 Sameer Hassamal1,2,3*
Sameer Hassamal1,2,3*- 1California University of Sciences and Medicine, Colton, CA, United States
- 2Clinicaltriallink, Los Angeles, CA, United States
- 3California Neuropsychiatric Institute, Ontario, CA, United States
In a subset of patients, chronic exposure to stress is an etiological risk factor for neuroinflammation and depression. Neuroinflammation affects up to 27% of patients with MDD and is associated with a more severe, chronic, and treatment-resistant trajectory. Inflammation is not unique to depression and has transdiagnostic effects suggesting a shared etiological risk factor underlying psychopathologies and metabolic disorders. Research supports an association but not necessarily a causation with depression. Putative mechanisms link chronic stress to dysregulation of the HPA axis and immune cell glucocorticoid resistance resulting in hyperactivation of the peripheral immune system. The chronic extracellular release of DAMPs and immune cell DAMP-PRR signaling creates a feed forward loop that accelerates peripheral and central inflammation. Higher plasma levels of inflammatory cytokines, most consistently interleukin IL-1β, IL-6, and TNF-α, are correlated with greater depressive symptomatology. Cytokines sensitize the HPA axis, disrupt the negative feedback loop, and further propagate inflammatory reactions. Peripheral inflammation exacerbates central inflammation (neuroinflammation) through several mechanisms including disruption of the blood–brain barrier, immune cellular trafficking, and activation of glial cells. Activated glial cells release cytokines, chemokines, and reactive oxygen and nitrogen species into the extra-synaptic space dysregulating neurotransmitter systems, imbalancing the excitatory to inhibitory ratio, and disrupting neural circuitry plasticity and adaptation. In particular, microglial activation and toxicity plays a central role in the pathophysiology of neuroinflammation. Magnetic resonance imaging (MRI) studies most consistently show reduced hippocampal volumes. Neural circuitry dysfunction such as hypoactivation between the ventral striatum and the ventromedial prefrontal cortex underlies the melancholic phenotype of depression. Chronic administration of monoamine-based antidepressants counters the inflammatory response, but with a delayed therapeutic onset. Therapeutics targeting cell mediated immunity, generalized and specific inflammatory signaling pathways, and nitro-oxidative stress have enormous potential to advance the treatment landscape. Future clinical trials will need to include immune system perturbations as biomarker outcome measures to facilitate novel antidepressant development. In this overview, we explore the inflammatory correlates of depression and elucidate pathomechanisms to facilitate the development of novel biomarkers and therapeutics.
1. Introduction
Depression is quickly becoming a public health crisis despite advances in diagnostics and therapeutics. At the height of the COVID-19 pandemic, the prevalence of depression tripled from 10 to 30% in the U.S. (1). The sharp rise in depression has been attributed to chronic stress stemming from social isolation and global economic uncertainty amid the largest land war in Europe since World War Two (2). Moreover, COVID-19 infection itself triggers an inflammatory cascade leaving the population primed for the development of neuroinflammation by an interplay between physiologic and psychological stress. Persistent stress exposure particularly among susceptible individuals with adverse childhood experiences induces chronic low-grade inflammation and increases the risk for emotional disturbances. Several lines of evidence support a link between chronic stress, low-grade inflammation, and depressive symptoms (3). Thus far, the evidence has been observational in nature and does not necessarily establish causality.
Stress induced hyperactivation of immunological processes represents a distinct pathway underlying depression. Innate and adaptive immune responses compromise the integrity of the blood brain barrier propagating bidirectional inflammatory signals between the periphery and central nervous system (4). Neuroinflammation, defined as activation of the central innate immune system, results in a depressive phenotype manifested with severe symptomatology and greater morbidity and mortality (5). A large body of evidence supports a positive association between higher serum and cerebrospinal fluid (CSF) concentrations of inflammatory cytokines, depression severity, and treatment resistance (6). Normalizing immune signaling, cytokine expression, and the nitro-oxidative balance reduces the severity of depression (7). The precise mechanistic pathways linking inflammation and depression remains to be elucidated, however, a plausible explanation is the disruption of neurotransmitters and neural networks distinct to depression.
Neuroinflammation affects 1 in 4 patients with depression and is associated with a poor prognosis, treatment resistance, and a reduced health related quality of life. There is no unifying diagnostic criteria for depression associated with inflammation, however a peripheral C-reactive protein (CRP) level exceeding 3 mg/L is commonly used as a marker of subtyping inflammation (8). Peripheral CRP levels strongly correlate with CSF CRP levels and are a reliable marker of neuroinflammation (9). Peripheral CRP levels exceeding 3 mg/l are associated with a specific depressive phenotype resembling “sickness behavior”: anhedonia, apathy, decreased appetite, fatigue, sleepiness, pain, suicidality, and cognitive impairments (5). These symptoms are present across disorders suggesting neuroinflammation underlies the shared variance between neuropsychiatric disorders. Exploring the inflammatory correlates of depression will elucidate pathomechanisms and facilitate the development of novel biomarkers and therapeutics. In this overview, we summarize the literature on putative mechanisms underlying stress dysregulation of the immune system, neuroinflammation, and depression (Figure 1).
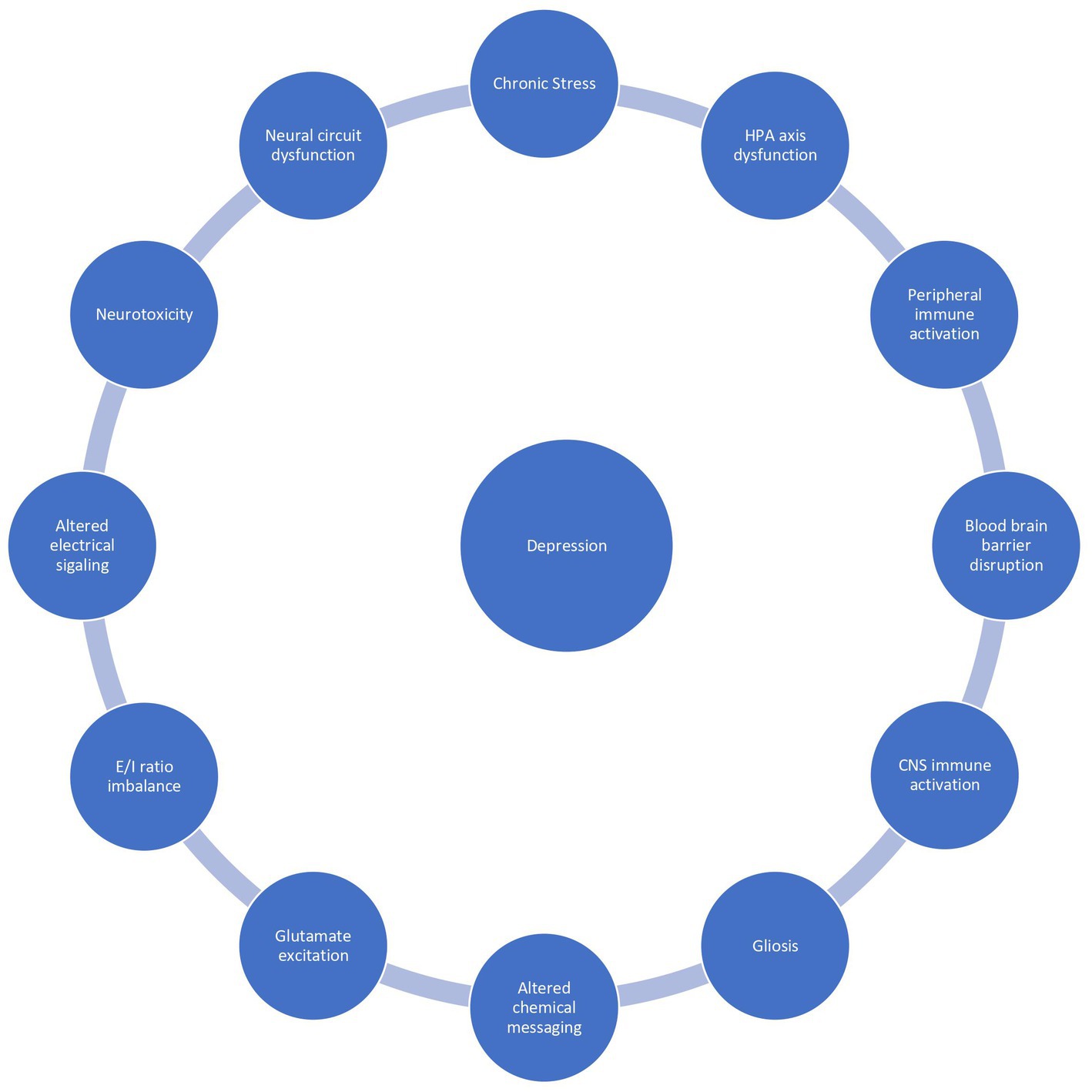
Figure 1. The vicious reciprocal relationship between chronic stress, inflammation, and depression.
2. The link between stress, inflammation, and depression
Preclinical data laid the foundation for understanding the relationship between stress, inflammation, and depression. Various forms of stress exposure (chronic mild stress, learned helplessness, repeated social defeat stress) induce a melancholic phenotype of depression, increase insulin insensitivity and levels of pro-inflammatory cytokines. Peripheral levels of IL-1β, IL-6, and TNF-α are consistently upregulated in susceptible animal models (10). Hodes and colleagues found that peripheral levels of IL-6 were 27 times higher in susceptible compared to resilient mice after repeated social defeat stress. Administering an IL-6 monoclonal antibody 5 min prior to repeated social defeat stress prevented the development of social avoidance suggesting that IL-6 contributes to stress-induced vulnerability (11). In a similar study, stress susceptible mice exhibited depressive-like behaviors that were interrelated with adrenocortical activation, increased IL-1 β levels in the hippocampus, and reduced hippocampal neurogenesis. Removing the adrenal glands abolished the depressive behaviors. In contrast, administering corticosterone amplified the depressive behaviors and reduced neurogenesis in mice with and without the IL-1R. The data suggests that adrenocortical activation mediates the relationship between IL-1 and stress-induced depression (12). Additional preclinical studies have established a relationship between cortisol dysregulation and depression. Lu and colleagues found that deletion of the astrocytic glucocorticoid receptor NR3C1 gene induced social avoidance and downregulated ATP release via the PI3K-AKT signaling pathway. The PI3K-AKT signaling pathway regulates physiological processes important for cell survival (13). Translational research has demonstrated that differential epigenetic NR3C1 gene hypermethylation patterns in individuals with a history of adverse childhood experiences contributes to a stress sensitive HPA axis response (14). Overall, the data suggest that aberrant glucocorticoid signaling mediates the relationship between the stress response, inflammation, and depression (15).
3. Hypothalamic–pituitary–adrenal axis
The initial mechanism linking chronic stress, inflammation, and depression can be explained by the dysregulation of the HPA axis and activation of immune response patterns. Typically, under normal conditions, cortisol decreases inflammation. However, persistent stress overstimulates the HPA axis causing an excess release of cortisol. Consequently, a dysregulation of the glucocorticoid negative feedback loop occurs due to glucocorticoid receptor resistance resulting in immune cell pro-inflammatory cytokine release (16). The dexamethasone suppression test (DST) is commonly used to assess HPA axis functioning. Low dose dexamethasone suppresses the release of cortisol and failure to do so is diagnostic of HPA axis hyperactivity and hypercortisolism (17). Dexamethasone fails to suppress adrenocorticotrophic hormone (ACTH) and cortisol in a subset of patients with melancholic depression suggesting underlying HPA axis dysfunction. Higher serum levels of cortisol and adrenocorticotrophic hormone (ACTH) have been reported in patients with severe and melancholic depression. In addition, urinary free cortisol levels are approximately two times higher in depressed patients compared to non-depressed patients (18). Overall, the sensitivity of the DST in diagnosing very severe cases of melancholic depression is approximately 70% (19).
Targeting HPA axis overactivity is a focus of antidepressant drug development. Pharmacotherapies antagonizing the GR and inhibiting cortisol synthesis have been developed, however, their efficacy has been stymied by phenotypic and genotypic diagnostic heterogeneities (20). More recent work has focused on arginine-vasopressin (AVP) and the vasopressin v1b receptor (V1BR) as primary factors contributing to HPA axis dysregulation. AVP-V1BR binding stimulates the pituitary release of ACTH and the secretion of cortisol. Multiple studies have reported that AVP levels are increased in the plasma and brain nuclei of depressed patients, indicating that overactivity of AVP-V1bR dysregulates the HPA axis (21). Thus far, the antidepressant effects of V1bR antagonists in small clinical trials are mixed but show efficacy signals in patients with overactive HPA activity (22) (Figure 2).
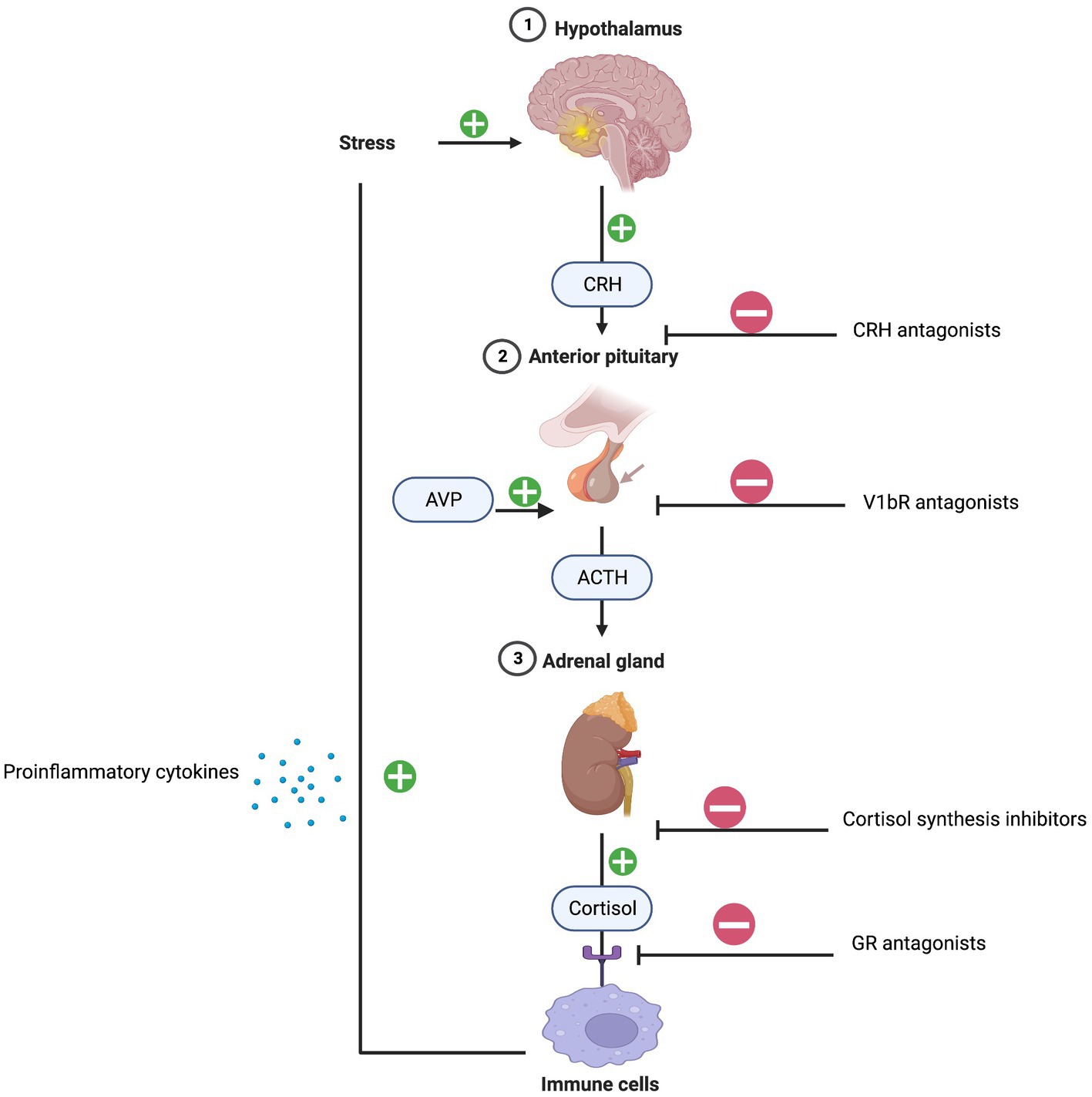
Figure 2. HPA-axis and inflammation. Created with BioRender.com.
