 Atsushi Sato
Atsushi Sato Hiroko Kotajima-Murakami
Hiroko Kotajima-Murakami Miho Tanaka2,3
Miho Tanaka2,3 Yoshihisa Katoh
Yoshihisa Katoh Kazutaka Ikeda
Kazutaka Ikeda- 1Department of Pediatrics, The University of Tokyo Hospital, Tokyo, Japan
- 2Addictive Substance Project, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan
- 3Department of Psychiatry, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
- 4Department of Obstetrics and Gynecology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
Autism spectrum disorder (ASD) affects reciprocal social interaction and produces abnormal repetitive, restrictive behaviors and interests. The diverse causes of ASD are divided into genetic alterations and environmental risks. The prevalence of ASD has been rising for several decades, which might be related to environmental risks as it is difficult to consider that the prevalence of genetic disorders related to ASD would increase suddenly. The latter includes (1) exposure to medications, such as valproic acid (VPA) and selective serotonin reuptake inhibitors (SSRIs) (2), maternal complications during pregnancy, including infection and hypertensive disorders of pregnancy, and (3) high parental age. Epidemiological studies have indicated a pathogenetic role of prenatal exposure to VPA and maternal inflammation in the development of ASD. VPA is considered to exert its deleterious effects on the fetal brain through several distinct mechanisms, such as alterations of γ-aminobutyric acid signaling, the inhibition of histone deacetylase, the disruption of folic acid metabolism, and the activation of mammalian target of rapamycin. Maternal inflammation that is caused by different stimuli converges on a higher load of proinflammatory cytokines in the fetal brain. Rodent models of maternal exposure to SSRIs generate ASD-like behavior in offspring, but clinical correlations with these preclinical findings are inconclusive. Hypertensive disorders of pregnancy and advanced parental age increase the risk of ASD in humans, but the mechanisms have been poorly investigated in animal models. Evidence of the mechanisms by which environmental factors are related to ASD is discussed, which may contribute to the development of preventive and therapeutic interventions for ASD.
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder with two symptomatic domains: impairments in reciprocal social interaction and repetitive and restrictive behaviors and interests (1). The prevalence of ASD is estimated to be 0.76% worldwide (2), varying across geographic regions, from 0.5% in Asia (3) to 2.5% in the United States (4). Compared with other psychiatric disorders, such as attention-deficit/hyperactivity disorder, ASD has many unanswered questions. There is no evidence of ASD remission in adulthood (2), and there are no effective pharmacotherapies for core symptoms of ASD. Understanding the pathomechanisms of ASD is necessary to develop preventive and therapeutic interventions.
The causes of ASD are divided into genetic alterations and environmental risk factors. Early studies of monozygotic and dizygotic twins estimated that the heritability of ASD is as high as 90% (5). However, subsequent research with improved methodologies showed heritability of 50–60%. Most of this heritability was attributed to common genetic variations, whereas the contribution of rare inherited mutations is lower (6). At the time of this writing, more than 1,200 genes and 2,200 copy number variations are listed in the Autism Database (7) as implicated in ASD (8).
Some of these genes cause congenital disorders, such as Fragile X syndrome and tuberous sclerosis complex. Both of these disorders have been extensively studied in both humans and animal models (9). Genetic analyses of familial ASD cases with no other specific manifestation have also unveiled many copy number variations and genetic mutations (10–12). The introduction of these genetic alterations in animals has contributed to our understanding of common neuronal mechanisms of ASD. For example, brains from mouse models of tuberous sclerosis complex (13, 14) and PTEN tumor hamartoma syndrome (15) exhibited the hyperactivity of mammalian target of rapamycin (mTOR) complex 1 (mTORC1). The inhibition of mTORC1 by rapamycin reversed ASD-related behavioral abnormalities (13–15). Mutations of SHANK2, SHANK3, NLGN4, and NRXN1α are considered to cause ASD by altering synaptic connectivity and neuronal excitability, resulting in excitatory/inhibitory (E/I) imbalance (16). A recent large-scale exome sequencing study found significant mutations of 102 genes, most of which were enriched in the excitatory and inhibitory neuronal lineage (17). Investigation of these genetic models has revealed the neuropathological findings in ASD (16). The brain of neuron-specific Pten knockout mice exhibited hypertrophy of hippocampal neurons (15). Synaptic density was increased in the cerebellum of Tsc1-deficient Purkinje mice (13) and stem cells carrying ASD-related NLGN4 mutations (16). Lower neuronal excitability was found in Tsc1-deficient Purkinje cells (13) and SHANK3-deficient neurons (16), whereas neuronal excitability was enhanced in CNTN5+/− and EHMT2+/− cells (16).
The aforementioned evidence, however, does not necessarily explain the increasing prevalence of ASD over recent decades (i.e., from 0.4/1,000 to 2/1,000 in the 1970s to 1.5% or higher in recent studies) (18–20). According to recent studies, environmental risk factors account for the remaining 40–50% of ASD cases (6, 21), which might partially explain the increase in ASD prevalence. Meta-analyses have indicated several significant risk factors, such as advanced parental age, prenatal exposure to antidepressants, and complications during pregnancy (21–23). In contrast to epidemiological evidence, some of these risks are difficult to be reproduced without causing deleterious alterations in rodents, such as premature birth (24, 25) and low birth weight (26, 27), which hampers our understanding of whether and how these perinatal problems might contribute to ASD. The environment-induced ASD models discussed in this review also exhibit neuropathological features similar to those found in genetic models of ASD. However, the relation between each environmental risk and specific feature appears to be complicated. For example, prenatal exposure to valproic acid (VPA) results in ASD through different mechanisms, including disrupted γ-aminobutyric acid (GABA) signaling and mTORC1 hyperactivity. Shorter dendrites and reduced synapse pruning are observed in different ASD models, such as prenatal exposure to selective serotonin transporter inhibitors (SSRIs) and maternal inflammation.
The present review focuses on three groups of environmental risks of ASD: prenatal exposure to drugs, maternal complications during pregnancy, and advanced parental age. We discuss how each of these leads to ASD to better understand the mechanisms, with the goal of mitigating risk and developing potential therapeutics (Figure 1).
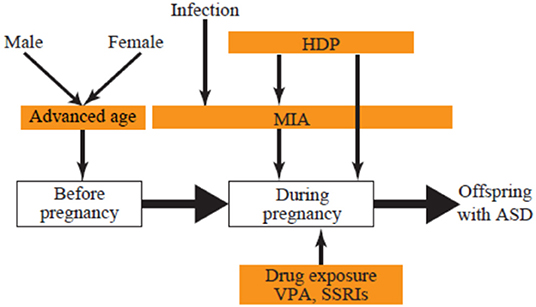
Figure 1. Developmental stage and environmental ASD risks. ASD, autism spectrum disorder; HDP, hypertensive disorders of pregnancy; MIA, maternal immune activation; SSRI, selective serotonin reuptake inhibitor; VPA, valproic acid.
Prenatal Exposure to Drugs
Exposure to medications during pregnancy is a well-known risk factor for ASD (21–23). Considering the abundance of research on mechanisms, VPA and SSRIs are discussed in this review.
VPA
VPA has been clinically used as an antiepileptic drug for more than 50 years, and it is listed as a first-line drug for generalized epilepsy (28). Other indications of VPA include the prevention of migraine (29) and treatment of acute mania (30). It has a simple molecular structure (2-propylepntanoic acid) and appears to exert its clinical effects through various mechanisms, including the inhibition of GABA transaminase, voltage-gated Na+ channels, and T-type Ca2+ channels (29).
Despite its excellent clinical efficacy, congenital malformations and neurodevelopmental problems have been observed in children of mothers who took VPA during pregnancy (31). Congenital malformations that are related to VPA include neural tube defects, congenital heart disease, and cleft palate (32). Prenatal exposure to VPA increases the risk of ASD, but other antiepileptic drugs, such as carbamazepine and lamotrigine, do not (33). Children of mothers who used high-dose VPA (>800 mg/day) during pregnancy had a lower IQ at 6 years of age. Low-dose VPA (800 mg/day or less) did not affect IQ in children but was associated with impairments in verbal ability compared with other antiepileptic drugs (34). Prenatal exposure to carbamazepine or clonazepam did not affect the prevalence of ASD in offspring (33), implying that the inhibition of Na+ channels or potentiation of GABAergic signaling (i.e., effects of VPA) is insufficient to cause ASD. As discussed below, other pharmacological effects of VPA, either alone or combined, may be required. Based on these data, women who are considering becoming pregnant are now recommended to avoid using high-dose VPA by switching to other antiepileptic drugs or reducing VPA to dose to 600 mg/day or less before pregnancy (35). By contrast, those who are found to be pregnant while taking VPA are not advised to switch from VPA to other medications in order to avoid the risk of worsening epilepsy.
In accordance with the human data, rodents that were exposed to VPA in utero exhibited similar congenital anomalies and cognitive deficits (36). Rodent models of VPA-induced ASD are generated by administering higher doses of VPA after conception (see Table 1), whereas human mothers have an increased risk of having offspring with ASD when they become pregnant while taking VPA (>800 mg/day ≈15 mg/kg/day for increasing the risk of ASD). The methodology by which VPA is used to induce ASD-like behavior is well established, which sheds light on several pathways that lead to ASD.
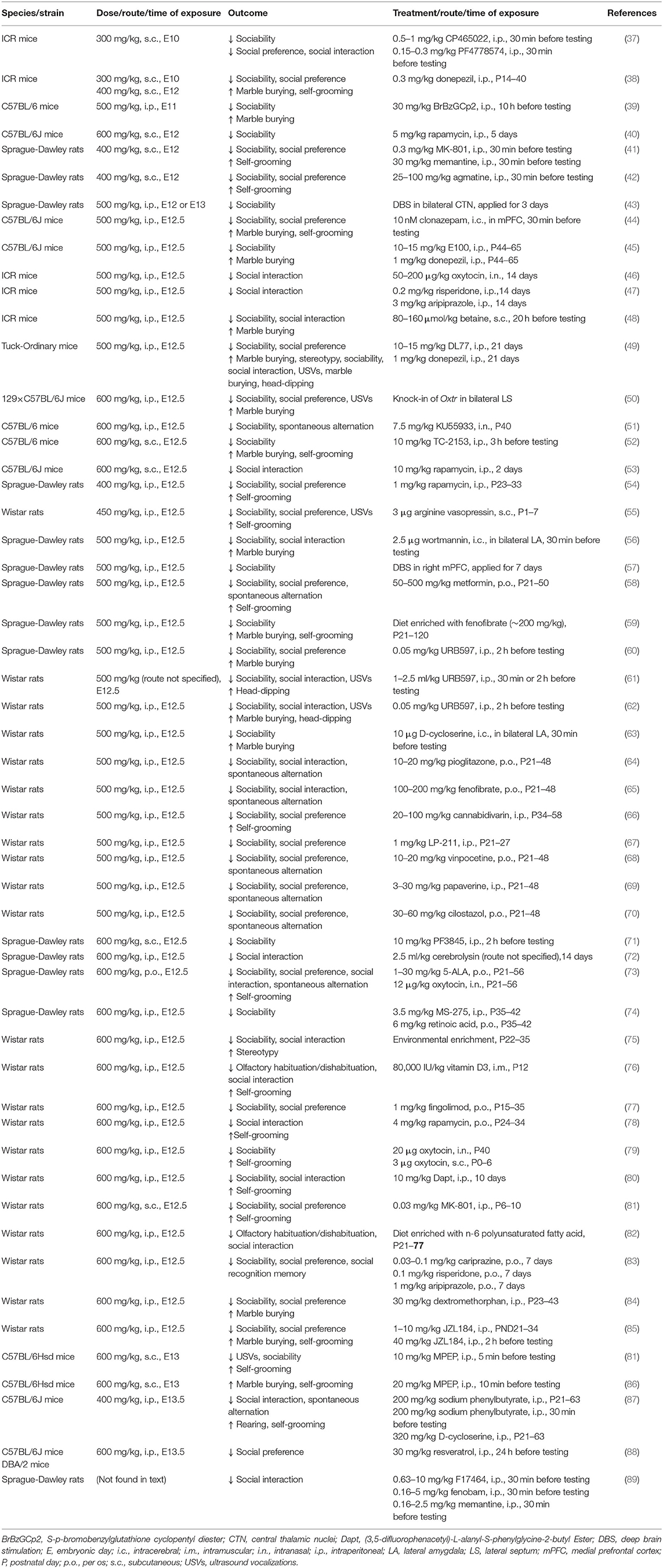
Table 1. Postnatal treatment in ASD rodent models of prenatal exposure to VPA.
Alterations of GABAergic Signaling
Although it is one of the main mechanisms of action by which clinical effects manifest, remaining unclear is whether the inhibition of GABA transaminase by VPA is critical for disturbing the development of fetal brains. GABA is synthesized by glutamic acid decarboxylase (GAD), which converts glutamate to GABA, and degraded by GABA transaminase. GABA exerts its action by binding to specific receptors, GABAA or GABAB receptors, which results in the opening of Cl− channels. GABA stimulates the influx of Cl− ions in the mature brain, resulting in a decrease in intracellular transmembrane potential (90). However, neurons in the immature brain have higher concentrations of intracellular Cl− because of higher activity of Na-K-Cl cotransporter 1 (NKCC1) that passes Cl− into neurons. The binding of GABA to GABA receptors in immature neurons causes the efflux of Cl− and depolarization of neurons (91). This excitatory signaling via GABA is important for normal development of the immature brain, including neurogenesis and synapse formation (92, 93).
The mechanism by which VPA potentiates GABAergic signaling in the fetal brain may be related to the development of ASD. Prenatal treatment with a single dose of VPA, mainly on embryonic day (E) 12.5 in mice (44) and rats (94, 95) but as late as E17 in mice (96) reduced GAD expression in different brain regions that suggests a lower number of GABAergic neurons (94–96), and reduced GABA concentrations and the number of GABA receptors (44). These changes have been associated with impairments in fear conditioning, object recognition (94), and ASD-like social deficits (94, 95). Interestingly, the prenatal inhibition of GABAergic signaling with GABA receptor antagonists, such as picrotoxin administration on E10-12, suppressed neurogenesis in the fetal brain (97) and resulted in ASD-like social deficits in offspring (97, 98). These findings suggest that GABA signaling should be controlled within a limited range to maintain normal development of the fetal brain.
Another mechanism may involve the excitatory-to-inhibitory shift of GABAergic signaling during delivery. This shift may be triggered by oxytocin. Oxytocin blockade on the day before delivery resulted in persistent excitatory GABAergic signaling in the postnatal brain (99). Pups that were born to rats that received VPA on E12 also exhibited diminished excitatory-to-inhibitory shift of GABAergic signaling on postnatal day (P) 15. Maternal pretreatment with bumetanide, an inhibitor of NKCC1, restored the inhibitory GABAergic signaling, and pups of bumetanide-treated mothers exhibited the normalization of isolation-induced ultrasound vocalizations (USVs) (99). In an immature neuron model that utilized cultured cortical neurons that were prepared from P1 rats, 3-day VPA treatment immediately after preparation of the neurons reduced vesicular GABA transporter (VGAT) expression and the number of GABAergic synapses, which persisted for 10 days after cessation of VPA exposure (100). However, VPA did not reduce VGAT expression when treatment began after cultivating the neurons for 8 days. Considering that P1 in rats corresponds to approximately 23 weeks gestation in humans, and P8 around delivery (101), these findings suggest that mid-gestational exposure to VPA produces excitatory-dominant E/I imbalance by blocking the excitatory-to-inhibitory shift of GABAergic signaling or reducing VGAT expression and decreasing GABA synthesis in the postnatal brain. This is further supported by the therapeutic effect of the postnatal restoration of E/I balance for ASD. Postnatal intracerebral administration of the GABAA receptor agonist clonazepam rescued deficient social novelty in mice that were exposed to VPA on E12.5, whereas the GABAB receptor agonist baclofen did not (44). The postnatal suppression of glutamatergic signaling using the N-methyl-D-aspartate receptor antagonist MK-801 or metabotropic glutamate receptor 5 antagonist MPEP was similarly effective in restoring GABAergic signaling in rodents that were exposed to VPA on E12-13 (41, 81, 86, 102). Prenatal exposure to VPA may cause persistent excitatory-dominant E/I imbalance and ASD, which might be reversed by the postnatal correction of E/I balance.
