 Reza Rahimian
Reza Rahimian Claudia Belliveau
Claudia Belliveau Rebecca Chen
Rebecca Chen Naguib Mechawar
Naguib Mechawar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 16 June 2022
Sec. Molecular Psychiatry
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.871997
This article is part of the Research TopicNeuroinflammation, Metabolism, and Psychiatric DisordersView all 12 articles
Increasing evidence supports the notion that neuroinflammation plays a critical role in the etiology of major depressive disorder (MDD), at least in a subset of patients. By virtue of their capacity to transform into reactive states in response to inflammatory insults, microglia, the brain’s resident immune cells, play a pivotal role in the induction of neuroinflammation. Experimental studies have demonstrated the ability of microglia to recognize pathogens or damaged cells, leading to the activation of a cytotoxic response that exacerbates damage to brain cells. However, microglia display a wide range of responses to injury and may also promote resolution stages of inflammation and tissue regeneration. MDD has been associated with chronic priming of microglia. Recent studies suggest that altered microglial morphology and function, caused either by intense inflammatory activation or by senescence, may contribute to depression and associated impairments in neuroplasticity. In this context, modifying microglia phenotype by tuning inflammatory pathways might have important translational relevance to harness neuroinflammation in MDD. Interestingly, it was recently shown that different microglial phenotypes are associated with distinct metabolic pathways and analysis of the underlying molecular mechanisms points to an instrumental role for energy metabolism in shaping microglial functions. Here, we review various canonical pro-inflammatory, anti-inflammatory and metabolic pathways in microglia that may provide new therapeutic opportunities to control neuroinflammation in brain disorders, with a strong focus on MDD.
An association between inflammation and major depressive disorder (MDD) has long been hypothesized based on investigations using various approaches. Studies have reported elevated levels of both peripheral (1) and central (2–7) pro-inflammatory cytokines in depressed patients, supporting the hypothesis of an immune-mediated etiology of MDD (8–10). Indeed, subsets of MDD patients have increased concentrations of circulating cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin (IL)-6 (11) and increased expression of innate immune-related genes in the blood. Also, bidirectional relationships between depression and inflammatory or autoimmune disorders exist. Namely, there is a high incidence of co-morbid inflammatory disease and rheumatoid arthritis (RA) in MDD patients (12).
One of the most important cell types involved in regulating neuroinflammation are microglia, which can modulate immunological responses and play a fundamental role in maintaining homeostatic brain functions. The implication of microglia in normal brain physiology includes, but is not limited to, synaptic pruning, phagocytosis, oligodendrocyte maturation and neurogenesis (10, 13). Functionally, microglia are one of the most diverse cell types in the central nervous system (CNS), as they dynamically adapt, at both the cellular and molecular levels, to their ever-changing environment (10, 14). The heterogeneous nature of microglia has been highlighted by high-throughput approaches like single cell RNA-sequencing. Indeed, factors such as brain region, sex, age and type of pathology can significantly affect microglial phenotype, including gene expression signatures and secretory profiles (15, 16). Under physiological and pathological conditions, microglia display spatial heterogeneity in density, morphology, turnover rate, pruning, metabolism and molecular signature (17–19).
Microglial activation occurs through inflammatory insult or slight alterations in brain homeostasis. This activation is dependent on the context and the type of stressor or pathology. Microglia determine the pathological outcome of stressors through secretion of cytokines, chemokines and growth factors and psychopathologies have repeatedly been associated with long-lasting priming and sensitization of cerebral microglia (10, 20, 21). Microglia also modulate communication between the nervous and the immune system in response to different physiological, psychological and immunological stressors. They are in fact considered to be responsible for the decreased neuroplasticity observed in depression (22) and recent findings have associated microglial abnormalities with neuropsychiatric disorders such as MDD, which have been termed microgliopathies by some (23–25).
Manipulating the microglial phenotype is an intriguing strategy for developing new therapeutics for MDD. Particular attention is currently given to exploiting alternative microglial polarization as a potential therapeutic option in a wide range of neurological and neuropsychiatric disorders (15). To achieve this, canonical pathways that govern tuning of the microglial phenotype have been investigated including transforming growth factor β (TGF-β), IL-4 receptor and peroxisome proliferator-activated receptors-gamma (PPAR-γ) (26, 27). We and others previously published reviews on the roles played by microglia in psychiatric disorders (10, 13, 21, 28). However, a review focused on different canonical microglial pro-inflammatory, anti-inflammatory and metabolic pathways and their translational value in drug discovery for MDD is lacking. We aim to fill the gap.
The role(s) played by the innate immune response in MDD has been the object of several experimental and clinical studies in which microglia have increasingly become the focus of investigation. Microglia have been studied in different psychiatric disorders using various approaches, including postmortem investigations in humans as well as experimental studies in animal models (21, 29, 30). The term neuroinflammation, denoting inflammatory processes in the CNS, is a rather general notion that could include both peripheral and central components of inflammation. Interestingly, a number of studies have suggested the involvement of peripheral inflammation in the pathogenesis of depression. Menard et al. associated a reduced expression of the endothelial cell tight junction protein claudin-5 with abnormal blood vessel morphology in the nucleus accumbens of stress-susceptible but not resilient mice (31). In a more recent study, the same group showed that epigenetic regulation of claudin-5 is associated with stress resilience. Indeed, they identified nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signaling pathway and histone deacetylase 1 as mediators of stress susceptibility. Pharmacological inhibition of histone deacetylase 1 rescued claudin-5 expression in the nucleus accumbens and promoted resilience (32). It is to be noted, however, that blood–brain barrier (BBB) disruption is not a common phenomenon in animal models of depression (31) and that the mechanism underlying this region-specific BBB abnormality remains to be clarified.
Recently, neuroinflammation has been used to explain functional microglial abnormalities observed in psychopathologies. A developed mouse model of obsessive-compulsive disorder serves as an interesting example of the link between microglial abnormalities and mental illness. Mutant HOXB8 mice display unexpected behavior manifested by compulsive grooming and hair removal (33). These actions directly mirror trichotillomania seen in humans with obsessive-compulsive spectrum disorder (33). Chen et al. reported that, in the brain, the HOXB8 cell lineage exclusively labels bone marrow-derived microglia. This finding strongly fosters the theory that the excessive grooming behavior observed in HOXB8 mutant mice is a consequence of defective microglia, thus relating hematopoietic function to mouse behavior (33). Another interesting example is the effect of CX3CR1 deficiency on behavior relevant to post-traumatic stress disorder. Neuronal CX3CL1 and its microglial target CX3CR1 play an essential role in synaptic plasticity and a correlation between CX3CR1 deficiency and increased fear behavior as well as an anxiolytic-like phenotype have been reported by Schubert et al. (34). However, Milior et al. observed a contradicting finding in which microglial CX3CR1 knock-out mice were resilient to chronic unpredictable stress (CUS) suggesting microglia-neuron communication may be at the interplay of resilience or susceptibility to a depression-like phenotype (35). Colony-stimulating factor 1 receptor signaling was also shown to control cerebellar microglia and to be essential for motor function and social interaction (36).
Despite a strong correlation between microglial activation and depression in pre-clinical and clinical studies, it remains unclear whether microglial abnormities play a causal role in depression (37). Clinical findings indicate a strong correlation between disease etiology and inflammation (37). Subsets of MDD patients consistently display increased levels of pro-inflammatory cytokines such as TNF-α and IL-6 (11, 30). A previous postmortem investigation by our group has shown that the percentage of primed microglia is increased in the dorsal anterior cingulate cortex (ACC) of depressed suicides compared to matched controls (38). This observation is consistent with independent reports of microglial activation in the ACC of MDD patients (39).
There is a growing body of literature showing increased microglial activation in inflammatory and non-inflammatory rodent models of depression. The most studied inflammatory model is that of lipopolysaccharide (LPS) injection. Systematic LPS administration not only triggers peripheral immune responses but also activates microglia in the brain (37, 40). Following LPS challenge, pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 are upregulated in different brain areas (41, 42). These inflammatory changes are accompanied by decreased sucrose preference and increased immobility in the forced swim test (43). Moreover, animals that receive acute or chronic non-inflammatory stress also show microglial activation along with morphological changes and increased levels of pro-inflammatory cytokine release in different brain regions including the hippocampus, thalamus and prefrontal cortex. For example, mice subjected to chronic social defeat (CSD) exhibit increased numbers of CD68-expressing microglia that have increased phagocytic capacity. Several other groups have reported microglial dysregulation following CSD stress (44–49). Lehmann et al. have reported that depressive-like behavior throughout and following CSD involves microglia-derived reactive oxygen species (ROS). Using colony stimulating factor receptor antagonist PLX5622 to deplete microglia before and during the 14-day CSD procedure, mice were protected from the effects of stress as measured by light/dark and social interaction paradigms (10, 50). More evidence indicating the involvement of microglial activation in depression comes from minocycline studies. Minocycline is an anti-inflammatory tetracycline that inhibits microglial activation and subsequent neuroinflammation (37, 51). Minocycline treatment does not have anti-depressive behavioral effects in naïve mice (52), however, it elicits significant anti-depressive effects in the rat model of chronic unpredictable mild stress (CUMS) (53). Intriguingly, combinatorial therapy of minocycline with antidepressants provides better clinical outcomes in some MDD patients, implying the contribution of neuroinflammation and microglial activation in a subset of patients afflicted with this psychopathology (54).
In the following sections, we aim to highlight the potential roles played by microglia in the pathogenesis of MDD, with a focus on the roles played by the canonical pro-inflammatory, anti-inflammatory and metabolic pathways.
TNF-α is a trimeric cytokine that is expressed either in a 27 kDa transmembrane form or a 17 kDa soluble form processed by TNF-α converting enzyme (55–57). TNF-α exerts its pleiotropic effects by binding to two primary receptors: TNF-α Receptor 1 (TNFR1) and TNF-α Receptor 2 (TNFR2). Soluble TNF-α typically binds TNFR1 after clustering at the cell membrane (58, 59). Transmembrane TNF-α preferentially binds TNFR2 as a ligand and can serve as a receptor for cell-to-cell contact (56, 59). Regardless of form, after a TNF-α ligand binds, TNFR1 and TNFR2 from homodimers to induce downstream signaling for cellular processes such as defense from foreign pathogens, enhancing inflammation and promoting cell survival or apoptosis (57, 60, 61).
TNF-α is produced by different cell types in the CNS, including neurons, astrocytes, microglia and endothelial cells (62, 63). However, monocytic immune cells like microglia are the dominant secretors and targets of TNF-α (57). TNF-α is now well understood as a critical pro-inflammatory cytokine that has an instrumental roles in the CNS, including innate immunity, sleep regulation, neuronal activity and necrotic and apoptotic cell death (58, 64). Under physiological conditions TNF expression is induced by basal activity in microglia, neurons and astrocytes. This cytokine is essential for regulating neuronal function including synaptic activity. Neurons constitutively express TNF-α receptors, which are important for mediating neuroprotection against neurotoxic stimuli (65).
Microglia-derived TNF-α is critical in innate immune responses within the CNS (66). Previous studies have documented the ability of TNF-α to influence microglial function in response to neuroinflammatory insults. For instance, to achieve a swifter recognition of foreign pathogens, TNF-α binds to its microglial receptors and upregulates the expression of toll-like receptor (TLR) 2, a pattern recognition receptor specialized for bacteria, enhancing microglia’s overall immune response (67). Aside from priming microglia for enhanced pathogen detection, this cytokine also enhances natural killer cells’ and macrophages’ ability to kill cells and phagocytose, respectively (58, 67).
TNFR1 and TNFR2 notably have varying functions within the brain (Table 1), some of which are region-specific. It was previously observed that the reparative function of TNF-α on neurons in the striatum is reliant upon TNFR1 (68). At the same time, similar capabilities of TNF-α in the hippocampus depend upon TNFR2 but the receptors’ expressions were equal in both regions (68). A more recent study found similar effects when investigating TNFR1 and TNFR2 single nucleotide polymorphisms (69). These results indicated that TNF-α regulation of striatal morphology was predominated by TNFR1 signaling while regulation of hippocampal morphology was shown to rely primarily on TNFR2 (69). It is thought that this region-specificity may be due to the two receptors’ differential impacts on cell survival since TNFR1’s downstream processes promote apoptosis and TNFR2’s pathways are more anti-apoptotic (68).
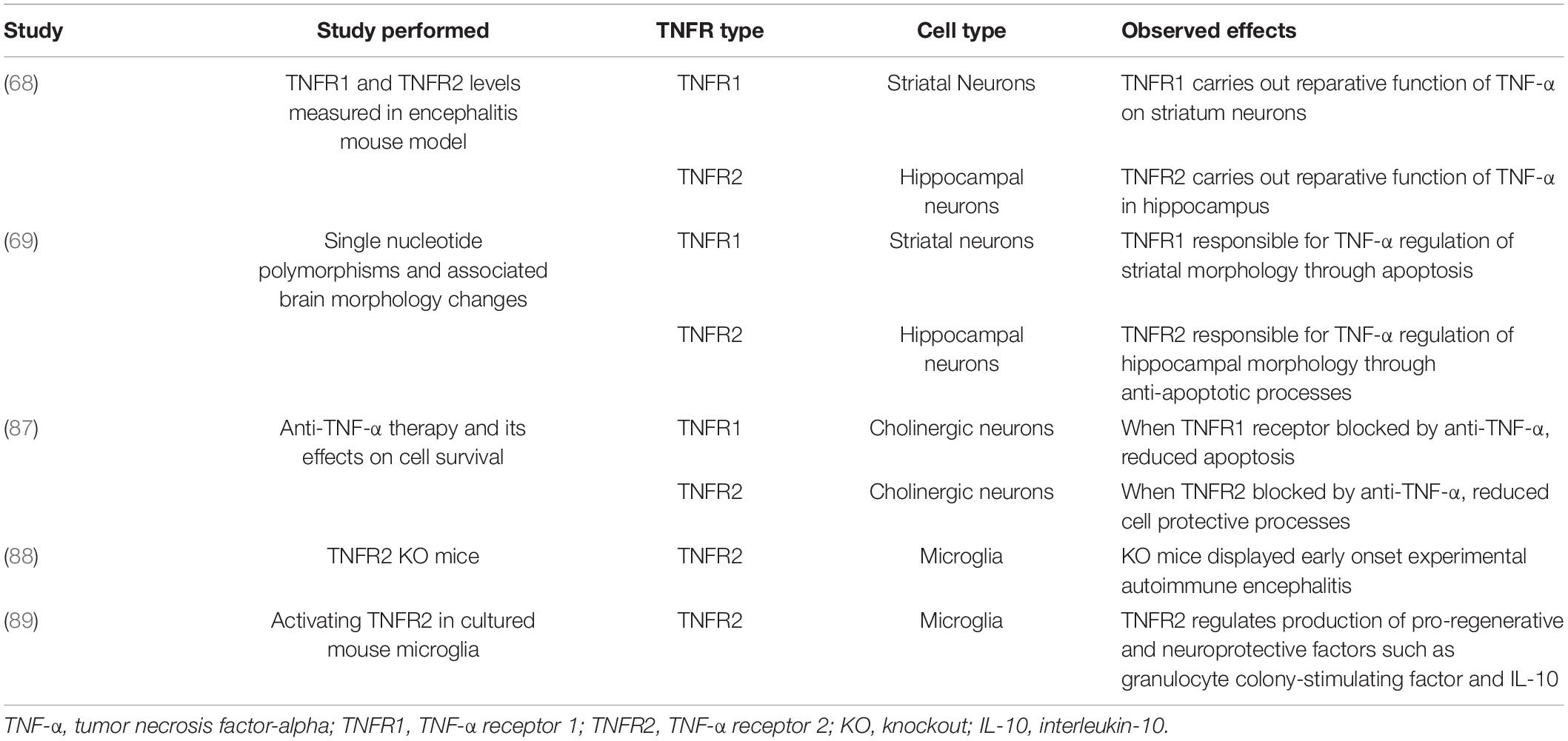
Table 1. Overview of studies on TNFR1 and TNFR2’s differential processes within the brain.
The distinct effects of these receptors on cell survival and inflammation can be understood through their different recruitment of signaling complexes after a ligand binds (Figure 1). Active TNFR1 homodimers allow four different complexes to form to engage various cellular processes (70). Complexes I, IIa, and IIb similarly activate NF-κB and mitogen-activated protein kinases (MAPK) to promote cell survival, cell proliferation, immune defense and inflammation (70). Complexes IIa and IIb are additionally responsible for activating the caspase apoptotic pathway (70). The last complex, complex IIc, also plays a role in inflammation but is most notable for its role in necroptosis (70). Unlike TNFR1 pathways, the current understanding of TNFR2 is not as comprehensive (71). However, previous studies with TNFR2 knockout mice have shown its importance in anti-inflammatory and cell-protective processes (71). Furthermore, it is theorized that active TNFR2 homodimers recruit adapter protein TNF receptor-associated factor 2 to activate NF-κB pathways (71). Despite TNFRs sharing similar structures and ligands there is a clear heterogeneity in their downstream effects.
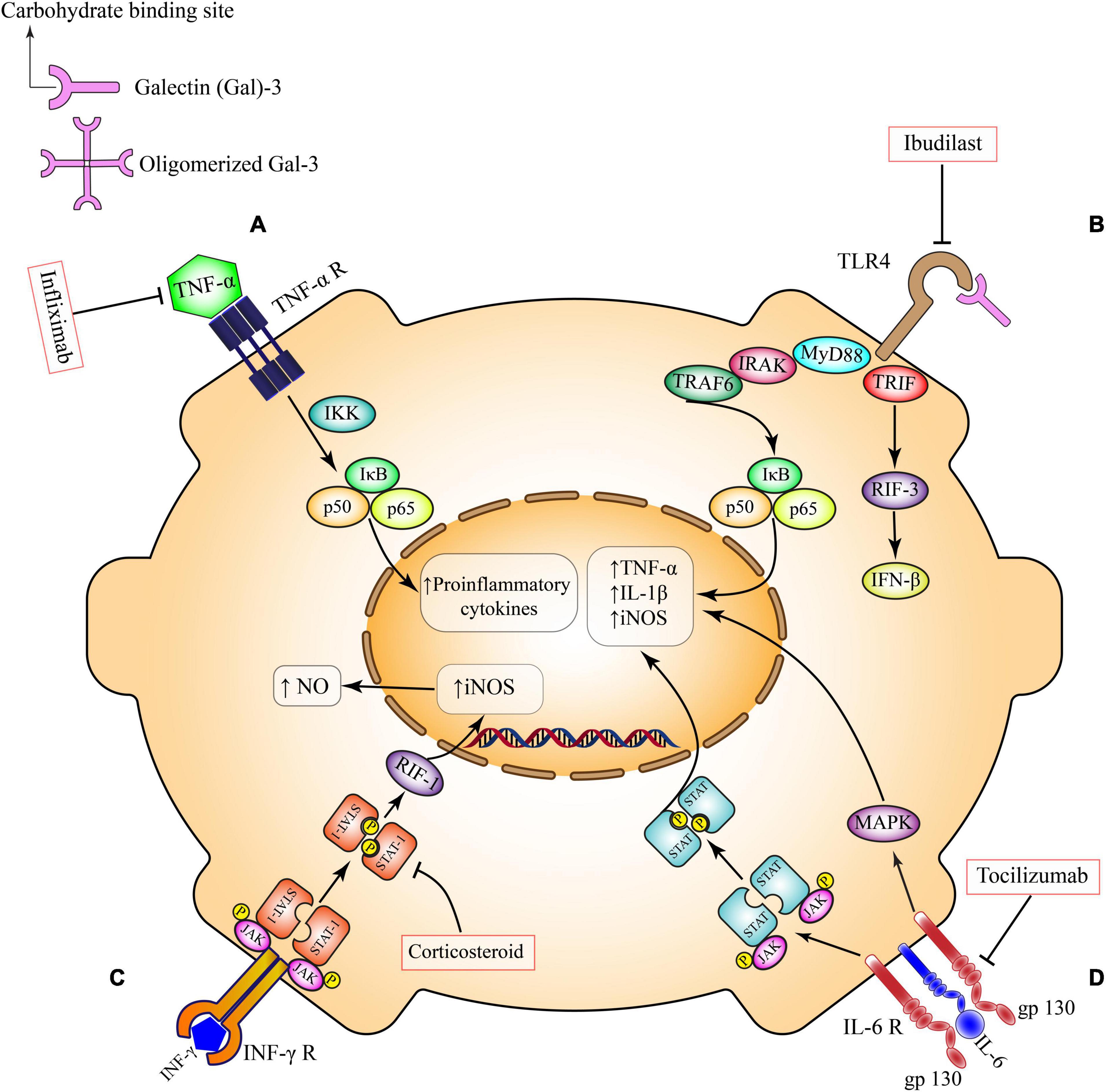
Figure 1. Pro-inflammatory pathways in microglia. (A) TNF-α receptor activation, induces the canonical pro-inflammatory transcriptional factors such as NFκB and subsequent production of inflammatory mediators. This pathway can be inhibited by Infliximab. (B) TLR4 ligands and secreted Gal-3 directly bind to TLR4 on the microglial surface and exacerbates inflammatory responses through induction of different cytokines and chemokines. This pathway can be inhibited by Ibudilast. (C) Activation of INF-γ receptor on microglia triggers the overexpression of inducible nitric oxide synthase (iNOS) and overproduction of nitric oxide via Janus kinase (JAK)/signal transducer and activator of transcription (STAT)/RIF-1 pathway. Corticosteroids can suppress this pathway at the level of STAT factors. (D) IL-6 trans-signaling occurs in brain cell types that have membrane bound gp130, including microglia. IL-6 bound to soluble IL-6R activates signaling through membrane bound gp130. This trans-signaling is thought to be pro-inflammatory via the induction of JAK/STAT and MAPK signaling pathways. Tocilizumab inhibits this pro-inflammatory pathway. TNF-α, tumor necrosis factor-α; IKK, the IκB kinase; NO, nitric oxide; iNOS, inducible nitric oxide synthase; IL-1β, interleukin 1 beta; IL-6, interleukin 6; JAK, Janus kinase; STAT, signal transducer and activator of transcription; RIF-1, replication timing regulatory factor 1; MAPK, a mitogen-activated protein kinase; TLR4, toll-like receptor 4; MYD88, myeloid differentiation primary response 88; TRIF, TIR-domain-containing adapter-inducing interferon-β; IRAK, interleukin 1 receptor associated kinase; TRAF, TNF receptor associated factor; IFN-β, interferon-β; IFN-γ, interferon gamma.
TNF-α has long been implicated in several peripheral and central inflammatory conditions (58, 72, 73). Meanwhile, research on the role of TNF-α in psychiatric disorders is evolving. In this context, TNF-α’s pro-inflammatory functions may exacerbate or contribute to depressive symptoms (74). Both TNFR1 and TNFR2 pathways can modulate inflammatory pathways through downstream NFkB signaling (60). Additionally, TNF-α can induce glutamate-mediated excitotoxicity. This cytokine facilitates crosstalk between microglia and astrocytes to promote the release of astrocytic glutamate, the formation of excitatory synapses and the release of more TNF-α from microglia (75). Therefore, TNF-α mediated pathways can lead to extraneous inflammation and cell death contributing to the worsening of MDD.
In accordance, several studies have seen higher levels of serum TNF-α associated with depressive symptoms and in MDD patients compared to matched controls (76–78). A few studies measuring cytokine profiles in cerebrospinal fluid (CSF) have not found differences in TNF-α levels between depressed patients and healthy controls (79). However, there are only few such studies of CSF cytokine levels and the results seem rather inconsistent as compared to studies of peripheral (plasma) cytokines (79). In addition, plasma cytokine profiles do not necessarily represent cytokine levels within the CNS (80).
In a pioneer study, Ohgidani et al. showed that acute stress induces TNF-α secretion from hippocampal microglia resulting in mouse working memory deficits. These authors observed that morphological changes in hippocampal microglia did not occur (81). Furthermore, etanercept, a TNF-α inhibitor, rescued the working memory impairment accompanied by a reduction in hippocampal TNF-α (81). Indeed, maladaptive microglial activation may be linked to MDD and modulating microglial activation seems a promising therapeutic target for depression.
