 Rachel Jun
Rachel Jun Wen Zhang
Wen Zhang Nicholas J. Beacher
Nicholas J. Beacher Yan Zhang
Yan Zhang Yun Li
Yun Li Da-Ting Lin
Da-Ting Lin- 1Intramural Research Program, National Institute on Drug Abuse, National Institutes of Health, Baltimore, MD, United States
- 2National Institute on Drug Dependence and Beijing Key Laboratory of Drug Dependence, Peking University, Beijing, China
- 3Department of Zoology and Physiology, University of Wyoming, Laramie, WY, United States
Schizophrenia is a psychiatric disorder characterized by hallucinations, anhedonia, disordered thinking, and cognitive impairments. Both genetic and environmental factors contribute to schizophrenia. Dysbindin-1 (DTNBP1) and brain-derived neurotrophic factor (BDNF) are both genetic factors associated with schizophrenia. Mice lacking Dtnbp1 showed behavioral deficits similar to human patients suffering from schizophrenia. DTNBP1 plays important functions in synapse formation and maintenance, receptor trafficking, and neurotransmitter release. DTNBP1 is co-assembled with 7 other proteins into a large protein complex, known as the biogenesis of lysosome-related organelles complex-1 (BLOC-1). Large dense-core vesicles (LDCVs) are involved in the secretion of hormones and neuropeptides, including BDNF. BDNF plays important roles in neuronal development, survival, and synaptic plasticity. BDNF is also critical in maintaining GABAergic inhibitory transmission in the brain. Two studies independently showed that DTNBP1 mediated activity-dependent BDNF secretion to maintain inhibitory transmission. Imbalance of excitatory and inhibitory neural activities is thought to contribute to schizophrenia. In this mini-review, we will discuss a potential pathogenetic mechanism for schizophrenia involving DTNBP1, BDNF, and inhibitory transmission. We will also discuss how these processes are interrelated and associated with a higher risk of schizophrenia development.
Introduction
Schizophrenia is a neurodevelopmental disorder with a life-time prevalence of ~0.4% (1). It is characterized by positive symptoms (presence of auditory and visual hallucinations, delusions, disorganized behaviors), negative symptoms (loss of motivation, anhedonia, and blunted affect), and cognitive deficits (impairments in learning and problem solving) (2, 3). A combination of different genetic and environmental factors can increase the risk for schizophrenia (4). Studies have shown that schizophrenia has a heritability of ~80%, and twin studies established that genetics play an important role in the development of schizophrenia (5, 6). Further studies suggest significant associations between individuals with high schizophrenia polygenic risk scores and comorbidity with cognitive disorders, respiratory illness, and digestive diseases (7). Genetic influences to the development of schizophrenia are numerous and complex. In this mini-review, we will summarize evidence for the dysfunctions in dysbindin-1 (DTNBP1), a protein coding gene regularly implicated in schizophrenia as well as brain-derived neurotrophic factor (BDNF), and GABAergic circuit function in relation to schizophrenia.
DYSBINDIN-1
Dysbindin-1 (DTNBP1) is a protein encoded by dystrobrevin-binding protein 1 gene (DTNBP1), located at chromosome 6, position 22.3 (8). It is a coiled-coil-containing protein and found in various brain regions (8, 9). A study on Drosophila's neuromuscular junction using electrophysiological screening revealed that DTNBP1 was required for presynaptic retrograde and homeostatic regulation of neurotransmission downstream or independently of calcium influx (10). Another electrophysiological study reported that DTNBP1 loss led to decreases in readily releasable pool in the calyx of Held synapses and could be related to the cognitive impairment in schizophrenia (11). In addition, a recent study has shown that DTNBP1 plays an important part in the axonal mitochondrial movement which further affects calcium homeostasis in presynaptic terminals (12). The null protein mutation of Dtnbp1 in sandy (sdy) mice (13) displayed schizophrenia-like behaviors and deficits in dopaminergic, glutamatergic, and GABAergic neurotransmission (14–21). Patients with schizophrenia have significantly lower levels of DTNBP1 mRNA in the dorsolateral prefrontal cortex, hippocampus, and nucleus accumbens compared to healthy controls (22). In the post-mortem brain, presynaptic DTNBP1 is reduced in synaptic terminals of hippocampal formations, which may contribute to cognitive deficits commonly seen in schizophrenia (9). Genetic studies have also provided evidence of DTNBP1 impacting susceptibility to schizophrenia. Genome-wide association studies have identified multiple single nucleotide polymorphisms (SNPs) of DTNBP1 as potential risk factors for schizophrenia (23). Several studies conducted in Japanese, Irish, and Chinese populations suggest that genetic variation in DTNBP1 is associated with schizophrenia (8, 24, 25). DTNBP1 is also shown to be involved in initiating an immune response to environmental stimuli, which might explain the increased vulnerability of schizophrenia due to environmental impact in combination with genetic influence (26).