4. Damage-associated molecular patterns
Stress induces cells to release damage-associated molecular patterns (DAMPs), endogenous danger signals, into the extracellular space. DAMPs bind to pattern-recognition receptors (PRRs) on immune cells and activate the innate immune response. Several DAMPs have been implicated in depression, including high mobility group box-1 (HMGB-1), extracellular ATP, purine bases and metabolites, heat shock proteins (HSPs), S100 proteins, and galectin- 3 (Gal-3) (23). Studies have focused on S100 proteins, HSPs, and Gal-3 because of their pathogenic immunoregulatory roles in comorbid inflammatory diseases (24).
4.1. S100B
The S100B protein is highly expressed in neuronal cells, especially astrocytes. S100 B can either trigger trophic or inflammatory responses depending on the extracellular concentration. S100 B induces neurogenesis at nanomolar concentrations and inflammation at micromolar concentrations (25). Wang and colleagues reported that 43 days of chronic unpredictable mild stress increased hippocampal S100b expression in susceptible rats. Venlafaxine reduced hippocampal S100b expression and reversed depressive-like behavior suggesting that high levels of S100b predict the risk for depression and antidepressant response (26). In addition, S100B has been linked to suicide, and Dogan and colleagues found that CSF levels of S100B were higher in completed suicide autopsy cases compared to controls (27). Several clinical studies reported that serum levels of S100B are correlated with depression severity and recurrence. A study examining soldiers during intense combat training found that higher serum levels of S100 B correlated with greater distress levels and pro-inflammatory cytokines. The clinical data clearly support an association but not necessarily a causal link between S100 B, inflammation, and depression (28).
S100B is linked to comorbid inflammatory conditions, such as diabetes and metabolic syndrome. Adipose tissue contains S100B, which predisposes obese patients to depression. Diabetes, hypertension, high cholesterol, and metabolic syndrome (29). In a nested case–control study, patients with metabolic syndrome were 1.5 times more likely to use antidepressants compared to patients without metabolic syndrome (30). Higher levels of S100B are correlated with lower levels of serum-insulin one hour after an oral glucose tolerance test, suggesting that insulin tightly regulates S100B secretion from adipocytes (31). A 12-week randomized, placebo-controlled study found that pioglitazone, an insulin-sensitizing drug, improved glucose control and reduced chronic depression in patients with type II diabetes (28). Furthermore, emerging data supports that glucagon-like peptide-1 (GLP-1) agonists, such as liraglutide, have pro-cognitive, neuroprotective, and antidepressant effects (32). Overall, regulating S100B and glucose metabolism is likely to alleviate depression in patients with metabolic disorders.
4.2. HSPs
HSP’s are a molecular class of chaperones that help client proteins fold correctly and are pivotal to maintaining cellular integrity during stress. HSPs are upregulated in response to stress and prevent intracellular proteins from misfolding and aggregating (33). Exogenous administration of HSP70 exerts neuroprotective effects and protects against neurodegeneration in animal models (34). However, under stressful conditions, HSP70 is passively and actively released into the extracellular space triggering inflammation (35). In vitro studies have demonstrated that HSP70 and HSP90 stimulate macrophages to release the inflammatory mediators IL-1β, TNF-α -, IL-12, and granulocyte-macrophage colony-stimulating factor. In clinical studies, the relationship between serum Hsp70 and depression is not precise with variable findings between serum levels of HSP70 and depressive symptoms (36). Based on the preclinical data, HSP70 appears to be a promising target for antidepressant therapeutic development.
The unfolded protein response (UPR) plays a prominent role in the etiopathogenesis of depression. The function of the endoplasmic reticulum (ER) is to properly fold proteins and traffic them to various organelles and the cell membrane (37). Stress increases the accumulation of unfolded and misfolded proteins in the ER and in response the UPR signals HSPs, such as Grp94 and GRP78, to refold the proteins. IRE-1 detects when these mechanisms are insufficient to recover protein folding and activates apoptotic and NF-κB signaling pathways leading to the transcription of inflammatory cytokines (38). Timberlake and colleagues found that cortisol levels and GRP78 and GRP94 gene expression were higher in the hippocampus of rats with learned helplessness compared to rats without learned helplessness (39). Moreover, Kim and colleagues found that macrophage cell lines treated with tunicamycin increased the expression of ER stress markers, GRP94 and GRP78, and secretion of IL-6 (40). The preclinical data supports that stress induced activation of the UPR system in the ER is associated with inflammatory cytokines and depressive behaviors. Drug development targeting ER stress has the potential to ameliorate depression. ER membranes express sigma-1 receptors (sig-1R), and preliminary preclinical data suggest that induction of sig-1R alleviates ER stress and represses apoptotic signaling pathways (41). Therapeutics directed at sig-1R are currently under development for a variety of neuropsychiatric disorders.
4.3. Gal-3
Gal-3 is a multifunctional protein expressed in peripheral and central immune cells that regulates crucial cellular processes such as growth, proliferation, differentiation, and apoptosis. Gal-3 is released into the extracellular space during the differentiation of monocytes into macrophages (42). Gal-3 activates microglia and innate immune responses, accelerating the secretion of pro-inflammatory cytokines (43). The role of Gal-3 in depression has primarily been studied in patients with comorbid medical conditions. Observational studies in diabetic and obese patients have found that depression is linked to higher levels of gal-3, inflammatory cytokines, and parallel dysregulation of glucose metabolism (44). Moreover, King and colleagues reported that higher levels of Gal-3 were associated with greater depressive symptoms in a community-based sample of patients with heart failure and stroke (45). Emerging literature supports that treatment with hypoglycemic medications, such as metformin, are associated with reduced depressive symptoms and correspondingly lower levels of Gal-3. Moreover, in preclinical studies, glucagon-like peptide-1 agonists, which are utilized in the treatment of type 2 diabetes and obesity, have been shown to reduce Gal-3 and correspondingly ameliorate depression indirectly through neurogenesis (46). Based on the preliminary evidence, Gal-3 is a promising biomarker for monitoring and treating depression in chronic inflammatory states.
5. Pattern-recognition receptors
Pattern-recognition receptors (PRRs) are proteins expressed primarily on macrophages, dendritic cells, and microglial cells. Immune cells expressing PRRs detect DAMPs and activate peripheral and neuroinflammation. DAMP-PRR signaling is central to the initiation and perpetuation of the innate immune system. Several PRRs, toll-like receptor-4 (TLR-4) and NLR family pyrin domain containing 3 (NLRP3), have been implicated in depression (47).
5.1. NLRP3
NLRP3 is an intracellular PRR that activates downstream signaling cascades leading to activation of inflammasome components. Activated NLRP3 oligomerizes with adaptor proteins and pro-caspase-1 resulting in the formation of the NLRP3 inflammasome (48). The NLR3P inflammasome mediates proteolytic cleave of pro-caspase-1 leading to activation of caspase-1, which cleaves pro-interleukin 1β and 18 into their active cytokine forms (47). IL-1β cytokines have a role in stress responses and depression. IL-1β inhibits hippocampal neurogenesis and contributes to diminishing hedonic responses in susceptible animal models (49). Reversing the effects of IL-1β by means of antagonizing the IL-1R restores hippocampal neurogenesis and reduces depressive-like behaviors (50). Clinical studies have corroborated that depressed patients have higher serum levels of IL-1β compared to non-depressed patients. Moreover, Ferentinos and colleagues reported that depression severity correlated with higher serum levels of IL-1β (51).
In addition, IL-18 has been linked to inflammation and depression. IL-18 binds to the IL-18R and stimulates the production of IFN-γ and pro-inflammatory cytokines. IL-18 has been extensively studied in post-stroke mice with the evidence supporting that IL-18 predicts the risk of developing depression. Moreover, Wu and colleagues found that injecting recombinant mouse IL-18 into the amygdala increased depressive-like behaviors, which was reversed with an IL-18R antagonist (52). The preclinical data strongly support a central role for IL-18 in the development of depression post brain injury. A longitudinal study following post stroke patients reported that plasma levels of IL-18 were independently associated with the development of minor and major depression (53). The depressogenic effects of IL-18 might be secondary to disruption of functional neural networks. Peripheral levels of IL-18 correlate with degree centrality of the left posterior cingulate gyrus and decreased connectivity between the posterior cingulate cortex and the bilateral caudate in depressed patients (54, 55) The imaging data support that IL-18 affects neural networks that modulate motivation and reward processing in depression.
5.2. TLR-4
TLR-4 is a membrane bound receptor primarily found on macrophages and microglia. Homodimerization of TLR-4 receptors leads to the activation of the enzyme glycogen synthase kinase (GSK-3) and the transcription factor NF-κB, which induce the production of the pro-inflammatory cytokines IL-1β, TNF-α, IL-6, and CXCL10 (56). Repeated restraint or acoustic stress exposure activates the TLR-4 pathway and increases inflammatory cytokines in the prefrontal cortex of mice. Inhibiting TLR-4 prior to restraint or acoustic stress prevents activation of NF-κB and proinflammatory enzymes (57). Similarly, Zhang and colleagues found that chronic social defeat stress increased TLR-4 protein levels in the hippocampus and induced behavioral despair, social avoidance, and anxiety-like behaviors in susceptible mice. Administering fluoxetine reduced the expression of TLR-4 and reversed the depressive phenotype (58). Corroborating the preclinical data, observational studies have reported that depressed patients express higher levels of TLR-4 and NF-κB in peripheral blood mononuclear cells compared to non-depressed patients (59). Additionally, postmortem studies have found that mRNA expression of TLR-3 and TLR-4 is significantly increased in the dorsolateral prefrontal cortex of depressed patients compared to controls (60). These findings support that TLR-4 is a promising treatment target for inflammation in depressed patients (Figure 3).
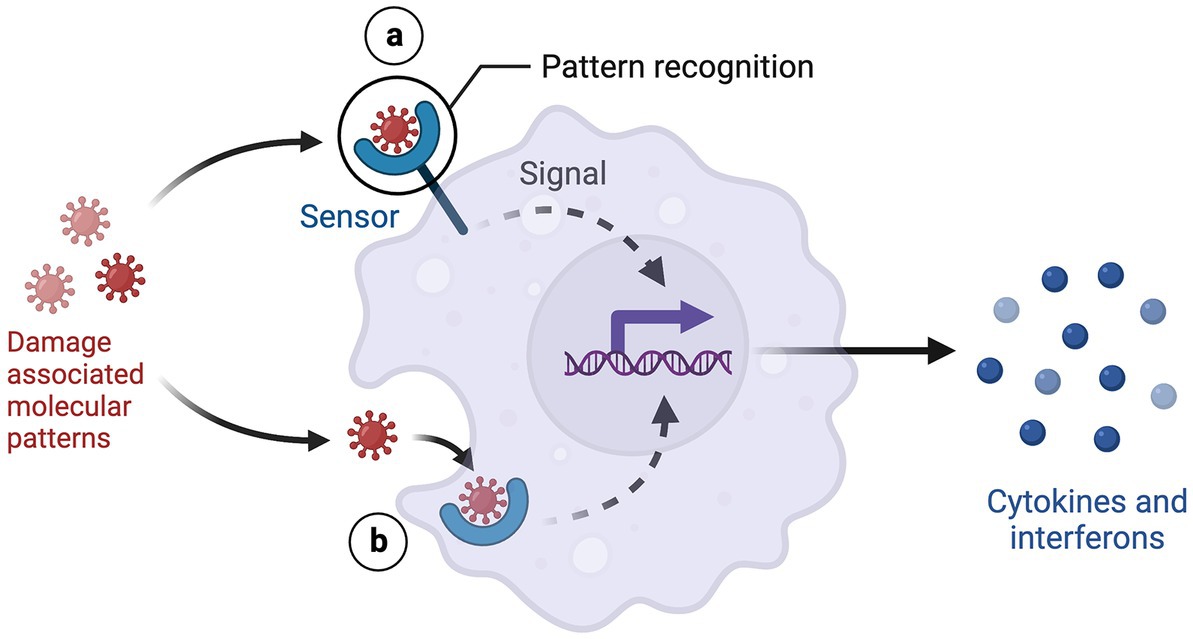
Figure 3. DAMP-PRR signaling. Created with BioRender.com.
6. Cytokines
Activation of the innate and adaptive immune system has been described in depressed patients. Accumulating evidence implicates cytokines in the pathogenesis of depression particularly in patients with chronic systemic conditions. Cytokines are signaling molecules that regulate the immune system and can either activate or inhibit inflammation. Pro-inflammatory cytokines cross the BBB or are produced by neuronal cells dysregulating neurotransmission and neurocircuitry (61). An imbalance between pro- and anti-inflammatory cytokines represents a distinct pathway for depression. A meta-analysis reported higher levels of serum pro-inflammatory cytokines (IL-1β, IL-2, IL-6, IL-12, TNF-α) and reduced anti-inflammatory cytokines (IL-4, IL-10, transforming growth factor [TGF]-β1) in depressed compared to non-depressed patients (62).
Exogenous IFN-α induced depression provided the earliest support that cytokines contribute to clinical depression. Approximately 30% of patients with hepatitis C develop major depression within the first 3 months of receiving IFN-α (63). Yamano and colleagues reversed INF-α induced stress susceptibility in mice with a corticotropin-releasing hormone receptor antagonist in a dose dependent fashion, suggesting that overactivity of the HPA axis mediates the cytokine depression response (64).
TNF-α is a potent proinflammatory cytokine secreted by activated macrophages directly. TNF-α is critical for activating a host of innate and adaptive immune responses. In particular, TNF-α induces cellular apoptosis and the extracellular release of DAMPs. DAMP-PRR signaling activates macrophages and microglial cells to release interleukins, chemokines, and reactive nitrogen and oxygen species (23). In addition, dendritic cells present DAMP associated antigens to T cell receptors differentiating naïve CD4+ T cells into T regulatory cells (Treg) and T helper cells (Th). Treg cells suppress the immune response whereas Th cells trigger immunological responses (65). Elevated serum levels of Th1 and Th17 cells along with a decrease in Treg cells have been reported in depression (66).
Th1 cells release IL-2 and IFN-γ initiating a constellation of inflammatory responses through activation of macrophages, CD8+ T cells, and B lymphocytes. Observational studies have reported an increase in circulating Th1 cells in patients with depression, and effective antidepressant treatment is associated with a reduction in circulating Th1 cells (67). Moreover, Myint and colleagues reported higher ratios of the Th1 cytokine IFN-γ/Il-4 and IFN-γ / TGF-β1 in depressed patients compared to non-depressed patients (68). Therapeutics downregulating IFN-γ have the potential to ameliorate inflammation and depression. Kubera and colleagues reported that monoamine-based antidepressants increased IL-10, a negative immunoregulator of IFN-γ, suggesting that the therapeutic efficacy of antidepressants may be related to their immunoregulatory effects on pro-inflammatory cytokines (69).