A previous study with monoclonal antibody against TNF-α revealed that this intervention quelled symptoms of anhedonia but did not affect depression scores significantly compared to placebo groups (82). Current data seem to support the effectiveness of anti-inflammatory agents as antidepressants only in patients with increased peripheral inflammation (83). This subgroup may include MDD patients with increased inflammatory markers or those with medical conditions characterized by increased levels of peripheral inflammation (84). Specifically, anti-TNF-α therapy was found not to be effective in all treatment-resistant depression patients, but it did improve depressive symptoms in those with higher baselines of inflammatory markers (85). One major reason for such failure is the double-edged role of microglial TNF-α in fundamental physiological processes such as the neuroinflammatory response to tissue damage, neuronal circuit formation, synaptic plasticity and myelin degeneration and repair (86). Indeed, any positive impact from blocking TNFR1 receptors would be nullified by blocking TNFR2 activation, adding another complex layer upon possible therapies for MDD (87). It has been shown that the TNF-α mediated activation of microglial TNFR2 is instrumental for the protective functions of these cells (86). For instance, microglia-specific TNFR2 knockout mice display early onset of experimental autoimmune encephalitis (88). In this context, it has been shown that TNFR2 regulates the production of pro-regenerative and neuroprotective factors including granulocyte colony-stimulating factor and IL-10 in microglia (89). It is noteworthy that protective aspects of TNF-α signaling in microglia have been greatly overlooked with respect to the pro-inflammatory ones. Thus, more investigations are needed to decipher the mechanisms regulating the balance between TNFR1 and TNFR2 pathways in microglia in order to limit the detrimental immune responses without blocking the protective ones (86).
Over the years, the cytokine IL-6 has been linked to stress-related disorders such as depression and anxiety (90). This cytokine is a small multifunctional protein (91) that can be produced by several cell types including endothelial cells, epithelial cells, astrocytes, microglia and neurons (92, 93). IL-6 belongs to a family of proteins that utilize glycoprotein 130 (gp130) as a signal transducer (Figure 1). Depending on the presence of IL-6 receptor (IL-6R) or membrane bound gp130 which are expressed differently in different cell types, IL-6 has pro- or anti-inflammatory properties (93) resulting in either inflammatory or anti-inflammatory cascades (90).
According to a few major meta-analyses, IL-6 is one of the most consistently elevated cytokines in the blood of patients with MDD (11, 94, 95). Remarkably, IL-6 blood levels might have a predictive value as a biomarker. Moreover, peripheral levels of IL-6 correlate with symptom severity of antidepressant non-responders (96). In addition, increased levels of IL-6 have been reported in the CSF of patients with MDD as well as in suicide attempters (3, 97). Unfortunately, research addressing the role of microglial derived IL-6 are lacking in human postmortem studies. Only one pioneer study indicates a non-inflammatory phenotype of microglia in MDD following single-cell mass cytometry of microglia (98). The authors performed single-cell analysis of microglia from four different postmortem brain regions including frontal lobe, temporal lobe, thalamus and subventricular zone of medicated individuals with MDD and they found no evidence for the induction of canonical pro-inflammatory (IL-1β, IL-6, and TNF-α) and anti-inflammatory cytokines such as IL-10 (98).
Various pre-clinical studies have investigated the role of microglial IL-6 in the context of stress. For instance, increased IL-6 mRNA is found in microglia isolated directly from the brains of mice that have undergone repeated social defeat (RSD) stress (99) and treatment with the antidepressant imipramine inhibits social avoidance behavior and diminished microglia IL-6 in mice exposed to stress (90, 99). In another study, Aniszewska et al. found that stress induced a significant increase in the number of IL-6-immunoreactive microglia in the hippocampus, cortex and brain stem (100).
Blocking IL-6R-mediated pathways (e.g., tocilizumab) or neutralizing IL-6 function (e.g., sirukumab) might have clinical value in a subset of MDD patients, especially in treatment-resistant cases or in patients with peripheral inflammatory diseases (101). Interestingly, the efficacy of interleukin-6 neutralizing antibodies on symptoms of MDD patients with RA has been reported (102). However, BBB penetration and adverse effects may limit their use in MDD patients without the history of peripheral inflammatory diseases such as RA (103). In addition, there are two types of IL-6 signaling: a classical anti-inflammatory signaling and a trans-signaling proinflammatory signaling. It means that general targeting of IL-6 pathways in MDD either with IL-6R inhibitors or IL-6 blocker is not an optimal choice (101, 103). A more selective intervention seems more promising for future drug discovery targeting IL-6 signaling (101).
TLR4 is one of nine members in the TLR family of pathogen-specific pattern recognition receptors dedicated to responding to unique structural components of foreign microbial agents to trigger immune responses (104, 105). As such, this receptor is highly involved in regulating brain innate immune responses in pathophysiological conditions. TLR4 is notably the most researched receptor within the TLR family. It is well known to be primarily responsible in the reaction against Gram-negative bacteria by binding to LPS, its pathogen-associated molecular pattern (106). However, TLR4 has also been shown to recognize damage-associated molecular patterns (DAMPs) and xenobiotics (107). Additionally, TLR4 can be activated by other non-bacterial TLR4 agonists naturally present within the body such as saturated fatty acids (108). TLR4s are generally expressed in myeloid lineage cells like macrophages and other non-immune cells like endothelial cells (106). Within the CNS, TLR4 is expressed primarily by microglia but can also be found in astrocytes, oligodendrocytes and neurons (107).
NF-κB refers to a group of transcription factors that serve as significant regulators of pro-inflammatory genes and has been of particular interest as a target for pharmacological treatments in inflammatory diseases (109). They exist as two subfamilies of inducible dimers, made up of either DNA binding proteins from the ‘Nκ-kB’ or ‘Rel’ family, which can then form homodimers or heterodimers (110). Alternatively, the term NF-κB can also describe p50-RelA heterodimer, the predominant NF-κB dimer present in many cells (111). Regardless, all NF-κB dimers are constitutively inhibited by IkB proteins that are degraded to activate and translocate the transcription factor into the nucleus (109). NF-κB activity is rapidly induced in microglia following inflammatory insults (112).
During activation with LPS, the pathogen-associated molecular pattern forms a multimolecular complex with TLR4 and its accessory molecules which readies the receptor for dimerization (113). The formed dimer then induces downstream effects (Figure 1) either through the myeloid differentiation primary response 88 (MyD88)-dependent pathway or the MyD88-independent pathway (106, 114). The MyD88-dependent pathway leads to activation of transcription factors that induce the expression of pro-inflammatory cytokine genes (114). The MyD88-independent pathway, also known as the TIR-domain-containing adapter-inducing interferon-β dependent pathway, also activates transcription factors but mediates the induction of type 1 interferon-inducible genes (114). In both cases NF-κB is activated (115).
TLR4 and NF-κB are of particular interest in pathophysiological conditions due to their significant roles in innate immunity. Accordingly, many of the following studies have implicated their dysregulation within various neurodegenerative diseases and psychiatric disorders (116–118). Elevated TLR4 expression and associated overactive microglia were observed within a transgenic mouse model of Alzheimer’s Disease (AD) leading to cognitive impairment (119, 120). Furthermore, previous investigations have also characterized an increase in NF-κB activation within the CNS of animal models and patients with neurodegenerative disorders (117). Interestingly, a study of postmortem brains from patients with schizophrenia revealed that TLR4 levels are increased within the cerebellum but decreased within the prefrontal cortex (PFC) suggesting that general dysregulation, rather than upregulation, could lead to harmful effects (118). In the same study, NF-κB levels were notably altered inversely to TLR4 levels, with increased levels within the PFC and decreases in the cerebellum (118). Another investigation demonstrated increases in NF-κB levels, especially in microglia, as a neuroinflammatory mechanism in autism spectrum conditions (116). The studies above support the idea that TLR4, NF-κB and their combined pathway likely play significant roles in neuroinflammatory response in MDD. More specifically, the dysregulation of TLR4 and NF-κB inflammatory processes has been suggested to be involved in MDD. TLR4 single gene polymorphisms were associated with suicide and anxiety scores in MDD patients, while methylation levels of TLR4-associated CpGs were related to the severity of depressive symptoms (121, 122). In general, increased TLR4 expression and decreased expression of its inhibitor TNFAIP3 were associated with depressive symptoms (107). Similarly, increased NF-κB activity also appears to play a role in depression. In a rat early-life stress model, the depression-susceptible animals generally displayed more activated NF-κB. Inversely, the subgroup showing resilient phenotype had more inactivated NF-κB (123). Another notable study supported this idea by demonstrating improved depressive-like behaviors in mice when increased NF-κB expression was inhibited (124). Other studies have specifically investigated TLR4-NF-κB pathways in depression. A supporting study indicated that suppressing the TLR4-NFκB signaling pathway inhibits depression-like behavior in mice (125). These studies sparked further interest in investigating these pathways to develop new antidepressant candidates for MDD patients (Table 2).
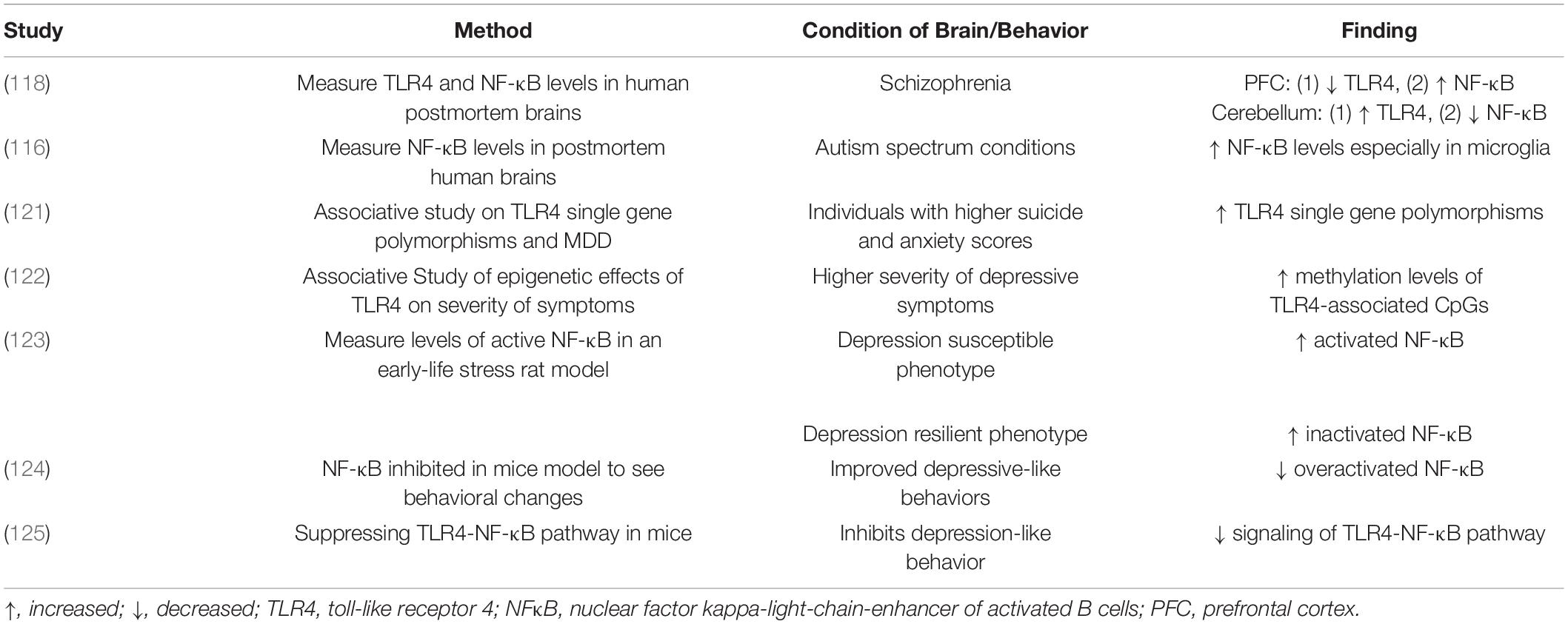
Table 2. Overview of studies performed to investigate TLR4 and NF-κB’s roles in neuropsychiatric disorders.
The current literature strongly supports the existence and importance of TLR4, NF-κB and their conjoined roles in MDD. Although TLR4 activation seems to play a role in MDD, the underlying mechanisms are unclear. Much of the previous work implicating TLR4 in MDD have primarily focused on the role of TLR4 following activation by LPS in bacterial infections (114). However, as noted previously, TLR4 can be triggered by many different ligands; thus, current perspectives are limiting and require further investigation from novel angles (106–108). More specifically, it may be useful to consider the immune signaling that coincides with other events aside from infection, such as during the critical period of development or epigenetic modifications (126, 127). These are important time points that may impart predisposing factors on individuals if TLR4 signaling is altered (126, 127). Furthermore, these deleterious inflammatory states associated with altered TLR4 pathways may also be attributed to other mechanisms such as neuroendocrine signaling and dysregulated gut microbiota (127). Therefore, to improve the efficacy of future drugs targeting TLR4 in MDD, research into more diverse mechanistic facets should be conducted. A recent study used this approach to propose polyphenols as a potential group of drug candidates for future consideration (128).
Interferon-gamma (IFN-γ) is a pleiotropic soluble cytokine that is produced by different immune cell types including lymphocytes, B cells and antigen-presenting cells. In the CNS, different cells such as neurons, microglia and astrocytes produce this cytokine and express its receptors. The activation of the IFN-γ receptor induces several canonical downstream pathways (Figure 1) such as the janus kinase (JAK) 1 and 2, signal transducer and activator of transcription (STAT) 1 and the extracellular-signal-regulated-kinase (ERK) 1/2. Many genes, as well as micro RNAs and long non-coding RNAs, are activated following IFN-γ receptor stimulation (129). Neuroinflammation mediated by IFN-γ has been reported in neurological disorders. However, the effects of IFN-γ on behavior in the context of stress are mostly unknown (129). Intriguingly, IFN-γ knockout mice show decreased anxiety- and depressive-like behaviors (129). These effects are accompanied by elevation of serotonergic and noradrenergic activity in the central amygdaloid nucleus, together with increased baseline plasma corticosterone, decreased neurogenesis in the hippocampus and decreased levels of nerve growth factors in the PFC, indicating that IFN-γ modulates anxiety and depressive states and is involved in CNS plasticity (130–132). In an intriguing report, Zhang et al. showed that intracerebroventricular injection of IFN-γ in mice causes impairment of adult hippocampal neurogenesis, behavioral despair, anhedonia and cognitive loss. Furthermore, IFN-γ induces microglial activation that is associated with morphological changes and upregulation of phagocytic marker CD68 and pro-inflammatory cytokines (IL-1β, TNF-α, and IL-6) (133). Inhibition of the JAK/STAT1 pathway, downstream of IFN-γ receptor, suppresses microglial-mediated neuroinflammation, diminishes depressive-like behaviors and improves memory (133).
An array of studies has demonstrated the possible effects of antidepressants, particularly selective serotonin reuptake inhibitors (SSRIs), on inflammatory responses and microglial function (10, 134). Besides their known therapeutic mechanism involving the modulation of the serotonergic system, SSRIs can regulate the activation state and secretory profile of microglia (10). In this context, Horikawa et al. reported that paroxetine and sertraline prevent microglial activation by inhibiting IFN-γ-induced elevation of intracellular calcium (135). Interestingly, Alboni et al. found that quality of the environment effects the mechanism of action of Fluoxetine. Enriched environments coincident with Fluoxetine administration induced pro-inflammatory microglial profiles while a stressful environment resulted in anti-inflammatory secretory profiles (134).
It is established that microglia adopt reactive states in response to an inflammatory insult. However, at both transcriptional and functional levels, microglia appear to be more complex and dynamic than anticipated. This might explain why engagement of microglia can be either neuroprotective or neurotoxic, leading to attenuation or exacerbation of disease progression (10, 15, 136) depending on the context. According to the traditional classification of macrophages/microglia, during microglial activation following an inflammatory insult, cell morphology is altered either to M1, the typically activated phenotype, or to M2, an alternative activated phenotype; and this phenotypic switch depends on the type of insult. M1 microglia are considered proinflammatory and produce mediators such as TNF-α and IL-1β. It has been shown that INF is a canonical cytokine that can polarize microglia toward M1. It is noteworthy that the classification of the M1 and M2 phenotypes have been challenged (10, 136, 137). The reason is that such classification has been defined mainly based on in vitro studies of peripheral macrophages and that M1 and M2 states fail to emerge in brain resident microglia. It is now accepted that activated microglia co-express canonical gene products associated with both M1 and M2 states. Indeed, following brain injury, microglia do not simply switch to a polarized “M1-only” or “M2-only” phenotype but rather display a mixed phenotype due to the complex signaling cascades surrounding them (27, 136, 137). However, in the context of this review, we will continue to use the broad categories of activated pro-inflammatory and anti-inflammatory unless the study mentioned investigated more dynamic phenotypes.
The choroid plexus (ChP), a highly vascularized tissue that produces CSF and lacks a BBB, is an interface between peripheral and central immune responses (138). Our group previously investigated the cellular and molecular inflammatory profile of the ChP of the lateral ventricle in depressed suicides and healthy controls (138). We measured the content of several pro- and anti-inflammatory transcripts as well as the density of Iba1+ macrophages associated with the ChP epithelial cell layer. The levels of pro-inflammatory markers, ICAM1 (a protein implicated in immune cell trafficking) and Iba1, were measured to be significantly downregulated in depressed suicides as compared to controls (138). Intriguingly IFN-γ signaling has been shown as a selective key regulator of immune cell trafficking across the ChP epithelium under physiological conditions of CNS immune surveillance and following neuroinflammatory insult (139). This unique mechanism could be harnessed to adjust the interplay between the peripheral immune system and microglia in affective disorders.
The alpha-7 nicotinic acetylcholine receptor (α7 nAChR) is a ligand-gated ion channel expressed by macrophage/microglia (140, 141) and has proved to be a promising target in pharmacotherapy of psychiatric disorders. α7 nAChR agonists or partial agonists are known to improve cognitive dysfunction by regulating microglial activation through inhibition of canonical pro-inflammatory transcriptional factors such as NFkB and induction of anti-inflammatory signaling pathways such as nuclear factor-erythroid factor 2-related factor 2 (Nrf2) (142–144). In fact, there is ample evidence that the cholinergic system plays a fundamental role in regulating central inflammation and glial activation via homomeric α7 nAChRs (145–147). The α7 nAChRs consist of five α subunits and are expressed by neuronal and glial cells (148, 149). These ligand-gated ion channels allow for calcium influx and subsequent ultra-rapid desensitization (150, 151). α7 nAChRs are widely expressed in the brain, including in regions such as the PFC, hippocampus and other limbic areas (150). Microglial α7 nAChRs play important roles in regulating inflammatory processes in the CNS (148, 150). Stimulation of α7 nAChR leads to a reduction in glial activation and decreases in proinflammatory cytokine levels in different brain regions (152–154).
The microglial α7 nAChRs have dual ionotropic/metabotropic properties and their intracellular signaling pathways that modulate inflammation do not only depend on transient ion influx (145, 155, 156). Indeed, neuronal α7 nAChRs mainly have an ionotropic function (145). The downstream metabotropic signaling pathways of microglial α7 nAChRs are different from neuronal α7 nAChRs (157). Activation of microglial α7 nAChRs induces phospholipase C and enhanced calcium release from intercellular stores which are sensitive to inositol trisphosphate (153). This process results in the inhibition of NF-κB transcriptional activity (Figure 2) (158). As a result of this inhibition the levels of pro-inflammatory cytokines are decreased (154).
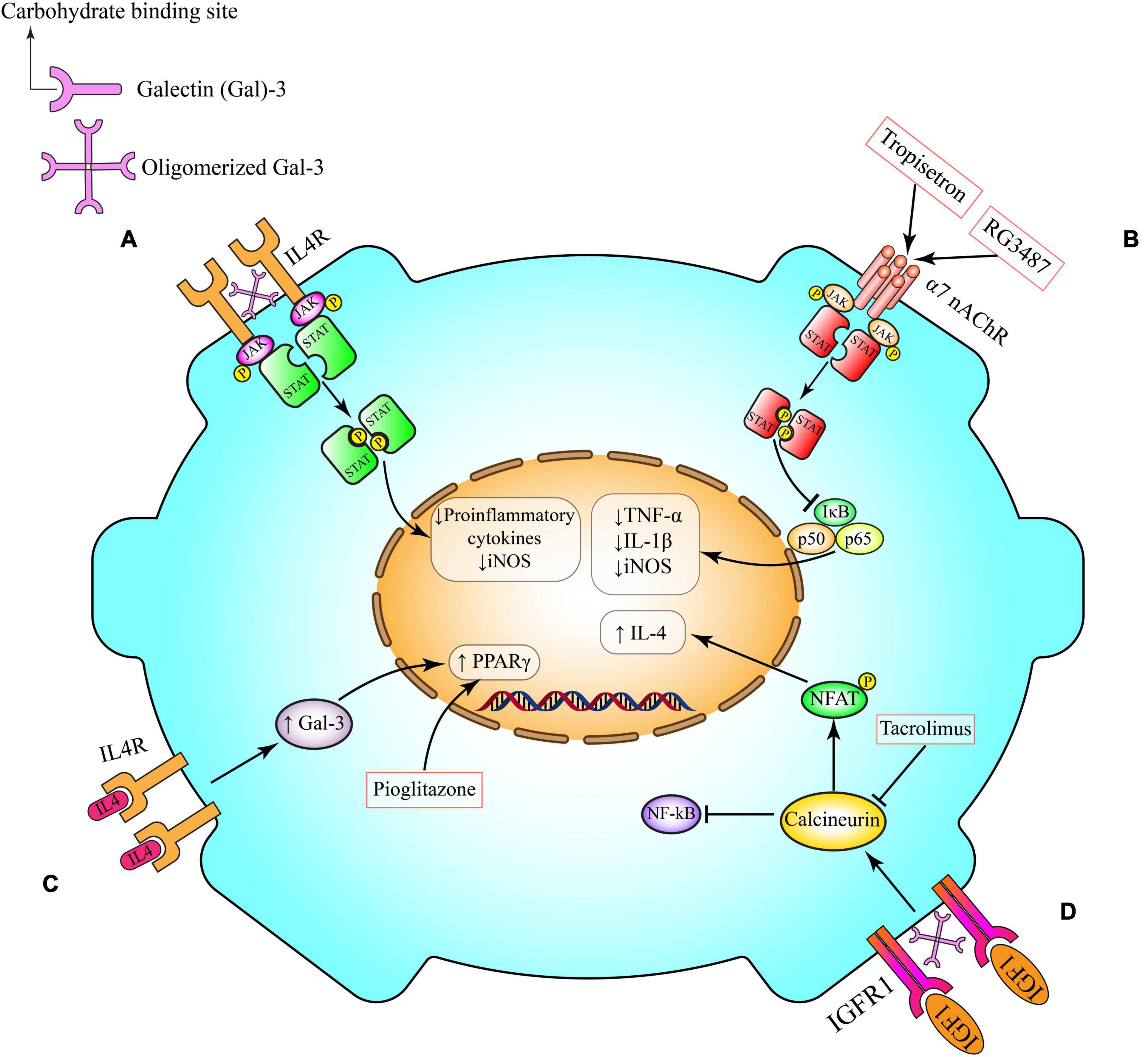
Figure 2. Anti-inflammatory pathways in microglia. (A) Gal-3 induces alternative microglia activation through interaction with IL-4 receptor (IL4R). Following Gal-3 lattice formation the carbohydrate-binding site of Gal-3 molecules interacts with glycosylated IL4R and prevents their endocytosis and also over activation of IL4R and its anti-inflammatory signaling. (B) Activation of α7 nAChR on microglia triggers anti-inflammatory cascades, including Janus kinase (JAK)/signal transducer and activator of transcription (STAT) and PI3K/Akt, which potentiate the activity of transcriptional factor Nrf2 and its downstream pathways (HO-1 and CAT), and inhibits the canonical proinflammatory protein NFκB, which governs the production of proinflammatory cytokines (e.g., TNF-α) and enzymes (e.g., iNOS and COX-2) involved in neuroinflammation. This pathway can be induced by tropisetron and RG3487. (C) IL-4 can interact with its tyrosine kinase IL4R on microglia cell surface. This interaction might activate one of the canonical transcriptional factors that are involved in microglia polarization such as PPAR-γ. Pioglitazone activates PPAR-γ. (D) By crosslinking insulin-like growth factor 1 receptor (IGFR-1), secreted Gal-3 will prevent early endocytosis and over-activate the Janus kinase (JAK)/signal transducer and activator of transcription STAT pathway and the transcription of genes needed for production of anti-inflammatory cytokines such as IL-4. IL-4, interleukin 4; iNOS, inducible nitric oxide synthase; TNF-α, tumor necrosis factor-α; JAK, Janus kinase; STAT, signal transducer and activator of transcription; NFAT, nuclear factor of activated T-cells; NF-κB, nuclear factor kappa B; α7 nAChR, α7 nicotinic acetylcholine receptors; Gal-3, galectin-3; IGFR, insulin-like growth factor 1 (IGF-1) receptor; PPAR-γ, peroxisome proliferator- activated receptor-gamma.
