 Cong Hu
Cong Hu Heli Li
Heli Li Jinhui Li
Jinhui Li Xiaoping Luo
Xiaoping Luo Yan Hao
Yan Hao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 17 November 2022
Sec. Autism
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.958661
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by variable impairment of social communication and repetitive behaviors, highly restricted interests, and/or sensory behaviors beginning early in life. Many individuals with ASD have dysfunction of microglia, which may be closely related to neuroinflammation, making microglia play an important role in the pathogenesis of ASD. Mounting evidence indicates that microglia, the resident immune cells of the brain, are required for proper brain function, especially in the maintenance of neuronal circuitry and control of behavior. Dysfunction of microglia will ultimately affect the neural function in a variety of ways, including the formation of synapses and alteration of excitatory–inhibitory balance. In this review, we provide an overview of how microglia actively interact with neurons in physiological conditions and modulate the fate and functions of synapses. We put a spotlight on the multi-dimensional neurodevelopmental roles of microglia, especially in the essential influence of synapses, and discuss how microglia are currently thought to influence ASD progression.
Autism spectrum disorder (ASD) is a group of neurodevelopmental disorders characterized by impairments in social communication and restricted or repetitive behaviors or interests (1). The worldwide prevalence of ASD has increased, across all 11 Autism and Developmental Disabilities Monitoring (ADDM) sites in the United States, 1/44 has been estimated to have ASD of 8 years old children reported by the American Centers for Disease Control and Prevention (CDC) in 2021, and incidence and median age varied widely from site to site (2). Accumulating evidence has revealed that both genetic (e.g., de novo variants, copy number variations, and large deletions) (3) and environmental factors [e.g., perinatal events (4) and maternal obesity (5)] are potential risk factors for ASD (6). Less is known about the physiological pathology of ASD, but it may involve a number of systemic connections, nerves, biochemistry, cellular, and molecular characteristics (5). It has been recognized that complex interactions and combinations of genetic, environmental factors, and immune dysfunction can play a potential role in its development (7, 8). In particular, previous research has highlighted that ASD is an imbalance of central nervous system homeostasis caused by chronic inflammatory responses, which are often accompanied by the activation of microglia (9, 10).
Neurodevelopmental abnormalities in early life are an essential mechanism of ASD development, in which microglia work as critical regulators. Specifically, neurons have been examined to be over-produced than required for the proper function of the development of the cerebral cortex during fetal neurogenesis (11). Microglia probe their micro-environment and correlate with the activity of individual synapses (12, 13). To maintain environmental stability, when neurogenesis is nearing completion, microglia limit the overproduction of neurons by swallowing precursor neural cells. In addition, they shape the connections between synapses through synaptic pruning during postnatal brain development (11, 14). Once neurodevelopment is completed, microglia act as innate immune cells in the CNS by monitoring the microenvironment and becoming activated when irritated by injury, infection, or disease (15, 16). In the early stages of brain development, chronic neuroinflammation could activate microglia, which could affect the length, orientation, and assignment of dendritic spines on neurons, especially excitatory and inhibitory neuron assignment, inducing the impairment of behavioral and cognitive (17) or social communication impairment in ASD (18, 19). For example, it has been observed in mice that increased synaptic density was caused by inhibited microglial autophagy, ultimately leading to decreased sociability (20).
In summary, recent studies have shown that activation of microglia can influence the structure and function of synapses and the process of neurodevelopment. For instance, synaptic defects disrupt the excitatory and inhibitory balance and synaptic pruning, and the accurate mechanisms in the neurodevelopment of microglia in cellular and molecular ASD remain to be fully elucidated. The main idea of this review is that microglial activation is associated with ASD and may have a significant impact on synaptic function. This study will summarize the factors and pathways that may be involved in microglia and synaptic function, thus providing a new understanding of the role of microglia in ASD.
As a vital role in regulating brain development, neuronal networks, and injury repair, microglia are the primary cells that maintain defense stability throughout the brain parenchyma (21). Recent studies highlight that microglia are a type of phagocytic cell in the brain that can eliminate entire cells or substructures of cellular, especially synapses both in humans and mice (16, 22).
The origin of microglia is shown concretely in mouse studies. During the first trimester after pregnancy in murine, the fate-mapping analysis revealed that adult microglia derive from C-KIT+/CD41+ erythromyeloid myeloid progenitors that arise before embryonic day 8 (E8.0) in the developing yolk sac (23). During fetal development, microglia progenitor cells migrate and colonize in the brain before cerebral vascular branching is completed and the blood–brain barrier (BBB) is fully shaped (24, 25). In mice, these precursors migrate into the embryonic brain around E9.5 and are restricted by the fully completed BBB as an autonomous, long-lived cell population that maintains the ability to divide and self-renew throughout the lifespan (26–28).
During neurogenesis of early brain development, excess synaptic connections will be removed to maintain proper connections by synaptic sculpting, which is important for maintaining normal neural function (29). Microglia exert a vital role in regulating immature synapses during development viaengulfing synaptic structures and synaptic pruning (30). For example, in mice with the defective function of microglia, the spine density and the frequency of miniature excitatory postsynaptic currents were increased (20, 31). Therefore, it could be speculated that the aberrant function of microglia influenced by risk factors of genetics and environment, could lead to neurodevelopmental disorder in ASD.
Nowadays, neuroinflammation has increasingly gained interest as a target to explore the mechanism of neurodevelopment (32). The occurrence and spread of neuroinflammation are closely related to the interaction between microglia and neurons (33). While certain features of closely regulated proinflammatory activity are necessary for healthy neural development, uncontrolled early inflammation may alter the programming of the microglial population itself, thereby perpetuating neuroinflammatory damage produced early in life (34).
Microglia are thought to be “quiescent” but recent evidence suggests that they continuously scan the brain environment and make contact with synapses in the normal brain (29, 35). Microglia are easily affected by environmental high-risk factors, especially in pregnancy and prenatal development. Interestingly, in the early pathological process of many diseases, microglia are rapidly activated, which is characterized by increased somatic cell size and pro-inflammatory cytokines. Reactive microglia could have an important impact on different processes of neuroinflammation. They can not only act directly via the abundantly expressed or release the molecules or mediators, such as interleukin-6 (IL-6), interleukin-1β (IL-1β), tumor necrosis factor -α (TNF-α), NO, C-X-C chemokine receptor type 4 (CXCL-4), and toll-like receptors (TLRs) (36), but also indirectly act as catalysts and amplify cellular and molecular responses, influencing neurogenesis, and BBB permeability (37, 38). Microglia activation would lead to abnormal neurogenesis and changes in synapse pruning, then resulting in structural dysfunction of neurons in adults and neuronal–microglia interaction disorders in the subsequent process (29, 39, 40).
Additionally, specific histone marks deposited in genomic regions associated with inflammatory pathways maintain microglia priming and long-lasting memory following initial exposure to inflammatory stimuli in animal studies. In subsequent immune stimulation, aberrant activation of inflammatory pathways in microglia leads to a loss of immune homeostasis (41). Therefore, if there is an immune challenge early in life, microglia with enhanced activation may be left in the brains of offspring (42, 43). This may be an important mechanism by which children exposed to environmental risk factors early in life are prone to neuroinflammation and abnormal synapse formation.
Recent evidence also indicates that the ongoing process of neuroinflammation suffered by children with ASD may come from intestinal microbiota dysfunction, resulting in microglial activation in different brain areas (44). When microglia are activated continuously for a period, mediators will be constantly produced and then lead to the diminution of synaptic connections and neuronal cell death (45).
Several studies have shown that there are obvious abnormalities of microglia both in the morphological characteristics and the functions in the brains of patients with ASD, which would cause defects in social interaction and communication (46, 47). Brain tissues obtained from 11 patients with ASD have demonstrated microglia were consistently activated in all brain regions, especially in the cerebellum (40). Cerebral cortex of patients with ASD exhibited altered microglial activity as evidenced by morphological changes, including the increased microglial soma size and extension of filopodia (46). In addition, the 18 kDa translocator protein (TSPO) is highly responsive to inflammatory stimulation, which seemed as an in vivo marker of microglia activation (48, 49). Recent studies have sought to detect activated microglia in patients using positron emission tomography for the TSPO (48), and shreds of evidence suggested TSPO was broadly increased in different brain regions of ASD, including pre-frontal, temporal, cerebellar, and anterior cingulate cortices (50).
