 Kang Chen
Kang Chen Kun Wang
Kun Wang Tianhui Wang1,2*
Tianhui Wang1,2*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 20 September 2022
Sec. Aging Psychiatry
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.967683
This article is part of the Research TopicReviews in Psychiatry 2022: Aging PsychiatryView all 5 articles
Despite being one of the greatest global challenges for health and social care in the 21st century, Alzheimer’s disease (AD) lacks specific medicine. Irisin, an exercise-generated muscle factor, emerges as a potential hormone for AD prevention and treatment because of its role in promoting the browning of white adipose tissue, accelerating energy expenditure, regulating energy metabolism, and improving insulin resistance. The study reviews classic hallmarks of AD and irisin’s physiology before discussing the possible mechanism by which irisin protects against AD in terms of its effects related to molecular biology and cellular biology. Results reveal that irisin sharpens learning memory by inducing the production of brain-derived neurotrophic factor (BDNF), lowers the production of inflammatory factors, protects neurology through astrocytes, and ameliorates AD symptoms by improving insulin resistance. The review aims to facilitate future experimental studies and clinical applications of irisin in preventing and treating AD.
Alzheimer’s disease, as a neurodegenerative disease, impacts not only patients and their families but also social security (1, 2). World Health Organization revealed that over 55 million people (8.1% of women and 5.4% of men over the age of 65) worldwide suffered from dementia in 2021. AD is the first to blame when it comes to disability in the elderly, and its incidence grows exponentially with age. It also requires higher treatment costs compared with heart diseases and cancers, increasing the financial burden on the global economy and patients’ families (3). Dr. Alois Alzheimer first reported AD and its features of progressive memory loss and other cognitive impairments as early as 1907 (4), but no definitive therapy was discovered.
The neurodegenerative disease has complicated and diverse causes represented by genetic factors and aging (5), and largely unknown mechanisms (6). Multiple studies proved the effectiveness of exercise in reducing the risk factors for AD (7–9). A growing body of evidence shows the higher risk of AD caused by inadequate physical exercise, obesity, high blood pressure, and diabetes, and interventions in such factors prevent or delay 40% of dementia (10). Klodian et al. stated the close relationship between risk factors and incidence (11). Exercise helps reduce common risk factors among patients with AD and exhibits higher adherence than medication interference because of its fewer side effects (7, 8). The two main pharmacological treatments for AD, namely, cholinesterase inhibitors and N-methyl-D-aspartate amino receptor blockers in clinical trials, fail to improve symptoms significantly (12, 13), indicating the importance of alternative forms of treatment and prevention methods such as exercise. Alzheimer’s Disease International calls for 150-min of moderate aerobic exercise or 75-min of vigorous aerobic exercise each week among adults for normal heart function, blood circulation, weight management, and mental health. There is also evidence that exercise can be an effective intervention in early AD (7, 14, 15).
Recently, many studies have discovered that irisin produced by exercise can improve cognition through various pathways. Specifically, irisin promotes neurogenesis by BDNF, increases neuronal survival and synaptic plasticity (16), reduces the release of inflammatory factors IL-6 and IL-1β, decreases Aβ protein and tau protein formation, and undermines insulin resistance (15, 17). These factors indicate that irisin could potentially play a significant role in the treatment and prevention of AD. This review discusses irisin’s role in AD, highlighting irisin’s mechanism of action in relation to AD pathology and assessing the current evidence base for the use of irisin as a treatment for AD.
Aβ protein deposition serves as an essential part of classic hallmarks for AD, and Aβ refers to a polypeptide formed by amyloid precursor protein hydrolysis by β and γ secretases. According to animal experiments, rats, which were injected with Aβ and underwent the Morris water maze test, performed decreased spatial learning and memory of Aβ. Hardy and Higgins first proposed the protein cascade hypothesis in 1992, which regarded amyloid deposition as the primary risk factor for AD. Aβ deposition triggers the dysfunction and death of neurons in the brain, crippling normal brain function. In particular, the imbalance of Aβ protein metabolism directly stimulates the production of inflammatory factors in neurons, leading to neuroinflammation and oxidative stress. Meanwhile, Aβ protein activates microglia and astrocytes around senile plaques, releasing pro-inflammatory factors such as TNF-α or IL-1-β (18). Prolonged neuroinflammation leads to neurosynaptic dysfunction, thus accelerating neuronal degeneration, exacerbating brain damage, and causing lesions. The interaction between Aβ and glial cells is supposedly mixed. On the one hand, glial cells reduce Aβ protein deposition through phagocytic clearance and degradation to protect neurons and prevent amyloid plaque formation in the brain. On the other hand, Aβ protein activates glial cells and facilitates the sustained release of inflammatory cytokines, which promotes Aβ protein deposition and neuroinflammatory responses, incurs immune responses, and accelerates the AD pathological process. In addition, Aβ protein deposition decreases the level of proteins that specialize in the long-term potentiation effect in the hippocampus, which explains the progressively impaired learning memory capacity of patients with AD. Recent studies have also found that Aβ protein aggregation can affect the mitochondrial function of neuronal cells, thereby causing mitochondrial damage and leading to oxidative stress (19). Excessive oxidative stress not only hinders mitochondrial electron transfer but also damages mitochondrial DNA and related synapses. Notably, serious mitochondrial damage accelerates the apoptosis of associated neuronal cells and worsens the condition. In addition, Aβ protein deposition also decreases CBF (20), which is assumed to inhibit mitochondrial ATP production. Studies that supported the hypothesis proved the accumulation of soluble Aβ protein in the cerebral vasculature and the following decline in CBF and vasoconstriction, which compromises neuronal effort. Overall, by stimulating nerve cells, Aβ releases inflammatory factors that promotes neuronal dysfunction and death and affects the normal function of mitochondria and CBF, thus denting the normal function of the brain.