Disturbances in Folic Acid Metabolism
The second mechanism by which VPA is implicated in ASD is the association between VPA and folic acid deficiency. Neural tube defects (i.e., a congenital malformation of the spinal cord in which the neural tube fails to close during embryogenesis and results in exposure of the “unclosed” spinal cord, also called spina bifida) were shown to be related to vitamin deficiencies, including folic acid deficiency (103). A randomized control study found that daily folic acid supplementation reduced the risk of neural tube defects, but other vitamins did not (104). Unexpectedly, children who were exposed to VPA in utero had a higher risk of neural tube defects (32) and ASD (33), similar to children who were born to women with folic acid deficiency during pregnancy. These observations raised the possibility that folic acid supplementation may be useful for the prevention of VPA-related conditions, such as neural tube defects and ASD. Folic acid supplementation before and during pregnancy significantly reduced the risk of ASD in children who were exposed to antiepileptic drugs, including VPA (105). Interestingly, folic acid supplementation lowered the risk of ASD in offspring to less than half, regardless of the concomitant use of antiepileptic drugs (106).
Despite these clinical findings that strongly suggest a similarity between prenatal exposure to VPA and folic acid deficiency in the development of ASD, little is known about the underlying mechanisms by which these two conditions result in ASD or the mechanisms by which folic acid prevents ASD. In a study of human placentas from women without epilepsy, VPA perfusion for 3 h reduced placental concentration of folic acid by ~30% and suppressed mRNA levels of the FOLR1 gene, which encodes folate receptor α (107). VPA is also a non-competitive inhibitor of high-affinity folate receptors, such as folate receptor α (108), and may disturb folic acid metabolism (109). The preventive effect of maternal folic acid supplementation was recapitulated in rodents with regard to neural tube defects. VPA administration in pregnant ICR mice on E8 caused neural tube defects, which were prevented by oral folic acid administration prior to the VPA injection (110). A recent study investigated the effect of folic acid supplementation on VPA-induced ASD. Neuropathological changes in offspring that were prenatally exposed to VPA on E12.5 included an increase in dendritic spine density, an increase in the expression of vesicular glutamate transporter 1 (VGLUT1) and postsynaptic density 95 (PSD95; i.e., markers of excitatory neurons), and a decrease in the expression of GAD65 and gephyrin (i.e., markers of inhibitory neurons) (111), suggesting a net result of excitatory-dominant E/I imbalance. The offspring also exhibited ASD-like behavioral deficits that were dose-dependently prevented by maternal folic acid supplementation from E1 to E12.5 (111), which was consistent with the lower prevalence of ASD that correlates with maternal folic acid supplementation (33). Further studies should investigate how folic acid normalizes VPA-induced alterations in the fetal brain.
Inhibition of Histone Deacetylase
Two other mechanisms may also link in utero VPA exposure to ASD in offspring, although they seem less relevant to clinical effects: inhibition of histone deacetylase (HDAC) and activation of the mTORC1 signaling pathway. Trichostatin A, an established HDAC inhibitor, was teratogenic in Xenopus embryos similarly to VPA (112). VPA and trichostatin A also reduced VGAT expression in cortical neurons obtained from P1 rats (100). This change is considered to result in lower extracellular GABA levels and the disruption of E/I balance toward predominately excitatory transmission, which was observed in another study that employed a rat model of prenatal VPA exposure (111). Moreover, maternal exposure to VPA and trichostatin A on E12.5 decreased USVs and sociability (113). These alterations that are associated with VPA and trichostatin A appear to be caused by HDAC inhibition. Valpromide, an analog of VPA that lacks HDAC inhibitor activity, failed to recapitulate phenotypic changes that were induced by VPA. At the histological level, valpromide administration did not reduce VGAT expression in cortical neurons (100) or global gene expression in the embryonic telencephalon (114). The number of Nissl-positive cells was comparable in the prefrontal and somatosensory cortices when valpromide was administered (115). No congenital malformation (112), deficits in social interaction, anxiety-related behavior, or learning and memory deficits (115) were observed in rodent offspring that were exposed to valpromide in utero. These findings emphasize that the HDAC inhibition by VPA affects normal embryogenesis and neuronal development that leads to ASD.
Activation of mTORC1
Another mechanism of VPA-induced ASD is activation of the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mTORC1 pathway (116). Briefly, the mTORC1 signaling pathway is stimulated by growth factors and altered energy levels, and activates mTORC1 that results in stimulation of cell growth and proliferation (117). These effects are mediated by the phosphorylation of downstream signaling molecules, including ribosomal protein S6 kinases (S6Ks) that regulate global protein synthesis, ULK-51-like kinase 1 (ULK1) that is involved in initiating macroautophagy, and eukaryotic translation initiation factor-4E (eIF4E)-binding proteins (4E-BPs) that control cap-dependent translation (117). mTORC1 hyperactivity underlies ASD in human diseases that are caused by mutations of genes that are upstream of mTORC1, such as tuberous sclerosis complex (13, 14) and PTEN tumor hamartoma syndrome (15). Rodent models in which genes that are downstream of mTORC1 were deleted also exhibited ASD-like behavioral changes that were associated with excessive protein synthesis, impairments in autophagy, and alterations of the gene translation profile (118). In summary, alterations of cell growth and proliferation that are under the control of mTORC1 result in impairments in social behaviors that are relevant to ASD.
Both mice (40, 53) and rats (54) that were exposed to VPA on E12 exhibited an increase in mTORC1 activity in the brain and ASD-like behaviors, such as a decrease in social interaction and increase in self-grooming (40, 53, 54). With regard to developmental delays in children who are prenatally exposed to VPA (34), VPA-exposed mice exhibited deficits in early postnatal development, including eye opening, the righting reflex, and performance in the hanging wire test (53). Related to these behavioral changes, postnatal brains of these animals exhibited signs of the suppression of autophagy (40, 54), one of the main consequences of mTORC1 hyperactivity (117, 118). The expression of a set of genes was also changed, including Fyb, which functions downstream of the mTORC1 signaling pathway (53). Deficits in behavior, autophagy, and gene expression were all reversed by postnatal treatment with the mTORC1 inhibitor rapamycin (40, 53, 54). The constitutive activation of mTORC1 is likely a treatable mechanism by which social behavior is disrupted in VPA-induced ASD.
SSRIs
SSRIs, Serotonin Metabolism, and ASD in Humans
Serotonin (5-hydroxytryptamine [5-HT]) is a neurotransmitter that is involved in diverse brain functions, including motor activity and emotion. When released from presynaptic vesicles, 5-HT binds to post- and presynaptic receptors. Serotonin is then either degraded or collected by the serotonin transporter (SERT). Serotonin plays a critical role in neurodevelopment, including the development of serotonergic fibers (119). SSRIs are a group of drugs that inhibit the SERT and elevate extracellular 5-HT levels in the brain (120). SSRIs are used as a first-line treatment of major depressive disorder (121). The prevalence of depression during pregnancy is estimated to be as high as 10%, and its treatment is often indicated during pregnancy (122, 123).
The role of alterations of serotoninergic signaling in ASD was suggested by the observation that approximately 30% of individuals with ASD have high mean blood levels of 5-HT (124). The maternal use of SSRIs is speculated to elevate maternal 5-HT levels, which might affect fetal brain development and result in a higher risk of ASD in offspring. Case-control studies found a 2-fold increase in the risk of ASD in children of mothers who reported antidepressant use during pregnancy (125, 126). This effect was particularly strong when antidepressants were used during the first trimester of pregnancy, regardless of whether or not the mothers had depression (125). In another cohort study, the use of SSRIs during the second and third trimesters of pregnancy doubled the risk of ASD in offspring, regardless of a maternal history of depression (127). However, later studies reported inconsistent findings with regard to the association between antidepressant use during pregnancy and ASD in offspring (128). A meta-analysis reported an association between the pre-pregnancy maternal use of antidepressants and ASD in offspring and not during pregnancy (23). Another case-control study found that the risk of neurodevelopmental disorders, including ASD, was associated with maternal psychiatric conditions but not the maternal use of SSRIs during pregnancy (129). A nationwide cohort study in Finland reported the influence of prenatal SSRI use on a higher incidence of depression but not ASD (130). In summary, the relationship between ASD and maternal conditions, such as psychiatric comorbidity and antidepressant use, remains elusive and requires further epidemiological studies.
SSRIs, Serotonin Metabolism, and ASD in Animal Models
Investigations of pregnant rodents have improved our understanding of 5-HT dynamics and its alterations by SSRIs during pregnancy. Analyses of Pet1 knockout mice, in which most dorsal raphe neurons lacked 5-HT, revealed that 5-HT in the fetal forebrain was of placental and not maternal or fetal origin (131). The blockade of SERTs using the serotonin-norepinephrine transporter inhibitor venlafaxine by gavage from E8 to E20 decreased placental weight and SERT expression in the placenta in rats (132). The inhibition of 5-HT signaling with the 5-HT2 receptor antagonist ketanserin by gavage from E15 to E20 reduced placental weight and placental blood flow in rats (133). SSRI use during pregnancy could result in lifelong consequences on the brain in offspring (134), but the underlying mechanism is complex, including fetal exposure to SSRIs and alterations of 5-HT supply from the placenta. Decreases in placental weight and blood flow that are associated with maternal treatment with SSRIs could also lower the supply of oxygen and nutrients to the fetus and result in lower offspring weight, which might affect fetal brain development.
In contrast to VPA, few studies have investigated how prenatal exposure to SSRIs affects social behavior in offspring (Table 2). At the behavioral level, fluoxetine administration in mice that began before pregnancy or from early gestation to late-gestation or delivery produced ASD-like behavioral deficits in offspring, decreased USVs in pups, disrupted social interaction, enhanced social dominance, and increased tactile hypersensitivity (135–137). Citalopram, an SSRI with particularly high specificity for blocking SERT compared with dopamine and norepinephrine transporters, also altered behavior in mouse offspring, decreased sociability, decreased social preference, decreased locomotor activity, and increased anxiety-related behavior when given during late gestation (138). In fetal brains that were exposed to fluoxetine, neurons in the prefrontal cortex exhibited a reduction of the frequency of inhibitory synaptic currents, and interneurons exhibited an increase in intrinsic and serotonin-induced excitability (136). Prefrontal cortex tissue from the fluoxetine-exposed brain exhibited high mRNA levels of 5-HT2A receptor (136). Striatal extracts from mice that were prenatally exposed to citalopram expressed higher levels of NMDAR1 and CaMKIIα, which were associated with morphological changes in the striatal neurons and decreases in dendritic length, number, and branch patterns (138). Prenatal exposure to SSRIs is suggested to result in excessive 5-HT signaling and altered E/I balance.
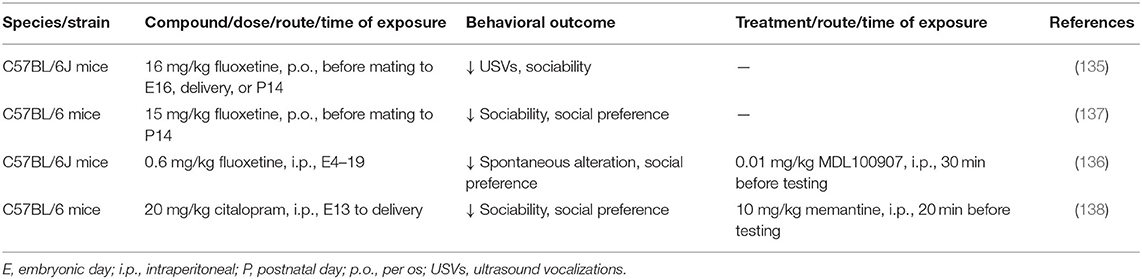
Table 2. Rodent models of prenatal exposure to SSRIs.
The effects of therapeutic interventions in these models also appear to be consistent with these findings. High 5-HT level in the brain that are caused by reexposure to fluoxetine in adulthood recovered tactile hypersensitivity (135). The 5-HT2A receptor antagonist MDL100907 suppressed abnormal excitability in neurons in the prefrontal cortex and reversed social preference in a model of fluoxetine-induced ASD (136). In a citalopram model, high levels of NMDAR1 and CaMKIIα were normalized by postnatal treatment with memantine, which was associated with the recovery of sociability and social preference (138). More research is required to determine whether and how exposure to SSRIs in utero affects fetal brain development and causes social deficits.
The aforementioned interventions can be partially replicated by manipulating genes that are involved in 5-HT neurotransmission because 5-HT levels are consistently changed in these models (i.e., either elevated or depleted). The most extensively investigated models are SERT knockout mice and rats (139). In these animals, extracellular 5-HT levels are several times higher, and consequently the density of 5-HT1A and 5-HT1B receptors is decreased (140–144). Diverse behavioral phenotypes are observed in SERT knockout rodents, such as an increase in anxiety and fear (139), that may affect social behavior in SERT knockout animals. Intact sociability was observed in Sert −/− and Sert +/− mice, reflected by the time spent sniffing a novel mouse or a novel object (145). The heterozygous loss of Sert aggravated deficient sociability in Pten+/− mice, a genetic model of ASD that is associated with the activation of mTORC1 activity (146). Reciprocal social interaction in the resident-intruder paradigm was unaffected in Sert −/− and Sert+/− rats (147). A recent study reported deficits in social interaction, sociability, and social novelty in Sert −/− and Sert +/− mice (148). This was associated with high levels of 5-HT in the brain in Sert −/− mice but not in Sert +/− mice. Impairments in social interaction were ameliorated by restricting the dietary intake of tryptophan (i.e., the precursor of 5-HT), which lowers 5-HT levels in the brain (148). Constitutively elevated 5-HT levels are thus considered to disrupt social behavior.
Reducing 5-HT concentrations in the brain, opposite to SERT deletion, may also give rise to ASD. One method to reduce 5-HT levels is to introduce a gain-of-function mutation of the SERT gene. SERT Ala56 mice that expressed an ASD-associated variant in humans exhibited elevations of 5-HT clearance in the brain and ASD-related social impairments and repetitive behavior, but forebrain 5-HT levels did not change in the mutants (149). This increase in 5-HT clearance was reversed by MW150, a p38α mitogen-activated protein kinase inhibitor, which also normalized social dominance in the tube test (150). Brain 5-HT levels can also be depleted by deleting the tryptophan hydroxylase 2 (TPH2) gene, which is essential for synthesizing 5-HT in the brain. TPH2 knockout mice exhibited diverse ASD-related behaviors, including impairments in social interaction, an increase in marble burying, and deficient early developmental milestones (151). Female TPH2 knockout mice exhibited high levels of aggression against co-housed mice and an increase in defensive behavior when paired with a knockout mouse (152). These studies suggest that prenatal increases and decreases in fetal 5-HT levels may result in the subsequent development of ASD.
Maternal Immune Activation
Another possible environmental risk factor for ASD is maternal inflammation that is induced by infection during pregnancy. Associations between ASD and prenatal infection with specific pathogens, such as rubella and cytomegalovirus, have been repeatedly reported (153). Meta-analyses revealed a mild but significant increase in the risk of ASD in children of mothers who experienced infection during pregnancy, regardless of the pathogen (154, 155). One study estimated that maternal infection accounts for 12–17% of ASD cases (155). The risk of ASD appears to correlate with the severity of maternal infection, in which the risk was further elevated when mothers required hospitalization because of the infection (154). In another meta-analysis, maternal fever during pregnancy, regardless of whether it was caused by infection, increased the risk of neurodevelopmental disorders (156). The invasion of pathogens does not appear to be essential for the development of ASD in offspring; instead, the maternal inflammatory response itself seems sufficient.
Maternal immune activation (MIA) and cytokine production following infection are likely central mechanisms that link maternal infection and ASD in offspring (157). Briefly, MIA-induced ASD appears to begin with the selective activation of Th1 cells, consequently resulting in high interleukin-6 (IL-6) levels, but other mechanisms may also mediate the development of ASD. IL-6 activates retinoic acid receptor-related orphan nuclear receptor γt (RORγt) in naive CD4+ T cells, which stimulates its differentiation into Th17 cells, Activated Th17 cells produce cytokines, including IL-17A that may be critically involved in the development of ASD (158–160). Microglia are local macrophages in the brain that mediate MIA and neurodevelopment. Normal functions of microglia include neurogenesis and synapse pruning, playing an important role in synaptic plasticity (161). The production of proinflammatory cytokines by MIA, such as IL-6 and IL-17, results in microglial activation, which suppresses synapse pruning particularly in the hippocampus and disrupts synapse function (162). The relation between MIA and the development of ASD in offspring has been demonstrated in both humans and in rodents.