Activated macrophages release IL-23, which stimulates the development and differentiation of Th17 cells. Serum levels of IL-17, the signature cytokine of Th17 cells, are higher in depressed patients compared to non-depressed patients, and positively correlate with depressive symptoms (70). IL-17 exacerbates neuronal loss in basal ganglia neuroanatomic circuits important for mood regulation in depression (71). Treatment with a bupropion-SSRI combination reverses dopaminergic neuronal loss and ameliorates depression indicating that IL-17 predicts bupropion-SSRI combination response (72). IL-17 is weakly linked to suicide based on results from the AMAGINE-1 and AMAGINE-2 studies which evaluated the safety and efficacy of Brodalumab, a monoclonal antibody targeting the IL-17RA, for psoriasis. Four patients receiving Brodalumab in the open-label treatment arm completed suicide leading to a black box warning for suicidal ideation and behavior. Schiweck and colleagues postulated that blocking the IL-17RA led to higher circulating IL-17 levels and an increased risk for suicidal behaviors (73). A thorough investigation later revealed there was no causal link between Brodalumab and suicidal behaviors (74). Treatments neutralizing IL-17 have the potential to reduce neurotoxicity and improve depression and suicidality. In animal models, anti-IL-17 treatment rescues depression and anxiety behaviors in mice exposed to cumulative mild stress (75). IL-17 and IL-23 are central neuroinflammatory mediators, and clinical trials are currently underway examining the efficacy and safety of monoclonal antibodies to IL-17 and 23 in treatment resistant depression.
NF-κB is a central pleiotropic mediator of inflammation and induces the expression of multiple cytokines and chemokines. NF-κB has dichotomous effects on inflammation, and depending on the phosphorylation activation site, can either promote or inhibit neuronal growth (76). Wang and colleagues found that chronic unpredictable mild stress increased NF-κB expression and pro-inflammatory cytokines in the frontal cortex and hippocampus of susceptible mice. The depressive-like behaviors were reversed with a TLR4 antagonist suggesting that hyperactive TLR4-NF-κB signaling is involved in the pathogenesis of depression (77). Similarly, Munhoz and colleagues found that chronic unpredictable stress increased LPS activated NF-κB binding in the frontal cortex and hippocampus. NF-κB signaling was blunted with a glucocorticoid receptor antagonist supporting the central proinflammatory role of glucocorticoids in depression (78). In clinical studies, post-endotoxin depressed mood is predicted by higher baseline activity of the NF-κB transcription factor in peripheral blood mononuclear cells (79). Modulation of NF-κB signaling is currently being investigated as an approach to treat major depression (80).
7. Blood brain barrier
Proinflammatory cytokine mediated disruption of the BBB is the primary link connecting peripheral and neuroinflammation (81). Under normal conditions, peripheral inflammatory molecules are unable to pass through the blood–brain barrier (BBB). However, during chronic stress and inflammation, the BBB becomes increasingly permeable to immune cells, cytokines, and nitro-oxidative stress molecules. The BBB primarily consists of brain microvascular endothelial cells (BMECs) and astrocytic end feet. BMECs are connected to each other by tight junctions, GAP junctions, and adherens junctions (82). Claudin and occludin are the major transmembrane proteins of tight junctions and are connected to the actin cytoskeleton with zo 1 and zo 2 adaptor proteins. Pro-inflammatory cytokines and astrocytic release of VEGF-A decrease the expression and assembly of claudin and occludin and increase the paracellular permeability of peripheral immune cells (83). Invading macrophages secrete reactive oxygen species (ROS) and matrix metalloproteinases (MMP-1 and MMP-2) further degrading the BBB and perpetuating the cycle of immune cell migration and inflammation. Several pathways to cross the BBB have been described: (1) Entering the brain parenchyma through leaky capillaries in the circumventricular organs and choroid plexus, (2) Direct transportation into the CNS, (3) Binding to vagal afferents and entering the brain, and (4) Direct cellular trafficking (84).
The neurobiology of BBB permeability was first elucidated in animal models. Menard and colleagues found reduced expression of claudin-5, an endothelial cell tight junction protein, and abnormal blood vessel morphology in the nucleus accumbens promoted the passage of IL-6 across the BBB. The infiltration of peripheral IL-6 into the brain parenchyma led to the development of depression-like behaviors. Chronic administration of imipramine normalized claudin-5 mRNA expression in the nucleus accumbens and reversed depressive behaviors in stress susceptible mice (81). Similarly, confirmatory studies reported that the expression of claudin-5, occludin, and zo-1 were reduced in the frontal cortex and hippocampus of stress susceptible rats (85). These findings support that claudin-5 is the major gate keeper of the BBB. Treatment with lithium, haloperidol and chlorpromazine significantly increase levels of claudin-5 protein in mice brain microvascular endothelial cells suggesting that psychotropic efficacy might be in part due to improvements in BBB integrity (86) (Figure 4).
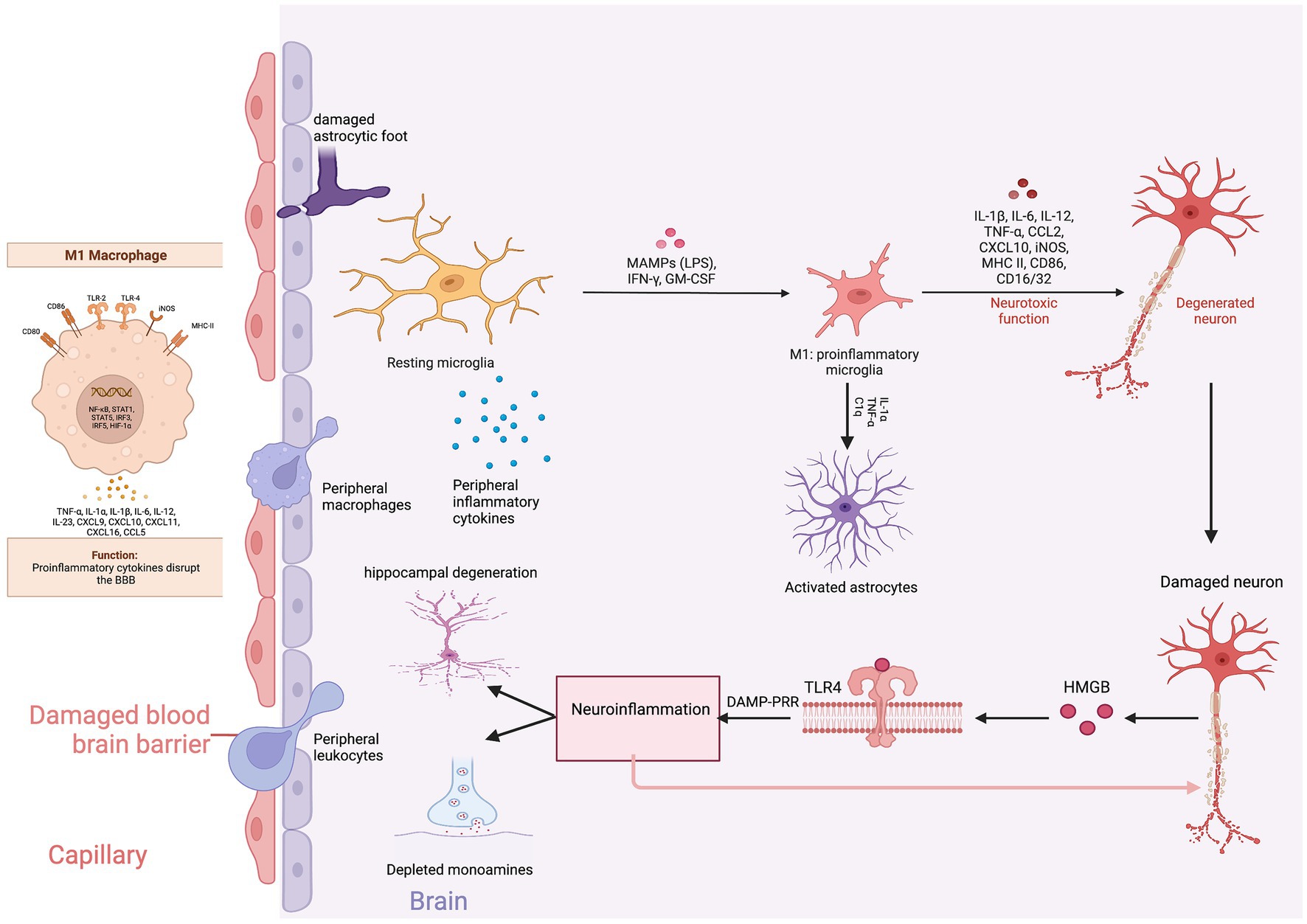
Figure 4. Neuroinflammation and depression. Created with BioRender.com.
8. Gliosis
The infiltrating peripheral immune cells release inflammatory cytokines and monocyte chemotactic protein-1 (MCP-1) inducing the migration of macrophages and monocytes into the CNS. Macrophages produce phosphodiesterase-4 (PDE4) leading to the production of pro-inflammatory cytokines (87). The proinflammatory cytokines and chemokines trigger gliosis, a neuro-immune response manifested by the proliferation and hypertrophy of glial cells (astrocytes, microglia, and oligodendrocytes) (88). Mounting evidence supports that gliosis is associated with increased levels of proinflammatory cytokines and reactive oxygen and nitrogen species (RONS) in subcortical limbic brain regions implicated in depression (89).
Depending on the phenotypic expression, microglia can either induce neurotoxicity (M1) or provide neuroprotection (M2). Therefore, the balance between M1 and M2 microglia is critical for maintaining homeostatic synaptic plasticity. During inflammation, microglia polarize toward the M1 phenotype, releasing proinflammatory cytokines and excessive glutamate (90). Translocator protein 18 kDa (TSPO) is expressed on the outer mitochondrial membrane of M1 microglia and is an indirect measure of microglial activation and neuroinflammation (91). PET imaging studies have found an increased uptake of TSPO ligands in the prefrontal cortex, anterior cingulate cortex, and the hippocampal formation in depressed compared to non-depressed patients indicating that microglial activation leads to neuroinflammation (92). Furthermore, a meta-analysis of molecular neuroimaging studies estimated that depressed patients had an 18% increase of TSPO availability compared to non-depressed patients (93). Minocycline, an antibiotic that reduces inflammation through inhibition of microglia, improves depression with effect sizes as high as 0.71 compared to placebo (94). Targeting M1 microglial activation may pave the way for the development of novel therapeutic interventions.
M1 microglia induced astrocytic neurotoxicity is a critical factor contributing to depression. Astrocytes in the hippocampus are crucial for regulating glucocorticoid-mediated feedback to the HPA axis and maintaining cortisol tone. Glial fibrillary acidic protein (GFAP), an astrocytic structural intermediate filament protein, is commonly measured to distinguish astrocytic activity from other glial cells. Mice exposed to chronic daily social defeat expressed reduced GFAP in stress-related brain regions associated with depressive-like behaviors (95). Administering fluoxetine reversed the loss of GFAP-positive astrocytes and ameliorated depressive behaviors (96). Similarly, postmortem studies have confirmed a reduced density of GFAP-immunoreactive astrocytes in brain tissues of patients with a history of MDD or who have completed suicide compared to controls (97). The preliminary data support that astrocytic dysfunction is associated with depression and suicide. Furthermore, a retrospective study found increased GFAP concentrations in the CSF of patients with unipolar depression compared to controls, indicative of astroglia injury (98). Steinacker and colleagues found that serum GFAP levels, at a cut off 130 pg./ml, distinguished MDD from bipolar disorder and schizophrenia with a sensitivity and specificity of 87 and 70%, respectively (99). Therefore, serum GFAP levels might be a useful biomarker candidate to distinguish and monitor the severity of major depressive disorder.
9. Oxidative stress
In addition to IL mediated inflammation, an imbalance between free radicals and antioxidants has been implicated in depression. The brain’s high consumption of oxygen makes it particularly vulnerable to oxidative stress compared to other organs and tissues (100). Activated macrophages and microglia release ROS that disrupt cell membranes, cellular DNA, and protein synthesis (101). The high levels of ROS overwhelm the capacity of antioxidant enzymes and disturb redox homeostasis. In addition to the overproduction of ROS, reduced detoxification capacity accelerates cellular damage. Reduced plasma levels of antioxidants (vitamin E, zinc, coenzyme Q10, glutathione) and enzymes (xanthine oxidase and superoxide dismutase) are interrelated with worsening depressive symptoms (102).
Evidence of a pro-oxidative state has been validated in patients with depression. Studies have reported higher levels of oxidative stress byproducts and lower levels of antioxidant enzymes (103). Moreover, correlative studies have found a positive interrelation between oxidative stress by products and pro-inflammatory cytokines suggesting a bidirectional association with the progression of depression. Rybka and colleagues found higher concentrations of malondialdehyde and IL-6 and lower levels of glutathione peroxidase and erythrocyte superoxide dismutase (SOD-1) in depressed compared to non-depressed patients. The authors concluded that the oxidative inflammatory cytokine response represents a molecular pathway underpinning depression (104). Moreover, greater symptoms of depression are positively correlated with higher urinary levels of oxidatively damaged DNA and RNA products (105). Oxidative associated depression is also characterized by IgM-related autoimmune responses directed against neoepitopes from damaged lipids, proteins, and DNA. These autoimmune responses dysregulate key cellular functions and are thought to play a role in the neuroprogression of inflammation and depression (106).
Effective antidepressant treatment reverses the effects of oxidative damage. Khanzode and colleagues reported that SSRIs reduced serum levels of malondialdehyde and increased plasma ascorbic acid levels, which correlated with improvements on the Hamilton Depression Rating Scale (107). Brain Derived Neurotrophic Factor (BDNF) regulates the antioxidant gene transcription factor erythroid 2-related factor 2 (Nrf2) (108). Reduced BDNF inhibits Nrf2 transcription of antioxidant enzymes and increases susceptibility to depressive behaviors in animal models (109). Correspondingly, post-mortem studies have found decreased expression of Nrf2 in the prefrontal cortex of depressed compared to non-depressed patients (110). Administering nrf-2 activators, such as sulforaphane, reduces serum corticosterone, IL-6, and TNF-α, and subsequently, reverses depressive behaviors in mice (111). Therefore, increasing the antioxidant capacity is an effective strategy to reduce inflammation and ameliorate depression. A systematic review reported that diets rich in antioxidant vitamins (A, C, and E) and nutrients (beta carotene, selenium, copper, zinc) reduced the risk of developing depression by about 33% (112).
10. Reactive nitrogen species
Nitrosative stress occurs when there is an abnormal increase in the level of nitric oxide (NO) produced by the inducible isoenzymes of NO synthase: neuronal, inducible, and endothelial (113). NO reacts rapidly with superoxide (O2−) to produce peroxynitrite (ONOO−), a potent ER stress oxidant inducing agent. ONOO− inhibits energy metabolism, damages DNA, alters cytoskeletal organization, and impairs cell signal transduction (114). ONOO− nitrates tyrosine residues on proteins, generating 3-nitrotyrosine (3-NT), a biomarker of NO-dependent nitrosative stress. Urine levels of 3-NT are higher in depressed compared to non-depressed patients, implying that nitrosative stress is a major contributor to cytokine induced depression (115).
NO is synthesized from oxygen dependent l-arginine by nNOS in glial cells (116). NO is a second messenger with pleiotropic effects, and at excessive concentrations, overstimulates the NMDA receptor precipitating excitotoxicity (117). Narsapur and colleagues reported that treatment with methylene blue, a NO scavenger, improved depressive symptoms in patients resistant to lithium (118). Similarly, paroxetine inhibits nNOS and subsequently reduces serum levels of NO and improves depression (119). To maintain homeostasis, the enzyme nitric oxide reductase (NOR) inactivates and catalyzes NO to nitrous oxide (N2O). Preliminary evidence supports that inhaled N2O improves treatment-resistant depressive symptoms (120). Therefore, modulation of NO signaling is a potential therapeutic target for MDD. Nagele and colleagues found that inhaled N2O at a concentration of either 25% or 50% significantly reduced scores on the Hamilton Depression Rating Scale (HDRS) at week 2 in patients with treatment resistant depression. The antidepressant effects of N2O are mediated through antagonism of the metabotropic glutamatergic NMDA receptors (121). Similarly, preliminary findings in animal models support that inhalation of sub-anesthetic doses of xenon, a potent antiglutamatergic anesthetic, reduces the severity of depressive disorders (122).
nNOS is differentially expressed throughout the brain in patients with MDD. Oliveira and colleagues found increased nNOS CA1 hippocampal immunoreactivity whereas Bernstein and colleagues found reduced nNOS locus coeruleus immunoreactivity in postmortem samples of depressed patients (123). The data suggest that an imbalance in nNOS activity is associated with depression. Preclinical work supports that administration of nNOS inhibitors, such as 7-nitroindole, reduces depressive-like behaviors in obese mice (124). Moreover, Zhou and colleagues found that mice exposed to chronic stress exhibited behavioral despair, which correlated with nNOS overexpression and impaired neurogenesis in the hippocampus. Administering inhibitors of nNOS reversed the effects of chronic stress and increased neurogenesis (125). The data suggest that inhibition of nNOS is a promising novel antidepressant target.