One of the pathological features of AD is the presence of neurofibrillary tangles (NFTs) in neuronal cells, whose main component is the abnormally phosphorylated tau protein. It refers to a phosphorylated glycoprotein closely related to microtubule structure and is essential for the synthesis and maintenance of the neuronal skeleton and intracellular transmission of substances and information between neurons. Over-phosphorylation of tau protein, however, disrupts neuronal microtubule structure, reduces the binding to microtubule proteins, and hinders microtubule formation and microtubule stability (21, 22), which results in the formation of NFTs and AD development. NFTs impair axonal transport function, causing synaptic loss, cytoskeletal and mitochondrial dysfunction, and memory loss. Therefore, hyperphosphorylated soluble tau protein may be responsible for AD neurotoxicity (23). The quantity and distribution of NFTs in the brain of patients with AD are higher than that of normal elderly, and NFTs climb with AD development and positively correlate with the degree of clinical dementia, which explains the general application of NFTs accumulation in the patient’s brain as one of the critical indicators of AD development degree in clinical tests (24). Some new therapeutic approaches, such as combined drugs targeting tau protein and Aβ, are vital for tau-related pathological processes in AD (25). In summary, tau protein phosphorylation in the brain of patients with AD leads to NETs that deepen the degree of AD pathology by affecting the transport function of neurons and the normal function of mitochondria.
Clinical studies have shown that neuroinflammation is a key pathological mechanism in AD (26). Neuroinflammation and activation of microglia and astrocytes are critical to the pathogenesis of AD (27). Shen et al. explored the difference in cerebrospinal fluid (CSF) or peripheral blood inflammatory markers between patients with AD and MCI and proved the higher level of interleukin-6 (IL-6), interleukin-10 (IL-10), and soluble tumor necrosis factor receptor 1 (sTNFR1) in the brains of patients with AD compared with patients with MCI (28). In addition, peripheral inflammation is one of the contributors to neuroinflammation in the brain, and pro-inflammatory cytokines have been shown to cross the blood-brain barrier, which in turn triggers a central inflammatory response. Damage to the blood-brain barrier also exacerbates central inflammation, allowing the entry of peripheral immune cells into the brain and promoting inflammation there. Besides, obesity is a critical factor in peripheral systemic inflammation, as studies have proved that excess saturated fatty acids in obese patients can cross the blood-brain barrier to directly affect neuroglia activation in the brain, and type 2 diabetes caused by obesity accelerates memory dysfunction and neuroinflammation in mouse models of AD (29). Neuroinflammation and β-amyloid deposition are causal, and even to the extent that neuroinflammation determines the pathophysiological process of β-amyloid. β-amyloid deposition in the brain results in continuous activation of glial cells, followed by neuroinflammatory factor release and inflammatory response. Dysfunctional energy metabolism in the brain also induces neuroinflammation. Mitochondrial dysfunction not only generates high levels of reactive oxygen species followed by microglia activation but also discourages the neuroprotective function of astrocytes, the latter of which comes down to astrocytes’ reliance on functional mitochondria for energy (30, 31). The neuroinflammation accordingly plays a pivotal role in AD. Several epidemiological studies have confirmed the role of non-steroidal anti-inflammatory drugs (NSAIDs) in preventing AD. Specifically, patients who used NSAIDs for more than 2 years had a 60% lower risk of AD, and those for less than 2 years had a 35% lower risk (32). Another study discovered an approximately 55% risk reduction in patients who had used NSAIDs for more than 2 years (33). In summary, neuroinflammation can increase the risk factors for AD, which should be tackled with inflammation reduction in the future.
Insulin refers to a peptide secreted by the pancreas whose regular expression benefits cognition. Insulin resistance, however, is prevalent in the brain of patients with AD (34). Insulin regulates glucose metabolism in peripheral tissues, enhances synaptic viability and dendritic spine formation, and increases neurotransmitter turnover through brain bioenergetics. Besides, its downstream signaling molecules are mainly distributed in the cerebral cortex and hippocampus (35). Insulin in the brain controls glucose and lipid metabolism, affects neurodevelopment, and sways learning and memory processes (34), while insulin/IR signaling in the central nervous system improves cognitive function, including learning and memory. The impairment of hippocampal insulin signaling in the brain of patients with AD undermines memory and other executive functions because of the concomitant decline in insulin signaling and insulin resistance. Decreasing insulin/IGF-1 levels and insulin/IGF-1 receptor deficiency has been found in models of aging and AD (36). Peripheral insulin resistance decreases insulin signaling in the central nervous system, that is, central insulin resistance, which disrupts brain metabolism and increases Aβ toxicity. Furthermore, insulin resistance prompts tau hyperphosphorylation, oxidative stress, and neuroinflammation, which ultimately results in neurodegeneration. Autopsy analysis of AD brains found increased dystrophic synapses and Aβ plaques but decreased levels of IGF-1 neurons. Some studies indicated the decline in energy metabolism that patients with AD experienced before cognitive dysfunction. Furthermore, the degeneration and loss of neurons in the brain of patients with AD aggravate abnormal glucose metabolism (36). Therefore, modulation of energy metabolism may be an important target for AD treatment.