It has been shown that chronic restraint stress (CRS) alters central cholinergic signaling in brain regions that have been implicated in MDD (159). Namely, CRS induces hippocampal choline acetyltransferase protein expression and decreases nuclear STAT3 signaling. CRS also augments signaling activity, IL-1β and TNF-α expression and microglial activation. Intriguingly, cholinergic stimulation with a selective α7 nAChR agonist significantly diminishes CRS-induced depressive-like behavior, neuroinflammation and neuronal damage. Moreover, activation of α7 nAChRs restores central cholinergic signaling function, inhibits TLR4-mediated inflammatory signaling and microglial activity and increases the number of regulatory T-cells in the hippocampus following stress (159).
α7 nAChR activation induces the transcriptional activity of Nrf2 (143). Previous studies especially by the Lopez group indicate that α7 nAChR mediated activation of Nrf2 elicits anti-inflammatory mechanisms in microglia (160–165). This anti-inflammatory axis might play an instrumental role in antidepressant aspects of α7 nAChR modulators. In this context, the efficacy of selective or promiscuous ligands that can activate α7 nAChR-Nrf2 pathway have been shown in depressive disorders (144, 164). One interesting example for the promiscuous ligand is Tropisetron. This ligand is a 5-HT3 receptor antagonist and α7 nAChR partial agonist. This serotonergic ligand has shown a great efficacy in a wide range of psychiatric disorders including MDD and schizophrenia in both experimental models and clinical trials (144, 166). The other example is RG3487 (C15H19ClN4O), the novel 5-HT3 antagonist with α7 nAChR partial agonist properties. This ligand significantly improves attentional performance in experimental models and has shown promising results in clinical trials for cognitive impairment associated with schizophrenia (142, 167).
IL-4 is a multifunctional cytokine secreted by Th2 cells, mast cells, eosinophils and basophils (168, 169). IL-4 is a crucial molecule for microglia and macrophage polarization and it plays pivotal roles in brain function following neuroinflammatory insult (169). The effects of IL-4 are mediated through the IL-4 receptor α-chain. Following binding to its ligand, IL-4 receptor α-chain dimerizes either with the common γ-chain to produce the type-1 signaling complex located mainly on hematopoietic cells, or with the IL-13 receptor α 1 to produce the type-2 complex, which is expressed also on non-hematopoietic cells. The type-1 signaling complex (Figure 2) is pivotal for alternatively activated macrophages (168). Upon activation, the type-1 complex signals through JAK1 and JAK3, which phosphorylate and create docking sites for the transcription factor STAT6. This transcriptional factor then dimerizes and translocates to the cell nucleus to regulate the expression of several genes (168).
IL-4 might be protective against depression due to its ability to harness inflammation and to inhibit serotonin transporter activity. Wachholz et al. demonstrated that a decreased IL-4 responsiveness of microglia is specifically related to the development of depressive-like behavior. IL-4 deficient mice show notable augmentation of depressive-like behavior in the forced swim and tail suspension test (170). In experimental models of stress, the decline in IL-4 levels in the locus coeruleus may be involved in anxiety-like behavior and an inverse relationship between IL-4 secretion and hypothalamic-pituitary-adrenal (HPA)/sympathetic-adrenal-medullary-axes activation has been reported (171). These findings suggest that modulation of the IL-4 receptor signaling pathway is required to adapt to homeostatic mechanisms in response to stressful events (171). In addition, it has been shown that microglial IL-4 receptor pathway modulates cognitive function following neuroinflammation (172).
It is well established that adult neurogenesis in the dentate gyrus of the hippocampus is regulated by specific microglia population and potentially implicated in MDD (10, 173). Very recently, Zhang et al. showed in rodents that IL-4 driven microglia modulate stress resilience through BDNF-dependent neurogenesis (173). Their findings indicated that IL-4 driven microglia are characterized by a high expression of Arg1 which is critical in maintaining hippocampal neurogenesis and stress resistance. Decreasing Arg1+ microglia in the hippocampus by knocking down the microglial IL-4 receptor inhibited hippocampal neurogenesis and enhanced stress vulnerability. Indeed, Increasing Arg1+ microglia in the hippocampus by enhancing IL4 signaling restored hippocampal neurogenesis and the resilience to stress-induced depression (173).
Following an inflammatory insult, endogenous IL-4 can interact with its tyrosine kinase IL-4 receptor on the microglia cell surface (Figure 2). This interaction induces the production of Galectin-3 (Gal-3), prostaglandin (PG) J2 and activates STAT6. Production or activation of these molecules ultimately leads to activation of canonical transcriptional factors for microglia polarization such as PPAR-γ. The transcriptional activity of PPAR-γ can induce microglia alternative activation by decreasing the production of ROS, pro-inflammatory cytokines and suppressing the activity of NF-κB (15, 27, 174). PPAR-γ is an important and canonical transcriptional factor in the induction of anti-inflammatory signaling pathways (discussed in detail in the next section).
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors of the nuclear hormone receptor superfamily. PPARs exist as three isoforms (α, γ, and δ/β). PPARs have a ligand binding domain and a DNA binding domain. When their endogenous or exogenous ligands bind to PPARs, they create a heterodimeric complex which recruits other co-activators including PPAR coactivator-1, PPAR-interacting protein, PPAR-binding protein, steroid receptor co-activator-1 and CREB binding protein. This complex binds to the promoter regions of specific genes that contain a regulatory element known as the peroxisome proliferator response element which either activates or transrepresses the target genes (175). In the mammalian body, PPARs control glucose metabolism, cell proliferation and differentiation (176). The fact that PPAR-γ is involved in the modulation of macrophage differentiation and activation in peripheral tissues led to studying of the role of PPAR-γ in CNS resident microglia. Several investigations indicate that PPAR-γ endogenous ligand and synthetic agonists might influence brain inflammation by inhibiting different functions related to microglial activation, such as production of inflammatory cytokines, chemokines, nitric oxide and prostaglandins (PGs) (177).
PPARγ ligands elicit anti-inflammatory and neuroprotective actions in various experimental models of neurodegenerative diseases (178). Indeed, PPARγ activation inhibits the activity of transcription factors including NF-κB, AP1, and STAT. Some studies indicate that IL-4 receptor signaling increases the endogenous level of PPAR-γ ligands such as PGJ2 and subsequently amplifies transcriptional activity of PPAR-γ which might polarize microglia phenotype toward the anti-inflammatory one (179).
The PPARγ-mediated pathway has been the subject of several pre-clinical and human studies of MDD. A low PPARγ level in the hippocampus and PFC has been associated with depressive-like behavior in mice (180). Selective agonists of PPAR-γ already have FDA approval for the treatment of type 2 diabetes and their potential antidepressant effects, through modulation of metabolism and inflammation, have been investigated in different models (181). Interestingly, the efficacy of PPAR-γ ligands has been shown in metabolic disorder to be associated with depressive-like behavior in rodents. Namely, obesity in rats results in downregulation of PPARγ in the PFC (182), meanwhile chronic treatment with pioglitazone reversed depressive-like behaviors associated with obesity in CUMS mouse model (183). There is an increased risk for obese patients with chronic low-grade inflammation to develop depression (184, 185). Also, obesity induces microglial activation and neuroinflammation that play crucial roles in the pathogenesis of depression (186).
In an interesting report Qin et al. demonstrated that CUMS can induce severe depressive-like behaviors, neuroinflammation and reduced expression of PPARγ in leptin-deficient (ob/ob) mice as compared to wild type mice. Administration of a selective PPARγ agonist, pioglitazone rectified the behavioral abnormalities and alleviated microglial pro-inflammatory cytokine levels and NF-κB activation in PFC and hippocampus (187). Other studies also investigated anti-inflammatory and antidepressant effects of PPAR-γ agonists. Li et al. aimed to explore the effects of pioglitazone on depressive-like behaviors of mice treated with LPS and elucidated the underlying mechanisms. Their findings indicated that PPAR-γ activation induces PI3K/AKT/JNK/p38 signaling pathway and counteract LPS mediated apoptosis in mice PFC (188).
Studies have provided evidence that the antidepressant-like effect of pioglitazone in the forced swim test is mediated partly through N-methyl-D-aspartate (NMDA) receptor signaling and nitric oxide pathway (189, 190). Cognitive impairment is a feature of both AD and psychiatric disorders. The PPARγ agonist, rosiglitazone improves hippocampus-dependent cognitive deficits. Its cognitive enhancement partly occurs through the induction of ERK cascade, a critical mediator of memory consolidation. Jahrling et al. showed that PPARγ agonism facilitated recruitment of PPARγ to pERK during memory consolidation (191). Other investigations have pointed to the involvement of NMDA receptor and nitric oxide pathway in the memory improving effects of PPARγ ligands (192, 193). These findings highlight the fact that PPARγ ligands might have therapeutic implication in MDD specifically in the patients that have memory impairments. Namely, Sepanjnia et al. showed that pioglitazone, a selective PPAR-γ agonist, is an effective and safe short-term add-on therapy to Citalopram in non-diabetic patients with MDD and was associated with a high rate of early improvement and remission (194). Taken together, these findings showcase the potential for developing new interventions that target the brain’s innate immune responses in different psychiatric disorders. However, much is still unknown about role of microglia in psychiatric disorders and why neuroinflammation is not a common phenomenon in all MDD patients (21).
Galectins are a family of soluble β-galactoside-binding proteins found in all multicellular organisms. They act as both DAMPs in innate immunity and/or as pattern-recognition receptors that bind to pathogen-associated molecular patterns. Gal-3 has recently been implicated in studies of neuroinflammatory diseases (195). This lectin is involved in cell-cell adhesion, modulation of the brain’s innate immune response and microglial activation patterns in both physiological and pathophysiological settings. Gal-3 also mediates cell proliferation and migration (196, 197). Several studies using different approaches and methods have demonstrated both protective and deleterious effects of Gal-3 in neuroinflammatory diseases making Gal-3 an attractive target in drug discovery. Among different galectin family members, Gal-3 is unique in that in addition to the carbohydrate recognition domain, it possesses a proline and glycine-rich N-terminal domain through which it forms oligomers (195, 198). Gal-3 is expressed in epithelial cells, endothelial cells, neurons and immune cells where it is synthesized as a cytosolic protein. It can be released or secreted into the extracellular space where several bind to cell surface glycoproteins (199). Originally identified as a marker of activated macrophages, there is increasing evidence suggesting its role as a modulator of microglial phenotypes in neuroinflammation (200).
Gal-3 plays important extracellular physiological roles. It uses IL-4 dependent mechanisms to mediate microglial arborization (195). In vitro studies highlight the importance of the carbohydrate-binding site of extracellular Gal-3 in microglia motility and ramification. Microglia pruning of axons and synaptic terminals might involve Gal-3 (18). Intracellular Gal-3 also holds distinct roles in physiological and pathological conditions. Following inflammatory insult, endogenous IL-4 interacts with microglial IL-4 receptors, thereby increasing the production of Gal-3 and subsequently inducing the canonical transcriptional factor PPAR-γ leading to anti-inflammatory signaling (Figure 2), as described in Section “Peroxisome Proliferator-Activated Receptor Gamma Mediated Pathway” (15, 201). In neuroinflammatory events Gal-3 elicits time-dependent protective actions. For instance, a Gal-3 feedback loop is critical for IL-4-mediated alternative polarization of peripheral macrophages (200, 202). It was also shown that following neuroinflammatory insult induction of Gal-3 in proliferating resident microglia is neuroprotective (27). Additionally, Gal-3 positive proliferating microglia are the major contributing cells of neurotrophic molecules such as insulin-like growth factor 1 (IGF-1) (200) which can also enhance the effects of trophic factors such as IGF-1 through inhibition of IGF-1 endocytosis (Figure 2) (195, 200). In 2019, Rahimian et al. studied time- and context-dependent effects of Gal-3 as a neuroprotective mediator following neuroinflammation (27). We showed that Gal-3 induces an anti-inflammatory microglial phenotype through IL-4 receptor pathway. It is likely that the polarization following neuroinflammatory insults is influenced by Gal-3 binding to glycans attached to IL-4 receptors (27).
In addition to its protective actions, Gal-3 also plays a pro-inflammatory role as reported in different animal and human studies especially in neurodegenerative disorders (195). Literature suggests that microglia-derived Gal-3 is detrimental in certain neuroinflammatory conditions. Indeed, comprehensive single-cell RNA analyses of CNS immune cells in neurodegenerative conditions including AD have led to the discovery of disease-associated microglia (DAM). This subpopulation of microglia displays a distinct transcriptional and functional signature (17). Boza-Serrano et al. showed that expression of Gal-3 in DAM in a mouse model of familial AD (5xFAD). They demonstrated that in 5xFAD mice Gal-3 is expressed solely in microglia associated with amyloid-β plaques and its deletion both decreases amyloid-β burden and improves memory function. Moreover, Gal-3 was found to be a TREM2 endogenous ligand binding through its carbohydrate-binding domain (203). Gal-3 direct interaction with TLR4 receptor may be an additional mechanism by which it regulates the severity of inflammation. Burguillos et al. showed that interaction of Gal-3 with TLR4 receptors in acute phase of neuroinflammation exacerbates neural cell death and prolongs inflammation, while its ablation elicits anti-inflammatory and neuroprotective effects (204).
Emerging experimental and clinical evidence indicates that Gal-3 may also play a role in MDD (205). Recently, Stajic et al. investigated the role of Gal-3 in modulation of anxiety levels in mice (206). The finding of this study revealed contradictory effects of Gal-3 on anxiety levels in the physiological condition and following acute inflammatory challenge with LPS. Gal-3 deficiency showed clear anxiogenic effect in basal conditions that is accompanied with lower expression of brain-derived neurotrophic factor (BDNF) and GABAA receptors. Gal-3 deficiency was also associated with anxiolytic response following acute administration of LPS (206). Intriguingly, the relationship between the novel inflammatory aspect of Gal-3 and depression symptom severity has been studied. In a large sample size, King et al. demonstrated higher Gal-3 levels were associated with higher levels of depressive symptoms. Their findings suggest that Gal-3 may be a new and useful inflammatory biomarker associated with depression (207). Another interesting clinical investigation showed that depression in type 1 diabetes is associated with high levels of circulating Gal-3 (208).
The endogenous cannabinoid (endocannabinoid) system has been implicated in synaptic communication and influences anxiety and cognition, metabolism, growth and development and response to internal and external immune insults via an array of actions mediated by their receptors (209, 210). In the CNS, endocannabinoids such as anandamide (AEA) and 2-arachidonoylglycerol (2-AG) regulate several physiological functions via two main G-protein-coupled cannabinoid receptors 1 and 2 (CB1 and CB2) (211). Endocannabinoids can also interact with several extracellular and intracellular targets such as G-protein-coupled receptor 55 (GPR55), PPARs and transient receptor potential vanilloid 1 (212). CB1 receptors are expressed in the cortex, hippocampus, cerebellum, basal ganglia and brainstem, usually at presynaptic terminals or on axons (210). They have also been reportedly found on glial cells (213, 214). CB2 receptors are much less expressed in the CNS compared to CB1 receptors, however, they have been found in the brainstem, cerebellum and hippocampus among other areas (212) and are primarily found on immune cells, astrocytes and less commonly in neurons (215). Endocannabinoids are generally synthesized post-synaptically after Ca2+ influx or activation of Gq/11-linked G-protein-coupled receptors; they act in a retrograde fashion influencing presynaptic cell firing (210, 215).
AEA is synthesized by phospholipase D catalyzed hydrolysis of N-acylphosphatidylethanolamine (216), meanwhile, 2-AG synthesis from membrane phospholipids is catalyzed by phospholipase C and diacylglycerol lipase (217). AEA degradation occurs predominantly by the enzyme fatty acid amide hydrolase (FAAH) or by cyclooxygenase (COX)-2 oxidation creating PGs (210) while 2-AG degradation occurs mostly through monoacylglycerol lipase (MAGL) but it can sometimes be oxidized by COX-2 or hydrolyzed by FAAH (210, 218). Although some processes occur due to crosstalk between cell types (219), microglia contain the complete machinery required for a functional endocannabinoid system. Rodent microglia are known to express both CB1 and CB2 (220, 221). The presence of CB1 in human microglia is controversial (211, 222), however, a few studies describe CB1 microglial expression in active multiple sclerosis plaques of postmortem human brain samples (223, 224). Microglia also produce the enzymes responsible for hydrolysis and inactivation of AEA and 2-AG.
The endocannabinoid system has recently been implicated as a regulator of microglial migration and activity which points to cannabinoids being a useful target for modifying microglia in pathological conditions. Reusch et al. showed that the CB2 receptor is necessary for TLR-mediated microglia activation through p38 MAPK signaling (225). Other studies have revealed that the CB2 receptor is instrumental to induce the anti-inflammatory phenotype in microglia (226, 227). Tao et al. (227) found that JWH133, a selective CB2 receptor agonist promotes the anti-inflammatory phenotype in microglia through CB2 receptor stimulated cAMP/PKA pathway (227). We and others have showed that CB2 activation can trigger the activity of canonical anti-inflammatory transcriptional factors such as PPAR-γ (Figure 3) (228–230). As discussed in previous sections, the transcriptional activity of PPAR-γ is pivotal for microglia alternative activation by diminishing the production of pro-inflammatory cytokines and inhibiting the activity of NF-κB. Following neuroinflammation, CB2 receptors are upregulated (212) which has been shown to trigger microglia migration to the site of injury/lesion (231, 232). Experimental studies elucidate that neuroinflammation produces adenosine triphosphate (ATP) (discussed in the Purinergic Signaling section), which causes 2–AG production, commencing microglia migration through activation of the CB2 receptors at the microglial leading edge (233).
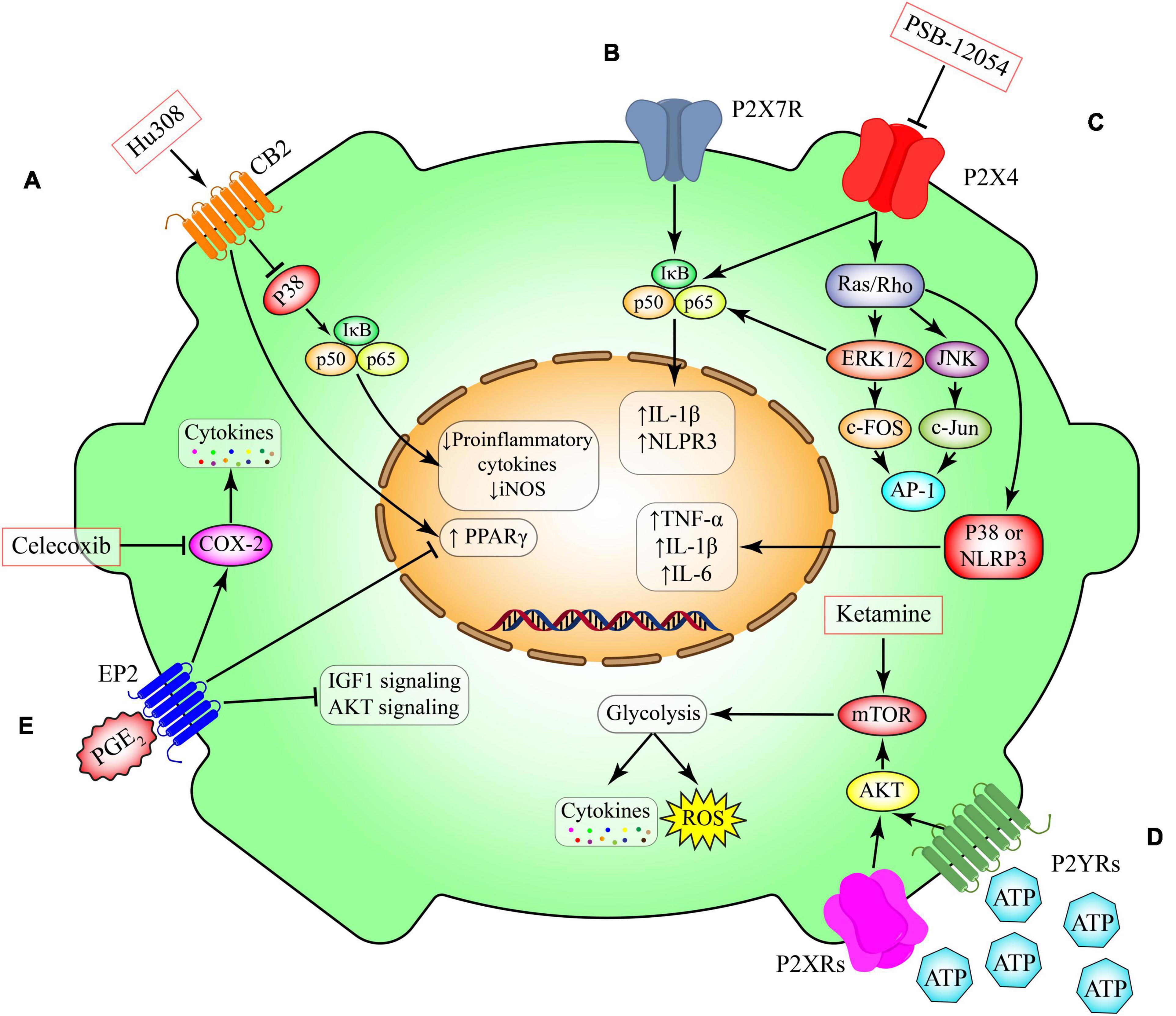
Figure 3. Metabolic pathways in microglia. (A) Activation of CB2 receptors that are expressed in non-neural cells including microglia promotes anti-inflammatory cascades through inhibition of NFκB or induction of anti-inflammatory transcriptional factor such as peroxisome proliferator-activated receptor gamma (PPAR-γ). This pathway can be induced by Hu308. (B) P2X7 receptor activation, induces the canonical pro-inflammatory transcriptional factors such as NFκB and subsequent production of inflammatory mediators such as IL-1 beta and NLPR3. (C) Activation of ligand gated ion channel P2X4 triggers the switching on two canonical pathways including NFκB and Ras/ERK/JNK. These proteins induce the production of several cytokines such as TNF-α, IL-beta and IL-6. This pathway can be inhibited by PSB-12054. (D) Induction of the G-protein coupled receptors P2YRs have essential roles in modulating the expression of metabolic pathways such as mTOR and their downstream glucose metabolism. The induction of glycolysis through mTOR has been implicated in production of several cytokines and chemokines. Ketamine triggers the mTOR pathways leading to induction of glycolysis. (E) Prostaglandin E2 (PGE 2) is a lipid mediator derived from the fatty acid arachidonic acid. Its interaction with the microglial G-protein coupled receptor EP2 induces the activity of cyclooxygenase-2 (COX-2) and inhibits several intracellular pathways including PPAR-γ, AKT and IGF1. Celecoxib is s selective COX-2 inhibitor. CB2, cannabinoid type 2 (CB2) receptor; P2X4, P2X purinoceptor 4; P2X7, P2X purinoceptor 7; P2YR, purinergic receptor P2Y; EP2, prostaglandin E2 receptor 2; PGE2, prostaglandin E2; IGF1, insulin-like growth factor 1; AKT, RAC(Rho family)-alpha serine/threonine-protein kinase; COX-2, cyclooxygenase-2; TNF-α, tumor necrosis factor-α; iNOS, inducible nitric oxide synthase; IL-1β, interleukin 1 beta; IL-6, interleukin 6; PPAR-γ, peroxisome proliferator- activated receptor-gamma; mTOR, mammalian target of rapamycin; AP-1, activator protein 1; JNK, c-Jun N-terminal kinases; NLRP3, NLR family pyrin domain containing 3; ERK, extracellular-signal-regulated-kinase; IκB, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor; ROS, reactive oxygen species.