11. Neurotransmitters
Pro-inflammatory cytokines reduce the bioavailability of neurotransmitters critical for synaptic communication (61). Pro-inflammatory cytokines decrease monoamine availability through induction of mitogen activated protein kinase (MAPK) signaling, depletion of the cofactor Tetrahydrobiopterin (BH4), and activation of indolamine 2,3 dioxygenase (IDO). Activation of MAPK increases the expression of reuptake pumps for serotonin, norepinephrine, and dopamine, subsequently decreasing the availability of monoamines at the synaptic cleft (126). Inflammatory cytokines also increase the utilization of BH4, a cofactor necessary for nNOS to produce NO. Decreased availability of BH4, a cofactor for the rate limiting enzymes tryptophan hydroxylase and tyrosine hydroxylase, reduces the synthesis of dopamine and serotonin (127). In addition, IDO metabolizes tryptophan, an amino acid precursor to serotonin, thus depleting the availability of serotonin (128).
11.1. Quinolinic acid
Activated microglia upregulate IL-18, which increases glycogen synthase kinase (GSK-3β) and IFNγ-dependent IDO expression in glial cells (129). IDO catabolizes tryptophan, a serotonin precursor, to kynurenine. Kynurenine is further converted to either kynurenic acid (KA) or quinolinic acid (QUIN) in astrocytes or microglia, respectively (130). QUIN has neurotoxic effects, and at high concentrations, overstimulates the NMDA receptor resulting in glutamatergic excitotoxicity (131). Moreover, QUIN increases the release and blocks the reuptake of glutamate in astrocytes, thus increasing glutamatergic extracellular concentrations (132). Bay-Richter and colleagues found increased CSF levels of QUIN and decreased levels of KA in suicide attempters. As expected, lower KA levels correlated with severe depressive symptoms and higher IL-6 levels suggesting dysregulation of the kynurenine pathway increases the risk for suicidality (133). QUIN induces neurotoxicity primarily in the striatum, the pallidal formation and the hippocampus (134). This is consistent with morphological and functional findings demonstrating decreased grey matter in the frontotemporal area and reduced connectivity between brain regions linked to reward processing (135).
Meta-analyses have confirmed elevated plasma levels of QUIN in depressed patients compared to non-depressed patients (136). Similarly, post-mortem studies have reported elevated GSK-3β mRNA levels in the hippocampus of patients with major depression. Treatment with GSK-3β inhibitors reduces the production of QUIN and subsequently alleviates depressive, anxious, and aggressive behaviors in animal models (137). Moreover, Verdonk and colleagues reported that decreases in QUIN after a ketamine infusion strongly predicted improvements in MADRS scores in patients with treatment resistant depression (138). The data support that QUIN mediates inflammatory neurotoxicity and disrupts neural circuitry in depression.
11.2. Neurotoxicity
Pro-inflammatory cytokines disrupt neurocircuitry implicated in the pathogenesis of depression. fMRI studies have associated higher peripheral levels of IFN-α and TNF- α with increased glutamatergic activity in the basal ganglia and anterior cingulate cortex regions involved in emotion regulation and reward processing (139). Similarly, Capuron and colleagues examined PET changes in patients with HCV treated with IFN-α and found decreased blood-oxygen level dependent responses in the ventral striatal regions during a hedonic gambling reward task. Reduced activity in the ventral striatum was interrelated with reduced motivation, fatigue, and depressed mood on the neurotoxicity scale (140). These findings suggest that cytokines disrupt neurocircuitry likely involved in the pathogenesis of depression.
Neuroinflammatory induced synaptic loss and functional connectivity deficits are considered a final common pathway for depression. The synaptic vesicle glycoprotein 2A (SV2A), a membrane protein expressed primarily in GABAergic containing neurons, is a proxy for estimating synaptic density. Greater symptoms of depression are associated with reduced radioligand binding to SV2A in the dorsolateral prefrontal cortex, anterior cingulate cortex, and hippocampal regions (141). Furthermore, the severity of depression is inversely correlated with neuronal SV2A density and reduced GABAergic inhibitory neurotransmission (142). Garcia-Oscos and colleagues found that administration of IL-6 to cortical rat slice preparations decreased GABAergic inhibition shifting the balance to glutamatergic central hyperexcitability (143). Altered cortical inhibition is implicated in depressive disorders.
Neuronal loss of SV2A results in a global imbalance between glutamatergic excitation and GABAergic inhibition. GABAergic neurons constitute 15 to 25% of the total neuronal population and reduced densities in the prefrontal cortices of mice is associated with major depression (144). Specifically, reduced dendritic inhibition from somatostatin expressing GABAergic interneurons is linked to glutamatergic overexcitability and treatment resistant major depressive disorder. Furthermore, proton magnetic resonance spectroscopy shows reductions of GABA in the occipital cortex, anterior cingulate, and prefrontal cortex in patients with treatment resistant depression (145). Confirmatory post-mortem studies in depressed patients have found reduced somatostatin gene expression in the subgenual anterior cingulate cortex (146). Somatostatin regulates ACTH secretion and loss of somatostatin-expressing interneurons increases glucocorticoid signaling and glutamate neurotransmission in the hippocampus (147). Synchronization of the excitatory to inhibitory (E/I) ratio is important for balancing the signal-to-noise ratio in neural circuits, and reduced inhibition leads to noisier circuits and less efficient information processing (148).
Neuronal circuits are composed of a combination of excitatory and inhibitory neurons. Most neurons are excitatory, and their function is to propagate depolarizing electrical signals to activate downstream neurons. Alternatively, inhibitory neurons hyperpolarize neurons and constrain excitatory neurotransmission (149). Excitatory and inhibitory neurotransmission is critical for regulating the transmission of information between brain areas that control behavior, thought, and emotion. An imbalance between cortical E/I neurotransmission leads to asynchronous and irregular network functioning related to depression. Resting-state functional magnetic resonance imaging has demonstrated reduced functional connectivity between the salience network and right and middle inferior temporal gyrus in depressed patients compared to non-depressed patients. The salience network is important for regulating emotion and cognition, and functional deficits correlate with impairments of cognition and emotion regulation in depression (150). Newer treatments are focusing on restoring the glutamate and GABAergic neurotransmission balance (151). SV2A antidepressant ligand treatments are currently under development to restore the GABAergic tone (152). Similarly, drug discovery is focusing on developing differentiated Kv7 potassium channel openers that generate a hyperpolarizing M-current and reduce hyperexcitability (153) (Table 1).
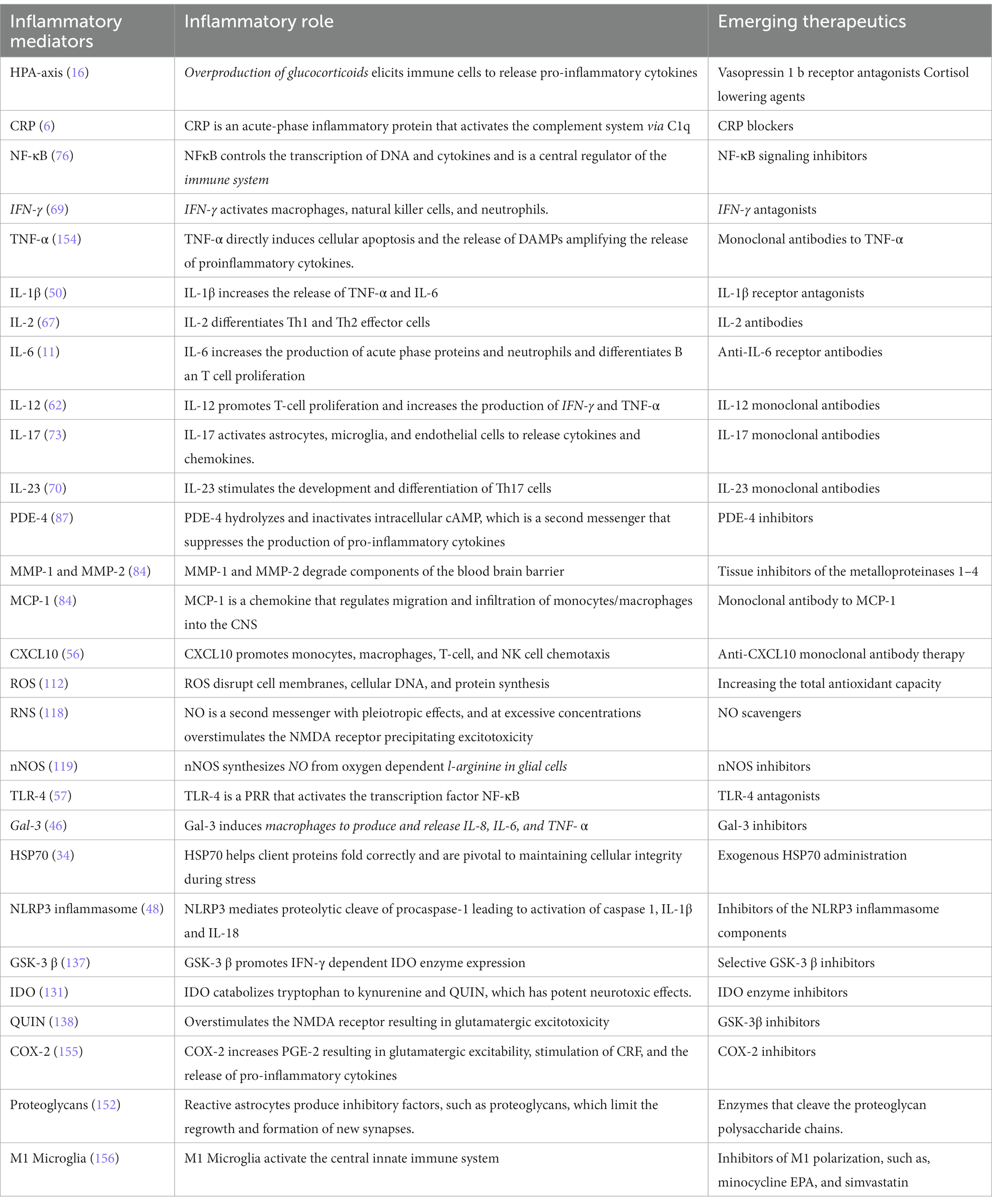
Table 1. Emerging inflammatory biomarkers and treatment targets for depression.
12. Systemic anti-inflammatories
The efficacy and safety of systemic anti-inflammatories has been studied in depressive disorders. Limitations of data interpretation include the small number of studies, moderate to large heterogeneity, variations in diagnostic and treatment planning, and inclusion of patients without evidence of inflammation. Overall, the literature does support that NSAIDs, statins, omega-3 fatty acids, N-acetylcysteine, and COX-2 inhibitors have a small to moderate antidepressant effect sizes over the course of 4 to 12 weeks of treatment. Adjunctive treatment has a larger antidepressant effect size than monotherapy in part due to synergy between monoamine-based treatments and anti-inflammatories. Augmentation with an anti-inflammatory appears to be 52% more effective in reducing symptom severity compared to placebo. Of the anti-inflammatories, NSAIDs, omega-3 fatty acids, and statins, are the most effective at reducing depressive symptoms compared to placebo (157).
12.1. NSAIDs
COX-1 and COX-2 enzymes are necessary for the synthesis of prostaglandins involved in inflammation. Prostaglandins increase the release of pro-inflammatory cytokines, suppress Th2 cells, and induce the synthesis of chemokines (158). The COX 2 enzyme is largely induced in the CNS by LPS and proinflammatory cytokines. COX-2 overexpression increases PGE-2 resulting in glutamatergic excitability, stimulation of corticotropin-releasing hormone, and the release of pro-inflammatory cytokines (159). Preclinical studies confirm that chronic unpredictable stress increases the uptake of PET radioligands targeting the COX-2 enzyme and cerebral PGE-2 concentrations in susceptible compared to resilient mice (160).
These findings have led to an increasing amount of research on the potential role of COX-2 inhibitors in the treatment of depression (161). Administering celecoxib, a COX-2 inhibitor, reverses the effects of chronic unpredictable stress and reduces COX-2 expression in a dose-dependent manner (162). A randomized, double-blind, placebo-controlled trial found that the addition of celecoxib to sertraline over 4 weeks improved response and remission rates suggesting a synergistic relationship between celecoxib and sertraline (163). Similarly, Kohler and colleagues found that patients prescribed celecoxib compared to placebo were 7 times more likely to respond to an antidepressant with no increased risks of GI or CV effects (155).
12.2. Omega-3 fatty acids
Omega-3 fatty acids, specifically, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), have been studied in depression. EPA and DHA ameliorate inflammation by inhibiting leukocyte chemotaxis, adhesion molecule expression, and the production of inflammatory cytokines and prostaglandins (164). The anti-inflammatory effects are mediated by the production of a distinct class of metabolites known as specialized pro-resolving mediators. Intracranial injection of specialized pro-resolving mediators in mice exposed to chronic unpredictable stress or LPS inhibits astrocyte and microglia activation, reduces the expression of IL-1β and TNF-α in the hippocampus, and reduces the degradation of synapsin (165).
Clinical studies have validated the relationship between omega-3 fatty acids and depression. Chang and colleagues found that depressed patients with hepatitis C treated with IFN- α had lower serum EPA levels and higher scores on neurotoxicity rating scale (NRS) for somatic symptoms compared to non-depressed patients (166). Pretreatment with EPA may reduce the risk of developing depression in patients with hepatitis C treated with INF- α (167). A dose finding study comparing 1 g, 2 g, or 4 g of omega-fatty acid capsules with a 4:1 ratio of EPA:DPA reported that high-doses (4 g/per day) were required for antidepressant efficacy in overweight or obese patients with inflammation (156). Overall, the data indicate that high doses of EPA are required to achieve anti-depressant efficacy.
12.3. Statins
Inhibitors of 3-hydroxy-3-methylglutaryl-Coenzyme A (HMG-CoA) reductase commonly known as statins have been studied in depression due to their broad anti-inflammatory effects. Simvastatin treatment in high fat diet or streptozotocin-induced diabetic mice has been shown to rescue depressive-like behaviors, and restore the expression of 5-HT receptors, BDNF, and anti-inflammatory cytokines in the hippocampus (168). Similarly, Na and colleagues reported that administration of atorvastatin to streptozotocin-induced diabetic mice rescued depressive behaviors, attenuated microglial activation in the prefrontal cortex, and reduced the expression of IL-1β, TNF-α, and NF-κB p65 expression in the prefrontal cortex. Overall, the data in animal models supports that statins reduce stress induced depression through amelioration of inflammation (169).
A meta-analysis of RCTs found that the addition of a statin to an antidepressant had a moderate effect on reducing depressive scores compared to placebo at week 8 and 12 with no report of serious adverse events across all trials (170). Statins have varied lipophilic profiles, and the ability to cross the BBB affects antidepressant efficacy and activity. Simvastatin is more lipophilic than other statins, and a study comparing antidepressant outcomes in patients post CABG found that simvastatin had greater antidepressant effects than atorvastatin (171). A large Swedish national cohort study confirmed that statins reduced the odds of depression by 8% compared to placebo, but a subgroup analysis found that only simvastatin significantly reduced the odds of depression (172). Based on the body of evidence, simvastatin appears to be the most promising statin treatment for reducing neuroinflammation in depression.