Irisin is a muscle factor discovered in 2012 by Bostrom et al. They found that exercise upregulated the gene expression of fibronectin type III domain-containing protein 5 (FNDC5) in skeletal muscle. FNDC5 was expressed and modified into a new small molecule, irisin, and released into the blood. Jedrychowski et al. identified and measured human irisin in plasma using mass spectrometry, determining its level in circulating blood to be 3.6–4.3°ng/ml (37). Ruan confirmed the higher level of irisin in the elderly and men than in the young and women, respectively, among an average population. Irisin in the cerebrospinal fluid was also positively correlated with BDNF and Aβ (38, 39). Irisin, a glycosylated protein hormone that consists of 112 amino acid residues, is highly conserved structurally and 100% homologous in mice and humans (40). Huh et al. scanned the distribution of FNDC5 in humans using tissue qPCR analysis microarrays and revealed the high expression of FNDC5 in skeletal muscle (41). Xie et al. detected FNDC5 gene expression in 47 human tissues, such as cardiac and smooth muscle, endothelium, adipose tissue, the liver, kidney, and pancreas, among which, after exercise, cardiac FDNC5 was highly expressed, and more irisin was produced in cardiac muscle than in skeletal muscle (42). Meanwhile, irisin is vital for the human body, and exercise-produced irisin has been proven to intervene in AD in several ways (7). Specifically, irisin can accelerate metabolism (represented by irisin browning of white fat and improving glucose homeostasis), cross the blood-brain barrier, and initiate neuroprotective genetic programs in the hippocampus, which incurs higher expression of BDNF and promotes synaptogenesis and neuronal cell survival (43). In addition, the multiple neuroprotective effects of irisin against injury in AD models have been proven. Recent studies confirmed the association between irisin in the risk factor of AD and the lower irisin level in the hippocampus and cerebrospinal fluid (CSF) of patients with AD (39). In contrast, the relation between higher irisin levels in the CSF and better cognitive function, and less amyloid-β pathology is observed in both patients with AD and non-AD (38). The relationship between blood levels of irisin and cognitive function, however, remains unknown given the identity of irisin as a hormone. Tsai et al. explored the circulating level of irisin and neurocognitive performance of thirty-two individuals with a family history of AD (ADFH) and obesity (ADFH-obesity group) and 32 controls (ADFH-non-obesity group) during a visuospatial working memory task and discovered the direct proportion between serum irisin levels and cognitive function in cognitively normal individuals, which indicates the protective role of irisin in the brain (44). In contrast, subsequent studies obtained the opposite results that serum irisin levels did not differ between cognitively normal and AD individuals (45). The above suggests the different effects of irisin on brain cells in the AD brain. The varied roles of irisin in different degrees of AD validate its effective intervention in AD.
Exercise-induced irisin can cross the blood-brain barrier with the help of peripheral transport before entering the central nervous system and inducing BDNF expression (46). The receptor of irisin in the neonatal brain is reported to be an integrin αV/β5 heterodimer while that in the adult brain remains untouched (47). Huang revealed that irisin could promote BDNF expression through an experiment in which diabetic rats were divided into four groups, namely, control, model, irisin (irisin overexpression), and irisin-short hairpin (sh)-RNA (irisin interference). The results confirmed the significantly lower levels of irisin and BDNF in the model and irisin-shRNA groups, and the much higher level in the irisin group. Importantly, irisin and BDNF performed at higher levels in the irisin group compared with the model group, and their expressions were slashed in the irisin-shRNA group compared with the model group (48). The same finding was obtained by Wrann et al., who employed siRNA to interfere with FNDC5/irisin expression in cortical neurons and found a decrease in BDNF expression (49). Meanwhile, one study found that recombinant irisin could promote BDNF formation by stimulating the cAMP→PKA→CREB→BDNF pathway, which was shared by other studies that confirmed the role of irisin in enhancing BDNF expression by increasing cAMP and phosphorylating CREB (pCREB). Therefore, there is much evidence that irisin can induce BDNF production directly in the brain or via the cAMP→PKA→CREB→BDNF pathway, which protects the brain (50).