The human endocannabinoid system has been implicated in MDD (234, 235). A 2019 meta-analysis revealed a very strong association of CB2rs2501432 polymorphism with depressive disorder, but not CB1rs1049353 polymorphism (236). Moreover, peripheral serum levels of AEA and 2-AG are significantly reduced in women diagnosed with MDD (237). Few studies have investigated the endocannabinoid system in postmortem human brain of psychiatric cases. Hungund et al. (238) found that CB1 receptor protein is increased in the dorsolateral PFC of depressed suicides. Moreover, using [35S]GTPgammaS binding assays which assesses coupling of G-proteins to G-protein-coupled receptors in postmortem brain, the authors found that CB1 cannabinoid signaling was increased in the same region when compared to healthy controls (238) implicating cannabinoid signaling in both depression and suicide.
The role of the endocannabinoid system has also been studied in different stress paradigms (Table 3) (239, 240). RSD of mice has been used to model depression and anxiety. This paradigm not only has stress related behavioral outcomes and impaired fear extinction but also an increase in inflammation both peripherally and in the brain. Lisboa et al. showed that stimulating CB1/2 by injecting WIN55,212-2, a non-selective agonist, daily before the RSD paradigm, reduced IL-1β mRNA in the brain but specifically in CD68+ activated microglia. Moreover, activation of the CB1/2 receptors before RSD repaired fear extinction and stress-related behavioral deficits (240). Although interesting, this study begs the question of whether these protective effects were mediated through the CB1 or CB2 receptor. In 2011, Zoppi et al. showed that daily pre-stress administration of arachidonyl-2′-chloroethylamide (ACEA), a selective CB1 receptor agonist, prevented upregulation of pro-inflammatory markers in the PFC of wild type mice; but not in CB1–/– knockouts (239). In another study by García-Gutiérrez et al. overexpression of CB2 receptor in mice had a protective effect, providing resilience to chronic mild stress and decreased depressive-like behaviors measured by the forced swim test and novelty-suppressed feeding test. Interestingly, chronic (4-week) administration of the CB2 receptor antagonist AM630 had anti-depressant like effects in wild type mice but not those that overexpressed CB2 receptor (241). More recently, a CDS stress paradigm was used in both CB1–/– knockouts and wild type littermates. Beins et al. found that CB1 knockouts mice were much more susceptible to CSD stress and mild CSD showing significant stress behaviors. Moreover, these stressed CB1–/– mice had dysregulated HPA axes with insufficient glucocorticoid signaling and hyper-activated microglia (242). Interestingly, at baseline, CB1–/– mice have increased expression of Fkbp5, a negative regulator of glucocorticoid signaling and a gene already implicated in depression (243). Overall, it seems that the endocannabinoid system serves a protective role in counteracting neuroinflammation by induction of anti-inflammatory profiles in microglia.

Table 3. Summary of cannabinoid receptors involvement in stress response.
Rescuing cellular function as a treatment for MDD has been mainly considered for neurons but not for glial cells. However, many investigations have demonstrated the functional impairment in glia cells as mentioned throughout this review. Microglial malfunctions have been studied in many different neuroinflammatory settings (244). Research has shown that different aspects of microglia such as phagocytic activity, secretory profile and metabolic pathways can be affected by neuroinflammation. Due to this complexity, selective modulators rather than general anti-inflammatory agents might be needed to rescue microglial functions following different types of inflammatory insults (10). One interesting and promising target could be endocannabinoid system. Fine tuning of this system in microglia can open a new avenue of research in pharmacotherapy of depression although several experimental and clinical studies should be performed long before being able to design cell-specific interventions for treating MDD.
The PGs are a class of eicosanoids that are formed by the liberation of arachidonic acid from phospholipids and a 2-step conversion by COX, the rate-limiting enzymes (245). Two main isoforms of COX exist, COX-1 and COX-2. COX-1 is traditionally considered as a constitutive enzyme while COX-2 is inducible. However, such classification is not very precise especially in the brain where constitutive expression of COX-2 has been reported (245). One of the canonical PGs in the CNS is PGE2. It interacts with different G-protein-coupled receptors including EP1, EP2, EP3, and EP4 (Figure 3) (246). Intriguingly, elevated PGE2 in the saliva, serum and CSF of depressed patients has previously been reported (247–250). Clinical investigations also revealed that adjunctive therapy with non-steroidal anti-inflammatory drugs, known as COX inhibitors, might have therapeutic effects in a subset of MDD patients (251, 252).
Human studies have shown decreased dopamine metabolites in the CSF of MDD patients. Furthermore, acute treatment with antidepressants induces dopamine release in the medial PFC (253, 254). These findings imply that activation of the mesocortical dopaminergic pathway has anti-depressive properties. In this context, the role of PGE2 has been studied in several experimental paradigms. It has been shown that EP1-deficient mice showed hyperdopaminergic activity, leading to impulsive behaviors under acute social and environmental stress (255). It is noteworthy that EP1 is located on GABAergic terminals on midbrain dopamine neurons and electrophysiological recording indicates that EP1 stimulation potentiates inhibitory synaptic inputs to these neurons (256). These findings suggest that PGE2-EP1 signaling suppresses midbrain dopamine neurons and regulates impulsive behaviors under acute stress (257). However, the mechanisms underlying involvement of the mesocortical dopaminergic pathway in vulnerability to repeated stress is unknown. Indeed, PGE2-EP1 signaling attenuates mesocortical dopaminergic pathway, leading to susceptibility of mice to RSD (254). Analyses of c-Fos expression of ventral tegmental area dopamine neurons and dopamine turnover in medial PFC showed that the mesocortical dopaminergic pathway is activated upon social defeat and attenuated with repetition of social defeat in wild-type mice. EP1 deficiency abolished such repeated stress-induced attenuation of mesocortical dopaminergic pathway (254).
Intriguingly, PGE2 acting on striatal medium spiny neurons has been suggested to elicit a negative affective state in response to inflammatory or social stress (254, 258). Our knowledge about the source of PGE2 was limited until recently. Klawonn et al. revealed that microglial PG signaling as critical for inflammation-induced aversion and is a potential mechanism by which different types of stressors may converge to produce a negative affective state and potentially depression (259). Indeed, these findings demonstrated that striatal microglial activation induces negative affect and both IL-6 and PG dependent signaling in microglia is critical for inflammation induced aversion. Chemogenetic activation of striatal microglia induces an aversive affective state while chemogenetic inhibition of microglia blocks inflammation induced aversion. Microglial IL-6 signaling and PG synthesis regulate affective state and finally PGE2 from activated microglia reduces the excitability of striatal neurons (259). In agreement with these findings, different investigations indicate that low-dose aspirin, which primarily inhibits COX-1 and consequently PG production, reduces the risk of depression (260). The inducible form of COX, COX2, might also be involved in the MDD pathology, since the COX-2 selective inhibitor celecoxib has beneficial effects in subset of depressed patients (83, 261). Since strong microglial COX-1 expression is complemented by COX-2 in response to chronic inflammation and stress (262, 263), both enzymes could contribute to depressive symptoms at different stages of the disease or in distinct patient groups (259).
The mammalian target of rapamycin (mTOR), the evolutionarily conserved serine/threonine protein kinase, may be activated by phosphorylation in response to growth factors (such as BDNF), mitogens and stress (264, 265). The mTOR signaling pathway plays a fundamental role in the regulation of protein synthesis, energy metabolism, lipid metabolism, cell growth and autophagy (266). In the CNS, mTOR is also involved in axonal sprouting, axonal regeneration and myelination, ionic and receptor channel expression, dendritic spine growth, as well as astrocyte migration and proliferation. mTOR-regulated processes in the brain influence neuronal excitability, neuronal survival, synaptic and behavioral plasticity, cognition, feeding, and control of circadian rhythm (265). In recent years, special attention has been given to the role of mTOR signaling in MDD. Several investigations have reported decreased brain mTOR activation in animal models of depression (267). One of the most studied models is CUS, which mimics several behavioral and neurochemical alterations that occur in depressed individuals (268). Rodents exposed to CUS exhibit depressive-like behaviors associated with a reduction in phosphorylation levels of mTOR and its downstream signaling components, such as phosphor-p70S6K, in the PFC, hippocampus and amygdala (268, 269). Regarding the anti-depressive role of the mTOR pathway, an elegant study by Li et al. revealed a single dose of ketamine can activate mTOR, resulting in increased PFC synaptic protein expression within 2 h and increased dendritic spine density and synaptic activity within 24 h (267). Importantly, clinical evidence also confirms the role of mTOR signaling in MDD pathology (270). It has been shown that mTOR, p70S6K, eIF4B, and p-eIF4B protein expression in PFC of deceased MDD subjects were reduced when compared with controls, indicating a deficit in mTOR-dependent signaling leading to impairment in its downstream targets that control translation of synaptic proteins (270).
Intriguingly, mTOR signaling can regulate several aspects of microglial function such as phagocytosis and cell survival (271). For instance, inhibition of mTOR diminishes the viability of primary cultured microglia (272), whereas induction of mTOR activity by inhibiting its upstream suppressor, tuberous sclerosis 1, enhanced phagocytosis in microglia (271, 273). Furthermore, microglial-specific inhibition of mTOR pathway decreases proinflammatory cytokines and chemokines (274). More recently, it has been shown that mTOR-mediated metabolic reprogramming shapes distinct microglial functions in response to LPS and ATP (271). Hu et al. showed that both LPS and ATP induced rapid activation of mTOR and glycolysis in microglia. Blocking either glucose metabolism or mTOR activity inhibits glycolysis significantly and mitigates LPS-induced production of proinflammatory cytokines, indicating that mTOR-driven glycolysis is required for the proinflammatory responses of LPS-primed microglia (271). Additionally, blocking mTOR activity not only inhibits glycolysis but also suppresses BDNF and TNF-α production in ATP-activated microglia, suggesting the critical role of mTOR in tuning microglia function (271). Better understanding of the metabolic regulation of microglia help us to manipulate and control the activity of microglia following different neuroinflammatory insults. The distinct metabolic adaptation in microglia in response to different stressors may provide diverse approaches to target microglia at different states and restrain microglia-triggered neuroinflammation in neuropsychiatric disorders (271).
Our group provided the first evidence of increased microglial activation in dorsal ACC white matter of depressed suicides. Although total density of Iba1+ microglia remained unchanged between depressed suicides and matched controls, the ratio of primed to ramified microglia was significantly increased in depressed suicides (10, 38). The mechanisms underlying microglial priming are unknown. However, it has been shown that the mTOR signaling pathway plays a crucial role in microglial priming during aging. Keane et al. were the first to show that microglia from aged mice have upregulated mTOR complex 1 signaling controlling translation and protein levels of inflammatory mediators (275). Genetic ablation of mTOR signaling in mouse microglia caused an NF-κB–dependent upregulation of priming genes at the mRNA level. However, mice displayed reduced cytokine protein levels, lessened microglial activation and milder sickness behavior. Similar changes were present in aged human microglia revealing that upregulation of mTOR-dependent translation is an essential aspect of microglia priming in aging (275). It is possible that abnormalities in microglial mTOR signaling are involved in the emergence of the primed microglial phenotype in MDD.
ATP, adenosine di-phosphate (ADP) and adenosine are molecules that are involved in purinergic signaling through P1, P2X, and P2Y receptors. G-protein-coupled P1 receptors are selective for adenosine and there are four subtypes A1, A2A, A2B, and A3. Meanwhile P2 receptors are activated by both ATP and ADP. There are two types of P2 receptors, namely P2X ionotropic channels and P2Y G-protein-coupled receptors (Figure 3). P2X receptors have seven subtypes while P2Y have eight subtypes. Microglia express several purinergic receptors (276) including: P2X1, P2X4, P2X7 (277), P2Y4, P2Y7, P2Y6, P2Y11 P2Y12, P2Y13, A1, A2A and A2B (278). Notably, adenosine binding to A2A has been shown to mediate microglial process retraction (279). Besides microglial motility, purinergic signaling is important in many other processes such as neurodevelopment and neuron-glial crosstalk and inflammation (280). Interestingly, many aspects of the purinergic signaling system have been implicated in depression (281) and here, we will focus on the receptors expressed by microglia (Table 4).
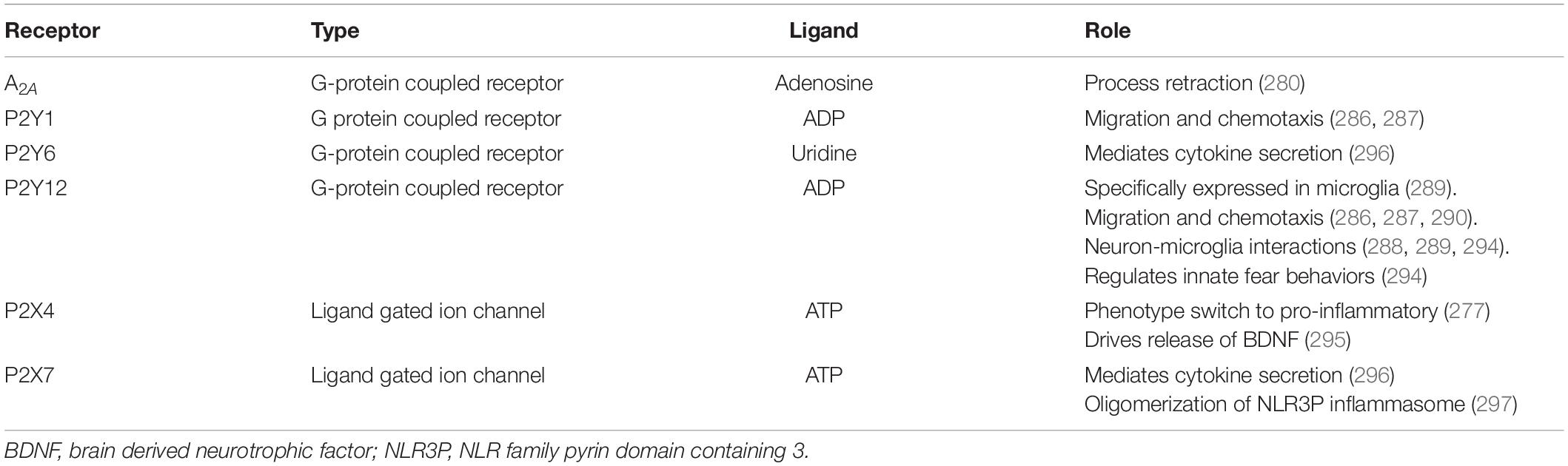
Table 4. Summary of microglial purinergic signal transduction.
ATP is released by injured cells into the extracellular space acting as DAMPs and a chemoattractant for microglial processes (233, 282, 283). Experimentally injected ATP causes microglia chemotaxis and process extension toward the site of injection (284). This process occurs through the G-protein-coupled P2Y (P2Y1R and P2Y12R) receptors (285, 286). P2Y12R is involved in long-range communication between neurons and microglia (287, 288), as well as microglia chemotaxis preceding phagocytosis (288). Additionally, P2Y12R acts as a marker for healthy microglia and is downregulated in the active pro-inflammatory phenotype in mice (289). There seems to be a species difference in the expression of P2Y12R as human brain resection from epileptic temporal lobe shows PY212R in both ramified and amoeboid microglia (290). Furthermore, the same tissue shows ADP stimulating process retraction (291) conversely to process extension in mouse microglia (284, 289). Although similar methods of comparative investigation have not yet been applied to all purinergic signaling, these findings alone suggest that they may differ greatly between species, providing necessary context to rodent studies. Loss of P2Y12R is associated with alteration in recognition and social memory as well as anxiety-like behavior in adult mice (292). Also, microglial P2Y12R regulates neural excitability and fear behaviors in developing and adult mice (293). Intriguingly, a human postmortem study found increased levels of P2Y12R protein in MDD microglia from four different brain regions, suggesting more resting state microglia in MDD and contradicting the neuroinflammation hypothesis (98).
Activation of microglial P2X4R by ATP (Figure 3) leads to a phenotype switch to pro-inflammatory state (276). Interestingly, in an ischemic stroke model, deletion of P2X4R from myeloid cells was shown to be protective in mice by inhibition of excessive pro-inflammatory cytokine release. However, after 30 days, the mice developed depressive-like phenotypes possibly due to missing P2X4R’s crucial role in BDNF release (294).
Once microglia are activated, cytokine secretion is mediated by P2Y6R (295) and P2X7R. Activation of P2X7R by ATP acting as DAMPs signal leads to oligomerization of NLR3P inflammasomes (Figure 3) (296). NLR3P inflammasome cleavage of pro-caspase 1 into caspase 1 results in the production of IL1-β from these activated microglia in situations of neuroinflammation such as in depression (297–299). In fact, a 2017 study showed that a 3-week CUS paradigm induced a depressive phenotype, increased extracellular ATP leading to P2X7R activation; this was inhibited by chronic treatment with P2X7R antagonists Brilliant Blue G (BBG) and A438079. Furthermore, P2X7R null mice that underwent the CUS did not develop a depressive phenotype irrespective of sex (300). In fact, the antidepressant phenotype of P2X7R deletion (301, 302) might be mediated by increased neurogenesis and serotonin bioavailability in the hippocampus (303). In humans, studies have suggested that polymorphisms Gln460Arg (rs2230912) (304) and His155Tyr (rs208294) in the P2RX7 gene are associated with depression and other mood disorders (305, 306). However, a 2014 meta-analysis has contradicted these findings (307).
In the healthy brain, P2X7R is not activated by physiological ATP levels, in fact, P2X7R activation only occurs when ATP acts as DAMPs (308). Intriguingly, P2X7R antagonists that cross the BBB are in clinical trials with the hopes of using them as adjunct or monotherapy for depression caused by neuroinflammation (309, 310). Interestingly, cholesterol and other lipids like phosphatidylglycerol and sphingomyelin modulate the function of P2X7R (311). Our lab and others have found altered brain lipids in psychopathology (312) and depression (313–315) suggesting lipid control of P2X7R should be further investigated. Generally, lipids in the cell membrane organize position and function of proteins. They can be also released from the membrane acting as messengers transmitting signals (316). Lipid composition of the brain affects cognition, perception and mood most likely by influencing neuroinflammation and neurogenesis (317). Moreover, decreased polyunsaturated fatty acids seem to be the most frequently implicated lipid change in depression (313, 317) and they are known in regulating CB1 receptor (317) and P2X7R (311, 318). Interestingly, dietary supplementation with fish oil rich in polyunsaturated fats decreases depressive-like behavior in LPS treated mice and suppresses activation of the NLRP3 inflammasome and P2X7R (318). Meanwhile, sphingolipids have been implicated in both the etiology of MDD and the beneficial effects of antidepressants (316, 319). In addition to diet (320), lipid composition in the brain can be altered by environmental factors including exercise (321) and medications (322), making them promising targets for treatment (319).
To our knowledge, there is a paucity of studies on the role of microglial adenosine receptors in depression. However, A1 receptor activation generally inhibits serotonin release in the hippocampus, while A2 receptor activation promotes the release of this neurotransmitter (323). Moreover, A2 receptors have been found to be involved in the dysregulation of the HPA axis observed in depression (324). Furthermore, a 2021 study showed that 14 days of restraint stress induced depression and anxiety in the mice with a neuroinflammatory phenotype. The authors discovered increased expression of P2X7R and A2A receptor in the hippocampus and prefrontal cortex. Next, they showed that BBG a P2X7R-selective antagonist and caffeine an A2A receptor antagonist attenuated the depressive phenotype (325).
Iron metabolism is crucial for normal functioning of the brain, including myelination by oligodendrocytes and neurotransmitter synthesis (serotonin, dopamine, and norepinephrine) (326). Transferrin, a glycoprotein is mainly responsible for the movement of iron through the body and has high affinity to Fe3+ (ferric iron) (327). Movement of iron from the periphery into the brain occurs through the BBB. Transferrin receptor 1 (TfR1) on endothelial cells mediates the uptake of transferrin bound iron (Tf-Fe) into the brain, while also regulating the return of iron-depleted transferrin back to the blood (326, 328). Tf-Fe is packaged into endosomes and then released into the brain through ferroportin 1 (Fp1) as Fe2+ where it is oxidized to Fe3+ through ferroxidases hephaestin or ceruloplasmin (Cp) (326, 329). Fp1 levels are controlled by hepcidin (HepC) which binds to Fp1 leading to its internalization and subsequent degradation (330). The metabolism of iron in the brain is highly controlled as it is extremely important for proper cell functioning, however, iron overload can be extremely toxic and cause cell death through oxidative stress (329). In the brain, iron is distributed heterogeneously with concentration varying by region (331, 332) and cell type (333). Iron is mainly distributed in the hippocampus (331) and the basal ganglia stored in ferritin (334) or neuromelanin (335). Presumably all cell types can uptake iron through either Tf-Fe or non-transferrin bound pathways. Neurons and astrocytes uptake Tf-Fe through TfR1 and divalent metal transporter 1 (DMT1). Oligodendrocytes uptake iron through the TfR1/DMT1 pathway only in immature states; once they mature, iron enters through heavy (H-) chain ferritin binding to H-ferritin receptor on the cell membrane (336). Oligodendrocytes have very high iron necessity for their normal functioning and proper myelination (333, 336). Microglia have two different ways of regulating iron influx; the first being through Tf-Fe uptake via the TfR1/DMT1 pathway while the second is uptake of non-Tf-Fe through transferrin-independent mechanisms (326). In healthy brain, iron usually travels bound to transferrin, which is produced by oligodendrocytes and ChP, however, it is only secreted by the latter (337).
Pathogens such as bacteria utilize iron from the host to replicate. To protect the host, immune cells are programmed to reduce iron availability to prevent further infiltration by the invader (338). This happens through increased production of HepC in hepatocytes, stimulated by IL-6 release from immune cells (339) which reduces the export of iron into the blood by negative regulation of ferroportin (340). It has been shown that different activation status in microglia prefer one iron intake pathway over the other (341). In neuroinflammatory conditions, iron is sequestered in pro-inflammatory microglia and neurons through non-transferrin bound pathways (342). In vitro LPS stimulation of microglia results in downregulation of TfR1 (338, 343) and ferroportin (338) and upregulation of DMT1 and ferritin (344). Under this condition, iron accumulates intracellularly and secretion of inflammatory cytokines and metalloproteinases is increased (343). Conversely, when microglia are stimulated with IL-4, an anti-inflammatory phenotype of microglia prefers Tf-Fe uptake and increases cellular transferrin receptor levels (344).
The WHO estimates that 37% of females are iron deficient globally, while males are rarely diagnosed (345). Both iron deficiency and depression were in the top disorders for most years lived with disability globally (346). Interestingly, females are twice as more likely to be diagnosed with MDD. Recent literature has implicated iron dysregulation, specifically iron deficiency, in the pathology of depression (347–351). Indeed, a recent study found that depressed individuals are 3 times more likely than healthy individuals to have hypoferritinemia (low iron storage) and acquired hypotransferrinemia (decreased levels of protein transferrin) (351). The fact that iron is a co-factor for rate limiting steps of serotonin, dopamine and norepinephrine synthesis and neuronal uptake further implicates involvement of iron metabolism in the etiology of depression (351, 352). Whether iron deficiency detected in the periphery is adequately representative of iron deficiency in the brain is still under speculation due to the tight control of iron metabolism in the brain (352). However, a 2007 study by Vostrikov et al., reported significantly reduced numbers of oligodendrocytes in layer 3 of BA9 in the PFC of postmortem samples from MDD patients (353). Given that oligodendrocytes hold the most iron in the brain, it is reasonable to infer decreased iron levels in postmortem MDD brain. Moreover, research from our lab has implicated oligodendrocyte precursor cell dysregulation with MDD (354). Iron deficiency has been shown to lead to hypomyelination in both animals and humans (355). A 2018 study revealed that developmental iron deficiency kept not only oligodendrocyte lineage cells in immature states but also other glia cells. Interestingly, iron deficiency inhibits microglial inflammatory cytokine secretion after LPS stimulation (356). Additionally, it has been shown that microglia constitute the primary source of iron to oligodendrocyte precursor cells during myelination (357).
