoid leukemia (ALL), which is the most common childhood cancer that represents inherited genotoxicity and has previously been linked with community cannabis exposure (188), it was shown that a key driving mutation occurs in the GATA3 24 enhancer, which changes chromatin conformation and gene expression (300). GATA3 is a pioneer factor that recruits the SMARCA4 (SWI/SNF-related, matrix-associated, actin-dependent chromatin regulator, subfamily A, member 4) 25 complex to open up the genome and sets in train a GATA3/CRLF2 26/JAK2 27/STAT5 28 signaling pathway to leukaemogenesis (300). SMARCAs perform energy-dependent repositioning of nucleosomes and increase the accessibility of genes to the transcription machinery. GATA activation induces a state switch in the nuclear synthetic compartments (B (silent) to A (active transcription) compartment switching) for many genes. GATA3 overexpression induced enhancer hijacking (300). GATA3 activation has also been identified in many other hematologic malignancies, such as the Reed–Sternberg cells in Hodgkin's disease (300). Interestingly, GATA3-binding sites were located near the Philadelphia-like chromosome break point. However, this study could not demonstrate a causal link related to this issue. Widespread B to A compartment switching was also identified in another study of acute lymphoid leukemia (299). Importantly, the rs3824662 risk variant in the GATA3 promoter is inheritable (300).
Of further importance, there were 127 hits for GATA in the Schrott epigenomic cannabis screen (11). There were over 28 DMRs for actin-related proteins in the Schrott dataset (11). Seven DMRs were identified for SMARCAs 1, 2, 4, and 5 (11). Since SMARCAs are both ATP- and actin-dependent, and since cannabinoids disrupt both actin production and ATP synthesis as well as SMARCAs themselves, it follows that nucleosomal positioning and gene transcription are necessarily disrupted. SMARCAs have also been shown to be of pivotal importance in enhancer-addicted prostate cancer (302).
Therefore, to observe that cannabis significantly disrupts both CTCF as the fundamental boundary element defining transcription regions and the machinery and motors that drive chromosomal loop extrusion and orchestrate gene transcription is to necessarily point to a major disruption of the fundamental process of gene transcription.
It should also be observed that normal genomic processes can induce DNA breaks, including DNA transcription and duplication, base excision repair, and active DNA demethylation (314, 315).
3.2. Epigenomic memory
It has also been shown that many cell types record histories of past exposures in the highly complex post-translational codes in their epigenome, especially their histone codes (145, 146). These codes form memories. They are also advantageous in that should an inflammatory or toxic insult recur, gene cassettes are often poised for rapid reactivation and usually have a modified response, which may be either potentiated in the case of an infective insult (145) or ameliorated in the case of pancreatitis (316–318).
3.3. Cannabinoid impacts on epigenomic machinery
The study of Schrott and colleagues (11) also described the manner in which cannabis dependence and withdrawal disrupt the basic machinery of epigenetic regulation, including DNA methylation writers and erasers (DNMT1/329 and TETs30), histone methylation and acetylation writers and erasers (KMTs31, KDMs32, KATs33, HDACs34, and sirtuins35), stem cell regenerative transcription factors, key elements of the polycomb repressive machinery, major ATP-dependent factors that reposition nucleosomes and enable new genes to be transcribed (SMARCA2/436), and coordinators of epigenetic processes, including DNA methylation and histone post-translational modifications (UHRF137). UHRF1 is also a key regulator of cell growth. Growth inhibition explains some of the growth-inhibitory actions of cannabis, as described in studies involving babies' heads, brains, and hearts (191, 195, 196, 201, 203, 205, 207, 319–321).
From this analysis and concise review, it can be observed that cannabis broadly disrupts the fundamental epigenomic machinery and necessarily disrupts the basic machinery of gene transcription, thereby disrupting normal promoter–enhancer interactions. Deleterious effects on neurodevelopment, patterns of congenital anomalies, and cancerogenesis, including heritable cancerogenesis, should be the expected outcomes and are indeed also the observed outcomes.
Through the induction of genome-wide relative DNA methylation (9, 12, 13), single- and double-stranded DNAs and chromosomal breaks (2–8), inhibition of mitochondrial metabolism by diverse pathways (55, 56, 66, 256–260, 322), and within the context of widespread epigenomic disruption and interference with the basic gene looping mechanism of gene transcription, cannabis will necessarily reorganize nuclear pathophysiology. This reorganization can lead to genomic instability, numerous adverse congenital outcomes, including neurodevelopmental outcomes, and cellular aging, according to recent epigenomic pathophysiologic descriptions (11, 22, 294, 323, 324).
It is also of interest to consider the overlap between genes described in certain syndromes and those known to be epigenomically perturbed by cannabis use. Some of the largest gene databases in the existing literature have been intersected in this way with the epigenomic cannabis screen of Schrott and colleagues. This has produced the data shown in Table 1.
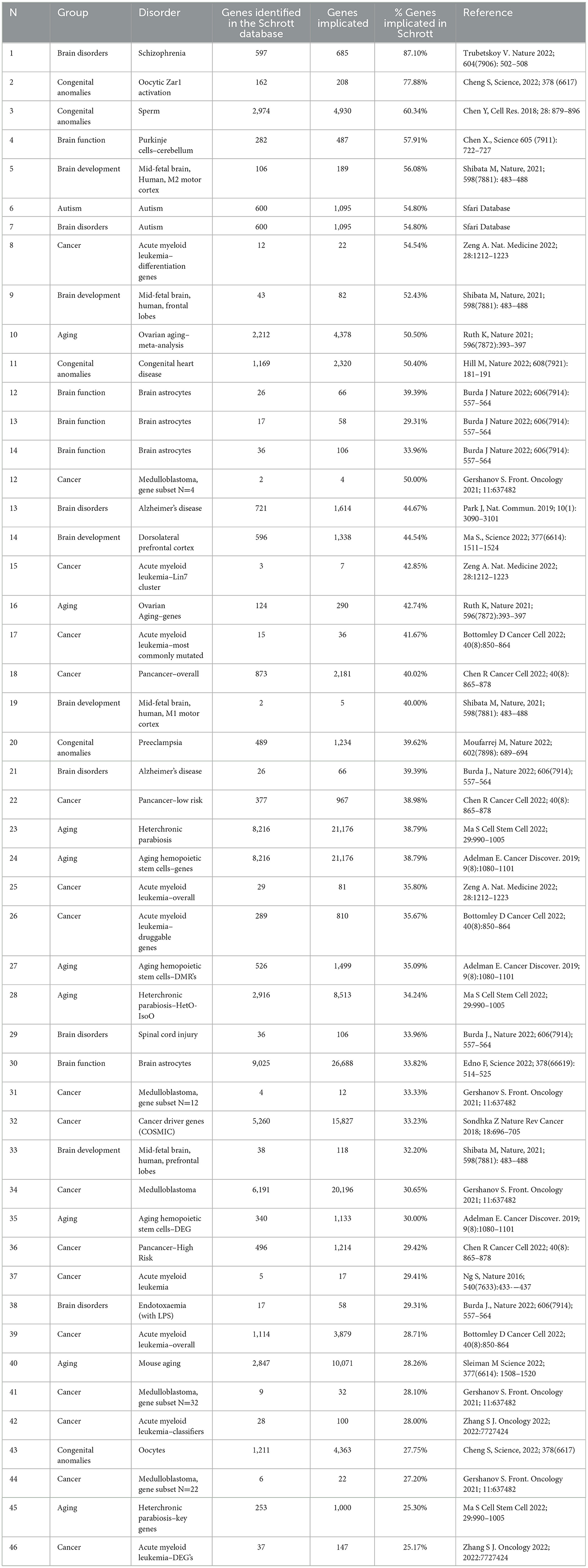
Table 1. Syndromic genes identified in the schrott cannabis epigenomic screen (11).
From Table 1, it can be observed that the overlap runs from 25.17% for acute myeloid leukemia and 25.3% in aging to 77.9% for congenital anomalies and 87.1% for schizophrenia. The autism screen is also of particular interest. The dataset used for the assessment was the Sfari database, which contains 1,095 genes and is the world's largest autism gene set database (325). The common intersected fraction identified with the Schrott epigenomic screen with the autism dataset was 54.8%.
3.4. Exponentiation
Substantial experimental evidence points toward the conclusion that the effects of cannabinoids are exponential and that it must be assumed that this is a normal class effect in the low micromolar range. This exponentiation applies to both its genotoxic (8, 113, 326–334) and metabolic effects (53–58). Since these epigenomics and metabolomics are closely related, this implies that this exponentiation is compounded in this case.
The low micromolar serum level is readily reached in patients who consume cannabis either regularly or daily (335). This issue is exacerbated by the accumulation of cannabinoids in tissues and their generally long tissue half-life (335).
The issue of exponential dose-response effects is of great importance in the public health context. When legislation exists, which attaches penalties to cannabis use, cannabis use is naturally discouraged. However, under decriminalized or legalized legislative frameworks, cannabis use has been shown many times to increase (336–341), along with an increase in the potency of the THC or cannabidiol products consumed. This rise is accompanied by the number of individuals who consume cannabis on a relatively intense or daily basis. Clearly, this places a significant number of people in the community into a high cannabis exposure zone relatively abruptly, where adverse genotoxic and neurotoxic outcomes become more commonplace.
For these reasons, it is envisaged that the triple confluence of rising cannabis prevalence rates, intensity of use rates, and cannabinoid potency will manifest relatively abruptly as steep rises in adverse mental health, as well as teratologic, carcinogenic, and age-related outcomes, as are indeed being observed and documented in several jurisdictions (16, 25, 27–32, 179–187, 189–205, 207, 232, 234–237, 321, 342–345).
3.4.1. Fetal alcohol syndrome–fetal cannabinoid syndrome
The incidence of fetal alcohol syndrome (FAS) is increasing in many places. Indeed, a recent space–time and quantitative causal inference study in Europe showed that FAS was rising in association with increased cannabis use (201). This result went beyond merely reporting an association because it has currently been well established that FAS is mediated largely via the CB1R cannabinoid receptor (111, 114–117, 346–355), with GABAergic neurons shown to be particularly susceptible (114). This effect is also mediated by the sonic hedgehog receptor (shh), where cannabinoids bind to the shh-smoothened receptor (113, 114, 116, 118).
Indeed, a remarkably close phenotypic resemblance between infants exposed antenatally to cannabis and alcohol has been noted by many investigators (113–115).
Moreover, cannabis and alcohol compound the foetotoxic effects of each other so that their combined effect is potentiated (111, 113–115, 347, 351, 356). A corollary of this is that multisystem foetotoxic effects manifest at otherwise subthreshold doses (113).
Importantly, multisystem VACTERL (vertebral, anal, cardiac, tracheo-esophageal, renal, and limb) disorder has also been shown to be more common across Europe and has been formally causally related to cannabis exposure (201). As noted, this is a multisystem disease, and sonic hedgehog interference has been implicated in its pathoaetiology (357–359). Since cannabis is known to interfere with sonic hedgehog signaling both directly (111, 114–117, 346–355) and epigenomically (11), this further implicates cannabis in the teratology of these seven systems.
It has also been noted that teratologic syndromes otherwise uncharacterized have arisen across space and time in a manner causally related to cannabis exposure in Europe across the same period (201).
Importantly, the effects of alcohol have been shown to be mediated in part by the endocannabinoid system and associated epigenomic changes to the DNA methylation, histone structure, and chromatin architecture (360, 361). This implicates cannabinoids in the full spectrum of fetal alcohol spectrum disorders (FASD) in adults and young adults, in addition to their increasingly recognized role in developmental and congenital disruptions (360, 361).
3.4.2. Daily cannabis use
Since much of the evidence points to high-dose cannabis use as being of utmost concern, it is of interest to quantify and define this key variable that is of the highest relevance to genotoxic and neurotoxic outcomes. As the best dataset for doing this is in the USA, the USA will be the nation of interest.
The most recent data on national drug use rates in the USA is available from the National Survey of Drug Use and Health conducted annually by the Substance Abuse and Mental Health Services Administration (362). Accessing the Public Use Data Analysis System website38 and running the data input code MRJMDAYS allows one to study the rates of daily or near-daily cannabis use39 across the whole population of individuals older than 12 years on an annual basis. The rate of near-daily cannabis use across the entire adult US population rose, as shown in Table 2, Figure 1A. This indicates that the rate of growth of cannabis devotees who smoked almost daily rose 265.5% nationally during 2002–2020. It should also be pointed out that the largest group in the survey comprised those who did not use cannabis at all, which in 2020 was 88.5%. Figure 1B shows the rate of near-daily use in each of the pregnancy trimesters. Figure 1C shows the rate of daily cannabis use summed across the three pregnancy trimesters.
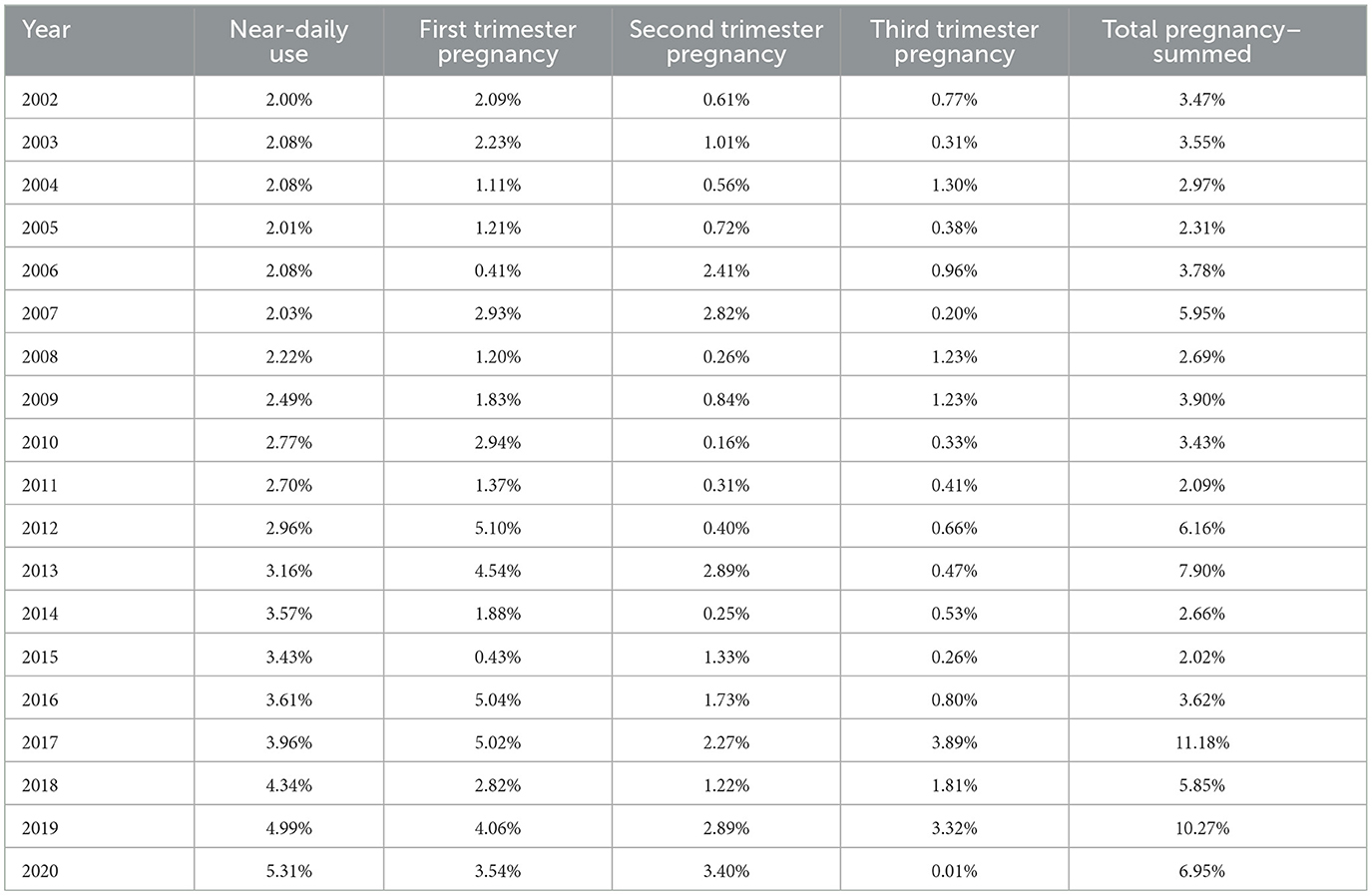
Table 2. Daily cannabis use in the entire community and by pregnancy trimesters.
