 Paraskevi Krashia
Paraskevi Krashia Elena Spoleti
Elena Spoleti Marcello D'Amelio
Marcello D'Amelio
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Psychiatry , 17 October 2022
Sec. Aging Psychiatry
Volume 13 - 2022 | https://doi.org/10.3389/fpsyt.2022.1039725
This article is part of the Research Topic The Affective Side of Alzheimer’s Disease (AD): Neuropsychiatric Symptoms as Early Sentinel of Cognitive Decline and Pathogenetic Factors in Disease Progression View all 5 articles
Neuropsychiatric symptoms (NPS) occur in nearly all patients with Alzheimer's Disease (AD). Most frequently they appear since the mild cognitive impairment (MCI) stage preceding clinical AD, and have a prognostic importance. Unfortunately, these symptoms also worsen the daily functioning of patients, increase caregiver stress and accelerate the disease progression from MCI to AD. Apathy and depression are the most common of these NPS, and much attention has been given in recent years to understand the biological mechanisms related to their appearance in AD. Although for many decades these symptoms have been known to be related to abnormalities of the dopaminergic ventral tegmental area (VTA), a direct association between deficits in the VTA and NPS in AD has never been investigated. Fortunately, this scenario is changing since recent studies using preclinical models of AD, and clinical studies in MCI and AD patients demonstrated a number of functional, structural and metabolic alterations affecting the VTA dopaminergic neurons and their mesocorticolimbic targets. These findings appear early, since the MCI stage, and seem to correlate with the appearance of NPS. Here, we provide an overview of the recent evidence directly linking the dopaminergic VTA with NPS in AD and propose a setting in which the precocious identification of dopaminergic deficits can be a helpful biomarker for early diagnosis. In this scenario, treatments of patients with dopaminergic drugs might slow down the disease progression and delay the impairment of daily living activities.
Alzheimer's Disease (AD) is the prevailing form of age-associated dementia, accounting for approximately 60–80% of cases (1–3). The neuropathology of AD and the progressive cognitive decline are highly linked to the presence amyloid-beta (Aβ) and neurofibrillary tangles, whose deposition in the brain first appears in neocortical regions involved in cognition—mainly the entorhinal cortex and the hippocampal formation—and later spreads to neural hubs that underlie motor and sensory structures, planning, emotion and spatial navigation (4–10).
Neuropsychiatric symptoms (NPS) are also particularly common in the course of the disease, affecting up to half of patients with Mild Cognitive Impairment (MCI) and nearly all patients with AD-dementia (11–14). These symptoms mainly include depression, anxiety, psychosis, agitation, irritability and sleep disturbances, although apathy—described as loss of motivation, decreased interest and lack of emotional reactivity (15)—is the most common of these symptoms, with a prevalence in approximately 50% of MCI and 80% of AD patients (16–20).
Due to their appearance since the MCI stage, apathy and other NPS have an important prognostic value, as they help to predict the incidence of dementia, conversion from MCI to AD, and the acceleration in disease progression and cognitive decline (13, 21–25). However, the manifestation of these symptoms is not to be underestimated as they contribute significantly to worsening the daily functioning and disability of patients, they accelerate and prolong hospitalization and financial burden, and they worsen caregiver distress due to increased reliance of patients for everyday living (26, 27). Of note, in line with the faster disease progression in patients with NPS, recently an association was observed between apathy and an increased risk of mortality in dementia and elderly people (28, 29). Thus, interventions focused on treating NPS could have an important impact on patients, caregivers, and society. As such, an increased knowledge of the biological mechanisms underlying apathy and other symptoms in AD would result in a better perception of the disease and its etiology, and could lead to earlier and more efficient treatments and care planning.
The rising interest in NPS observed in MCI and early AD patients led to a number of investigations of brain structure in post-mortem tissue and in metabolic and neuroimaging studies, to correlate brain alterations with NPS, with main focus on apathy and depression. These studies appear to highlight abnormalities/deficits mainly in the medial frontal cortex, as well as in other brain regions such as the posterior and anterior cingulate cortex, medial orbitofrontal cortex and the ventral striatum for the development of apathetic or depressive behavior in subjects with MCI and AD (30–39). Of note, all of these regions are interconnected. More importantly still, each region receives monosynaptic dopaminergic projections from the ventral tegmental area (VTA) of the midbrain. The widespread involvement, in apathy and depression, of these brain regions modulated by dopamine perhaps makes it unsurprising that these NPS are considered, at least in part, hypodopaminergic symptoms (33, 40).
In this Perspective, we will examine in more detail the particular connections between the dopaminergic VTA and brain areas involved in the appearance of NPS and other relevant cognitive symptoms in AD. We will also describe the most recent preclinical and clinical studies that argue in favor of a precocious dysfunction of VTA dopaminergic neurons in AD and its contribution to the early appearance of NPS. Finally, we will propose a scenario in which the prompt identification of dopaminergic deficits since the MCI stage and an early dopaminergic-based pharmacological treatment of patients might be particularly beneficial for improvement of NPS, and therefore particularly relevant to test the effectiveness for slowing down the disease progression.