Considering that microglia are involved in oligodendrocyte differentiation and myelin repair (358), it is a logical next step to investigate the role of iron metabolism in microglia and the relationship to depression. Few studies have investigated the role of microglia iron metabolism and depression and the ones that examined this relationship in animals found a different trend than the aforementioned human studies. Gao et al. showed that CSD stress resulted in depressive phenotype, increased microglial activation, increased iron, decreased Fp1 and increased ferritin, DMT1 and HepC in the hippocampus (359). Similarly, Jiao et al. found that mice with CUMS-induced behavioral despair were found to have increased brain iron, increased inflammation and Fe2+ levels in the hippocampus. Genes involved in ferroptosis were differentially regulated; specifically, glutathione peroxidase (GPX4, necessary for repairing oxidative damage) was downregulated after CUMS suggesting a role of ferroptosis in the phenotype of depression. In addition, this study also showed that fluoxetine not only resolved depressive behaviors but also restored normal ferroptosis signaling (360). Experimental models also display an increase in iron load in the brain which results in anxiety symptoms. A study by Texel et al. shows that Cp knockout mice have iron overload in the periphery, but lower levels of iron in the hippocampus accompanied by decreased levels of 5-HT, norepinephrine and BDNF (361). In another study, Pellegrino et al. showed that knockout of transferrin receptor 2 leads to iron overload in the brain, dysregulation of microglia activation status and increased anxiety levels in the animals (362). Overall, regulation of iron metabolism by microglia is an important factor in brain physiology and pathophysiology. Future studies are needed to investigate the postmortem brain of depressed individuals to better understand the link between iron metabolism abnormalities in brain innate immune system and MDD pathology.
In recent years, special attention has also been given to the sexually dimorphic role of microglia in both healthy neurodevelopmental and diseased neurodegenerative brains (15). Sex differences have been reported in distribution, structure, function, transcriptomic and proteomic profiles of microglia in both physiological and neuroinflammatory conditions (10). Evidence demonstrates that microglia play an instrumental role in the sexually dimorphic differentiation of the developing brain (363). Interestingly, the number and phenotype of microglia differ in many regions of the brain when comparing male and female rodents (364). Brain areas such as the preoptic area, cerebral cortex and the amygdala and hippocampus have been reported to have microglial sexual dissimilarity (363–366). Importantly, the postnatal sexual dimorphism persists into the adult brain. These marked differences may lead to development of distinct, sex-dependent microglia inflammatory responses in pathological conditions. In fact, sex differences in microglial activation patterns following neuroinflammation have been reported by different groups (367, 368). However, whether the observed differences are exclusively hormone-dependent and/or stem from distinct developmental mechanisms remains to be elucidated. The analyses of the different anti- and pro-inflammatory signaling events reveal that they differ in males and females.
Several observations suggest marked sex differences in the microglial activation patterns following stress. In a pioneer study, Bollinger et al. revealed differential effects of stress on microglial activation in female and male medial PFC (369). Importantly, dysfunction of this brain area is implicated in MDD pathology. It has been shown that chronic stress affects the medial PFC in a sex-dependent manner and impairs prefrontal mediated behaviors in males and females (369). Unstressed female rats show a greater proportion of primed to ramified microglia relative to males, alongside increased CX3CL1-CX3CR1 levels. In addition, acute stress and CRS diminished the ratio of primed to ramified microglia and microglial CD40 expression in females but not in males (369). Another intriguing study revealed that the expression of genes related to cellular stress, neuroimmune state and neuron-microglia communication varied between unstressed male and female rats in a region-specific manner (370). Namely, in the dorsal hippocampus, chronic stress increased immune markers expression in males but not females. It is noteworthy that the type of stress can also mediate sex-specific innate immune response. For instance, acute stress increased microglia-associated transcripts in basolateral amygdala in males, whereas chronic stress altered immune factor expression in basolateral amygdala more broadly in females (370). It was recently shown that chronic stress induces different neurobiological adaptations in the PFC of male and female mice. CUS causes behavioral changes and microglia-mediated neuronal remodeling only in the frontal cortex of male mice (371). Fluorescence-activated cell sorting and gene expression analyses by Woodburn et al. indicate that CUS increased expression of markers of phagocytosis only in male PFC microglia (371). Overall, these findings suggest that sex differences might impact microglia pro- and anti-inflammatory signaling pathways, leading to different outcomes.
Cytokine research in MDD faces several difficulties, such as conflicting results and high variability of cytokine levels within samples. Several studies have only reported serum or plasma cytokine levels without taking into account potential confounding factors such as age, body weight, smoking, alcohol consumption and medication, which can all influence the plasma content of cytokines (372, 373). These limitations imply that serum levels of cytokines may not reflect their levels in the brain and, therefore, may not reflect pathology (373, 374). Direction of causality is another important factor that is not clear in the cytokine research in psychiatric disorders. Although there is strong evidence for the involvement of cytokines in the pathology of MDD, the direction of this relationship has not yet been clarified with certainty. For instance, the alteration of cytokine levels might also be the consequence of a psychiatric disorder. Factors such as antidepressants or body mass index (that could change as a result of the disorder) might cause fluctuation in cytokine levels (373, 375).
An association between neuroinflammation and MDD has been reported by multiple studies (10) and it is well-established that inflammatory markers are increased in a subset of MDD patients (84, 85, 376) but not all (10, 21). Although microglial abnormalities have been reported in MDD, no link has been made with specific diagnostic categories. It is noteworthy that recent attempts at targeting inflammation as a new therapy for MDD have not been very promising. In fact, it appears that anti-inflammatory treatment options may only be effective in patients who show signs of increased peripheral inflammation along with depressive behaviors (83, 84). Namely, it has been shown that antagonizing TNF-α as a therapy may only prove useful in patients with high baseline inflammatory biomarkers (85). With similar uncertainty, general immunosuppressants and anti-inflammatory agents such as minocycline and non-steroidal anti-inflammatory drugs that were promising in animal studies (10) have failed in treating neuropsychiatric disorders probably due to their non-selective dampening of the immune response. Targeting of pro- or anti-inflammatory brain cytokines for developing new pharmacotherapeutics is not easy. As discussed in detail in this review, these cytokines have essential physiological actions and blockade of their signaling might influence several crucial processes such as cell survival and neural plasticity. Furthermore, the diversity of brain cell types that produce cytokines makes it difficult to target their signaling in a neuroinflammatory context. Finally, different receptor subtypes have different roles following neuroinflammation (e.g., TNF-α and IL-6 receptors).
It is becoming increasingly obvious that microglia act as a double-edged sword in the etiopathology of MDD. On one hand, some experimental studies both in vitro and in vivo have linked neuronal damage with the inflammatory phenotype of microglia releasing neurotoxic mediators and ROS (377). On the other hand, other investigations have reported the beneficial roles of microglial anti-inflammatory phenotype in neuronal regeneration and neurogenesis. Whether microglia play a positive or negative role depends on several factors including type of neuroinflammatory insult, brain region, time, sex and age. Indeed, heterogeneity of microglia and their context-dependent properties warrant more research to gain a clearer understanding of this dynamic cell type before new therapeutics can be successfully developed for psychiatric disorders such as MDD.
RR, CB, and NM conceived the review. RR, CB, and RC wrote the manuscript. NM supervised the project and revised the manuscript. All authors read and agreed with the final version of the manuscript.
This work was funded by a CIHR grant to NM (PJT-156346). RR is an FRQ-S fellow and CB the recipient of an FRQ-S doctoral scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MDD, major depressive disorder; TNF- α, tumor necrosis factor-alpha; IL, interleukin; RA, rheumatoid arthritis; TGF- β, transforming growth factor β; PPAR- γ, peroxisome proliferator-activated receptors-gamma; CNS, central nervous system; NF κ B, nuclear factor kappa-light-chain-enhancer of activated B cells; BBB, blood–brain barrier; CUS, chronic unpredictable stress; TNF- α, tumor necrosis factor-alpha; ACC, anterior cingulate cortex; LPS, lipopolysaccharide; CSD, chronic social defeat; ROS, reactive oxygen species; CUMS, chronic unpredictable mild stress; TLR, toll-like receptor; TNFR1, TNF- α Receptor 1; TNFR2, TNF- α Receptor 2; MAPK, mitogen-activated protein kinases; CSF, cerebrospinal fluid; HPA, hypothalamic-pituitary-adrenal; gp130, glycoprotein 130; RSD, repeated social defeat; IL-6R, IL-6 receptor; DAMPs, danger-associated molecular patterns; MyD88, myeloid differentiation primary response 88; AD, Alzheimer’s disease; PFC, prefrontal cortex; IFN- γ, interferon-gamma; JAK, janus kinase; STAT, signal transducer and activator of transcription; ERK, extracellular-signal-regulated-kinase; SSRIs, selective serotonin reuptake inhibitors; ChP, choroid plexus; α 7 nAChR, Alpha-7 nicotinic acetylcholine receptor; Nrf2, nuclear factor-erythroid factor 2-related factor 2; CRS, chronic restraint stress; Gal-3, galectin-3; PG, prostaglandin; PPARs, peroxisome proliferator-activated receptors; NMDA, N-methyl-D-aspartate; IGF-1, insulin-like growth factor 1; DAM, disease-associated microglia; BDNF, brain-derived neurotrophic factor; endocannabinoid, endogenous cannabinoid; AEA, anandamide; 2-AG, 2-arachidonoylglycerol; CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2; GPR55, G-protein -coupled receptor 55; FAAH, fatty acid amide hydrolase; COX, cyclooxygenase; MAGL, monoacylglycerol lipase; ATP, adenosine triphosphate; ACEA, arachidonyl-2 ′ -chloroethylamide; mTOR, mammalian target of rapamycin; ADP, adenosine di-phosphate; BBG, Brilliant Blue G; Tf-Fe, transferrin bound iron; Fp1, ferroportin 1; Cp, ceruloplasmin; HepC, hepcidin.
1. Young JJ, Bruno D, Pomara N. A review of the relationship between proinflammatory cytokines and major depressive disorder. J Affect Disord. (2014) 169:15–20. doi: 10.1016/j.jad.2014.07.032
2. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. (2006) 27:24–31. doi: 10.1016/j.it.2005.11.006
3. Lindqvist D, Janelidze S, Hagell P, Erhardt S, Samuelsson M, Minthon L, et al. Interleukin-6 is elevated in the cerebrospinal fluid of suicide attempters and related to symptom severity. Biol Psychiatry. (2009) 66:287–92. doi: 10.1016/j.biopsych.2009.01.030
4. Brundin L, Sellgren CM, Lim CK, Grit J, Pålsson E, Landén M, et al. An enzyme in the kynurenine pathway that governs vulnerability to suicidal behavior by regulating excitotoxicity and neuroinflammation. Transl Psychiatry. (2016) 6:e865–865. doi: 10.1038/tp.2016.133
5. Felger JC, Li Z, Haroon E, Woolwine BJ, Jung MY, Hu X, et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. (2016) 21:1358–65. doi: 10.1038/mp.2015.168
6. Haroon E, Fleischer CC, Felger JC, Chen X, Woolwine BJ, Patel T, et al. Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol Psychiatry. (2016) 21:1351–7. doi: 10.1038/mp.2015.206
7. Haroon E, Chen X, Li Z, Patel T, Woolwine BJ, Hu XP, et al. Increased inflammation and brain glutamate define a subtype of depression with decreased regional homogeneity, impaired network integrity, and anhedonia. Transl Psychiatry. (2018) 8:189. doi: 10.1038/s41398-018-0241-4
8. Maes M, Meltzer HY, Bosmans E, Bergmans R, Vandoolaeghe E, Ranjan R, et al. Increased plasma concentrations of interleukin-6, soluble interleukin-6, soluble interleukin-2 and transferrin receptor in major depression. J Affect Disord. (1995) 34:301–9. doi: 10.1016/0165-0327(95)00028-L
9. Maes M, Bosmans E, De Jongh R, Kenis G, Vandoolaeghe E, Neels H. Increased serum Il-6 and Il-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. (1997) 9:853–8. doi: 10.1006/cyto.1997.0238
10. Rahimian R, Wakid M, O’Leary LA, Mechawar N. The emerging tale of microglia in psychiatric disorders. Neurosci Biobehav Rev. (2021) 131:1–29. doi: 10.1016/j.neubiorev.2021.09.023
11. Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. (2010) 67:446–57. doi: 10.1016/j.biopsych.2009.09.033
12. Marrie RA, Walld R, Bolton JM, Sareen J, Patten SB, Singer A, et al. Psychiatric comorbidity increases mortality in immune-mediated inflammatory diseases. Gen Hosp Psychiatry. (2018) 53:65–72. doi: 10.1016/j.genhosppsych.2018.06.001
13. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. (2017) 23:1018–27. doi: 10.1038/nm.4397
14. Tan Y-L, Yuan Y, Tian L. Microglial regional heterogeneity and its role in the brain. Mol Psychiatry. (2020) 25:351–67. doi: 10.1038/s41380-019-0609-8
15. Rahimian R, Cordeau P Jr, Kriz J. Brain response to injuries: when microglia go sexist. Neuroscience. (2019) 405:14–23. doi: 10.1016/j.neuroscience.2018.02.048
16. Bordt EA, Ceasrine AM, Bilbo SD. Microglia and sexual differentiation of the developing brain: a focus on ontogeny and intrinsic factors. Glia. (2020) 68:1085–99. doi: 10.1002/glia.23753
17. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. (2017) 169:1276–90.e1217. doi: 10.1016/j.cell.2017.05.018
18. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. (2019) 50:253–71.e256. doi: 10.1016/j.immuni.2018.11.004
19. Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. (2019) 566:388–92. doi: 10.1038/s41586-019-0924-x
20. Mechawar N, Savitz J. Neuropathology of mood disorders: do we see the stigmata of inflammation? Transl Psychiatry. (2016) 6:e946. doi: 10.1038/tp.2016.212
21. Mondelli V, Vernon AC, Turkheimer F, Dazzan P, Pariante CM. Brain microglia in psychiatric disorders. Lancet Psychiatry. (2017) 4:563–72. doi: 10.1016/s2215-0366(17)30101-3
22. Singhal G, Baune BT. Microglia: an interface between the loss of neuroplasticity and depression. Front Cell Neurosci. (2017) 11:270. doi: 10.3389/fncel.2017.00270
23. Yirmiya R, Rimmerman N, Reshef R. Depression as a microglial disease. Trends Neurosci. (2015) 38:637–58. doi: 10.1016/j.tins.2015.08.001
24. Tay TL, Béchade C, D’Andrea I, St-Pierre M-K, Henry MS, Roumier A, et al. Microglia gone rogue: impacts on psychiatric disorders across the lifespan. Front Mol Neurosci. (2018) 10:421. doi: 10.3389/fnmol.2017.00421
25. Deng SL, Chen JG, Wang F. Microglia: a central player in depression. Curr Med Sci. (2020) 40:391–400. doi: 10.1007/s11596-020-2193-1
26. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat Neurosci. (2014) 17:131–43. doi: 10.1038/nn.3599
27. Rahimian R, Lively S, Abdelhamid E, Lalancette-Hebert M, Schlichter L, Sato S, et al. Delayed galectin-3-mediated reprogramming of microglia after stroke is protective. Mol Neurobiol. (2019) 56:6371–85. doi: 10.1007/s12035-019-1527-0
28. Dietz AG, Goldman SA, Nedergaard M. Glial cells in schizophrenia: a unified hypothesis. Lancet Psychiatry. (2020) 7:272–81. doi: 10.1016/S2215-0366(19)30302-5
29. Wang Q, Timberlake MA II, Prall K, Dwivedi Y. The recent progress in animal models of depression. Prog Neuropsychopharmacol Biol Psychiatry. (2017) 77:99–109. doi: 10.1016/j.pnpbp.2017.04.008
30. Enache D, Pariante CM, Mondelli V. Markers of central inflammation in major depressive disorder: a systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav Immun. (2019) 81:24–40. doi: 10.1016/j.bbi.2019.06.015
31. Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, et al. Social stress induces neurovascular pathology promoting depression. Nat Neurosci. (2017) 20:1752–60. doi: 10.1038/s41593-017-0010-3
32. Dudek KA, Dion-Albert L, Lebel M, LeClair K, Labrecque S, Tuck E, et al. Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. Proc Natl Acad Sci U.S.A. (2020) 117:3326–36. doi: 10.1073/pnas.1914655117
33. Chen SK, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, et al. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell. (2010) 141:775–85. doi: 10.1016/j.cell.2010.03.055
34. Schubert I, Ahlbrand R, Winter A, Vollmer L, Lewkowich I, Sah R. Enhanced fear and altered neuronal activation in forebrain limbic regions of CX3CR1-deficient mice. Brain Behav Immun. (2018) 68:34–43. doi: 10.1016/j.bbi.2017.09.013
35. Milior G, Lecours C, Samson L, Bisht K, Poggini S, Pagani F, et al. Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain Behav Immun. (2016) 55:114–25. doi: 10.1016/j.bbi.2015.07.024
36. Kana V, Desland FA, Casanova-Acebes M, Ayata P, Badimon A, Nabel E, et al. CSF-1 controls cerebellar microglia and is required for motor function and social interaction. J Exp Med. (2019) 216:2265–81. doi: 10.1084/jem.20182037
37. Jia X, Gao Z, Hu H. Microglia in depression: current perspectives. Sci China Life Sci. (2021) 64:911–25. doi: 10.1007/s11427-020-1815-6
38. Torres-Platas SG, Cruceanu C, Chen GG, Turecki G, Mechawar N. Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav Immun. (2014) 42:50–9. doi: 10.1016/j.bbi.2014.05.007
39. Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation. (2011) 8:94. doi: 10.1186/1742-2094-8-94
40. Boutej H, Rahimian R, Thammisetty SS, Béland LC, Lalancette-Hébert M, Kriz J. Diverging mRNA and protein networks in activated microglia reveal SRSF3 suppresses translation of highly upregulated innate immune transcripts. Cell Rep. (2017) 21:3220–33. doi: 10.1016/j.celrep.2017.11.058
41. Dillingh MR, van Poelgeest EP, Malone KE, Kemper EM, Stroes ESG, Moerland M, et al. Characterization of inflammation and immune cell modulation induced by low-dose LPS administration to healthy volunteers. J Inflamm. (2014) 11:28. doi: 10.1186/s12950-014-0028-1
42. Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation. (2015) 12:114. doi: 10.1186/s12974-015-0332-6
43. Guan YF, Huang GB, Xu MD, Gao F, Lin S, Huang J, et al. Anti-depression effects of ketogenic diet are mediated via the restoration of microglial activation and neuronal excitability in the lateral habenula. Brain Behav Immun. (2020) 88:748–62. doi: 10.1016/j.bbi.2020.05.032
44. Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner L, Tonia M, et al. β-adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. (2011) 31:6277. doi: 10.1523/JNEUROSCI.0450-11.2011
45. Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology. (2012) 37:1491–505. doi: 10.1016/j.psyneuen.2012.02.003
46. Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, et al. Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry. (2014) 75:970–81. doi: 10.1016/j.biopsych.2013.11.029
47. Wohleb ES, Patterson JM, Sharma V, Quan N, Godbout JP, Sheridan JF. Knockdown of interleukin-1 receptor type-1 on endothelial cells attenuated stress-induced neuroinflammation and prevented anxiety-like behavior. J Neurosci. (2014) 34:2583. doi: 10.1523/JNEUROSCI.3723-13.2014
48. McKim DB, Niraula A, Tarr AJ, Wohleb ES, Sheridan JF, Godbout JP. Neuroinflammatory dynamics underlie memory impairments after repeated social defeat. J Neurosci. (2016) 36:2590. doi: 10.1523/JNEUROSCI.2394-15.2016
49. Ramirez K, Sheridan JF. Antidepressant imipramine diminishes stress-induced inflammation in the periphery and central nervous system and related anxiety- and depressive- like behaviors. Brain Behav Immunity. (2016) 57:293–303. doi: 10.1016/j.bbi.2016.05.008
50. Lehmann ML, Weigel TK, Poffenberger CN, Herkenham M. The behavioral sequelae of social defeat require microglia and are driven by oxidative stress in mice. J Neurosci. (2019) 39:5594. doi: 10.1523/JNEUROSCI.0184-19.2019
51. Maes M, Yirmyia R, Noraberg J, Brene S, Hibbeln J, Perini G, et al. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis. (2009) 24:27–53. doi: 10.1007/s11011-008-9118-1
52. Vogt MA, Mallien AS, Pfeiffer N, Inta I, Gass P, Inta D. Minocycline does not evoke anxiolytic and antidepressant-like effects in C57BL/6 mice. Behav Brain Res. (2016) 301:96–101. doi: 10.1016/j.bbr.2015.12.015
53. Zhang C, Zhang YP, Li YY, Liu BP, Wang HY, Li KW, et al. Minocycline ameliorates depressive behaviors and neuro-immune dysfunction induced by chronic unpredictable mild stress in the rat. Behav Brain Res. (2019) 356:348–57. doi: 10.1016/j.bbr.2018.07.001
54. Miyaoka T, Wake R, Furuya M, Liaury K, Ieda M, Kawakami K, et al. Minocycline as adjunctive therapy for patients with unipolar psychotic depression: an open-label study. Prog Neuropsychopharmacol Biol Psychiatry. (2012) 37:222–6. doi: 10.1016/j.pnpbp.2012.02.002
55. Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. (1996) 334:1717–25. doi: 10.1056/nejm199606273342607
56. Horiuchi T, Mitoma H, Harashima S-I, Tsukamoto H, Shimoda T. Transmembrane TNF-α: structure, function and interaction with anti-TNF agents. Rheumatology. (2010) 49:1215–28. doi: 10.1093/rheumatology/keq031
57. Parameswaran N, Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr. (2010) 20:87–103. doi: 10.1615/critreveukargeneexpr.v20.i2.10
58. Idriss HT, Naismith JH. TNFα and the TNF receptor superfamily: structure-function relationship(s). Microsc Res Tech. (2000) 50:184–95. doi: 10.1002/1097-0029(20000801)50:33.0.CO;2-H
59. Subedi L, Lee SE, Madiha S, Gaire BP, Jin M, Yumnam S, et al. Phytochemicals against TNFα-mediated neuroinflammatory diseases. Int J Mol Sci. (2020) 21:764. doi: 10.3390/ijms21030764
60. Guadagno J, Xu X, Karajgikar M, Brown A, Cregan SP. Microglia-derived TNFα induces apoptosis in neural precursor cells via transcriptional activation of the Bcl-2 family member Puma. Cell Death Dis. (2013) 4:e538. doi: 10.1038/cddis.2013.59
61. Uzzan S, Azab AN. Anti-TNF-α compounds as a treatment for depression. Molecules. (2021) 26:2368. doi: 10.3390/molecules26082368
62. Spriggs DR, Deutsch S, Kufe DW. Genomic structure, induction, and production of TNF-alpha. Immunol Ser. (1992) 56:3–34.
63. Muhammad M. Tumor Necrosis Factor Alpha: A Major Cytokine of Brain Neuroinflammation. London: IntechOpen (2020).
64. To SQ, Knower KC, Clyne CD. Origins and actions of tumor necrosis factor α in postmenopausal breast cancer. J Interferon Cytokine Res. (2013) 33:335–45. doi: 10.1089/jir.2012.0155
65. Probert L. TNF and its receptors in the CNS: the essential, the desirable and the deleterious effects. Neuroscience. (2015) 302:2–22. doi: 10.1016/j.neuroscience.2015.06.038
66. Yang I, Han SJ, Kaur G, Crane C, Parsa AT. The role of microglia in central nervous system immunity and glioma immunology. J Clin Neurosci. (2010) 17:6–10. doi: 10.1016/j.jocn.2009.05.006
67. Syed MM, Phulwani NK, Kielian T. Tumor necrosis factor-alpha (TNF-α) regulates toll-like receptor 2 (TLR2) expression in microglia. J Neurochem. (2007) 103:1461–71. doi: 10.1111/j.1471-4159.2007.04838.x
68. Rodriguez M, Zoecklein L, Papke L, Gamez J, Denic A, Macura S, et al. Tumor necrosis factor alpha is reparative via TNFR2 [corrected] in the hippocampus and via TNFR1 [corrected] in the striatum after virus-induced encephalitis. Brain Pathol. (2009) 19:12–26. doi: 10.1111/j.1750-3639.2008.00151.x
69. Stacey D, Redlich R, Büschel A, Opel N, Grotegerd D, Zaremba D, et al. TNF receptors 1 and 2 exert distinct region-specific effects on striatal and hippocampal grey matter volumes (VBM) in healthy adults. Genes Brain Behav. (2017) 16:352–60. doi: 10.1111/gbb.12318
70. Jang D-I, Lee AH, Shin H-Y, Song H-R, Park J-H, Kang T-B, et al. The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in therapeutics. Int J Mol Sci. (2021) 22:2719. doi: 10.3390/ijms22052719
71. Medler J, Wajant H. Tumor necrosis factor receptor-2 (TNFR2): an overview of an emerging drug target. Exp Opin Therap Targets. (2019) 23:295–307. doi: 10.1080/14728222.2019.1586886
72. Vasanthi P, Nalini G, Rajasekhar G. Role of tumor necrosis factor-alpha in rheumatoid arthritis: a review. APLAR J Rheumatol. (2007) 10:270–4. doi: 10.1111/j.1479-8077.2007.00305.x
73. Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin. (2008) 29:1275–88. doi: 10.1111/j.1745-7254.2008.00889.x
74. Troubat R, Barone P, Leman S, Desmidt T, Cressant A, Atanasova B, et al. Neuroinflammation and depression: a review. Eur J Neurosci. (2021) 53:151–71.