The functional regulation of microglia by many similar genes emphasizes their important roles in ASD. Children with Rett syndrome (RTT) may display many autism-like features, such as impairment of social communication and skills, reduced eye contact as well as restricted interests, and they initially may be diagnosed with autism (51). RTT is attributed to the mutations of the MECP2 gene (52). To better study the pathogenesis of RTT, the MECP2-knockout mice model is often used, which exhibits similar behavioral characteristics to patients with RTT. Previous studies have shown that the autophagy activity of microglia is significantly reduced in MECP2-knockout mice (51). Specific expression of MECP2 in cells of microglia could partially rescue the mouse phenotype in MECP2-knockout mice (29). In addition, disease progression can be prevented by the implantation of bone-derived myeloid cells with microglial phenotype into the brain parenchyma through wild-type bone marrow transplantation into Mecp2-null hosts under irradiation conditioned. However, when cranial irradiation is blocked by a lead shield, and microglial engraftment is prevented, the disease will not be arrested (53). The report confirms the strong neurotoxic activity of glutamate in conditioned culture medium (CM) obtained from Mecp2 deficient microglia, but not astrocytes. Hippocampal neurons treated with CM from Mecp2 deficient microglia showed abnormal development and bead-like dendritic morphology over 24 h, as well as signs of microtubule destruction and damage to postsynaptic glutamate energy components (54). Evidence demonstrates that microglia are involved in pathogenesis with synaptic loss through excessively engulfing, thereby eliminating presynaptic inputs at the end stages of disease (≥P56 Mecp2-null mice) (55). Therefore, the appropriate microglial activity may be critical for the development or maintenance of neuronal circuits.
Some genes have been validated in mouse models and behavioral testing for the possibility of pathogenicity in ASD. For example, evidence shows that exaggerated translation of eIF4E in microglia, but not astrocytes or other neurons, could lead to autism-like behaviors in male mice. Translation of mRNAs requires binding of a translation initiation factor eIF4E with cap (56). Elevating eIF4E translation in males, increases microglial density and size, shifting the function to enhanced phagocytic capacity and altered synapse formation (57). Atg7-deficient microglia resulted in social behavioral defects and repetitive behaviors, characteristic features of ASD. It has shown increasing defects in synaptic refinement, which is significantly correlated with the function of microglia in synaptic pruning (20).
There are several environmental factors that contribute to placental inflammatory histological changes and the production of pro-inflammatory cytokines, including maternal obesity, depression, and smoking (58, 59). For example, the expression of TLR4 mRNA in placental immune and non-immune cells increased 3–9-fold in obese mothers, which correlated with IL-6 expression in placental, leading to microglial activation in offspring (60, 61).
On the one hand, we can find that a variety of ASD-related genes could cause changes in microglia morphology or function, leading to the onset and development of the disease. On the other hand, heterogeneous environmental states, including maternal asthma, gestational diabetes, pre-eclampsia, and air pollutants exposure, are inducing microglial activation and increasing the risk of ASD (62–64). Each proinflammatory state may have multiple mechanisms of action, and one of the important mechanisms is that the activation microglia regulate metabolic stress, oxidative stress, and neuroendocrine mechanisms to affect neural development (65, 66). The assumption is that microglia are particularly important in the etiopathogenesis of genetic and environmental factors in ASD. Therefore, it is essential to improve our comprehension of how microglia affect neural development.
Several environmental risk factors increase the risk of ASD, such as lifestyle, prenatal or maternal exposure, including maternal smoking, toxins, gestational diabetes mellitus (GDM), thyroidism alteration, and infections (viral or bacterial) (4, 67). For instance, exposure to maternal GDM diagnosed by 26 weeks of gestation was linked to an increased risk of ASD in offspring in a large, multiethnic clinical cohort of singletons (68). Epidemiological studies reported a significant association between allergic diseases and ASD risk both in maternal and infants, including asthma, eczema, atopic dermatitis, allergic rhinitis, and food allergies (69–71). One study has demonstrated the activation of spinal microglia in adult asthma and atopic dermatitis models (72), whereas another study shows that allergic immune activation in prenatal maternal attenuated microglial activation in rats (73). A meta-analysis by Jiang et al. shows that maternal infections during the first or second trimester of pregnancy, whether bacterial, viral, or otherwise, were associated with a significantly increased risk of ASD in offspring (64). Two meta-analyses conducted by Chen et al. and Wu et al. have found that maternal autoimmune illness is associated with a significant, precise, and consistent increase in the risk of ASD in the offspring (62, 74). Maternal immune dysregulation during gestation is a high-risk factor for autism (75). In cohort studies, infections during pregnancy, such as rubella or influenza viruses, have been shown to have a significant impact on neurodevelopmental processes by causing immune disruptions and cytokine production in the mother (75). Those factors contributing to neurodevelopmental disorders may correlate with altered immune status characterized by microglial activation in various parts of the brain, which is also related to genetic and epigenetic-related effects, neurotransmitter alterations and abnormalities in signaling pathways, and endocrine disruption (4, 20, 76–78).
Maternal immune activation (MIA) models of monkeys and rodents have attracted much attention nowadays. A prenatal polyinosinic polycytidylic acid (poly I:C) model in rhesus monkeys represented increased repetitive behaviors, abnormal communication, and impaired social interactions. First-trimester MIA offspring showed atypical social behavior by inappropriately approaching or remaining in immediate proximity to an unfamiliar animal (79). Extensive work by Dr. Paul Patterson and other highly influential researchers has elucidated part of the mechanisms by which viral infection or viral mimetic MIA in rodents models, which can alter offspring immune function persistently and modify fetal brain development, ultimately lead to the phenotype of autism-like behaviors (42, 80, 81).
Additionally, research in both human and animal models suggested that MIA, during crucial times for neurodevelopment and immune system development, programs the fetal brain and immune system through inflammatory and epigenetic mechanisms (42). MIA models are identified to be related to altered immune status, increased oxidative stress, and an active neuroinflammatory process characterized by microglial activation in various brain regions (82, 83). There are a lot of cytokines that are related to the microglia involved in this progress. MIA could promote the release of pro-inflammatory cytokines, such as IL-6, IL-1β, and TNF-α. In addition, these cytokines may cross the placenta directly into fetal circulation (84) and promote the activation of microglia (85, 86). Then, activated microglia could secrete several cytokines, including IL-6, IL-1β, and TNF-α, to regulate neuronal function and neural plasticity (87).
The vital effect of those cytokines in fetal neurodevelopment of the MIA mice models has been well established (42, 88, 89). IL-6, one of the key factors, was enhanced in the maternal serum, as well as in the placenta and fetal brain in MIA models. Autism-like behavioral changes were not seen in the offspring of IL-6 knockout mice after MIA treatment (90). The combined use of anti-IL-6 antibodies in pregnant mice exposed to poly (I:C) prevented behavioral defects and normalized variations in brain gene expression (90). Wei and his colleagues have confirmed that IL-6 overexpression could impair and facilitate the formation of excitatory synapses between mouse cerebellar (91). IL-1β could inhibit long-term potentiation induction which is involved in reducing synaptic strength (92), as well as modulating memory and learning (93). TNF-α is another immunomodulatory molecule that could produce by glial cells in the CNS. Chronic increase of TNF-α may potentially hinder learning and memory in ASD by scaling up synapses during prolonged activity blockade (94).
Proinflammatory cytokines that cross the disruption BBB (driven by increased proliferation of microglia overexpressing of cyclooxygenase-2 in the fetal brain) may initiate a neuroinflammation cascade, which promotes microglial overactivation and behavioral alterations of ASD life span (95). Immune activation during pregnancy has been considered a potential cause of dysfunctional synaptic pruning and abnormal microglia-mediated neurogenesis (96, 97), which will be discussed soon.
As described by Peter in the early 1990s, early brain development was characterized by marked changes in synaptic connections. In the human cortex, synaptic density rises significantly during the first 1–2 years of life, after which competitive and activity-dependent abolition of synapses reduces synaptic connection density by approximately 50% (98). That prolonged pruning process was crucial for the proper development of brain circuits and cognitive functions. In autism, the long-term pruning process was crucial not only in shaping brain circuits by increasing dendritic spine densities but also in the normal development of cognitive function in the process of neural development (99). Besides, To keep a steadily internal environment homeostasis, cells are needed to be equipped with helpers capable of regulating synaptic exfoliation and remodeling, scavenging abnormal proteins, invading pathogens, and damaging tissue fragments (100). Activation of microglia can respond to invading pathogens and local fragments or proteins in a neuroprotective manner (101–103). There are similar pathophysiological characteristics can be identified in the brain between the neurodevelopment diseases and neurodegenerative diseases (104). These actions ensure appropriate signal transduction to regulate clearance processes and transition the response to one of resolution and repair, providing nutritional support for the release of various growth factors (104). Any deficiency in the ability of microglia, either a reduction in cell number or the loss of function, could alter the normal structure and function of brain health (104). Abnormal microglia function plays a constructive role in the early occurrence of those diseases (105, 106). As will be mentioned later, activated microglia cloud induce pathways that impacted synapse function and ultimately neuronal function.