Studies have established the important role of DTNBP1 in intracellular protein trafficking, which affects various neuronal functions, including synapse formation and maintenance, receptor trafficking, and transmitter release (27–32). In neurons, DTNBP1 is located in the cytoplasm and can be assembled with several other proteins into a large protein complex, known as the biogenesis of lysosome-related organelles complex-1 (BLOC-1). BLOC-1 contains eight proteins: DTNBP1, cappuccino, pallidin, muted, snapin, and BLOS1-3 (33). In the central nervous system, BLOC-1 subunits, including DTNBP-1A, -1B, and -1C, are found in multiple brain regions, including hippocampal formation (HF) and are associated with synaptic vesicles or postsynaptic densities (34, 35). Previous studies reported DTNBP-1A as playing an important role in neuron development and spine growth (36–40). The DTNBP-1B subunit is present in humans, but not in mice (34). In studies involving genetically engineered mice, DTNBP-1B forms aggresomes at perinuclear regions in order to separate aggregated proteins produced by misfolded protein (41, 42). However, in humans, DTNBP-1B is diffused within the neuronal nuclei and axon terminals (34). Both DTNBP-1B and -1C isoforms are reduced in schizophrenic HF (34, 35) and significant reduction of DTNBP-1C was found in dorsolateral prefrontal cortex in schizophrenia (43). An experiment involving sdy mice with mutations in both DTNBP-1A and -1C indicated that decreases in DTNBP-1C led to decreased hilar mossy cells of dendate gyrus (35). This indicates the role of DTNBP-1C in maturation of newborn neurons in the dendate gyrus in a BLOC-1 independent manner (35). In addition, systematic investigation of BLOC-1 genes in schizophrenia patients revealed a significant association between the BLOC1S3 gene and schizophrenia (44). During embryonic and early postnatal development, BLOC-1 is expressed more abundantly (45), which implicates an important role of BLOC-1 during early-life neural development. Another role of the BLOC-1 complex is the biogenesis of melanosomes and platelet-dense granules through self-assembly and interaction with actin cytoskeleton (46). Actin polymerization complex is a necessary organelle for synaptic function, and the expression of this actin cytoskeleton was reduced in DTNBP1-deficient cells (47). Other studies have shown that DTNBP1 recruited BLOC-1 is important for the regulation of oxytocin, metabotropic glutamate receptor, synaptic NMDA receptors, serotonin transmission, activity-dependent synaptic vesicle recycling, and synaptic plasticity (17, 48–55). Disruption of the BLOC-1 complex can lead to changes in the formation of large dense-core vesicles (LDCVs), which are involved in the secretion of hormones and neuropeptides. Studies have shown that LDCVs mediate the release of monoamines (Serotonin, Dopamine, and Noradrenaline) and peptides, including substance P, BDNF, and oxytocin (56). Of BLOC-1 complex proteins, loss of DTNBP1 or muted both led to the enlargement of LDCVs, and loss of DTNBP1 alone led to reduced LDCV numbers in cells from mice while muted deletion did not change the LDCV number (57). The release of LDCVs in neurons is also activity-dependently regulated in the central nervous system. For example, deletion of Munc13, a classic presynaptic protein involved in anchoring and activity-dependent release of synaptic vesicles, led to a reduction of LDCV release (58). BLOC-1 also contributes to the activity-dependent vesicle release. For example, it is shown that mutations of pallidin, whose encoded protein directly binds with DTNBP1 in BLOC-1, disrupted activity-dependent synaptic vesicle recycling (54). Therefore, DTNBP1 plays an essential role in activity-dependent neurotransmitter release, and DTNBP1 downregulations or genetic variations may contribute to schizophrenia by decreasing the size of readily releasable pool of synaptic vesicles which regulates synaptic transmission.
BDNF
Another gene known to be associated with schizophrenia is brain-derived neurotrophic factor (BDNF) located in chromosome 11p13 (59). BDNF has been known to play important roles in neuronal development, survival, and synaptic plasticity (60). A single nucleotide polymorphism rs6265 in BDNF gene, known as Val66Met, impacts intracellular trafficking and activity-dependent secretion of BDNF (61), which can subsequently lead to memory impairment (62). A case-control association study of the Han Chinese population revealed a positive correlation between rs6265 and schizophrenia (63).
DTNBP1 is involved in the secretion of BDNF from pyramidal neurons in the cortex (64). Super-resolution imaging showed that DTNBP1 was located close to BDNF in the cytoplasm of neurons, possibly on LDCVs (65). Deletion of Dtnbp1 in primary cultured neurons did not change spontaneous BDNF release but did change activity-dependent BDNF secretion (65, 66). These results suggest that DTNBP1 binds to LDCVs containing BDNF and regulates its activity-dependent secretion. The activity-dependent secretion of BDNF exerts a profound effect on synapse formation and maintenance as well as circuit function (67, 68). Activity-dependent production and secretion of BDNF exhibits a synapse-type specific effect on inhibitory synapses of hippocampal neurons. For example, disruption of activity-dependent transcription of BDNF selectively affects soma-targeting GABAergic synapses of hippocampal CA1 neurons, but not other types of GABAergic synapses or excitatory synapses of these neurons (69). This study suggests that activity-dependent BDNF production and its subsequent secretion from pyramidal neurons selectively affects the maintenance of those soma-targeting inhibitory synapses. Consistent with synapse-specific regulation by the activity-dependent secretion of BDNF, deletion of Dtnbp1 in mouse prefrontal cortex (PFC) pyramidal neurons exclusively affected GABAergic inhibitory synaptic transmission, but not excitatory transmission (65), which has also been observed in cultured neurons (66). The resulting reduction in inhibition showed similar synapse-type specificity, as those located on the soma of pyramidal neurons were most affected. In contrast, GABAergic synapses targeting distal dendrites showed normal functions (65). The selective deletion of DTNBP1 also induced a behavioral deficit in pre-pulse-inhibition (PPI) that was rescued by direct administration of BDNF into the affected brain region (65). PPI deficiency is useful for modeling schizophrenia in animals (70) and has been used for decades to assess pharmaceutical efficacy in human patients suffering from schizophrenia (71). PPI behavioral deficits that were reversed by local infusion of BDNF in Dtnbp1 knockout mice with schizophrenia-like behavior underscores the importance of DTNBP1 in regulating BDNF secretion and its role in the development and treatment of schizophrenia (65).