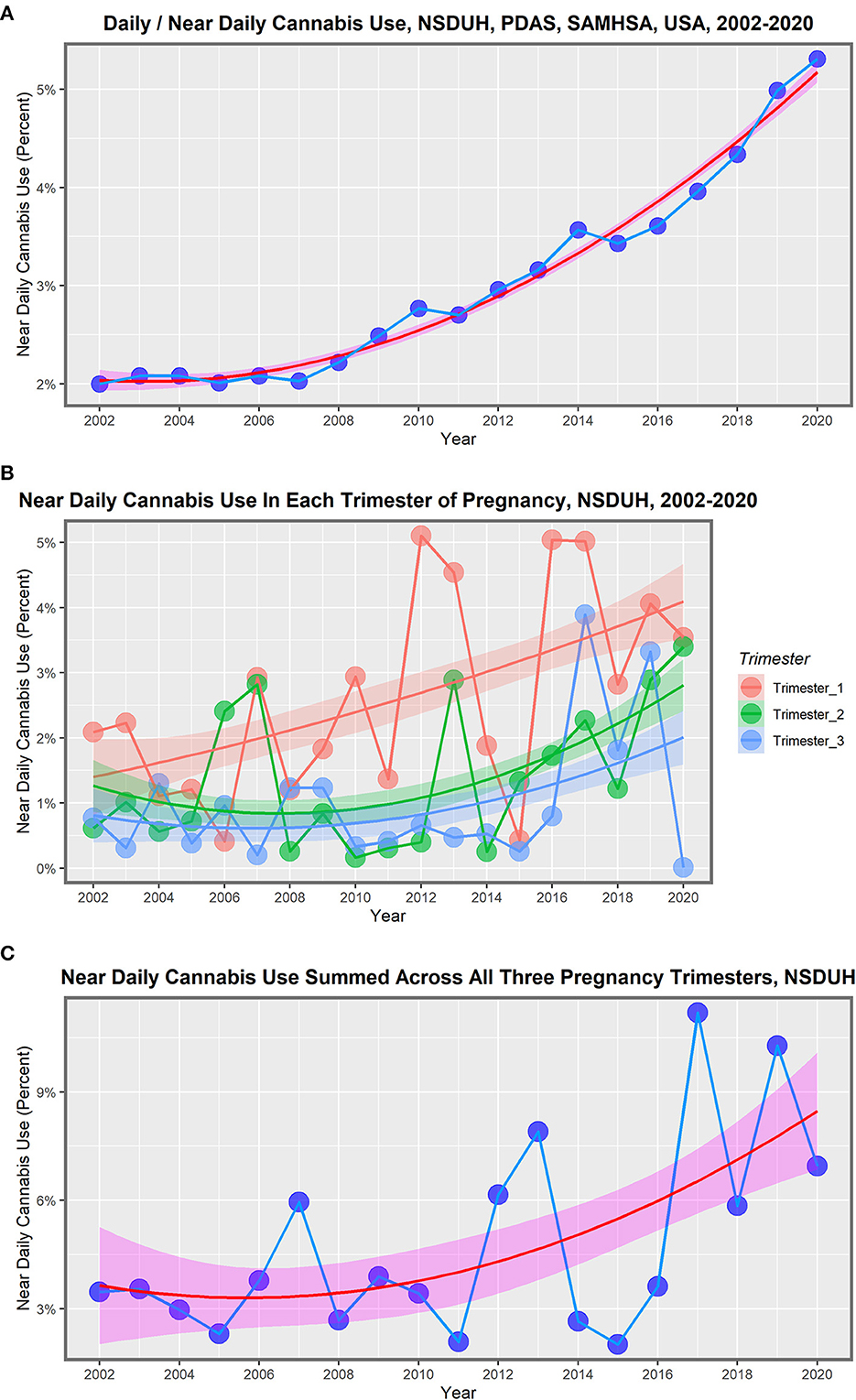
Figure 1. Daily cannabis use, the USA, 2002–2020. (A) Near daily cannabis use as reported at the national levels by NSDUH annual PDAS. (B) Near daily cannabis use in each of the trimesters of pregnancy by NSDUH. (C) Near daily cannabis use summed across all three pregnancy trimesters.
These lines show a high degree of year-on-year variation. If one uses simple mathematical smoothing on these data and the quadratic polynomial, which is the most appropriate of the common models in the predict function in R, the data presented in Table 3 for smoothed modeled values is derived. If one compares the first period 2002–2010 to the second decade 2010–2020, the rise in the rate is clear in all cases. Daily cannabis use rose 24.9% in the first trimester and then 103.4% in the second trimester. The use in the second- and third-trimester use rose from −22.1% to 209.4% and −14.6% to 191.0%, respectively. The sum across all three pregnancy trimesters rose from 3.5% in the first period to 124.1% in the second period. Hence, these data demonstrate a greater rise across the board nationwide in near-daily cannabis use in all metrics and trimesters in the second decade. The first trimester is the only exception, where the relationship showed a linear modeled response across the whole period. These lines are all graphed in Figure 2.
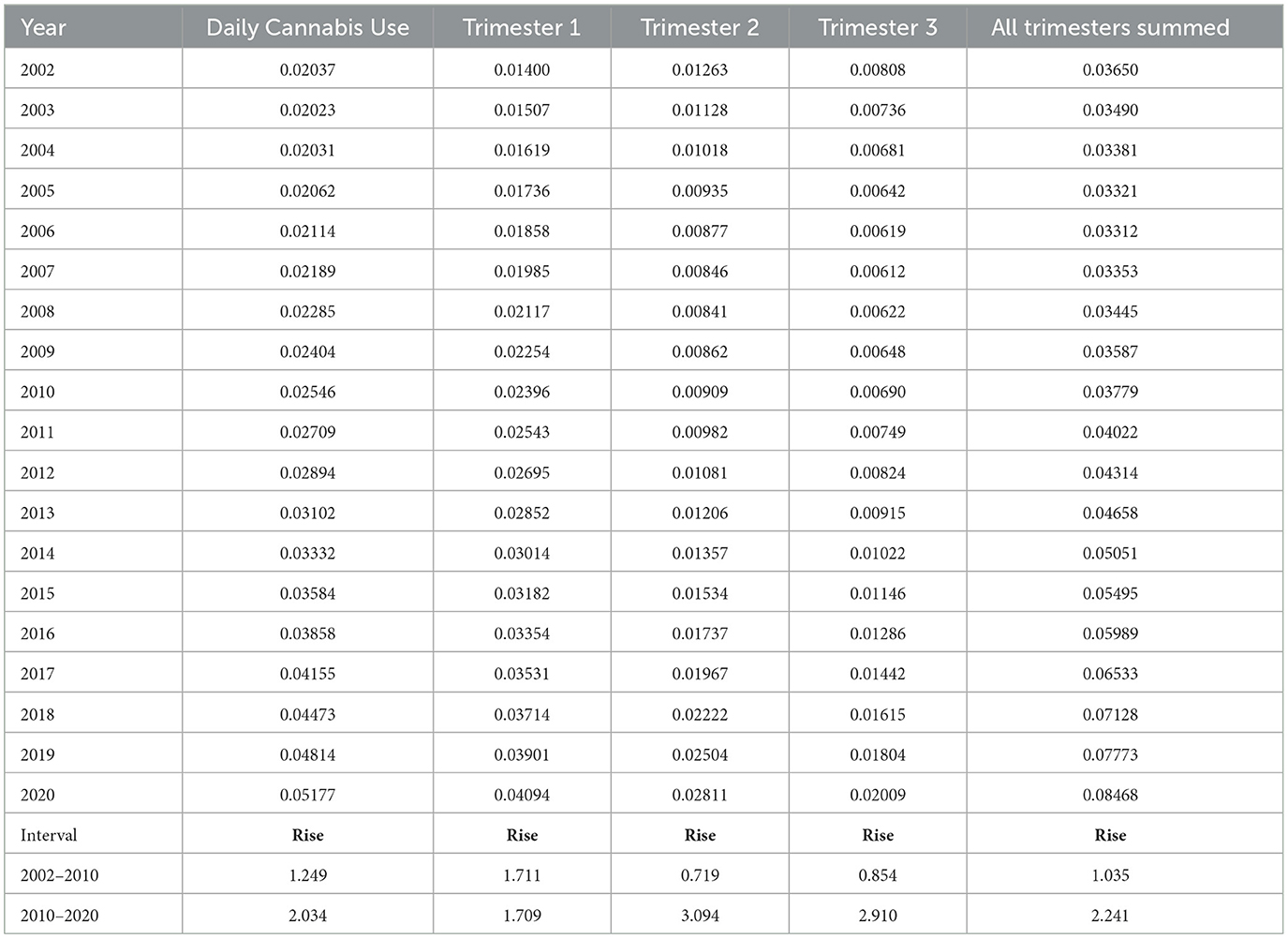
Table 3. Modeled daily cannabis use (smoothed data).
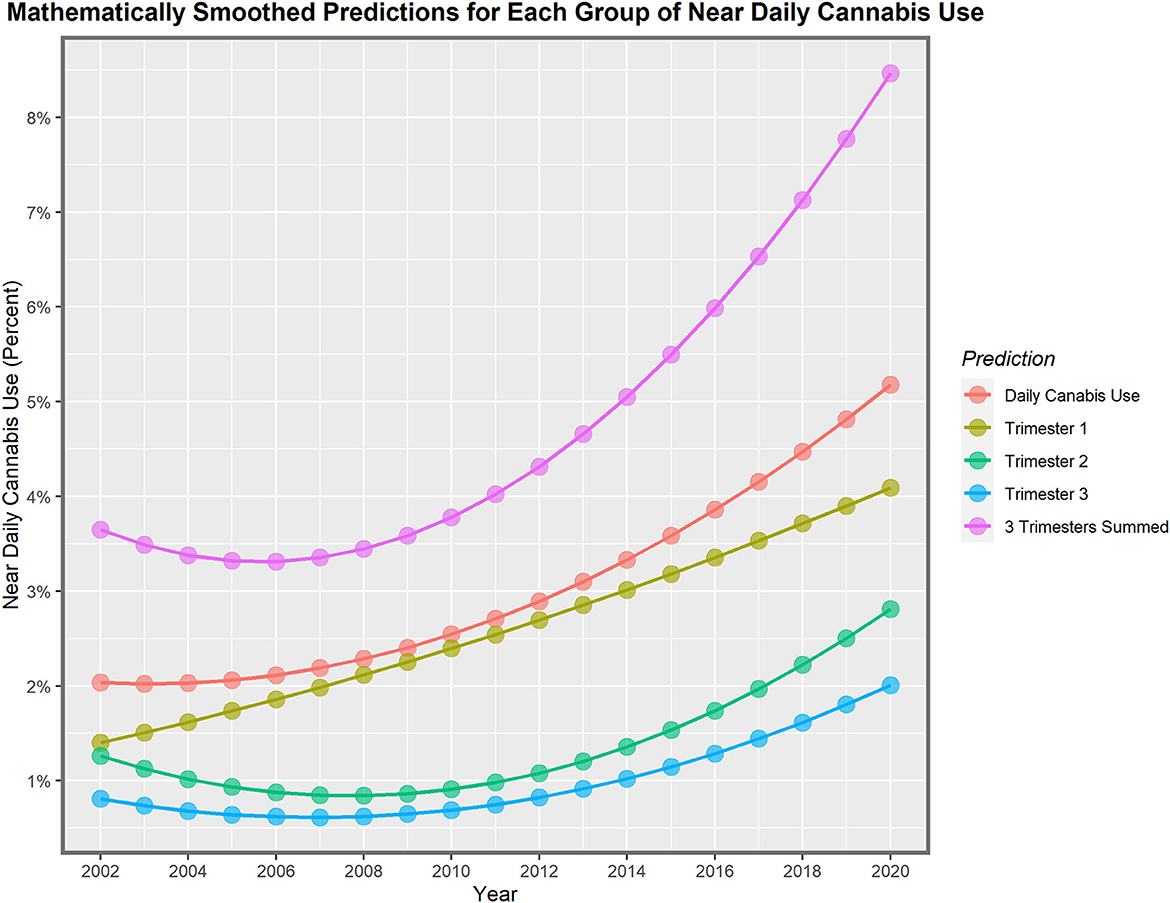
Figure 2. Modeled daily cannabis use.
3.5. Epitranscriptomic metabolomics
RNA is subject to over 200 post-transcriptional modifications. The most common of these is m6 adenosine methylation (m6A). It has been shown that the m6A modification is applied to METTL16 40 uniquely in leukaemic stem cells (LSCs), which creates a particular therapeutic vulnerability of LSCs. METTL16 deposits an m6A mark on the first and second codons for branched-chain amino acids (BCAA, including valine, leucine, and isoleucine) transaminases (BCAT1/2), which stabilizes the BCAA mRNAs and establishes them as a fundamental metabolic fuel for LSCs (363). Thus, the pathway is the METTL16/m6A/BCAT1/2/BCAA axis. Cancer-associated metabolic reprogramming has been shown to profoundly affect gene expression, differentiation, and tumor progression and is an emerging hallmark of malignancy. BCAT1/2 upregulation has been shown to be a marker of tumor aggressiveness across many tumor types. BCAAs are requisite to protein synthesis; they replenish TCA 41 intermediates and act as a nitrogen source for nucleotide synthesis via the glutamine–glutamate pathway. Therefore, the upregulation of BCAAs metabolically reprograms oxidative phosphorylation, the citric acid cycle, and nucleotide synthesis to fuel the rapid growth of malignant cells. AML cells are known to be addicted to BCAAs. METTL16 inhibition has been shown to drop LSC frequency 10-200-fold (363).
Some researchers worked with a standard model of acute myeloid leukemia and found that the mRNA for IGF2BP242 is an m6A reader. This m6A reader stabilized the m6A modification of PRMT643, which post-translationally modified histone H3R2me2a44. This modification suppressed the lipid transporter MFSD2A45, thereby reducing the lipid transport into LSCs (364). Indeed, approximately 60% of m6A targets were only observed in LSCs. It was also noted that m6A mRNA targets are enriched in immune checkpoint targets, which might be a key explanation of how LSCs avoid or subvert immunosurveillance (364).
4. Conclusion
The above considerations clearly demonstrate the salience and centrality of the epigenome, including the three-dimensional architecture of the nucleus, for determining gene expression and its major perturbation by cannabis exposure. Well-documented rising rates of daily cannabis use, cannabis use in pregnancy, and the currently amply demonstrated exponential cannabis genotoxic dose-response relationship imply that such studies are of primary importance and are a major research priority for addiction medicine, neuropsychiatric understandings, and public health management. These issues are pursued further in Part 2, which examines the metabolic and immunomic underpinning of these features and the manner in which these issues apply to neuronal toxicity and epigenotoxicity, along with the disruption of key events at the synapse. Specifically, these investigations elegantly demonstrate the importance and relevance of all of the considered levels of cellular machinery dysregulation.
Author contributions
AR conceived the idea, performed the literature review, and wrote the first draft. GH added meaningful intellectual input, edited the first draft, provided project supervision and support, curated resources, and supervised the conduct of the project. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ATP, Adenosine triphosphate; cAMP, Cyclin adenosine monophosphate; CB1R, Cannabinoid type 1 receptor; CB2R, Cannabinoid type 2 receptor; CCL2, Chemokine motif ligand 2; CCL20, Chemokine motif ligand 20; CDKN1A, Cyclin-dependent kinase 1A; CRLF2, Cytokine receptor-like factor 2; CTCF, CCCTF-binding factor; DDR, DNA damage repair; DNMT, DNA methyltransferases; DMG, Differentially methylated genes; DSB, Double-stranded break in DNA; ECS, Endocannabinoid system; GABAAR, Gamma-aminobutyric acid A receptor; GATA3, GATA-binding protein 3; GPCR, G-protein coupled receptor; GPR, G-protein receptor; H3K27, Histone 3 lysine 27; H3K4, Histone 3 lysine 4; H3K4me3, Histone 3 lysine 4 trimethylation; H3K56, Histone 3 lysine 56; H3R2me2a, Histone 3 arginine 2 demethylation; HDAC, Histone deacetylases; HDAC1, Histone deacetylase 1; 5HT3R, 5-hydroxytryptamine receptor; IAP, Inhibitors of apoptosis; ICE, Inducible changes to the epigenome; IGF2BP2, Insulin-like growth factor 2 binding partner 2; JAK2, Janus kinase 2; KAT, Lysine acetyl transferases; KDM, Lysine demethylase; KMT, Lysine methyltransferase; LINE1, Long interspersed nuclear elements 1; METTL16, Methyltransferase 16, N6-methyladenosine; MFSD2A, MFSD2 lysolipid transporter A, lysophospholipid; NMDAR, N-methyl D-aspartate receptor; OPC, Oligodendroglial progenitor cells; PARP1, Poly (ADP-ribose) polymerase−1; PPARα, Peroxisome proliferator activator potential receptor α; PPARγ, Peroxisome proliferator activator potential receptor γ; PRMT, Protein arginine methyltransferase 6; RAD51, Recombinase/recombination protein A/RAD51 (S. cerevisiae) homolog (E coli RecA homolog); SIRT, Silencer of information transfer; Sirtuins, Silencer of information transfer /histone/lysine deacetylases; SMARCA, SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A; SMARCA4, SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, Member 4; SMC, Structural maintenance of chromosomes; STAG, Stromal antigen. STAG1 is also called SCC1. STAG2 is also called SCC3B. STAG3 is also called SCC3H3.; STAT5, Signal transducer and activator of transcription 5; TCA, Tricarboxylic acid cycle, also known as citric acid cycle and Krebs cycle; TET, Ten eleven translocation DNA methylcytosine dioxygenase; TRPA1, Transient receptor potential ankyrin 1; TRPV1, Transient receptor potential cation channel V member 1; TRPV4, Transient receptor potential cation channel V member 4; TRPV5, Transient receptor potential cation channel V member 5; UHRF1, Ubiquitin-like with PHD and ring finger domains 1; VDAC, Voltage-dependent anion selective channel 1.