Abnormal cortical circuit development can be found in many neurodevelopmental disorders and neuropsychiatric diseases, which may be a striking feature, including ASD, schizophrenia, and intellectual disability (107, 108). And deregulation of synaptic plasticity in ASD has been approved (109). The function of microglia in neural circuits and synaptic plasticity has received extensive attention (110, 111). In Tsc2+/− ASD mice and patDp/+ mice that it can be found elevated spined densities in the temporal cortex and cerebellum and defected adolescent pruning (47, 112). In neurons, axon guidance, vesicle release, dendritic spine structure, spine pruning, and synaptic plasticity were closely related to microglia function (113). Microglia dysfunction impaired synaptic pruning and led to deficits in social behavior (20). For example, microglia were found to play an important role in the development of synaptic plasticity. Atg7 is necessary for the formation of autophagic vesicles to transport substances to lysosomes. Atg7-deficient mice showed autism-like behaviors and increasing dendritic spine density. Atg7-deficient microglia co-cultured with neurons showed defective synaptosome breakdown, confirming that microglia malfunction causes aberrant synaptic pruning (20). The spine numbers were also increased in mice deficient in microglial Atg7 (114, 115). Ptenm3m4/m3m4 mice without nuclear PTEN have shown autism-like behaviors and active microglia with increased Iba1 and C1q expression, which identifies microglial activation has an etiological role in ASD via regulating synaptic pruning (116).
In the mammalian cerebral cortex, there is a dynamic process of simultaneous formation and elimination/pruning in postnatal synaptic development (117). There are excessive synapses produced in early life which is critical for the remodeling of neural circuits. Synaptic pruning is continuous from childhood to adolescence, and alters the density of dendritic spines peaks in early childhood, followed by a sharp decline to adult levels in later life, culminating in the formation of normal neural network relationships (118). Therefore, we list a number of elements and mechanisms that demonstrate the significance of microglia in synaptic function (Table 1).

Table 1. Related mechanisms in microglia acting on synapses.
Gamma-aminobutyric acid (GABA), an essential neurotransmitter, could promote neural stem cell proliferation, neuron migration, axon growth, synapse formation, and circuit perfection. In addition, as the major inhibitory neurotransmitter in the mature brain, it also could mediate the transmission of nerve signals (119). Emerging evidence suggested that it is a common pathophysiological phenomenon that imbalance between inhibitory and excitatory transmission of neurons in children with ASD (157). An imbalance of E/I in neural circuits has been postulated as a key neurobiological characteristic of ASD (158). Notably, evidence has shown abnormal expression of GABA receptors in the postmortem brain of patients with ASD (159).
Microglia express receptors for sensing synaptic activity, such as GABA and glutamate (120). GABA can initiate a transcriptional synaptic remodeling program within microglia to shape inhibitory connections without affecting excitatory synapses. Loss of function of GABA receptors in microglia disrupts this process and leads to abnormal behavior (121). Microglia depletion or GABA receptors ablation may lead to superabundant synapses (121). Co-culture with activated microglia impaired synapse formation and synaptic GABA release of induced pluripotent stem cells (iPSCs) (122). In mice, blocking GABA transport abolished stimulation-induced microglial responses (120). Furthermore, activation of the toll-like receptors 4 (TLR4) signaling pathway following maternal LPS exposure induced the abnormal activation of microglia, leading to lower social and exploration behavior, and more repetitive behaviors in offspring (160, 161). LPS can activate microglia through TLR4 and release the proinflammatory cytokines, IL-1β, which subsequently inhibits GABA receptor activity at postsynaptic sites and reduces GABA synthesis at presynaptic sites (123, 162).
The underlying process affecting aberrant synaptic plasticity is that E/I imbalance can be mediated not only by GABA but also by Glutamate (Glu) (124). Dysregulation of glutamatergic and dysconnectivity of functional in ASD are arising from the alterations of glutamatergic and GABAergic which changes brain functional connectivity and ultimately contribute to behavioral disabilities (163–165).
There are three main types of glutamate receptors called n-methyl-d-aspartate receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs), and metabotropic glutamate receptors (mGluRs) (125). NMDARs mediated synaptic overexpression led to the amplification of postsynaptic plasticity of neurons (126). Established evidence suggested that activity-dependent AMPARs insertion and removal from the postsynaptic membrane played an important role in the long-term plasticity of excitatory synaptic transmission (127). Much evidence supported that both NMDARs and AMPARs have been closely associated with ASD children (128). Moreover, the depletion of microglia could damage the function activity both in AMPARs and NMDARs (129).
Decreased glutamate concentrations in the cortical were linked with the severity of social impairments (125). Nlgn3KO mice, exhibiting observably autism-like behaviors, have markedly increased mGlu1 receptor expression in the brain, which is resulted from mGlu1 receptor blockade (130–132). RNA-Seq data were indicating that microglia from male MG4E mice (expressed highly eIF4E) upregulated the expression of several cytokines (C-X-C motif chemokine 10 (CXCL10), CXCL16, IL-1β). These cytokines might upregulate microglia expression neuroligins through post-translational modification, alter glutamate receptor function, and lead to mEPSCs amplitude increasing (57). Finally, E/I imbalance was often affected by an increase in excitatory synaptic function, as well as an increase in excitatory synapses density (166), which resulted in autism-like symptoms (57).
Brain-derived neurotrophic factor (BDNF) is the major principal neurotrophic cytokine in the CNS. It contributes to the development of the prenatal and postnatal brain, which can not only promote neuronal survival, growth, and differentiation (167) but also profound impact on synaptic and structural plasticity (133). The near meta-analysis showed that higher peripheral BDNF was consistent with several neurological and psychological theories on the etiology and core manifestations of ASD (167).
BDNF acting on microglia could increase the phosphorylation of neuronal tropomyosin-related kinase receptor B, an important mediator of synaptic plasticity (134). BDNF is occurred both at presynaptic and postsynaptic sites, promoting neurotransmitter releasing, promoting the function of ion-transmitters and NMDARs, and accelerating the potency NMDARs and ion-transmitters (135). Overall, BDNF appeared to enhance excitatory synapses and weakened inhibitory synapses, leading to an imbalance of E/I (45). Similar conclusions were reached in gene-depleted microglia BDNF (134).
BDNF was found to be highly expressed in children with ASD (136), BDNF may lead to increased synthesis of synaptic proteins associated with autism and participate in the development of autism (137), which may enhance synaptic plasticity (138)or increase the density of dendritic spine (139). Besides, BDNF mRNA levels can be decreased by increased levels of the pro-inflammatory cytokine IL-1β, which is mainly expressed by activated microglia (140).
Together, these findings showed a crucial link between higher BDNF levels in the neurons of people with ASD (133), inducing synapse formation, altering functional connectivity and myelination, and modulating behavioral performance (141).
DNAX-activating protein of 12 kDa (DAP12) is a signaling protein expressed by a variety of cells for general function, which act in immune responses (108). Triggering receptors expressed on myeloid cells (TREMs) are a family of cell surface receptors expressed broadly on myeloid cells, TREM2 stimulates the protein tyrosine kinase ERK via DAP12, and TREM2-DAP2 is a complex expressed exclusively in microglia in the CNS (142), sustaining many transcriptional programs (108, 143). TREM2 is anchored on the surface of microglia and interacts with DAP12 to initiate signal transduction pathways that promote microglia activation, apoptosis, phagocytosis, and survival. The defective TREM2-DAP12 function is identified to be involved in the pathogenesis of ASD (144).
Interestingly, TREM2 is highly expressed in resting (unstimulated) microglia and downregulated after LPS or IFN-γ inflammatory stimulation (135). Among the many complex processes that happened during brain development, the TREM2-DAP12 axis is critical for maintaining central nervous system tissue homeostasis by modulating microglia phagocytosis and the overall fitness of microglia throughout their life cycle (107). The expression of TREM2 in microglia decreased the ability of phagocytic membrane fragments and increased the gene transcription of proinflammatory cytokines (107). Notably, long-term circuit hyperexcitability and decreased functional connectivity were observed in Trem2 (KO) mice, which displayed modifications in social behavior and repetitive behaviors resembling the phenotype of autism in humans (144).