13. Anti-cytokine therapies
A limitation of systemic anti-inflammatories is that they are not specific to cell mediated immunity. To address this limitation, the antidepressant efficacy of targeted monoclonal antibodies to inflammatory cytokines has been studied in randomized clinical trials primarily as a secondary outcome in patients with autoimmune and rheumatological conditions. A meta-analysis of seven randomized controlled trials reported that anti-cytokine treatment had a moderate effect size (Cohen’s d = 0.40) on depression compared to placebo among patients with chronic inflammatory conditions independent of improvements in physical symptoms. Specifically, antibodies against TNF-alpha (Adalimumab, etanercept, infliximab) and IL-6 (tocilizumab) showed statistically significant improvements in depressive symptoms (173). Antibodies to TNF-α are the most studied anti-cytokine therapeutics in depression. A double-blind, placebo-controlled, randomized clinical trial in treatment resistant depression found that infusions of infliximab significantly reduced scores on the HAM-D at week 12 in patients with a baseline CRP concentration greater than 5 mg/L (154). The literature supports that anti-cytokine treatments might be useful as a treatment option for depression associated with an inflammatory component.
Novel pipeline drugs focusing on antibodies to IL-6 and IL-17 show some promise. Sun and colleagues reported that antibodies to IL-6, sirukumab and siltuximab, significantly improved depressive symptoms in patients with rheumatoid arthritis or multicentric Castleman’s disease even after adjusting for disease severity, pain, and physical functioning (174). A study analyzing the efficacy of 3 randomized, double-blind, controlled trials in patients with moderate-to-severe plaque psoriasis, found that ixekizumab, an anti-IL-17 antibody, reduced scores on the QIDS-SR-16 and improved depression at week 12. Moreover, 45% of patients treated with ixekizumab remitted compared to 18% on placebo. The improvement in depressive symptoms correlated with reductions in CRP and the psoriasis area index (175). To date, there is a paucity of published literature on the antidepressant efficacy of monoclonal antibodies to inflammatory cytokines in patients without autoimmune conditions. Additional safety and efficacy studies are needed in treatment resistant patients without a history of autoimmune and rheumatological conditions.
14. Transforming growth factor beta (TGF- β)
TGF-β is a multifunctional set of pleiotropic cytokines that control cell proliferation, differentiation, and motility of neural cells. Canonical TGF-β signaling exerts anti-inflammatory neuroprotection and influences memory formation and synaptic plasticity (176). TGF-β exists in three isoforms, of which TGFB1 is the most widely studied in depression. TGFβ1 binds to the TGF beta receptor 1 (TGFBR1) on microglial cells and inhibits the production and release of cytokines, chemokines, adhesion molecules, and ROS through activation of p38 MAPK and Akt phosphorylation (176). Silencing the TGFBR1 in microglia abolishes the neuroprotective and anti-inflammatory properties of TGF-β1 in animal models. Moreover, a study found that inhibiting p38 MAPK prevented TGF-β1 from inhibiting microglial activation and 1-methyl-4-phenylpyridinium (MPP+)-induced dopaminergic neuronal toxicity. The preclinical data suggest that impaired TGFβ1 signaling increases microglial activation and inflammation in depression (177). Moreover, a growing body of research suggests that the antidepressant effects underlying r-ketamine are dependent on microglial TGFβ1. Zhang and colleagues reported that administering neutralizing antibodies to TGF-β1 blocked the antidepressant effects of (R)-ketamine in chronic social defeat stress mouse models (178). Further supporting the immunoregulatory role of TGFβ, pre-clinical research has found that inhalation of recombinant TGF-β1 elicits neurogenesis and reduces depressive behaviors in chronic social defeat stress (CSDS) models (179).
More recent developments have focused on understanding the clinical application of TGFβ1 in depression. Studies have not shown a consistent relationship between plasma levels of TGFβ1 and depression, nonetheless, TGFβ1 appears to regulate inflammatory processes underlying MDD. Preliminary evidence supports that childhood maltreatment mediates a higher concentration of TGFβ1 in adults with major depressive disorder suggesting an epigenetic role for TGFβ1 in stress related disorders (180). Caraci and colleagues reported that TGF-β1 plasma levels were reduced in patients with major depression and correlated with depression severity and treatment resistance. Administering sertraline and venlafaxine increased serum levels of TGF-β1 and reduced depression (181). In contrast, despite a response, Rush and colleagues found that ECT did not normalize reduced levels of TGF-β in patients with melancholic depression (182). Larger and well-designed studies are needed to determine the prognostic and therapeutic potential of TGF-B1 in depression.
15. Neurotrophic factors
Neurotrophic factors are key molecules that play important functions in the regulation of immune cell activation and neuro-survival. The primary neurotrophic factors (BDNF, NGF, and NT) reduce neuroinflammation through activation of specific tyrosine kinase receptors (TrkA TrkB, TrkC) and downstream signaling of the PKC pathway, Ras/MAPK pathway, and PI3 pathway resulting in neuronal remodeling and formation of novel synapses (183). The activation of these multiple signaling cascades results in neuronal remodeling and adaptive circuitry functioning countering inflammation mediated neurotoxicity (184).
Neurotrophic factors and downstream signaling effects play a critical role in synaptogenesis and plasticity. Therefore, modulating neurotrophic cascade signaling is an effective strategy to ameliorate symptoms of depression rapidly and effectively. As such, innovative treatments to boost neurotrophic factors have been developed to counteract stress and inflammation. To date, drug discovery has primarily focused on indirectly boosting endogenous neurotrophic factors by antagonizing N-methyl-D-aspartate (NMDA) or potentiating alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) (185, 186).
Direct administration of exogenous neurotrophic factors has been limited by pharmacokinetic factors as well as safety and efficacy concerns. Exogenous administration of neurotrophic factors is limited by blood–brain-barrier (BBB) permeability, poor half-life, and rapid degradation (183). Moreover, the preliminary data has been inconsistent as neurotrophic factors exert multiple pleiotropic off-target effects potentially interfering with restoration of synaptogenesis. To overcome these limitations, low molecular weight dimeric dipeptides that mimic BDNF loop 4 have been designed to specifically activate the tyrosine kinase B receptor. Two BDNF mimickers currently under investigation are dipeptide N-monosuccinyl-L-seryl-L-lysine and 7,8-Dihydroxyflavone (187). Novel therapeutics specific to the neurotrophic downstream signaling cascade effects are currently under development for depression (Table 2).
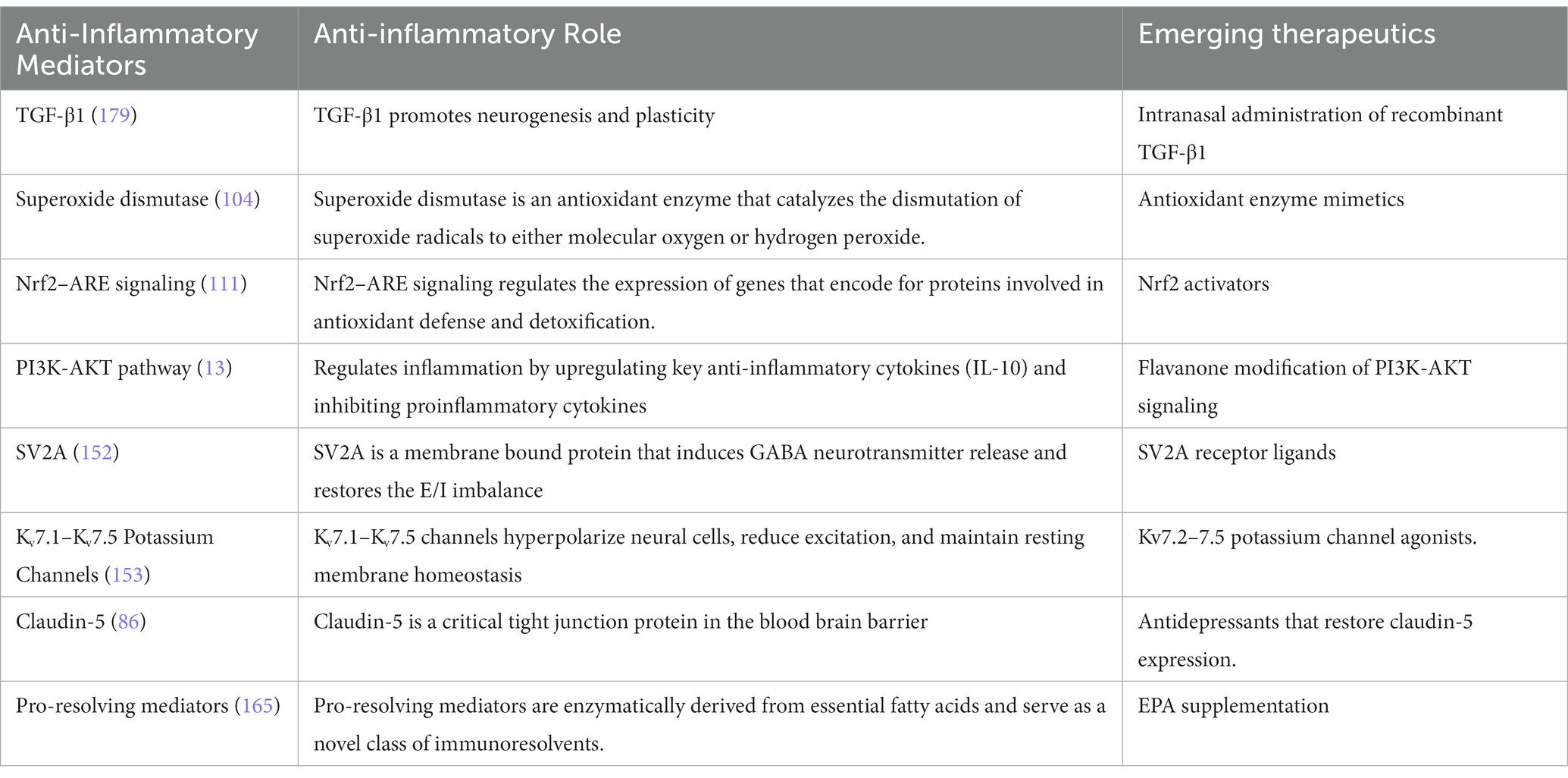
Table 2. Novel anti-inflammatory drug candidates for depression.
16. Conclusion
Inflammation mediates the relationship between chronic stress and the evolution and progression of depression and comorbid conditions. Persistent stress creates a disruptive cycle between allostatic loading, dysregulated glucocorticoid functioning, overactivation of the immune system, and the excessive release of inflammatory molecules, tilting the balance toward a pro-inflammatory state. The extracellular release of DAMPs and DAMP-PRR signaling sets in motion a self-perpetuating cycle of inflammation. The perpetual flux of inflammatory mediators disrupts the blood–brain barrier allowing for cellular and cytokine trafficking into the CNS and activation of glial cells. Glial cell cross talk with peripheral immune cells and cytokines is vital for orchestrating a bi-directional inflammatory network between the periphery and central nervous system. The cascade of pro-inflammatory cytokines and oxidative stress induces neurotoxicity, perturbs neurotransmitter functioning, and globally disrupts the E/I balance within neural circuits related to depression. Reduced gray matter volumes in frontolimbic brain structures and aberrant functional connectivity in reward pathways underlie the biological basis of neuroinflammatory depression. Current monoamine-based treatments reduce neuroinflammation through multiple mechanisms however their effects are limited by delayed therapeutic responses. The diversity of cell mediated immunities, cytokines, and nitro-oxidative stress contributing to depression provides an opportunity to discover stable diagnostic and prognostic biomarkers for neuroinflammatory depressive phenotypes. The relationship between neuroinflammation and depression sets the stage for the development of novel pharmaceuticals. Randomized controlled trials have predominantly focused on the efficacy of systemic anti-inflammatories with preliminary findings demonstrating improvements in depressive symptoms. Systemic anti-inflammatories appear to be promising, however, tailoring treatments to the specific neurobiological mechanism has the potential to robustly improve antidepressant outcomes. Targeted therapies have been limited to animal models or patients with concomitant autoimmune and rheumatological conditions. Future clinical trials should focus on testing therapeutics that regulate the HPA axis, modulate immune cells responses, antagonize inflammatory cytokines, reduce nitro-oxidative stress, and mimic neurotrophic factors with the goal of restoring the E/I imbalance and neural circuitry functioning.