Footnotes
1. ^CB1R, Cannabinoid type 1 receptor.
2. ^CB2R, Cannabinoid type 2 receptor.
3. ^TRPV, Transient receptor potential voltage-gated channels.
4. ^PPAR, Peroxisome proliferator activator receptor.
5. ^GPCR, G-protein coupled receptor; GPR, G-protein receptor.
6. ^NMDAR, N-methyl-D-aspartate receptor.
7. ^GABAAR, γ-amino-butyric acid receptor.
8. ^5HT3R, 5-hydroxytryptamine (serotonin) 3 receptor.
9. ^CCL2, C-C motif chemokine ligand 2 (CCL2). Also known as monocyte chemoattractant protein 1 (MCP1).
10. ^CCL20, C-C motif chemokine ligand 2 (CCL20). Also known as liver activation regulated chemokine (LARC) or macrophage inflammatory protein 3 (MIP3A).
11. ^LINE1, Long interspersed nuclear elements 1.
12. ^H3K27ac, Histone 3 lysine 27 acetylation.
13. ^H3K4me3, Histone 3 lysine 4 trimethylation.
14. ^Histone 3 lysine 9 trimethylation.
15. ^Histone 3 lysine 27 trimethylation.
16. ^CDKN1A, Cyclin-dependent kinase 1A.
17. ^SIRT, Silencer of information transfer–a major sirtuin class silencing gene expression. Sirtuins also possess HDAC activity and together constitute a major class of HDACs.
18. ^HDAC1, Histone deacetylase 1.
19. ^PARP1, Poly (ADP-ribose) polymerase−1.
20. ^CTCF, CCCTF-binding factor.
21. ^SMC, Structural maintenance of chromosomes.
22. ^RAD51, Recombinase/recombination protein A/RAD51 (S. cerevisiae) Homolog (E coli RecA homolog).
23. ^STAG, Stromal antigen. STAG1 is also called SCC1. STAG2 is also called SCC3B. STAG3 is also called SCC3H3.
24. ^GATA3, GATA binding Protein 3.
25. ^SMARCA4, SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 4.
26. ^CRLF2, Cytokine receptor-like factor 2.
27. ^JAK2, Janus kinase 2.
28. ^STAT5, Signal transducer and activator of transcription 5.
29. ^DNMT, DNA methyltransferases.
30. ^TET, Ten-eleven translocation DNA methylcytosine dioxygenase.
31. ^KMT, Lysine methyltransferase.
32. ^KDM, Lysine demethylase.
33. ^KAT, Lysine acetyl transferases.
34. ^HDAC, Histone deacetylases.
35. ^Sirtuins, Silencer of information transfer/histone/lysine deacetylases.
36. ^SMARCA, SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A.
37. ^UHRF1, Ubiquitin-like with PHD and ring finger domains 1.
38. ^URL: https://pdas.samhsa.gov/#/ (accessed January 27th, 2023).
39. ^Use of cannabis 20-30 days per month.
40. ^METTL16, Methyltransferase 16, N6-methyladenosine.
41. ^TCA, Tricarboxylic acid cycle, also known as citric acid cycle and Krebs cycle.
42. ^IGF2BP2, Insulin-like growth factor 2 binding partner 2.
43. ^PRMT, Protein arginine methyltransferase 6.
44. ^H3R2me2a, Histone 3 arginine 2 demethylation.
45. ^MFSD2A, MFSD2 lysolipid transporter A, lysophospholipi.
References
1. Stewart E, McEvoy J, Wang H, Chen X, Honnell V, Ocarz M, et al. Identification of therapeutic targets in rhabdomyosarcoma through integrated genomic, epigenomic, and proteomic analyses. Cancer Cell. (2018) 34:411–26.e19. doi: 10.1016/j.ccell.2018.07.012
2. Leuchtenberger C, Leuchtenberger R. Morphological and cytochemical effects of marijuana cigarette smoke on epithelioid cells of lung explants from mice. Nature. (1971) 234:227–9. doi: 10.1038/234227a0
3. Leuchtenberger C, Leuchtenberger R, Schneider A. Effects of marijuana and tobacco smoke on human lung physiology. Nature. (1973) 241:137–9. doi: 10.1038/241137a0
4. Stenchever MA, Kunysz TJ, Allen MA. Chromosome breakage in users of marihuana. Am J Obstet Gynecol. (1974) 118:106–13. doi: 10.1016/S0002-9378(16)33653-5
5. Nahas GG, Morishima A, Desoize B. Effects of cannabinoids on macromolecular synthesis and replication of cultured lymphocytes. Fed Proc. (1977) 36:1748–52.
6. Dalterio S, Badr F, Bartke A, Mayfield D. Cannabinoids in male mice: effects on fertility and spermatogenesis. Science. (1982) 216:315–6. doi: 10.1126/science.6801767
7. Dalterio SL, deRooij DG. Maternal cannabinoid exposure. Effects on spermatogenesis in male offspring. Int. J. Andrology. (1986) 9:250–8. doi: 10.1111/j.1365-2605.1986.tb00888.x
8. Russo C, Ferk F, Mišík M, Ropek N, Nersesyan A, Mejri D, et al. Low doses of widely consumed cannabinoids (cannabidiol and cannabidivarin) cause DNA damage and chromosomal aberrations in human-derived cells. Arch Toxicol. (2019) 93:179–88. doi: 10.1007/s00204-018-2322-9
9. Watson CT, Szutorisz H, Garg P, Martin Q, Landry JA, Sharp A, et al. Genome-wide DNA methylation profiling reveals epigenetic changes in the rat nucleus accumbens associated with cross-generational effects of adolescent THC exposure. Neuropsychopharmacology. (2015) 40:2993–3005. doi: 10.1038/npp.2015.155
10. Reece AS, Hulse GK. Cannabinoid exposure and altered DNA methylation in rat and human sperm. Epigenetics. (2018) 13:1208–21. doi: 10.1080/15592294.2018.1554521
11. Schrott R, Murphy SK, Modliszewski JL, King DE, Hill B, Itchon-Ramos N, et al. Refraining from use diminishes cannabis-associated epigenetic changes in human sperm. Environmental Epigenetics. (2021) 7:1–10. doi: 10.1093/eep/dvab009
12. Schrott R, Greeson KW, King D, Symosko Crow KM, Easley CAt, Murphy SK. Cannabis alters DNA methylation at maternally imprinted and autism candidate genes in spermatogenic cells. Syst Biol Reprod Med. (2022) 2022:1–13. doi: 10.1080/19396368.2022.2073292
13. Schrott R, Modliszewski JL, Hawkey AB, Grenier C, Holloway Z, Evans J, et al. Sperm DNA methylation alterations from cannabis extract exposure are evident in offspring. Epigenetics Chromatin. (2022) 15:33. doi: 10.1186/s13072-022-00466-3
14. Allen JP, Danoff JS, Costello MA, Hunt GL, Hellwig AF, Krol KM, et al. Lifetime marijuana use and epigenetic age acceleration: a 17-year prospective examination. Drug Alcohol Depend. (2022) 233:109363. doi: 10.1016/j.drugalcdep.2022.109363
15. Reece AS, Norman A, Hulse GK. Cannabis exposure as an interactive cardiovascular risk factor and accelerant of organismal ageing: a longitudinal study. BMJ Open. (2016) 6:e011891–901. doi: 10.1136/bmjopen-2016-011891
16. Phillips KT, Pedula KL, Choi NG, Tawara K-AK, Simiola V, Satre DD, et al. Chronic health conditions, acute health events, and healthcare utilization among adults over age 50 in Hawai'i who use cannabis: A matched cohort study. Drug Alcohol Depend. (2022) 234:109387. doi: 10.1016/j.drugalcdep.2022.109387
17. DiNieri JA, Wang X, Szutorisz H, Spano SM, Kaur J, Casaccia P, et al. Maternal cannabis use alters ventral striatal dopamine D2 gene regulation in the offspring. Biol Psychiatry. (2011) 70:763–9. doi: 10.1016/j.biopsych.2011.06.027
18. Szutorisz H, DiNieri JA, Sweet E, Egervari G, Michaelides M, Carter JM, et al. Parental THC exposure leads to compulsive heroin-seeking and altered striatal synaptic plasticity in the subsequent generation. Neuropsychopharmacology. (2014) 39:1315–23. doi: 10.1038/npp.2013.352
19. Szutorisz H, Hurd YL. Epigenetic effects of cannabis exposure. Biol Psychiatry. (2016) 79:586–94. doi: 10.1016/j.biopsych.2015.09.014
20. Ellis RJ, Bara A, Vargas CA, Frick J, Loh E, Landry J, et al. Prenatal Δ(9)-tetrahydrocannabinol exposure in males leads to motivational disturbances related to striatal epigenetic dysregulation. Biol Psychiatry. (2021) 92:127–38. doi: 10.1016/j.biopsych.2021.09.017
21. Mon MJ, Haas AE, Stein JL, Stein GS. Influence of psychoactive and nonpsychoactive cannabinoids on cell proliferation and macromolecular biosynthesis in human cells. Biochem Pharmacol. (1981) 30:31–43. doi: 10.1016/0006-2952(81)90282-3
22. Yang J-H, Hayano M, Griffin PT, Amorim JA, Bonkowski MS, Apostolides JK, et al. Loss of epigenetic information as a cause of mammalian aging. Cell. (2023) 186:305–26.e27. doi: 10.1016/j.cell.2022.12.027
23. Bar-Ziv R, Bolas T, Dillin A. Systemic effects of mitochondrial stress. EMBO Rep. (2020) 21:e50094. doi: 10.15252/embr.202050094
24. Zhu D, Li X, Tian Y. Mitochondrial-to-nuclear communication in aging: an epigenetic perspective. Trends Biochem Sci. (2022) 47:645–59. doi: 10.1016/j.tibs.2022.03.008
25. Warburg O. On respiratory impairment in cancer cells. Science. (1956) 124:269–70. doi: 10.1126/science.124.3215.269
26. Wang J, Yuan W, Li MD. Genes and pathways co-associated with the exposure to multiple drugs of abuse, including alcohol, amphetamine/methamphetamine, cocaine, marijuana, morphine, and/or nicotine: a review of proteomics analyses. Mol Neurobiol. (2011) 44:269–86. doi: 10.1007/s12035-011-8202-4
27. Reece AS, Hulse GK. Chromothripsis and epigenomics complete causality criteria for cannabis- and addiction-connected carcinogenicity, congenital toxicity and heritable genotoxicity. Mutat Res. (2016) 789:15–25. doi: 10.1016/j.mrfmmm.2016.05.002
28. Reece AS, Hulse G. epidemiological overview of multidimensional chromosomal and genome toxicity of cannabis exposure in congenital anomalies and cancer development. Scientific Rep. (2021) 11:13892–912. doi: 10.1038/s41598-021-93411-5
29. Reece AS, Hulse GK. Cannabis- and substance- related epidemiological patterns of chromosomal congenital anomalies in europe: geospatiotemporal and causal inferential study. Int J Environ Res Public Health. (2022). 19:11208–58. doi: 10.3390/ijerph191811208
30. Reece AS. Disruption of interlocking synchrony between metabolome and epigenome key to understanding widespread embyrotoxicity and carcinogenicity of diverse cannabinoids. Br Med J. (2022) 377:o1567. doi: 10.1136/bmj.o1567
31. Reece AS, Hulse GK. Epigenomic and other evidence for cannabis-induced aging contextualized in a synthetic epidemiologic overview of cannabinoid-related teratogenesis and cannabinoid-related carcinogenesis. Int J Environ Res Public Health. (2022) 19:16721 - 16776. doi: 10.3390/ijerph192416721
32. Reece AS, Hulse GK. Novel insights into potential cannabis-related cancerogenesis from recent key whole epigenome screen of cannabis dependence and withdrawal: epidemiological comment and explication of Schrott et.al. Genes (Basel). (2022) 14:1–14. doi: 10.3390/genes14010032
33. de Pagter MS, van Roosmalen MJ, Baas AF, Renkens I, Duran KJ, van Binsbergen E, et al. Chromothripsis in healthy individuals affects multiple protein-coding genes and can result in severe congenital abnormalities in offspring. Am J Hum Genet. (2015) 96:651–6. doi: 10.1016/j.ajhg.2015.02.005
34. Forment JV, Kaidi A, Jackson SP. Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer. (2012) 12:663–70. doi: 10.1038/nrc3352
35. Kloosterman WP, Guryev V, van Roosmalen M, Duran KJ, de Bruijn E, Bakker SCM, et al. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum Mol Genet. (2011) 20:1916–24. doi: 10.1093/hmg/ddr073
36. Kloosterman WP, Hoogstraat M, Paling O, Tavakoli-Yaraki M, Renkens I, Vermaat JS, et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol. (2011) 12:R103. doi: 10.1186/gb-2011-12-10-r103
37. Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage in micronuclei. Nature. (2015) 522:179–84. doi: 10.1038/nature14493
38. Nissen L, Casciano F, Babini E, Gianotti A. Beneficial metabolic transformations and prebiotic potential of hemp bran and its alcalase hydrolysate, after colonic fermentation in a gut model. Sci Rep. (2023) 13:1552. doi: 10.1038/s41598-023-27726-w
39. Wan X, Eguchi A, Qu Y, et al. Gut-microbiota-brain axis in the vulnerability to psychosis in adulthood after repeated cannabis exposure during adolescence. Eur Arch Psychiatry Clin Neurosci. (2022) 272:1297–309. doi: 10.1007/s00406-022-01437-1
40. Al-Ghezi ZZ, Busbee PB, Alghetaa H, Nagarkatti PS, Nagarkatti M. Combination of cannabinoids, delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD), mitigates experimental autoimmune encephalomyelitis (EAE) by altering the gut microbiome. Brain Behav Immun. (2019) 82:25–35. doi: 10.1016/j.bbi.2019.07.028
41. Zhang X, Cong R, Geng T, Zhang J, Liu D, Tian Q, et al. Assessment of the causal effect of IgG N-glycosylation level on risk of dementia: a 2-sample mendelian randomization study. J Alzheimers Dis. (2022) 88:1435–41. doi: 10.3233/JAD-220074
42. Wang B, Liu D, Song M, Wang W, Guo B, Wang Y. Immunoglobulin G N-glycan, inflammation and type 2 diabetes in East Asian and European populations: a Mendelian randomization study. Mol Med. (2022) 28:114. doi: 10.1186/s10020-022-00543-z
43. Liu D, Dong J, Zhang J, Xu X, Tian Q, Meng X, et al. Genome-wide mapping of plasma IgG N-glycan quantitative trait loci identifies a potentially causal association between IgG N-glycans and rheumatoid arthritis. J Immunol. (2022) 208:2508–14. doi: 10.4049/jimmunol.2100080
44. Adua E, Afrifa-Yamoah E, Peprah-Yamoah E, et al. Multi-block data integration analysis for identifying and validating targeted N-glycans as biomarkers for type II diabetes mellitus. Sci Rep. (2022) 12:10974. doi: 10.1038/s41598-022-15172-z
45. Zhang X, Yuan H, Lyu J, Meng X, Tian Q, Li Y, et al. Association of dementia with immunoglobulin G N-glycans in a Chinese Han population. npj Aging and Mechanisms of Disease. (2021) 7:3. doi: 10.1038/s41514-021-00055-w
46. Wu Z, Pan H, Liu D, Zhou D, Tao L, Zhang J, et al. Variation of IgG N-linked glycosylation profile in diabetic retinopathy. J Diabetes. (2021) 13:672–80. doi: 10.1111/1753-0407.13160
47. Wang X, Zhong Z, Balmer L, Wang W. Glycosylation profiling as a biomarker of suboptimal health status for chronic disease stratification. Adv Exp Med Biol. (2021) 1325:321–39. doi: 10.1007/978-3-030-70115-4_16
48. Russell A, Wang W. The rapidly expanding nexus of immunoglobulin g n-glycomics, suboptimal health status, and precision medicine. Exp Suppl. (2021) 112:545–64. doi: 10.1007/978-3-030-76912-3_17
49. Li X, Wang H, Zhu Y, Cao W, Song M, Wang Y, et al. Heritability enrichment of immunoglobulin G N-glycosylation in specific tissues. Front Immunol. (2021) 12:741705. doi: 10.3389/fimmu.2021.741705
50. Hou H, Yang H, Liu P, Huang C, Wang M, Li Y, et al. Profile of immunoglobulin G N-glycome in COVID-19 patients: a case-control study. Front Immunol. (2021) 12:748566. doi: 10.3389/fimmu.2021.748566
51. COVID-19 and Preparing Planetary Health for Future Ecological Crises: Hopes from Glycomics for Vaccine Innovation. OMICS. (2021) 25:234–41. doi: 10.1089/omi.2021.0011
52. Xia X, Chen X, Wu G, et al. Three-dimensional facial-image analysis to predict heterogeneity of the human ageing rate and the impact of lifestyle. Nature Metabolism. (2020) 2:946–57. doi: 10.1038/s42255-020-00270-x
53. Fisar Z, Singh N, Hroudova J. Cannabinoid-induced changes in respiration of brain mitochondria. Toxicol Lett. (2014) 231:62–71. doi: 10.1016/j.toxlet.2014.09.002
54. Morimoto S, Tanaka Y, Sasaki K, Tanaka H, Fukamizu T, Shoyama Y, et al. Identification and characterization of cannabinoids that induce cell death through mitochondrial permeability transition in Cannabis leaf cells. J Biol Chem. (2007) 282:20739–51. doi: 10.1074/jbc.M700133200
55. Sarafian TA, Habib N, Oldham M, Seeram N, Lee R-P, Lin L, et al. Inhaled marijuana smoke disrupts mitochondrial energetics in pulmonary epithelial cells in vivo. Am J Physiology. (2006) 290:L1202–9. doi: 10.1152/ajplung.00371.2005