The CX3CL1 (fractalkine)-CX3CR1 signaling pathway is the most important communication channel between neurons and microglia. Neurons' expression of the CX3CL1 and connection with CX3CR1 expression on the surface of microglia, their combined interaction plays an important role in regulating neurons maturation and function (108).
CX3CR1, encoded by the Cx3cr1 gene, is a Gi-protein coupled receptor that is mainly expressed by microglia in the CNS (168). CX3CL1 is mainly expressed in neurons, combined with CX3CR1 (169), and is thought to be a shutdown signal, maintaining microglia in a resting situation (168, 170). On the one hand, soluble fractalkine might act to promote microglia migration into the brain or proliferation during development; on the other hand, tethered or locally released fractalkine might be critical for microglia recognition of synapses before or during engulfment, in which case the density of microglia might be normal but the efficiency of engulfment might be reduced (11).
In the Cx3cr1-deficient mice, a transient decrease in microglia density was founded at different stages of development (Postnatal days 8, 15, 28), which impaired its ability to phagocytose synaptic material (11). In the mice hippocampus, altered phagocytosis causes a transient increase in dendritic spine density and a raising in Psd95 protein detected in microglial activation (11). Immature connections enhanced long-term depression (LTD), and impaired function of excitatory synaptic networks during the development of the hippocampal, especially the postnatal area, leads to altered neural function (145). Both young and adult mice exhibited deficits in social communication and repetitive behaviors increasing, which were closely linked to decreased functional connectivity and reduced synaptic transmission in the CNS (11, 146). CX3CL1 can also affect synaptic plasticity. For example, upregulation of CX3CL1 expression in the hippocampus is associated with processes that shape memory-related synaptic plasticity (147). Yet, a detectable reduction in long-term potentiation (LTP) correlates with synaptic function in Cx3cr1-deficient mice (148). In conclusion, once the microglia-specific Cx3cr1 gene was knocked out, it resulted in a transient decrease of microglia, defects in synaptic pruning, and autism-like behavior (146).
The synapses morphogenesis was relying on the signal of CX3CL1-CX3CR1 induced by microglia (11), and this signaling is also a key pathway for neuron-microglia interactions (149).
Microglia play vital roles in eliminating excess synapses through the process of synapse pruning (150). This process requires involving of complement, for instance, labeling of unnecessary synapses with complement 3 (C3), and subsequent recognition of C3 by microglia C3 receptors (151).
Complement 4 (C4) promotes the activation of C3, making C3 firmly attached to its target, mediating the phagocytosis of the labeled target by microglia (152). Therefore, C4 deficiency led to synaptic elimination aberrant (153). In a complement-focused study, patients with ASD have significantly higher degrees of C4B variant deficiency, compared with controls (154), it could also find that C1q, C3, and C4 mRNA levels were visibly reduced in the prefrontal cortex (155). Inhibition of C1q, C3, or CR3 reduces the number of phagocyte microglia and the degree of early synaptic loss (156).
Collectively, interactions between complements may affect synaptic recognition and phagocytosis by microglia. Future research on C4 risk variants and other complementary systems is needed to uncover the possible mechanisms for psychiatric disorders and may provide new ideas for treatment (22).
Taken together, functional mechanisms of microglia affecting ASD appear to be increasing. Evidence supports a role for microglia influenced by genetics and environment is crucial in ASD pathogenesis. According to the similar pathophysiological characteristics which have been identified in both neurodegenerative diseases and ASD, we assume that microglia have irreplaceable roles in shaping neuronal connectivity by regulating synaptic plasticity, and E/I balance. We summarize the essential cytokines and pathways related to microglia dysfunction which leads to impairments of behaviors. Suggested microglia are essential for neurodevelopment in ASD, far beyond current research understanding (Figure 1). We believe that exploring the mechanisms of action of microglia in ASD is critical for future prevention and treatment.
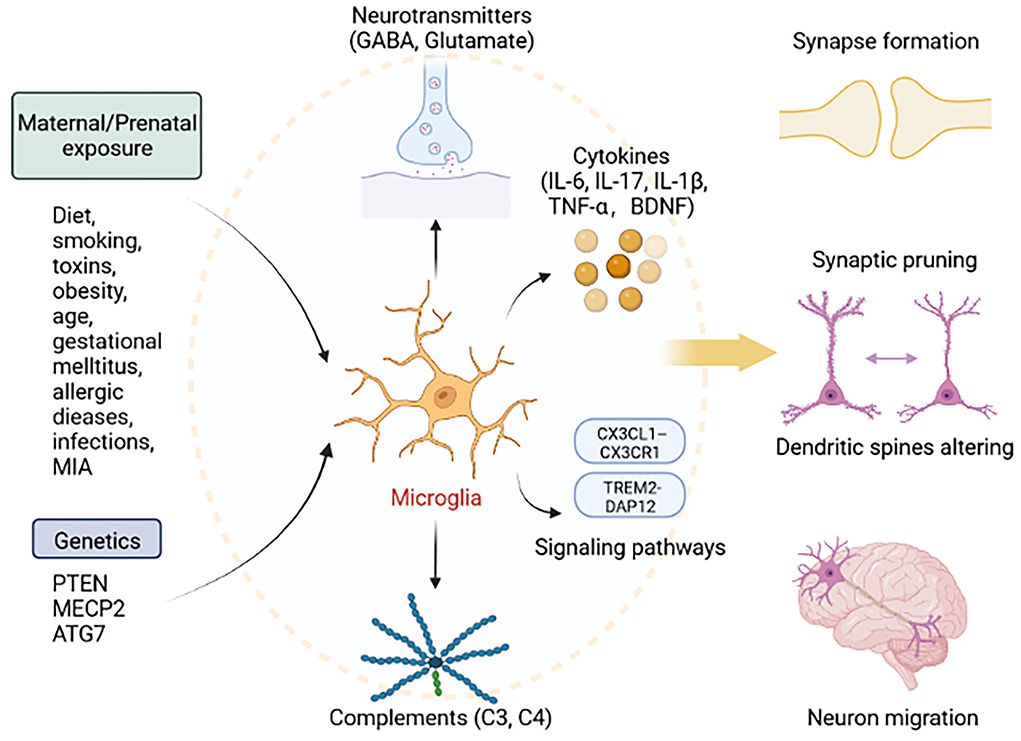
Figure 1. Roles of microglia in ASD. Exposure to factors during pregnancy, including diet, smoking, toxins, obesity, age, gestational mellitus, allergic diseases, infections, MIA, and genetic factors, including mutations in PTEN, MECP2, and ATG7, can lead to abnormal microglial function. Microglia can secrete cytokines (IL-6, IL-17, IL-1β, TNF-α, and BDNF), neurotransmitters (GABA, Glutamate), and complements (C3 and C4), and activate signaling pathway (TREM2-DAP12 and CX3CL1–CX3CR1). Thus, activated microglia lead to the abnormal development of synapse formation, synaptic pruning, dendritic spines altering, and neuron migration. This figure was created with BioRender.com.
CH and JL made manuscript planning and structuring. HL and CH did literature searching and drafted the manuscript. YH and XL revised the manuscript and edited the final manuscript. All authors have read and approved the final version of the manuscript.
This work was supported by Huazhong University of Science and Technology Emergency Technology Research Project Response to COVID-19 (Grant Number 2020kfyXGYJ020) and Key Project of Independent Innovation Research Fund of Huazhong University of Science and Technology (Grant Number 2017KFYXJJ100).