Brain-derived neurotrophic factor, as a neurotrophic factor, significantly promotes synaptogenesis and plasticity, neuronal cell survival, migration, and dendritic branching. Mainly released by microglia and astrocytes, it is abundantly expressed in those areas of the brain associated with cognition, including the hippocampus, cerebral cortex, amygdala, and cerebellum. Animal models observed the exercise-induced BDNF in different brain regions, the most obvious of which is the hippocampus. Notably, decreased expression levels of BDNF were found in different areas of the brain of patients with AD (51). Other studies have reported the association of BDNF with the mesocortical limbic system in the brain and the importance of BDNF in brain circuits. Besides, hippocampal neurogenesis and hippocampal neural circuits are inseparable from BDNF, which matters much for LTP and learning memory functions. Mychael et al., who supported the finding, injected C57BL/6 mice intraperitoneally with lentiviruses containing two shRNAs, which effectively knocked down FNDC5. The results revealed impaired hippocampal long-term potentiation and memory maintenance in a novel object recognition task (50). Moreover, BDNF underlies neuronal stress resistance and synaptic plasticity by facilitating the differentiation and maturation of developing neurons and enhancing synaptic transmission and plasticity in mature neurons, which contributes to memory formation and learning (52). Recent studies have also found the three approaches adopted by BDNF to regulate synapse formation: increasing axonal and dendritic branching, inducing the formation of axonal and dendritic buckles, and stabilizing existing synapses. Notably, BDNF can promote hippocampal cell proliferation, and so can irisin by increasing the release of BDNF, which regulates the STAT3 signaling pathway. Such a process ultimately leads to a reduced risk of AD. One study found that 100°nmol/L irisin promoted hippocampal cell proliferation by 70–80%, and BDNF expression in FNDC5-transduced primary cortical neurons was increased fourfold compared to the control group (49). Several other studies have also found that irisin may induce the STAT3 signaling pathway through the motor-irisin-BDNF axis to proliferate hippocampal cells (17), supporting irisin as a therapeutic target of AD (53). In summary, irisin passes through the blood-brain barrier with the help of peripheral circulation and eventually enters the brain, which then induces BDNF production directly or indirectly, allowing BDNF to exert protective effects on brain function through various pathways.
Irisin exerts its anti-inflammatory effects by promoting the expression of BDNF, which binds to its receptor TrkB before the activation of AKT and ERK1/2 signaling pathways and reduces the role of IκBα phosphorylation and NF-κb (54). In addition, irisin curtails the release of inflammatory factors through its action on astrocytes (15). Altered roles of BDNF/TrkB cause neurodegenerative changes, while lower BDNF/TrkB levels incur neuroinflammation. Moreover, irisin promotes the release of inflammatory cytokines IL-1β and IL-6 and triggers the JAK2/STAT3 signaling pathway, which upregulates the C/EBPβ/AEP pathway. Such a process finally results in Aβ precursor protein breakage, followed by Aβ and tau protein and cognitive dysfunction (55). In a word, irisin deficiency decreases BDNF/TrkB levels and induces the formation of Aβ and tau protein, thus exacerbating cognitive dysfunction. The role of irisin on astrocytes by decreasing pro-inflammatory factor release also deserves attention.
Astrocytes (also astroglia), the most numerous and prominent cells in the nervous system, contribute much to neuronal metabolism, the repair of injuries to the brain and spinal cord, phagocytosis and isolation of synapses, and the metabolism of neurons passing through astrocytes. Irisin protects cultured neurons by modulating astrocytes and preventing neuronal cells from Aβ. One study found that the treatment of astrocyte-conditioned cultures with irisin for approximately 12 h protects the neurons from the toxic effects of Aβ (56), which is shared by an in vitro study. Despite a potential neuroprotective impact of irisin on primary neuronal cultures after Aβ injury, no protection of neuronal cells was observed when cultured neurons were treated directly with irisin or Aβ. The cells, however, were under protection when they were treated with irisin and Aβ protein and co-cultured with astrocytes. Several other studies also demonstrated that irisin significantly increased neuronal cell viability after treatment with astrocyte-conditioned cultures, which achieves its neuroprotective effect by reducing the release of IL-6 and IL-1β from cultured astrocytes and decreasing COX-2 expression and AKT phosphorylation. For example, Wang et al. found that irisin improved memory and cognition in diabetic mice by decreasing IL-6 and IL-1β expression in their hippocampus (56). Given its protection of neurons by reducing the release of pro-inflammatory factors through various pathways, irisin may be a new target for AD prevention or treatment.
Multiple studies have found that irisin can improve insulin resistance and glucose homeostasis through pathways of PI3K/Akt signaling and p38 mitogen-activated protein kinase (p38MAPK). Animal experiments proved the role of overexpressed FNDC5/irisin in mice fed a high-fat diet in improving insulin resistance and lowering blood glucose, a finding supported by Tang, who treated mouse hepatocytes cultured with irisin at high glucose levels and confirmed that irisin could decrease intracellular insulin resistance, increase glycogen synthesis, and increase the number of surviving cells (57). Liu verified the ability of irisin in promoting BDNF expression, followed by the activation of the PI3K/Akt signaling pathway, which controls neuronal glucose homeostasis, mitochondrial biogenesis, and integrity. Higher insulin resistance emerges finally (58). The p38MAPK signaling pathway is also adopted by irisin to improve insulin resistance, and the inhibition or knockdown of p38MAPK inhibits irisin-induced glucose uptake and promotes insulin resistance. Exercise increases reactive oxygen species (ROS) levels, which activates p38MAPK. PGC-1α, regulated by p38MAPK, increases irisin secretion by promoting FNDC5 expression, and irisin acts on white adipocytes and stimulates UCP1 expression, which promotes betatrophin secretion and β-cell regeneration and reduces insulin resistance. Therefore, the ROS→p38MAPK→PGC-1α→irisin→betatrophin→β-cell regeneration pathway may reduce insulin resistance. Ye verified the dependence of irisin-mediated reversal of IR on p38MAPK and that the inhibition of p38MAPK via SB203580 significantly reduced irisin-mediated glucose uptake, whereas inhibition of the ERK MAPK pathway via U0126 produced a slight blocking effect (59). Therefore, irisin can improve insulin resistance via the signaling pathways of PI3K/Akt and p38MAPK and enhance the AD brain’s glucose metabolism and the role of insulin.