56. Sarafian TA, Kouyoumjian S, Khoshaghideh F, Tashkin DP, Roth MD. Delta 9-tetrahydrocannabinol disrupts mitochondrial function and cell energetics. American journal of physiology. (2003) 284:L298–306. doi: 10.1152/ajplung.00157.2002
57. Singh N, Hroudova J, Fisar Z. Cannabinoid-induced changes in the activity of electron transport chain complexes of brain mitochondria. J Mol Neurosci. (2015) 56:926–31. doi: 10.1007/s12031-015-0545-2
58. Tahir SK, Zimmerman AM. Influence of marihuana on cellular structures and biochemical activities. Pharmacol Biochem Behav. (1991) 40:617-23. doi: 10.1016/0091-3057(91)90372-9
59. Bino T, Chari-Bitron A, Shahar A. Biochemical effects and morphological changes in rat liver mitochondria exposed to 1 -tetrahydrocannabinol. Biochim Biophys Acta. (1972) 288:195–202. doi: 10.1016/0005-2736(72)90238-6
60. Ryan D, Drysdale AJ, Lafourcade C, Pertwee RG, Platt B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J Neurosci. (2009) 29:2053–63. doi: 10.1523/JNEUROSCI.4212-08.2009
61. Wu HY, Huang CH, Lin YH, Wang CC, Jan TR. Cannabidiol induced apoptosis in human monocytes through mitochondrial permeability transition pore-mediated ROS production. Free Radic Biol Med. (2018) 124:311–8. doi: 10.1016/j.freeradbiomed.2018.06.023
62. Winklmayr M, Gaisberger M, Kittl M, Fuchs J, Ritter M, Jakab M. Dose-dependent cannabidiol-induced elevation of intracellular calcium and apoptosis in human articular chondrocytes. J Orthop Res. (2019) 37:2540–9. doi: 10.1002/jor.24430
63. Olivas-Aguirre M, Torres-López L, Pottosin I, Dobrovinskaya O. Phenolic compounds cannabidiol, curcumin and quercetin cause mitochondrial dysfunction and suppress acute lymphoblastic leukemia cells. Int J Mol Sci. (2020) 22:1. doi: 10.3390/ijms22010204
64. Chan JZ, Duncan RE. Regulatory effects of cannabidiol on mitochondrial functions: a review. Cells. (2021) 10:1251. doi: 10.3390/cells10051251
65. Drummond-Main CD, Ahn Y, Kesler M, Gavrilovici C, Kim DY, Kiroski I, et al. cannabidiol impairs brain mitochondrial metabolism and neuronal integrity. Cannabis Cannabinoid Res. (2022) 8:283–98. doi: 10.1089/can.2022.0011
66. Rupprecht A, Theisen U, Wendt F, Frank M, Hinz B. The combination of Δ(9)-tetrahydrocannabinol and cannabidiol suppresses mitochondrial respiration of human glioblastoma cells via downregulation of specific respiratory chain proteins. Cancers (Basel). (2022) 14:13 doi: 10.3390/cancers14133129
67. Chinta SJ, Poksay KS, Kaundinya G, Hart M, Bredesen DE, Andersen JK, et al. Endoplasmic reticulum stress-induced cell death in dopaminergic cells: effect of resveratrol. J Mol Neurosci. (2009) 39:157–68. doi: 10.1007/s12031-008-9170-7
68. Coope A, Milanski M, Arruda AP, Ignacio-Souza LM, Saad MJ, Anhê GF, et al. Chaperone insufficiency links TLR4 protein signaling to endoplasmic reticulum stress. J Biol Chem. (2012) 287:15580–9. doi: 10.1074/jbc.M111.315218
69. Miani M, Barthson J, Colli ML, Brozzi F, Cnop M, Eizirik DL. Endoplasmic reticulum stress sensitizes pancreatic beta cells to interleukin-1beta-induced apoptosis via Bim/A1 imbalance. Cell Death Dis. (2013) 4:e701. doi: 10.1038/cddis.2013.236
70. Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci U S A. (2011) 108:2939–44. doi: 10.1073/pnas.1006875108
71. Yao S, Yang N, Song G, Sang H, Tian H, Miao C, et al. Minimally modified low-density lipoprotein induces macrophage endoplasmic reticulum stress via toll-like receptor 4. Biochim Biophys Acta. (2012) 1821:954–63. doi: 10.1016/j.bbalip.2012.03.003
72. Zhou J, Mao B, Zhou Q, Ding D, Wang M, Guo P, et al. Endoplasmic reticulum stress activates telomerase. Aging Cell. (2013) 13:197–200. doi: 10.1111/acel.12161
73. Li X, Yang Y, Zhang B, Lin X, Fu X, An Y, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther. (2022) 7:305. doi: 10.1038/s41392-022-01151-3
74. Yang Z, Yan C, Ma J, Peng P, Ren X, Cai S, et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nature Metabolism. (2023). doi: 10.1038/s42255-022-00710-w
75. Sun S, Hu F, Wu J, Zhang S. Cannabidiol attenuates OGD/R-induced damage by enhancing mitochondrial bioenergetics and modulating glucose metabolism via pentose-phosphate pathway in hippocampal neurons. Redox Biol. (2017) 11:577–85. doi: 10.1016/j.redox.2016.12.029
76. Zehra A, Burns J, Liu CK, Manza P, Wiers CE, Volkow ND, Wang GJ. Cannabis addiction and the brain: a review. J Neuroimmune Pharmacol. (2018) 13:438–52. doi: 10.1007/s11481-018-9782-9
77. Volkow ND, Wang G-J, Telang F, Fowler JS, Alexoff D, Logan J, et al. Decreased dopamine brain reactivity in marijuana abusers is associated with negative emotionality and addiction severity. Proc Natl Acad Sci U S A. (2014) 111:E3149–56. doi: 10.1073/pnas.1411228111
78. Wiers CE, Shokri-Kojori E, Wong CT, Abi-Dargham A, Demiral SB, Tomasi D, et al. Cannabis abusers show hypofrontality and blunted brain responses to a stimulant challenge in females but not in males. Neuropsychopharmacology. (2016) 41:2596–605. doi: 10.1038/npp.2016.67
79. Hanus C, Ehrensperger MV, Triller A. Activity-dependent movements of postsynaptic scaffolds at inhibitory synapses. J Neurosci. (2006) 26:4586-95. doi: 10.1523/JNEUROSCI.5123-05.2006
80. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
81. Philpot BD, Zukin RS. Synapse-specific metaplasticity: to be silenced is not to silence 2B. Neuron. (2010) 66:814–6. doi: 10.1016/j.neuron.2010.06.014
82. Zamberletti E, Gabaglio M, Grilli M, Prini P, Catanese A, Pittaluge A, et al. Long-term hippocampal glutamate synapse and astrocyte dysfunctions underlying the altered phenotype induced by adolescent THC treatment in male rats. Pharmacol Res. (2016) 111:459–70. doi: 10.1016/j.phrs.2016.07.008
83. Dejanovic B, Huntley MA, De Mazière A, Meilandt WJ, Wu T, Srinivasan K, et al. Changes in the synaptic proteome in tauopathy and rescue of Tau-Induced Synapse Loss By C1q antibodies. Neuron. (2018) 100:1322–36 e7. doi: 10.1016/j.neuron.2018.10.014
84. Awasthi A, Ramachandran B, Ahmed S, Benito E, Shinoda Y, Nitzan N, et al. Synaptotagmin-3 drives AMPA receptor endocytosis, depression of synapse strength, and forgetting. Science. (2019) 363:eaav1483. doi: 10.1126/science.aav1483
85. Huang H, Liu S, Kornberg TB. Glutamate signaling at cytoneme synapses. Science. (2019) 363:948–55. doi: 10.1126/science.aat5053
86. Holler S, Köstinger G, Martin KAC, Schuhknecht GFP, Stratford KJ. Structure and function of a neocortical synapse. Nature. (2021) 591:111–6. doi: 10.1038/s41586-020-03134-2
87. Bernard C, Exposito-Alonso D, Selten M, Sanalidou S, Hanusz-Godoy A, Aguilera A, et al. Cortical wiring by synapse type-specific control of local protein synthesis. Science. (2022) 378:eabm7466. doi: 10.1126/science.abm7466
88. Muniyappa R, Sable S, Ouwerkerk R, Mari A, Gharib AM, Walter M, et al. Metabolic effects of chronic cannabis smoking. Diabetes Care. (2013) 36:2415–22. doi: 10.2337/dc12-2303
89. Volkow ND, Swanson JM, Evins AE, DeLisi LE, Meier MH, Gonzalez R, et al. Effects of cannabis use on human behavior, including cognition, motivation, and psychosis: a review. JAMA Psychiatry. (2016) 73:292–7. doi: 10.1001/jamapsychiatry.2015.3278
90. van de Giessen E, Weinstein JJ, Cassidy CM, Haney M, Dong Z, Ghazzaoui R, et al. Deficits in striatal dopamine release in cannabis dependence. Mol Psychiatry. (2017) 22:68–75. doi: 10.1038/mp.2016.21
91. Hirvonen J, Goodwin RS, Li C-T, Terry GE, Zoghbi SS, Morse C, et al. Reversible and regionally selective downregulation of brain cannabinoid CB1 receptors in chronic daily cannabis smokers. Mol Psychiatry. (2012) 17:642–9. doi: 10.1038/mp.2011.82
92. Tomasi D, Wang GJ, Volkow ND. Balanced modulation of striatal activation from D2/D3 receptors in caudate and ventral striatum: Disruption in cannabis abusers. Hum Brain Mapp. (2015) 36:3154–66. doi: 10.1002/hbm.22834
93. Klein TW, Newton C, Larsen K, Lu L, Perkins I, Nong L, et al. The cannabinoid system and immune modulation. J Leukocyte Biol. (2003) 74:486–96. doi: 10.1189/jlb.0303101
94. Greineisen WE, Turner H. Immunoactive effects of cannabinoids: considerations for the therapeutic use of cannabinoid receptor agonists and antagonists. Int Immunopharmacol. (2010) 10:547–55. doi: 10.1016/j.intimp.2010.02.012
95. Rieder SA, Chauhan A, Singh U, Nagarkatti M, Nagarkatti P. Cannabinoid-induced apoptosis in immune cells as a pathway to immunosuppression. Immunobiology. (2010) 215:598–605. doi: 10.1016/j.imbio.2009.04.001
96. Robinson RH, Meissler JJ, Breslow-Deckman JM, Gaughan J, Adler MW, Eisenstein TK. Cannabinoids inhibit T-cells via cannabinoid receptor 2 in an in vitro assay for graft rejection, the mixed lymphocyte reaction. J Neuroimmune Pharmacol. (2013) 8:1239–50. doi: 10.1007/s11481-013-9485-1
97. Alshaarawy O, Anthony JC. Cannabis smoking and serum C-reactive protein: a quantile regressions approach based on NHANES 2005-2010. Drug Alcohol Depend. (2015) 147:203–7. doi: 10.1016/j.drugalcdep.2014.11.017
98. Chandra LC, Kumar V, Torben W, Stouwe CV, Winsauer P, Amedee A, et al. Chronic administration of Delta9-tetrahydrocannabinol induces intestinal anti-inflammatory microRNA expression during acute simian immunodeficiency virus infection of rhesus macaques. J Virol. (2015) 89:1168–81. doi: 10.1128/JVI.01754-14
99. Eisenstein TK, Meissler JJ. Effects of Cannabinoids on T-cell Function and Resistance to Infection. J Neuroimmune Pharmacol. (2015) 10:204–16. doi: 10.1007/s11481-015-9603-3
100. Zumbrun EE, Sido JM, Nagarkatti PS, Nagarkatti M. Epigenetic regulation of immunological alterations following prenatal exposure to marijuana cannabinoids and its long term consequences in offspring. J Neuroimmune Pharmacol. (2015) 10:245–54. doi: 10.1007/s11481-015-9586-0
101. Chiurchiu V. Endocannabinoids and immunity. Cannabis Cannabinoid Res. (2016) 1:59–66. doi: 10.1089/can.2016.0002
102. Bayazit H, Selek S, Karababa IF, Cicek E, Aksoy N. Evaluation of oxidant/antioxidant status and cytokine levels in patients with cannabis use disorder. Clin Psychopharmacol Neurosci. (2017) 15:237–42. doi: 10.9758/cpn.2017.15.3.237
103. Marinelli C, Bertalot T, Zusso M, Skaper SD, Giusti P. Systematic Review of Pharmacological Properties of the Oligodendrocyte Lineage. Front Cell Neurosci. (2016) 10:27. doi: 10.3389/fncel.2016.00027
104. Ilyasov AA, Milligan CE, Pharr EP, Howlett AC. The endocannabinoid system and oligodendrocytes in health and disease. Front Neurosci. (2018) 12:733. doi: 10.3389/fnins.2018.00733
105. Sanchez-Rodriguez MA, Gomez O, Esteban PF, Garcia-Ovejero D, Molina-Holgado E. The endocannabinoid 2-arachidonoylglycerol regulates oligodendrocyte progenitor cell migration. Biochem Pharmacol. (2018) 157:180–8. doi: 10.1016/j.bcp.2018.09.006
106. Iram T, Kern F, Kaur A, Myneni S, Morningstar AR, Shin H, et al. Young CSF restores oligodendrogenesis and memory in aged mice via Fgf17. Nature. (2022) 605:509–15. doi: 10.1038/s41586-022-04722-0
107. Zalesky A, Solowij N, Yücel M, Lubman DI, Takagi M, Harding IH, et al. Effect of long-term cannabis use on axonal fibre connectivity. Brain. (2012) 135:2245–55. doi: 10.1093/brain/aws136
108. Khara LS, Ali DW. The endocannabinoid system's involvement in motor development relies on cannabinoid receptors, TRP channels, and Sonic Hedgehog signaling. Physiol Rep. (2023) 11:e15565. doi: 10.14814/phy2.15565
109. Malheiro RF, Carmo H, Carvalho F, Silva JP. Cannabinoid-mediated targeting of mitochondria on the modulation of mitochondrial function and dynamics. Pharmacol Res. (2023) 187:106603. doi: 10.1016/j.phrs.2022.106603
110. Lian J, Casari I, Falasca M. Modulatory role of the endocannabinoidome in the pathophysiology of the gastrointestinal tract. Pharmacol Res. (2022) 175:106025. doi: 10.1016/j.phrs.2021.106025
111. Khaliullina H, Bilgin M, Sampaio JL, Shevchenko A, Eaton S. Endocannabinoids are conserved inhibitors of the Hedgehog pathway. Proc Natl Acad Sci U S A. (2015) 112:3415–20. doi: 10.1073/pnas.1416463112
112. Fernández-Moncada I, Marsicano G. Astroglial CB1 receptors, energy metabolism, and gliotransmission: an integrated signaling system? Essays Biochem. (2023) 67:49–61. doi: 10.1042/EBC20220089
113. Fish EW, Murdaugh LB, Zhang C, Boschen KE, Boa-Amponsem O, Mendoza-Romero HN, et al. Cannabinoids exacerbate alcohol teratogenesis by a CB1-hedgehog interaction. Sci Rep. (2019) 9:16057–75. doi: 10.1038/s41598-019-52336-w
114. Boa-Amponsem O, Zhang C, Burton D, Williams KP, Cole GJ. Ethanol and cannabinoids regulate zebrafish GABAergic neuron development and behavior in a sonic hedgehog and fibroblast growth factor-dependent mechanism. Alcohol Clin Exp Res. (2020) 44:1366–77. doi: 10.1111/acer.14383
115. Boa-Amponsem O, Zhang C, Mukhopadhyay S, Ardrey I, Cole GJ. Ethanol and cannabinoids interact to alter behavior in a zebrafish fetal alcohol spectrum disorder model. Birth Defects Res. (2019) 111:775–88. doi: 10.1002/bdr2.1458
116. Burton DF, Zhang C, Boa-Amponsem O, Mackinnon S, Cole GJ. Long-term behavioral change as a result of acute ethanol exposure in zebrafish: Evidence for a role for sonic hedgehog but not retinoic acid signaling. Neurotoxicol Teratol. (2017) 61:66–73. doi: 10.1016/j.ntt.2017.01.006
117. Loucks EJ, Ahlgren SC. Deciphering the role of Shh signaling in axial defects produced by ethanol exposure. Birth Defects Res A Clin Mol Teratol. (2009) 85:556–67. doi: 10.1002/bdra.20564
118. Zhang C, Ojiaku P, Cole GJ. Forebrain and hindbrain development in zebrafish is sensitive to ethanol exposure involving agrin, Fgf, and sonic hedgehog function. Birth Defects Res A Clin Mol Teratol. (2013) 97:8–27. doi: 10.1002/bdra.23099
119. Zhang C, Turton QM, Mackinnon S, Sulik KK, Cole GJ. Agrin function associated with ocular development is a target of ethanol exposure in embryonic zebrafish. Birth Defects Res A Clin Mol Teratol. (2011) 91:129–41. doi: 10.1002/bdra.20766
120. Winters BL, Vaughan CW. Mechanisms of endocannabinoid control of synaptic plasticity. Neuropharmacology. (2021) 197:108736. doi: 10.1016/j.neuropharm.2021.108736
121. Balezina OP, Tarasova EO, Gaydukov AE. Noncanonical activity of endocannabinoids and their receptors in central and peripheral synapses. Biochemistry (Mosc). (2021) 86:818–32. doi: 10.1134/S0006297921070038
122. Mahmoud AM, Kostrzewa M, Marolda V, Cerasuolo M, Maccarinelli F, Coltrini D, et al. Cannabidiol alters mitochondrial bioenergetics via VDAC1 and triggers cell death in hormone-refractory prostate cancer. Pharmacol Res. (2023) 189:106683. doi: 10.1016/j.phrs.2023.106683
123. Cutando L, Maldonado R, Ozaita A. Microglial activation and cannabis exposure. In: Preedy V, editor. Handbook of Cannabis and Related Pathologies: Biology, Pharmacology, Diagnosis and Treatment. New York: Academic Press. (2017) p. 401–412.