Maternal Infection, Immune Activation, and ASD in Humans
Findings are limited about markers that are suggestive that children with ASD were exposed to MIA in utero. The Early markers for Autism study found that high levels of IL-4, IL-5 and interferon-γ (IFN-γ) in maternal serum at 15–19 weeks of gestation were associated with a 50% higher risk of ASD, whereas high levels of IL-2, IL-4 and IL-6 was associated with developmental delay in the absence of ASD (163). Using a larger sample set, a subsequent analysis found high levels of many cytokines, including IL-1α and IL-6, in mothers of children with ASD and developmental delay compared with children with ASD without developmental delay and controls (164). The cytokine profiles of neonates that were exposed to MIA may reflect immunological alterations in their mothers, but the presence of neonatal cytokines was determined using dried blood spots from the neonates, and cytokines in these samples might be unstable. A study did not find changes in these cytokines, such as increase in IL-6; instead, Th1 and Th2 cytokine levels decreased (165) or were comparable between ASD children and controls (166). High IL-4 levels were associated with a higher risk of severe ASD (odds ratio o = 1.4), and higher IL-1β were associated with mild or moderate ASD (odds ratio = 3.02) (167). Children with ASD had high levels of IL-6 and IL-8 during neonatal periods compared with controls (168). No differences were found in cytokine levels between children with developmental delay in the absence of ASD and controls (167, 168).
High levels of proinflammatory cytokines may persist beyond the neonatal period. Two-to 5-year-old children with ASD had high plasma levels of the proinflammatory cytokine IL-6 compared with age-matched typically developing controls and those with developmental disabilities other than ASD (169). Brains of ASD patients also had high cytokine levels, including IL-6 and the Th1 cytokine IFN-γ, whereas levels of Th2 cytokines (i.e., IL-4, IL-5, and IL-10) were not elevated in brains in ASD patients (170). An increase in serum IL-17A levels was also detected in 6- to 11-year-old ASD children (171). The phytohemagglutinin-induced stimulation of peripheral blood mononuclear cells resulted in the production of IL-17 but not Th2 cytokines IL-4 and IL-13 in 2- to 5-year-old ASD children, whereas cytokine levels at baseline were comparable between ASD children and controls (172). Plasma levels of IL-17 and IL-1β remained elevated in 7- to 15-year-old ASD individuals (173). Heavy cytokine burden may be related to a more severe ASD phenotype. Children with the regressive form of ASD exhibited a significant increase in IL-1β and IL-6 levels, whereas those without the regressive form did not (169). Most children who had high serum IL-17A levels had severe ASD, based on the Childhood Autism Rating Scale (171). Another study, however, did not find a correlation between the cytokine profile and clinical variables, including the severity of ASD, was not found (173). Altogether, exposure to IL-6 and IL-17, rather than direct effects of pathogens, may be important for MIA-induced ASD.
Modeling Maternal Immune Activation in Animals
To investigate the mechanisms of MIA-related ASD in disease models, two approaches have been utilized. One is to inoculate human pathogens, and the other is to administer proinflammatory compounds (Table 3).
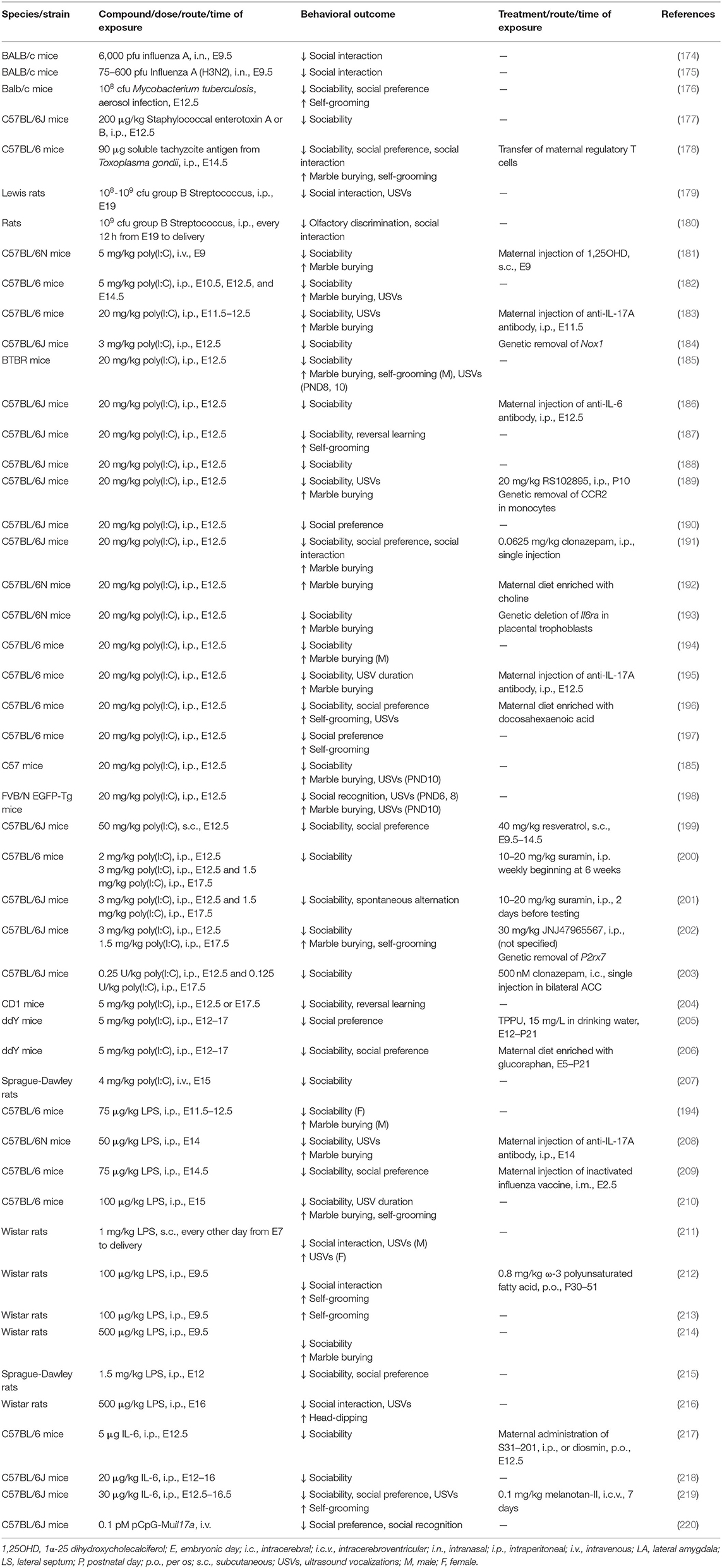
Table 3. Maternal immune activation models of ASD in rodents.
Pathogens that are administered in pregnant rodents range from viruses to bacteria and parasites, and their effects on ASD-related behavioral changes have been examined. Human influenza virus infection in pregnant mice on E9 reduced social interaction (174, 175). Offspring that were exposed to high virus titers exhibited lower 5-HT levels through an increase in metabolism and decrease in levels of oxytocin (175). An injection of group B Streptococcus, a major bacterial pathogen in pregnant women and neonates, impaired social behavior, decreased social interaction, and decreased USVs when inoculated in late gestation in rats (179, 180). These deficits were accompanied by white matter damage in the external capsule and corpus callosum (179, 180). Mice that were born to mothers that were infected with Mycobacterium tuberculosis on E12.5 exhibited deficits in approach to social novelty and social preference, and these effects were associated with high plasma IL-6 and IL-17A levels (176). A maternal injection of the soluble tachyzoite antigen of Toxoplasma gondii on E14.5 also elicited MIA and produced ASD-like behavior, such as impairments in social approach, an increase in self-grooming, and an increase in marble-burying behavior in offspring (178). Thus, maternal infection in mid- to late-gestation is considered to lead to ASD, regardless of the specific pathogen.
Given that different pathogens cause ASD-like behavioral alterations in rodents, common downstream mechanisms have been sought. Substances that induce MIA include the synthetic double-strand RNA polyinosine-polycytidylic acid (poly(I:C)), which mimics viral infection, lipopolysaccharide (LPS), which mimics bacterial infection, and ILs that are induced by inflammation, such as IL-6. Double-strand RNA is produced by most viruses during replication. A poly(I:C) injection causes the production of proinflammatory cytokines through Toll-like receptor 3 (221). A maternal injection of poly(I:C) on E12.5 promoted an ASD-like phenotype in offspring, including an increase in USVs, a decrease in sociability, and disorganized cortical cytoarchitecture (195). Offspring from LPS-injected mothers on E14 exhibited a decrease in USVs, an increase in marble burying behavior, and a decrease in interest in a novel mouse (208). These findings suggest that MIA models that use agents that mimic MIA contribute to revealing the detailed mechanisms of MIA-related ASD.
Infection or poly(I:C) induces the production of IL-6, which can cross the placenta and affect the fetal brain (222, 223). A maternal injection of IL-6 on E12.5 was sufficient to cause behavioral alterations of preference for social novelty in offspring. The co-administration of anti-IL-6 antibody ameliorated these alterations, but anti-IFN-γ antibody did not. Moreover, maternal poly(I:C) treatment did not disrupt social behavior in IL-6 knockout offspring (186). Parasitic MIA also resulted in deficient social recognition memory that was associated with IL-6 upregulation. IL-6 increased at both the mRNA and protein levels in the brain, and serum IL-6 levels were higher in response to Toxoplasma gondii antigen (178). Excessive maternal levels of IL-6 may thus play a role in triggering a cascade of events that lead to ASD in offspring.
Supporting the association between ASD and such cytokines as IL-6 and IL-17, experimental models of MIA-induced ASD have elucidated the role of IL-17 in the development of ASD. A poly(I:C) injection stimulated IL-17A production in dams, followed by higher IL-17A mRNA expression in the fetal brain, an ASD-like phenotype, and cerebral pathological changes in offspring (195). An injection of LPS elevated IL-6 and IL-17A levels only in pregnant mice but exerted ASD-like behaviors in offspring, including a decrease in USVs and lower interest in a novel mouse (208). Similar increases in IL-6 and IL-17A levels were observed in a mouse model of Mycobacterium tuberculosis infection, accompanied by behavioral alterations in offspring and increases in the mRNA expression of NRXN1 and NLGN1 (176).
Further studies revealed the critical role of fetal exposure to IL-17A but not IL-6 in MIA-associated ASD. The administration of IL-6 in pregnant mice on E12.5 altered social behavior in offspring (186), but an intraventricular injection of IL-6 into the fetal brain on E14.5 did not produce an ASD-like phenotype (195). Instead, an intraventricular injection of IL-17A in the fetal brain on E14.5 caused social deficits and cortical disorganization in wildtype pups (195). The MIA-associated phenotype was blocked by abolishing IL-17A secretion by deletion of the RORγt gene from Th17 cells (195), maternal pretreatment with an anti-IL-17A antibody (208), and deletion of the IL-17Ra gene in pups (195). The mechanisms of elevations of IL-17A that lead to ASD may involve alterations of the function of regulatory T (Treg) cells. Mice that were exposed to Toxoplasma gondii-induced MIA exhibited higher percentages of Th1 and Th17 cells but a lower percentage of Treg cells. The adoptive cell transfer of Treg cells from MIA mothers at 8 weeks of age largely reversed the abnormal behavioral phenotypes of offspring as early as at 9 weeks (178).
Microglial activation following MIA may be also be involved in ASD development, but few studies have investigated direct effect of alteration of microglial function on ASD-related behavioral deficits in offspring. Morphological changes suggest that microglial activation is not fully consistent in mouse models. Daily injection of IL-6 from E12.5 to delivery resulted in morphological changes of microglia, though not associated with ASD-like behavioral deficits (224). Poly(I:C) administration on E12 and E15 impaired direct social interaction and increased velocity of microglial process motility, but morphological changes was not observed (189). Another study showed that single injection of poly(I:C) effectively produced an ASD-like decrease in sociability and increase in stereotypy, but the superimposition of postnatal hypoxia/ ischemia (HI) insult was needed to alter microglial morphology (225). Microglial infiltration in the brain may be difficult to detect. In two models that produced ASD-like behavioral deficits following MIA [i.e., mice that were exposed to two doses of poly(I:C) on E12.5 and E17.5 (211) and rats that were exposed to daily injections of LPS from E7 to delivery (210)], microglial infiltration in the brain was not detected. In Chen et al., HI superimposition on MIA induced microglial infiltration in the hippocampus (225).
Despite the aforementioned limited histopathological observations of microglia, synapses and neurons that are exposed to MIA exhibit alterations that suggest microglial activation. An increase in spine density that was attributable to a reduction of spine pruning was found in the dentate gyrus in mice given LPS on E15 (226) and in the hippocampus in mice that were given 2 doses of poly(I:C) on E12.5 and E17.5 (211). Subchronic IL-6 exposure from E12.5 to delivery in mice delayed the migration of GABAergic progenitors (224). Parvalbumin-positive neurons in the hippocampus were entrapped in perineuronal nets following MIA alone (225), which may impair synaptic plasticity. A single injection of poly(I:C) on E12 reduced parvalbumin-positive cells and impaired GABAergic signaling in the dentate gyrus, which was associated with social withdrawal and deficient spatial memory (227). In this model, spatial memory and abnormal histology were restored by minocycline, which inhibits microglial activity (227). In the combined MIA/HI mouse model of ASD, the pharmacological inhibition of monocyte infiltration after HI insult prevented ASD-like behaviors (225). Interestingly, MIA-associated behaviors, including ASD-like social impairments, were recovered by exercise in adulthood, which was correlated with the normalization of synapse density and suggested intact microglial function in mice (211). Microglial activation and infiltration in the MIA-exposed brain may link high cytokine levels and the development of ASD, and MIA-induced ASD in offspring could be ameliorated by prenatal or postnatal immunological interventions.
Hypertensive Disorders of Pregnancy
Hypertensive disorders of pregnancy (HDP) comprise a collection of hypertensions in pregnant women. They are divided into (a) hypertension that arises de novo at or after 20 weeks of gestation and (b) hypertension that is known before pregnancy or present in the first 20 weeks of gestation (228). Hypertensive disorders of pregnancy that are accompanied by proteinuria or other maternal organ dysfunction, such as acute kidney injury, are referred to as pre-eclampsia. HDPs occur in ~5–8% of all pregnancies (229). Epidemiological studies reported an association between HDP and ASD. Both maternal chronic hypertension before pregnancy and gestational hypertension are associated with an ~40% increase in the odds of ASD in offspring (23, 230). Pre-eclampsia also increases the risk of ASD (231), but the presence of organ dysfunction in pre-eclampsia does not appear to be an additional risk factor (23, 230). Research on HDP has been performed by generating animal models. The central focus of HDP research has been on maternal pathophysiology and fetal growth failure. In contrast, there are sparse findings on how HDP affects fetal brain development and results in ASD-like social deficits in offspring.
Reduced Uteroplacental Pressure
Animal models of HDP were first established in rabbits, dogs, and monkeys by constricting the terminal aorta. This method is called reduced uteroplacental pressure (RUPP) (232). Later, this method was applied to rats and mice (233). The procedure was also improved so that uterine arteries were occluded instead of the abdominal aorta, resulting in the avoidance of hindlimb paraplegia in classic RUPP models (234). In addition to main manifestations of HDP, such as maternal hypertension, proteinuria, and fetal growth restriction (232–234), fetal brains also exhibited changes in response to HDP exposure, but their direct relevance to ASD remains unclear. Alteration of the brains of guinea pigs born to RUPP mothers included smaller volumes of the basal ganglia and impairments in prepulse inhibition at 12 weeks (235). Pregnant rats that underwent RUPP also exhibited immunological changes that are similar to MIA, including high levels of IL-6 and IL-17A that are produced by CD4+ T cells (236) and an increase in the number of Th17 cells (237). IL-17 recombinant receptor C was used to suppress Th17 cells and resulted in blunted hypertension and the recovery of pup and placenta weight (237). Placental hypoperfusion in animal models of RUPP-induced HDP may mediate morphological and functional changes that are related to MIA-induced ASD in offspring.
Angiotensin II
Numerous clinical observations have indicated a relationship between HDP and angiotensin (AT). Patients with HDP exhibited greater vascular reactivity to AT II (238), and serum from pregnant patients with HDP but not those with essential hypertension contained AT1 autoantibodies (AAs) that could stimulate AT II receptor 1 (239). AT1-autoantibodies that were isolated from women with HDP reproduce key features of HDP in mice (240), and their pharmacological blockade normalized maternal blood pressure during pregnancy (241). Exposure to LPS during pregnancy gave rise to features that are relevant to HDP in rats offspring, which was prevented by treatment with the AT II receptor inhibitor losartan (242). Infusions of AT II in pregnant mice stimulated the production of soluble fms-like tyrosine kinase-1, a circulating antagonist of vascular endothelial growth factor and placental growth factor (243), and IL-6 in the placenta (244). In summary, AT II infusion is sufficient to induce HDP and IL-6 production, and AT II inhibition is sufficient to prevent LPS-induced HDP, suggesting that the stimulation of AT II signaling underlies MIA and HDP. Thus, excessive AT II activity in HDP might give rise to ASD through an increase in cytokine production as observed in MIA. Further research is required to elucidate the neurological phenotype that is induced by AT II in these models.