In this review, we highlighted several remarks concerning the potential beneficial role of irisin in AD. For example, irisin can enhance cognitive AD by promoting BDNF production, mitigate AD by reducing Aβ protein deposition, reduce neuroinflammation by inhibiting pro-inflammatory cytokine expression, and protect against AD by activating the Akt/ERK1/2 signaling pathway to inhibit oxidative stress or ameliorate cardiovascular disease (Figure 1). The mechanism of irisin against AD facilitates the development and testing of drugs for the treatment or prevention of AD, and further physical benefits to patients with AD who can no longer exercise due to health or locomotor conditions. Studies on irisin intervention in AD, however, still face many challenges, one of which is that many current studies are based on experimental studies despite some registered clinical trials aiming to elucidate irisin’s effects on humans (Table 1). Therefore, the exploration of the relationships between irisin and cognition requires extensive clinical studies and long-term follow-up, and the analysis and comparison of animal experiments and clinical studies are necessary, which will deepen the understanding of the differences in the effects of FNDC5/irisin in mice and humans.
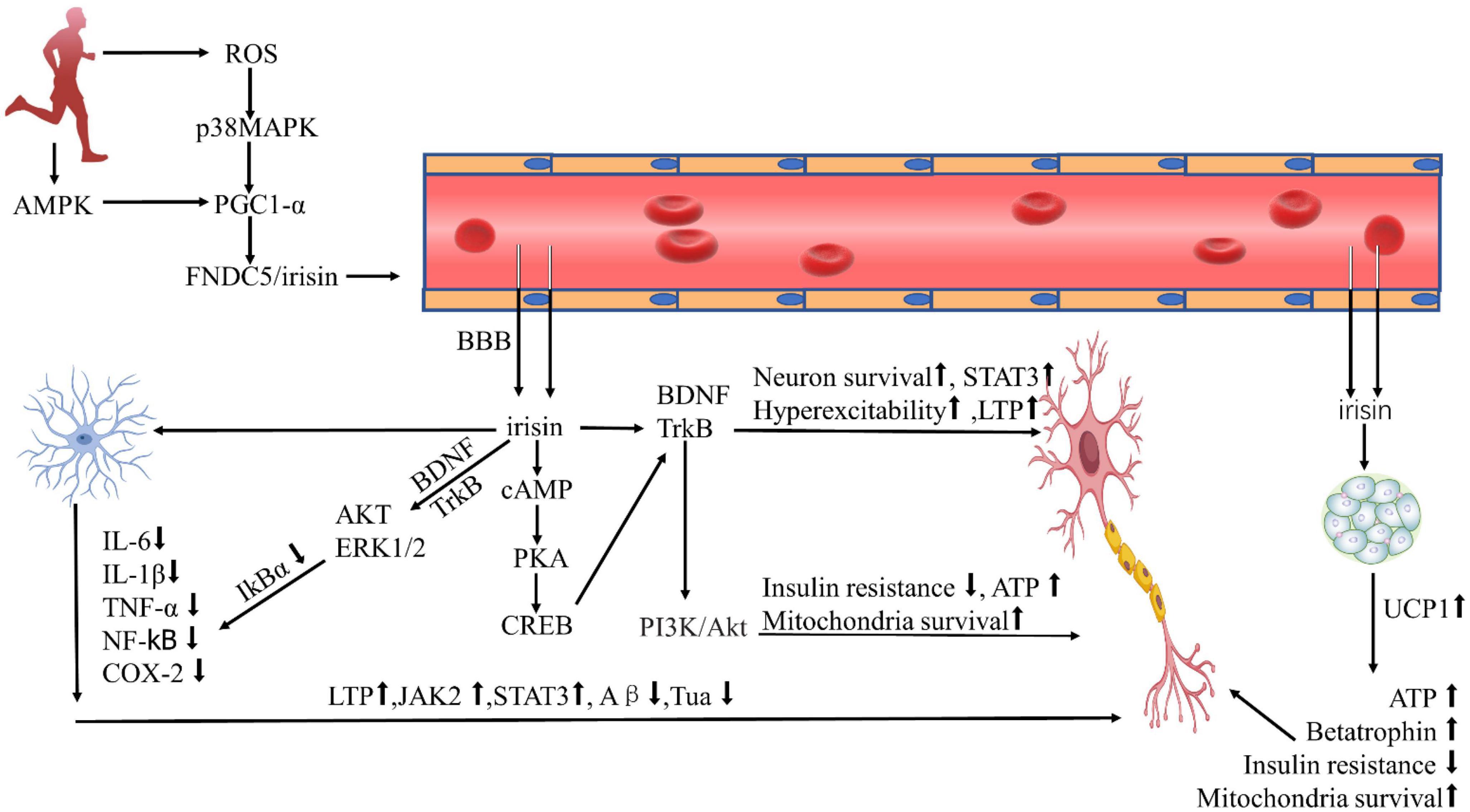
Figure 1. Protective effect of irisin against Alzheimer’s disease. ROS, reactive oxygen species; AMPK, AMP-activated protein kinase; PGC-1α, PPARγ coactivator 1α; FNDC5, fibronectin type III domain-containing protein-5; BDNF, brain-derived neurotrophic factor; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; CREB, cAMP-response element binding protein; P13K, phosphatidylinositol 3 kinase; Akt, protein kinase B; ERK1/2, extracellular signal-regulated kinase1/2; IL-6, interleukin-6; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor-α; NF-κb, nuclear factor κb; COX-2, cyclooxygenase-2; LTP, long-term potentiation; STAT3, signal transducer and activator of transcription-3; UCP1, uncoupling protein-1; BBB, blood-brain barrier; ATP, adenosine triphosphate.