124. Chiu P, Karler R, Craven C, Olsen DM, Turkanis SA. The influence of delta9-tetrahydrocannabinol, cannabinol and cannabidiol on tissue oxygen consumption. Res Commun Chem Pathol Pharmacol. (1975) 12:267–86.
125. Koch M, Varela L, Kim JG, Kim JD, Hernández-Nuño F, Simonds SE, et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. (2015) 519:45–50. doi: 10.1038/nature14260
126. Hebert-Chatelain E, Desprez T, Serrat R, Bellachio L, Soria-Gomez E, Garcia AB, et al. A cannabinoid link between mitochondria and memory. Nature. (2016) 539:555–9. doi: 10.1038/nature20127
127. Harkany T, Horvath TL. (S)Pot on mitochondria: cannabinoids disrupt cellular respiration to limit neuronal activity. Cell Metab. (2017) 25:8–10. doi: 10.1016/j.cmet.2016.12.020
128. Gómez-Cañas M, Rodríguez-Cueto C, Satta V, Hernández-Fisac I, Navarro E, Fernández-Ruiz J. Endocannabinoid-binding receptors as drug targets. Methods Mol Biol. (2023) 2576:67–94. doi: 10.1007/978-1-0716-2728-0_6
129. Przybycień P, Gasior-Perczak D, Placha W. Cannabinoids and PPAR ligands: the future in treatment of polycystic ovary syndrome women with obesity and reduced fertility. Cells. (2022) 11:16. doi: 10.3390/cells11162569
130. Parksepp M, Haring L, Kilk K, Koch K, Uppin K, Kangro R, et al. The Expanded Endocannabinoid System Contributes to Metabolic and Body Mass Shifts in First-Episode Schizophrenia: A 5-Year Follow-Up Study. Biomedicines. (2022) 10:2. doi: 10.3390/biomedicines10020243
131. Mock ED, Gagestein B, van der Stelt M. Anandamide and other N-acylethanolamines: A class of signaling lipids with therapeutic opportunities. Prog Lipid Res. (2022) 2022:101194. doi: 10.1016/j.plipres.2022.101194
132. Lott J, Jutkiewicz EM, Puthenveedu MA. The synthetic cannabinoid WIN55,212-2 can disrupt the golgi apparatus independent of cannabinoid receptor-1. Mol Pharmacol. (2022) 101:371–80. doi: 10.1124/molpharm.121.000377
133. Liu YH, Liu Y, Zhang X, Fang L, Zhao BL, Wang NP. Activation of the endocannabinoid system mediates cardiac hypertrophy induced by rosiglitazone. Acta Pharmacol Sin. (2022) 43:2302–12. doi: 10.1038/s41401-022-00858-x
134. Lee HS, Tamia G, Song HJ, Amarakoon D, Wei CI, Lee SH. Cannabidiol exerts anti-proliferative activity via a cannabinoid receptor 2-dependent mechanism in human colorectal cancer cells. Int Immunopharmacol. (2022) 108:108865. doi: 10.1016/j.intimp.2022.108865
135. Hirao-Suzuki M, Takayuki K, Takiguchi M, Peters JM, Takeda S. Cannabidiolic acid activates the expression of the PPARβ/δ target genes in MDA-MB-231 cells. Arch Biochem Biophys. (2022) 731:109428. doi: 10.1016/j.abb.2022.109428
136. Volkow ND, Gillespie H, Mullani N, Tancredi L, Grant C, Valentine A, et al. Brain glucose metabolism in chronic marijuana users at baseline and during marijuana intoxication. Psychiatry Res. (1996) 67:29–38. doi: 10.1016/0925-4927(96)02817-X
137. Callén L, Moreno E, Barroso-Chinea P, Moreno-Delgado D, Cortés A, Mallol J, et al. Cannabinoid receptors CB1 and CB2 form functional heteromers in brain. J Biol Chem. (2012) 287:20851–65. doi: 10.1074/jbc.M111.335273
138. Chien KR, Karsenty G. Longevity and lineages: toward the integrative biology of degenerative diseases in heart, muscle, and bone. Cell. (2005) 120:533–44. doi: 10.1016/j.cell.2005.02.006
139. Borowska M, Czarnywojtek A, Sawicka-Gutaj N, Woliński K, Płazińska MT, Mikołajczak P, et al. The effects of cannabinoids on the endocrine system. Endokrynol Pol. (2018) 69:705–19. doi: 10.5603/EP.a2018.0072
140. Reece AS, Thomas MR, Norman A, Hulse GK. Dramatic acceleration of reproductive aging, contraction of biochemical fecundity and healthspan-lifespan implications of opioid-induced endocrinopathy-FSH/LH ratio and other interrelationships. Reprod Toxicol. (2016) 66:20–30. doi: 10.1016/j.reprotox.2016.09.006
141. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. (2005) 120:483–95. doi: 10.1016/j.cell.2005.02.001
142. Guarente L, Picard F. Calorie restriction–the SIR2 connection. Cell. (2005) 120:473–82. doi: 10.1016/j.cell.2005.01.029
143. Hadley EC, Lakatta EG, Morrison-Bogorad M, Warner HR, Hodes RJ. The future of aging therapies. Cell. (2005) 120:557–67. doi: 10.1016/j.cell.2005.01.030
144. Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW, et al. repair, genome stability, and aging. Cell. (2005) 120:497–512. doi: 10.1016/j.cell.2005.01.028
145. de Laval B, Maurizio J, Kandalla PK, et al. C/EBPβ-dependent epigenetic memory induces trained immunity in hematopoietic stem cells. Cell Stem Cell. (2020) 26:657–674.e8. doi: 10.1016/j.stem.2020.01.017
146. Gonzales KAU, Polak L, Matos I, Tierney MT, Gola A, Wong E, et al. Stem cells expand potency and alter tissue fitness by accumulating diverse epigenetic memories. Science. (2021) 374:eabh2444. doi: 10.1126/science.abh2444
147. Pucci M, Rapino C, Di Francesco A, Dainese E, D'Addario C, Maccarrone M. Epigenetic control of skin differentiation genes by phytocannabinoids. Br J Pharmacol. (2013) 170:581–91. doi: 10.1111/bph.12309
148. Gobbi G, Atkin T, Zytynski T, Wang S, Askari S, Boruff J, et al. Association of cannabis use in adolescence and risk of depression, anxiety, and suicidality in young adulthood: a systematic review and meta-analysiscannabis use in adolescence and risk of depression, anxiety, and suicidality in young adulthoodcannabis use in adolescence and risk of depression, anxiety, and suicidality in young adulthood. JAMA Psychiatry. (2019) 76:426–34. doi: 10.1001/jamapsychiatry.2018.4500
149. Wanner NM, Colwell M, Drown C, Faulk C. Developmental cannabidiol exposure increases anxiety and modifies genome-wide brain DNA methylation in adult female mice. Clin Epigenetics. (2021) 13:4. doi: 10.1186/s13148-020-00993-4
150. Mok PLH, Pedersen CB, Springate D, Astrup A, Kapur N, Antonsen S, et al. Parental psychiatric disease and risks of attempted suicide and violent criminal offending in offspring: a population-based cohort study. JAMA Psychiatry. (2016) 73:1015–22. doi: 10.1001/jamapsychiatry.2016.1728
151. Huckins LM. Linking cannabis use to depression and suicidal thoughts and behaviours. The lancet Psychiatry. (2017) 4:654–6. doi: 10.1016/S2215-0366(17)30311-5
152. Smolkina M, Morley KI, Rijsdijk F, Agrawal A, Bergin JE, Nelson EC, et al. Cannabis and depression: a twin model approach to co-morbidity. Behav Genet. (2017) 47:394–404. doi: 10.1007/s10519-017-9848-0
153. Troup LJ, Torrence RD, Andrzejewski JA, Braunwalder JT. Effects of cannabis use and subclinical depression on the P3 event-related potential in an emotion processing task. Medicine (Baltimore). (2017) 96:e6385. doi: 10.1097/MD.0000000000006385
154. Dierker L, Selya A, Lanza S, Li R, Rose J. Depression and marijuana use disorder symptoms among current marijuana users. Addict Behav. (2018) 76:161–8. doi: 10.1016/j.addbeh.2017.08.013
155. Filbey FM, Aslan S, Lu H, Peng SL. Residual effects of THC via novel measures of brain perfusion and metabolism in a large group of chronic cannabis users. Neuropsychopharmacology. (2018) 43:700–7. doi: 10.1038/npp.2017.44
156. Lisboa SF, Gomes FV, Terzian AL, Aguair DC, Moreira FA, Resstel LBM, et al. The endocannabinoid system and anxiety. Vitam Horm. (2017) 103:193–279. doi: 10.1016/bs.vh.2016.09.006
157. Otten R, Huizink AC, Monshouwer K, Creemers HE, Onrust S. Cannabis use and symptoms of anxiety in adolescence and the moderating effect of the serotonin transporter gene. Addict Biol. (2017) 22:1081–9. doi: 10.1111/adb.12372
158. Duperrouzel J, Hawes SW, Lopez-Quintero C, Pacheco-Colon I, Comer J, Gonzalez R. The association between adolescent cannabis use and anxiety: a parallel process analysis. Addict Behav. (2018) 78:107–13. doi: 10.1016/j.addbeh.2017.11.005
159. Ecker AH, Buckner JD. Cannabis-related problems and social anxiety: the mediational role of post-event processing. Subst Use Misuse. (2018) 53:36–41. doi: 10.1080/10826084.2017.1322984
160. Kvitland LR, Ringen PA, Aminoff SR, Demmo C, Hellvin T, Lagerberg TV, et al. Duration of untreated illness in first-treatment bipolar I disorder in relation to clinical outcome and cannabis use. Psychiatry Res. (2016) 246:762–8. doi: 10.1016/j.psychres.2016.07.064
161. Weinstock LM, Gaudiano BA, Wenze SJ, Epstein-Lubow G, Miller IW. Demographic and clinical characteristics associated with comorbid cannabis use disorders (CUDs) in hospitalized patients with bipolar I disorder. Compr Psychiatry. (2016) 65:57–62. doi: 10.1016/j.comppsych.2015.10.003
162. Hanna RC, Perez JM, Ghose S. Cannabis and development of dual diagnoses: a literature review. Am J Drug Alcohol Abuse. (2017) 43:442–55. doi: 10.1080/00952990.2016.1213273
163. Marwaha S, Winsper C, Bebbington P, Smith D. Cannabis use and hypomania in young people: a prospective analysis. Schizophr Bull. (2018) 44:1267–74. doi: 10.1093/schbul/sbx158
164. Bartoli F, Crocamo C, Carra G. Cannabis use disorder and suicide attempts in bipolar disorder: a meta-analysis. Neurosci Biobehav Rev. (2019) 103:14–20. doi: 10.1016/j.neubiorev.2019.05.017
165. Dennen CA, Blum K, Bowirrat A, Khalsa J, Thanos PK, Baron D, et al. Neurogenetic and epigenetic aspects of cannabinoids. Epigenomes. (2022) 6:3. doi: 10.3390/epigenomes6030027
166. Fusar-Poli L, Pries L-K, van Os J, Radhakrishnan R, Pençe AY, Erzin G, et al. The association between cannabis use and facial emotion recognition in schizophrenia, siblings, and healthy controls: Results from the EUGEI study. Eur Neuropsychopharmacol. (2022) 63:47–59. doi: 10.1016/j.euroneuro.2022.08.003
167. Greco LA, Reay WR, Dayas CV, Cairns MJ. Pairwise genetic meta-analyses between schizophrenia and substance dependence phenotypes reveals novel association signals with pharmacological significance. Transl Psychiatry. (2022) 12:403. doi: 10.1038/s41398-022-02186-4
168. Ibarra-Lecue I, Unzueta-Larrinaga P, Barrena-Barbadillo R, Villate A, Horrillo I, Mendivil B, et al. Cannabis use selectively modulates circulating biomarkers in the blood of schizophrenia patients. Addict Biol. (2022) 27:e13233. doi: 10.1111/adb.13233
169. Kayir H, Ruffolo J, McCunn P, Khokhar JY. The relationship between cannabis, cognition, and schizophrenia: it's complicated. Curr Top Behav Neurosci. (2022) 63:437–61. doi: 10.1007/7854_2022_396
170. Little R, D'Mello D. A cannabinoid hypothesis of schizophrenia: pathways to psychosis. Innov Clin Neurosci. (2022) 19:38–43.
171. Baranger DAA, Paul SE, Colbert SMC, Karcher NR, Johnson EC, Hatoum AS, et al. Association of mental health burden with prenatal cannabis exposure from childhood to early adolescence: longitudinal findings from the adolescent brain cognitive development (ABCD) study. JAMA Pediatr. (2022) 176:1261–5. doi: 10.1001/jamapediatrics.2022.3191
172. Reece AS, Hulse GK. Chapter 1: close parallels between cannabis use and deteriorating us mental health at four levels supports and extends the epidemiological salience of demonstrated causal mental health relationships: a geospatiotemporal study. In: Epidemiology of Cannabis: Genotoxicity and Neurotoxicity, Epigenomics and Aging. New York, USA: Elsevier; (2023).