GABAergic Transmission
The balance between excitatory and inhibitory transmission in the brain is vital for normal cognitive function. Studies have shown that dysfunction in GABAergic inhibitory circuits can lead to impaired cognition (72) and may contribute to schizophrenia (Figure 1). GABAergic interneurons are a heterogeneous group of neurons often categorized into at least three groups: parvalbumin-positive (PV+), somatostatin-positive (SST+), and 5-hydroxytryptamine 3a receptor-positive (5HT3R+) interneurons (73, 74). PV+ interneurons are one of the most abundant GABAergic interneurons in the cortex, comprising roughly 30% of interneurons in the cortex. PV+ neurons express parvalbumin, a Ca2+-binding protein in the cytoplasm, which could regulate the short-term plasticity of PV+ neurons (75). The majority of PV+ neurons emerge from Medial Ganglionic Eminence during development and migrate to the cortex (76). Mature PV+ neurons showed extensive axon arbors and connections to adjacent pyramidal neurons, which are considered non-selective (77). The axons of PV+ neurons mostly target soma and axon initial segment regions of the post-synaptic neurons; The majority of PV+ neurons show a high-frequency firing pattern when excited (76). These properties combined with their unique axon terminal location render PV+ neurons the major inhibitory driving force in the cortex (76). Moreover, PV+ neurons are also interconnected via electrical synapses, or gap junctions, making them more capable of synchronizing the inhibition of adjacent neurons (78). Of the other types of GABAergic interneurons, SST+ neurons mainly target the dendrites and soma of post-synaptic neurons, while 5HT3R+ neurons more likely target the distal dendrites of post-synaptic neurons (79, 80). The axon targeting specificity of these interneurons, together with differences in synaptic strength, enables diverse functions of neuronal micro-circuits. For example, in a simple circuit configuration of PV+, SST+, and excitatory neurons, the overall effect of SST+ neuron activities on excitatory neurons could be either inhibition or dis-inhibition when the microcircuit was located in different cortical regions (81, 82).
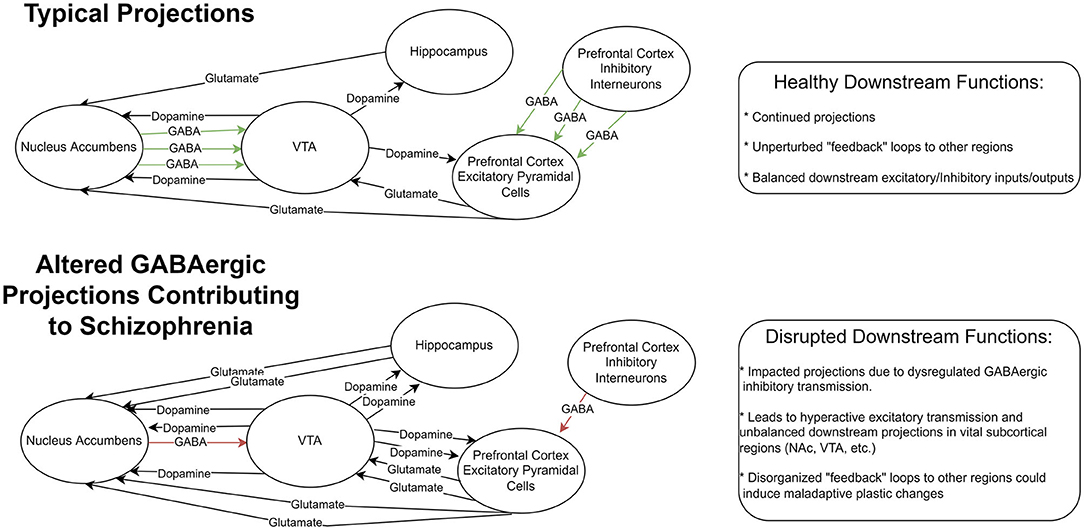
Figure 1. Representation of GABAergic circuitry in prefrontal cortex. Top row: A sample diagram depicting typical projections in a healthy system with functioning feedback loops and balanced excitatory and inhibitory inputs and outputs. Botom row: a sample diagram of this system altered by GABAegic cell changes seen in Schizophrenia. This altered system includes reduced inhibitory GABAergic signaling resulting in downstream hyperactive excitatory transmission. These could result in disrupted feedback systems and maladaptive plastic changes in vital subcortical regions.
GABAergic transmission is regulated by the activity of glutamic acid decarboxylase (GAD) that participates in GABA synthesis (72). Several studies indicated that decreased expression of 67 kD isoform of GAD (GAD67) is associated with schizophrenia and bipolar disorders (83, 84). Analysis of GAD immunoreactivity in post-mortem brains from schizophrenic patients also reveal reduced GAD in patients compared to controls, indicating GABAergic dysfunction in schizophrenia (85). Interestingly, DTNBP1 emerges as one of the candidates for regulating inhibitory synapse strength. In vitro recordings of Dtnbp1 deficient mice revealed significant decreases in GABAergic transmission at both pre- and post-synaptic levels and decreased parvalbumin markers (86). Deletion of DTNBP1 specifically in pyramidal neurons reduced the soma-targeting inhibitory synapse density without alterations in dendritic inhibitory synapses or excitatory transmission (65). Consequently, application of BDNF into extracellular space rescued this type-specific reduction of inhibitory synapses (65). Taken together, these results indicate that DTNBP1 plays several important roles in regulating inhibitory transmission and pathogenic mechanisms of schizophrenia, at least in part via BDNF secretion.