Vasopressin
In contrast to the AT-related HDP model, pregnant women with preeclampsia are reported to exhibit lower activity of the renin-angiotensin system (245). Women with HDP exhibited higher levels of copeptin, a pro-segment of arginine vasopressin (AVP) (246), raising the hypothesis that high AVP levels during pregnancy play a role in the pathogenesis of HDP. Infusions of AVP during pregnancy resulted in an HDP phenotype in mice (247). The simultaneous inhibition of AVP receptors 1 and 2 abolished the AVP-induced elevation of blood pressure, whereas blocking each receptor alone resulted in the insufficient normalization of blood pressure in mid- and late-pregnancy (247). Infusions of AVP during pregnancy increased IL-17 levels in maternal plasma and the placenta (248), suggesting that IL-17 may also affect fetal brain development following AVP loading. To date, however, only one study has reported neurological and behavioral consequence of AVP-infused HDP in offspring (249). Both male and female offspring that were exposed to AVP on P7 had a smaller neocortex, but caudate-putamen volume decreased in males only and increased relative to the neocortex in females. At the behavioral level, male offspring that were exposed to AVP exhibited an increase in anxiety-like behavior in the elevated plus maze test, but females did not. Social behavior was analyzed in males only, in which an increase in sociability was observed in AVP-exposed males compared with controls (249). Further research on ASD-like phenotypes in the AVP infusion model of HDP is necessary to determine whether AVP-induced HDP recapitulates the higher risk of ASD in humans.
Serotonin Metabolism in the Placenta
Women with HDP have high 5-HT levels in blood (250) and the placenta (251). Such a hyperserotonemic state is attributable to a reduction of activity of monoamine oxidase A and not a defect in 5-HT transport in the placenta (251, 252). Serotonin levels correlated with the severity of maternal hypertension (251), suggesting a causal role for hyperserotonemia in HDP. Serotonin exerts a contraction effect on placental vascular smooth muscle, which may be a primary way 5-HT plays a role in HDP (253, 254). Chorionic arteries from HDP women exhibited a lower contraction response to the in vitro perfusion of 5-HT (255), presumably because of chronic exposure to excessive 5-HT during pregnancy. Ketanserin, a 5-HT2A receptor antagonist, attenuated arterial contraction that was stimulated by 5-HT in both normal and HDP samples (255). Moreover, 5-HT administration in normal pregnant rats produced HDP-like alterations, such as a reduction of growth of the placenta and fetus (256). Continuous hyperserotonemia in Sert−/− mice, which was 2-fold higher than controls, produced smaller placentas, marked cell death, and necrotic lesions that were similar to placentas from HDP women (257). Undetectable 5-HT levels in Tph1−/− mice also resulted in smaller placentas and cell death, although these effects were much milder than in Sert−/− mice (257). These similar and contrasting findings from hyperserotonemic and hyposerotonemic models suggest a more deleterious effect of hyperserotonemia on the placenta and possibly on the fetus during pregnancy. As discussed in Section SSRIs above, the relationship between high 5-HT levels and ASD has been demonstrated in different animal models. Investigations of these models in view of HDP may improve our understanding of how HDP causes ASD in offspring.
Advanced Parental Age
Advanced parental age does not appear to accompany the visible effects that are discussed above, such as the effects of medication use and the inflammatory response during pregnancy. A detailed population study sought to identify prenatal and perinatal risks of ASD and found higher parental ages of ASD children compared with non-ASD children (258), prompting further studies of the association between parental age and the risk of ASD.
Effects of Paternal and Maternal Age and Their Interaction on ASD
The risk of higher paternal age for ASD have been consistently reported. In a study in Israel, the odds ratio (OR) of ASD in offspring marginally increased for paternal ages of 30–39 years and significantly increased (OR = 5.75) for paternal ages of 40 years and older when adjusted for maternal age and compared with paternal ages of <30 years (259). A cohort study in Sweden also reported an association between paternal age and the risk of ASD, which began to increase at the paternal age of 30 years (260). The authors then confirmed this association in a meta-analysis of Western and non-Western cohorts, which revealed a dosage effect on the OR of ASD in offspring (1.22 for paternal age 30–39 years, 1.58 for paternal age 40–49 years, and 2.66 for paternal age 50 years and older) (260). A similar linear increase in the risk of ASD according to paternal age was also found in a large-scale cohort study (261). A recent meta-analysis showed that high paternal age was associated with a 55% higher risk of ASD (262).
Maternal age also appears to affect the risk of ASD in offspring, though in a different manner compared with paternal age. In a population study, logistic regression analysis was conducted that included maternal but not paternal age as regression coefficients. This study found that having children with ASD was related to higher maternal age (258). However, the effect of advanced maternal age was marginal when adjusted for parental age (259). Later studies found that the risk of ASD in offspring increased in mothers who were 30 of age or older (261) and 35 years of age or older (263). When the ages of both mothers and fathers were adjusted, the risk of ASD in offspring began to increase in mothers who were 30–39 years old and evident in mothers who were 40 years old (264).
In contrast to consistent findings that advanced parental age elevates the risk of ASD in offspring, remaining to be clarified is whether lower parental age heightens the risk of ASD in children. A cohort study in Sweden found that the risk of ASD for younger mothers was comparable to a reference group (29 years old), whereas children of younger fathers had a lower risk of ASD (261). Another cohort study of European and non-European countries reported a higher risk of ASD in children of mothers who were younger than 20 years old, whereas no association was observed for younger fathers (264). According to a meta-analysis from North America, Europe, Asia, and Oceania, the lowest paternal and maternal age categories were associated with a lower risk of ASD (262). Lower parental age might have a protective effect against the risk of ASD in offspring, which should be investigated further.
Remaining to be determined are which maternal and paternal ages have the greatest impact on the risk of ASD. A Swedish population study reported a higher OR of ASD, particularly ASD with intellectual disability, that was associated with advanced maternal age (2.04 for mothers aged 40–45 years and 1.18 for fathers aged 40–44 years) (261). Another study reported a higher risk of ASD for fathers aged 40–49 years (OR = 1.52) compared with mothers of the same age group (OR = 1.15) (264). A recent meta-analysis found a comparable risk of ASD for the highest age group (OR = 1.41 for mothers and 1.55 for fathers) (262). Further research is needed to distinguish the effects of maternal and paternal ages.
In summary, both advanced maternal and paternal age are likely related to a higher risk of ASD. Data have been insufficient or inconsistent with regard to differences in the impact of maternal and paternal ages and their interaction on ASD.
Modeling Advanced Parental Age in Rodents
The influence of advanced parental age has been investigated in rodent models, mainly for older fathers. Paternal age did not affect the general health of pups, such as the number of pups, body weight, and early mortality (265). Mice from older fathers exhibited alterations of behavioral phenotypes. These offspring took longer to attain the righting reflex, exhibited a decrease in spontaneous motor activity, deficient memory retention in the passive avoidance test (265), and decreases in ambulatory distance and prepulse inhibition (266). ASD-related behavior was also found in advanced paternal age models. Male mice from older fathers exhibited impairments in social preference (267), the deficient discrimination of social novelty (267, 268), an increase in self-grooming (268), and increases in or alterations of USVs that are suggestive of ASD (268, 269). The magnitude of social deficits correlated with paternal age. Lower social interaction was observed in offspring of 40-week-old fathers and further decreased in offspring of 48-week-old fathers (270). Autism spectrum disorder-like social deficits that are caused by advanced paternal age may transmit to the second generation. When both parents were born to fathers that were 12 months of age or older, the offspring exhibited a decrease in sociability, an increase in repetitive behavior, and anxiety-like behavior (267, 268).
Little is understood about what accompanies behavioral changes that are observed in offspring of older fathers. Advanced paternal age may produce morphological changes in the brain in offspring, but the findings are inconsistent. Male mouse offspring of 12- to 18-month-old fathers had a higher rostral cortical volume and lower lateral ventricle volume, but social interaction was unaffected (271). A recent study that used male mice that were older than 12 months of age found that their offspring emitted abnormal USVs. This finding was associated with a smaller cortical thickness of layer 6 of the primary motor cortex, suggesting that alterations in the motor cortex impair vocal communication (269). A possible mechanism of paternal age-induced ASD may be related to alterations of sperm DNA methylation. Older fathers and their offspring shared hypomethylation in regions that flank CpG island promotors, which was not found in younger fathers and their offspring (266). Whole-genome DNA methylome analyses of sperm identified hypomethylated genomic regions that were enriched in RE1-silencing transcription factor/neuron-restrictive silencer factor binding motifs (265). The treatment of young male mice with the DNA-demethylating drug T5-Aza reproduced DNA hypomethylation in sperm, and their offspring exhibited alterations of USV patterns (269). Alterations of DNA methylation patterns in sperm may contribute to the effect of advanced paternal age on ASD in offspring.
Analyzing advanced maternal age in animal models is challenging because aged females may exhibit changes in nursing behavior. For example, 30-week-old female mice exhibited enhanced nest-building performance during pregnancy and lactation (272). Cesarean section and cross-fostering were applied to avoid confounding effects of alterations of nursing behavior in 15- to 18-month-old female mice, but their pups still exhibited more USVs on P8 and an increase in anxiety-related behavior in the elevated plus maze (273). Associated with the behavioral changes, pups that were born to aged female mice exhibited high hippocampal mRNA expression of genes that are related to ASD, such as Ada (which influences ASD development) and Egr2 (which influences ASD severity). Several other genes were enriched in the Gene Ontology analysis, including “protein folding” and “protein post-translational modification” (273). Younger maternal age in mice (32–35 weeks) also affected cognitive functions in offspring, including impairments in learning in the passive avoidance test, spatial memory in the Morris water maze test, and memory in the novel object recognition test (274). Interestingly, a marked decrease in expression of the vitamin D receptor gene was found in the placenta in aged female mice and their offspring (274). When these aged female mice received vitamin D supplementation before pregnancy, their offspring exhibited intact learning and memory (275). Advanced maternal age likely affects cognitive function in offspring, and possible ASD-like social deficits in these models should also be investigated.
Concluding Remarks
We discussed the various contributions of environmental risk factors to the development of ASD. Prenatal exposure to VPA and MIA have been extensively investigated in epidemiological and biological studies. The findings indicate the critical role of these risk factors in producing ASD. Hypertensive disorders of pregnancy and advanced maternal age elevate the risk of ASD, but remaining unclear is how these risk factors give rise to ASD-like cognitive dysfunction. The association between maternal SSRI use and ASD in offspring is inconclusive, but rodent models that show alterations of 5-HT metabolism provide a plausible rationale for this association. Future research is expected to develop therapeutic interventions for ASD that target environmental risk factors.
Author Contributions
AS wrote the manuscript. HK-M, MT, YK, and KI critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the Japan Society for the Promotion of Science (JSPS) KAKENHI (no. 21H03028).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RK declared a shared affiliation with two of the authors, MT and YK, to the handling editor at the time of review.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Mr. Michael Arends for English proofreading.
References
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Publishing (2013).
2. Baxter AJ, Brugha TS, Erskine HE, Scheurer RW, Vos T, Scott JG. The epidemiology and global burden of autism spectrum disorders. Psychol Med. (2015) 45:601–13. doi: 10.1017/S003329171400172X
3. Qiu S, Lu Y, Li Y, Shi J, Cui H, Gu Y, et al. Prevalence of autism spectrum disorder in Asia: a systematic review and meta-analysis. Psychiatry Res. (2020) 284:112679. doi: 10.1016/j.psychres.2019.112679
4. Xu G, Strathearn L, Liu B, Bao W. Prevalence of autism spectrum disorder among US children and adolescents, 2014-2016. JAMA. (2018) 319:81–2. doi: 10.1001/jama.2017.17812
5. Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: a decade of new twin studies. Am J Med Genet B Neuropsychiatr Genet. (2011) 156B:255–74. doi: 10.1002/ajmg.b.31159
6. Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. (2014) 46:881–5. doi: 10.1038/ng.3039