The authors would like to thank all participants for their important contributions to this review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Battle DE. Diagnostic and Statistical Manual of Mental Disorders (DSM). Codas. (2013) 25:191–2. doi: 10.1590/s2317-17822013000200017
2. Maenner MJ, Shaw KA, Bakian AV, Bilder DA, Durkin MS, Esler A, et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveill Summ. (2021) 70:1–16. doi: 10.15585/mmwr.ss7011a1
3. Wang T, Guo H, Xiong B, Stessman HAF, Wu H, Coe BP, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. (2016) 7:13316. doi: 10.1038/ncomms13316
4. Modabbernia A, Velthorst E, Reichenberg A. Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol Autism. (2017) 8:13. doi: 10.1186/s13229-017-0121-4
5. Bai D, Yip BHK, Windham GC, Sourander A, Francis R, Yoffe R, et al. Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry. (2019) 76:1035–43. doi: 10.1001/jamapsychiatry.2019.1411
6. Lord C, Brugha TS, Charman T, et al. Autism spectrum disorder. Nat Rev Dis Primers. (2020) 6:5. doi: 10.1038/s41572-019-0138-4
7. Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. (2014) 383:896–910. doi: 10.1016/S0140-6736(13)61539-1
8. Ormstad H, Bryn V, Saugstad OD, Skjeldal O. Maes M. Role of the Immune System in Autism Spectrum Disorders (ASD). CNS Neurol Disord Drug Targets. (2018) 17:489–95. doi: 10.2174/1871527317666180706123229
9. Bjorklund G, Saad K, Chirumbolo S, Kern JK, Geier DA, Geier MR, et al. Immune dysfunction and neuroinflammation in autism spectrum disorder. Acta Neurobiol Exp (Wars). (2016) 76:257–68. doi: 10.21307/ane-2017-025
10. Mattei D, Ivanov A, Ferrai C, Jordan P, Guneykaya D, Buonfiglioli A, et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry. (2017) 7:e1120. doi: 10.1038/tp.2017.80
11. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
12. Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. (2009) 29:3974–80. doi: 10.1523/JNEUROSCI.4363-08.2009
13. Tremblay MÈ, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. (2010) 8:e1000527. doi: 10.1371/journal.pbio.1000527
14. Lindholm D, Castrén E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-beta 1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol. (1992) 117:395–400. doi: 10.1083/jcb.117.2.395
15. Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. (2013) 155:1451–63. doi: 10.1016/j.cell.2013.11.024
16. Chen Z, Trapp BD. Microglia and neuroprotection. J Neurochem. (2016) 136 Suppl 1:10–7. doi: 10.1111/jnc.13062
17. Toscano C, Barros L, Lima AB, Nunes T, Carvalho HM, Gaspar JM. Neuroinflammation in autism spectrum disorders: exercise as a “pharmacological” tool. Neurosci Biobehav Rev. (2021) 129:63–74. doi: 10.1016/j.neubiorev.2021.07.023
18. Miyanishi K, Sato A, Kihara N, Utsunomiya R, Tanaka J. Synaptic elimination by microglia and disturbed higher brain functions. Neurochem Int. (2021) 142:104901. doi: 10.1016/j.neuint.2020.104901
19. Buffelli M, Burgess RW, Feng G, Lobe CG, Lichtman JW, Sanes JR. Genetic evidence that relative synaptic efficacy biases the outcome of synaptic competition. Nature. (2003) 424:430–4. doi: 10.1038/nature01844
20. Kim H-J, Cho M-H, Shim WH, Kim JK, Jeon E-Y, Kim D-H, et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry. (2017) 22:1576–84. doi: 10.1038/mp.2016.103
21. Koyama R, Ikegaya Y. Microglia in the pathogenesis of autism spectrum disorders. Neurosci Res. (2015) 100:1–5. doi: 10.1016/j.neures.2015.06.005
22. Bar E, Barak B. Microglia roles in synaptic plasticity and myelination in homeostatic conditions and neurodevelopmental disorders. Glia. (2019) 67:2125–41. doi: 10.1002/glia.23637
23. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
24. Theoharides TC, Zhang B. Neuro-inflammation, blood-brain barrier, seizures and autism. J Neuroinflammation. (2011) 8:168. doi: 10.1186/1742-2094-8-168
25. Esposito P, Chandler N, Kandere K, Basu S, Jacobson S, Connolly R, et al. Corticotropin-releasing hormone and brain mast cells regulate blood-brain-barrier permeability induced by acute stress. J Pharmacol Exp Ther. (2002) 303:1061–6. doi: 10.1124/jpet.102.038497
26. Ford TC, Crewther DP, Abu-Akel A. Psychosocial deficits across autism and schizotypal spectra are interactively modulated by excitatory and inhibitory neurotransmission. Autism. (2020) 24:364–73. doi: 10.1177/1362361319866030
27. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu1- and Irf8-dependent pathways. Nat Neurosci. (2013) 16:273–80. doi: 10.1038/nn.3318
28. Mosser CA, Baptista S, Arnoux I, Audinat E. Microglia in CNS development: shaping the brain for the future. Prog Neurobiol. (2017) 149-150:1-20. doi: 10.1016/j.pneurobio.2017.01.002
29. Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. (2013) 77:10–8. doi: 10.1016/j.neuron.2012.12.023
30. Cornell J, Salinas S, Huang HY, Zhou M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen Res. (2022) 17:705–16. doi: 10.4103/1673-5374.322423
31. Lee EJ, Choi SY, Kim E, NMDA. receptor dysfunction in autism spectrum disorders. Curr Opin Pharmacol. (2015) 20:8–13. doi: 10.1016/j.coph.2014.10.007
32. Skaper SD, Facci L, Giusti P. Mast cells, glia and neuroinflammation: partners in crime. Immunology. (2014) 141:314–27. doi: 10.1111/imm.12170
33. Dong H, Zhang X, Qian Y. Mast cells and neuroinflammation. Med Sci Monit Basic Res. (2014) 20:200–6. doi: 10.12659/MSMBR.893093
34. Cowan M, Petri WA Jr. Microglia: immune regulators of neurodevelopment. Front Immunol. (2018) 9:2576. doi: 10.3389/fimmu.2018.02576
35. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
36. Zhang S, Dong H, Zhang X, Li N, Sun J, Qian Y. Cerebral mast cells contribute to postoperative cognitive dysfunction by promoting blood brain barrier disruption. Behav Brain Res. (2016) 298(Pt B): 158–66. doi: 10.1016/j.bbr.2015.11.003
37. Na KS, Jung HY, Kim YK. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. (2014) 48:277–86. doi: 10.1016/j.pnpbp.2012.10.022
38. Zhu Q, Enkhjargal B, Huang L, Zhang T, Sun C, Xie Z, et al. Aggf1 attenuates neuroinflammation and BBB disruption via PI3K/Akt/NF-κB pathway after subarachnoid hemorrhage in rats. J Neuroinflammation. (2018) 15:178. doi: 10.1186/s12974-018-1211-8
39. Hendriksen E, van Bergeijk D, Oosting RS, Redegeld FA. Mast cells in neuroinflammation and brain disorders. Neurosci Biobehav Rev. (2017) 79:119–33. doi: 10.1016/j.neubiorev.2017.05.001
40. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. (2005) 57:67–81. doi: 10.1002/ana.20315
41. Rodriguez RM, Suarez-Alvarez B, Lopez-Larrea C. Therapeutic epigenetic reprogramming of trained immunity in myeloid cells. Trends Immunol. (2019) 40:66–80. doi: 10.1016/j.it.2018.11.006
42. Han VX, Patel S, Jones HF, Dale RC. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat Rev Neurol. (2021) 17:564–79. doi: 10.1038/s41582-021-00530-8
43. Meyer U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry. (2014) 75:307–15. doi: 10.1016/j.biopsych.2013.07.011
44. Heiss CN, Olofsson LE. The role of the gut microbiota in development, function and disorders of the central nervous system and the enteric nervous system. J Neuroendocrinol. (2019) 31:e12684. doi: 10.1111/jne.12684