Table 1. Preclinical and clinical studies on irisin and Alzheimer’s disease and Alzheimer’s disease-related dementia.
KC: writing, original draft preparation, conceptualization, and editing. KW: supervision, reviewing, validation, and editing. TW: supervision, methodology, reviewing, resources, and validation. All authors contributed to the article and approved the submitted version.
This study has been supported by the grant BWS21J001.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wimo A, Guerchet M, Ali GC, Wu YT, Prina AM, Winblad B, et al. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimers Dement. (2017) 13:1–7. doi: 10.1016/j.jalz.2016.07.150
2. El-Hayek YH, Wiley RE, Khoury CP, Daya RP, Ballard C, Evans AR, et al. Tip of the iceberg: assessing the global socioeconomic costs of Alzheimer’s Disease and related dementias and strategic implications for stakeholders. J Alzheimers Dis. (2019) 70:323–41. doi: 10.3233/JAD-190426
3. Kosaner Kliess M, Martins R, Connolly MP. Major cost drivers in assessing the economic burden of Alzheimer’s Disease: a structured, rapid review. J Prev Alzheimers Dis. (2021) 8:362–70. doi: 10.14283/jpad.2021.17
4. Soria Lopez JA, Gonzalez HM, Leger GC. Alzheimer’s disease. Handb Clin Neurol. (2019) 167:231–55. doi: 10.1016/B978-0-12-804766-8.00013-3
5. Belloy ME, Napolioni V, Greicius MDA. Quarter century of APOE and Alzheimer’s Disease: progress to date and the path forward. Neuron. (2019) 101:820–38. doi: 10.1016/j.neuron.2019.01.056
6. Knopman DS, Amieva H, Petersen RC, Chetelat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Primers. (2021) 7:33. doi: 10.1038/s41572-021-00269-y
7. De la Rosa A, Olaso-Gonzalez G, Arc-Chagnaud C, Millan F, Salvador-Pascual A, Garcia-Lucerga C, et al. Physical exercise in the prevention and treatment of Alzheimer’s disease. J Sport Health Sci. (2020) 9:394–404. doi: 10.1016/j.jshs.2020.01.004
8. Valenzuela PL, Castillo-Garcia A, Morales JS, de la Villa P, Hampel H, Emanuele E, et al. Exercise benefits on Alzheimer’s disease: state-of-the-science. Ageing Res Rev. (2020) 62:101108. doi: 10.1016/j.arr.2020.101108
9. Yu F, Vock DM, Zhang L, Salisbury D, Nelson NW, Chow LS, et al. Cognitive effects of aerobic exercise in Alzheimer’s Disease: a pilot randomized controlled trial. J Alzheimers Dis. (2021) 80:233–44. doi: 10.3233/JAD-201100
10. Baranowski BJ, Marko DM, Fenech RK, Yang AJT, MacPherson REK. Healthy brain, healthy life: a review of diet and exercise interventions to promote brain health and reduce Alzheimer’s disease risk. Appl Physiol Nutr Metab. (2020) 45:1055–65. doi: 10.1139/apnm-2019-0910
11. Dhana K, Franco OH, Ritz EM, Ford CN, Desai P, Krueger KR, et al. Healthy lifestyle and life expectancy with and without Alzheimer’s dementia: population based cohort study. BMJ. (2022) 377:e068390. doi: 10.1136/bmj-2021-068390
12. McShane R, Westby MJ, Roberts E, Minakaran N, Schneider L, Farrimond LE, et al. Memantine for dementia. Cochrane Database Syst Rev. (2019) 3:CD003154. doi: 10.1002/14651858.CD003154.pub6
13. Huang LK, Chao SP, Hu CJ. Clinical trials of new drugs for Alzheimer disease. J Biomed Sci. (2020) 27:18. doi: 10.1186/s12929-019-0609-7
14. Cui MY, Lin Y, Sheng JY, Zhang X, Cui RJ. Exercise intervention associated with cognitive improvement in Alzheimer’s Disease. Neural Plast. (2018) 2018:9234105. doi: 10.1155/2018/9234105
15. Madhu LN, Somayaji Y, Shetty AK. Promise of irisin to attenuate cognitive dysfunction in aging and Alzheimer’s disease. Ageing Res Rev. (2022) 78:101637. doi: 10.1016/j.arr.2022.101637
16. Islam MR, Young MF, Wrann CD. The Role of FNDC5/Irisin in the Nervous System and as a Mediator for Beneficial Effects of Exercise on the Brain. In: B Spiegelman editor. Hormones, Metabolism and the Benefits of Exercise. Cham: Springer (2017). p. 93–102.