The VTA (area A10 in humans) is comprised of a group of notoriously heterogeneous neurons located near the midline in the midbrain, in close proximity to the more lateral substantia nigra pars compacta (SNpc, A9). In addition to dopamine neurons, the VTA contains GABAergic and glutamatergic neurons, while some dopaminergic neurons were also shown to co-release dopamine together with glutamate or GABA (41, 42).
The mesocorticolimbic dopaminergic pathway consists of cell bodies of dopamine neurons located in the VTA, that send ascending long-projection fibers rostrally toward limbic and cortical regions through the medial forebrain bundle, respectively forming the mesolimbic and mesocortical branches of the mesocorticolimbic system (Figure 1, Panel A). In recent years, the use of advanced tracing and optogenetics techniques in animals, and the improvement in imaging methods, have helped to well–define the circuit organization of the mesocorticolimbic system (41, 43–50). The most dense mesolimbic innervation from the dopaminergic VTA is at the level of the nucleus accumbens (NAc); these fibers then either remain in the NAc, innervating the core and shell subdomains, or they diverge to reach their terminal targets by extending toward the dorsal striatum and other limbic regions such as the amygdala and hippocampus. The mesocortical fibers instead, after going along the medial forebrain bundle, either bypass the NAc to extend dorsally toward the prefrontal cortex (PFC), or cross the NAc and dorsal striatum en route to the PFC. Of note, the mesocorticolimbic wiring is one of the last neuronal circuits of the brain to reach maturity. As observed both in rodents and non-human primates, this circuitry changes significantly not only during embryonic life and early postnatal development but also later on, to finally reach the mature form during adolescence (51–54). This peculiar feature of the mesocorticolimbic system is perhaps the basis for the increased vulnerability of this system to early-life stress and the many different positive or negative experiences that can drastically, and permanently, shape its development and function (55–58).
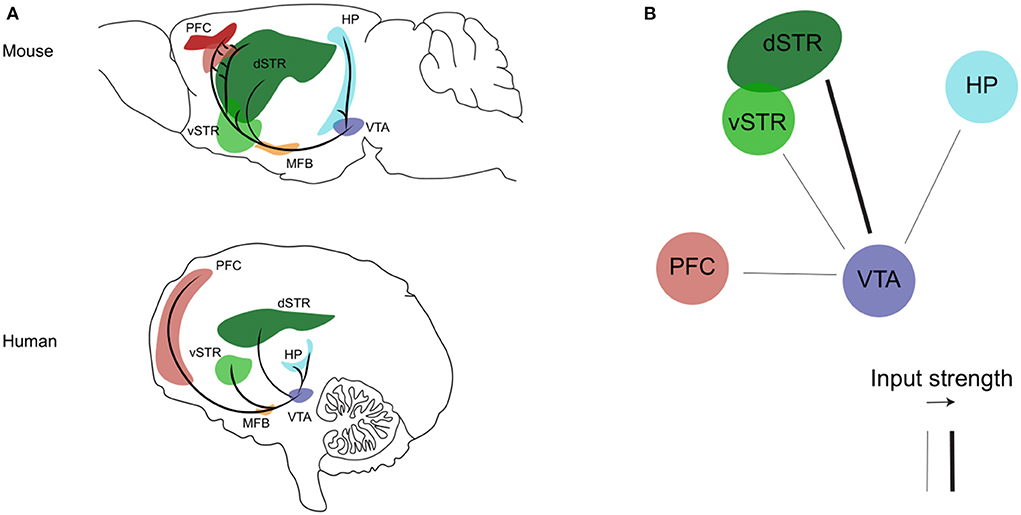
Figure 1. The mesocorticolimbic dopaminergic system. (A) Sagittal view of the mesocorticolimbic dopamine pathway and its principal target nuclei in the mouse (upper) and human (bottom) brain. (B) Representation of the impaired (thin line) dopaminergic inputs from the VTA described so far during the progression of AD (see text for details). Representations are adapted from the mouse and human brain atlas (Allen Institute). PFC, prefrontal cortex; HP, hippocampus; dSTR, dorsal striatum; vSTR, ventral striatum; MFB, medial forebrain bundle; VTA, ventral tegmental area.
The vast connections of the mesocorticolimbic dopamine system with the various cortical, subcortical and limbic regions reflects the heterogeneity of its many functions, further enhanced by the functional and molecular heterogeneity of the VTA neurons themselves. In fact, the mesolimbic and mesocortical dopamine pathways have been shown to differ in their molecular markers, electrophysiological properties, anatomical organization and response to various stimuli (41, 59–63). This intrinsic variability and extensive neuronal connections of the mesocorticolimbic system allow for the vast range of its functions: indeed this system participates, amongst others, in brain functions like fear, aversion and reward, positive and negative reinforcement, motivated behavior, working-, place- and reward-associated memory, object recognition, decision-making, cognitive and executive functions, goal-directed behaviors, temporal control and food-intake (64–73). It is therefore not surprising that the mesocorticolimbic system has been highly implicated in a number of mood and neuropsychiatric disorders, ranging from anxiety, schizophrenia and depression to food and drug addiction, attention-deficit disorders and dementia (66, 72, 74–80).