173. Reece AS, Hulse GK. Co-occurrence across time and space of drug- and cannabinoid- exposure and adverse mental health outcomes in the National Survey of Drug Use and Health: combined geotemporospatial and causal inference analysis. BMC Public Health. (2020) 20:1655–69. doi: 10.1186/s12889-020-09748-5
174. Mukhopadhyay B, Schuebel K, Mukhopadhyay P, Cinar R, Godlewski G, Xiong K, et al. Cannabinoid receptor 1 promotes hepatocellular carcinoma initiation and progression through multiple mechanisms. Hepatology. (2015) 61:1615–26. doi: 10.1002/hep.27686
175. Mukhopadhyay B, Cinar R, Yin S, Liu J, Tam J, Godlewski G, et al. Hyperactivation of anandamide synthesis and regulation of cell-cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proc Natl Acad Sci U S A. (2011) 108:6323–8. doi: 10.1073/pnas.1017689108
176. Yang Y-Y, Lin H-C, Huang Y-T, Lee T-Y, Hou M-C, Wang Y-W, et al. Effect of chronic CB1 cannabinoid receptor antagonism on livers of rats with biliary cirrhosis. Clin Sci (Lond). (2007) 112:533–42. doi: 10.1042/CS20060260
177. Morishima A. Effects of cannabis and natural cannabinoids on chromosomes and ova. NIDA Res Monogr. (1984) 44:25–45.
178. Huang HFS, Nahas GG, Hembree WC. Effects of Marijuana Inhalation on Spermatogenesis of the Rat. In: Nahas GG, Sutin KM, Harvey DJ, Agurell S, editors. Marijuana in Medicine. Totowa, New York: Human Press. (1999) p. 359–366.
179. Reece AS, Hulse GK. Cannabis Genotoxicity and Cancer Incidence: A Highly Concordant Synthesis of European and USA Datasets. In: Preedy. V., Patel V, editors. Cannabis, Cannabinoids and Endocannabinoids. London: Elsevier. (2023) p. 93–112.
180. Reece AS, Hulse GK. Geotemporospatial and causal inferential epidemiological overview and survey of USA cannabis, cannabidiol and cannabinoid genotoxicity expressed in cancer incidence 2003–2017: Part 1 – continuous bivariate analysis. Archives of Public Health. (2022) 80:99–133. doi: 10.1186/s13690-022-00811-8
181. Reece AS, Hulse GK. Geotemporospatial and causal inferential epidemiological overview and survey of USA cannabis, cannabidiol and cannabinoid genotoxicity expressed in cancer incidence 2003–2017: Part 2—categorical bivariate analysis and attributable fractions. Archives of Public Health. (2022) 80:100–35. doi: 10.1186/s13690-022-00812-7
182. Reece AS, Hulse GK. Geotemporospatial and causal inferential epidemiological overview and survey of USA cannabis, cannabidiol and cannabinoid genotoxicity expressed in cancer incidence 2003–2017: Part 3—spatiotemporal, multivariable and causal inferential pathfinding and exploratory analyses of prostate and ovarian cancers. Archives of Public Health. (2022) 80:100–36. doi: 10.1186/s13690-022-00813-6
183. Reece AS, Hulse GK. Geospatiotemporal and causal inference study of cannabis and other drugs as risk factors for female breast cancer USA 2003–2017. Environ Epigenetics. (2022). 8:1–22. doi: 10.1093/eep/dvac006
184. Reece AS, Hulse GK. State trends of cannabis liberalization as a causal driver of increasing testicular cancer rates across the USA. Int J Environ Res Public Health. (2022). 19:12759 – 12796. doi: 10.3390/ijerph191912759
185. Reece AS, Hulse GK. Clinical epigenomic explanation of the epidemiology of cannabinoid genotoxicity manifesting as transgenerational teratogenesis, cancerogenesis and aging acceleration. Int J Environ Res Public Health. (2023) 20:3360–3383. doi: 10.3390/ijerph20043360
186. Reece AS, Hulse GK, A. geospatiotemporal and causal inference epidemiological exploration of substance and cannabinoid exposure as drivers of rising US pediatric cancer rates. BMC Cancer. (2021) 21:197–230. doi: 10.1186/s12885-021-07924-3
187. Reece AS, Hulse GK. Causal inference multiple imputation investigation of the impact of cannabinoids and other substances on ethnic differentials in US testicular cancer incidence. BMC Pharmacol Toxicol. (2021) 22:40–71. doi: 10.1186/s40360-021-00505-x
188. Reece AS, Hulse GK. Cannabinoid exposure as a major driver of pediatric acute lymphoid Leukaemia rates across the USA: combined geospatial, multiple imputation and causal inference study. BMC Cancer. (2021) 21: 984–1017. doi: 10.1186/s12885-021-08598-7
189. Reece AS, Hulse GK. Cannabis- and substance-related carcinogenesis in europe: a lagged causal inferential panel regression modelling study. J Xenobiotics. (2023). 13:3232–385. doi: 10.3390/jox13030024
190. Reece AS, Hulse GK. Cannabis could be the missing environmental pancreatic carcinogen hiding in plain view. Gastroenterology. (2023) 23:S0016-5085(23)00587-5. doi: 10.1053/j.gastro.2023.02.050
191. Reece AS, Hulse GK. Cannabinoid- and substance- relationships of european congenital anomaly patterns: a space-time panel regression and causal inferential study. Environmental Epigenetics. (2022) 8:1–40. doi: 10.1093/eep/dvab015
192. Reece AS, Hulse GK. Cannabinoid genotoxicity and congenital anomalies: a convergent synthesis of european and USA datasets. In: Preedy V, Patel V, editors. Cannabis, Cannabinoids and Endocannabinoids. London, UK: Elsevier. (2023) p. 71–92.
193. Reece AS, Hulse GK. Congenital anomaly epidemiological correlates of δ8thc across USA 2003–2016: panel regression and causal inferential study. Environmental Epigenetics. (2022) 8:1–17. doi: 10.1093/eep/dvac012
194. Reece AS, Hulse GK. European epidemiological patterns of cannabis- and substance- related congenital body wall anomalies: geospatiotemporal and causal inferential study. Int J Environ Res Public Health. (2022) 19:9027–64. doi: 10.3390/ijerph19159027
195. Reece AS, Hulse GK. European epidemiological patterns of cannabis- and substance- related congenital cardiovascular anomalies: geospatiotemporal and causal inferential study. Environmental Epigenetics. (2022) 8:1–55. doi: 10.1093/eep/dvac015
196. Reece AS, Hulse GK. European epidemiological patterns of cannabis- and substance- related congenital neurological anomalies: geospatiotemporal and causal inferential study. Int J Environ Res Public Health. (2022) 20:441–75. doi: 10.3390/ijerph20010441
197. Reece AS, Hulse GK. Effects of cannabis on congenital limb anomalies in 14 european nations: a geospatiotemporal and causal inferential study. Environmental Epigenetics. (2022) 8:1–34. doi: 10.1093/eep/dvac016
198. Reece AS, Hulse GK. European epidemiological patterns of cannabis- and substance- related congenital uronephrological anomalies: geospatiotemporal and causal inferential study. Int J Environ Res Public Health. (2022) 19:13769–828. doi: 10.3390/ijerph192113769
199. Reece AS, Hulse GK. Geospatiotemporal and causal inferential study of european epidemiological patterns of cannabis- and substance- related congenital orofacial anomalies. J Xenobiotics. (2023) 13:42–74. doi: 10.3390/jox13010006
200. Reece AS, Hulse GK. Congenital gastrointestinal anomalies in europe 2010-2019: a geospatiotemporal and causal inferential study of epidemiological patterns in relationship to cannabis- and substance- exposure. Gastrointestinal Insights. (2023) 14:64–109. doi: 10.3390/gastroent14010007
201. Reece AS, Hulse GK. Patterns of cannabis- and substance- related congenital general anomalies in europe: a geospatiotemporal and causal inferential study. Pediatric Reports. (2023). 15:69–121. doi: 10.3390/pediatric15010009
202. Reece AS, Hulse GK. Chapter 3: Geospatiotemporal and Causal Inferential Analysis of United States Congenital Anomalies as a Function of Multiple Cannabinoid- and Substance- Exposures: Phenocopying Thalidomide and Hundred Megabase-Scale Genotoxicity. In: Epidemiology of Cannabis: Genotoxicity and Neurotoxicity, Epigenomics and Aging. New York, USA: Elsevier; (2023). In Press:2500.
203. Reece AS, Hulse GK. Cannabis teratology explains current patterns of coloradan congenital defects: the contribution of increased cannabinoid exposure to rising teratological trends. Clin Pediatr (Phila). (2019) 58:1085–123. doi: 10.1177/0009922819861281
204. Reece AS, Hulse GK. Canadian cannabis consumption and patterns of congenital anomalies: an ecological geospatial analysis. J Addict Med. (2020) 14:e195–210. doi: 10.1097/ADM.0000000000000638
205. Reece AS, Hulse GK. Geotemporospatial and causal inference epidemiological analysis of US survey and overview of cannabis, cannabidiol and cannabinoid genotoxicity in relation to congenital anomalies 2001–2015. BMC Pediatr. (2022) 22:47–124. doi: 10.1186/s12887-021-02996-3
206. Reece AS, Hulse GK. Contemporary epidemiology of rising atrial septal defect trends across USA 1991-2016: a combined ecological geospatiotemporal and causal inferential study. BMC Pediatr. (2020) 20:539–50. doi: 10.1186/s12887-020-02431-z
207. Forrester MB, Merz RD. Risk of selected birth defects with prenatal illicit drug use, Hawaii, 1986-2002. J Toxicol Environm Health. (2007) 70:7–18. doi: 10.1080/15287390600748799
208. Hussein NAEM, El-Toukhy MAE-F, Kazem AH, et al. Protective and therapeutic effects of cannabis plant extract on liver cancer induced by dimethylnitrosamine in mice. Alexandria Journal of Medicine. (2014) 50:241–51. doi: 10.1016/j.ajme.2014.02.003
209. Blevins RD, Regan JD. delta-9-Tetrahydrocannabinol: effect on macromolecular synthesis in human and other mammalian cells. Archives of Toxicol. (1976).35:127–35. doi: 10.1007/BF00372766
210. Mon MJ, Jansing RL, Doggett S, Stein JL, Stein GS. Influence of delta9-tetrahydrocannabinol on cell proliferation and macromolecular biosynthesis in human cells. Biochemical Pharmacol. (1978) 27:1759–65. doi: 10.1016/0006-2952(78)90553-1
211. McClean DK, Zimmerman AM. Action of delta 9-tetrahydrocannabinol on cell division and macromolecular synthesis in division-synchronized protozoa. Pharmacology. (1976) 14:307–21. doi: 10.1159/000136610
212. Libro R, Scionti D, Diomede F, Marchisio M, Grassi G, Pollastro F, et al. Cannabidiol modulates the immunophenotype and inhibits the activation of the inflammasome in human gingival mesenchymal stem cells. Front Physiol. (2016) 7:559. doi: 10.3389/fphys.2016.00559
213. Kozela E, Juknat A, Gao F, Kaushansky N, Coppola G, Vogel Z. Pathways and gene networks mediating the regulatory effects of cannabidiol, a nonpsychoactive cannabinoid, in autoimmune T cells. J Neuroinflammation. (2016) 13:136. doi: 10.1186/s12974-016-0603-x
214. Chen J, Hou C, Chen X, Wang D, Yang P, He X, et al. Protective effect of cannabidiol on hydrogen peroxideinduced apoptosis, inflammation and oxidative stress in nucleus pulposus cells. Mol Med Rep. (2016) 14:2321–7. doi: 10.3892/mmr.2016.5513
215. Benamar K, Geller EB, Adler MW. First in vivo evidence for a functional interaction between chemokine and cannabinoid systems in the brain. J Pharmacol Exp Ther. (2008) 325:641–5. doi: 10.1124/jpet.107.135053
216. Benamar K, Yondorf M, Geller EB, Eisenstein TK, Adler MW. Physiological evidence for interaction between the HIV-1 co-receptor CXCR4 and the cannabinoid system in the brain. Br J Pharmacol. (2009) 157:1225–31. doi: 10.1111/j.1476-5381.2009.00285.x
217. Heller JE, Baty DE, Zhang M, Li H, Adler M, Ganea D, et al. The combination of selective inhibition of the cannabinoid CB1 receptor and activation of the cannabinoid CB2 receptor yields improved attenuation of motor and autonomic deficits in a mouse model of spinal cord injury. Clin Neurosurg. (2009).56:84–92. doi: 10.1227/01.NEU.0000333487.18324.36
218. Hser YI, Kagihara J, Huang D, Evans E, Messina N. Mortality among substance-using mothers in California: a 10-year prospective study. Addiction. (2012) 107:215–22. doi: 10.1111/j.1360-0443.2011.03613.x
219. Frost L, Mostofsky E, Rosenbloom JI, Mukamal KJ, Mittleman MA. Marijuana use and long-term mortality among survivors of acute myocardial infarction. Am Heart J. (2013) 165:170–5. doi: 10.1016/j.ahj.2012.11.007
220. Daling JR, Doody DR, Sun X, Trabert BL, Weiss NS, Chen C, et al. Association of marijuana use and the incidence of testicular germ cell tumors. Cancer. (2009) 115:1215–23. doi: 10.1002/cncr.24159
221. Fridell M, Bäckström M, Hesse M, Krantz P, Perrin S, Nyhlén A. Prediction of psychiatric comorbidity on premature death in a cohort of patients with substance use disorders: a 42-year follow-up. BMC Psychiatry. (2019) 19:150. doi: 10.1186/s12888-019-2098-3
222. Desai R, Patel U, Sharma S, Amin P, Bhuva R, Patel MS, et al. Recreational marijuana use and acute myocardial infarction: insights from nationwide inpatient sample in the United States. Cureus. (2017) 9:e1816. doi: 10.7759/cureus.1816
223. Arendt M, Munk-Jorgensen P, Sher L, Jensen SO. Mortality among individuals with cannabis, cocaine, amphetamine, MDMA, and opioid use disorders: a nationwide follow-up study of Danish substance users in treatment. Drug Alcohol Depend. (2011) 114:134–9. doi: 10.1016/S0924-9338(11)71719-9
224. Callaghan RC, Cunningham JK, Verdichevski M, Sykes J, Jaffer SR, Kish SJ. All-cause mortality among individuals with disorders related to the use of methamphetamine: a comparative cohort study. Drug Alcohol Depend. (2012) 125:290–4. doi: 10.1016/j.drugalcdep.2012.03.004
225. Davstad I, Allebeck P, Leifman A, Stenbacka M, Romelsjo A. Self-reported drug use and mortality among a nationwide sample of Swedish conscripts - a 35-year follow-up. Drug Alcohol Depend. (2011) 118:383–90. doi: 10.1016/j.drugalcdep.2011.04.025
226. Calabria B, Degenhardt L, Hall W, Lynskey M. Does cannabis use increase the risk of death? Systematic review of epidemiological evidence on adverse effects of cannabis use. Drug Alcohol Rev. (2010) 29:318–30. doi: 10.1111/j.1465-3362.2009.00149.x
227. Pavarin RM, Berardi D. Mortality risk in a cohort of subjects reported by authorities for cannabis possession for personal use. Results of a longitudinal study. Epidemiol Prev. (2011) 35:89–93.
228. DeFilippis EM, Singh A, Divakaran S, Gupta A, Collins BL, Biery D, et al. Cocaine and marijuana use among young adults presenting with myocardial infarction: the partners YOUNG-MI Registry. J Am College of Cardiol. (2018) 71:2540–51. doi: 10.1016/j.jacc.2018.02.047
229. von Greiff N, Skogens L, Berlin M, Bergmark A. Mortality and cause of death-a 30-year follow-up of substance misusers in Sweden. Substance Use Misuse. (2018) 2018:1–9. doi: 10.1080/10826084.2018.1452261
230. Reece AS, Hulse GK. Chapter 5: Multivalent Cannabinoid Epigenotoxicities and Multigenerational Aging. In: Epidemiology of Cannabis: Genotoxicity, Neurotoxicity, Epigenomics and Aging. New York, USA: Elsevier. (2023).
231. Ruth KS, Day FR, Hussain J, Martínez-Marchal A, Aiken CE, Azad A, et al. Genetic insights into biological mechanisms governing human ovarian ageing. Nature. (2021) 596:393–7. doi: 10.1038/s41586-021-03779-7
232. Reece A.S., Hulse G.K. Chapter 4: Geospatiotemporal and Causal Inferential Epidemiological Survey and Exploration of Cannabinoid- and Substance- Related Carcinogenesis in USA 2003-2017. In: Epidemiology of Cannabis: Genotoxicity and Neurotoxicity, Epigenomics and Aging. New York, USA: Elsevier. (2023).