17. Kim OY, Song J. The role of irisin in Alzheimer’s Disease. J Clin Med. (2018) 7:407. doi: 10.3390/jcm7110407
18. Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener. (2020) 15:40. doi: 10.1186/s13024-020-00391-7
19. Agrawal I, Jha S. Mitochondrial dysfunction and Alzheimer’s Disease: role of microglia. Front Aging Neurosci. (2020) 12:252. doi: 10.3389/fnagi.2020.00252
20. Solis E Jr, Hascup KN, Hascup ER. Alzheimer’s Disease: the link between amyloid-beta and neurovascular dysfunction. J Alzheimers Dis. (2020) 76:1179–98. doi: 10.3233/JAD-200473
21. Silva MVF, Loures CMG, Alves LCV, de Souza LC, Borges KBG, Carvalho MDG. Alzheimer’s disease: risk factors and potentially protective measures. J Biomed Sci. (2019) 26:33. doi: 10.1186/s12929-019-0524-y
22. Drummond E, Pires G, MacMurray C, Askenazi M, Nayak S, Bourdon M, et al. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain. (2020) 143:2803–17. doi: 10.1093/brain/awaa223
23. Gao Y, Tan L, Yu JT, Tan L. Tau in Alzheimer’s Disease: mechanisms and therapeutic strategies. Curr Alzheimer Res. (2018) 15:283–300. doi: 10.2174/1567205014666170417111859
24. Long JM, Holtzman DM. Alzheimer Disease: an update on pathobiology and treatment strategies. Cell. (2019) 179:312–39. doi: 10.1016/j.cell.2019.09.001
25. Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. (2018) 14:399–415. doi: 10.1038/s41582-018-0013-z
26. Uddin MS, Hasana S, Ahmad J, Hossain MF, Rahman MM, Behl T, et al. Anti-neuroinflammatory potential of polyphenols by inhibiting NF-kappaB to halt Alzheimer’s Disease. Curr Pharm Des. (2021) 27:402–14. doi: 10.2174/1381612826666201118092422
27. Fu WY, Wang X, Ip NY. Targeting neuroinflammation as a therapeutic strategy for Alzheimer’s Disease: mechanisms, drug candidates, and new opportunities. ACS Chem Neurosci. (2019) 10:872–9. doi: 10.1021/acschemneuro.8b00402
28. Shen XN, Niu LD, Wang YJ, Cao XP, Liu Q, Tan L, et al. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: a meta-analysis and systematic review of 170 studies. J Neurol Neurosurg Psychiatry. (2019) 90:590–8. doi: 10.1136/jnnp-2018-319148
29. Pugazhenthi S, Qin L, Reddy PH. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1037–45. doi: 10.1016/j.bbadis.2016.04.017
30. Sivanandam TM, Thakur MK. Traumatic brain injury: a risk factor for Alzheimer’s disease. Neurosci Biobehav Rev. (2012) 36:1376–81. doi: 10.1016/j.neubiorev.2012.02.013
31. Hunter R, Ojha U, Bhurtel S, Bing G, Choi DY. Lipopolysaccharide-induced functional and structural injury of the mitochondria in the nigrostriatal pathway. Neurosci Res. (2017) 114:62–9. doi: 10.1016/j.neures.2016.09.007
32. Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. (1997) 48:626–32. doi: 10.1212/wnl.48.3.626
33. Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC, et al. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology. (2002) 59:880–6. doi: 10.1212/wnl.59.6.880
34. Sedzikowska A, Szablewski L. Insulin and insulin resistance in Alzheimer’s Disease. Int J Mol Sci. (2021) 22:9987. doi: 10.3390/ijms22189987
35. Rorbach-Dolata A, Piwowar A. neurometabolic evidence supporting the hypothesis of increased incidence of type 3 diabetes mellitus in the 21st century. Biomed Res Int. (2019) 2019:1435276. doi: 10.1155/2019/1435276
36. Sohrabi M, Floden AM, Manocha GD, Klug MG, Combs CK. IGF-1R inhibitor ameliorates neuroinflammation in an Alzheimer’s Disease transgenic mouse model. Front Cell Neurosci. (2020) 14:200. doi: 10.3389/fncel.2020.00200
37. Jedrychowski MP, Wrann CD, Paulo JA, Gerber KK, Szpyt J, Robinson MM, et al. Detection and quantitation of circulating human irisin by tandem mass spectrometry. Cell Metab. (2015) 22:734–40. doi: 10.1016/j.cmet.2015.08.001
38. Lourenco MV, Ribeiro FC, Sudo FK, Drummond C, Assuncao N, Vanderborght B, et al. Cerebrospinal fluid irisin correlates with amyloid-beta, BDNF, and cognition in Alzheimer’s disease. Alzheimers Dement. (2020) 12:e12034. doi: 10.1002/dad2.12034
39. Ruan Q, Huang Y, Yang L, Ruan J, Gu W, Zhang X, et al. The effects of both age and sex on irisin levels in paired plasma and cerebrospinal fluid in healthy humans. Peptides. (2019) 113:41–51. doi: 10.1016/j.peptides.2019.01.004
40. Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. (2012) 481(7382):463–8. doi: 10.1038/nature10777
41. Huh JY, Panagiotou G, Mougios V, Brinkoetter M, Vamvini MT, Schneider BE, et al. FNDC5 and irisin in humans: I. predictors of circulating concentrations in serum and plasma and II. mRNA expression and circulating concentrations in response to weight loss and exercise. Metabolism. (2012) 61:1725–38. doi: 10.1016/j.metabol.2012.09.002