Author contributions
SH prepared and supervised the draft, contributed to the article, and approved the submitted version.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ettman, CK, Cohen, GH, Abdalla, SM, Sampson, L, Trinquart, L, Castrucci, BC, et al. Persistent depressive symptoms during COVID-19: a national, population-representative, longitudinal study of U.S. adults. Lancet Reg Health Am. (2022) 5:100091. doi: 10.1016/j.lana.2021.100091
2. Chaaya, C, Thambi, VD, Sabuncu, Ö, Abedi, R, Osman, AOA, Uwishema, O, et al. Ukraine–Russia crisis and its impacts on the mental health of Ukrainian young people during the COVID-19 pandemic. Annal Med Surg. (2022) 79:104033. doi: 10.1016/j.amsu.2022.104033
3. Danese, A, Pariante, CM, Caspi, A, Taylor, A, and Poulton, R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci U S A. (2007) 104:1319–24. doi: 10.1073/pnas.0610362104
4. Medina-Rodriguez, EM, and Beurel, E. Blood brain barrier and inflammation in depression. Neurobiol Dis. (2022) 175:105926. doi: 10.1016/j.nbd.2022.105926
5. Maes, M, Berk, M, Goehler, L, Song, C, Anderson, G, Gałecki, P, et al. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. (2012) 10:1–19. doi: 10.1186/1741-7015-10-66
6. Osimo, EF, Baxter, LJ, Lewis, G, Jones, PB, and Khandaker, GM. Prevalence of low-grade inflammation in depression: a systematic review and meta-analysis of CRP levels. Psychol Med. (2019) 49:1958–70. doi: 10.1017/S0033291719001454
7. Gazal, M, Souza, LD, Fucolo, BA, Wiener, CD, Silva, RA, Pinheiro, RT, et al. The impact of cognitive behavioral therapy on IL-6 levels in unmedicated women experiencing the first episode of depression: a pilot study. Psychiatry Res. (2013) 209:742–5. doi: 10.1016/j.psychres.2013.03.002
8. Pariante, CM. Increased inflammation in depression: a little in all, or a lot in a few? Am J Psychiatry. (2021) 178:1077–9. doi: 10.1176/appi.ajp.2021.21101043
9. Felger, JC, Haroon, E, Patel, TA, Goldsmith, DR, Wommack, EC, Woolwine, BJ, et al. What does plasma CRP tell us about peripheral and central inflammation in depression? Mol Psychiatry. (2020) 25:1301–11. doi: 10.1038/s41380-018-0096-3
10. Hodes, GE, Kana, V, Menard, C, Merad, M, and Russo, SJ. Neuroimmune mechanisms of depression. Nat Neurosci. (2015) 18:1386–93. doi: 10.1038/nn.4113
11. Hodes, GE, Pfau, ML, Leboeuf, M, Golden, SA, Christoffel, DJ, et al. Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci U S A. (2014) 111:16136–41. doi: 10.1073/pnas.1415191111
12. Goshen, I, Kreisel, T, Ben-Menachem-Zidon, O, Licht, T, Weidenfeld, J, Ben-Hur, T, et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry. (2008) 13:717–28. doi: 10.1038/sj.mp.4002055
13. Lu, CL, Ren, J, Mo, JW, Fan, J, Guo, F, Chen, LY, et al. Glucocorticoid receptor-dependent astrocytes mediate stress vulnerability. Biol Psychiatry. (2022) 92:204–15. doi: 10.1016/j.biopsych.2021.11.022
14. van der Knaap, LJ, Oldehinkel, AJ, Verhulst, FC, van Oort, FV, and Riese, H. Glucocorticoid receptor gene methylation and HPA-axis regulation in adolescents. The TRAILS study. Psychoneuroendocrinology. (2015) 58:46–50. doi: 10.1016/j.psyneuen.2015.04.012
15. Unemura, K, Kume, T, Kondo, M, Maeda, Y, Izumi, Y, and Akaike, A. Glucocorticoids decrease astrocyte numbers by reducing glucocorticoid receptor expression in vitro and in vivo. J Pharmacol Sci. (2012) 119:30–9. doi: 10.1254/jphs.12047fp
16. Mikulska, J, Juszczyk, G, Gawronska-Grzywacz, M, and Herbet, M. HPA Axis in the Pathomechanism of depression and schizophrenia: new therapeutic strategies based on its participation. Brain Sci. (2021) 11:1298. doi: 10.3390/brainsci11101298
17. Alacreu-Crespo, A, Olie, E, Guillaume, S, Girod, C, Cazals, A, Chaudieu, I, et al. Dexamethasone suppression test May predict more severe/violent suicidal behavior. Front Psych. (2020) 11:97. doi: 10.3389/fpsyt.2020.00097
18. Nandam, LS, Brazel, M, Zhou, M, and Jhaveri, DJ. Cortisol and major depressive disorder-translating findings from humans to animal models and Back. Front Psych. (2019) 10:974. doi: 10.3389/fpsyt.2019.00974
19. The dexamethasone suppression test: The dexamethasone suppression test: an overview of its current status in psychiatry. The APA task force on laboratory tests in psychiatry. Am J Psychiatry. (1987) 144:1253–62. doi: 10.1176/ajp.144.10.1253
20. Miller, AH, and Raison, CL. Burning down the house: reinventing drug discovery in psychiatry for the development of targeted therapies. Mol Psychiatry. (2022) 28:68–75. doi: 10.1038/s41380-022-01887-y
21. Meynen, G, Unmehopa, UA, van Heerikhuize, JJ, Hofman, MA, Swaab, DF, and Hoogendijk, WJ. Increased arginine vasopressin mRNA expression in the human hypothalamus in depression: a preliminary report. Biol Psychiatry. (2006) 60:892–5. doi: 10.1016/j.biopsych.2005.12.010
22. Chaki, S. Vasopressin V1B receptor antagonists as potential antidepressants. Int J Neuropsychopharmacol. (2021) 24:450–63. doi: 10.1093/ijnp/pyab013
23. Fries, GR, Saldana, VA, Finnstein, J, and Rein, T. Molecular pathways of major depressive disorder converge on the synapse. Mol Psychiatry. (2023) 28:284–97. doi: 10.1038/s41380-022-01806-1
24. Roh, JS, and Sohn, DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. (2018) 18:e27. doi: 10.4110/in.2018.18.e27
25. Franklin, TC, Xu, C, and Duman, RS. Depression and sterile inflammation: essential role of danger associated molecular patterns. Brain Behav Immun. (2018) 72:2–13. doi: 10.1016/j.bbi.2017.10.025
26. Wang, CH, Gu, JY, Zhang, XL, Dong, J, Yang, J, Zhang, YL, et al. Venlafaxine ameliorates the depression-like behaviors and hippocampal S100B expression in a rat depression model. Behav Brain Funct. (2016) 12:34. doi: 10.1186/s12993-016-0116-x
27. Dogan, KH, Unaldi, M, and Demirci, S. Evaluation of postmortem cerebrospinal fluid S100B protein and serotonin levels: comparison of suicidal versus nonsuicidal deaths in Konya, Turkey. J Forensic Sci. (2016) 61:1285–91. doi: 10.1111/1556-4029.13124
28. Li, X, Wilder-Smith, CH, Kan, ME, Lu, J, Cao, Y, and Wong, RK. Combat-training stress in soldiers increases S100B, a marker of increased blood-brain-barrier permeability, and induces immune activation. Neuro Endocrinol Lett. (2014) 35:58–63.
29. Kheirouri, S, Ebrahimi, E, and Alizadeh, M. Association of S100B serum levels with metabolic syndrome and its components. Acta Medica Port. (2018) 31:201–6. doi: 10.20344/amp.9073
30. Imaizumi, T, Toda, T, Maekawa, M, Sakurai, D, Hagiwara, Y, Yoshida, Y, et al. Identifying high-risk population of depression: association between metabolic syndrome and depression using a health checkup and claims database. Sci Rep. (2022) 12:18577. doi: 10.1038/s41598-022-22048-9
31. Steiner, J, Bernstein, HG, Schiltz, K, Haase, T, Meyer-Lotz, G, Dobrowolny, H, et al. Decrease of serum S100B during an oral glucose tolerance test correlates inversely with the insulin response. Psychoneuroendocrinology. (2014) 39:33–8. doi: 10.1016/j.psyneuen.2013.10.001
32. Detka, J, and Glombik, K. Insights into a possible role of glucagon-like peptide-1 receptor agonists in the treatment of depression. Pharmacol Rep. (2021) 73:1020–32. doi: 10.1007/s43440-021-00274-8
33. Dubrez, L, Causse, S, Borges Bonan, N, Dumetier, B, and Garrido, C. Heat-shock proteins: chaperoning DNA repair. Oncogene. (2020) 39:516–29. doi: 10.1038/s41388-019-1016-y
34. Bobkova, NV, Garbuz, DG, Nesterova, I, Medvinskaya, N, Samokhin, A, Alexandrova, I, et al. Therapeutic effect of exogenous hsp70 in mouse models of Alzheimer’s disease. J Alzheimers Dis. (2014) 38:425–35. doi: 10.3233/JAD-130779
35. Hulina, A, Grdic Rajkovic, M, Jaksic Despot, D, Jelic, D, Dojder, A, Cepelak, I, et al. Extracellular Hsp70 induces inflammation and modulates LPS/LTA-stimulated inflammatory response in THP-1 cells. Cell Stress Chaperones. (2018) 23:373–84. doi: 10.1007/s12192-017-0847-0
36. Zhong, JM, Wu, SY, Bai, J, Guo, Q, Tao, J, Chen, H, et al. Antidepressant effect of geranylgeranylacetone in a chronic mild stress model of depression and its possible mechanism. Exp Ther Med. (2012) 4:627–32. doi: 10.3892/etm.2012.669
37. Hetz, C, Zhang, K, and Kaufman, RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. (2020) 21:421–38. doi: 10.1038/s41580-020-0250-z
38. Chipurupalli, S, Samavedam, U, and Robinson, N. Crosstalk between ER stress, autophagy and inflammation. Front Med. (2021) 8:758311. doi: 10.3389/fmed.2021.758311
39. Timberlake, MA 2nd, and Dwivedi, Y. Altered expression of endoplasmic reticulum stress associated genes in Hippocampus of learned helpless rats: relevance to depression pathophysiology. Front Pharmacol. (2015) 6:319. doi: 10.3389/fphar.2015.00319
40. Kim, HM, Do, CH, and Lee, DH. Combined effects of multiple endoplasmic reticulum stresses on cytokine secretion in macrophage. Biomol Ther (Seoul). (2012) 20:346–51. doi: 10.4062/biomolther.2012.20.3.346
41. Shi, M, Chen, F, Chen, Z, Yang, W, Yue, S, Zhang, J, et al. Sigma-1 receptor: a potential therapeutic target for traumatic brain injury. Front Cell Neurosci. (2021) 15:685201. doi: 10.3389/fncel.2021.685201
42. Slack, RJ, Mills, R, and Mackinnon, AC. The therapeutic potential of galectin-3 inhibition in fibrotic disease. Int J Biochem Cell Biol. (2021) 130:105881. doi: 10.1016/j.biocel.2020.105881
43. Burguillos, MA, Svensson, M, Schulte, T, Boza-Serrano, A, Garcia-Quintanilla, A, Kavanagh, E, et al. Microglia-secreted Galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. (2015) 10:1626–38. doi: 10.1016/j.celrep.2015.02.012
44. Melin, EO, Dereke, J, Thunander, M, and Hillman, M. Depression in type 1 diabetes was associated with high levels of circulating galectin-3. Endocr Connect. (2018) 7:819–28. doi: 10.1530/EC-18-0108
45. King, DR, Salako, DC, Arthur-Bentil, SK, Rubin, AE, Italiya, JB, Tan, JS, et al. Relationship between novel inflammatory biomarker galectin-3 and depression symptom severity in a large community-based sample. J Affect Disord. (2021) 281:384–9. doi: 10.1016/j.jad.2020.12.050
46. Kim, YK, Kim, OY, and Song, J. Alleviation of depression by glucagon-like peptide 1 through the regulation of Neuroinflammation, neurotransmitters, neurogenesis, and synaptic function. Front Pharmacol. (2020) 11:1270. doi: 10.3389/fphar.2020.01270
47. Guo, H, Callaway, JB, and Ting, JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
48. Kelley, N, Jeltema, D, Duan, Y, and He, Y. The NLRP3 Inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:328. doi: 10.3390/ijms20133328
49. Koo, JW, and Duman, RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A. (2008) 105:751–6. doi: 10.1073/pnas.0708092105
50. Koo, JW, and Duman, RS. Evidence for IL-1 receptor blockade as a therapeutic strategy for the treatment of depression. Curr Opin Investigat Drugs. (2009) 10:664.
51. Ferentinos, P, Maratou, E, Antoniou, A, Serretti, A, Smyrnis, N, and Moutsatsou, P. Interleukin-1 Beta in peripheral blood mononuclear cell lysates as a longitudinal biomarker of response to antidepressants: a pilot study. Front Psych. (2021) 12:801738. doi: 10.3389/fpsyt.2021.801738
52. Wu, D, Zhang, G, Zhao, C, Yang, Y, Miao, Z, and Xu, X. Interleukin-18 from neurons and microglia mediates depressive behaviors in mice with post-stroke depression. Brain Behav Immun. (2020) 88:411–20. doi: 10.1016/j.bbi.2020.04.004
53. Kang, HJ, Bae, KY, Kim, SW, Kim, JT, Park, MS, Cho, KH, et al. Effects of interleukin-6, interleukin-18, and statin use, evaluated at acute stroke, on post-stroke depression during 1-year follow-up. Psychoneuroendocrinology. (2016) 72:156–60. doi: 10.1016/j.psyneuen.2016.07.001
54. Bluhm, R, Williamson, P, Lanius, R, Theberge, J, Densmore, M, Bartha, R, et al. Resting state default-mode network connectivity in early depression using a seed region-of-interest analysis: decreased connectivity with caudate nucleus. Psychiatry Clin Neurosci. (2009) 63:754–61. doi: 10.1111/j.1440-1819.2009.02030.x
55. Du, X, Zou, S, Yue, Y, Fang, X, Wu, Y, Wu, S, et al. Peripheral Interleukin-18 is negatively correlated with abnormal brain activity in patients with depression: a resting-state fMRI study. BMC Psychiatry. (2022) 22:531. doi: 10.1186/s12888-022-04176-8
56. Kalkman, HO. Inhibition of microglial GSK3beta activity is common to different kinds of antidepressants: a proposal for an in vitro screen to detect novel antidepressant principles. Biomedicine. (2023) 11:806. doi: 10.3390/biomedicines11030806
57. Garate, I, Garcia-Bueno, B, Madrigal, JL, Caso, JR, Alou, L, Gomez-Lus, ML, et al. Stress-induced neuroinflammation: role of the toll-like receptor-4 pathway. Biol Psychiatry. (2013) 73:32–43. doi: 10.1016/j.biopsych.2012.07.005
58. Zhang, K, Lin, W, Zhang, J, Zhao, Y, Wang, X, and Zhao, M. Effect of toll-like receptor 4 on depressive-like behaviors induced by chronic social defeat stress. Brain Behav. (2020) 10:e01525. doi: 10.1002/brb3.1525
59. Keri, S, Szabo, C, and Kelemen, O. Expression of toll-like receptors in peripheral blood mononuclear cells and response to cognitive-behavioral therapy in major depressive disorder. Brain Behav Immun. (2014) 40:235–43. doi: 10.1016/j.bbi.2014.03.020
60. Pandey, GN, Rizavi, HS, Ren, X, Bhaumik, R, and Dwivedi, Y. Toll-like receptors in the depressed and suicide brain. J Psychiatr Res. (2014) 53:62–8. doi: 10.1016/j.jpsychires.2014.01.021