Discussion
Via regulating activity-dependent BDNF secretion, DTNBP1 could regulate both neural development and inhibitory circuit function. It is also likely that malfunctioning of other DTNBP1 related processes could induce impairments that lead to the development of schizophrenia. While it is not yet known exactly how DTNBP1 regulates the functionality of different neural circuits in brain regions involved in schizophrenia, more studies are necessary to identify whether such a circuit change contributes to its role in the pathogenesis of schizophrenia. Modern in vivo imaging techniques can be harnessed to study how these different biological markers contribute to schizophrenia in animal models. For example, mouse models displaying schizophrenia-like behavior can be imaged with miniaturized microscopes (miniscopes) (87–93) to examine longitudinal changes in activity of excitatory and inhibitory neurons in brain regions implicated in schizophrenia. Simultaneous optogenetic manipulations of circuitry are also possible using miniscopes (94, 95) to explore how in vivo manipulation of circuitry impacts behavior using animal models for schizophrenia. These tools will further advance schizophrenia research in animals and help develop potential therapeutic interventions in human patients.
Author Contributions
RJ and WZ wrote the manuscript with inputs from NB, YZ, YL, and D-TL. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Intramural Research Program, National Institute on Drug Abuse, National Institutes of Health, and National Institutes of Health Grants 5P20GM121310, R61NS115161, and UG3NS115608 to YL, and National Key R&D Program of China (Nos. 2019YFA0706201 and 2021ZD0202900), National Natural Science Foundation of China (No. 32170960) to WZ.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bijl RV, Ravelli A, van Zessen G. Prevalence of psychiatric disorder in the general population: results of the netherlands mental health survey and incidence study (Nemesis). Soc Psychiatry Psychiatr Epidemiol. (1998) 33:587–95. doi: 10.1007/s001270050098
2. Khavari B, Cairns MJ. Epigenomic dysregulation in schizophrenia: in search of disease etiology and biomarkers. Cells. (2020) 9:837. doi: 10.3390/cells9081837
3. Correll CU, Schooler NR. Negative symptoms in schizophrenia: a review and clinical guide for recognition, assessment, and treatment. Neuropsychiatr Dis Treat. (2020) 16:519–34. doi: 10.2147/NDT.S225643
4. Ayhan Y, McFarland R, Pletnikov MV. Animal models of gene-environment interaction in schizophrenia: a dimensional perspective. Prog Neurobiol. (2016) 136:1–27. doi: 10.1016/j.pneurobio.2015.10.002
5. Riley B, Kendler KS. Molecular genetic studies of schizophrenia. Eur J Hum Genet. (2006) 14:669–80. doi: 10.1038/sj.ejhg.5201571
6. Xu T, Wang Y, Li Z, Huang J, Lui SS, Tan SP, et al. Heritability and familiality of neurological soft signs: evidence from healthy twins, patients with schizophrenia and non-psychotic first-degree relatives. Psychol Med. (2016) 46:117–23. doi: 10.1017/S0033291715001580
7. Zhang R, Sjolander A, Ploner A, Lu D, Bulik CM, Bergen SE. Novel disease associations with schizophrenia genetic risk revealed in ~400,000 Uk Biobank participants. Mol Psychiatry. (2022) 27:1448–54. doi: 10.1038/s41380-021-01387-5
8. Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, et al. Genetic variation in the 6p223 gene Dtnbp1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. (2002) 71:337–48. doi: 10.1086/341750
9. Talbot K, Eidem WL, Tinsley CL, Benson MA, Thompson EW, Smith RJ, et al. Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J Clin Invest. (2004) 113:1353–63. doi: 10.1172/JCI200420425
10. Dickman DK, Davis GW. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science. (2009) 326:1127–30. doi: 10.1126/science.1179685
11. Hu H, Wang X, Li C, Li Y, Hao J, Zhou Y, et al. Loss of dysbindin implicates synaptic vesicle replenishment dysregulation as a potential pathogenic mechanism in schizophrenia. Neuroscience. (2021) 452:138–52. doi: 10.1016/j.neuroscience.2020.10.020
12. Suh BK, Lee SA, Park C, Suh Y, Kim SJ, Woo Y, et al. Schizophrenia-associated dysbindin modulates axonal mitochondrial movement in cooperation with P150(Glued). Mol Brain. (2021) 14:14. doi: 10.1186/s13041-020-00720-3
13. Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O'Brien EP, et al. Hermansky-pudlak syndrome type 7 (Hps-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (Bloc-1). Nat Genet. (2003) 35:84–9. doi: 10.1038/ng1229
14. Talbot K. The Sandy (Sdy) mouse: a dysbindin-1 mutant relevant to schizophrenia research. Prog Brain Res. (2009) 179:87–94. doi: 10.1016/S0079-6123(09)17910-4