42. Xie C, Zhang Y, Tran TD, Wang H, Li S, George EV, et al. Irisin controls growth, intracellular Ca2+ signals, and mitochondrial thermogenesis in cardiomyoblasts. PLoS One. (2015) 10:e0136816. doi: 10.1371/journal.pone.0136816
43. Jodeiri Farshbaf M, Alvina K. Multiple roles in neuroprotection for the exercise derived myokine irisin. Front Aging Neurosci. (2021) 13:649929. doi: 10.3389/fnagi.2021.649929
44. Tsai CL, Pai MC. Circulating levels of Irisin in obese individuals at genetic risk for Alzheimer’s disease: correlations with amyloid-beta, metabolic, and neurocognitive indices. Behav Brain Res. (2021) 400:113013. doi: 10.1016/j.bbr.2020.113013
45. Conti E, Grana D, Stefanoni G, Corsini A, Botta M, Magni P, et al. Irisin and BDNF serum levels and behavioral disturbances in Alzheimer’s disease. Neurol Sci. (2019) 40:1145–50. doi: 10.1007/s10072-019-03781-y
46. Islam MR, Valaris S, Young MF, Haley EB, Luo R, Bond SF, et al. Exercise hormone irisin is a critical regulator of cognitive function. Nat Metab. (2021) 3:1058–70. doi: 10.1038/s42255-021-00438-z
47. Jackson TC, Gorse K, Herrmann JR, Kochanek PM. Hippocampal and prefrontal cortical brain tissue levels of irisin and GDF15 receptor subunits in children. Mol Neurobiol. (2021) 58:2145–57. doi: 10.1007/s12035-020-02250-4
48. Huang L, Yan S, Luo L, Yang L. Irisin regulates the expression of BDNF and glycometabolism in diabetic rats. Mol Med Rep. (2019) 19:1074–82. doi: 10.3892/mmr.2018.9743
49. Wrann CD, White JP, Salogiannnis J, Laznik-Bogoslavski D, Wu J, Ma D, et al. Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab. (2013) 18:649–59. doi: 10.1016/j.cmet.2013.09.008
50. Lourenco MV, Frozza RL, de Freitas GB, Zhang H, Kincheski GC, Ribeiro FC, et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat Med. (2019) 25:165–75. doi: 10.1038/s41591-018-0275-4
51. Budni J, Bellettini-Santos T, Mina F, Garcez ML, Zugno AI. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. (2015) 6:331–41. doi: 10.14336/AD.2015.0825
52. Di Carlo P, Punzi G, Ursini G. Brain-derived neurotrophic factor and schizophrenia. Psychiatr Genet. (2019) 29:200–10. doi: 10.1097/YPG.0000000000000237
53. Amidfar M, de Oliveira J, Kucharska E, Budni J, Kim YK. The role of CREB and BDNF in neurobiology and treatment of Alzheimer’s disease. Life Sci. (2020) 257:118020. doi: 10.1016/j.lfs.2020.118020
54. Li DJ, Li YH, Yuan HB, Qu LF, Wang P. The novel exercise-induced hormone irisin protects against neuronal injury via activation of the Akt and ERK1/2 signaling pathways and contributes to the neuroprotection of physical exercise in cerebral ischemia. Metabolism. (2017) 68:31–42. doi: 10.1016/j.metabol.2016.12.003
55. Wang ZH, Xiang J, Liu X, Yu SP, Manfredsson FP, Sandoval IM, et al. Deficiency in BDNF/TrkB neurotrophic activity stimulates delta-secretase by upregulating C/EBPbeta in Alzheimer’s Disease. Cell Rep. (2019) 28:655–69.e5. doi: 10.1016/j.celrep.2019.06.054
56. Wang K, Li H, Wang H, Wang JH, Song F, Sun Y. Irisin exerts neuroprotective effects on cultured neurons by regulating astrocytes. Mediators Inflamm. (2018) 2018:9070341. doi: 10.1155/2018/9070341
57. Tang H, Yu R, Liu S, Huwatibieke B, Li Z, Zhang W. Irisin inhibits hepatic cholesterol synthesis via AMPK-SREBP2 signaling. EBioMedicine. (2016) 6:139–48. doi: 10.1016/j.ebiom.2016.02.041
58. Liu TY, Shi CX, Gao R, Sun HJ, Xiong XQ, Ding L, et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin Sci. (2015) 129:839–50. doi: 10.1042/CS20150009
59. Ye X, Shen Y, Ni C, Ye J, Xin Y, Zhang W, et al. Irisin reverses insulin resistance in C2C12 cells via the p38-MAPK-PGC-1alpha pathway. Peptides. (2019) 119:170120. doi: 10.1016/j.peptides.2019.170120
60. Zarbakhsh S, Safari M, Aldaghi MR, Sameni HR, Ghahari L, Khaleghi Lagmouj Y, et al. Irisin protects the substantia nigra dopaminergic neurons in the rat model of Parkinson’s disease. Iran J Basic Med Sci. (2019) 22:722–8. doi: 10.22038/ijbms.2019.33444.7987
Keywords: irisin, Alzheimer’s disease, BDNF, neuroprotection, neuroinflammation
Citation: Chen K, Wang K and Wang T (2022) Protective effect of irisin against Alzheimer’s disease. Front. Psychiatry 13:967683. doi: 10.3389/fpsyt.2022.967683
Received: 13 June 2022; Accepted: 29 August 2022;
Published: 20 September 2022.
Edited by:
Rajesh Tampi, Creighton University, United StatesReviewed by:
Saeid Taheri, University of South Florida, United StatesCopyright © 2022 Chen, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun Wang, d2FuZ2swOUBxcS5jb20=; Tianhui Wang, d3lkbnk2NjhAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.