61. Miller, AH, Haroon, E, Raison, CL, and Felger, JC. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety. (2013) 30:297–306. doi: 10.1002/da.22084
62. Osimo, EF, Pillinger, T, Rodriguez, IM, Khandaker, GM, Pariante, CM, and Howes, OD. Inflammatory markers in depression: a meta-analysis of mean differences and variability in 5,166 patients and 5,083 controls. Brain Behav Immun. (2020) 87:901–9. doi: 10.1016/j.bbi.2020.02.010
63. Chiu, WC, Su, YP, Su, KP, and Chen, PC. Recurrence of depressive disorders after interferon-induced depression. Transl Psychiatry. (2017) 7:e1026. doi: 10.1038/tp.2016.274
64. Yamano, M, and Tohyama, M. Distribution of corticotropin-releasing factor and calcitonin gene-related peptide in the developing mouse cerebellum. Neurosci Res. (1994) 19:387–96. doi: 10.1016/0168-0102(94)90080-9
65. Zhu, J, Yamane, H, and Paul, WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
66. Ghosh, R, Kumar, PK, Mitra, P, Purohit, P, Nebhinani, N, and Sharma, P. Circulating T helper 17 and IFN-gamma positive Th17 cells in major depressive disorder. Behav Brain Res. (2020) 394:112811. doi: 10.1016/j.bbr.2020.112811
67. Martino, M, Rocchi, G, Escelsior, A, and Fornaro, M. Immunomodulation mechanism of antidepressants: interactions between serotonin/norepinephrine balance and Th1/Th2 balance. Curr Neuropharmacol. (2012) 10:97–123. doi: 10.2174/157015912800604542
68. Myint, AM, Leonard, BE, Steinbusch, HW, and Kim, YK. Th1, Th2, and Th3 cytokine alterations in major depression. J Affect Disord. (2005) 88:167–73. doi: 10.1016/j.jad.2005.07.008
69. Kubera, M, Lin, AH, Kenis, G, Bosmans, E, van Bockstaele, D, and Maes, M. Anti-inflammatory effects of antidepressants through suppression of the interferon-gamma/interleukin-10 production ratio. J Clin Psychopharmacol. (2001) 21:199–206. doi: 10.1097/00004714-200104000-00012
70. Milovanovic, J, Arsenijevic, A, Stojanovic, B, Kanjevac, T, Arsenijevic, D, Radosavljevic, G, et al. Interleukin-17 in chronic inflammatory neurological diseases. Front Immunol. (2020) 11:947. doi: 10.3389/fimmu.2020.00947
71. Davami, MH, Baharlou, R, Ahmadi Vasmehjani, A, Ghanizadeh, A, Keshtkar, M, Dezhkam, I, et al. Elevated IL-17 and TGF-beta serum levels: a positive correlation between T-helper 17 cell-related pro-inflammatory responses with major depressive disorder. Basic Clin Neurosci. (2016) 7:137–42. doi: 10.15412/J.BCN.03070207
72. Jha, MK, Minhajuddin, A, Gadad, BS, Greer, TL, Mayes, TL, and Trivedi, MH. Interleukin 17 selectively predicts better outcomes with bupropion-SSRI combination: novel T cell biomarker for antidepressant medication selection. Brain Behav Immun. (2017) 66:103–10. doi: 10.1016/j.bbi.2017.07.005
73. Schiweck, C, Aichholzer, M, Reif, A, and Thanarajah, SE. Targeting IL-17A signaling in suicidality, promise or the long arm of coincidence? Evidence in psychiatric populations revisited. J Affect Disord Rep. (2022) 100454
74. Lebwohl, M, Leonardi, C, Wu, JJ, Armstrong, A, Rawnsley, N, Merchant, M, et al. Two-year US Pharmacovigilance report on Brodalumab. Dermatol Ther (Heidelb). (2021) 11:173–80. doi: 10.1007/s13555-020-00472-x
75. Kim, J, Suh, YH, and Chang, KA. Interleukin-17 induced by cumulative mild stress promoted depression-like behaviors in young adult mice. Mol Brain. (2021) 14:11. doi: 10.1186/s13041-020-00726-x
76. Liu, D, Zhong, Z, and Karin, M. NF-κB: a double-edged sword controlling inflammation. Biomedicine. (2022) 10:1250. doi: 10.3390/biomedicines10061250
77. Wang, Y, Xu, J, Liu, Y, Li, Z, and Li, X. TLR4-NF-kappaB signal involved in depressive-like behaviors and cytokine expression of frontal cortex and Hippocampus in stressed C57BL/6 and Ob/Ob mice. Neural Plast. (2018) 2018:7254016. doi: 10.1155/2018/7254016
78. Munhoz, CD, Lepsch, LB, Kawamoto, EM, Malta, MB, Lima Lde, S, Avellar, MC, et al. Chronic unpredictable stress exacerbates lipopolysaccharide-induced activation of nuclear factor-kappaB in the frontal cortex and hippocampus via glucocorticoid secretion. J Neurosci. (2006) 26:3813–20. doi: 10.1523/JNEUROSCI.4398-05.2006
79. Cho, JH, Irwin, MR, Eisenberger, NI, Lamkin, DM, and Cole, SW. Transcriptomic predictors of inflammation-induced depressed mood. Neuropsychopharmacology. (2019) 44:923–9. doi: 10.1038/s41386-019-0316-9
80. Beurel, E, Toups, M, and Nemeroff, CB. The bidirectional relationship of depression and inflammation: double trouble. Neuron. (2020) 107:234–56. doi: 10.1016/j.neuron.2020.06.002
81. Menard, C, Pfau, ML, Hodes, GE, Kana, V, Wang, VX, Bouchard, S, et al. Social stress induces neurovascular pathology promoting depression. Nat Neurosci. (2017) 20:1752–60. doi: 10.1038/s41593-017-0010-3
82. Hawkins, BT, and Davis, TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. (2005) 57:173–85. doi: 10.1124/pr.57.2.4
83. Sun, Y, Koyama, Y, and Shimada, S. Inflammation from peripheral organs to the brain: how does systemic inflammation cause Neuroinflammation? Front Aging Neurosci. (2022) 14:903455. doi: 10.3389/fnagi.2022.903455
84. Quan, N. Immune-to-brain signaling: how important are the blood-brain barrier-independent pathways? Mol Neurobiol. (2008) 37:142–52. doi: 10.1007/s12035-008-8026-z
85. Santha, P, Veszelka, S, Hoyk, Z, Meszaros, M, Walter, FR, Toth, AE, et al. Restraint stress-induced morphological changes at the blood-brain barrier in adult rats. Front Mol Neurosci. (2015) 8:88. doi: 10.3389/fnmol.2015.00088
86. Greene, C, Kealy, J, Humphries, MM, Gong, Y, Hou, J, Hudson, N, et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol Psychiatry. (2018) 23:2156–66. doi: 10.1038/mp.2017.156
87. Yu, Y, Wang, J, and Huang, X. The anti-depressant effects of a novel PDE4 inhibitor derived from resveratrol. Pharm Biol. (2021) 59:418–23. doi: 10.1080/13880209.2021.1907422
88. Pekny, M, and Pekna, M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. (2014) 94:1077–98. doi: 10.1152/physrev.00041.2013
89. Afridi, R, and Suk, K. Neuroinflammatory basis of depression: learning from experimental models. Front Cell Neurosci. (2021) 15:691067. doi: 10.3389/fncel.2021.691067
90. Guo, S, Wang, H, and Yin, Y. Microglia polarization from M1 to M2 in neurodegenerative diseases. Front Aging Neurosci. (2022) 14:815347. doi: 10.3389/fnagi.2022.815347
91. Yao, R, Pan, R, Shang, C, Li, X, Cheng, J, Xu, J, et al. Translocator protein 18 kDa (TSPO) deficiency inhibits microglial activation and impairs mitochondrial function. Front Pharmacol. (2020) 11:986. doi: 10.3389/fphar.2020.00986
92. Setiawan, E, Wilson, AA, Mizrahi, R, Rusjan, PM, Miler, L, Rajkowska, G, et al. Role of Translocator protein density, a marker of Neuroinflammation, in the brain during major depressive episodes. JAMA Psychiat. (2015) 72:268–75. doi: 10.1001/jamapsychiatry.2014.2427
93. Eggerstorfer, B, Kim, JH, Cumming, P, Lanzenberger, R, and Gryglewski, G. Meta-analysis of molecular imaging of translocator protein in major depression. Front Mol Neurosci. (2022) 15:981442. doi: 10.3389/fnmol.2022.981442
94. Dean, OM, Maes, M, Ashton, M, Berk, L, Kanchanatawan, B, Sughondhabirom, A, et al. Protocol and rationale-the efficacy of minocycline as an adjunctive treatment for major depressive disorder: a double blind, randomised, placebo controlled trial. Clin Psychopharmacol Neurosci. (2014) 12:180–8. doi: 10.9758/cpn.2014.12.3.180
95. Araya-Callis, C, Hiemke, C, Abumaria, N, and Flugge, G. Chronic psychosocial stress and citalopram modulate the expression of the glial proteins GFAP and NDRG2 in the hippocampus. Psychopharmacology. (2012) 224:209–22. doi: 10.1007/s00213-012-2741-x
96. Shu, X, Sun, Y, Sun, X, Zhou, Y, Bian, Y, Shu, Z, et al. The effect of fluoxetine on astrocyte autophagy flux and injured mitochondria clearance in a mouse model of depression. Cell Death Dis. (2019) 10:577. doi: 10.1038/s41419-019-1813-9
97. Cobb, JA, O’Neill, K, Milner, J, Mahajan, GJ, Lawrence, TJ, May, WL, et al. Density of GFAP-immunoreactive astrocytes is decreased in left hippocampi in major depressive disorder. Neuroscience. (2016) 316:209–20. doi: 10.1016/j.neuroscience.2015.12.044
98. Michel, M, Fiebich, BL, Kuzior, H, Meixensberger, S, Berger, B, Maier, S, et al. Increased GFAP concentrations in the cerebrospinal fluid of patients with unipolar depression. Transl Psychiatry. (2021) 11:308. doi: 10.1038/s41398-021-01423-6
99. Steinacker, P, Al Shweiki, MR, Oeckl, P, Graf, H, Ludolph, AC, Schonfeldt-Lecuona, C, et al. Glial fibrillary acidic protein as blood biomarker for differential diagnosis and severity of major depressive disorder. J Psychiatr Res. (2021) 144:54–8. doi: 10.1016/j.jpsychires.2021.09.012
100. Sarandol, A, Sarandol, E, Eker, SS, Erdinc, S, Vatansever, E, and Kirli, S. Major depressive disorder is accompanied with oxidative stress: short-term antidepressant treatment does not alter oxidative-antioxidative systems. Hum Psychopharmacol. (2007) 22:67–73. doi: 10.1002/hup.829
101. Schieber, M, and Chandel, NS. ROS function in redox signaling and oxidative stress. Curr Biol. (2014) 24:R453–62. doi: 10.1016/j.cub.2014.03.034
102. Somani, A, Singh, AK, Gupta, B, Nagarkoti, S, Dalal, PK, and Dikshit, M. Oxidative and Nitrosative stress in major depressive disorder: a case control study. Brain Sci. (2022) 12:144. doi: 10.3390/brainsci12020144
103. Liu, T, Zhong, S, Liao, X, Chen, J, He, T, Lai, S, et al. A Meta-analysis of oxidative stress markers in depression. PLoS One. (2015) 10:e0138904. doi: 10.1371/journal.pone.0138904
104. Rybka, J, Kedziora-Kornatowska, K, Banas-Lezanska, P, Majsterek, I, Carvalho, LA, Cattaneo, A, et al. Interplay between the pro-oxidant and antioxidant systems and proinflammatory cytokine levels, in relation to iron metabolism and the erythron in depression. Free Radic Biol Med. (2013) 63:187–94. doi: 10.1016/j.freeradbiomed.2013.05.019
105. Jorgensen, A, Krogh, J, Miskowiak, K, Bolwig, TG, Kessing, LV, Fink-Jensen, A, et al. Systemic oxidatively generated DNA/RNA damage in clinical depression: associations to symptom severity and response to electroconvulsive therapy. J Affect Disord. (2013) 149:355–62. doi: 10.1016/j.jad.2013.02.011
106. Maes, M, Mihaylova, I, Kubera, M, Leunis, JC, Twisk, FN, and Geffard, M. IgM-mediated autoimmune responses directed against anchorage epitopes are greater in Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) than in major depression. Metab Brain Dis. (2012) 27:415–23. doi: 10.1007/s11011-012-9316-8
107. Khanzode, SD, Dakhale, GN, Khanzode, SS, Saoji, A, and Palasodkar, R. Oxidative damage and major depression: the potential antioxidant action of selective serotonin re-uptake inhibitors. Redox Rep. (2003) 8:365–70. doi: 10.1179/135100003225003393
108. Dinkova-Kostova, AT, Kostov, RV, and Kazantsev, AG. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. (2018) 285:3576–90. doi: 10.1111/febs.14379
109. Bouvier, E, Brouillard, F, Molet, J, Claverie, D, Cabungcal, JH, Cresto, N, et al. Nrf2-dependent persistent oxidative stress results in stress-induced vulnerability to depression. Mol Psychiatry. (2017) 22:1795. doi: 10.1038/mp.2016.211
110. Martin-Hernandez, D, Caso, JR, Javier Meana, J, Callado, LF, Madrigal, JLM, Garcia-Bueno, B, et al. Intracellular inflammatory and antioxidant pathways in postmortem frontal cortex of subjects with major depression: effect of antidepressants. J Neuroinflammation. (2018) 15:251. doi: 10.1186/s12974-018-1294-2
111. Wu, S, Gao, Q, Zhao, P, Gao, Y, Xi, Y, Wang, X, et al. Sulforaphane produces antidepressant- and anxiolytic-like effects in adult mice. Behav Brain Res. (2016) 301:55–62. doi: 10.1016/j.bbr.2015.12.030
112. Lassale, C, Batty, GD, Baghdadli, A, Jacka, F, Sanchez-Villegas, A, Kivimaki, M, et al. Healthy dietary indices and risk of depressive outcomes: a systematic review and meta-analysis of observational studies. Mol Psychiatry. (2019) 24:965–86. doi: 10.1038/s41380-018-0237-8
113. Forstermann, U, and Sessa, WC. Nitric oxide synthases: regulation and function. Eur Heart J. (2012) 33:829–37. doi: 10.1093/eurheartj/ehr304
114. Wang, F, Yuan, Q, Chen, F, Pang, J, Pan, C, Xu, F, et al. Fundamental mechanisms of the cell death caused by Nitrosative stress. Front Cell Dev Biol. (2021) 9:742483. doi: 10.3389/fcell.2021.742483
115. Nobis, A, Zalewski, D, Samaryn, E, Maciejczyk, M, Zalewska, A, and Waszkiewicz, N. Urine 3-Nitrotyrosine and serum HDL as potential biomarkers of depression. J Clin Med. (2023) 12:377. doi: 10.3390/jcm12010377
116. Gao, YT, Panda, SP, Roman, LJ, Martásek, P, Ishimura, Y, and Masters, BSS. Oxygen metabolism by neuronal nitric-oxide synthase. J Biol Chem. (2007) 282:7921–9. doi: 10.1074/jbc.M609814200
117. Liu, W, Li, Q, Ye, B, Cao, H, Shen, F, Xu, Z, et al. Repeated nitrous oxide exposure exerts antidepressant-like effects through neuronal nitric oxide synthase activation in the medial prefrontal cortex. Front Psych. (2020) 11:837. doi: 10.3389/fpsyt.2020.00837
118. Narsapur, SL, and Naylor, GJ. Methylene blue. A possible treatment for manic depressive psychosis. J Affect Disord. (1983) 5:155–61. doi: 10.1016/0165-0327(83)90008-3
119. Finkel, MS, Laghrissi-Thode, F, Pollock, BG, and Rong, J. Paroxetine is a novel nitric oxide synthase inhibitor. Psychopharmacol Bull. (1996) 32:653–8.