7. SFARI Gene. (2021). Available online at: https://gene.sfari.org/database/human-gene/ (accessed November 22, 2021).
8. Basu SN, Kollu R, Banerjee-Basu S. AutDB: a gene reference resource for autism research. Nucleic Acids Res. (2009) 37:D832–6. doi: 10.1093/nar/gkn835
9. Vorstman JAS, Parr JR, Moreno-De-Luca D, Anney RJL, Nurnberger JI Jr, Hallmayer JF. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet. (2017) 18:362–76. doi: 10.1038/nrg.2017.4
10. Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. (2013) 77:259–73. doi: 10.1016/j.neuron.2012.11.002
11. Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. (2014) 94:677–94. doi: 10.1016/j.ajhg.2014.03.018
12. Leppa VM, Kravitz SN, Martin CL, Andrieux J, Le Caignec C, Martin-Coignard D, et al. Rare inherited and de novo CNVs reveal complex contributions to ASD risk in multiplex families. Am J Hum Genet. (2016) 99:540–54. doi: 10.1016/j.ajhg.2016.06.036
13. Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. (2012) 488:647–51. doi: 10.1038/nature11310
14. Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, et al. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun. (2012) 3:1292. doi: 10.1038/ncomms2295
15. Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, et al. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. (2009) 29:1773–83. doi: 10.1523/JNEUROSCI.5685-08.2009
16. Culotta L, Penzes P. Exploring the mechanisms underlying excitation/inhibition imbalance in human iPSC-derived models of ASD. Mol Autism. (2020) 11:32. doi: 10.1186/s13229-020-00339-0
17. Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. (2020) 180:568–84.e23. doi: 10.1016/j.cell.2019.12.036
18. Elsabbagh M, Divan G, Koh YJ, Kim YS, Kauchali S, Marcín C, et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. (2012) 5:160–79. doi: 10.1002/aur.239
19. Wallace S, Fein D, Rosanoff M, Dawson G, Hossain S, Brennan L, et al. A global public health strategy for autism spectrum disorders. Autism Res. (2012) 5:211–7. doi: 10.1002/aur.1236
20. Fombonne E. Editorial: the rising prevalence of autism. J Child Psychol Psychiatry. (2018) 59:717–20. doi: 10.1111/jcpp.12941
21. Modabbernia A, Velthorst E, Reichenberg A. Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol Autism. (2017) 8:13. doi: 10.1186/s13229-017-0121-4
22. Chaste P, Leboyer M. Autism risk factors: genes, environment, and gene-environment interactions. Dialogues Clin Neurosci. (2012) 14:281–92. doi: 10.31887/DCNS.2012.14.3/pchaste
23. Kim JY, Son MJ, Son CY, Radua J, Eisenhut M, Gressier F, et al. Environmental risk factors and biomarkers for autism spectrum disorder: an umbrella review of the evidence. Lancet Psychiatry. (2019) 6:590–600. doi: 10.1016/S2215-0366(19)30181-6
24. Leavey A, Zwaigenbaum L, Heavner K, Burstyn I. Gestational age at birth and risk of autism spectrum disorders in Alberta, Canada. J Pediatr. (2013) 162:361–8. doi: 10.1016/j.jpeds.2012.07.040
25. Persson M, Opdahl S, Risnes K, Gross R, Kajantie E, Reichenberg A, et al. Gestational age and the risk of autism spectrum disorder in Sweden, Finland, and Norway: a cohort study. PLoS Med. (2020) 17:e1003207. doi: 10.1371/journal.pmed.1003207
26. Abel KM, Dalman C, Svensson AC, Susser E, Dal H, Idring S, et al. Deviance in fetal growth and risk of autism spectrum disorder. Am J Psychiatry. (2013) 170:391–8. doi: 10.1176/appi.ajp.2012.12040543
27. Korzeniewski SJ, Allred EN, Joseph RM, Heeren T, Kuban KCK, O'Shea TM, et al. Neurodevelopment at age 10 years of children born <28 weeks with fetal growth restriction. Pediatrics. (2017) 140:e20170697. doi: 10.1542/peds.2017-0697
28. Tomson T, Battino D, Perucca E. Valproic acid after five decades of use in epilepsy: time to reconsider the indications of a time-honoured drug. Lancet Neurol. (2016) 15:210–28. doi: 10.1016/S1474-4422(15)00314-2
29. Linde M, Mulleners WM, Chronicle EP, McCrory DC. Valproate (valproic acid or sodium valproate or a combination of the two) for the prophylaxis of episodic migraine in adults. Cochrane Database Syst Rev. (2013) 6:CD010611. doi: 10.1002/14651858.CD010611
30. McIntyre RS, Berk M, Brietzke E, Goldstein BI, López-Jaramillo C, Kessing LV, et al. Bipolar disorders. Lancet. (2020) 396:1841–56. doi: 10.1016/S0140-6736(20)31544-0
31. Kozma C. Valproic acid embryopathy: report of two siblings with further expansion of the phenotypic abnormalities and a review of the literature. Am J Med Genet. (2001) 98:168–75. doi: 10.1002/1096-8628(20010115)98:2<168::AID-AJMG1026>3.0.CO;2-O
32. Jentink J, Loane MA, Dolk H, Barisic I, Garne E, Morris JK, et al. Valproic acid monotherapy in pregnancy and major congenital malformations. N Engl J Med. (2010) 362:2185–93. doi: 10.1056/NEJMoa0907328
33. Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, et al. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA. (2013) 309:1696–703. doi: 10.1001/jama.2013.2270
34. Baker GA, Bromley RL, Briggs M, Cheyne CP, Cohen MJ, García-Fiñana M, et al. IQ at 6 years after in utero exposure to antiepileptic drugs: a controlled cohort study. Neurology. (2015) 84:382–90. doi: 10.1212/WNL.0000000000001182
35. Tomson T, Marson A, Boon P, Canevini MP, Covanis A, Gaily E, et al. Valproate in the treatment of epilepsy in girls and women of childbearing potential. Epilepsia. (2015) 56:1006–19. doi: 10.1111/epi.13021
36. Schneider T, Przewtocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal models of autism. Neuropsychopharmacology. (2005) 30:80–9. doi: 10.1038/sj.npp.1300518
37. Kim JW, Park K, Kang RJ, Gonzales ELT, Kim DG, Oh HA, et al. Pharmacological modulation of AMPA receptor rescues social impairments in animal models of autism. Neuropsychopharmacology. (2019) 44:314–23. doi: 10.1038/s41386-018-0098-5
38. Kim JW, Seung H, Kwon KJ, Ko MJ, Lee EJ, Oh HA, et al. Subchronic treatment of donepezil rescues impaired social, hyperactive, and stereotypic behavior in valproic acid-induced animal model of autism. PLoS ONE. (2014) 9:e104927. doi: 10.1371/journal.pone.0104927
39. Wang K, Li N, Xu M, Huang M, Huang F. Glyoxalase 1 inhibitor alleviates autism-like phenotype in a prenatal valproic acid-induced mouse model. ACS Chem Neurosci. (2020) 11:3786–92. doi: 10.1021/acschemneuro.0c00482
40. Lieberman OJ, Cartocci V, Pigulevskiy I, Molinari M, Carbonell J, Broseta MB, et al. mTOR suppresses macroautophagy during striatal postnatal development and is hyperactive in mouse models of autism spectrum disorders. Front Cell Neurosci. (2020) 14:70. doi: 10.3389/fncel.2020.00070
41. Kim KC, Lee DK, Go HS, Kim P, Choi CS, Kim JW, et al. Pax6-dependent cortical glutamatergic neuronal differentiation regulates autism-like behavior in prenatally valproic acid-exposed rat offspring. Mol Neurobiol. (2014) 49:512–28. doi: 10.1007/s12035-013-8535-2
42. Kim JW, Seung H, Kim KC, Gonzales ELT, Oh HA, Yang SM, et al. Agmatine rescues autistic behaviors in the valproic acid-induced animal model of autism. Neuropharmacology. (2017) 113:71–81. doi: 10.1016/j.neuropharm.2016.09.014
43. Lin TC, Lo YC, Lin HC, Li SJ, Lin SH, Wu HF, et al. MR imaging central thalamic deep brain stimulation restored autistic-like social deficits in the rat. Brain Stimul. (2019) 12:1410–20. doi: 10.1016/j.brs.2019.07.004
44. Yang JQ, Yang CH, Yin BQ. Combined the GABA-A and GABA-B receptor agonists attenuates autistic behaviors in a prenatal valproic acid-induced mouse model of autism. Behav Brain Res. (2021) 403:113094. doi: 10.1016/j.bbr.2020.113094
45. Eissa N, Azimullah S, Jayaprakash P, Jayaraj RL, Reiner D, Ojha SK, et al. The dual-active histamine H3 receptor antagonist and acetylcholine esterase inhibitor E100 alleviates autistic-like behaviors and oxidative stress in valproic acid induced autism in mice. Int J Mol Sci. (2020) 21:3996. doi: 10.3390/ijms21113996
46. Hara Y, Ago Y, Higuchi M, Hasebe S, Nakazawa T, Hashimoto H, et al. Oxytocin attenuates deficits in social interaction but not recognition memory in a prenatal valproic acid-induced mouse model of autism. Horm Behav. (2017) 96:130–6. doi: 10.1016/j.yhbeh.2017.09.013
47. Hara Y, Ago Y, Taruta A, Hasebe S, Kawase H, Tanabe W, et al. Risperidone and aripiprazole alleviate prenatal valproic acid-induced abnormalities in behaviors and dendritic spine density in mice. Psychopharmacology. (2017) 234:3217–28. doi: 10.1007/s00213-017-4703-9
48. Huang F, Chen X, Jiang X, Niu J, Cui C, Chen Z, et al. Betaine ameliorates prenatal valproic-acid-induced autism-like behavioral abnormalities in mice by promoting homocysteine metabolism. Psychiatry Clin Neurosci. (2019) 73:317–22. doi: 10.1111/pcn.12833
49. Eissa N, Jayaprakash P, Azimullah S, Ojha SK, Al-Houqani M, Jalal FY, et al. The histamine H3R antagonist DL77 attenuates autistic behaviors in a prenatal valproic acid-induced mouse model of autism. Sci Rep. (2018) 8:13077. doi: 10.1038/s41598-018-31385-7
50. Horiai M, Otsuka A, Hidema S, Hiraoka Y, Hayashi R, Miyazaki S, et al. Targeting oxytocin receptor (Oxtr)-expressing neurons in the lateral septum to restore social novelty in autism spectrum disorder mouse models. Sci Rep. (2020) 10:22173. doi: 10.1038/s41598-020-79109-0
51. Pizzamiglio L, Focchi E, Cambria C, Ponzoni L, Ferrara S, Bifari F, et al. The DNA repair protein ATM as a target in autism spectrum disorder. JCI Insight. (2021) 6:e133654. doi: 10.1172/jci.insight.133654
52. Chatterjee M, Singh P, Xu J, Lombroso PJ, Kurup PK. Inhibition of striatal-enriched protein tyrosine phosphatase (STEP) activity reverses behavioral deficits in a rodent model of autism. Behav Brain Res. (2020) 391:112713. doi: 10.1016/j.bbr.2020.112713
53. Kotajima-Murakami H, Kobayashi T, Kashii H, Sato A, Hagino Y, Tanaka M, et al. Effects of rapamycin on social interaction deficits and gene expression in mice exposed to valproic acid in utero. Mol Brain. (2019) 12:3. doi: 10.1186/s13041-018-0423-2
54. Qin L, Dai X, Yin Y. Valproic acid exposure sequentially activates Wnt and mTOR pathways in rats. Mol Cell Neurosci. (2016) 75:27–35. doi: 10.1016/j.mcn.2016.06.004
55. Wu J, Dai YC, Lan XY, Zhang HF, Bai SZ, Hu Y, et al. Postnatal AVP treatments prevent social deficit in adolescence of valproic acid-induced rat autism model. Peptides. (2021) 137:170493. doi: 10.1016/j.peptides.2021.170493
56. Wu HF, Chen PS, Chen YJ, Lee CW, Chen IT, Lin HC. Alleviation of N-methyl-D-aspartate receptor-dependent long-term depression via regulation of the glycogen synthase kinase-3β pathway in the amygdala of a valproic acid-induced animal model of autism. Mol Neurobiol. (2017) 54:5264–76. doi: 10.1007/s12035-016-0074-1
57. Wu HF, Chen YJ, Chu MC, Hsu YT, Lu TY, Chen IT, et al. Brain stimulation modified autism-like deficits via the serotonin system in a valproic acid-induced rat model. Int J Mol Sci. (2018) 19:2840. doi: 10.3390/ijms19092840
58. Ishola IO, Balogun AO, Adeyemi OO. Novel potential of metformin on valproic acid-induced autism spectrum disorder in rats: involvement of antioxidant defence system. Fundam Clin Pharmacol. (2020) 34:650–61. doi: 10.1111/fcp.12567
59. Scheggi S, Guzzi F, Braccagni G, De Montis MG, Parenti M, Gambarana C. Targeting PPARα in the rat valproic acid model of autism: focus on social motivational impairment and sex-related differences. Mol Autism. (2020) 11:62. doi: 10.1186/s13229-020-00358-x
60. Wu HF, Lu TY, Chu MC, Chen PS, Lee CW, Lin HC. Targeting the inhibition of fatty acid amide hydrolase ameliorate the endocannabinoid-mediated synaptic dysfunction in a valproic acid-induced rat model of autism. Neuropharmacology. (2020) 162:107736. doi: 10.1016/j.neuropharm.2019.107736
61. Servadio M, Melancia F, Manduca A, di Masi A, Schiavi S, Cartocci V, et al. Targeting anandamide metabolism rescues core and associated autistic-like symptoms in rats prenatally exposed to valproic acid. Transl Psychiatry. (2016) 6:e902. doi: 10.1038/tp.2016.182
62. Melancia F, Schiavi S, Servadio M, Cartocci V, Campolongo P, Palmery M, et al. Sex-specific autistic endophenotypes induced by prenatal exposure to valproic acid involve anandamide signalling. Br J Pharmacol. (2018) 175:3699–712. doi: 10.1111/bph.14435
63. Wu HF, Chen PS, Hsu YT, Lee CW, Wang TF, Chen YJ, et al. D-cycloserine ameliorates autism-like deficits by removing GluA2-containing AMPA receptors in a valproic acid-induced rat model. Mol Neurobiol. (2018) 55:4811–24. doi: 10.1007/s12035-017-0685-1
64. Mirza R, Sharma B. Benefits of fenofibrate in prenatal valproic acid-induced autism spectrum disorder related phenotype in rats. Brain Res Bull. (2019) 147:36–46. doi: 10.1016/j.brainresbull.2019.02.003
65. Mirza R, Sharma B. Beneficial effects of pioglitazone, a selective peroxisome proliferator-activated receptor-γ agonist in prenatal valproic acid-induced behavioral and biochemical autistic like features in Wistar rats. Int J Dev Neurosci. (2019) 76:6–16. doi: 10.1016/j.ijdevneu.2019.05.006
66. Zamberletti E, Gabaglio M, Woolley-Roberts M, Bingham S, Rubino T, Parolaro D. Cannabidivarin treatment ameliorates autism-like behaviors and restores hippocampal endocannabinoid system and glia alterations induced by prenatal valproic acid exposure in rats. Front Cell Neurosci. (2019) 13:367. doi: 10.3389/fncel.2019.00367
67. Khodaverdi M, Rahdar M, Davoudi S, Hajisoltani R, Tavassoli Z, Ghasemi Z, et al. 5-HT7 receptor activation rescues impaired synaptic plasticity in an autistic-like rat model induced by prenatal VPA exposure. Neurobiol Learn Mem. (2021) 183:107462. doi: 10.1016/j.nlm.2021.107462
68. Luhach K, Kulkarni GT, Singh VP, Sharma B. Vinpocetine amended prenatal valproic acid induced features of ASD possibly by altering markers of neuronal function, inflammation, and oxidative stress. Autism Res. (2021) 14:2270–86. doi: 10.1002/aur.2597
69. Luhach K, Kulkarni GT, Singh VP, Sharma B. Attenuation of neurobehavioural abnormalities by papaverine in prenatal valproic acid rat model of ASD. Eur J Pharmacol. (2021) 890:173663. doi: 10.1016/j.ejphar.2020.173663
70. Luhach K, Kulkarni GT, Singh VP, Sharma B. Cilostazol attenuated prenatal valproic acid-induced behavioural and biochemical deficits in a rat model of autism spectrum disorder. J Pharm Pharmacol. (2021) 73:1460–9. doi: 10.1093/jpp/rgab115
71. Kerr DM, Gilmartin A, Roche M. Pharmacological inhibition of fatty acid amide hydrolase attenuates social behavioural deficits in male rats prenatally exposed to valproic acid. Pharmacol Res. (2016) 113:228–35. doi: 10.1016/j.phrs.2016.08.033
72. Cuevas-Olguin R, Roychowdhury S, Banerjee A, Garcia-Oscos F, Esquivel-Rendon E, Bringas ME, et al. Cerebrolysin prevents deficits in social behavior, repetitive conduct, and synaptic inhibition in a rat model of autism. J Neurosci Res. (2017) 95:2456–68. doi: 10.1002/jnr.24072
73. Matsuo K, Yabuki Y, Fukunaga K. 5-Aminolevulinic acid inhibits oxidative stress and ameliorates autistic-like behaviors in prenatal valproic acid-exposed rats. Neuropharmacology. (2020) 168:107975. doi: 10.1016/j.neuropharm.2020.107975
74. Liu H, Tan M, Cheng B, Wang S, Xiao L, Zhu J, et al. Valproic acid induces autism-like synaptic and behavioral deficits by disrupting histone acetylation of prefrontal cortex ALDH1A1 in rats. Front Neurosci. (2021) 15:641284. doi: 10.3389/fnins.2021.641284
75. Schneider T, Turczak J, Przewłocki R. Environmental enrichment reverses behavioral alterations in rats prenatally exposed to valproic acid: issues for a therapeutic approach in autism. Neuropsychopharmacology. (2006) 31:36–46. doi: 10.1038/sj.npp.1300767
76. Du L, Zhao G, Duan Z, Li F. Behavioral improvements in a valproic acid rat model of autism following vitamin D supplementation. Psychiatry Res. (2017) 253:28–32. doi: 10.1016/j.psychres.2017.03.003
77. Wu H, Wang X, Gao J, Liang S, Hao Y, Sun C, et al. Fingolimod (FTY720) attenuates social deficits, learning and memory impairments, neuronal loss and neuroinflammation in the rat model of autism. Life Sci. (2017) 173:43–54. doi: 10.1016/j.lfs.2017.01.012
78. Zhang J, Liu LM, Ni JF. Rapamycin modulated brain-derived neurotrophic factor and B-cell lymphoma 2 to mitigate autism spectrum disorder in rats. Neuropsychiatr Dis Treat. (2017) 13:835–42. doi: 10.2147/NDT.S125088
79. Dai YC, Zhang HF, Schön M, Böckers TM, Han SP, Han JS, et al. Neonatal oxytocin treatment ameliorates autistic-like behaviors and oxytocin deficiency in valproic acid-induced rat model of autism. Front Cell Neurosci. (2018) 12:355. doi: 10.3389/fncel.2018.00355
80. Zhang Y, Xiang Z, Jia Y, He X, Wang L, Cui W. The Notch signaling pathway inhibitor Dapt alleviates autism-like behavior, autophagy and dendritic spine density abnormalities in a valproic acid-induced animal model of autism. Prog Neuropsychopharmacol Biol Psychiatry. (2019) 94:109644. doi: 10.1016/j.pnpbp.2019.109644
81. Gandal MJ, Edgar JC, Ehrlichman RS, Mehta M, Roberts TP, Siegel SJ. Validating γ oscillations and delayed auditory responses as translational biomarkers of autism. Biol Psychiatry. (2010) 68:1100–6. doi: 10.1016/j.biopsych.2010.09.031
82. Wang J, Zheng B, Zhou D, Xing J, Li H, Li J, et al. Supplementation of diet with different n-3/n-6 PUFA ratios ameliorates autistic behavior, reduces serotonin, and improves intestinal barrier impairments in a valproic acid rat model of autism. Front Psychiatry. (2020) 11:552345. doi: 10.3389/fpsyt.2020.552345
83. Román V, Adham N, Foley AG, Hanratty L, Farkas B, Lendvai B, et al. Cariprazine alleviates core behavioral deficits in the prenatal valproic acid exposure model of autism spectrum disorder. Psychopharmacology. (2021) 238:2381–92. doi: 10.1007/s00213-021-05851-6
84. Singla R, Mishra A, Joshi R, Kumar R, Sarma P, Sharma AR, et al. Inhibition of the ERK1/2 phosphorylation by dextromethorphan protects against core autistic symptoms in VPA induced autistic rats: in silico and in vivo drug repurposition study. ACS Chem Neurosci. (2021) 12:1749–67. doi: 10.1021/acschemneuro.0c00672
85. Zou M, Liu Y, Xie S, Wang L, Li D, Li L, et al. Alterations of the endocannabinoid system and its therapeutic potential in autism spectrum disorder. Open Biol. (2021) 11:200306. doi: 10.1098/rsob.200306
86. Mehta MV, Gandal MJ, Siegel SJ. mGluR5-antagonist mediated reversal of elevated stereotyped, repetitive behaviors in the VPA model of autism. PLoS ONE. (2011) 6:e26077. doi: 10.1371/journal.pone.0026077
87. Ryu YK, Park HY, Go J, Choi DH, Choi YK, Rhee M, et al. Sodium phenylbutyrate reduces repetitive self-grooming behavior and rescues social and cognitive deficits in mouse models of autism. Psychopharmacology. (2021) 238:1833–45. doi: 10.1007/s00213-021-05812-z
88. Hidema S, Kikuchi S, Takata R, Yanai T, Shimomura K, Horie K, et al. Single administration of resveratrol improves social behavior in adult mouse models of autism spectrum disorder. Biosci Biotechnol Biochem. (2020) 84:2207–14. doi: 10.1080/09168451.2020.1794783
89. Cosi C, Martel JC, Auclair AL, Collo G, Cavalleri L, Heusler P, et al. Pharmacology profile of F17464, a dopamine D3 receptor preferential antagonist. Eur J Pharmacol. (2021) 890:173635. doi: 10.1016/j.ejphar.2020.173635
90. Lee SE, Lee Y, Lee GH. The regulation of glutamic acid decarboxylases in GABA neurotransmission in the brain. Arch Pharm Res. (2019) 42:1031–9. doi: 10.1007/s12272-019-01196-z
91. Ben-Ari Y, Khalilov I, Kahle KT, Cherubini E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist. (2012) 18:467–86. doi: 10.1177/1073858412438697
92. Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. (2007) 87:1215–84. doi: 10.1152/physrev.00017.2006
93. Ben-Ari Y. The GABA excitatory/inhibitory developmental sequence: a personal journey. Neuroscience. (2014) 279:187–219. doi: 10.1016/j.neuroscience.2014.08.001
94. Hou Q, Wang Y, Li Y, Chen D, Yang F, Wang S. A developmental study of abnormal behaviors and altered GABAergic signaling in the VPA-treated rat model of autism. Front Behav Neurosci. (2018) 12:182. doi: 10.3389/fnbeh.2018.00182
95. Win-Shwe TT, Nway NC, Imai M, Lwin TT, Mar O, Watanabe H. Social behavior, neuroimmune markers and glutamic acid decarboxylase levels in a rat model of valproic acid-induced autism. J Toxicol Sci. (2018) 43:631–43. doi: 10.2131/jts.43.631
96. Wei R, Li Q, Lam S, Leung J, Cheung C, Zhang X, et al. A single low dose of valproic acid in late prenatal life alters postnatal behavior and glutamic acid decarboxylase levels in the mouse. Behav Brain Res. (2016) 314:190–8. doi: 10.1016/j.bbr.2016.08.006
97. Tochitani S, Furukawa T, Bando R, Kondo S, Ito T, Matsushima Y, et al. GABAA receptors and maternally derived taurine regulate the temporal specification of progenitors of excitatory glutamatergic neurons in the mouse developing cortex. Cereb Cortex. (2021) 31:4554–75. doi: 10.1093/cercor/bhab106
98. Kotajima-Murakami H, Hagihara H, Sato A, Hagino Y, Tanaka M, Katoh Y, et al. Exposure of GABAA receptor antagonist picrotoxin in pregnant mice causes autism-like behaviors and aberrant gene expression in offspring. Front Psychiatry. (2022) 13. doi: 10.3389/fpsyt.2022.821354. [Epub ahead of print].
99. Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. (2014) 343:675–9. doi: 10.1126/science.1247190
100. Kumamaru E, Egashira Y, Takenaka R, Takamori S. Valproic acid selectively suppresses the formation of inhibitory synapses in cultured cortical neurons. Neurosci Lett. (2014) 569:142–7. doi: 10.1016/j.neulet.2014.03.066
101. Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. (2013) 106–7:1–16. doi: 10.1016/j.pneurobio.2013.04.001
102. Mohammadi S, Asadi-Shekaari M, Basiri M, Parvan M, Shabani M, Nozari M. Improvement of autistic-like behaviors in adult rats prenatally exposed to valproic acid through early suppression of NMDA receptor function. Psychopharmacology. (2020) 237:199–208. doi: 10.1007/s00213-019-05357-2
103. Smithells RW, Sheppard S, Schorah CJ. Vitamin deficiencies and neural tube defects. Arch Dis Child. (1976) 51:944–50. doi: 10.1136/adc.51.12.944
104. MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet. (1991) 338:131–7. doi: 10.1016/0140-6736(91)90133-A
105. Bjørk M, Riedel B, Spigset O, Veiby G, Kolstad E, Daltveit AK, et al. Association of folic acid supplementation during pregnancy with the risk of autistic traits in children exposed to antiepileptic drugs in utero. JAMA Neurol. (2018) 75:160–8. doi: 10.1001/jamaneurol.2017.3897
106. Levine SZ, Kodesh A, Viktorin A, Smith L, Uher R, Reichenberg A, et al. Association of maternal use of folic acid and multivitamin supplements in the periods before and during pregnancy with the risk of autism spectrum disorder in offspring. JAMA Psychiatry. (2018) 75:176–84. doi: 10.1001/jamapsychiatry.2017.4050
107. Rubinchik-Stern M, Shmuel M, Bar J, Kovo M, Eyal S. Adverse placental effects of valproic acid: studies in perfused human placentas. Epilepsia. (2018) 59:993–1003. doi: 10.1111/epi.14078
108. Fathe K, Palacios A, Finnell RH. Brief report novel mechanism for valproate-induced teratogenicity. Birth Defects Res A Clin Mol Teratol. (2014) 100:592–7. doi: 10.1002/bdra.23277
109. Wegner C, Nau H. Alteration of embryonic folate metabolism by valproic acid during organogenesis: implications for mechanism of teratogenesis. Neurology. (1992) 42(4 Suppl. 5):17–24.
110. Bold J, Sakata-Haga H, Fukui Y. Spinal nerve defects in mouse embryos prenatally exposed to valproic acid. Anat Sci Int. (2018) 93:35–41. doi: 10.1007/s12565-016-0363-9
111. Di Y, Li Z, Li J, Cheng Q, Zheng Q, Zhai C, et al. Maternal folic acid supplementation prevents autistic behaviors in a rat model induced by prenatal exposure to valproic acid. Food Funct. (2021) 12:4544–55. doi: 10.1039/D0FO02926B
112. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. (2001) 276:36734–41. doi: 10.1074/jbc.M101287200
113. Moldrich RX, Leanage G, She D, Dolan-Evans E, Nelson M, Reza N, et al. Inhibition of histone deacetylase in utero causes sociability deficits in postnatal mice. Behav Brain Res. (2013) 257:253–64. doi: 10.1016/j.bbr.2013.09.049
114. Juliandi B, Tanemura K, Igarashi K, Tominaga T, Furukawa Y, Otsuka M, et al. Reduced adult hippocampal neurogenesis and cognitive impairments following prenatal treatment of the antiepileptic drug valproic acid. Stem Cell Rep. (2015) 5:996–1009. doi: 10.1016/j.stemcr.2015.10.012
115. Kataoka S, Takuma K, Hara Y, Maeda Y, Ago Y, Matsuda T. Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int J Neuropsychopharmacol. (2013) 16:91–103. doi: 10.1017/S1461145711001714
116. Gurpur PB, Liu J, Burkin DJ, Kaufman SJ. Valproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophy. Am J Pathol. (2009) 174:999–1008. doi: 10.2353/ajpath.2009.080537
117. Mizuguchi M, Ohsawa M, Kashii H, Sato A. Brain symptoms of tuberous sclerosis complex: pathogenesis and treatment. Int J Mol Sci. (2021) 22:6677. doi: 10.3390/ijms22136677
118. Sato A. mTOR, a potential target to treat autism spectrum disorder. CNS Neurol Disord Drug Targets. (2016) 15:533–43. doi: 10.2174/1871527315666160413120638
119. Garbarino VR, Gilman TL, Daws LC, Gould GG. Extreme enhancement or depletion of serotonin transporter function and serotonin availability in autism spectrum disorder. Pharmacol Res. (2019) 140:85–99. doi: 10.1016/j.phrs.2018.07.010
120. Sangkuhl K, Klein TE, Altman RB. Selective serotonin reuptake inhibitors pathway. Pharmacogenet Genomics. (2009) 19:907–9. doi: 10.1097/FPC.0b013e32833132cb
121. Clevenger SS, Malhotra D, Dang J, Vanle B, IsHak WW. The role of selective serotonin reuptake inhibitors in preventing relapse of major depressive disorder. Ther Adv Psychopharmacol. (2018) 8:49–58. doi: 10.1177/2045125317737264
122. Gavin NI, Gaynes BN, Lohr KN, Meltzer-Brody S, Gartlehner G, Swinson T. Perinatal depression: a systematic review of prevalence and incidence. Obstet Gynecol. (2005) 106:1071–83. doi: 10.1097/01.AOG.0000183597.31630.db
123. Ko JY, Rockhill KM, Tong VT, Morrow B, Farr SL. Trends in postpartum depressive symptoms: 27 states, 2004, 2008, and 2012. MMWR Morb Mortal Wkly Rep. (2017) 66:153–8. doi: 10.15585/mmwr.mm6606a1
124. Gabriele S, Sacco R, Persico AM. Blood serotonin levels in autism spectrum disorder: a systematic review and meta-analysis. Eur Neuropsychopharmacol. (2014) 24:919–29. doi: 10.1016/j.euroneuro.2014.02.004
125. Croen LA, Grether JK, Yoshida CK, Odouli R, Hendrick V. Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch Gen Psychiatry. (2011) 68:1104–12. doi: 10.1001/archgenpsychiatry.2011.73
126. Rai D, Lee BK, Dalman C, Golding J, Lewis G, Magnusson C. Parental depression, maternal antidepressant use during pregnancy, and risk of autism spectrum disorders: population based case-control study. BMJ. (2013) 346:f2059. doi: 10.1136/bmj.f2059
127. Boukhris T, Sheehy O, Mottron L, Bérard A. Antidepressant use during pregnancy and the risk of autism spectrum disorder in children. JAMA Pediatr. (2016) 170:117–24. doi: 10.1001/jamapediatrics.2015.3356
128. Fitton CA, Steiner MFC, Aucott L, Pell JP, Mackay DF, Fleming M, et al. In utero exposure to antidepressant medication and neonatal and child outcomes: a systematic review. Acta Psychiatr Scand. (2020) 141:21–33. doi: 10.1111/acps.13120
129. Ames JL, Ladd-Acosta C, Fallin MD, Qian Y, Schieve LA, DiGuiseppi C, et al. Maternal psychiatric conditions, treatment with selective serotonin reuptake inhibitors, and neurodevelopmental disorders. Biol Psychiatry. (2021) 90:253–62. doi: 10.1016/j.biopsych.2021.04.002
130. Malm H, Brown AS, Gissler M, Gyllenberg D, Hinkka-Yli-Salomäki S, McKeague IW, et al. Gestational exposure to selective serotonin reuptake inhibitors and offspring psychiatric disorders: a national register-based study. J Am Acad Child Adolesc Psychiatry. (2016) 55:359–66. doi: 10.1016/j.jaac.2016.02.013
131. Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC, et al. A transient placental source of serotonin for the fetal forebrain. Nature. (2011) 472:347–50. doi: 10.1038/nature09972
132. Laurent L, Huang C, Ernest SR, Berard A, Vaillancourt C, Hales BF. In utero exposure to venlafaxine, a serotonin-norepinephrine reuptake inhibitor, increases cardiac anomalies and alters placental and heart serotonin signaling in the rat. Birth Defects Res A Clin Mol Teratol. (2016) 106:1044–55. doi: 10.1002/bdra.23537
133. Furuhashi N, Tsujiei M, Kimura H, Yajima A. Effects of ketanserin—a serotonin receptor antagonist—on placental blood flow, placental weight and fetal weight of spontaneously hypertensive rats and normal Wistar Kyoto rats. Gynecol Obstet Invest. (1991) 32:65–7. doi: 10.1159/000292996
134. Rosenfeld CS. Placental serotonin signaling, pregnancy outcomes, and regulation of fetal brain development. Biol Reprod. (2020) 102:532–8. doi: 10.1093/biolre/ioz204
135. Maloney SE, Akula S, Rieger MA, McCullough KB, Chandler K, Corbett AM, et al. Examining the reversibility of long-term behavioral disruptions in progeny of maternal SSRI exposure. eNeuro. (2018) 5:ENEURO.0120-18.2018. doi: 10.1523/ENEURO.0120-18.2018
136. Yu W, Yen YC, Lee YH, Tan S, Xiao Y, Lokman H, et al. Prenatal selective serotonin reuptake inhibitor (SSRI) exposure induces working memory and social recognition deficits by disrupting inhibitory synaptic networks in male mice. Mol Brain. (2019) 12:29. doi: 10.1186/s13041-019-0452-5
137. Bond CM, Johnson JC, Chaudhary V, McCarthy EM, McWhorter ML, Woehrle NS. Perinatal fluoxetine exposure results in social deficits and reduced monoamine oxidase gene expression in mice. Brain Res. (2020) 1727:146282. doi: 10.1016/j.brainres.2019.06.001
138. Zahra A, Jiang J, Chen Y, Long C, Yang L. Memantine rescues prenatal citalopram exposure-induced striatal and social abnormalities in mice. Exp Neurol. (2018) 307:145–54. doi: 10.1016/j.expneurol.2018.06.003
139. Kalueff AV, Olivier JD, Nonkes LJ, Homberg JR. Conserved role for the serotonin transporter gene in rat and mouse neurobehavioral endophenotypes. Neurosci Biobehav Rev. (2010) 34:373–86. doi: 10.1016/j.neubiorev.2009.08.003
140. Fabre V, Beaufour C, Evrard A, Rioux A, Hanoun N, Lesch KP, et al. Altered expression and functions of serotonin 5-HT1A and 5-HT1B receptors in knock-out mice lacking the 5-HT transporter. Eur J Neurosci. (2000) 12:2299–310. doi: 10.1046/j.1460-9568.2000.00126.x
141. Mathews TA, Fedele DE, Coppelli FM, Avila AM, Murphy DL, Andrews AM. Gene dose-dependent alterations in extraneuronal serotonin but not dopamine in mice with reduced serotonin transporter expression. J Neurosci Methods. (2004) 140:169–81. doi: 10.1016/j.jneumeth.2004.05.017
142. Shen HW, Hagino Y, Kobayashi H, Shinohara-Tanaka K, Ikeda K, Yamamoto H, et al. Regional differences in extracellular dopamine and serotonin assessed by in vivo microdialysis in mice lacking dopamine and/or serotonin transporters. Neuropsychopharmacology. (2004) 29:1790–9. doi: 10.1038/sj.npp.1300476
143. Homberg JR, Olivier JD, Smits BM, Mul JD, Mudde J, Verheul M, et al. Characterization of the serotonin transporter knockout rat: a selective change in the functioning of the serotonergic system. Neuroscience. (2007) 146:1662–76. doi: 10.1016/j.neuroscience.2007.03.030
144. Olivier JD, Van Der Hart MG, Van Swelm RP, Dederen PJ, Homberg JR, Cremers T, et al. A study in male and female 5-HT transporter knockout rats: an animal model for anxiety and depression disorders. Neuroscience. (2008) 152:573–84. doi: 10.1016/j.neuroscience.2007.12.032
145. Moy SS, Nadler JJ, Young NB, Nonneman RJ, Grossman AW, Murphy DL, et al. Social approach in genetically engineered mouse lines relevant to autism. Genes Brain Behav. (2009) 8:129–42. doi: 10.1111/j.1601-183X.2008.00452.x
146. Page DT, Kuti OJ, Prestia C, Sur M. Haploinsufficiency for Pten and serotonin transporter cooperatively influences brain size and social behavior. Proc Natl Acad Sci USA. (2009) 106:1989–94. doi: 10.1073/pnas.0804428106
147. Homberg JR, Pattij T, Janssen MC, Ronken E, De Boer SF, Schoffelmeer AN, et al. Serotonin transporter deficiency in rats improves inhibitory control but not behavioural flexibility. Eur J Neurosci. (2007) 26:2066–73. doi: 10.1111/j.1460-9568.2007.05839.x
148. Tanaka M, Sato A, Kasai S, Hagino Y, Kotajima-Murakami H, Kashii H, et al. Brain hyperserotonemia causes autism-relevant social deficits in mice. Mol Autism. (2018) 9:60. doi: 10.1186/s13229-018-0243-3
149. Veenstra-VanderWeele J, Muller CL, Iwamoto H, Sauer JE, Owens WA, Shah CR, et al. Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc Natl Acad Sci USA. (2012) 109:5469–74. doi: 10.1073/pnas.1112345109
150. Robson MJ, Quinlan MA, Margolis KG, Gajewski-Kurdziel PA, Veenstra-VanderWeele J, Gershon MD, et al. p38α MAPK signaling drives pharmacologically reversible brain and gastrointestinal phenotypes in the SERT Ala56 mouse. Proc Natl Acad Sci USA. (2018) 115:E10245–54. doi: 10.1073/pnas.1809137115
151. Kane MJ, Angoa-Peréz M, Briggs DI, Sykes CE, Francescutti DM, Rosenberg DR, et al. Mice genetically depleted of brain serotonin display social impairments, communication deficits and repetitive behaviors: possible relevance to autism. PLoS ONE. (2012) 7:e48975. doi: 10.1371/journal.pone.0048975
152. Kästner N, Richter SH, Urbanik S, Kunert J, Waider J, Lesch KP, et al. Brain serotonin deficiency affects female aggression. Sci Rep. (2019) 9:1366. doi: 10.1038/s41598-018-37613-4
153. Shuid AN, Jayusman PA, Shuid N, Ismail J, Kamal Nor N, Mohamed IN. Association between viral infections and risk of autistic disorder: an overview. Int J Environ Res Public Health. (2021) 18:2817. doi: 10.3390/ijerph18062817
154. Jiang HY, Xu LL, Shao L, Xia RM, Yu ZH, Ling ZX, et al. Maternal infection during pregnancy and risk of autism spectrum disorders: a systematic review and meta-analysis. Brain Behav Immun. (2016) 58:165–72. doi: 10.1016/j.bbi.2016.06.005
155. Tioleco N, Silberman AE, Stratigos K, Banerjee-Basu S, Spann MN, Whitaker AH, et al. Prenatal maternal infection and risk for autism in offspring: a meta-analysis. Autism Res. (2021) 14:1296–316. doi: 10.1002/aur.2499
156. Antoun S, Ellul P, Peyre H, Rosenzwajg M, Gressens P, Klatzmann D, et al. Fever during pregnancy as a risk factor for neurodevelopmental disorders: results from a systematic review and meta-analysis. Mol Autism. (2021) 12:60. doi: 10.1186/s13229-021-00464-4
157. Jash S, Sharma S. In utero immune programming of autism spectrum disorder (ASD). Hum Immunol. (2021) 82:379–84. doi: 10.1016/j.humimm.2021.02.002
158. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. (2007) 8:967–74. doi: 10.1038/ni1488
159. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature. (2008) 453:236–40. doi: 10.1038/nature06878
160. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
161. Prins JR, Eskandar S, Eggen BJL, Scherjon SA. Microglia, the missing link in maternal immune activation and fetal neurodevelopment; and a possible link in preeclampsia and disturbed neurodevelopment? J Reprod Immunol. (2018) 126:18–22. doi: 10.1016/j.jri.2018.01.004
162. Han VX, Patel S, Jones HF, Dale RC. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat Rev Neurol. (2021) 7:564–79. doi: 10.1038/s41582-021-00530-8
163. Goines PE, Croen LA, Braunschweig D, Yoshida CK, Grether J, Hansen R, et al. Increased midgestational IFN-γ, IL-4 and IL-5 in women bearing a child with autism: a case-control study. Mol Autism. (2011) 2:13. doi: 10.1186/2040-2392-2-13
164. Jones KL, Croen LA, Yoshida CK, Heuer L, Hansen R, Zerbo O, et al. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol Psychiatry. (2017) 22:273–9. doi: 10.1038/mp.2016.77
165. Abdallah MW, Larsen N, Mortensen EL, Atladóttir HÓ, Nørgaard-Pedersen B, Bonefeld-Jørgensen EC, et al. Neonatal levels of cytokines and risk of autism spectrum disorders: an exploratory register-based historic birth cohort study utilizing the Danish Newborn Screening Biobank. J Neuroimmunol. (2012) 252:75–82. doi: 10.1016/j.jneuroim.2012.07.013
166. Zerbo O, Yoshida C, Grether JK, Van de Water J, Ashwood P, Delorenze GN, et al. Neonatal cytokines and chemokines and risk of autism spectrum disorder: the Early Markers for Autism (EMA) study: a case-control study. J Neuroinflammation. (2014) 11:113. doi: 10.1186/1742-2094-11-113