15. Cox MM, Tucker AM, Tang J, Talbot K, Richer DC, Yeh L, et al. Neurobehavioral abnormalities in the dysbindin-1 mutant, sandy, on a C57bl/6j genetic background. Genes Brain Behav. (2009) 8:390–7. doi: 10.1111/j.1601-183X.2009.00477.x
16. Papaleo F, Yang F, Garcia S, Chen J, Lu B, Crawley JN, et al. Dysbindin-1 modulates prefrontal cortical activity and schizophrenia-like behaviors via dopamine/D2 pathways. Mol Psychiatry. (2012) 17:85–98. doi: 10.1038/mp.2010.106
17. Bhardwaj SK, Ryan RT, Wong TP, Srivastava LK. Loss of dysbindin-1, a risk gene for schizophrenia, leads to impaired group 1 metabotropic glutamate receptor function in mice. Front Behav Neurosci. (2015) 9:72. doi: 10.3389/fnbeh.2015.00072
18. Feng YQ, Zhou ZY, He X, Wang H, Guo XL, Hao CJ, et al. Dysbindin deficiency in sandy mice causes reduction of snapin and displays behaviors related to schizophrenia. Schizophr Res. (2008) 106:218–28. doi: 10.1016/j.schres.2008.07.018
19. Hattori S, Murotani T, Matsuzaki S, Ishizuka T, Kumamoto N, Takeda M, et al. Behavioral abnormalities and dopamine reductions in sdy mutant mice with a deletion in Dtnbp1, a susceptibility gene for schizophrenia. Biochem Biophys Res Commun. (2008) 373:298–302. doi: 10.1016/j.bbrc.2008.06.016
20. Carr GV, Jenkins KA, Weinberger DR, Papaleo F. Loss of dysbindin-1 in mice impairs reward-based operant learning by increasing impulsive and compulsive behavior. Behav Brain Res. (2013) 241:173–84. doi: 10.1016/j.bbr.2012.12.021
21. Huang CCY, Muszynski KJ, Bolshakov VY, Balu DT. Deletion of Dtnbp1 in mice impairs threat memory consolidation and is associated with enhanced inhibitory drive in the amygdala. Transl Psychiatry. (2019) 9:132. doi: 10.1038/s41398-019-0465-y
22. Weickert CS, Straub RE, McClintock BW, Matsumoto M, Hashimoto R, Hyde TM, et al. Human dysbindin (Dtnbp1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch Gen Psychiatry. (2004) 61:544–55. doi: 10.1001/archpsyc.61.6.544
23. Tang J, Fan Y, Li H, Xiang Q, Zhang DF, Li Z, et al. Whole-genome sequencing of monozygotic twins discordant for schizophrenia indicates multiple genetic risk factors for schizophrenia. J Genet Genomics. (2017) 44:295–306. doi: 10.1016/j.jgg.2017.05.005
24. Numakawa T, Yagasaki Y, Ishimoto T, Okada T, Suzuki T, Iwata N, et al. Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet. (2004) 13:2699–708. doi: 10.1093/hmg/ddh280
25. Tang JX, Zhou J, Fan JB, Li XW, Shi YY, Gu NF, et al. Family-based association study of Dtnbp1 in 6p223 and schizophrenia. Mol Psychiatry. (2003) 8:717–8. doi: 10.1038/sj.mp.4001287
26. Al-Shammari AR, Bhardwaj SK, Musaelyan K, Srivastava LK, Szele FG. Schizophrenia-related dysbindin-1 gene is required for innate immune response and homeostasis in the developing subventricular zone. NPJ Schizophr. (2018) 4:15. doi: 10.1038/s41537-018-0057-5
27. Iizuka Y, Sei Y, Weinberger DR, Straub RE. Evidence that the Bloc-1 protein dysbindin modulates dopamine D2 receptor internalization and signaling but not D1 internalization. J Neurosci. (2007) 27:12390–5. doi: 10.1523/JNEUROSCI.1689-07.2007
28. Marley A, von Zastrow M. Dysbindin promotes the post-endocytic sorting of G protein-coupled receptors to lysosomes. PLoS ONE. (2010) 5:e9325. doi: 10.1371/journal.pone.0009325
29. Ji Y, Yang F, Papaleo F, Wang HX, Gao WJ, Weinberger DR, et al. Role of dysbindin in dopamine receptor trafficking and cortical gaba function. Proc Natl Acad Sci U S A. (2009) 106:19593–8. doi: 10.1073/pnas.0904289106
30. Tang TT, Yang F, Chen BS, Lu Y, Ji Y, Roche KW, et al. Dysbindin regulates hippocampal Ltp by controlling nmda receptor surface expression. Proc Natl Acad Sci U S A. (2009) 106:21395–400. doi: 10.1073/pnas.0910499106
31. Larimore J, Tornieri K, Ryder PV, Gokhale A, Zlatic SA, Craige B, et al. The schizophrenia susceptibility factor dysbindin and its associated complex sort Cargoes from cell bodies to the synapse. Mol Biol Cell. (2011) 22:4854–67. doi: 10.1091/mbc.e11-07-0592
32. Mullin AP, Sadanandappa MK, Ma W, Dickman DK, VijayRaghavan K, Ramaswami M, et al. Gene dosage in the dysbindin schizophrenia susceptibility network differentially affect synaptic function and plasticity. J Neurosci. (2015) 35:325–38. doi: 10.1523/JNEUROSCI.3542-14.2015
33. Lee HH, Nemecek D, Schindler C, Smith WJ, Ghirlando R, Steven AC, et al. Assembly and architecture of biogenesis of lysosome-related organelles complex-1 (Bloc-1). J Biol Chem. (2012) 287:5882–90. doi: 10.1074/jbc.M111.325746