233. Schrott R, Acharya K, Itchon-Ramos N, Hawkey AB, Pippen E, Mitchell JT, et al. Cannabis use is associated with potentially heritable widespread changes in autism candidate gene DLGAP2 DNA methylation in sperm. Epigenetics. (2019) 15:161–73. doi: 10.1080/15592294.2019.1656158
234. Reece AS, Hulse GK. Sociodemographically stratified exploration of pancreatic cancer incidence in younger us patients: implication of cannabis exposure as a risk factor. Gastroenterology Insights. (2023) 14:204–35. doi: 10.3390/gastroent14020016
235. Ugai T, Sasamoto N, Lee H-Y, Ando M, Song M, Tamimi RM, et al. Is early-onset cancer an emerging global epidemic? Nat Rev Clin Oncol. (2022) 19:656–73. doi: 10.1038/s41571-022-00672-8
236. Reece AS. cannabinoid genotoxic trifecta - cancerogenesis, clinical teratogenesis and cellular ageing. Br Med J. (2022) 376:n3114. doi: 10.1136/bmj.n3114
237. Reece AS. Rapid response: cannabinoid genotoxic trifecta - cancerogenesis, clinical teratogenesis and cellular ageing. BMJ. (2022) 376:n3114.
238. Aldington S, Harwood M, Cox B, Weatherall M, Beckert L, Hansell A, et al. Cannabis use and risk of lung cancer: a case-control study. Eur Respir J. (2008) 31:280–6. doi: 10.1183/09031936.00065707
239. Voirin N, Berthiller J, Benhaïm-Luzon V, Boniol M, Straif K, Ayoub WB, et al. Risk of lung cancer and past use of cannabis in Tunisia. J Thorac Oncol. (2006) 1:577–9. doi: 10.1097/01243894-200607000-00013
240. Berthiller J, Straif K, Boniol M, Voirin N, Benhaïm-Luzon V, Ayoub WB, et al. Cannabis smoking and risk of lung cancer in men: a pooled analysis of three studies in Maghreb. J Thorac Oncol. (2008) 3:1398–403. doi: 10.1097/JTO.0b013e31818ddcde
241. Zhang ZF, Morgenstern H, Spitz MR, Tashkin DP, Yu GP, Marshall JR, et al. Marijuana use and increased risk of squamous cell carcinoma of the head and neck. Cancer Epidemiol Biomarkers Prev. (1999) 8:1071–8.
242. Hashibe M, Ford DE, Zhang ZF. Marijuana smoking and head and neck cancer. J Clini Pharmacol. (2002) 42:103S−107S. doi: 10.1002/j.1552-4604.2002.tb06010.x
243. Sidney S, Quesenberry CP, Friedman GD, Tekawa IS. Marijuana use and cancer incidence (California, United States). Cancer Causes Control. (1997) 8:722–8. doi: 10.1023/A:1018427320658
244. Efird JT, Friedman GD, Sidney S, Klatsky A, Habel LA, Udaltsova NV, et al. The risk for malignant primary adult-onset glioma in a large, multiethnic, managed-care cohort: cigarette smoking and other lifestyle behaviors. J Neuro-Oncol. (2004) 68:57–69. doi: 10.1023/B:NEON.0000024746.87666.ed
245. Moiche Bokobo P, Atxa de la Presa MA, Cuesta Angulo J. Transitional cell carcinoma in a young heavy marihuana smoker. Archivos espanoles de urologia. (2001) 54:165-7.
246. Chacko JA, Heiner JG, Siu W, Macy M, Terris MK. Association between marijuana use and transitional cell carcinoma. Urology. (2006) 67:100–4. doi: 10.1016/j.urology.2005.07.005
247. Nieder AM, Lipke MC, Madjar S. Transitional cell carcinoma associated with marijuana: case report and review of the literature. Urology. (2006) 67:200. doi: 10.1016/j.urology.2005.08.006
248. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. (2018) 555:321–7. doi: 10.1038/nature25480
249. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. (2018) 555:371–6. doi: 10.1038/nature25795
250. Bluhm EC, Daniels J, Pollock BH, Olshan AF. Maternal use of recreational drugs and neuroblastoma in offspring: a report from the Children's Oncology Group (United States). Cancer Causes Control. (2006) 17:663-9. doi: 10.1007/s10552-005-0580-3
251. Hashibe M, Straif K, Tashkin DP, Morgenstern H, Greenland S, Zhang ZF. Epidemiologic review of marijuana use and cancer risk. Alcohol (Fayetteville, NY. (2005) 35:265–75. doi: 10.1016/j.alcohol.2005.04.008
252. Robison LL, Buckley JD, Daigle AE, Wells R, Benjamin D, Arthur DC, et al. Maternal drug use and risk of childhood nonlymphoblastic leukemia among offspring. An epidemiologic investigation implicating marijuana (a report from the Childrens Cancer Study Group). Cancer. (1989) 63:1904–11. doi: 10.1002/1097-0142(19890515)63:10<1904::AID-CNCR2820631006>3.0.CO;2-W
253. Hézode C, Roudot-Thoraval F, Nguyen S, Grenard P, Julien B, Zafrani E-S, et al. Daily cannabis smoking as a risk factor for progression of fibrosis in chronic hepatitis C. Hepatology. (2005) 42:63–71. doi: 10.1002/hep.20733
254. Patsenker E, Stickel F. Cannabinoids in liver diseases. Clin Liver Dis (Hoboken). (2016) 7:21–5. doi: 10.1002/cld.527
255. Patsenker E, Stoll M, Millonig G, Agaimy A, Wissniowski T, Schneider V, et al. Cannabinoid receptor type I modulates alcohol-induced liver fibrosis. Mol Med. (2011) 17:1285–94. doi: 10.2119/molmed.2011.00149
256. Costa B, Colleoni M. Changes in rat brain energetic metabolism after exposure to anandamide or Delta(9)-tetrahydrocannabinol. Eur J Pharmacol. (2000) 395:1–7. doi: 10.1016/S0014-2999(00)00170-9
257. Rossato M, Ion Popa F, Ferigo M, Clari G, Foresta C. Human sperm express cannabinoid receptor Cb1, the activation of which inhibits motility, acrosome reaction, and mitochondrial function. J Clin Endocrinol Metab. (2005) 90:984–91. doi: 10.1210/jc.2004-1287
258. Badawy ZS, Chohan KR, Whyte DA, Penefsky HS, Brown OM, Souid AK. Cannabinoids inhibit the respiration of human sperm. Fertil Steril. (2009) 91:2471–6. doi: 10.1016/j.fertnstert.2008.03.075
259. Adverse health effects of marijuana use. N Engl J Med. (2014) 371:878–79. doi: 10.1056/NEJMc1407928
260. Wolff V, Schlagowski A-I, Rouyer O, Charles A-L, Singh F, Auger C, et al. Tetrahydrocannabinol induces brain mitochondrial respiratory chain dysfunction and increases oxidative stress: a potential mechanism involved in cannabis-related stroke. Biomed Res Int. (2015) 2015:323706. doi: 10.1155/2015/323706
261. Zottola ACP, Severi I, Cannich A, Ciofi P, Cota D, Marsicano G, et al. Expression of Functional Cannabinoid Type-1 (CB(1)) Receptor in Mitochondria of White Adipocytes. Cells. (2022) 11(16) doi: 10.3390/cells11162582
262. Lamanna-Rama N, MacDowell KS, López G, Leza JC, Desco M, Ambrosio E, et al. Neuroimaging revealed long-lasting glucose metabolism changes to morphine withdrawal in rats pretreated with the cannabinoid agonist CP-55,940 during periadolescence. Eur Neuropsychopharmacol. (2023) 69:60–76. doi: 10.1016/j.euroneuro.2023.01.005
263. Ma L, Zhang H, Liu C, Liu M, Shangguan F, Liu Y, et al. A novel mechanism of cannabidiol in suppressing ovarian cancer through LAIR-1 mediated mitochondrial dysfunction and apoptosis. Environ Toxicol. (2023) 38:1118–32. doi: 10.1002/tox.23752
265. Elkon R, Agami R. Two-layer design protects genes from mutations in their enhancers. Nature. (2022) 609:477–8. doi: 10.1038/d41586-022-02341-3
266. Emerson DJ, Zhao PA, Cook AL, Barnett J, Klein K, Saulebekova D, et al. Cohesin-mediated loop anchors confine the locations of human replication origins. Nature. (2022) 606:812–9. doi: 10.1038/s41586-022-04803-0
267. Xu J, Song F, Lyu H, et al. Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature. (2022) 611:387–98. doi: 10.1038/s41586-022-05365-x
268. Hua P, Badat M, Hanssen LLP, Hentges LD, Crump N, Downes DJ, et al. Defining genome architecture at base-pair resolution. Nature. (2021) 595:125–9. doi: 10.1038/s41586-021-03639-4
269. Kloetgen A, Thandapani P, Ntziachristos P, Ghebrechristos Y, Nomikou S, Lazaris C, et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat Genet. (2020) 52:388–400. doi: 10.1038/s41588-020-0602-9
270. Zuin J, Roth G, Zhan Y, Cramard J, Redolfi J, Piskadlo E, et al. Nonlinear control of transcription through enhancer-promoter interactions. Nature. (2022) 604:571–7. doi: 10.1038/s41586-022-04570-y
271. Dequeker BJH, Scherr MJ, Brandão HB, Gassler J, Powell S, Gaspar I, et al. MCM complexes are barriers that restrict cohesin-mediated loop extrusion. Nature. (2022) 606:197–203. doi: 10.1038/s41586-022-04730-0
272. Loyfer N, Magenheim J, Peretz A, Cann G, Bredno J, Klochendler A, et al. A DNA methylation atlas of normal human cell types. Nature. (2023) 613:355–64. doi: 10.1038/s41586-022-05580-6
273. Asami M, Lam BYH, Ma MK, Rainbow K, Braun S, VerMilyea MD, et al. Human embryonic genome activation initiates at the one-cell stage. Cell Stem Cell. (2022) 29:209–216.e4. doi: 10.1016/j.stem.2021.11.012
274. Levo M, Raimundo J, Bing XY, Sisco Z, Batut PJ, Ryabichko S, et al. Transcriptional coupling of distant regulatory genes in living embryos. Nature. (2022) 605:754–60. doi: 10.1038/s41586-022-04680-7
275. Zhang J, Zhang Y, You Q, Huang C, Zhang T, Wang M, et al. Highly enriched BEND3 prevents the premature activation of bivalent genes during differentiation. Science. (2022) 375:1053–8. doi: 10.1126/science.abm0730
276. Batut PJ, Bing XY, Sisco Z, Raimundo J, Levo M, Levine MS. Genome organization controls transcriptional dynamics during development. Science. (2022) 375:566–70. doi: 10.1126/science.abi7178
277. Barter MJ, Bui C, Cheung K, Falk J, Gómez R, Skelton AJ, et al. DNA hypomethylation during MSC chondrogenesis occurs predominantly at enhancer regions. Sci Rep. (2020) 10:1169. doi: 10.1038/s41598-020-58093-5
278. Bergman DT, Jones TR, Liu V, Ray J, Jagoda E, Siraj L, et al. Compatibility rules of human enhancer and promoter sequences. Nature. (2022) 607:176–84. doi: 10.1038/s41586-022-04877-w
279. Winick-Ng W, Kukalev A, Harabula I, et al. Cell-type specialization is encoded by specific chromatin topologies. Nature. (2021) 599:684–91. doi: 10.1038/s41586-021-04081-2
280. Du J, Zheng L, Gao P, et al. A small-molecule cocktail promotes mammalian cardiomyocyte proliferation and heart regeneration. Cell Stem Cell (2022). 29:545-558.e13. doi: 10.1016/j.stem.2022.03.009
281. Hill MC, Kadow ZA, Long H, Morikawa Y, Martin TJ, Birks EJ, et al. Integrated multi-omic characterization of congenital heart disease. Nature. (2022) 608:181–91. doi: 10.1038/s41586-022-04989-3
282. Yang D, Gomez-Garcia J, Funakoshi S, Tran T, Fernandes I, Bader GD, et al. Modeling human multi-lineage heart field development with pluripotent stem cells. Cell Stem Cell. (2022). 29:1382–1401.e8. doi: 10.1016/j.stem.2022.08.007
283. Yao Z, Liu H, Xie F, et al. A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex. Nature. (2021) 598:103–10. doi: 10.1038/s41586-021-03500-8
284. Li YE, Preissl S, Hou X, Zhang Z, Zhang K, Qiu Y, et al. An atlas of gene regulatory elements in adult mouse cerebrum. Nature. (2021) 598:129–36. doi: 10.1038/s41586-021-03604-1
285. Zhang M, Eichhorn SW, Zingg B, Yao Z, Cotter K, Zeng H, et al. Spatially resolved cell atlas of the mouse primary motor cortex by MERFISH. Nature. (2021) 598:137–43. doi: 10.1038/s41586-021-03705-x
286. Berg J, Sorensen SA, Ting JT, Miller JA, Chartrand T, Buchin A, et al. Human neocortical expansion involves glutamatergic neuron diversification. Nature. (2021) 598:151–8. doi: 10.1038/s41586-021-03813-8
287. Zhang Z, Zhou J, Tan P, Pang Y, Rivkin AC, Kirchgessner MA, et al. Epigenomic diversity of cortical projection neurons in the mouse brain. Nature. (2021) 598:167–73. doi: 10.1038/s41586-021-03223-w
288. Bhaduri A, Sandoval-Espinosa C, Otero-Garcia M, Oh I, Yin R, Eze UC, et al. An atlas of cortical arealization identifies dynamic molecular signatures. Nature. (2021) 598:200–4. doi: 10.1038/s41586-021-03910-8
289. Ziffra RS, Kim CN, Ross JM, Wilfert A, Turner T, Haeusler M, et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature. (2021) 598:205–13. doi: 10.1038/s41586-021-03209-8
290. Kozareva V, Martin C, Osorno T, Rudolf S, Guo C, Vanderburg C, et al. A transcriptomic atlas of mouse cerebellar cortex comprehensively defines cell types. Nature. (2021) 598:214–9. doi: 10.1038/s41586-021-03220-z
291. Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denhittz M, et al. Non-coding transcription instructs chromatin folding and compartmentalization to dictate enhancer-promoter communication and T cell fate. Cell. (2017) 171:103–119.e18. doi: 10.1016/j.cell.2017.09.001
292. Yuan S, Stewart KS, Yang Y, Abdusselamoglu MD, Parigi SM, Feinberg TY, et al. Ras drives malignancy through stem cell crosstalk with the microenvironment. Nature. (2022) 612:555–63. doi: 10.1038/s41586-022-05475-6
293. Guan J, Wang G, Wang J, Zhang Z, Fu Y, Cheng L, et al. Chemical reprogramming of human somatic cells to pluripotent stem cells. Nature. (2022) 605:325–31. doi: 10.1038/s41586-022-04593-5
294. Lu Y, Brommer B, Tian X, Krishnan A, Meer M, Wang C, et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature. (2020) 588:124–9. doi: 10.1038/s41586-020-2975-4
295. Cañellas-Socias A, Cortina C, Hernando-Momblona X, Palomo-Ponce S, Mulholland EJ, Turon G, et al. Metastatic recurrence in colorectal cancer arises from residual EMP1(+) cells. Nature. (2022) 611:603–13. doi: 10.1038/s41586-022-05402-9
296. Isoda T, Morio T, Takagi M. Noncoding RNA transcription at enhancers and genome folding in cancer. Cancer Sci. (2019) 110:2328–36. doi: 10.1111/cas.14107
297. Lomakin A, Svedlund J, Strell C, Gataric M, Shmatko A, Rukhovich G, et al. Spatial genomics maps the structure, nature and evolution of cancer clones. Nature. (2022) 611:594–602. doi: 10.1038/s41586-022-05425-2
298. Wala J, Beroukhim RCANCER. The oncogene makes its escape. Science. (2016) 351:1398–9. doi: 10.1126/science.aaf5542
299. Diedrich JD, Dong Q, Ferguson DC, Bergeron BP, Autry RJ, Qian M, et al. Profiling chromatin accessibility in pediatric acute lymphoblastic leukemia identifies subtype-specific chromatin landscapes and gene regulatory networks. Leukemia. (2021) 35:3078–91. doi: 10.1038/s41375-021-01209-1
300. Yang H, Zhang H, Luan Y, Liu T, Yang W, Roberts KG, et al. Noncoding genetic variation in GATA3 increases acute lymphoblastic leukemia risk through local and global changes in chromatin conformation. Nat Genet. (2022) 54:170–9. doi: 10.1038/s41588-021-00993-x
301. Heide T, Househam J, Cresswell GD, Spiteri I, Lynn C, Mossner M, et al. The co-evolution of the genome and epigenome in colorectal cancer. Nature. (2022) 611:733–43. doi: 10.1038/s41586-022-05202-1