45. Das UN. Autism as a disorder of deficiency of brain-derived neurotrophic factor and altered metabolism of polyunsaturated fatty acids. Nutrition. (2013) 29:1175–85. doi: 10.1016/j.nut.2013.01.012
46. Tetreault NA, Hakeem AY, Jiang S, Williams BA, Allman E, Wold BJ, et al. Microglia in the cerebral cortex in autism. J Autism Dev Disord. (2012) 42:2569–84. doi: 10.1007/s10803-012-1513-0
47. Tang G, Gudsnuk K, Kuo S-H, Cotrina ML, Rosoklija G, Sosunov A, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. (2014) 83:1131–43. doi: 10.1016/j.neuron.2014.07.040
48. Nutma E, Ceyzériat K, Amor S, Tsartsalis S, Millet P, Owen DR, et al. Cellular sources of TSPO expression in healthy and diseased brain. Eur J Nucl Med Mol Imaging. (2021) 49:146–63. doi: 10.1007/s00259-020-05166-2
49. Wang W, Zhang L, Zhang X, Xue R, Li L, Zhao W, et al. Lentiviral-Mediated Overexpression of the 18 kDa Translocator Protein (TSPO) in the Hippocampal Dentate Gyrus Ameliorates LPS-Induced cognitive impairment in mice. Front Pharmacol. (2016) 7:384. doi: 10.3389/fphar.2016.00384
50. Simpson D, Gharehgazlou A, Da Silva T, Labrie-Cleary C, Wilson AA, Meyer JH, et al. In vivo imaging translocator protein (TSPO) in autism spectrum disorder. Neuropsychopharmacology. (2022) 47:1421–7. doi: 10.1038/s41386-022-01306-4
51. Glaze DG. Rett syndrome: of girls and mice–lessons for regression in autism. Ment Retard Dev Disabil Res Rev. (2004) 10:154–8. doi: 10.1002/mrdd.20030
52. Banerjee A, Miller MT, Li K, Sur M, Kaufmann WE. Towards a better diagnosis and treatment of Rett syndrome: a model synaptic disorder. Brain. (2019) 142:239–48. doi: 10.1093/brain/awy323
53. Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SBG, Guyenet PG, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. (2012) 484:105–9. doi: 10.1038/nature10907
54. Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. (2010) 30:5346–56. doi: 10.1523/JNEUROSCI.5966-09.2010
55. Schafer DP, Heller CT, Gunner G, Heller M, Gordon C, Hammond T, et al. Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. Elife. (2016) 5. doi: 10.7554/eLife.15224
56. Richter JD. Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. (2005) 433:477–80. doi: 10.1038/nature03205
57. Xu Z-X, Kim GH, Tan J-W, Riso AE, Sun Y, Xu EY, et al. Elevated protein synthesis in microglia causes autism-like synaptic and behavioral aberrations. Nat Commun. (2020) 11:1797. doi: 10.1038/s41467-020-15530-3
58. Keenan-Devlin LS, Ernst LM, Ross KM, Qadir S, Grobman WA, Holl JL, et al. Maternal income during pregnancy is associated with chronic placental inflammation at birth. Am J Perinatol. (2017) 34:1003–10. doi: 10.1055/s-0037-1601353
59. Altmäe S, Segura MT, Esteban FJ, Bartel S, Brandi P, Irmler M, et al. Maternal pre-pregnancy obesity is associated with altered placental transcriptome. PLoS ONE. (2017) 12:e0169223. doi: 10.1371/journal.pone.0169223
60. Koga K, Mor G. Expression and function of toll-like receptors at the maternal-fetal interface. Reprod Sci. (2008) 15:231–42. doi: 10.1177/1933719108316391
61. Yang X, Li M, Haghiac M, Catalano PM, O'Tierney-Ginn P, Hauguel-de Mouzon S. Causal relationship between obesity-related traits and TLR4-driven responses at the maternal-fetal interface. Diabetologia. (2016) 59:2459–66. doi: 10.1007/s00125-016-4073-6
62. Chen S-W, Zhong X-S, Jiang L-N, Zheng X-Y, Xiong Y-Q, Ma S-J, et al. Maternal autoimmune diseases and the risk of autism spectrum disorders in offspring: a systematic review and meta-analysis. Behav Brain Res. (2016) 296:61–9. doi: 10.1016/j.bbr.2015.08.035
63. Croen LA, Grether JK, Yoshida CK, Odouli R, Van de Water J. Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders: a case-control study. Arch Pediatr Adolesc Med. (2005) 159:151–7. doi: 10.1001/archpedi.159.2.151
64. Jiang H-Y, Xu L-L, Shao L, Xia R-M, Yu Z-H, Ling Z-X, et al. Maternal infection during pregnancy and risk of autism spectrum disorders: a systematic review and meta-analysis. Brain Behav Immun. (2016) 58:165–72. doi: 10.1016/j.bbi.2016.06.005
65. Jones HF, Han VX, Patel S, Gloss BS, Soler N, Ho A, et al. Maternal autoimmunity and inflammation are associated with childhood tics and obsessive-compulsive disorder: Transcriptomic data show common enriched innate immune pathways. Brain Behav Immun. (2021) 94:308–17. doi: 10.1016/j.bbi.2020.12.035
66. Han VX, Patel S, Jones HF, Nielsen TC, Mohammad SS, Hofer MJ, et al. Maternal acute and chronic inflammation in pregnancy is associated with common neurodevelopmental disorders: a systematic review. Transl Psychiatry. (2021) 11:71. doi: 10.1038/s41398-021-01198-w
67. Banik A, Kandilya D, Ramya S, Stünkel W, Chong YS, Dheen ST. Maternal factors that induce epigenetic changes contribute to neurological disorders in offspring. Genes (Basel). (2017) 8:150. doi: 10.3390/genes8060150
68. Xiang AH, Wang X, Martinez MP, Walthall JC, Curry ES, Page K, et al. Association of maternal diabetes with autism in offspring. JAMA. (2015) 313:1425–34. doi: 10.1001/jama.2015.2707
69. Chen M-H, Su T-P, Chen Y-S, Hsu J-W, Huang K-L, Chang W-H, et al. Is atopy in early childhood a risk factor for ADHD and ASD? a longitudinal study. J Psychosom Res. (2014) 77:316–21. doi: 10.1016/j.jpsychores.2014.06.006
70. Li Y, Missig G, Finger BC, Landino SM, Alexander AJ, Mokler EL, et al. Maternal immune conditions are increased in males with autism spectrum disorders and are associated with behavioural and emotional but not cognitive co-morbidity. Transl Psychiatry. (2020) 10:286. doi: 10.1038/s41398-020-00976-2
71. Saitoh B-Y, Tanaka E, Yamamoto N, van Kruining D, Iinuma K, Nakamuta Y, et al. Early postnatal allergic airway inflammation induces dystrophic microglia leading to excitatory postsynaptic surplus and autism-like behavior. Brain Behav Immun. (2021) 95:362–80. doi: 10.1016/j.bbi.2021.04.008
72. Yamasaki R, Fujii T, Wang B, Masaki K, Kido MA, Yoshida M, et al. Allergic inflammation leads to neuropathic pain via glial cell activation. J Neurosci. (2016) 36:11929–45. doi: 10.1523/JNEUROSCI.1981-16.2016
73. Lenz KM, Pickett LA, Wright CL, Galan A, McCarthy MM. Prenatal allergen exposure perturbs sexual differentiation and programs lifelong changes in adult social and sexual behavior. Sci Rep. (2019) 9:4837. doi: 10.1038/s41598-019-41258-2
74. Wu S, Ding Y, Wu F, Li R, Xie G, Hou J, et al. Family history of autoimmune diseases is associated with an increased risk of autism in children: a systematic review and meta-analysis. Neurosci Biobehav Rev. (2015) 55:322–32. doi: 10.1016/j.neubiorev.2015.05.004
75. Meltzer A, Van de Water J. The role of the immune system in autism spectrum disorder. Neuropsychopharmacology. (2017) 42:284–98. doi: 10.1038/npp.2016.158
76. Lauritsen MB, Pedersen CB, Mortensen PB. Effects of familial risk factors and place of birth on the risk of autism: a nationwide register-based study. J Child Psychol Psychiatry. (2005) 46:963–71. doi: 10.1111/j.1469-7610.2004.00391.x
77. Becker KG. Autism, asthma, inflammation, and the hygiene hypothesis. Med Hypotheses. (2007) 69:731–40. doi: 10.1016/j.mehy.2007.02.019
78. Blaylock RL, A. possible central mechanism in autism spectrum disorders, part 2: immunoexcitotoxicity. Altern Ther Health Med. (2009) 15:60–7.