167. Krakowiak P, Goines PE, Tancredi DJ, Ashwood P, Hansen RL, Hertz-Picciotto I, et al. Neonatal cytokine profiles associated with autism spectrum disorder. Biol Psychiatry. (2017) 81:442–51. doi: 10.1016/j.biopsych.2015.08.007
168. Heuer LS, Croen LA, Jones KL, Yoshida CK, Hansen RL, Yolken R, et al. An exploratory examination of neonatal cytokines and chemokines as predictors of autism risk: the Early Markers for Autism study. Biol Psychiatry. (2019) 86:255–64. doi: 10.1016/j.biopsych.2019.04.037
169. Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. (2011) 25:40–5. doi: 10.1016/j.bbi.2010.08.003
170. Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, et al. Elevated immune response in the brain of autistic patients. J Neuroimmunol. (2009) 207:111–6. doi: 10.1016/j.jneuroim.2008.12.002
171. Al-Ayadhi LY, Mostafa GA. Elevated serum levels of interleukin-17A in children with autism. J Neuroinflammation. (2012) 9:158. doi: 10.1186/1742-2094-9-158
172. Akintunde ME, Rose M, Krakowiak P, Heuer L, Ashwood P, Hansen R, et al. Increased production of IL-17 in children with autism spectrum disorders and co-morbid asthma. J Neuroimmunol. (2015) 286:33–41. doi: 10.1016/j.jneuroim.2015.07.003
173. Suzuki K, Matsuzaki H, Iwata K, Kameno Y, Shimmura C, Kawai S, et al. Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PLoS ONE. (2011) 6:e20470. doi: 10.1371/journal.pone.0020470
174. Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. (2003) 23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003
175. Miller VM, Zhu Y, Bucher C, McGinnis W, Ryan LK, Siegel A, et al. Gestational flu exposure induces changes in neurochemicals, affiliative hormones and brainstem inflammation, in addition to autism-like behaviors in mice. Brain Behav Immun. (2013) 33:153–63. doi: 10.1016/j.bbi.2013.07.002
176. Manjeese W, Mvubu NE, Steyn AJC, Mpofana T. Mycobacterium tuberculosis-induced maternal immune activation promotes autism-like phenotype in infected mice offspring. Int J Environ Res Public Health. (2021) 18:4513. doi: 10.3390/ijerph18094513
177. Glass R, Norton S, Fox N, Kusnecov AW. Maternal immune activation with staphylococcal enterotoxin A produces unique behavioral changes in C57BL/6 mouse offspring. Brain Behav Immun. (2019) 75:12–25. doi: 10.1016/j.bbi.2018.05.005
178. Xu Z, Zhang X, Chang H, Kong Y, Ni Y, Liu R, et al. Rescue of maternal immune activation-induced behavioral abnormalities in adult mouse offspring by pathogen-activated maternal Treg cells. Nat Neurosci. (2021) 24:818–30. doi: 10.1038/s41593-021-00837-1
179. Allard MJ, Bergeron JD, Baharnoori M, Srivastava LK, Fortier LC, Poyart C, et al. A sexually dichotomous, autistic-like phenotype is induced by Group B Streptococcus maternofetal immune activation. Autism Res. (2017) 10:233–45. doi: 10.1002/aur.1647
180. Bergeron JD, Deslauriers J, Grignon S, Fortier LC, Lepage M, Stroh T, et al. White matter injury and autistic-like behavior predominantly affecting male rat offspring exposed to group B streptococcal maternal inflammation. Dev Neurosci. (2013) 35:504–15. doi: 10.1159/000355656
181. Vuillermot S, Luan W, Meyer U, Eyles D. Vitamin D treatment during pregnancy prevents autism-related phenotypes in a mouse model of maternal immune activation. Mol Autism. (2017) 8:9. doi: 10.1186/s13229-017-0125-0
182. Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. (2012) 26:607–16. doi: 10.1016/j.bbi.2012.01.011
183. Lammert CR, Frost EL, Bolte AC, Paysour MJ, Shaw ME, Bellinger CE, et al. Cutting edge: critical roles for microbiota-mediated regulation of the immune system in a prenatal immune activation model of autism. J Immunol. (2018) 201:845–50. doi: 10.4049/jimmunol.1701755
184. Zhang X, Ibi M, Haga R, Iwata K, Matsumoto M, Asaoka N, et al. NOX1/NADPH oxidase affects the development of autism-like behaviors in a maternal immune activation model. Biochem Biophys Res Commun. (2021) 534:59–66. doi: 10.1016/j.bbrc.2020.11.070
185. Schwartzer JJ, Careaga M, Onore CE, Rushakoff JA, Berman RF, Ashwood P. Maternal immune activation and strain specific interactions in the development of autism-like behaviors in mice. Transl Psychiatry. (2013) 3:e240. doi: 10.1038/tp.2013.16
186. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. (2007) 27:10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
187. Amodeo DA, Lai CY, Hassan O, Mukamel EA, Behrens MM, Powell SB. Maternal immune activation impairs cognitive flexibility and alters transcription in frontal cortex. Neurobiol Dis. (2019) 125:211–8. doi: 10.1016/j.nbd.2019.01.025
188. Haida O, Al Sagheer T, Balbous A, Francheteau M, Matas E, Soria F, et al. Sex-dependent behavioral deficits and neuropathology in a maternal immune activation model of autism. Transl Psychiatry. (2019) 9:124. doi: 10.1038/s41398-019-0457-y
189. Ozaki K, Kato D, Ikegami A, Hashimoto A, Sugio S, Guo Z, et al. Maternal immune activation induces sustained changes in fetal microglia motility. Sci Rep. (2020) 10:21378. doi: 10.1038/s41598-020-78294-2
190. Garcia-Valtanen P, van Diermen BA, Lakhan N, Lousberg EL, Robertson SA, Hayball JD, et al. Maternal host responses to poly(I:C) during pregnancy leads to both dysfunctional immune profiles and altered behaviour in the offspring. Am J Reprod Immunol. (2020) 84:e13260. doi: 10.1111/aji.13260
191. Yang Y, Wang B, Zhong Z, Chen H, Ding W, Hoi MPM. Clonazepam attenuates neurobehavioral abnormalities in offspring exposed to maternal immune activation by enhancing GABAergic neurotransmission. Biochem Pharmacol. (2021) 192:114711. doi: 10.1016/j.bcp.2021.114711
192. Wu WL, Adams CE, Stevens KE, Chow KH, Freedman R, Patterson PH. The interaction between maternal immune activation and alpha 7 nicotinic acetylcholine receptor in regulating behaviors in the offspring. Brain Behav Immun. (2015) 46:192–202. doi: 10.1016/j.bbi.2015.02.005
193. Wu WL, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav Immun. (2017) 62:11–23. doi: 10.1016/j.bbi.2016.11.007
194. Xuan IC, Hampson DR. Gender-dependent effects of maternal immune activation on the behavior of mouse offspring. PLoS ONE. (2014) 9:e104433. doi: 10.1371/journal.pone.0104433
195. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. (2016) 351:933–9. doi: 10.1126/science.aad0314
196. Weiser MJ, Mucha B, Denheyer H, Atkinson D, Schanz N, Vassiliou E, et al. Dietary docosahexaenoic acid alleviates autistic-like behaviors resulting from maternal immune activation in mice. Prostaglandins Leukot Essent Fatty Acids. (2016) 106:27–37. doi: 10.1016/j.plefa.2015.10.005
197. Sunwoo JS, Jeon D, Lee ST, Moon J, Yu JS, Park DK, et al. Maternal immune activation alters brain microRNA expression in mouse offspring. Ann Clin Transl Neurol. (2018) 5:1264–76. doi: 10.1002/acn3.652
198. Pendyala G, Chou S, Jung Y, Coiro P, Spartz E, Padmashri R, et al. Maternal immune activation causes behavioral impairments and altered cerebellar cytokine and synaptic protein expression. Neuropsychopharmacology. (2017) 42:1435–46. doi: 10.1038/npp.2017.7
199. Ferreira FR, de Moura NSB, Hassib L, Pombo TR. Resveratrol ameliorates the effect of maternal immune activation associated with schizophrenia in adulthood offspring. Neurosci Lett. (2020) 734:135100. doi: 10.1016/j.neulet.2020.135100
200. Naviaux RK, Zolkipli Z, Wang L, Nakayama T, Naviaux JC, Le TP, et al. Antipurinergic therapy corrects the autism-like features in the poly(IC) mouse model. PLoS ONE. (2013) 8:e57380. doi: 10.1371/journal.pone.0057380
201. Naviaux JC, Schuchbauer MA, Li K, Wang L, Risbrough VB, Powell SB, et al. Reversal of autism-like behaviors and metabolism in adult mice with single-dose antipurinergic therapy. Transl Psychiatry. (2014) 4:e400. doi: 10.1038/tp.2014.33
202. Horváth G, Otrokocsi L, Beko K, Baranyi M, Kittel Á, Fritz-Ruenes PA, et al. P2X7 receptors drive poly(I:C) induced autism-like behavior in mice. J Neurosci. (2019) 39:2542–61. doi: 10.1523/JNEUROSCI.1895-18.2019
203. Okamoto K, Hitora-Imamura N, Hioki H, Ikegaya Y. GABAergic malfunction in the anterior cingulate cortex underlying maternal immune activation-induced social deficits. J Neuroimmunol. (2018) 321:92–6. doi: 10.1016/j.jneuroim.2018.06.006
204. Dabbah-Assadi F, Alon D, Golani I, Doron R, Kremer I, Beloosesky R, et al. The influence of immune activation at early vs late gestation on fetal NRG1-ErbB4 expression and behavior in juvenile and adult mice offspring. Brain Behav Immun. (2019) 79:207–15. doi: 10.1016/j.bbi.2019.02.002
205. Ma M, Ren Q, Yang J, Zhang K, Xiong Z, Ishima T, et al. Key role of soluble epoxide hydrolase in the neurodevelopmental disorders of offspring after maternal immune activation. Proc Natl Acad Sci USA. (2019) 116:7083–8. doi: 10.1073/pnas.1819234116
206. Fujita Y, Fujita A, Ishima T, Hirai A, Suzuki S, Suganuma H, et al. Dietary intake of glucoraphanin during pregnancy and lactation prevents the behavioral abnormalities in the offspring after maternal immune activation. Neuropsychopharmacol Rep. (2020) 40:268–74. doi: 10.1002/npr2.12112
207. Lins BR, Marks WN, Zabder NK, Greba Q, Howland JG. Maternal immune activation during pregnancy alters the behavior profile of female offspring of Sprague Dawley rats. eNeuro. (2019) 6:ENEURO.0437-18.2019. doi: 10.1523/ENEURO.0437-18.2019
208. Yasumatsu K, Nagao JI, Arita-Morioka KI, Narita Y, Tasaki S, Toyoda K, et al. Bacterial-induced maternal interleukin-17A pathway promotes autistic-like behaviors in mouse offspring. Exp Anim. (2020) 69:250–60. doi: 10.1538/expanim.19-0156
209. Wu Y, Qi F, Song D, He Z, Zuo Z, Yang Y, et al. Prenatal influenza vaccination rescues impairments of social behavior and lamination in a mouse model of autism. J Neuroinflammation. (2018) 15:228. doi: 10.1186/s12974-018-1252-z
210. Vojtechova I, Maleninska K, Kutna V, Klovrza O, Tuckova K, Petrasek T, et al. Behavioral alterations and decreased number of parvalbumin-positive interneurons in Wistar rats after maternal immune activation by lipopolysaccharide: sex matters. Int J Mol Sci. (2021) 22:3274. doi: 10.3390/ijms22063274
211. Andoh M, Shibata K, Okamoto K, Onodera J, Morishita K, Miura Y, et al. Exercise reverses behavioral and synaptic abnormalities after maternal inflammation. Cell Rep. (2019) 27:2817–25.e5. doi: 10.1016/j.celrep.2019.05.015
212. Fortunato JJ, da Rosa N, Martins Laurentino AO, Goulart M, Michalak C, Borges LP, et al. Effects of ω-3 fatty acids on stereotypical behavior and social interactions in Wistar rats prenatally exposed to lipopolysaccarides. Nutrition. (2017) 35:119–27. doi: 10.1016/j.nut.2016.10.019
213. Kirsten TB, Bernardi MM. Prenatal lipopolysaccharide induces hypothalamic dopaminergic hypoactivity and autistic-like behaviors: repetitive self-grooming and stereotypies. Behav Brain Res. (2017) 331:25–9. doi: 10.1016/j.bbr.2017.05.013
214. Lee GA, Lin YK, Lai JH, Lo YC, Yang YSH, Ye SY, et al. Maternal immune activation causes social behavior deficits and hypomyelination in male rat offspring with an autism-like microbiota profile. Brain Sci. (2021) 11:1085. doi: 10.3390/brainsci11081085
215. Talukdar PM, Abdul F, Maes M, Berk M, Venkatasubramanian G, Kutty BM, et al. A proof-of-concept study of maternal immune activation mediated induction of Toll-like receptor (TLR) and inflammasome pathways leading to neuroprogressive changes and schizophrenia-like behaviours in offspring. Eur Neuropsychopharmacol. (2021) 52:48–61. doi: 10.1016/j.euroneuro.2021.06.009
216. Vitor-Vieira F, Vilela FC, Giusti-Paiva A. Hyperactivation of the amygdala correlates with impaired social play behavior of prepubertal male rats in a maternal immune activation model. Behav Brain Res. (2021) 414:113503. doi: 10.1016/j.bbr.2021.113503
217. Parker-Athill E, Luo D, Bailey A, Giunta B, Tian J, Shytle RD, et al. Flavonoids, a prenatal prophylaxis via targeting JAK2/STAT3 signaling to oppose IL-6/MIA associated autism. J Neuroimmunol. (2009) 217:20–7. doi: 10.1016/j.jneuroim.2009.08.012
218. Washington J 3rd, Kumar U, Medel-Matus JS, Shin D, Sankar R, Mazarati A. Cytokine-dependent bidirectional connection between impaired social behavior and susceptibility to seizures associated with maternal immune activation in mice. Epilepsy Behav. (2015) 50:40–5. doi: 10.1016/j.yebeh.2015.05.040
219. Minakova E, Lang J, Medel-Matus JS, Gould GG, Reynolds A, Shin D, et al. Melanotan-II reverses autistic features in a maternal immune activation mouse model of autism. PLoS ONE. (2019) 14:e0210389. doi: 10.1371/journal.pone.0210389
220. Murakami Y, Imamura Y, Kasahara Y, Yoshida C, Momono Y, Fang K, et al. The effects of maternal interleukin-17A on social behavior, cognitive function, and depression-like behavior in mice with altered kynurenine metabolites. Int J Tryptophan Res. (2021) 14:11786469211026639. doi: 10.1177/11786469211026639
221. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by toll-like receptor 3. Nature. (2001) 413:732–8. doi: 10.1038/35099560
222. Dahlgren J, Samuelsson AM, Jansson T, Holmang A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res. (2006) 60:147–51. doi: 10.1203/01.pdr.0000230026.74139.18
223. Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. (2011) 25:604–15. doi: 10.1016/j.bbi.2010.12.017
224. Gumusoglu SB, Fine RS, Murray SJ, Bittle JL, Stevens HE. The role of IL-6 in neurodevelopment after prenatal stress. Brain Behav Immun. (2017) 65:274–83. doi: 10.1016/j.bbi.2017.05.015
225. Chen HR, Chen CW, Mandhani N, Short-Miller JC, Smucker MR, Sun YY, et al. Monocytic infiltrates contribute to autistic-like behaviors in a two-hit model of neurodevelopmental defects. J Neurosci. (2020) 40:9386–400. doi: 10.1523/JNEUROSCI.1171-20.2020
226. Fernández de Cossío L, Guzmán A, van der Veldt S, Luheshi GN. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain Behav Immun. (2017) 63:88–98. doi: 10.1016/j.bbi.2016.09.028
227. Xia Y, Zhang Z, Lin W, Yan J, Zhu C, Yin D, et al. Modulating microglia activation prevents maternal immune activation induced schizophrenia-relevant behavior phenotypes via arginase 1 in the dentate gyrus. Neuropsychopharmacology. (2020) 45:1896–908. doi: 10.1038/s41386-020-0743-7
228. Brown MA, Magee LA, Kenny LC, Karumanchi SA, McCarthy FP, Saito S, et al. The hypertensive disorders of pregnancy: ISSHP classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens. (2018) 13:291–310. doi: 10.1161/HYPERTENSIONAHA.117.10803
229. Umesawa M, Kobashi G. Epidemiology of hypertensive disorders in pregnancy: prevalence, risk factors, predictors and prognosis. Hypertens Res. (2017) 40:213–20. doi: 10.1038/hr.2016.126
230. Xu RT, Chang QX, Wang QQ, Zhang J, Xia LX, Zhong N, et al. Association between hypertensive disorders of pregnancy and risk of autism in offspring: a systematic review and meta-analysis of observational studies. Oncotarget. (2017) 9:1291–301. doi: 10.18632/oncotarget.23030
231. Dachew BA, Mamun A, Maravilla JC, Alati R. Pre-eclampsia and the risk of autism-spectrum disorder in offspring: meta-analysis. Br J Psychiatry. (2018) 212:142–7. doi: 10.1192/bjp.2017.27
232. Abitbol MM, Ober MB, Gallo GR, Driscoll SG, Pirani CL. Experimental toxemia of pregnancy in the monkey, with a preliminary report on renin and aldosterone. Am J Pathol. (1977) 86:573–90.