302. Xiao L, Parolia A, Qiao Y, Bawa P, Eyunni S, Mannan R, et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature. (2022) 601:434–9. doi: 10.1038/s41586-021-04246-z
303. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. (2013) 155:934–47. doi: 10.1016/j.cell.2013.09.053
304. Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell. (2015) 58:362–70. doi: 10.1016/j.molcel.2015.02.014
305. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. (2013) 153:307–19. doi: 10.1016/j.cell.2013.03.035
306. Luo Z, Xia M, Shi W, Zhao C, Wang J, Xin D, et al. Human fetal cerebellar cell atlas informs medulloblastoma origin and oncogenesis. Nature. (2022) 612:787–94. doi: 10.1038/s41586-022-05487-2
307. Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. (2013) 153:320–34. doi: 10.1016/j.cell.2013.03.036
308. Liu H, Zhou J, Tian W, Luo C, Bartlett A, Aldridge A, et al. DNA methylation atlas of the mouse brain at single-cell resolution. Nature. (2021) 598:120–8. doi: 10.1038/s41586-020-03182-8
309. O'Neill AC, Uzbas F, Antognolli G, Merino F, Draganova K, Jäck A, et al. Spatial centrosome proteome of human neural cells uncovers disease-relevant heterogeneity. Science. (2022) 376:eabf9088. doi: 10.1126/science.abf9088
310. Gupta M, Will B. SEPHguarding acute myeloid leukemia. Cell Stem Cell. (2022) 29:350–2. doi: 10.1016/j.stem.2022.02.007
311. Hnisz D, Weintraub AS, Day DS, Valton A-L, Bak RO, Li CH, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. (2016) 351:1454–8. doi: 10.1126/science.aad9024
312. Petrovic J, Zhou Y, Fasolino M, Goldman N, Schwartz GW, Mumbach MR, et al. Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol Cell. (2019). 73:1174–90.e12. doi: 10.1016/j.molcel.2019.01.006
313. Kon A, Shih L-Y, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. (2013) 45:1232–7. doi: 10.1038/ng.2731
314. Tang S, Stokasimov E, Cui Y, Pellman D. Breakage of cytoplasmic chromosomes by pathological DNA base excision repair. Nature. (2022) 606:930–6. doi: 10.1038/s41586-022-04767-1
315. Wang D, Wu W, Callen E, Pavani R, Zolnerowich N, Kodali S, et al. Active DNA demethylation promotes cell fate specification and the DNA damage response. Science. (2022) 378:983–9. doi: 10.1126/science.add9838
316. Poggetto ED, Ho I-L, Balestrieri C, Yen E-Y, Zhang S, Citron F, et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science. (2021) 373:eabj0486. doi: 10.1126/science.abj0486
317. Lan L, Evan T, Li H, Hussain A, Ruiz EJ, Thin MZ, et al. GREM1 is required to maintain cellular heterogeneity in pancreatic cancer. Nature. (2022) 607:163–8. doi: 10.1038/s41586-022-04888-7
318. Park W, Chawla A, O'Reilly EM. Pancreatic cancer: a review. Jama. (2021) 326:851–62. doi: 10.1001/jama.2021.13027
319. Volkow ND, Han B, Compton WM, Blanco C. Marijuana use during stages of pregnancy in the United States. Ann Intern Med. (2017) 166:763–4. doi: 10.7326/L17-0067
320. Volkow ND, Compton WM, Wargo EM. The risks of marijuana use during pregnancy. JAMA. (2017) 317:129–30. doi: 10.1001/jama.2016.18612
321. Reece AS, Hulse GK. Broad Spectrum epidemiological contribution of cannabis and other substances to the teratological profile of northern New South Wales: geospatial and causal inference analysis. BMC Pharmacol Toxicol. (2020) 21:75–103. doi: 10.1186/s40360-020-00450-1
322. Wolff V, Rouyer O, Schlagowski A, Zoll J, Raul JS, Marescaux C. Étude de l'effet du THC sur la respiration mitochondriale du cerveau de rat. Une piste de réflexion pour expliquer le lien entre la consommation de cannabis et la survenue d'infarctus cérébral chez l'homme Study of the effect of THC on mitochondrial respiration of the rat brain. One line of thought to explain the link between cannabis use and the occurrence of cerebral infarction in men Revue Neurologique. Neurolog Rev. (2014) 170:A19–A20. doi: 10.1016/j.neurol.2014.01.081
323. Mkrtchyan GV, Abdelmohsen K, Andreux P, Bagdonaite I, Barzilai N, Brunak S, et al. ARDD 2020: from aging mechanisms to interventions. Aging (Albany NY). (2020) 12:24484–503. doi: 10.18632/aging.202454
324. Schultz MB, Sinclair DA. When stem cells grow old: phenotypes and mechanisms of stem cell aging. Development. (2016) 143:3–14. doi: 10.1242/dev.130633
325. Group SG. Sfari Gene Portal. In: Sfari Gene Group. Available online at: https://gene.sfari.org/about-sfari-gene/ (accessed February 20, 2023).
326. Vela G, Martín S, García-Gil L, Crespo JA, Ruiz-Gayo M, Fernández-Ruiz JJ, et al. Maternal exposure to delta9-tetrahydrocannabinol facilitates morphine self-administration behavior and changes regional binding to central mu opioid receptors in adult offspring female rats. Brain Res. (1998) 807:101–9. doi: 10.1016/S0006-8993(98)00766-5
327. Hölzel BN, Pfannkuche K, Allner B, Allner HT, Hescheler J, Derichsweiler D, et al. Following the adverse outcome pathway from micronucleus to cancer using H2B-eGFP transgenic healthy stem cells. Arch Toxicol. (2020) 94:3265–80. doi: 10.1007/s00204-020-02821-3
328. Tahir SK, Trogadis JE, Stevens JK, Zimmerman AM. Cytoskeletal organization following cannabinoid treatment in undifferentiated and differentiated PC12 cells. Biochem Cell Biol. (1992) 70:1159–73. doi: 10.1139/o92-162
329. Busch FW, Seid DA, Wei ET. Mutagenic activity of marihuana smoke condensates. Cancer Lett. (1979) 6:319–24. doi: 10.1016/S0304-3835(79)80088-9
330. Koller VJ, Ferk F, Al-Serori H, Mišík M, Nersesyan A, Auwärter V, et al. Genotoxic properties of representatives of alkylindazoles and aminoalkyl-indoles which are consumed as synthetic cannabinoids. Food Chem Toxicol. (2015) 80:130–6. doi: 10.1016/j.fct.2015.03.004
331. Zimmerman AM, Raj AY. Influence of cannabinoids on somatic cells in vivo. Pharmacology. (1980) 21:277–87. doi: 10.1159/000137442
332. Shoyama Y, Sugawa C, Tanaka H, Morimoto S. Cannabinoids act as necrosis-inducing factors in Cannabis sativa. Plant Signal Behav. (2008) 3:1111–2. doi: 10.4161/psb.3.12.7011
333. Price PJ, Suk WA, Spahn GJ, Freeman AE. Transformation of Fischer rat embryo cells by the combined action of murine leukemia virus and (-)-trans- 9 -tetrahydrocannabinol. Proc Soc Exp Biol Med. (1972) 140:454–6. doi: 10.3181/00379727-140-36478
334. Koller VJ, Auwarter V, Grummt T, Moosmann B, Misik M, Knasmuller S. Investigation of the in vitro toxicological properties of the synthetic cannabimimetic drug CP-47,497-C8. Toxicol Appl Pharmacol. (2014) 277:164–71. doi: 10.1016/j.taap.2014.03.014
335. Wagner B, Gerletti P, Fürst P, Keuth O, Bernsmann T, Martin A, et al. Transfer of cannabinoids into the milk of dairy cows fed with industrial hemp could lead to Δ9-THC exposure that exceeds acute reference dose. Nature Food. (2022) 3:921–32. doi: 10.1038/s43016-022-00623-7
336. Cerda M, Wall M, Keyes KM, Galea S, Hasin D. Medical marijuana laws in 50 states: investigating the relationship between state legalization of medical marijuana and marijuana use, abuse and dependence. Drug Alcohol Depend. (2012) 120:22–7. doi: 10.1016/j.drugalcdep.2011.06.011
337. Hasin DS, Wall M, Keyes KM, Cerdá M, Schulenberg J, O'Malley PM, et al. Medical marijuana laws and adolescent marijuana use in the USA from 1991 to 2014: results from annual, repeated cross-sectional surveys. The lancet Psychiatry. (2015) 2:601–8. doi: 10.1016/S2215-0366(15)00217-5
338. Hasin DS, Sarvet AL, Cerdá M, Keyes KM, Stohl M, Galea S, et al. US adult illicit cannabis use, cannabis use disorder, and medical marijuana laws: 1991–1992 to 2012-−2013. JAMA Psychiatry. (2017) 74:579–88. doi: 10.1001/jamapsychiatry.2017.0724
339. Cerdá M, Sarvet AL, Wall M, Feng T, Keyes KM, Galea S, et al. Medical marijuana laws and adolescent use of marijuana and other substances: alcohol, cigarettes, prescription drugs, and other illicit drugs. Drug Alcohol Depend. (2018) 183:62–8. doi: 10.1016/j.drugalcdep.2017.10.021
340. Sarvet AL, Wall MM, Fink DS, Greene E, Le A, Boustead AE, et al. Medical marijuana laws and adolescent marijuana use in the United States: a systematic review and meta-analysis. Addiction. (2018) 113:1003–16. doi: 10.1111/add.14136
341. Compton WM, Volkow ND, Lopez MF. Medical marijuana laws and cannabis use: intersections of health and policy. JAMA Psychiatry. (2017) 74:559–60. doi: 10.1001/jamapsychiatry.2017.0723
342. Reece AS, Hulse GK. Cannabis consumption patterns explain the east-west gradient in canadian neural tube defect incidence: an ecological study. Glob Pediatr Health. (2019) 6:1–12. doi: 10.1177/2333794X19894798
343. Toumbourou JW, Lederhos C, Carolson B, Brown E, Reece AS, Brents L. Roundtable on “Is Cannabis Legalization Increasing Birth Defects and Cancers?”. In: Soc Prevention Res. Available online at: https://deakin.zoom.us/rec/share/XkFRt18GM5zh-XOOYKIb264J_twM1nHW_WuS908ESq3O0C34EFFzPA66r0VV2VIU.mcaSoNiICWMnU4pi (accessed February 20, 2023).
344. Reece AS, Hulse GK. Geospatiotemporal and Causal Inferential Epidemiological Survey and Exploration of Cannabinoid- and Substance- Related Carcinogenesis in USA 2003-2017. In: Epidemiology of Cannabis: Genotoxicity and Neurotoxicity, Epigenomics and Aging. New York, USA: Elsevier; In Press. (2500).
345. Reece AS, Hulse GK. Epidemiology of Δ8THC-Related Carcinogenesis in USA: A Panel Regression and Causal Inferential Study. Int J Environ Res Public Health. (2022) 19:7726–52. doi: 10.3390/ijerph19137726
347. Psychoyos D, Hungund B, Cooper T, Finnell RH, A. cannabinoid analogue of Delta9-tetrahydrocannabinol disrupts neural development in chick. Birth Defects Res B Dev Reprod Toxicol. (2008) 83:477–88. doi: 10.1002/bdrb.20166
348. Subbanna S, Shivakumar M, Psychoyos D, Xie S, Basavarajappa BS. Anandamide-CB1 receptor signaling contributes to postnatal ethanol-induced neonatal neurodegeneration, adult synaptic, and memory deficits. J Neurosci. (2013) 33:6350–66. doi: 10.1523/JNEUROSCI.3786-12.2013
349. Subbanna S, Nagre NN, Umapathy NS, Pace BS, Basavarajappa BS. Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int J Neuropsychopharmacol. (2014) 18:1–15. doi: 10.1093/ijnp/pyu028
350. Subbanna S, Psychoyos D, Xie S, Basavarajappa BS. Postnatal ethanol exposure alters levels of 2-arachidonylglycerol-metabolizing enzymes and pharmacological inhibition of monoacylglycerol lipase does not cause neurodegeneration in neonatal mice. J Neurochem. (2015) 134:276–87. doi: 10.1111/jnc.13120
351. Seleverstov O, Tobiasz A, Jackson JS, Sullivan R, Ma D, Sullivan JP, et al. Maternal alcohol exposure during mid-pregnancy dilates fetal cerebral arteries via endocannabinoid receptors. Alcohol (Fayetteville, NY. (2017) 61:51–61. doi: 10.1016/j.alcohol.2017.01.014
352. Subbanna S, Nagre NN, Shivakumar M, Joshi V, Psychoyos D, Kutlar A, et al. CB1R-mediated activation of caspase-3 causes epigenetic and neurobehavioral abnormalities in postnatal ethanol-exposed mice. Front Mol Neurosci. (2018) 11:45. doi: 10.3389/fnmol.2018.00045
353. Joshi V, Subbanna S, Shivakumar M, Basavarajappa BS. CB1R regulates CDK5 signaling and epigenetically controls Rac1 expression contributing to neurobehavioral abnormalities in mice postnatally exposed to ethanol. Neuropsychopharmacology. (2019) 44:514–25. doi: 10.1038/s41386-018-0181-y
354. Shivakumar M, Subbanna S, Joshi V, Basavarajappa BS. Postnatal ethanol exposure activates HDAC-mediated histone deacetylation, impairs synaptic plasticity gene expression and behavior in mice. Int J Neuropsychopharmacol. (2020) 23:324–38. doi: 10.1093/ijnp/pyaa017
355. Subbanna S, Basavarajappa BS. Postnatal ethanol-induced neurodegeneration involves CB1R-mediated β-catenin degradation in neonatal mice. Brain Sci. (2020) 10:5. doi: 10.3390/brainsci10050271
356. Hutchings DE, Dow-Edwards D. Animal models of opiate, cocaine, and cannabis use. Clin Perinatol. (1991) 18:1–22. doi: 10.1016/S0095-5108(18)30531-1
357. Hosptial GOS. VACTERL Association: Information for Families. Great Ormond St. London United Kingdom. (2021). Available online at: https://media.gosh.nhs.uk/documents/VACTERL_association_F1612_FINAL_Sep19.pdf (accessed February 20, 2023).
358. Hosptial GOS. VACTERL Association. National Health Service. Available online at: https://www.gosh.nhs.uk/conditions-and-treatments/conditions-we-treat/vacterl-association-0/ (accessed February 20, 2023).
359. Ngan ESW, Kim KH, Hui CC. Sonic hedgehog signaling and VACTERL association. Mol Syndromol. (2013) 4:32–45. doi: 10.1159/000345725
360. Basavarajappa BS. Endocannabinoid system and alcohol abuse disorders. Adv Exp med Biol. (2019) 1162:89–127. doi: 10.1007/978-3-030-21737-2_6
361. Basavarajappa BS, Subbanna S. Molecular insights into epigenetics and cannabinoid receptors. Biomolecules. (2022) 12:11. doi: 10.3390/biom12111560
362. Substance Abuse and Mental Health Network. Substance Abuse and Mental Health Data Archive (SAMHDA) 2018-2019. In: Substance Abuse and Mental Health Services Administration. Available online at: https://www.datafiles.samhsa.gov/ (accessed February 20, 2023).
363. Han L, Dong L, Leung K, Zhao Z, Li Y, Gao L, et al. METTL16 drives leukemogenesis and leukemia stem cell self-renewal by reprogramming BCAA metabolism. Cell Stem Cell. (2023) 30:52–68.e13. doi: 10.1016/j.stem.2022.12.006
Keywords: cannabis, cannabinoid, genotoxicity, epigenotoxicity, transgenerational inheritance
Citation: Reece AS and Hulse GK (2023) Perturbation of 3D nuclear architecture, epigenomic dysregulation and aging, and cannabinoid synaptopathy reconfigures conceptualization of cannabinoid pathophysiology: part 1–aging and epigenomics. Front. Psychiatry 14:1182535. doi: 10.3389/fpsyt.2023.1182535
Received: 08 March 2023; Accepted: 07 August 2023;
Published: 05 September 2023.
Edited by:
Ilya Blokhin, Jackson Memorial Hospital, United StatesReviewed by:
Balapal Basavarajappa, New York University, United StatesJohn Winston Toumbourou, Deakin University, Australia
Copyright © 2023 Reece and Hulse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Albert Stuart Reece, c3R1YXJ0LnJlZWNlQHV3YS5lZHUuYXU=