34. Talbot K, Louneva N, Cohen JW, Kazi H, Blake DJ, Arnold SE. Synaptic dysbindin-1 reductions in schizophrenia occur in an isoform-specific manner indicating their subsynaptic location. PLoS ONE. (2011) 6:e16886. doi: 10.1371/journal.pone.0016886
35. Wang H, Yuan Y, Zhang Z, Yan H, Feng Y, Li W. Dysbindin-1c is required for the survival of hilar mossy cells and the maturation of adult newborn neurons in dentate gyrus. J Biol Chem. (2014) 289:29060–72. doi: 10.1074/jbc.M114.590927
36. Fei E, Chen P, Zhang Q, Zhong Y, Zhou T. Protein kinase B/Akt1 phosphorylates dysbindin-1a at serine 10 to regulate neuronal development. Neuroscience. (2022) 490:66–78. doi: 10.1016/j.neuroscience.2022.01.025
37. Ito H, Morishita R, Shinoda T, Iwamoto I, Sudo K, Okamoto K, et al. Dysbindin-1, a schizophrenia-related molecule, is involved in the regulation of neuronal dendritic development. Mol Psychiatry. (2010) 15:969. doi: 10.1038/mp.2010.93
38. Ma X, Fei E, Fu C, Ren H, Wang G. Dysbindin-1, a Schizophrenia-related protein, facilitates neurite outgrowth by promoting the transcriptional activity of P53. Mol Psychiatry. (2011) 16:1105–16. doi: 10.1038/mp.2011.43
39. Jia JM, Zhao J, Hu Z, Lindberg D, Li Z. Age-dependent regulation of synaptic connections by dopamine D2 receptors. Nat Neurosci. (2013) 16:1627–36. doi: 10.1038/nn.3542
40. Jia JM, Hu Z, Nordman J, Li Z. The schizophrenia susceptibility gene dysbindin regulates dendritic spine dynamics. J Neurosci. (2014) 34:13725–36. doi: 10.1523/JNEUROSCI.0184-14.2014
41. Xu Y, Sun Y, Ye H, Zhu L, Liu J, Wu X, et al. Increased dysbindin-1b isoform expression in schizophrenia and its propensity in aggresome formation. Cell Discov. (2015) 1:15032. doi: 10.1038/celldisc.2015.32
42. Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. (1998) 143:1883–98. doi: 10.1083/jcb.143.7.1883
43. Tang J, LeGros RP, Louneva N, Yeh L, Cohen JW, Hahn CG, et al. Dysbindin-1 in dorsolateral prefrontal cortex of schizophrenia cases is reduced in an isoform-specific manner unrelated to dysbindin-1 Mrna expression. Hum Mol Genet. (2009) 18:3851–63. doi: 10.1093/hmg/ddp329
44. Morris DW, Murphy K, Kenny N, Purcell SM, McGhee KA, Schwaiger S, et al. Dysbindin (Dtnbp1) and the biogenesis of lysosome-related organelles complex 1 (Bloc-1): main and epistatic gene effects are potential contributors to schizophrenia susceptibility. Biol Psychiatry. (2008) 63:24–31. doi: 10.1016/j.biopsych.2006.12.025
45. Ghiani CA, Starcevic M, Rodriguez-Fernandez IA, Nazarian R, Cheli VT, Chan LN, et al. The dysbindin-containing complex (Bloc-1) in brain: developmental regulation, interaction with snare proteins and role in neurite outgrowth. Mol Psychiatry. (2010) 15:115:204–15. doi: 10.1038/mp.2009.58
46. Falcon-Perez JM, Starcevic M, Gautam R, Dell'Angelica EC. Bloc-1, a novel complex containing the pallidin and muted proteins involved in the biogenesis of melanosomes and platelet-dense granules. J Biol Chem. (2002) 277:28191–9. doi: 10.1074/jbc.M204011200
47. Gokhale A, Hartwig C, Freeman AH, Das R, Zlatic SA, Vistein R, et al. The proteome of Bloc-1 genetic defects identifies the Arp2/3 actin polymerization complex to function downstream of the schizophrenia susceptibility factor dysbindin at the synapse. J Neurosci. (2016) 36:12393–411. doi: 10.1523/JNEUROSCI.1321-16.2016
48. Wentzel C, Delvendahl I, Sydlik S, Georgiev O, Müller M. Dysbindin links presynaptic proteasome function to homeostatic recruitment of low release probability vesicles. Nat Commun. (2018) 9:267. doi: 10.1038/s41467-017-02494-0
49. Sinclair D, Cesare J, McMullen M, Carlson GC, Hahn CG, Borgmann-Winter KE. Effects of sex and Dtnbp1 (Dysbindin) null gene mutation on the developmental Glun2b-Glun2a switch in the mouse cortex and hippocampus. J Neurodev Disord. (2016) 8:14. doi: 10.1186/s11689-016-9148-7
50. Hidalgo S, Castro C, Zárate RV, Valderrama BP, Hodge JJL, Campusano JM. The behavioral and neurochemical characterization of a drosophila dysbindin mutant supports the contribution of serotonin to schizophrenia negative symptoms. Neurochem Int. (2020) 138:104753. doi: 10.1016/j.neuint.2020.104753
51. Furukubo-Tokunaga K, Kurita K, Honjo K, Pandey H, Ando T, Takayama K, et al. Disc1 causes associative memory and neurodevelopmental defects in fruit flies. Mol Psychiatry. (2016) 21:1232–43. doi: 10.1038/mp.2016.15
52. Furukubo-Tokunaga K, Kurita K, Honjo K, Pandey H, Ando T, Takayama K, et al. Visualization of Disc1-dysbindin interaction in glutamatergic synaptic termini in fruit flies. Mol Psychiatry. (2016) 21:1157. doi: 10.1038/mp.2016.142
53. Ferretti V, Maltese F, Contarini G, Nigro M, Bonavia A, Huang H, et al. Oxytocin signaling in the central amygdala modulates emotion discrimination in mice. Curr Biol. (2019) 29:1938–53.e6. doi: 10.1016/j.cub.2019.04.070