233. Intapad S, Warrington JP, Spradley FT, Palei AC, Drummond HA, Ryan MJ, et al. Reduced uterine perfusion pressure induces hypertension in the pregnant mouse. Am J Physiol Regul Integr Comp Physiol. (2014) 307:R1353–7. doi: 10.1152/ajpregu.00268.2014
234. Morton JS, Levasseur J, Ganguly E, Quon A, Kirschenman R, Dyck JRB, et al. Characterisation of the selective reduced uteroplacental perfusion (sRUPP) model of preeclampsia. Sci Rep. (2019) 9:9565. doi: 10.1038/s41598-019-45959-6
235. Rehn AE, Van Den Buuse M, Copolov D, Briscoe T, Lambert G, Rees S. An animal model of chronic placental insufficiency: relevance to neurodevelopmental disorders including schizophrenia. Neuroscience. (2004) 129:381–91. doi: 10.1016/j.neuroscience.2004.07.047
236. Wallace K, Richards S, Dhillon P, Weimer A, Edholm ES, Bengten E, et al. CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension. (2011) 57:949–55. doi: 10.1161/HYPERTENSIONAHA.110.168344
237. Cornelius DC, Hogg JP, Scott J, Wallace K, Herse F, Moseley J, et al. Administration of interleukin-17 soluble receptor C suppresses TH17 cells, oxidative stress, and hypertension in response to placental ischemia during pregnancy. Hypertension. (2013) 62:1068–73. doi: 10.1161/HYPERTENSIONAHA.113.01514
238. Gant NF, Daley GL, Chand S, Whalley PJ, MacDonald PC. A study of angiotensin II pressor response throughout primigravid pregnancy. J Clin Invest. (1973) 52:2682–9. doi: 10.1172/JCI107462
239. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jüpner A, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. (1999) 103:945–52. doi: 10.1172/JCI4106
240. Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, et al. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med. (2008) 14:855–62. doi: 10.1038/nm.1856
241. Duncan JW, Azubuike D, Booz GW, Fisher B, Williams JM, Fan F, et al. Angiotensin II type 1 receptor autoantibody blockade improves cerebral blood flow autoregulation and hypertension in a preclinical model of preeclampsia. Hypertens Pregnancy. (2020) 39:451–60. doi: 10.1080/10641955.2020.1833215
242. Doering TP, Haller NA, Montgomery MA, Freeman EJ, Hopkins MP. The role of AT1 angiotensin receptor activation in the pathogenesis of preeclampsia. Am J Obstet Gynecol. (1998) 178:1307–12. doi: 10.1016/S0002-9378(98)70337-0
243. Zhou CC, Ahmad S, Mi T, Xia L, Abbasi S, Hewett PW, et al. Angiotensin II induces soluble fms-like tyrosine kinase-1 release via calcineurin signaling pathway in pregnancy. Circ Res. (2007) 100:88–95. doi: 10.1161/01.RES.0000254703.11154.18
244. Shirasuna K, Karasawa T, Usui F, Kobayashi M, Komada T, Kimura H, et al. NLRP3 deficiency improves angiotensin II-induced hypertension but not fetal growth restriction during pregnancy. Endocrinology. (2015) 156:4281–92. doi: 10.1210/en.2015-1408
245. Shah DM. The role of RAS in the pathogenesis of preeclampsia. Curr Hypertens Rep. (2006) 8:144–52. doi: 10.1007/s11906-006-0011-1
246. Santillan MK, Santillan DA, Scroggins SM, Min JY, Sandgren JA, Pearson NA, et al. Vasopressin in preeclampsia: a novel very early human pregnancy biomarker and clinically relevant mouse model. Hypertension. (2014) 64:852–9. doi: 10.1161/HYPERTENSIONAHA.114.03848
247. Sandgren JA, Deng G, Linggonegoro DW, Scroggins SM, Perschbacher KJ, Nair AR, et al. Arginine vasopressin infusion is sufficient to model clinical features of preeclampsia in mice. JCI Insight. (2018) 3:e99403. doi: 10.1172/jci.insight.99403
248. Scroggins SM, Santillan DA, Lund JM, Sandgren JA, Krotz LK, Hamilton WS, et al. Elevated vasopressin in pregnant mice induces T-helper subset alterations consistent with human preeclampsia. Clin Sci. (2018) 132:419–36. doi: 10.1042/CS20171059
249. Gumusoglu SB, Chilukuri ASS, Hing BWQ, Scroggins SM, Kundu S, Sandgren JA, et al. Altered offspring neurodevelopment in an arginine vasopressin preeclampsia model. Transl Psychiatry. (2021) 11:79. doi: 10.1038/s41398-021-01205-0
250. Jelen I, Fananapazir L, Crawford TB. The possible relation between late pregnancy hypertension and 5-hydroxytryptamine levels in maternal blood. Br J Obstet Gynaecol. (1979) 86:468–71. doi: 10.1111/j.1471-0528.1979.tb10791.x
251. Gujrati VR, Shanker K, Vrat S, Chandravati, Parmar SS. Novel appearance of placental nuclear monoamine oxidase: biochemical and histochemical evidence for hyperserotonomic state in preeclampsia-eclampsia. Am J Obstet Gynecol. (1996) 175:1543–50. doi: 10.1016/S0002-9378(96)70104-7
252. Carrasco G, Cruz MA, Gallardo V, Miguel P, Dominguez A, González C. Transport and metabolism of serotonin in the human placenta from normal and severely pre-eclamptic pregnancies. Gynecol Obstet Invest. (2000) 49:150–5. doi: 10.1159/000010237
253. Steyn DW, Odendaal HJ. Serotonin antagonism and serotonin antagonists in pregnancy: role of ketanserin. Obstet Gynecol Surv. (2000) 55:582–9. doi: 10.1097/00006254-200009000-00024
254. Hoyer D, Hannon JP, Martin GR. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol Biochem Behav. (2002) 71:533–54. doi: 10.1016/S0091-3057(01)00746-8
255. Ugun-Klusek A, Tamang A, Loughna P, Billett E, Buckley G, Sivasubramaniam S. Reduced placental vascular reactivity to 5-hydroxytryptamine in pre-eclampsia and the status of 5HT2A receptors. Vascul Pharmacol. (2011) 55:157–62. doi: 10.1016/j.vph.2011.07.006
256. Salas SP, Giacaman A, Romero W, Downey P, Aranda E, Mezzano D, et al. Pregnant rats treated with a serotonin precursor have reduced fetal weight and lower plasma volume and kallikrein levels. Hypertension. (2007) 50:773–9. doi: 10.1161/HYPERTENSIONAHA.107.094540
257. Hadden C, Fahmi T, Cooper A, Savenka AV, Lupashin VV, Roberts DJ, et al. Serotonin transporter protects the placental cells against apoptosis in caspase 3-independent pathway. J Cell Physiol. (2017) 232:3520–9. doi: 10.1002/jcp.25812
258. Glasson EJ, Bower C, Petterson B, de Klerk N, Chaney G, Hallmayer JF. Perinatal factors and the development of autism: a population study. Arch Gen Psychiatry. (2004) 61:618–27. doi: 10.1001/archpsyc.61.6.618
259. Reichenberg A, Gross R, Weiser M, Bresnahan M, Silverman J, Harlap S, et al. Advancing paternal age and autism. Arch Gen Psychiatry. (2006) 63:1026–32. doi: 10.1001/archpsyc.63.9.1026
260. Hultman CM, Sandin S, Levine SZ, Lichtenstein P, Reichenberg A. Advancing paternal age and risk of autism: new evidence from a population-based study and a meta-analysis of epidemiological studies. Mol Psychiatry. (2011) 16:1203–12. doi: 10.1038/mp.2010.121
261. Idring S, Magnusson C, Lundberg M, Ek M, Rai D, Svensson AC, et al. Parental age and the risk of autism spectrum disorders: findings from a Swedish population-based cohort. Int J Epidemiol. (2014) 43:107–15. doi: 10.1093/ije/dyt262
262. Wu S, Wu F, Ding Y, Hou J, Bi J, Zhang Z. Advanced parental age and autism risk in children: a systematic review and meta-analysis. Acta Psychiatr Scand. (2017) 135:29–41. doi: 10.1111/acps.12666
263. Sandin S, Hultman CM, Kolevzon A, Gross R, MacCabe JH, Reichenberg A. Advancing maternal age is associated with increasing risk for autism: a review and meta-analysis. J Am Acad Child Adolesc Psychiatry. (2012) 51:477–86.e1. doi: 10.1016/j.jaac.2012.02.018
264. Sandin S, Schendel D, Magnusson P, Hultman C, Surén P, Susser E, et al. Autism risk associated with parental age and with increasing difference in age between the parents. Mol Psychiatry. (2016) 21:693–700. doi: 10.1038/mp.2015.70
265. García-Palomares S, Pertusa JF, Miñarro J, García-Pérez MA, Hermenegildo C, Rausell F, et al. Long-term effects of delayed fatherhood in mice on postnatal development and behavioral traits of offspring. Biol Reprod. (2009) 80:337–42. doi: 10.1095/biolreprod.108.072066
266. Milekic MH, Xin Y, O'Donnell A, Kumar KK, Bradley-Moore M, Malaspina D, et al. Age-related sperm DNA methylation changes are transmitted to offspring and associated with abnormal behavior and dysregulated gene expression. Mol Psychiatry. (2015) 20:995–1001. doi: 10.1038/mp.2014.84
267. Zhao WL, Gu NH, Li ZZ, Wang GS, Cheng CY, Sun F. Autism-like behaviors and abnormality of glucose metabolism in offspring derived from aging males with epigenetically modified sperm. Aging. (2020) 12:19766–84. doi: 10.18632/aging.104061
268. Sampino S, Juszczak GR, Zacchini F, Swiergiel AH, Modlinski JA, Loi P, et al. Grand-paternal age and the development of autism-like symptoms in mice progeny. Transl Psychiatry. (2014) 4:e386. doi: 10.1038/tp.2014.27
269. Yoshizaki K, Kimura R, Kobayashi H, Oki S, Kikkawa T, Mai L, et al. Paternal age affects offspring via an epigenetic mechanism involving REST/NRSF. EMBO Rep. (2021) 22:e51524. doi: 10.15252/embr.202051524
270. Janecka M, Manduca A, Servadio M, Trezza V, Smith R, Mill J, et al. Effects of advanced paternal age on trajectories of social behavior in offspring. Genes Brain Behav. (2015) 14:443–53. doi: 10.1111/gbb.12227
271. Foldi CJ, Eyles DW, McGrath JJ, Burne TH. Advanced paternal age is associated with alterations in discrete behavioural domains and cortical neuroanatomy of C57BL/6J mice. Eur J Neurosci. (2010) 31:556–64. doi: 10.1111/j.1460-9568.2010.07074.x
272. Lerch S, Brandwein C, Dormann C, Gass P, Chourbaji S. Mice age: does the age of the mother predict offspring behaviour? Physiol Behav. (2015) 147:157–62. doi: 10.1016/j.physbeh.2015.04.041
273. Sampino S, Stankiewicz AM, Zacchini F, Goscik J, Szostak A, Swiergiel AH, et al. Pregnancy at advanced maternal age affects behavior and hippocampal gene expression in mouse offspring. J Gerontol A Biol Sci Med Sci. (2017) 72:1465–73. doi: 10.1093/gerona/glx016
274. Mao WJ, Wu ZY, Yang ZH, Xu YW, Wang SQ. Advanced maternal age impairs spatial learning capacity in young adult mouse offspring. Am J Transl Res. (2018) 10:975–88.
Keywords: autism spectrum disorder, prenatal drug exposure, valproic acid, selective serotonin reuptake inhibitor, maternal immune activation, hypertensive disorders of pregnancy, advanced parental age
Citation: Sato A, Kotajima-Murakami H, Tanaka M, Katoh Y and Ikeda K (2022) Influence of Prenatal Drug Exposure, Maternal Inflammation, and Parental Aging on the Development of Autism Spectrum Disorder. Front. Psychiatry 13:821455. doi: 10.3389/fpsyt.2022.821455
Received: 24 November 2021; Accepted: 12 January 2022;
Published: 09 February 2022.
Edited by:
Hideo Matsuzaki, University of Fukui, JapanCopyright © 2022 Sato, Kotajima-Murakami, Tanaka, Katoh and Ikeda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atsushi Sato, c2F0b2EtcGVkQGgudS10b2t5by5hYy5qcA==