75. Olmos G, Lladó J. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Med Inflamm. (2014) 2014:1–12. doi: 10.1155/2014/861231
76. Karol DE, Criscione-Schreiber LG, Lin M, Clowse ME. Depressive symptoms and associated factors in systemic lupus erythematosus. Psychosomatics. (2013) 54:443–50. doi: 10.1016/j.psym.2012.09.004
77. Postal M, Lapa AT, Sinicato NA, de Oliveira Peliçari K, Peres FA, Costallat LTL, et al. Depressive symptoms are associated with tumor necrosis factor alpha in systemic lupus erythematosus. J Neuroinflammation. (2016) 13:1–7.
78. Zou W, Feng R, Yang Y. Changes in the serum levels of inflammatory cytokines in antidepressant drug-naïve patients with major depression. PLoS One. (2018) 13:e0197267. doi: 10.1371/journal.pone.0197267
79. Felger JC, Lotrich FE. Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience. (2013) 246:199–229. doi: 10.1016/j.neuroscience.2013.04.060
80. Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neurosci Res. (2014) 79:1–12. doi: 10.1016/j.neures.2013.10.004
81. Ohgidani M, Kato TA, Sagata N, Hayakawa K, Shimokawa N, Sato-Kasai M, et al. TNF-α from hippocampal microglia induces working memory deficits by acute stress in mice. Brain Behav Immun. (2016) 55:17–24. doi: 10.1016/j.bbi.2015.08.022
82. Miller AH. Beyond depression: the expanding role of inflammation in psychiatric disorders. World Psychiatry. (2020) 19:108–9. doi: 10.1002/wps.20723
83. Köhler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O, et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry. (2014) 71:1381–91. doi: 10.1001/jamapsychiatry.2014.1611
84. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. (2016) 16:22–34.
85. Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. (2013) 70:31–41. doi: 10.1001/2013.jamapsychiatry.4
86. Raffaele S, Lombardi M, Verderio C, Fumagalli M. TNF production and release from microglia via extracellular vesicles: impact on brain functions. Cells. (2020) 9:2145. doi: 10.3390/cells9102145
87. Dong Y, Fischer R, Naudé PJ, Maier O, Nyakas C, Duffey M, et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc Natl Acad Sci U.S.A. (2016) 113:12304–9.
88. Gao H, Danzi MC, Choi CS, Taherian M, Dalby-Hansen C, Ellman DG, et al. Opposing functions of microglial and macrophagic TNFR2 in the pathogenesis of experimental autoimmune encephalomyelitis. Cell Rep. (2017) 18:198–212. doi: 10.1016/j.celrep.2016.11.083
89. Veroni C, Gabriele L, Canini I, Castiello L, Coccia E, Remoli ME, et al. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol Cell Neurosci. (2010) 45:234–44. doi: 10.1016/j.mcn.2010.06.014
90. Hodes GE, Ménard C, Russo SJ. Integrating interleukin-6 into depression diagnosis and treatment. Neurobiol Stress. (2016) 4:15–22. doi: 10.1016/j.ynstr.2016.03.003
91. Tanaka T, Kishimoto T. The biology and medical implications of interleukin-6. Cancer Immunol Res. (2014) 2:288–94. doi: 10.1158/2326-6066.Cir-14-0022
92. Spooren A, Kolmus K, Laureys G, Clinckers R, De Keyser J, Haegeman G, et al. Interleukin-6, a mental cytokine. Brain Res Rev. (2011) 67:157–83. doi: 10.1016/j.brainresrev.2011.01.002
93. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. (2015) 16:448–57. doi: 10.1038/ni.3153
94. Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimäki M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav Immun. (2015) 49:206–15. doi: 10.1016/j.bbi.2015.06.001
95. Ng A, Tam WW, Zhang MW, Ho CS, Husain SF, McIntyre RS, et al. IL-1β, IL-6, TNF- α and CRP in elderly patients with depression or Alzheimer’s disease: systematic review and meta-analysis. Sci Rep. (2018) 8:12050. doi: 10.1038/s41598-018-30487-6
96. Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. (2000) 22:370–9. doi: 10.1016/s0893-133x(99)00134-7
97. Sasayama D, Hattori K, Wakabayashi C, Teraishi T, Hori H, Ota M, et al. Increased cerebrospinal fluid interleukin-6 levels in patients with schizophrenia and those with major depressive disorder. J Psychiatr Res. (2013) 47:401–6. doi: 10.1016/j.jpsychires.2012.12.001
98. Böttcher C, Fernández-Zapata C, Snijders GJL, Schlickeiser S, Sneeboer MAM, Kunkel D, et al. Single-cell mass cytometry of microglia in major depressive disorder reveals a non-inflammatory phenotype with increased homeostatic marker expression. Transl Psychiatry. (2020) 10:310. doi: 10.1038/s41398-020-00992-2
99. Ramirez K, Shea DT, McKim DB, Reader BF, Sheridan JF. Imipramine attenuates neuroinflammatory signaling and reverses stress-induced social avoidance. Brain Behav Immun. (2015) 46:212–20. doi: 10.1016/j.bbi.2015.01.016
100. Aniszewska A, Chłodzińska N, Bartkowska K, Winnicka MM, Turlejski K, Djavadian RL. The expression of interleukin-6 and its receptor in various brain regions and their roles in exploratory behavior and stress responses. J Neuroimmunol. (2015) 284:1–9. doi: 10.1016/j.jneuroim.2015.05.001
101. Maes M, Anderson G, Kubera M, Berk M. Targeting classical IL-6 signalling or IL-6 trans-signalling in depression? Expert Opin Ther Targets. (2014) 18:495–512. doi: 10.1517/14728222.2014.888417
102. Sun Y, Wang D, Salvadore G, Hsu B, Curran M, Casper C, et al. The effects of interleukin-6 neutralizing antibodies on symptoms of depressed mood and anhedonia in patients with rheumatoid arthritis and multicentric Castleman’s disease. Brain Behav Immun. (2017) 66:156–64. doi: 10.1016/j.bbi.2017.06.014
103. Ting EY, Yang AC, Tsai SJ. Role of interleukin-6 in depressive disorder. Int J Mol Sci. (2020) 21:2194. doi: 10.3390/ijms21062194
104. Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. (2003) 85:85–95. doi: 10.1016/S0165-2478(02)00228-6
105. Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. (2005) 3:36–46. doi: 10.1038/nrmicro1068
106. Płóciennikowska A, Hromada-Judycka A, Borzęcka K, Kwiatkowska K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. (2015) 72:557–81. doi: 10.1007/s00018-014-1762-5
107. Figueroa-Hall LK, Paulus MP, Savitz J. Toll-like receptor signaling in depression. Psychoneuroendocrinology. (2020) 121:104843. doi: 10.1016/j.psyneuen.2020.104843
108. Rocha DM, Caldas AP, Oliveira LL, Bressan J, Hermsdorff HH. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis. (2016) 244:211–5. doi: 10.1016/j.atherosclerosis.2015.11.015
109. Tripathi P, Aggarwal A. NF-kB transcription factor: a key player in the generation of immune response. Curr Sci. (2006) 90:519–31.
110. Kuriyan J, Thanos D. Structure of the NF-κB transcription factor: a holistic interaction with DNA. Structure. (1995) 3:135–41. doi: 10.1016/S0969-2126(01)00143-5
111. Gilmore TD. Introduction to NF-κB: players, pathways, perspectives. Oncogene. (2006) 25:6680–4. doi: 10.1038/sj.onc.1209954
112. Kaltschmidt C, Kaltschmidt B, Lannes-Vieira J, Kreutzberg GW, Wekerle H, Baeuerle PA, et al. Transcription factor NF-κB is activated in microglia during experimental autoimmune encephalomyelitis. J Neuroimmunol. (1994) 55:99–106. doi: 10.1016/0165-5728(94)90151-1
113. Lucas K, Maes M. Role of the toll like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol Neurobiol. (2013) 48:190–204. doi: 10.1007/s12035-013-8425-7
114. Lu Y-C, Yeh W-C, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. (2008) 42:145–51. doi: 10.1016/j.cyto.2008.01.006
115. Takeda K, Akira S. TLR signaling pathways. Semin Immunol. (2004) 16:3–9. doi: 10.1016/j.smim.2003.10.003
116. Young A, Campbell E, Lynch S, Suckling J, Powis S. Aberrant NF-kappaB expression in autism spectrum condition: a mechanism for neuroinflammation. Front Psychiatry. (2011) 2:27. doi: 10.3389/fpsyt.2011.00027
117. Bassani TB, Vital MA, Rauh LK. Neuroinflammation in the pathophysiology of Parkinson’s disease and therapeutic evidence of anti-inflammatory drugs. Arq Neuropsiquiatr. (2015) 73:616–23. doi: 10.1590/0004-282x20150057
118. MacDowell KS, Pinacho R, Leza JC, Costa J, Ramos B, García-Bueno B. Differential regulation of the TLR4 signalling pathway in post-mortem prefrontal cortex and cerebellum in chronic schizophrenia: relationship with SP transcription factors. Prog Neuro Psychopharmacol Biol Psychiatry. (2017) 79:481–92. doi: 10.1016/j.pnpbp.2017.08.005
119. Song M, Jin J, Lim J-E, Kou J, Pattanayak A, Rehman JA, et al. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J Neuroinflammation. (2011) 8:92. doi: 10.1186/1742-2094-8-92
120. Zhou Y, Chen Y, Xu C, Zhang H, Lin C. TLR4 targeting as a promising therapeutic strategy for Alzheimer disease treatment. Front Neurosci. (2020) 14:602508. doi: 10.3389/fnins.2020.602508
121. Wang J, Yang C, Liu Z, Li X, Liu M, Wang Y, et al. Association of the TLR4 gene with depressive symptoms and antidepressant efficacy in major depressive disorder. Neurosci Lett. (2020) 736:135292. doi: 10.1016/j.neulet.2020.135292
122. Rasmusson AJ, Gallwitz M, Soltanabadi B, Ciuculete DM, Mengel-From J, Christensen K, et al. Toll-like receptor 4 methylation grade is linked to depressive symptom severity. Transl Psychiatry. (2021) 11:371. doi: 10.1038/s41398-021-01481-w
123. Salim S, Liu H, Atrooz F. P-395 - Early life stress, stress-resilience/susceptibility and oxidative stress. Free Radic Biol Med. (2018) 120:S165. doi: 10.1016/j.freeradbiomed.2018.04.542
124. Olugbemide AS, Ben-Azu B, Bakre AG, Ajayi AM, Femi-Akinlosotu O, Umukoro S. Naringenin improves depressive- and anxiety-like behaviors in mice exposed to repeated hypoxic stress through modulation of oxido-inflammatory mediators and NF-kB/BDNF expressions. Brain Res Bull. (2021) 169:214–27. doi: 10.1016/j.brainresbull.2020.12.003
125. Fu S, Wang J, Hao C, Dang H, Jiang S. Tetramethylpyrazine ameliorates depression by inhibiting TLR4-NLRP3 inflammasome signal pathway in mice. Psychopharmacology. (2019) 236:2173–85. doi: 10.1007/s00213-019-05210-6
126. Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Front Neuroendocrinol. (2012) 33:267–86. doi: 10.1016/j.yfrne.2012.08.006
127. Liu J, Buisman-Pijlman F, Hutchinson MR. Toll-like receptor 4: innate immune regulator of neuroimmune and neuroendocrine interactions in stress and major depressive disorder. Front Neurosci. (2014) 8:309. doi: 10.3389/fnins.2014.00309
128. Rahimifard M, Maqbool F, Moeini-Nodeh S, Niaz K, Abdollahi M, Braidy N, et al. Targeting the TLR4 signaling pathway by polyphenols: a novel therapeutic strategy for neuroinflammation. Ageing Res Rev. (2017) 36:11–9.
129. Inserra A, Mastronardi CA, Rogers G, Licinio J, Wong ML. Neuroimmunomodulation in major depressive disorder: focus on caspase 1, inducible nitric oxide synthase, and interferon-gamma. Mol Neurobiol. (2019) 56:4288–305. doi: 10.1007/s12035-018-1359-3
130. Kustova Y, Sei Y, Morse HC Jr, Basile AS. The influence of a targeted deletion of the IFNgamma gene on emotional behaviors. Brain Behav Immun. (1998) 12:308–24. doi: 10.1006/brbi.1998.0546
131. Litteljohn D, Cummings A, Brennan A, Gill A, Chunduri S, Anisman H, et al. Interferon-gamma deficiency modifies the effects of a chronic stressor in mice: implications for psychological pathology. Brain Behav Immun. (2010) 24:462–73. doi: 10.1016/j.bbi.2009.12.001
132. Campos AC, Vaz GN, Saito VM, Teixeira AL. Further evidence for the role of interferon-gamma on anxiety- and depressive-like behaviors: involvement of hippocampal neurogenesis and NGF production. Neurosci Lett. (2014) 578:100–5. doi: 10.1016/j.neulet.2014.06.039
133. Zhang J, He H, Qiao Y, Zhou T, He H, Yi S, et al. Priming of microglia with IFN-γ impairs adult hippocampal neurogenesis and leads to depression-like behaviors and cognitive defects. Glia. (2020) 68:2674–92. doi: 10.1002/glia.23878
134. Alboni S, Poggini S, Garofalo S, Milior G, El Hajj H, Lecours C, et al. Fluoxetine treatment affects the inflammatory response and microglial function according to the quality of the living environment. Brain Behav Immun. (2016) 58:261–71. doi: 10.1016/j.bbi.2016.07.155
135. Horikawa H, Kato TA, Mizoguchi Y, Monji A, Seki Y, Ohkuri T, et al. Inhibitory effects of SSRIs on IFN-γ induced microglial activation through the regulation of intracellular calcium. Prog Neuropsychopharmacol Biol Psychiatry. (2010) 34:1306–16. doi: 10.1016/j.pnpbp.2010.07.015
136. Augusto-Oliveira M, Arrifano GP, Delage CI, Tremblay M, Crespo-Lopez ME, Verkhratsky A. Plasticity of microglia. Biol Rev Camb Philos Soc. (2022) 97:217–50. doi: 10.1111/brv.12797
137. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
138. Devorak J, Torres-Platas SG, Davoli MA, Prud’homme J, Turecki G, Mechawar N. Cellular and molecular inflammatory profile of the choroid plexus in depression and suicide. Front Psychiatry. (2015) 6:138. doi: 10.3389/fpsyt.2015.00138
139. Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T, et al. IFN-γ-dependent activation of the brain’s choroid plexus for CNS immune surveillance and repair. Brain. (2013) 136(Pt 11):3427–40. doi: 10.1093/brain/awt259
140. Fakhfouri G, Rahimian R, Ghia JE, Khan WI, Dehpour AR. Impact of 5-HT3 receptor antagonists on peripheral and central diseases. Drug Discov Today. (2012) 17:741–7. doi: 10.1016/j.drudis.2012.02.009
141. Seyedabadi M, Rahimian R, Ghia JE. The role of alpha7 nicotinic acetylcholine receptors in inflammatory bowel disease: involvement of different cellular pathways. Expert Opin Ther Targets. (2018) 22:161–76. doi: 10.1080/14728222.2018.1420166
142. Fakhfouri G, Mousavizadeh K, Mehr SE, Dehpour AR, Zirak MR, Ghia JE, et al. From chemotherapy-induced emesis to neuroprotection: therapeutic opportunities for 5-HT3 receptor antagonists. Mol Neurobiol. (2015) 52:1670–9. doi: 10.1007/s12035-014-8957-5
143. Khalifeh S, Fakhfouri G, Mehr SE, Mousavizadeh K, Dehpour AR, Khodagholi F, et al. Beyond the 5-HT3 receptors: a role for α7nACh receptors in neuroprotective aspects of tropisetron. Hum Exp Toxicol. (2015) 34:922–31. doi: 10.1177/0960327114562034
144. Fakhfouri G, Rahimian R, Dyhrfjeld-Johnsen J, Zirak MR, Beaulieu JM. 5-HT receptor antagonists in neurologic and neuropsychiatric disorders: the iceberg still lies beneath the surface. Pharmacol Rev. (2019) 71:383–412. doi: 10.1124/pr.118.015487
145. King JR, Gillevet TC, Kabbani N. A G protein-coupled α7 nicotinic receptor regulates signaling and TNF-α release in microglia. FEBS Open Bio. (2017) 7:1350–61. doi: 10.1002/2211-5463.12270
146. Patel H, McIntire J, Ryan S, Dunah A, Loring R. Anti-inflammatory effects of astroglial α7 nicotinic acetylcholine receptors are mediated by inhibition of the NF-κB pathway and activation of the Nrf2 pathway. J Neuroinflammation. (2017) 14:192. doi: 10.1186/s12974-017-0967-6
147. Alzarea S, Rahman S. Effects of alpha-7 nicotinic allosteric modulator PNU 120596 on depressive-like behavior after lipopolysaccharide administration in mice. Prog Neuropsychopharmacol Biol Psychiatry. (2018) 86:218–28. doi: 10.1016/j.pnpbp.2018.05.018
148. Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. (2006) 27:482–91. doi: 10.1016/j.tips.2006.07.004
149. Williams DK, Wang J, Papke RL. Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem Pharmacol. (2011) 82:915–30. doi: 10.1016/j.bcp.2011.05.001
150. de Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol. (2007) 151:915–29. doi: 10.1038/sj.bjp.0707264
151. Papke RL, Kem WR, Soti F, López-Hernández GY, Horenstein NA. Activation and desensitization of nicotinic alpha7-type acetylcholine receptors by benzylidene anabaseines and nicotine. J Pharmacol Exp Ther. (2009) 329:791–807. doi: 10.1124/jpet.108.150151
152. Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, et al. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem. (2004) 89:337–43. doi: 10.1046/j.1471-4159.2004.02347.x
153. Suzuki T, Hide I, Matsubara A, Hama C, Harada K, Miyano K, et al. Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res. (2006) 83:1461–70. doi: 10.1002/jnr.20850
154. Abbas M, Alzarea S, Papke RL, Rahman S. The α7 nicotinic acetylcholine receptor positive allosteric modulator attenuates lipopolysaccharide-induced activation of hippocampal IκB and CD11b gene expression in mice. Drug Discov Ther. (2017) 11:206–11. doi: 10.5582/ddt.2017.01038
155. Corradi J, Bouzat C. Understanding the bases of function and modulation of α7 nicotinic receptors: implications for drug discovery. Mol Pharmacol. (2016) 90:288–99. doi: 10.1124/mol.116.104240
156. Alzarea S, Rahman S. Alpha-7 nicotinic receptor allosteric modulator PNU120596 prevents lipopolysaccharide-induced anxiety, cognitive deficit and depression-like behaviors in mice. Behav Brain Res. (2019) 366:19–28. doi: 10.1016/j.bbr.2019.03.019
157. Rahman S, Alzarea S. Glial mechanisms underlying major depressive disorder: potential therapeutic opportunities. Prog Mol Biol Transl Sci. (2019) 167:159–78. doi: 10.1016/bs.pmbts.2019.06.010
158. Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U.S.A. (2003) 100:1920–5. doi: 10.1073/pnas.0438019100
159. Zhao D, Xu X, Pan L, Zhu W, Fu X, Guo L, et al. Pharmacologic activation of cholinergic alpha7 nicotinic receptors mitigates depressive-like behavior in a mouse model of chronic stress. J Neuroinflammation. (2017) 14:234. doi: 10.1186/s12974-017-1007-2
160. Martín-de-Saavedra MD, Budni J, Cunha MP, Gómez-Rangel V, Lorrio S, Del Barrio L, et al. Nrf2 participates in depressive disorders through an anti-inflammatory mechanism. Psychoneuroendocrinology. (2013) 38:2010–22. doi: 10.1016/j.psyneuen.2013.03.020
161. Parada E, Egea J, Buendia I, Negredo P, Cunha AC, Cardoso S, et al. The microglial α7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid Redox Signal. (2013) 19:1135–48. doi: 10.1089/ars.2012.4671
162. Egea J, Buendia I, Parada E, Navarro E, León R, Lopez MG. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol. (2015) 97:463–72. doi: 10.1016/j.bcp.2015.07.032
163. Freitas AE, Egea J, Buendia I, Gómez-Rangel V, Parada E, Navarro E, et al. Agmatine, by improving neuroplasticity markers and inducing Nrf2, prevents corticosterone-induced depressive-like behavior in mice. Mol Neurobiol. (2016) 53:3030–45. doi: 10.1007/s12035-015-9182-6
164. Navarro E, Gonzalez-Lafuente L, Pérez-Liébana I, Buendia I, López-Bernardo E, Sánchez-Ramos C, et al. Heme-oxygenase I and PCG-1α regulate mitochondrial biogenesis via microglial activation of alpha7 nicotinic acetylcholine receptors using PNU282987. Antioxid Redox Signal. (2017) 27:93–105. doi: 10.1089/ars.2016.6698
165. Cuadrado A, Manda G, Hassan A, Alcaraz MJ, Barbas C, Daiber A, et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: a systems medicine approach. Pharmacol Rev. (2018) 70:348–83. doi: 10.1124/pr.117.014753
166. Rahimian R, Fakhfouri G, Zirak MR. Pros and cons of 5-HT receptor antagonists in neuropsychiatric diseases. Biomed Pharmacother. (2019) 118:109301. doi: 10.1016/j.biopha.2019.109301
167. Rezvani AH, Kholdebarin E, Brucato FH, Callahan PM, Lowe DA, Levin ED. Effect of R3487/MEM3454, a novel nicotinic alpha7 receptor partial agonist and 5-HT3 antagonist on sustained attention in rats. Prog Neuropsychopharmacol Biol Psychiatry. (2009) 33:269–75. doi: 10.1016/j.pnpbp.2008.11.018
168. Gadani SP, Cronk JC, Norris GT, Kipnis J. IL-4 in the brain: a cytokine to remember. J Immunol. (2012) 189:4213–9. doi: 10.4049/jimmunol.1202246
169. Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai M, et al. Interleukin-4 is essential for microglia/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke. (2016) 47:498–504. doi: 10.1161/strokeaha.115.012079
170. Wachholz S, Knorr A, Mengert L, Plümper J, Sommer R, Juckel G, et al. Interleukin-4 is a participant in the regulation of depressive-like behavior. Behav Brain Res. (2017) 326:165–72. doi: 10.1016/j.bbr.2017.03.020
171. Lee HJ, Park HJ, Starkweather A, An K, Shim I. Decreased interleukin-4 release from the neurons of the locus coeruleus in response to immobilization stress. Med Inflamm. (2016) 2016:3501905. doi: 10.1155/2016/3501905
172. Pu H, Ma C, Zhao Y, Wang Y, Zhang W, Miao W, et al. Intranasal delivery of interleukin-4 attenuates chronic cognitive deficits via beneficial microglial responses in experimental traumatic brain injury. J Cereb Blood Flow Metab. (2021) 41:2870–86. doi: 10.1177/0271678x211028680
173. Zhang J, Rong P, Zhang L, He H, Zhou T, Fan Y, et al. IL4-driven microglia modulate stress resilience through BDNF-dependent neurogenesis. Sci Adv. (2021) 7:eabb9888. doi: 10.1126/sciadv.abb9888
174. Rahimian R, Lalancette-Hébert M, Weng YC, Sato S, Kriz J. Glucosamine-mediated immunomodulation after stroke is sexually dimorphic. Brain Behav Immun Health. (2020) 3:100041. doi: 10.1016/j.bbih.2020.100041
175. Vemuganti R. Therapeutic potential of PPARγ activation in stroke. PPAR Res. (2008) 2008:461981. doi: 10.1155/2008/461981
176. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. (2002) 53:409–35. doi: 10.1146/annurev.med.53.082901.104018
177. Bernardo A, Minghetti L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr Pharm Des. (2006) 12:93–109. doi: 10.2174/138161206780574579
178. Landreth GE, Heneka MT. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol Aging. (2001) 22:937–44. doi: 10.1016/s0197-4580(01)00296-2
179. Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, et al. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. (1999) 400:378–82. doi: 10.1038/22572
180. Zhou L, Yin J, Wang C, Liao J, Liu G, Chen L. Lack of seipin in neurons results in anxiety- and depression-like behaviors via down regulation of PPARγ. Hum Mol Genet. (2014) 23:4094–102. doi: 10.1093/hmg/ddu126
181. Colle R, de Larminat D, Rotenberg S, Hozer F, Hardy P, Verstuyft C, et al. PPAR-γ agonists for the treatment of major depression: a review. Pharmacopsychiatry. (2017) 50:49–55. doi: 10.1055/s-0042-120120
182. Youssef DA, El-Fayoumi HM, Mahmoud MF. Beta-caryophyllene alleviates diet-induced neurobehavioral changes in rats: the role of CB2 and PPAR-γ receptors. Biomed Pharmacother. (2019) 110:145–54. doi: 10.1016/j.biopha.2018.11.039
183. Kurhe Y, Mahesh R. Pioglitazone, a PPARγ agonist rescues depression associated with obesity using chronic unpredictable mild stress model in experimental mice. Neurobiol Stress. (2016) 3:114–21. doi: 10.1016/j.ynstr.2016.05.001
184. Shelton RC, Miller AH. Inflammation in depression: is adiposity a cause? Dialog Clin Neurosci. (2011) 13:41–53. doi: 10.31887/DCNS.2011.13.1/rshelton
185. Capuron L, Lasselin J, Castanon N. Role of adiposity-driven inflammation in depressive morbidity. Neuropsychopharmacology. (2017) 42:115–28. doi: 10.1038/npp.2016.123
186. Alexaki VI. The impact of obesity on microglial function: immune, metabolic and endocrine perspectives. Cells. (2021) 10:1584. doi: 10.3390/cells10071584
187. Qin X, Wang W, Wu H, Liu D, Wang R, Xu J, et al. PPARγ-mediated microglial activation phenotype is involved in depressive-like behaviors and neuroinflammation in stressed C57BL/6J and ob/ob mice. Psychoneuroendocrinology. (2020) 117:104674. doi: 10.1016/j.psyneuen.2020.104674
188. Li J, Xu B, Chen Z, Zhou C, Liao L, Qin Y, et al. PI3K/AKT/JNK/p38 signalling pathway-mediated neural apoptosis in the prefrontal cortex of mice is involved in the antidepressant-like effect of pioglitazone. Clin Exp Pharmacol Physiol. (2018) 45:525–35. doi: 10.1111/1440-1681.12918
189. Sadaghiani MS, Javadi-Paydar M, Gharedaghi MH, Fard YY, Dehpour AR. Antidepressant-like effect of pioglitazone in the forced swimming test in mice: the role of PPAR-gamma receptor and nitric oxide pathway. Behav Brain Res. (2011) 224:336–43. doi: 10.1016/j.bbr.2011.06.011
190. Salehi-Sadaghiani M, Javadi-Paydar M, Gharedaghi MH, Zandieh A, Heydarpour P, Yousefzadeh-Fard Y, et al. NMDA receptor involvement in antidepressant-like effect of pioglitazone in the forced swimming test in mice. Psychopharmacology (Berl). (2012) 223:345–55. doi: 10.1007/s00213-012-2722-0
191. Jahrling JB, Hernandez CM, Denner L, Dineley KT. PPARγ recruitment to active ERK during memory consolidation is required for Alzheimer’s disease-related cognitive enhancement. J Neurosci. (2014) 34:4054–63. doi: 10.1523/jneurosci.4024-13.2014
192. Allami N, Javadi-Paydar M, Rayatnia F, Sehhat K, Rahimian R, Norouzi A, et al. Suppression of nitric oxide synthesis by L-NAME reverses the beneficial effects of pioglitazone on scopolamine-induced memory impairment in mice. Eur J Pharmacol. (2011) 650:240–8. doi: 10.1016/j.ejphar.2010.10.007
193. Almasi-Nasrabadi M, Javadi-Paydar M, Mahdavian S, Babaei R, Sharifian M, Norouzi A, et al. Involvement of NMDA receptors in the beneficial effects of pioglitazone on scopolamine-induced memory impairment in mice. Behav Brain Res. (2012) 231:138–45. doi: 10.1016/j.bbr.2012.03.006
194. Sepanjnia K, Modabbernia A, Ashrafi M, Modabbernia MJ, Akhondzadeh S. Pioglitazone adjunctive therapy for moderate-to-severe major depressive disorder: randomized double-blind placebo-controlled trial. Neuropsychopharmacology. (2012) 37:2093–100. doi: 10.1038/npp.2012.58
195. Rahimian R, Béland LC, Sato S, Kriz J. Microglia-derived galectin-3 in neuroinflammation; a bittersweet ligand? Med Res Rev. (2021) 41:2582–9. doi: 10.1002/med.21784
196. Sato S, St-Pierre C, Bhaumik P, Nieminen J. Galectins in innate immunity: dual functions of host soluble beta-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs). Immunol Rev. (2009) 230:172–87. doi: 10.1111/j.1600-065X.2009.00790.x
197. Rahimian R, Béland LC, Kriz J. Galectin-3: mediator of microglia responses in injured brain. Drug Discov Today. (2018) 23:375–81. doi: 10.1016/j.drudis.2017.11.004
198. Nieminen J, Kuno A, Hirabayashi J, Sato S. Visualization of galectin-3 oligomerization on the surface of neutrophils and endothelial cells using fluorescence resonance energy transfer. J Biol Chem. (2007) 282:1374–83. doi: 10.1074/jbc.M604506200
199. Elola MT, Blidner AG, Ferragut F, Bracalente C, Rabinovich GA. Assembly, organization and regulation of cell-surface receptors by lectin-glycan complexes. Biochem J. (2015) 469:1–16. doi: 10.1042/bj20150461
200. Lalancette-Hébert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, et al. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. (2012) 32:10383–95. doi: 10.1523/jneurosci.1498-12.2012
201. Baek JH, Kim SJ, Kang HG, Lee HW, Kim JH, Hwang KA, et al. Galectin-3 activates PPARγ and supports white adipose tissue formation and high-fat diet-induced obesity. Endocrinology. (2015) 156:147–56. doi: 10.1210/en.2014-1374
202. MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, et al. Regulation of alternative macrophage activation by galectin-3. J Immunol. (2008) 180:2650–8. doi: 10.4049/jimmunol.180.4.2650
203. Boza-Serrano A, Ruiz R, Sanchez-Varo R, García-Revilla J, Yang Y, Jimenez-Ferrer I, et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. (2019) 138:251–73. doi: 10.1007/s00401-019-02013-z
204. Burguillos MA, Svensson M, Schulte T, Boza-Serrano A, Garcia-Quintanilla A, Kavanagh E, et al. Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. (2015) 10:1626–38. doi: 10.1016/j.celrep.2015.02.012
205. Srejovic I, Selakovic D, Jovicic N, Jakovljević V, Lukic ML, Rosic G. Galectin-3: roles in neurodevelopment, neuroinflammation, and behavior. Biomolecules. (2020) 10:798. doi: 10.3390/biom10050798
206. Stajic D, Selakovic D, Jovicic N, Joksimovic J, Arsenijevic N, Lukic ML, et al. The role of galectin-3 in modulation of anxiety state level in mice. Brain Behav Immun. (2019) 78:177–87. doi: 10.1016/j.bbi.2019.01.019
207. King DR, Salako DC, Arthur-Bentil SK, Rubin AE, Italiya JB, Tan JS, et al. Relationship between novel inflammatory biomarker galectin-3 and depression symptom severity in a large community-based sample. J Affect Disord. (2021) 281:384–9. doi: 10.1016/j.jad.2020.12.050
208. Melin EO, Dereke J, Thunander M, Hillman M. Depression in type 1 diabetes was associated with high levels of circulating galectin-3. Endocr Connect. (2018) 7:819–28. doi: 10.1530/ec-18-0108
209. Skaper SD, Di Marzo V. Endocannabinoids in nervous system health and disease: the big picture in a nutshell. Philos Trans R Soc Lond B Biol Sci. (2012) 367:3193–200. doi: 10.1098/rstb.2012.0313
210. Lu HC, Mackie K. An introduction to the endogenous cannabinoid system. Biol Psychiatry. (2016) 79:516–25. doi: 10.1016/j.biopsych.2015.07.028
211. Mecha M, Carrillo-Salinas FJ, Feliú A, Mestre L, Guaza C. Microglia activation states and cannabinoid system: therapeutic implications. Pharmacol Ther. (2016) 166:40–55. doi: 10.1016/j.pharmthera.2016.06.011
212. Roche M, Finn DP. Brain CB2 receptors: implications for neuropsychiatric disorders. Pharmaceuticals (Basel). (2010) 3:2517–53. doi: 10.3390/ph3082517
213. Rodriguez JJ, Mackie K, Pickel VM. Ultrastructural localization of the CB1 cannabinoid receptor in mu-opioid receptor patches of the rat Caudate putamen nucleus. J Neurosci. (2001) 21:823–33. doi: 10.1523/jneurosci.21-03-00823.2001
214. Molina-Holgado E, Vela JM, Arévalo-Martín A, Almazán G, Molina-Holgado F, Borrell J, et al. Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J Neurosci. (2002) 22:9742–53. doi: 10.1523/jneurosci.22-22-09742.2002
215. Kendall DA, Yudowski GA. Cannabinoid receptors in the central nervous system: their signaling and roles in disease. Front Cell Neurosci. (2016) 10:294. doi: 10.3389/fncel.2016.00294
216. Di Marzo V, De Petrocellis L, Sepe N, Buono A. Biosynthesis of anandamide and related acylethanolamides in mouse J774 macrophages and N18 neuroblastoma cells. Biochem J. (1996) 316(Pt 3):977–84. doi: 10.1042/bj3160977
217. Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. (1997) 388:773–8. doi: 10.1038/42015
218. Duffy SS, Hayes JP, Fiore NT, Moalem-Taylor G. The cannabinoid system and microglia in health and disease. Neuropharmacology. (2021) 190:108555. doi: 10.1016/j.neuropharm.2021.108555
219. Ativie F, Komorowska JA, Beins E, Albayram Ö, Zimmer T, Zimmer A, et al. Cannabinoid 1 receptor signaling on hippocampal GABAergic neurons influences microglial activity. Front Mol Neurosci. (2018) 11:295. doi: 10.3389/fnmol.2018.00295
220. Navarrete M, Araque A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron. (2010) 68:113–26. doi: 10.1016/j.neuron.2010.08.043
221. Schmöle AC, Lundt R, Gennequin B, Schrage H, Beins E, Krämer A, et al. Expression analysis of CB2-GFP BAC transgenic mice. PLoS One. (2015) 10:e0138986. doi: 10.1371/journal.pone.0138986
222. Stella N. Endocannabinoid signaling in microglial cells. Neuropharmacology. (2009) 56(Suppl. 1):244–53. doi: 10.1016/j.neuropharm.2008.07.037
223. Benito C, Romero JP, Tolón RM, Clemente D, Docagne F, Hillard CJ, et al. Cannabinoid CB1 and CB2 receptors and fatty acid amide hydrolase are specific markers of plaque cell subtypes in human multiple sclerosis. J Neurosci. (2007) 27:2396–402. doi: 10.1523/jneurosci.4814-06.2007
224. Zhang H, Hilton DA, Hanemann CO, Zajicek J. Cannabinoid receptor and N-acyl phosphatidylethanolamine phospholipase D–evidence for altered expression in multiple sclerosis. Brain Pathol. (2011) 21:544–57. doi: 10.1111/j.1750-3639.2011.00477.x
225. Reusch N, Ravichandran KA, Olabiyi BF, Komorowska-Müller JA, Hansen JN, Ulas T, et al. Cannabinoid receptor 2 is necessary to induce toll-like receptor-mediated microglial activation. Glia. (2022) 70:71–88. doi: 10.1002/glia.24089
226. Mecha M, Feliú A, Carrillo-Salinas FJ, Rueda-Zubiaurre A, Ortega-Gutiérrez S, de Sola RG, et al. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav Immun. (2015) 49:233–45. doi: 10.1016/j.bbi.2015.06.002
227. Tao Y, Li L, Jiang B, Feng Z, Yang L, Tang J, et al. Cannabinoid receptor-2 stimulation suppresses neuroinflammation by regulating microglial M1/M2 polarization through the cAMP/PKA pathway in an experimental GMH rat model. Brain Behav Immun. (2016) 58:118–29. doi: 10.1016/j.bbi.2016.05.020
228. O’Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. (2007) 152:576–82. doi: 10.1038/sj.bjp.0707423
229. Fakhfouri G, Ahmadiani A, Rahimian R, Grolla AA, Moradi F, Haeri A. WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-γ pathway. Neuropharmacology. (2012) 63:653–66. doi: 10.1016/j.neuropharm.2012.05.013
230. Payandemehr B, Ebrahimi A, Gholizadeh R, Rahimian R, Varastehmoradi B, Gooshe M, et al. Involvement of PPAR receptors in the anticonvulsant effects of a cannabinoid agonist, WIN 55,212-2. Prog Neuropsychopharmacol Biol Psychiatry. (2015) 57:140–5. doi: 10.1016/j.pnpbp.2014.11.005
231. Benito C, Núñez E, Tolón RM, Carrier EJ, Rábano A, Hillard CJ, et al. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J Neurosci. (2003) 23:11136–41. doi: 10.1523/jneurosci.23-35-11136.2003
232. Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, et al. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci. (2003) 23:1398–405. doi: 10.1523/jneurosci.23-04-01398.2003
233. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
234. Domschke K, Dannlowski U, Ohrmann P, Lawford B, Bauer J, Kugel H, et al. Cannabinoid receptor 1 (CNR1) gene: impact on antidepressant treatment response and emotion processing in major depression. Eur Neuropsychopharmacol. (2008) 18:751–9. doi: 10.1016/j.euroneuro.2008.05.003
235. Onaivi ES, Ishiguro H, Gong JP, Patel S, Meozzi PA, Myers L, et al. Functional expression of brain neuronal CB2 cannabinoid receptors are involved in the effects of drugs of abuse and in depression. Ann N Y Acad Sci. (2008) 1139:434–49. doi: 10.1196/annals.1432.036
236. Kong X, Miao Q, Lu X, Zhang Z, Chen M, Zhang J, et al. The association of endocannabinoid receptor genes (CNR1 and CNR2) polymorphisms with depression: a meta-analysis. Medicine (Baltimore). (2019) 98:e17403. doi: 10.1097/md.0000000000017403
237. Hill MN, Miller GE, Carrier EJ, Gorzalka BB, Hillard CJ. Circulating endocannabinoids and N-acyl ethanolamines are differentially regulated in major depression and following exposure to social stress. Psychoneuroendocrinology. (2009) 34:1257–62. doi: 10.1016/j.psyneuen.2009.03.013
238. Hungund BL, Vinod KY, Kassir SA, Basavarajappa BS, Yalamanchili R, Cooper TB, et al. Upregulation of CB1 receptors and agonist-stimulated [35S]GTPgammaS binding in the prefrontal cortex of depressed suicide victims. Mol Psychiatry. (2004) 9:184–90. doi: 10.1038/sj.mp.4001376
239. Zoppi S, Pérez Nievas BG, Madrigal JL, Manzanares J, Leza JC, García-Bueno B. Regulatory role of cannabinoid receptor 1 in stress-induced excitotoxicity and neuroinflammation. Neuropsychopharmacology. (2011) 36:805–18. doi: 10.1038/npp.2010.214
240. Lisboa SF, Niraula A, Resstel LB, Guimaraes FS, Godbout JP, Sheridan JF. Repeated social defeat-induced neuroinflammation, anxiety-like behavior and resistance to fear extinction were attenuated by the cannabinoid receptor agonist WIN55,212-2. Neuropsychopharmacology. (2018) 43:1924–33. doi: 10.1038/s41386-018-0064-2
241. García-Gutiérrez MS, Pérez-Ortiz JM, Gutiérrez-Adán A, Manzanares J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB receptors. Br J Pharmacol. (2010) 160:1773–84. doi: 10.1111/j.1476-5381.2010.00819.x
242. Beins EC, Beiert T, Jenniches I, Hansen JN, Leidmaa E, Schrickel JW, et al. Cannabinoid receptor 1 signalling modulates stress susceptibility and microglial responses to chronic social defeat stress. Transl Psychiatry. (2021) 11:164. doi: 10.1038/s41398-021-01283-0
243. Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. (2009) 34(Suppl. 1):S186–95. doi: 10.1016/j.psyneuen.2009.05.021
244. De Schepper S, Crowley G, Hong S. Understanding microglial diversity and implications for neuronal function in health and disease. Dev Neurobiol. (2021) 81:507–23. doi: 10.1002/dneu.22777
245. Hein AM, O’Banion MK. Neuroinflammation and memory: the role of prostaglandins. Mol Neurobiol. (2009) 40:15–32. doi: 10.1007/s12035-009-8066-z
246. Narumiya S. Physiology and pathophysiology of prostanoid receptors. Proc Jpn Acad Ser B Phys Biol Sci. (2007) 83:296–319. doi: 10.2183/pjab/83.296
247. Linnoila M, Whorton AR, Rubinow DR, Cowdry RW, Ninan PT, Waters RN. CSF prostaglandin levels in depressed and schizophrenic patients. Arch Gen Psychiatry. (1983) 40:405–6. doi: 10.1001/archpsyc.1983.01790040059008
248. Ohishi K, Ueno R, Nishino S, Sakai T, Hayaishi O. Increased level of salivary prostaglandins in patients with major depression. Biol Psychiatry. (1988) 23:326–34. doi: 10.1016/0006-3223(88)90283-1
249. Nishino S, Ueno R, Ohishi K, Sakai T, Hayaishi O. Salivary prostaglandin concentrations: possible state indicators for major depression. Am J Psychiatry. (1989) 146:365–8. doi: 10.1176/ajp.146.3.365
250. Müller N. Immunology of major depression. Neuroimmunomodulation. (2014) 21:123–30. doi: 10.1159/000356540
251. Mendlewicz J, Kriwin P, Oswald P, Souery D, Alboni S, Brunello N. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. (2006) 21:227–31. doi: 10.1097/00004850-200607000-00005
252. Akhondzadeh S, Jafari S, Raisi F, Nasehi AA, Ghoreishi A, Salehi B, et al. Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depress Anxiety. (2009) 26:607–11. doi: 10.1002/da.20589
253. Tanda G, Carboni E, Frau R, Di Chiara G. Increase of extracellular dopamine in the prefrontal cortex: a trait of drugs with antidepressant potential? Psychopharmacology (Berl). (1994) 115:285–8. doi: 10.1007/bf02244785
254. Tanaka K, Furuyashiki T, Kitaoka S, Senzai Y, Imoto Y, Segi-Nishida E, et al. Prostaglandin E2-mediated attenuation of mesocortical dopaminergic pathway is critical for susceptibility to repeated social defeat stress in mice. J Neurosci. (2012) 32:4319–29. doi: 10.1523/jneurosci.5952-11.2012
255. Matsuoka Y, Furuyashiki T, Yamada K, Nagai T, Bito H, Tanaka Y, et al. Prostaglandin E receptor EP1 controls impulsive behavior under stress. Proc Natl Acad Sci U.S.A. (2005) 102:16066–71. doi: 10.1073/pnas.0504908102
256. Tanaka Y, Furuyashiki T, Momiyama T, Namba H, Mizoguchi A, Mitsumori T, et al. Prostaglandin E receptor EP1 enhances GABA-mediated inhibition of dopaminergic neurons in the substantia nigra pars compacta and regulates dopamine level in the dorsal striatum. Eur J Neurosci. (2009) 30:2338–46. doi: 10.1111/j.1460-9568.2009.07021.x
257. Furuyashiki T, Narumiya S. Stress responses: the contribution of prostaglandin E and its receptors. Nat Rev Endocrinol. (2011) 7:163–75. doi: 10.1038/nrendo.2010.194
258. Fritz M, Klawonn AM, Nilsson A, Singh AK, Zajdel J, Wilhelms DB, et al. Prostaglandin-dependent modulation of dopaminergic neurotransmission elicits inflammation-induced aversion in mice. J Clin Invest. (2016) 126:695–705. doi: 10.1172/jci83844
259. Klawonn AM, Fritz M, Castany S, Pignatelli M, Canal C, Similä F, et al. Microglial activation elicits a negative affective state through prostaglandin-mediated modulation of striatal neurons. Immunity. (2021) 54:225–34.e226. doi: 10.1016/j.immuni.2020.12.016
260. Kessing LV, Rytgaard HC, Gerds TA, Berk M, Ekstrøm CT, Andersen PK. New drug candidates for depression – a nationwide population-based study. Acta Psychiatr Scand. (2019) 139:68–77. doi: 10.1111/acps.12957
261. Müller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Müller B, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. (2006) 11:680–4. doi: 10.1038/sj.mp.4001805
262. Lehmann ML, Weigel TK, Cooper HA, Elkahloun AG, Kigar SL, Herkenham M. Decoding microglia responses to psychosocial stress reveals blood-brain barrier breakdown that may drive stress susceptibility. Sci Rep. (2018) 8:11240. doi: 10.1038/s41598-018-28737-8
263. Zabala A, Vazquez-Villoldo N, Rissiek B, Gejo J, Martin A, Palomino A, et al. P2X4 receptor controls microglia activation and favors remyelination in autoimmune encephalitis. EMBO Mol Med. (2018) 10:e8743. doi: 10.15252/emmm.201708743
264. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
265. Réus GQJ, Rodrigues AL. mTOR signaling in the neuropathophysiology of depression: current evidence. J Recept Ligand Channel Res. (2015) 8:65–74. doi: 10.2147/JRLCR.S70351
266. Bockaert J, Marin P. mTOR in brain physiology and pathologies. Physiol Rev. (2015) 95:1157–87. doi: 10.1152/physrev.00038.2014
267. Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. (2010) 329:959–64. doi: 10.1126/science.1190287
268. Zhong P, Wang W, Pan B, Liu X, Zhang Z, Long JZ, et al. Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology. (2014) 39:1763–76. doi: 10.1038/npp.2014.24
269. Chandran A, Iyo AH, Jernigan CS, Legutko B, Austin MC, Karolewicz B. Reduced phosphorylation of the mTOR signaling pathway components in the amygdala of rats exposed to chronic stress. Prog Neuropsychopharmacol Biol Psychiatry. (2013) 40:240–5. doi: 10.1016/j.pnpbp.2012.08.001
270. Jernigan CS, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. (2011) 35:1774–9. doi: 10.1016/j.pnpbp.2011.05.010
271. Hu Y, Mai W, Chen L, Cao K, Zhang B, Zhang Z, et al. mTOR-mediated metabolic reprogramming shapes distinct microglia functions in response to lipopolysaccharide and ATP. Glia. (2020) 68:1031–45. doi: 10.1002/glia.23760
272. Dello Russo C, Lisi L, Tringali G, Navarra P. Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem Pharmacol. (2009) 78:1242–51. doi: 10.1016/j.bcp.2009.06.097
273. Zhao X, Liao Y, Morgan S, Mathur R, Feustel P, Mazurkiewicz J, et al. Noninflammatory changes of microglia are sufficient to cause epilepsy. Cell Rep. (2018) 22:2080–93. doi: 10.1016/j.celrep.2018.02.004
274. Li D, Wang C, Yao Y, Chen L, Liu G, Zhang R, et al. mTORC1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from M1 type to M2 type. FASEB J. (2016) 30:3388–99. doi: 10.1096/fj.201600495R
275. Keane L, Antignano I, Riechers SP, Zollinger R, Dumas AA, Offermann N, et al. mTOR-dependent translation amplifies microglia priming in aging mice. J Clin Invest. (2021) 131:e132727. doi: 10.1172/jci132727
276. Calovi S, Mut-Arbona P, Sperlágh B. Microglia and the purinergic signaling system. Neuroscience. (2019) 405:137–47. doi: 10.1016/j.neuroscience.2018.12.021
277. Xiang Z, Burnstock G. Expression of P2X receptors on rat microglial cells during early development. Glia. (2005) 52:119–26. doi: 10.1002/glia.20227