79. Haddad FL, Patel SV, Schmid S. Maternal immune activation by poly i:c as a preclinical model for neurodevelopmental disorders: a focus on autism and schizophrenia. Neurosci Biobehav Rev. (2020) 113:546–67. doi: 10.1016/j.neubiorev.2020.04.012
80. Xu Z, Zhang X, Chang H, Kong Y, Ni Y, Liu R, et al. Rescue of maternal immune activation-induced behavioral abnormalities in adult mouse offspring by pathogen-activated maternal T(reg) cells. Nat Neurosci. (2021) 24:818–30. doi: 10.1038/s41593-021-00837-1
81. Reed MD, Yim YS, Wimmer RD, Kim H, Ryu C, Welch GM, et al. IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature. (2020) 577:249–53. doi: 10.1038/s41586-019-1843-6
82. Goines PE, Ashwood P. Cytokine dysregulation in autism spectrum disorders (ASD): possible role of the environment. Neurotoxicol Teratol. (2013) 36:67–81. doi: 10.1016/j.ntt.2012.07.006
83. Frustaci A, Neri M, Cesario A, Adams JB, Domenici E, Bernardina BD, et al. Oxidative stress-related biomarkers in autism: systematic review and meta-analyses. Free Radic Biol Med. (2012) 52:2128–41. doi: 10.1016/j.freeradbiomed.2012.03.011
84. Zawadzka A, Cieślik M, Adamczyk A. The role of maternal immune activation in the pathogenesis of autism: a review of the evidence, proposed mechanisms and implications for treatment. Int J Mol Sci. (2021) 22:1516. doi: 10.3390/ijms222111516
85. Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol. (2012) 71:444–57. doi: 10.1002/ana.22620
86. Saghazadeh A, Ataeinia B, Keynejad K, Abdolalizadeh A, Hirbod-Mobarakeh A, Rezaei N, et al. meta-analysis of pro-inflammatory cytokines in autism spectrum disorders: Effects of age, gender, and latitude. J Psychiatr Res. (2019) 115:90–102. doi: 10.1016/j.jpsychires.2019.05.019
87. Koo JW, Wohleb ES. How stress shapes neuroimmune function: implications for the neurobiology of psychiatric disorders. Biol Psychiatry. (2021) 90:74–84. doi: 10.1016/j.biopsych.2020.11.007
88. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. (2016) 351:933–9. doi: 10.1126/science.aad0314
89. Heuer LS, Croen LA, Jones KL, Yoshida CK, Hansen RL, Yolken R, et al. An Exploratory Examination of neonatal cytokines and chemokines as predictors of autism risk: the early markers for autism study. Biol Psychiatry. (2019) 86:255–64. doi: 10.1016/j.biopsych.2019.04.037
90. Smith SE Li J, Garbett K, Mirnics K. Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. (2007) 27:10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
91. Wei H, Zou H, Sheikh AM, Malik M, Dobkin C, Brown WT, et al. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflammation. (2011) 8:52. doi: 10.1186/1742-2094-8-52
92. Bellinger FP, Madamba S, Siggins GR. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. (1993) 628:227–34. doi: 10.1016/0006-8993(93)90959-Q
93. Goshen I, Kreisel T, Ounallah-Saad H, Renbaum P, Zalzstein Y, Ben-Hur T, et al. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology. (2007) 32:1106–15. doi: 10.1016/j.psyneuen.2007.09.004
94. Steinmetz CC, Turrigiano GG. Tumor necrosis factor-α signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci. (2010) 30:14685–90. doi: 10.1523/JNEUROSCI.2210-10.2010
95. Zhao Q, Dai W, Chen HY, Jacobs RE, Zlokovic BV, Lund BT, et al. Prenatal disruption of blood-brain barrier formation via cyclooxygenase activation leads to lifelong brain inflammation. Proc Natl Acad Sci U S A. (2022) 119:e2113310119. doi: 10.1073/pnas.2113310119
96. Maldonado-Ruiz R, Garza-Ocañas L, Camacho A. Inflammatory domains modulate autism spectrum disorder susceptibility during maternal nutritional programming. Neurochem Int. (2019) 126:109–17. doi: 10.1016/j.neuint.2019.03.009
97. Estes ML, McAllister AK. Maternal immune activation: Implications for neuropsychiatric disorders. Science. (2016) 353:772–7. doi: 10.1126/science.aag3194
98. PR. Morphometric study of human cerebral cortex development. Neuropsychologia. (1990) 28:517–27. doi: 10.1016/0028-3932(90)90031-I
99. Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. (2010) 1309:83–94. doi: 10.1016/j.brainres.2009.09.120
100. Hickman S, Izzy S, Sen P, Morsett L. El Khoury J. Microglia in neurodegeneration. Nat Neurosci. (2018) 21:1359–69. doi: 10.1038/s41593-018-0242-x
101. Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. (2004) 173:3916–24. doi: 10.4049/jimmunol.173.6.3916
102. Glezer I, Lapointe A, Rivest S. Innate immunity triggers oligodendrocyte progenitor reactivity and confines damages to brain injuries. FASEB J. (2006) 20:750–2. doi: 10.1096/fj.05-5234fje
103. Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. (2007) 147:867–83. doi: 10.1016/j.neuroscience.2007.02.055
104. Harry GJ. Microglia during development and aging. Pharmacol Ther. (2013) 139:313–26. doi: 10.1016/j.pharmthera.2013.04.013
105. Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. (2009) 118:475–85. doi: 10.1007/s00401-009-0556-6
106. Konishi H, Kiyama H. Non-pathological roles of microglial TREM2/DAP12: TREM2/DAP12 regulates the physiological functions of microglia from development to aging. Neurochem Int. (2020) 141:104878. doi: 10.1016/j.neuint.2020.104878
107. Neumann H, Takahashi K. Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J Neuroimmunol. (2007) 184:92–9. doi: 10.1016/j.jneuroim.2006.11.032
108. Mecca C, Giambanco I, Donato R, Arcuri C. Microglia and aging: the role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int J Mol Sci. (2018) 19:318. doi: 10.3390/ijms19010318
109. Hansel C. Deregulation of synaptic plasticity in autism. Neurosci Lett. (2019) 688:58–61. doi: 10.1016/j.neulet.2018.02.003
110. Südhof TC. Towards an understanding of synapse formation. Neuron. (2018) 100:276–93. doi: 10.1016/j.neuron.2018.09.040
111. Neniskyte U, Gross CT. Errant gardeners: glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat Rev Neurosci. (2017) 18:658–70. doi: 10.1038/nrn.2017.110
112. Piochon C, Kloth AD, Grasselli G, Titley HK, Nakayama H, Hashimoto K, et al. Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nat Commun. (2014) 5:5586. doi: 10.1038/ncomms6586
113. Fujita Y, Yamashita T. Mechanisms and significance of microglia-axon interactions in physiological and pathophysiological conditions. Cell Mol Life Sci. (2021) 78:3907–19. doi: 10.1007/s00018-021-03758-1
114. Lieberman OJ, McGuirt AF, Tang G, Sulzer D. Roles for neuronal and glial autophagy in synaptic pruning during development. Neurobiol Dis. (2019) 122:49–63. doi: 10.1016/j.nbd.2018.04.017
115. Wilton DK, Dissing-Olesen L. Stevens B. Neuron-glia signaling in synapse elimination. Annu Rev Neurosci. (2019) 42:107–27. doi: 10.1146/annurev-neuro-070918-050306
116. Sarn N, Jaini R, Thacker S, Lee H, Dutta R, Eng C. Cytoplasmic-predominant Pten increases microglial activation and synaptic pruning in a murine model with autism-like phenotype. Mol Psychiatry. (2021) 26:1458–71. doi: 10.1038/s41380-020-0681-0
117. Rakic P, Bourgeois JP, Eckenhoff MF, Zecevic N, Goldman-Rakic PS. Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science. (1986) 232:232–5. doi: 10.1126/science.3952506
118. Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. (2011) 14:285–93. doi: 10.1038/nn.2741
119. Salmon CK, Pribiag H, Gizowski C, Farmer WT, Cameron S, Jones EV, et al. Depolarizing GABA transmission restrains activity-dependent glutamatergic synapse formation in the developing hippocampal circuit. Front Cell Neurosci. (2020) 14:36. doi: 10.3389/fncel.2020.00036
120. Logiacco F, Xia P, Georgiev SV, Franconi C, Chang Y-J, Ugursu B, et al. Microglia sense neuronal activity via GABA in the early postnatal hippocampus. Cell Rep. (2021) 37:110128. doi: 10.1016/j.celrep.2021.110128
121. Favuzzi E, Huang S, Saldi GA, et al. GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell. (2021) 184:4048–4063.e32. doi: 10.1016/j.cell.2021.06.018
122. Park G-H, Noh H, Shao Z, Ni P, Qin Y, Liu D, et al. Activated microglia cause metabolic disruptions in developmental cortical interneurons that persist in interneurons from individuals with schizophrenia. Nat Neurosci. (2020) 23:1352–64. doi: 10.1038/s41593-020-00724-1
123. Wahab DYAA, Gau CH, Zakaria R, Karuppan MKM, A-Rahbi BS, Abdullah Z, et al. Review on cross talk between neurotransmitters and neuroinflammation in striatum and cerebellum in the mediation of motor behaviour. Biomed Res Int. (2019) 2019:1767203. doi: 10.1155/2019/1767203
124. Fatemi SH. The hyperglutamatergic hypothesis of autism. Prog Neuropsychopharmacol Biol Psychiatry. (2008) 32:911. doi: 10.1016/j.pnpbp.2007.11.005