54. Chen X, Ma W, Zhang S, Paluch J, Guo W, Dickman DK. The Bloc-1 subunit pallidin facilitates activity-dependent synaptic vesicle recycling. eNeuro. (2017) 4:ENEURO.0335-16.2017. doi: 10.1523/ENEURO.0335-16.2017
55. Banerjee A, Wang HY, Borgmann-Winter KE, MacDonald ML, Kaprielian H, Stucky A, et al. Src kinase as a mediator of convergent molecular abnormalities leading to nmdar hypoactivity in schizophrenia. Mol Psychiatry. (2015) 20:1091–100. doi: 10.1038/mp.2014.115
56. Trueta C, De-Miguel FF. Extrasynaptic exocytosis and its mechanisms: a source of molecules mediating volume transmission in the nervous system. Front Physiol. (2012) 3:319. doi: 10.3389/fphys.2012.00319
57. Hao Z, Wei L, Feng Y, Chen X, Du W, Ma J, et al. Impaired maturation of large dense-core vesicles in muted-deficient adrenal chromaffin cells. J Cell Sci. (2015) 128:1365–74. doi: 10.1242/jcs.161414
58. van de Bospoort R, Farina M, Schmitz SK, de Jong A, de Wit H, Verhage M, et al. Munc13 controls the location and efficiency of dense-core vesicle release in neurons. J Cell Biol. (2012) 199:883–91. doi: 10.1083/jcb.201208024
59. Balu DT, Coyle JT. Neuroplasticity signaling pathways linked to the pathophysiology of schizophrenia. Neurosci Biobehav Rev. (2011) 35:848–70. doi: 10.1016/j.neubiorev.2010.10.005
60. Ikegame T, Bundo M, Murata Y, Kasai K, Kato T, Iwamoto K, et al. Methylation of the Bdnf gene and its relevance to psychiatric disorders. J Hum Genet. (2013) 58:434–8. doi: 10.1038/jhg.2013.65
61. Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The Bdnf Val66met polymorphism affects activity-dependent secretion of bdnf and human memory and hippocampal function. Cell. (2003) 112:257–69. doi: 10.1016/S0092-8674(03)00035-7
62. Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, et al. Variant brain-derived neurotrophic factor (Bdnf) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type Bdnf in neurosecretory cells and cortical neurons. J Neurosci. (2004) 24:4401–11. doi: 10.1523/JNEUROSCI.0348-04.2004
63. Fu X, Wang J, Du J, Sun J, Baranova A, Zhang F. Bdnf gene's role in schizophrenia: from risk allele to methylation implications. Front Psychiatry. (2020) 11:564277. doi: 10.3389/fpsyt.2020.564277
64. Dieni S, Matsumoto T, Dekkers M, Rauskolb S, Ionescu MS, Deogracias R, et al. Bdnf and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J Cell Biol. (2012) 196:775–88. doi: 10.1083/jcb.201201038
65. Zhang W, Daly KM, Liang B, Zhang L, Li X, Li Y, et al. Bdnf rescues prefrontal dysfunction elicited by pyramidal neuron-specific Dtnbp1 deletion in vivo. J Mol Cell Biol. (2017) 9:117–31. doi: 10.1093/jmcb/mjw029
66. Yuan Q, Yang F, Xiao Y, Tan S, Husain N, Ren M, et al. Regulation of brain-derived neurotrophic factor exocytosis and gamma-aminobutyric acidergic interneuron synapse by the schizophrenia susceptibility gene dysbindin-1. Biol Psychiatry. (2016) 80:312–22. doi: 10.1016/j.biopsych.2015.08.019
67. Alsina B, Vu T, Cohen-Cory S. Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by Bdnf. Nat Neurosci. (2001) 4:1093–101. doi: 10.1038/nn735
68. Cohen-Cory S. The developing synapse: construction and modulation of synaptic structures and circuits. Science. (2002) 298:770–6. doi: 10.1126/science.1075510
69. Bloodgood BL, Sharma N, Browne HA, Trepman AZ, Greenberg ME. The activity-dependent transcription factor Npas4 regulates domain-specific inhibition. Nature. (2013) 503:121–5. doi: 10.1038/nature12743
70. Swerdlow NR, Braff DL, Taaid N, Geyer MA. Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry. (1994) 51:139–54. doi: 10.1001/archpsyc.1994.03950020063007
71. Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology. (1978) 15:339–43. doi: 10.1111/j.1469-8986.1978.tb01390.x
72. Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. (2012) 35:57–67. doi: 10.1016/j.tins.2011.10.004
73. DeFelipe J, Lopez-Cruz PL, Benavides-Piccione R, Bielza C, Larranaga P, Anderson S, et al. New insights into the classification and nomenclature of cortical gabaergic interneurons. Nat Rev Neurosci. (2013) 14:202–16. doi: 10.1038/nrn3444
74. Petilla Interneuron Nomenclature G, Ascoli GA, Alonso-Nanclares L, Anderson SA, Barrionuevo G, Benavides-Piccione R, et al. Petilla terminology: nomenclature of features of gabaergic interneurons of the cerebral cortex. Nat Rev Neurosci. (2008) 9:557–68. doi: 10.1038/nrn2402
75. Caillard O, Moreno H, Schwaller B, Llano I, Celio MR, Marty A. Role of the calcium-binding protein parvalbumin in short-term synaptic plasticity. Proc Natl Acad Sci U S A. (2000) 97:13372–7. doi: 10.1073/pnas.230362997