278. Burnstock G. Introduction to purinergic signaling. Methods Mol Biol. (2020) 2041:1–15. doi: 10.1007/978-1-4939-9717-6_1
279. Orr AG, Orr AL, Li XJ, Gross RE, Traynelis SF. Adenosine A(2A) receptor mediates microglial process retraction. Nat Neurosci. (2009) 12:872–8. doi: 10.1038/nn.2341
280. Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. (2005) 63:191–270. doi: 10.1016/s0074-7742(05)63007-3
281. Cheffer A, Castillo ARG, Corrêa-Velloso J, Gonçalves MCB, Naaldijk Y, Nascimento IC, et al. Purinergic system in psychiatric diseases. Mol Psychiatry. (2018) 23:94–106. doi: 10.1038/mp.2017.188
282. Tanaka K, Choi J, Cao Y, Stacey G. Extracellular ATP acts as a damage-associated molecular pattern (DAMP) signal in plants. Front Plant Sci. (2014) 5:446. doi: 10.3389/fpls.2014.00446
283. Vénéreau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol. (2015) 6:422. doi: 10.3389/fimmu.2015.00422
284. Iida T, Yoshikawa T, Kárpáti A, Matsuzawa T, Kitano H, Mogi A, et al. JNJ10181457, a histamine H3 receptor inverse agonist, regulates in vivo microglial functions and improves depression-like behaviours in mice. Biochem Biophys Res Commun. (2017) 488:534–40. doi: 10.1016/j.bbrc.2017.05.081
285. De Simone R, Niturad CE, De Nuccio C, Ajmone-Cat MA, Visentin S, Minghetti L. TGF-β and LPS modulate ADP-induced migration of microglial cells through P2Y1 and P2Y12 receptor expression. J Neurochem. (2010) 115:450–9. doi: 10.1111/j.1471-4159.2010.06937.x
286. Vultaggio-Poma V, Sarti AC, Di Virgilio F. Extracellular ATP: a feasible target for cancer therapy. Cells. (2020) 9:2496. doi: 10.3390/cells9112496
287. Yu T, Zhang X, Shi H, Tian J, Sun L, Hu X, et al. P2Y12 regulates microglia activation and excitatory synaptic transmission in spinal lamina II neurons during neuropathic pain in rodents. Cell Death Dis. (2019) 10:165. doi: 10.1038/s41419-019-1425-4
288. Lin SS, Tang Y, Illes P, Verkhratsky A. The safeguarding microglia: central role for P2Y(12) receptors. Front Pharmacol. (2020) 11:627760. doi: 10.3389/fphar.2020.627760
289. Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. (2006) 9:1512–9. doi: 10.1038/nn1805
290. Milior G, Morin-Brureau M, Chali F, Le Duigou C, Savary E, Huberfeld G, et al. Distinct P2Y receptors mediate extension and retraction of microglial processes in epileptic and peritumoral human tissue. J Neurosci. (2020) 40:1373–88. doi: 10.1523/jneurosci.0218-19.2019
291. Morin-Brureau M, Milior G, Royer J, Chali F, Le Duigou C, Savary E, et al. Microglial phenotypes in the human epileptic temporal lobe. Brain. (2018) 141:3343–60. doi: 10.1093/brain/awy276
292. Lowery RL, Mendes MS, Sanders BT, Murphy AJ, Whitelaw BS, Lamantia CE, et al. Loss of P2Y12 has behavioral effects in the adult mouse. Int J Mol Sci. (2021) 22:1868. doi: 10.3390/ijms22041868
293. Peng J, Liu Y, Umpierre AD, Xie M, Tian DS, Richardson JR, et al. Microglial P2Y12 receptor regulates ventral hippocampal CA1 neuronal excitability and innate fear in mice. Mol Brain. (2019) 12:71. doi: 10.1186/s13041-019-0492-x
294. Verma R, Cronin CG, Hudobenko J, Venna VR, McCullough LD, Liang BT. Deletion of the P2X4 receptor is neuroprotective acutely, but induces a depressive phenotype during recovery from ischemic stroke. Brain Behav Immun. (2017) 66:302–12. doi: 10.1016/j.bbi.2017.07.155
295. Yang X, Lou Y, Liu G, Wang X, Qian Y, Ding J, et al. Microglia P2Y6 receptor is related to Parkinson’s disease through neuroinflammatory process. J Neuroinflammation. (2017) 14:38. doi: 10.1186/s12974-017-0795-8
296. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. (2015) 6:262. doi: 10.3389/fphar.2015.00262
297. Iwata M, Ota KT, Li XY, Sakaue F, Li N, Dutheil S, et al. Psychological stress activates the inflammasome via release of adenosine triphosphate and stimulation of the purinergic type 2X7 receptor. Biol Psychiatry. (2016) 80:12–22. doi: 10.1016/j.biopsych.2015.11.026
298. He Y, Taylor N, Fourgeaud L, Bhattacharya A. The role of microglial P2X7: modulation of cell death and cytokine release. J Neuroinflammation. (2017) 14:135. doi: 10.1186/s12974-017-0904-8
299. Kaufmann FN, Costa AP, Ghisleni G, Diaz AP, Rodrigues ALS, Peluffo H, et al. NLRP3 inflammasome-driven pathways in depression: clinical and preclinical findings. Brain Behav Immun. (2017) 64:367–83. doi: 10.1016/j.bbi.2017.03.002
300. Yue N, Huang H, Zhu X, Han Q, Wang Y, Li B, et al. Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J Neuroinflammation. (2017) 14:102. doi: 10.1186/s12974-017-0865-y
301. Basso AM, Bratcher NA, Harris RR, Jarvis MF, Decker MW, Rueter LE. Behavioral profile of P2X7 receptor knockout mice in animal models of depression and anxiety: relevance for neuropsychiatric disorders. Behav Brain Res. (2009) 198:83–90. doi: 10.1016/j.bbr.2008.10.018
302. Boucher AA, Arnold JC, Hunt GE, Spiro A, Spencer J, Brown C, et al. Resilience and reduced c-Fos expression in P2X7 receptor knockout mice exposed to repeated forced swim test. Neuroscience. (2011) 189:170–7. doi: 10.1016/j.neuroscience.2011.05.049
303. Csölle C, Baranyi M, Zsilla G, Kittel A, Gölöncsér F, Illes P, et al. Neurochemical changes in the mouse hippocampus underlying the antidepressant effect of genetic deletion of P2X7 receptors. PLoS One. (2013) 8:e66547. doi: 10.1371/journal.pone.0066547
304. Nagy G, Ronai Z, Somogyi A, Sasvari-Szekely M, Rahman OA, Mate A, et al. P2RX7 Gln460Arg polymorphism is associated with depression among diabetic patients. Prog Neuropsychopharmacol Biol Psychiatry. (2008) 32:1884–8. doi: 10.1016/j.pnpbp.2008.08.021
305. McQuillin A, Bass NJ, Choudhury K, Puri V, Kosmin M, Lawrence J, et al. Case-control studies show that a non-conservative amino-acid change from a glutamine to arginine in the P2RX7 purinergic receptor protein is associated with both bipolar- and unipolar-affective disorders. Mol Psychiatry. (2009) 14:614–20. doi: 10.1038/mp.2008.6
306. Soronen P, Mantere O, Melartin T, Suominen K, Vuorilehto M, Rytsälä H, et al. P2RX7 gene is associated consistently with mood disorders and predicts clinical outcome in three clinical cohorts. Am J Med Genet B Neuropsychiatr Genet. (2011) 156b:435–47. doi: 10.1002/ajmg.b.31179
307. Feng WP, Zhang B, Li W, Liu J. Lack of association of P2RX7 gene rs2230912 polymorphism with mood disorders: a meta-analysis. PLoS One. (2014) 9:e88575. doi: 10.1371/journal.pone.0088575
308. Stokes L, Spencer SJ, Jenkins TA. Understanding the role of P2X7 in affective disorders-are glial cells the major players? Front Cell Neurosci. (2015) 9:258. doi: 10.3389/fncel.2015.00258
309. Bhattacharya A, Drevets WC. Role of neuro-immunological factors in the pathophysiology of mood disorders: implications for novel therapeutics for treatment resistant depression. Curr Top Behav Neurosci. (2017) 31:339–56. doi: 10.1007/7854_2016_43
310. Bhattacharya A. Recent advances in CNS P2X7 physiology and pharmacology: focus on neuropsychiatric disorders. Front Pharmacol. (2018) 9:30. doi: 10.3389/fphar.2018.00030
311. Karasawa A, Michalski K, Mikhelzon P, Kawate T. The P2X7 receptor forms a dye-permeable pore independent of its intracellular domain but dependent on membrane lipid composition. Elife. (2017) 6:e31186. doi: 10.7554/eLife.31186
312. Perlman K, Chouinard-Watkins R, Tanti A, Cisbani G, Orri M, Turecki G, et al. Fatty acid dysregulation in the anterior cingulate cortex of depressed suicides with a history of child abuse. Transl Psychiatry. (2021) 11:535. doi: 10.1038/s41398-021-01657-4
313. Yary T, Aazami S. The association between polyunsaturated fatty acids and depression among Iranian postgraduate students in Malaysia. Lipids Health Dis. (2011) 10:151. doi: 10.1186/1476-511x-10-151
314. Mühle C, Reichel M, Gulbins E, Kornhuber J. Sphingolipids in psychiatric disorders and pain syndromes. Handb Exp Pharmacol. (2013) 216:431–56. doi: 10.1007/978-3-7091-1511-4_22
315. Walther A, Cannistraci CV, Simons K, Durán C, Gerl MJ, Wehrli S, et al. Lipidomics in major depressive disorder. Front Psychiatry. (2018) 9:459. doi: 10.3389/fpsyt.2018.00459
316. Müller CP, Reichel M, Mühle C, Rhein C, Gulbins E, Kornhuber J. Brain membrane lipids in major depression and anxiety disorders. Biochim Biophys Acta. (2015) 1851:1052–65. doi: 10.1016/j.bbalip.2014.12.014
317. Bazinet RP, Layé S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat Rev Neurosci. (2014) 15:771–85. doi: 10.1038/nrn3820
318. Dang R, Zhou X, Tang M, Xu P, Gong X, Liu Y, et al. Fish oil supplementation attenuates neuroinflammation and alleviates depressive-like behavior in rats submitted to repeated lipopolysaccharide. Eur J Nutr. (2018) 57:893–906. doi: 10.1007/s00394-016-1373-z
319. Schneider M, Levant B, Reichel M, Gulbins E, Kornhuber J, Müller CP. Lipids in psychiatric disorders and preventive medicine. Neurosci Biobehav Rev. (2017) 76(Pt B):336–62. doi: 10.1016/j.neubiorev.2016.06.002
320. Chianese R, Coccurello R, Viggiano A, Scafuro M, Fiore M, Coppola G, et al. Impact of dietary fats on brain functions. Curr Neuropharmacol. (2018) 16:1059–85. doi: 10.2174/1570159x15666171017102547
321. Liśkiewicz A, Przybyła M, Wojakowska A, Marczak Ł, Bogus K, Nowacka-Chmielewska M, et al. Physical activity reduces anxiety and regulates brain fatty acid synthesis. Mol Brain. (2020) 13:62. doi: 10.1186/s13041-020-00592-7
322. Tkachev A, Stekolshchikova E, Bobrovskiy DM, Anikanov N, Ogurtsova P, Park DI, et al. Long-term fluoxetine administration causes substantial lipidome alteration of the juvenile macaque brain. Int J Mol Sci. (2021) 22:8089. doi: 10.3390/ijms22158089
323. Okada M, Nutt DJ, Murakami T, Zhu G, Kamata A, Kawata Y, et al. Adenosine receptor subtypes modulate two major functional pathways for hippocampal serotonin release. J Neurosci. (2001) 21:628–40. doi: 10.1523/jneurosci.21-02-00628.2001
324. Batalha VL, Pego JM, Fontinha BM, Costenla AR, Valadas JS, Baqi Y, et al. Adenosine A(2A) receptor blockade reverts hippocampal stress-induced deficits and restores corticosterone circadian oscillation. Mol Psychiatry. (2013) 18:320–31. doi: 10.1038/mp.2012.8
325. Dias L, Lopes CR, Gonçalves FQ, Nunes A, Pochmann D, Machado NJ, et al. Crosstalk between ATP-P(2X7) and adenosine A(2A) receptors controlling neuroinflammation in rats subject to repeated restraint stress. Front Cell Neurosci. (2021) 15:639322. doi: 10.3389/fncel.2021.639322
326. Qian ZM, Ke Y. Brain iron transport. Biol Rev Camb Philos Soc. (2019) 94:1672–84. doi: 10.1111/brv.12521
327. Ogun AS, Adeyinka A. Biochemistry, transferrin. In: StatPearls. Treasure Island, FL: StatPearls Publishing (2022).
328. Moos T, Rosengren Nielsen T, Skjørringe T, Morgan EH. Iron trafficking inside the brain. J Neurochem. (2007) 103:1730–40. doi: 10.1111/j.1471-4159.2007.04976.x
329. Mills E, Dong XP, Wang F, Xu H. Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Future Med Chem. (2010) 2:51–64. doi: 10.4155/fmc.09.140
330. Ganz T, Nemeth E. The hepcidin-ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematol Am Soc Hematol Educ Prog. (2011) 2011:538–42. doi: 10.1182/asheducation-2011.1.538
331. Erikson KM, Pinero DJ, Connor JR, Beard JL. Regional brain iron, ferritin and transferrin concentrations during iron deficiency and iron repletion in developing rats. J Nutr. (1997) 127:2030–8. doi: 10.1093/jn/127.10.2030
332. Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. (2014) 13:1045–60. doi: 10.1016/s1474-4422(14)70117-6
333. Reinert A, Morawski M, Seeger J, Arendt T, Reinert T. Iron concentrations in neurons and glial cells with estimates on ferritin concentrations. BMC Neurosci. (2019) 20:25. doi: 10.1186/s12868-019-0507-7
334. Quintana C, Bellefqih S, Laval JY, Guerquin-Kern JL, Wu TD, Avila J, et al. Study of the localization of iron, ferritin, and hemosiderin in Alzheimer’s disease hippocampus by analytical microscopy at the subcellular level. J Struct Biol. (2006) 153:42–54. doi: 10.1016/j.jsb.2005.11.001
335. Haining RL, Achat-Mendes C. Neuromelanin, one of the most overlooked molecules in modern medicine, is not a spectator. Neural Regen Res. (2017) 12:372–5. doi: 10.4103/1673-5374.202928
336. Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. (1996) 17:83–93. doi: 10.1002/(sici)1098-1136(199606)17:23.0.Co;2-7
337. Leitner DF, Connor JR. Functional roles of transferrin in the brain. Biochim Biophys Acta. (2012) 1820:393–402. doi: 10.1016/j.bbagen.2011.10.016
338. Ward RJ, Crichton RR, Taylor DL, Della Corte L, Srai SK, Dexter DT. Iron and the immune system. J Neural Transm (Vienna). (2011) 118:315–28. doi: 10.1007/s00702-010-0479-3
339. Wessling-Resnick M. Iron homeostasis and the inflammatory response. Annu Rev Nutr. (2010) 30:105–22. doi: 10.1146/annurev.nutr.012809.104804
340. Lokken KL, Tsolis RM, Bäumler AJ. Hypoferremia of infection: a double-edged sword? Nat Med. (2014) 20:335–7. doi: 10.1038/nm.3526
341. Nnah IC, Wessling-Resnick M. Brain iron homeostasis: a focus on microglial iron. Pharmaceuticals (Basel). (2018) 11:129. doi: 10.3390/ph11040129
342. Thomsen MS, Andersen MV, Christoffersen PR, Jensen MD, Lichota J, Moos T. Neurodegeneration with inflammation is accompanied by accumulation of iron and ferritin in microglia and neurons. Neurobiol Dis. (2015) 81:108–18. doi: 10.1016/j.nbd.2015.03.013
343. Mairuae N, Connor JR, Cheepsunthorn P. Increased cellular iron levels affect matrix metalloproteinase expression and phagocytosis in activated microglia. Neurosci Lett. (2011) 500:36–40. doi: 10.1016/j.neulet.2011.06.001
344. McCarthy RC, Sosa JC, Gardeck AM, Baez AS, Lee CH, Wessling-Resnick M. Inflammation-induced iron transport and metabolism by brain microglia. J Biol Chem. (2018) 293:7853–63. doi: 10.1074/jbc.RA118.001949
345. Pasricha SR, Tye-Din J, Muckenthaler MU, Swinkels DW. Iron deficiency. Lancet. (2021) 397:233–48. doi: 10.1016/s0140-6736(20)32594-0
346. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet. (2018) 392:1789–858. doi: 10.1016/s0140-6736(18)32279-7
347. Hidese S, Saito K, Asano S, Kunugi H. Association between iron-deficiency anemia and depression: a web-based Japanese investigation. Psychiatry Clin Neurosci. (2018) 72:513–21. doi: 10.1111/pcn.12656
348. Knyszyńska A, Radecka A, Zabielska P, Łuczak J, Karakiewicz B, Lubkowska A. The role of iron metabolism in fatigue, depression, and quality of life in multiple sclerosis patients. Int J Environ Res Public Health. (2020) 17:6818. doi: 10.3390/ijerph17186818
349. Lee HS, Chao HH, Huang WT, Chen SC, Yang HY. Psychiatric disorders risk in patients with iron deficiency anemia and association with iron supplementation medications: a nationwide database analysis. BMC Psychiatry. (2020) 20:216. doi: 10.1186/s12888-020-02621-0
350. Shah HE, Bhawnani N, Ethirajulu A, Alkasabera A, Onyali CB, Anim-Koranteng C, et al. Iron deficiency-induced changes in the hippocampus, corpus striatum, and monoamines levels that lead to anxiety, depression, sleep disorders, and psychotic disorders. Cureus. (2021) 13:e18138. doi: 10.7759/cureus.18138
351. Berthou C, Iliou JP, Barba D. Iron, neuro-bioavailability and depression. eJHaem. (2022) 3:263–75. doi: 10.1002/jha2.321
352. Hare D, Ayton S, Bush A, Lei P. A delicate balance: iron metabolism and diseases of the brain. Front Aging Neurosci. (2013) 5:34. doi: 10.3389/fnagi.2013.00034
353. Vostrikov VM, Uranova NA, Orlovskaya DD. Deficit of perineuronal oligodendrocytes in the prefrontal cortex in schizophrenia and mood disorders. Schizophr Res. (2007) 94:273–80. doi: 10.1016/j.schres.2007.04.014
354. Nagy C, Maitra M, Tanti A, Suderman M, Théroux JF, Davoli MA, et al. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat Neurosci. (2020) 23:771–81. doi: 10.1038/s41593-020-0621-y
355. Beard JL, Connor JR. Iron status and neural functioning. Annu Rev Nutr. (2003) 23:41–58. doi: 10.1146/annurev.nutr.23.020102.075739
356. Rosato-Siri MV, Marziali L, Guitart ME, Badaracco ME, Puntel M, Pitossi F, et al. Iron availability compromises not only oligodendrocytes but also astrocytes and microglial cells. Mol Neurobiol. (2018) 55:1068–81. doi: 10.1007/s12035-016-0369-2
357. Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR. Oligodendrocytes and myelination: the role of iron. Glia. (2009) 57:467–78. doi: 10.1002/glia.20784
358. Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One. (2011) 6:e26317. doi: 10.1371/journal.pone.0026317
359. Gao W, Wang W, Liu G, Zhang J, Yang J, Deng Z. Allicin attenuated chronic social defeat stress induced depressive-like behaviors through suppression of NLRP3 inflammasome. Metab Brain Dis. (2019) 34:319–29. doi: 10.1007/s11011-018-0342-z
360. Jiao H, Yang H, Yan Z, Chen J, Xu M, Jiang Y, et al. Traditional Chinese formula xiaoyaosan alleviates depressive-like behavior in CUMS mice by regulating PEBP1-GPX4-mediated ferroptosis in the hippocampus. Neuropsychiatr Dis Treat. (2021) 17:1001–19. doi: 10.2147/ndt.S302443
361. Texel SJ, Camandola S, Ladenheim B, Rothman SM, Mughal MR, Unger EL, et al. Ceruloplasmin deficiency results in an anxiety phenotype involving deficits in hippocampal iron, serotonin, and BDNF. J Neurochem. (2012) 120:125–34. doi: 10.1111/j.1471-4159.2011.07554.x
362. Pellegrino RM, Boda E, Montarolo F, Boero M, Mezzanotte M, Saglio G, et al. Transferrin receptor 2 dependent alterations of brain iron metabolism affect anxiety circuits in the mouse. Sci Rep. (2016) 6:30725. doi: 10.1038/srep30725
363. Lenz KM, McCarthy MM. A starring role for microglia in brain sex differences. Neuroscientist. (2015) 21:306–21. doi: 10.1177/1073858414536468
364. Caplan HW, Cox CS, Bedi SS. Do microglia play a role in sex differences in TBI? J Neurosci Res. (2017) 95:509–17. doi: 10.1002/jnr.23854
365. Mouton PR, Long JM, Lei DL, Howard V, Jucker M, Calhoun ME, et al. Age and gender effects on microglia and astrocyte numbers in brains of mice. Brain Res. (2002) 956:30–5. doi: 10.1016/s0006-8993(02)03475-3
366. Schwarz JM, Sholar PW, Bilbo SD. Sex differences in microglial colonization of the developing rat brain. J Neurochem. (2012) 120:948–63. doi: 10.1111/j.1471-4159.2011.07630.x
367. Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, et al. Sex-specific features of microglia from adult mice. Cell Rep. (2018) 23:3501–11. doi: 10.1016/j.celrep.2018.05.048
368. Hui CW, Vecchiarelli HA, Gervais É, Luo X, Michaud F, Scheefhals L, et al. Sex differences of microglia and synapses in the hippocampal dentate gyrus of adult mouse offspring exposed to maternal immune activation. Front Cell Neurosci. (2020) 14:558181. doi: 10.3389/fncel.2020.558181
369. Bollinger JL, Bergeon Burns CM, Wellman CL. Differential effects of stress on microglial cell activation in male and female medial prefrontal cortex. Brain Behav Immun. (2016) 52:88–97. doi: 10.1016/j.bbi.2015.10.003
370. Bollinger JL, Collins KE, Patel R, Wellman CL. Behavioral stress alters corticolimbic microglia in a sex- and brain region-specific manner. PLoS One. (2017) 12:e0187631. doi: 10.1371/journal.pone.0187631
371. Woodburn SC, Bollinger JL, Wohleb ES. Synaptic and behavioral effects of chronic stress are linked to dynamic and sex-specific changes in microglia function and astrocyte dystrophy. Neurobiol Stress. (2021) 14:100312. doi: 10.1016/j.ynstr.2021.100312
372. Yamamoto I, Eyanagi R. Positive skin reaction induced by 4,4’-azoxybenzenedisulfonamide in relationship to the sulfanilamide allergy. Int Arch Allergy Appl Immunol. (1977) 54:538–41. doi: 10.1159/000231874
373. Himmerich H, Patsalos O, Lichtblau N, Ibrahim MAA, Dalton B. Cytokine research in depression: principles, challenges, and open questions. Front Psychiatry. (2019) 10:30. doi: 10.3389/fpsyt.2019.00030
374. Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. (2011) 130:226–38. doi: 10.1016/j.pharmthera.2011.01.014
375. Himmerich H, Minkwitz J, Kirkby KC. Weight gain and metabolic changes during treatment with antipsychotics and antidepressants. Endocr Metab Immune Disord Drug Targets. (2015) 15:252–60. doi: 10.2174/1871530315666150623092031
376. Miller AH, Raison CL. Are anti-inflammatory therapies viable treatments for psychiatric disorders?: where the rubber meets the road. JAMA Psychiatry. (2015) 72:527–8. doi: 10.1001/jamapsychiatry.2015.22
Keywords: microglia, neuroinflammation, metabolic pathway, major depressive disorder, anti-inflammatory pathway, pro-inflammatory pathway, microglial pathways as therapeutic targets
Citation: Rahimian R, Belliveau C, Chen R and Mechawar N (2022) Microglial Inflammatory-Metabolic Pathways and Their Potential Therapeutic Implication in Major Depressive Disorder. Front. Psychiatry 13:871997. doi: 10.3389/fpsyt.2022.871997
Received: 09 February 2022; Accepted: 23 May 2022;
Published: 16 June 2022.
Edited by:
Brenda Penninx, Academic Medical Center, NetherlandsCopyright © 2022 Rahimian, Belliveau, Chen and Mechawar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naguib Mechawar, bmFndWliLm1lY2hhd2FyQG1jZ2lsbC5jYQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.