120. Guimarães, MC, Guimarães, TM, Hallak, JE, Abrão, J, and Machado-de-Sousa, JP. Nitrous oxide as an adjunctive therapy in major depressive disorder: a randomized controlled double-blind pilot trial. Brazilian J Psychiatry. (2021) 43:484–93. doi: 10.1590/1516-4446-2020-1543
121. Nagele, P, Palanca, BJ, Gott, B, Brown, F, Barnes, L, Nguyen, T, et al. A phase 2 trial of inhaled nitrous oxide for treatment-resistant major depression. Sci Transl Med. (2021) 13:eabe1376. doi: 10.1126/scitranslmed.abe1376
122. Shao, J, Meng, L, Yang, Z, Yu, P, Song, L, Gao, Y, et al. Xenon produces rapid antidepressant-and anxiolytic-like effects in lipopolysaccharide-induced depression mice model. Neuroreport. (2020) 31:387–93. doi: 10.1097/WNR.0000000000001415
123. Joca, SRL, Sartim, AG, Roncalho, AL, Diniz, CFA, and Wegener, G. Nitric oxide signalling and antidepressant action revisited. Cell Tissue Res. (2019) 377:45–58. doi: 10.1007/s00441-018-02987-4
124. Wang, P, Kong, FZ, Hong, XH, Zhang, L, Zhao, WH, Yang, JC, et al. Neuronal nitric oxide synthase regulates depression-like behaviors in shortening-induced obese mice. Nutrients. (2022) 14:4302. doi: 10.3390/nu14204302
125. Zhou, QG, Hu, Y, Hua, Y, Hu, M, Luo, CX, Han, X, et al. Neuronal nitric oxide synthase contributes to chronic stress-induced depression by suppressing hippocampal neurogenesis. J Neurochem. (2007) 103:1843–54. doi: 10.1111/j.1471-4159.2007.04914.x
126. Miller, AH. Mechanisms of cytokine-induced behavioral changes: psychoneuroimmunology at the translational interface. Brain Behav Immun. (2009) 23:149–58. doi: 10.1016/j.bbi.2008.08.006
127. Fanet, H, Capuron, L, Castanon, N, Calon, F, and Vancassel, S. Tetrahydrobioterin (BH4) pathway: from metabolism to neuropsychiatry. Curr Neuropharmacol. (2021) 19:591–609. doi: 10.2174/1570159X18666200729103529
128. Haroon, E, Raison, CL, and Miller, AH. Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology. (2012) 37:137–62. doi: 10.1038/npp.2011.205
129. Sutinen, EM, Korolainen, MA, Häyrinen, J, Alafuzoff, I, Petratos, S, Salminen, A, et al. Interleukin-18 alters protein expressions of neurodegenerative diseases-linked proteins in human SH-SY5Y neuron-like cells. Front Cell Neurosci. (2014) 8:214. doi: 10.3389/fncel.2014.00214
130. Campbell, BM, Charych, E, Lee, AW, and Möller, T. Kynurenines in CNS disease: regulation by inflammatory cytokines. Front Neurosci. (2014) 8:12. doi: 10.3389/fnins.2014.00012
131. da Silveira, TL, Zamberlan, DC, Arantes, LP, Machado, ML, da Silva, TC, de Freitas Câmara, D, et al. Quinolinic acid and glutamatergic neurodegeneration in Caenorhabditis elegans. Neurotoxicology. (2018) 67:94–101. doi: 10.1016/j.neuro.2018.04.015
132. Lugo-Huitron, R, Ugalde Muniz, P, Pineda, B, Pedraza-Chaverri, J, Rios, C, and Perez-de la Cruz, V. Quinolinic acid: an endogenous neurotoxin with multiple targets. Oxidative Med Cell Longev. (2013) 2013:104024. doi: 10.1155/2013/104024
133. Bay-Richter, C, Linderholm, KR, Lim, CK, Samuelsson, M, Traskman-Bendz, L, Guillemin, GJ, et al. A role for inflammatory metabolites as modulators of the glutamate N-methyl-D-aspartate receptor in depression and suicidality. Brain Behav Immun. (2015) 43:110–7. doi: 10.1016/j.bbi.2014.07.012
134. Schwarcz, R, and Kohler, C. Differential vulnerability of central neurons of the rat to quinolinic acid. Neurosci Lett. (1983) 38:85–90. doi: 10.1016/0304-3940(83)90115-5
135. Kandilarova, S, Stoyanov, D, Sirakov, N, Maes, M, and Specht, K. Reduced grey matter volume in frontal and temporal areas in depression: contributions from voxel-based morphometry study. Acta Neuropsychiatr. (2019) 31:252–7. doi: 10.1017/neu.2019.20
136. Ogyu, K, Kubo, K, Noda, Y, Iwata, Y, Tsugawa, S, Omura, Y, et al. Kynurenine pathway in depression: a systematic review and meta-analysis. Neurosci Biobehav Rev. (2018) 90:16–25. doi: 10.1016/j.neubiorev.2018.03.023
137. Duda, P, Hajka, D, Wójcicka, O, Rakus, D, and Gizak, A. GSK3β: a master player in depressive disorder pathogenesis and treatment responsiveness. Cells. (2020) 9:727. doi: 10.3390/cells9030727
138. Verdonk, F, Petit, A-C, Abdel-Ahad, P, Vinckier, F, Jouvion, G, de Maricourt, P, et al. Microglial production of quinolinic acid as a target and a biomarker of the antidepressant effect of ketamine. Brain Behav Immun. (2019) 81:361–73. doi: 10.1016/j.bbi.2019.06.033
139. Han, K-M, and Ham, B-J. How inflammation affects the brain in depression: a review of functional and structural MRI studies. J Clin Neurol. (2021) 17:503–15. doi: 10.3988/jcn.2021.17.4.503
140. Capuron, L, Pagnoni, G, Drake, DF, Woolwine, BJ, Spivey, JR, Crowe, RJ, et al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry. (2012) 69:1044–53. doi: 10.1001/archgenpsychiatry.2011.2094
141. Holmes, SE, Scheinost, D, Finnema, SJ, Naganawa, M, Davis, MT, Della Gioia, N, et al. Lower synaptic density is associated with depression severity and network alterations. Nat Commun. (2019) 10:1–10. doi: 10.1038/s41467-019-09562-7
142. Rossi, R, Arjmand, S, Baerentzen, SL, Gjedde, A, and Landau, AM. Synaptic vesicle glycoprotein 2A: features and functions. Front Neurosci. (2022) 16:864514. doi: 10.3389/fnins.2022.864514
143. Garcia-Oscos, F, Salgado, H, Hall, S, Thomas, F, Farmer, GE, Bermeo, J, et al. The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biol Psychiatry. (2012) 71:574–82. doi: 10.1016/j.biopsych.2011.11.018
144. Ma, K, Xu, A, Cui, S, Sun, MR, Xue, YC, and Wang, JH. Impaired GABA synthesis, uptake and release are associated with depression-like behaviors induced by chronic mild stress. Transl Psychiatry. (2016) 6:e910. doi: 10.1038/tp.2016.181
145. Yao, HK, Guet-McCreight, A, Mazza, F, Moradi Chameh, H, Prevot, TD, Griffiths, JD, et al. Reduced inhibition in depression impairs stimulus processing in human cortical microcircuits. Cell Rep. (2022) 38:110232. doi: 10.1016/j.celrep.2021.110232
146. Kecskes, M, Henn-Mike, N, Agocs-Laboda, A, Szocs, S, Petyko, Z, and Varga, C. Somatostatin expressing GABAergic interneurons in the medial entorhinal cortex preferentially inhibit layer(III-V) pyramidal cells. Commun Biol. (2020) 3:754. doi: 10.1038/s42003-020-01496-x
147. Popoli, M, Yan, Z, McEwen, BS, and Sanacora, G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. (2011) 13:22–37. doi: 10.1038/nrn3138
148. Sohal, VS, and Rubenstein, JL. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry. (2019) 24:1248–57. doi: 10.1038/s41380-019-0426-0
149. Sukenik, N, Vinogradov, O, Weinreb, E, Segal, M, Levina, A, and Moses, E. Neuronal circuits overcome imbalance in excitation and inhibition by adjusting connection numbers. Proc Natl Acad Sci U S A. (2021) 118:9118. doi: 10.1073/pnas.2018459118
150. Huang, H, Chen, C, Rong, B, Wan, Q, Chen, J, Liu, Z, et al. Resting-state functional connectivity of salience network in schizophrenia and depression. Sci Rep. (2022) 12:11204. doi: 10.1038/s41598-022-15489-9
151. Duman, RS, Sanacora, G, and Krystal, JH. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron. (2019) 102:75–90. doi: 10.1016/j.neuron.2019.03.013
152. Yang, X, Bognar, J Jr, He, T, Mohammed, M, Niespodziany, I, Wolff, C, et al. Brivaracetam augments short-term depression and slows vesicle recycling. Epilepsia. (2015) 56:1899–909. doi: 10.1111/epi.13223
153. Friedman, AK, Juarez, B, Ku, SM, Zhang, H, Calizo, RC, Walsh, JJ, et al. KCNQ channel openers reverse depressive symptoms via an active resilience mechanism. Nat Commun. (2016) 7:1–7. doi: 10.1038/ncomms11671
154. Raison, CL, Rutherford, RE, Woolwine, BJ, Shuo, C, Schettler, P, Drake, DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiat. (2013) 70:31–41. doi: 10.1001/2013.jamapsychiatry.4
155. Kohler, O, Benros, ME, Nordentoft, M, Farkouh, ME, Iyengar, RL, Mors, O, et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiat. (2014) 71:1381–91. doi: 10.1001/jamapsychiatry.2014.1611
156. Mischoulon, D, Dunlop, BW, Kinkead, B, Schettler, PJ, Lamon-Fava, S, Rakofsky, JJ, et al. Omega-3 fatty acids for major depressive disorder with high inflammation: a randomized dose-finding clinical trial. J Clin Psychiatry. (2022) 83:14074. doi: 10.4088/JCP.21m14074
157. Bai, S, Guo, W, Feng, Y, Deng, H, Li, G, Nie, H, et al. Efficacy and safety of anti-inflammatory agents for the treatment of major depressive disorder: a systematic review and meta-analysis of randomised controlled trials. J Neurol Neurosurg Psychiatry. (2020) 91:21–32. doi: 10.1136/jnnp-2019-320912
158. Leonard, BE. Inflammation and depression: a causal or coincidental link to the pathophysiology? Acta Neuropsychiatr. (2018) 30:1–16. doi: 10.1017/neu.2016.69
159. Yang, H, and Chen, C. Cyclooxygenase-2 in synaptic signaling. Curr Pharm Des. (2008) 14:1443–51. doi: 10.2174/138161208784480144
160. Prabhakaran, J, Molotkov, A, Mintz, A, and Mann, JJ. Progress in PET imaging of Neuroinflammation targeting COX-2 enzyme. Molecules. (2021) 26:3208. doi: 10.3390/molecules26113208
161. He, Y, Han, Y, Liao, X, Zou, M, and Wang, Y. Biology of cyclooxygenase-2: an application in depression therapeutics. Front Psych. (2022) 13:1037588. doi: 10.3389/fpsyt.2022.1037588
162. Guo, JY, Li, CY, Ruan, YP, Sun, M, Qi, XL, Zhao, BS, et al. Chronic treatment with celecoxib reverses chronic unpredictable stress-induced depressive-like behavior via reducing cyclooxygenase-2 expression in rat brain. Eur J Pharmacol. (2009) 612:54–60. doi: 10.1016/j.ejphar.2009.03.076
163. Majd, M, Hashemian, F, Hosseini, SM, Vahdat Shariatpanahi, M, and Sharifi, A. A randomized, double-blind, placebo-controlled trial of Celecoxib augmentation of sertraline in treatment of drug-naive depressed women: a pilot study. Iran J Pharm Res. (2015) 14:891–9.
164. Calder, PC. Marine omega-3 fatty acids and inflammatory processes: effects, mechanisms and clinical relevance. Biochim Biophys Acta. (2015) 1851:469–84. doi: 10.1016/j.bbalip.2014.08.010
165. Giacobbe, J, Benoiton, B, Zunszain, P, Pariante, CM, and Borsini, A. The anti-inflammatory role of Omega-3 polyunsaturated fatty acids metabolites in pre-clinical models of psychiatric, neurodegenerative, and neurological disorders. Front Psych. (2020) 11:122. doi: 10.3389/fpsyt.2020.00122
166. Chang, JP, Lai, HC, Yang, HT, Su, WP, Peng, CY, Galecki, P, et al. Polyunsaturated fatty acids levels and initial presentation of somatic symptoms induced by interferon-alpha therapy in patients with chronic hepatitis C viral infection. Nutr Neurosci. (2017) 20:291–6. doi: 10.1080/1028415X.2015.1123378
167. Su, KP, Lai, HC, Yang, HT, Su, WP, Peng, CY, Chang, JP, et al. Omega-3 fatty acids in the prevention of interferon-alpha-induced depression: results from a randomized, controlled trial. Biol Psychiatry. (2014) 76:559–66. doi: 10.1016/j.biopsych.2014.01.008
168. Wu, H, Lv, W, Pan, Q, Kalavagunta, PK, Liu, Q, Qin, G, et al. Simvastatin therapy in adolescent mice attenuates HFD-induced depression-like behavior by reducing hippocampal neuroinflammation. J Affect Disord. (2019) 243:83–95. doi: 10.1016/j.jad.2018.09.022
169. Hai-Na, Z, Xu-Ben, Y, Cong-Rong, T, Yan-Cheng, C, Fan, Y, Lei-Mei, X, et al. Atorvastatin ameliorates depressive behaviors and neuroinflammatory in streptozotocin-induced diabetic mice. Psychopharmacology. (2020) 237:695–705. doi: 10.1007/s00213-019-05406-w
170. De Giorgi, R, De Crescenzo, F, Rizzo Pesci, N, Martens, M, Howard, W, Cowen, PJ, et al. Statins for major depressive disorder: a systematic review and meta-analysis of randomized controlled trials. PLoS One. (2021) 16:e0249409. doi: 10.1371/journal.pone.0249409
171. Abbasi, SH, Mohammadinejad, P, Shahmansouri, N, Salehiomran, A, Beglar, AA, Zeinoddini, A, et al. Simvastatin versus atorvastatin for improving mild to moderate depression in post-coronary artery bypass graft patients: a double-blind, placebo-controlled, randomized trial. J Affect Disord. (2015) 183:149–55. doi: 10.1016/j.jad.2015.04.049
172. Redlich, C, Berk, M, Williams, LJ, Sundquist, J, Sundquist, K, and Li, X. Statin use and risk of depression: a Swedish national cohort study. BMC Psychiatry. (2014) 14:348. doi: 10.1186/s12888-014-0348-y
173. Kappelmann, N, Lewis, G, Dantzer, R, Jones, P, and Khandaker, G. Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol Psychiatry. (2018) 23:335–43. doi: 10.1038/mp.2016.167
174. Sun, Y, Wang, D, Salvadore, G, Hsu, B, Curran, M, Casper, C, et al. The effects of interleukin-6 neutralizing antibodies on symptoms of depressed mood and anhedonia in patients with rheumatoid arthritis and multicentric Castleman’s disease. Brain Behav Immun. (2017) 66:156–64. doi: 10.1016/j.bbi.2017.06.014
175. Griffiths, CEM, Fava, M, Miller, AH, Russell, J, Ball, SG, Xu, W, et al. Impact of Ixekizumab treatment on depressive symptoms and systemic inflammation in patients with moderate-to-severe psoriasis: an integrated analysis of three phase 3 clinical studies. Psychother Psychosom. (2017) 86:260–7. doi: 10.1159/000479163
176. Rustenhoven, J, Aalderink, M, Scotter, EL, Oldfield, RL, Bergin, PS, Mee, EW, et al. TGF-beta1 regulates human brain pericyte inflammatory processes involved in neurovasculature function. J Neuroinflammation. (2016) 13:37. doi: 10.1186/s12974-016-0503-0
177. Liu, Z, Chen, HQ, Huang, Y, Qiu, YH, and Peng, YP. Transforming growth factor-beta1 acts via TbetaR-I on microglia to protect against MPP(+)-induced dopaminergic neuronal loss. Brain Behav Immun. (2016) 51:131–43. doi: 10.1016/j.bbi.2015.08.006
178. Zhang, K, Yang, C, Chang, L, Sakamoto, A, Suzuki, T, Fujita, Y, et al. Essential role of microglial transforming growth factor-β1 in antidepressant actions of (R)-ketamine and the novel antidepressant TGF-β1. Transl Psychiatry. (2020) 10:1–12. doi: 10.1038/s41398-020-0733-x
179. Wei, Y, Chang, L, and Hashimoto, K. Intranasal administration of transforming growth factor-beta1 elicits rapid-acting antidepressant-like effects in a chronic social defeat stress model: a role of TrkB signaling. Eur Neuropsychopharmacol. (2021) 50:55–63. doi: 10.1016/j.euroneuro.2021.04.010
180. Jovanovic, AM, Mitkovic-Voncina, M, Kostic, M, Jeremic, M, Todorovic, J, Popadic, D, et al. Childhood maltreatment correlates with higher concentration of transforming growth factor beta (TGF-beta) in adult patients with major depressive disorder. Psychiatry Res. (2021) 301:113987. doi: 10.1016/j.psychres.2021.113987
181. Caraci, F, Spampinato, SF, Morgese, MG, Tascedda, F, Salluzzo, MG, Giambirtone, MC, et al. Neurobiological links between depression and AD: the role of TGF-β1 signaling as a new pharmacological target. Pharmacol Res. (2018) 130:374–84.
182. Rush, G, O’Donovan, A, Nagle, L, Conway, C, McCrohan, A, O’Farrelly, C, et al. Alteration of immune markers in a group of melancholic depressed patients and their response to electroconvulsive therapy. J Affect Disord. (2016) 205:60–8. doi: 10.1016/j.jad.2016.06.035
183. Houlton, J, Abumaria, N, Hinkley, SFR, and Clarkson, AN. Therapeutic potential of Neurotrophins for repair after brain injury: a helping hand from biomaterials. Front Neurosci. (2019) 13:790. doi: 10.3389/fnins.2019.00790
184. Mitre, M, Mariga, A, and Chao, MV. Neurotrophin signalling: novel insights into mechanisms and pathophysiology. Clin Sci. (2017) 131:13–23. doi: 10.1042/CS20160044
185. Olivares, D, Deshpande, VK, Shi, Y, Lahiri, DK, Greig, NH, Rogers, JT, et al. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr Alzheimer Res. (2012) 9:746–58. doi: 10.2174/156720512801322564
186. Tian, H, Hu, Z, Xu, J, and Wang, C. The molecular pathophysiology of depression and the new therapeutics. MedComm. (2022) 3:e156. doi: 10.1002/mco2.156
Keywords: depression, stress neuroinflammation, cortisol, HPA axis, neuroplasticity Glutamate, GABA
Citation: Hassamal S (2023) Chronic stress, neuroinflammation, and depression: an overview of pathophysiological mechanisms and emerging anti-inflammatories. Front. Psychiatry. 14:1130989. doi: 10.3389/fpsyt.2023.1130989
Edited by:
Stefania Schiavone, University of Foggia, ItalyReviewed by:
Karina S. MacDowell, Complutense University of Madrid, SpainAnna Brancato, University of Palermo, Italy
Copyright © 2023 Hassamal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sameer Hassamal, c2hhc3NhbWFAZ21haWwuY29t