76. Lim L, Mi D, Llorca A, Marin O. Development and functional diversification of cortical interneurons. Neuron. (2018) 100:294–313. doi: 10.1016/j.neuron.2018.10.009
77. Packer AM, Yuste R. Dense, unspecific connectivity of neocortical parvalbumin-positive interneurons: a canonical microcircuit for inhibition? J Neurosci. (2011) 31:13260–71. doi: 10.1523/JNEUROSCI.3131-11.2011
78. Meyer AH, Katona I, Blatow M, Rozov A, Monyer H. In vivo labeling of parvalbumin-positive interneurons and analysis of electrical coupling in identified neurons. J Neurosci. (2002) 22:7055–64. doi: 10.1523/JNEUROSCI.22-16-07055.2002
79. Kubota Y, Kawaguchi Y. Dependence of gabaergic synaptic areas on the interneuron type and target size. J Neurosci. (2000) 20:375–86. doi: 10.1523/JNEUROSCI.20-01-00375.2000
80. Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ. Hippocampal gabaergic inhibitory interneurons. Physiol Rev. (2017) 97:1619–747. doi: 10.1152/physrev.00007.2017
81. Xu H, Jeong HY, Tremblay R, Rudy B. Neocortical somatostatin-expressing gabaergic interneurons disinhibit the thalamorecipient layer 4. Neuron. (2013) 77:155–67. doi: 10.1016/j.neuron.2012.11.004
82. Zhang W, Zhang L, Liang B, Schroeder D, Zhang ZW, Cox GA, et al. Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat Neurosci. (2016) 19:557–9. doi: 10.1038/nn.4257
83. Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, et al. Decrease in reelin and glutamic acid decarboxylase67 (Gad67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. (2000) 57:1061–9. doi: 10.1001/archpsyc.57.11.1061
84. Subburaju S, Benes FM. Induction of the gaba cell phenotype: an in vitro model for studying neurodevelopmental disorders. PLoS ONE. (2012) 7:e33352. doi: 10.1371/journal.pone.0033352
85. Forster A, Model V, Gos T, Frodl T, Schiltz K, Dobrowolny H, et al. Reduced gabaergic neuropil and interneuron profiles in schizophrenia: complementary analysis of disease course-related differences. J Psychiatr Res. (2021) 145:50–9. doi: 10.1016/j.jpsychires.2021.11.028
86. Trantham-Davidson H, Lavin A. Loss of Dysbindin-1 Affects gabaergic transmission in the Pfc. Psychopharmacology. (2019) 236:3291–300. doi: 10.1007/s00213-019-05285-1
87. Ghosh KK, Burns LD, Cocker ED, Nimmerjahn A, Ziv Y, El Gamal A, et al. Miniaturized integration of a fluorescence microscope. Nat Methods. (2011) 8:871–8. doi: 10.1038/nmeth.1694
88. Cai DJ, Aharoni D, Shuman T, Shobe J, Biane J, Song W, et al. A shared neural ensemble links distinct contextual memories encoded close in time. Nature. (2016) 534:115–8. doi: 10.1038/nature17955
89. Zhang Y, Denman AJ, Liang B, Werner CT, Beacher NJ, Chen R, et al. Detailed mapping of behavior reveals the formation of prelimbic neural ensembles across operant learning. Neuron. (2021) 110:674–85. doi: 10.1016/j.neuron.2021.11.022
90. Zhang L, Liang B, Barbera G, Hawes S, Zhang Y, Stump K, et al. Miniscope grin lens system for calcium imaging of neuronal activity from deep brain structures in behaving animals. Curr Protoc Neurosci. (2019) 86:e56. doi: 10.1002/cpns.56
91. Barbera G, Liang B, Zhang L, Li Y, Lin DT. A wireless miniscope for deep brain imaging in freely moving mice. J Neurosci Methods. (2019) 323:56–60. doi: 10.1016/j.jneumeth.2019.05.008
92. Liang B, Zhang L, Barbera G, Fang W, Zhang J, Chen X, et al. Distinct and dynamic on and off neural ensembles in the prefrontal cortex code social exploration. Neuron. (2018) 100:700–14.e9. doi: 10.1016/j.neuron.2018.08.043
93. Barbera G, Liang B, Zhang L, Gerfen CR, Culurciello E, Chen R, et al. Spatially compact neural clusters in the dorsal striatum encode locomotion relevant information. Neuron. (2016) 92:202–13. doi: 10.1016/j.neuron.2016.08.037
94. de Groot A, van den Boom BJ, van Genderen RM, Coppens J, van Veldhuijzen J, Bos J, et al. Ninscope, a versatile miniscope for multi-region circuit investigations. Elife. (2020) 9:e49987. doi: 10.7554/eLife.49987
Keywords: schizophrenia, dysbindin-1, BDNF, GABAergic transmission, vesicular trafficking, activity-dependent release
Citation: Jun R, Zhang W, Beacher NJ, Zhang Y, Li Y and Lin D-T (2022) Dysbindin-1, BDNF, and GABAergic Transmission in Schizophrenia. Front. Psychiatry 13:876749. doi: 10.3389/fpsyt.2022.876749
Received: 15 February 2022; Accepted: 30 May 2022;
Published: 22 June 2022.
Edited by:
Konrad Talbot, Loma Linda University, United StatesReviewed by:
Kuan Hong Wang, University of Rochester, United StatesCopyright © 2022 Jun, Zhang, Beacher, Zhang, Li and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Zhang, emhhbmd3QGhzYy5wa3UuZWR1LmNu; Rachel Jun, cmFjaGVsLmp1bkBuaWguZ292