125. Antoni S, Spatuzza M, Bonaccorso CM, Musumeci SA, Ciranna L, et al. Dysregulation of group-I metabotropic glutamate (mGlu) receptor mediated signalling in disorders associated with Intellectual Disability and Autism. Neurosci Biobehav Rev. (2014) 46 Pt 228–241. doi: 10.1016/j.neubiorev.2014.02.003
126. Rinaldi T, Kulangara K, Antoniello K, Markram H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc Natl Acad Sci U S A. (2007) 104:13501–6. doi: 10.1073/pnas.0704391104
127. Huganir RL. Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. (2013) 80:704–17. doi: 10.1016/j.neuron.2013.10.025
128. Marotta R, Risoleo MC, Messina G, Parisi L, Carotenuto M, Vetri L, et al. The neurochemistry of autism. Brain Sci. (2020) 10:163. doi: 10.3390/brainsci10030163
129. Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, Inokuchi K. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci. (2012) 32:10413–22. doi: 10.1523/JNEUROSCI.4533-11.2012
130. McFarlane HG, Kusek GK, Yang M, Phoenix JL, Bolivar VJ, Crawley JN. Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav. (2008) 7:152–63. doi: 10.1111/j.1601-183X.2007.00330.x
131. Baudouin SJ, Gaudias J, Gerharz S, Hatstatt L, Zhou K, Punnakkal P, et al. Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science. (2012) 338:128–32. doi: 10.1126/science.1224159
132. Silverman JL, Smith DG, Rizzo SJS, Karras MN, Turner SM, Tolu SS, et al. Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci Transl Med. (2012) 4: 131ra51. doi: 10.1126/scitranslmed.3003501
133. Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. (2013) 14:7–23. doi: 10.1038/nrn3379
134. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates 3rd JR, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. (2013) 155:1596–609. doi: 10.1016/j.cell.2013.11.030
135. Saghazadeh A, Rezaei N. Brain-derived neurotrophic factor levels in autism: a systematic review and meta-analysis. J Autism Dev Disord. (2017) 47:1018–29. doi: 10.1007/s10803-016-3024-x
136. Ricci S, Businaro R, Ippoliti F, Vasco VRL, Massoni F, Onofri E, et al. Altered cytokine and BDNF levels in autism spectrum disorder. Neurotox Res. (2013) 24:491–501. doi: 10.1007/s12640-013-9393-4
137. Zaslavsky K, Zhang W-B, McCready FP, Rodrigues DC, Deneault E, Loo C, et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat Neurosci. (2019) 22:556–64. doi: 10.1038/s41593-019-0365-8
138. Oberman LM, Pascual-Leone A. Hyperplasticity in autism spectrum disorder confers protection from Alzheimer's disease. Med Hypotheses. (2014) 83:337–42. doi: 10.1016/j.mehy.2014.06.008
139. Kulkarni VA, Firestein BL. The dendritic tree and brain disorders. Mol Cell Neurosci. (2012) 50:10–20. doi: 10.1016/j.mcn.2012.03.005
140. Li M, Li C, Yu H, Cai X, Shen X, Sun X, et al. Lentivirus-mediated interleukin-1β (IL-1β) knock-down in the hippocampus alleviates lipopolysaccharide (LPS)-induced memory deficits and anxiety- and depression-like behaviors in mice. J Neuroinflammation. (2017) 14:190. doi: 10.1186/s12974-017-0964-9
141. Barrientos RM, Sprunger DB, Campeau S, Higgins EA, Watkins LR, Rudy JW, et al. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience. (2003) 121:847–53. doi: 10.1016/S0306-4522(03)00564-5
142. Zhong L, Zhang Z-L, Li X, Liao C, Mou P, Wang T, et al. TREM2/DAP12 complex regulates inflammatory responses in microglia via the JNK signaling pathway. Front Aging Neurosci. (2017) 9:204. doi: 10.3389/fnagi.2017.00204
143. Sessa G, Podini P, Mariani M, Meroni A, Spreafico R, Sinigaglia F, et al. Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur J Neurosci. (2004) 20:2617–28. doi: 10.1111/j.1460-9568.2004.03729.x
144. Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, et al. The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity. (2018) 48:979–91.e8. doi: 10.1016/j.immuni.2018.04.016
145. Arnoux I, Audinat E. Fractalkine signaling and microglia functions in the developing brain. Neural Plast. (2015) 2015:689404. doi: 10.1155/2015/689404
146. Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. (2014) 17:400–6. doi: 10.1038/nn.3641
147. Sheridan GK, Murphy KJ. Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol. (2013) 3:130181. doi: 10.1098/rsob.130181
148. Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. (2011) 31:16241–50. doi: 10.1523/JNEUROSCI.3667-11.2011
149. Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. (1998) 95:10896–901. doi: 10.1073/pnas.95.18.10896
150. Riccomagno MM, Kolodkin AL. Sculpting neural circuits by axon and dendrite pruning. Annu Rev Cell Dev Biol. (2015) 31:779–805. doi: 10.1146/annurev-cellbio-100913-013038
151. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
152. Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia. (2012) 60:993–1003. doi: 10.1002/glia.22331
153. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. (2016) 530:177–83. doi: 10.1038/nature16549
154. Odell D, Maciulis A, Cutler A, Warren L, McMahon WM, Coon H, et al. Confirmation of the association of the C4B null allelle in autism. Hum Immunol. (2005) 66:140–5. doi: 10.1016/j.humimm.2004.11.002
155. Fagan K, Crider A, Ahmed AO, Pillai A. Complement C3 expression is decreased in autism spectrum disorder subjects and contributes to behavioral deficits in rodents. Mol Neuropsychiatry. (2017) 3:19–27. doi: 10.1159/000465523
156. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
157. Sears SM, Hewett SJ. Influence of glutamate and GABA transport on brain excitatory/inhibitory balance. Experimental Biol Med (Maywood, NJ). (2021) 246:1069–83. doi: 10.1177/1535370221989263
158. Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron. (2015) 87:684–98. doi: 10.1016/j.neuron.2015.07.033
159. Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. (2011) 477:171–8. doi: 10.1038/nature10360
160. Xiao L, Yan J, Feng D, Ye S, Yang T, Wei H, et al. Critical role of TLR4 on the microglia activation induced by maternal LPS exposure leading to ASD-like behavior of offspring. Front Cell Dev Biol. (2021) 9:634837. doi: 10.3389/fcell.2021.634837
161. Fan LW, Tien LT, Zheng B, et al. Dopaminergic neuronal injury in the adult rat brain following neonatal exposure to lipopolysaccharide and the silent neurotoxicity. Brain Behav Immun. (2011) 25:286–97. doi: 10.1016/j.bbi.2010.09.020
162. Yan X, Jiang E, Weng HR. Activation of toll like receptor 4 attenuates GABA synthesis and postsynaptic GABA receptor activities in the spinal dorsal horn via releasing interleukin-1 beta. J Neuroinflammation. (2015) 12:222. doi: 10.1186/s12974-014-0222-3
163. Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, et al. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun. (2016) 7:11459. doi: 10.1038/ncomms11459
164. Hegarty JP II, Weber DJ, Cirstea CM, Beversdorf DQ. Cerebro-cerebellar functional connectivity is associated with cerebellar excitation-inhibition balance in autism spectrum disorder. J Autism Dev Disord. (2018) 48:3460–73. doi: 10.1007/s10803-018-3613-y
165. Siegel-Ramsay JE, Romaniuk L, Whalley HC, Roberts N, Branigan H, Stanfield AC, et al. Glutamate and functional connectivity - support for the excitatory-inhibitory imbalance hypothesis in autism spectrum disorders. Psychiatry Res Neuroimaging. (2021) 313:111302. doi: 10.1016/j.pscychresns.2021.111302
166. de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. (2016) 22:345–61. doi: 10.1038/nm.4071
167. CC, Huang TL. Brain-derived neurotrophic factor and mental disorders. Biomed J. (2020) 43:134–42. doi: 10.1016/j.bj.2020.01.001
168. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. (2006) 9:917–24. doi: 10.1038/nn1715
169. Tarozzo G, Bortolazzi S, Crochemore C, Chen S-C, Lira AS, Abrams JS, et al. Fractalkine protein localization and gene expression in mouse brain. J Neurosci Res. (2003) 73:81–8. doi: 10.1002/jnr.10645
Keywords: autism (ASD), microglia, neurodevelopmental, synapse, synaptic plasticity
Citation: Hu C, Li H, Li J, Luo X and Hao Y (2022) Microglia: Synaptic modulator in autism spectrum disorder. Front. Psychiatry 13:958661. doi: 10.3389/fpsyt.2022.958661
Received: 31 May 2022; Accepted: 28 October 2022;
Published: 17 November 2022.
Edited by:
Lan Xiong, McGill University, CanadaReviewed by:
Akira Monji, Saga University, JapanCopyright © 2022 Hu, Li, Li, Luo and Hao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Hao, aGFveWFuZXJAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.