It is well–known that the dopaminergic systems undergo several changes during normal aging. For example, decreased release of dopamine from its mesocorticolimbic terminals, reduced dopamine receptor expression, and in particular D2 receptors, or reduced dopamine transporter (DAT) expression in areas like the putamen, hippocampus and PFC are all features that are commonly observed in the human brain during physiological aging (80–83). Given the link between aging and dementia, it was not surprising that dopamine has intrigued neuroscientists also in relevance to AD (84–87). Thus, a number of earlier works provided evidence of reduced expression of dopamine receptors, DAT and tyrosine hydroxylase (TH; the rate-limiting enzyme necessary for dopamine synthesis) in post-mortem brains of people with AD, particularly in mesocorticolimbic areas such as the PFC, NAc and hippocampus (88–94). Additionally, these works demonstrated that at least 40% of AD cases also show pathological changes in the VTA and its dopamine neurons including Aβ plaques, tangles and decreased dopamine content (95–101). Although indirect, the evidence provided by these works support the notion that the mesocorticolimbic dopamine system undergoes changes in AD. Yet, such works could not answer the question of whether the deficits in the dopaminergic midbrain are an early event, or one of the many devastating consequences of brain damage occurring while the disease progresses.
Direct evidence supporting the contribution of dopaminergic system dysfunctions in early AD came from a preclinical study using the Tg2576 mouse model of AD. This mouse model overexpresses the human APPswe mutant (K670M/M671L) allele of familial AD (102). With more than 1,000 papers published over the last 20 years, this transgenic model is characterized by a particularly slow disease progression and has thus been essential for disentangling the early mechanisms underlying the disease (102–104). Indeed, Tg2576 mice develop early synaptic alterations in the hippocampus and cortex, reduced spine density of hippocampal pyramidal neurons, changes in hippocampal synaptic plasticity and deficits in memory and cognition that start since about 3 months of age (102–110).
The evidence linking the dopaminergic VTA with these early deficits in Tg2576 mice came after the observation of substantial apoptotic cell death of VTA dopamine neurons in these mice, accompanied by significant levels of local neuroinflammation, starting between 2–3 months of age and progressively worsening with age (111). The work by Nobili, et al., and subsequent papers, showed that the progressive degeneration of VTA TH+ neurons results in lower dopamine outflow in the hippocampus and correlates temporally with impairments in hippocampal neuronal function, synaptic plasticity, spatial memory and fear-associated memory performance (111–113). On the other hand, other brain regions containing TH+ neurons, in particular the SNpc and locus coeruleus (LC), are intact in Tg2576 mice at least until 6 months of age, suggesting that the early degeneration is selective for mesocorticolimbic neurons in the VTA and can account for the AD-like memory symptoms in these mice (114). Importantly, subchronic in-vivo treatment of Tg2576 mice with levodopa (L-DOPA; the natural dopamine precursor) or selegiline (an inhibitor of dopamine degradation by monoamine oxidase B) can completely rescue synaptic plasticity, pyramidal neuron excitability and memory deficits (111, 112), in line with previous evidence using different dopaminergic treatments (115–121). Similarly, a pro-autophagic treatment targeting the VTA dopamine neurons in Tg2576 mice, effective in delaying the neurodegenerative process, could also ameliorate the AD phenotype (122).
Since the early work in Tg2576 mice, alterations in the VTA, reductions of DAT in the hippocampus and loss of TH+ neurons were also observed in other mouse models of AD (3×Tg-AD, APPswe/PS11E9 and 5xFAD) (123–126). Overall, these works highlighted the dopaminergic VTA as a brain region associated with early AD, showing that the reduction of the mesocorticolimbic dopamine input precedes degeneration, inflammation or plaque formation in target areas, indicating also that hippocampal-related memory dysfunction in AD is secondary to VTA dopamine neuron degeneration. Of note, given the fact that the Tg2576 mouse overexpresses the APPswe mutation, one can assume that the early degeneration of the dopaminergic VTA can be associated with the higher levels of soluble and neurotoxic Aβ oligomers in this mouse, at a stage when insoluble Aβ (Aβ plaque deposition) is still undetectable (111). Indeed, Aβ oligomers have been found to impair autophagy and mitochondrial dynamics in AD, as demonstrated in cellular models and in dopaminergic neurons of the Tg2576 mouse model, thus contributing to cell death (122, 127, 128).
Importantly, the paper by Nobili et al., could explain not only memory deficits but also the occurrence of NPS in AD (111–114). This conclusion mainly derived from the evidence that the degeneration of VTA dopamine neurons in Tg2576 mice, and the sebsequent reduction of dopaminergic innervation in limbic brain areas related to reward, stress and emotional processing (129)—such as the NAc and ventral hippocampus—were associated with deficits during consumption of palatable food, affecting not only the ability of Tg2576 animals to favor an agreeable food such as chocolate over normal chow, but also reducing the overall levels of consumption. This depressive-like behavior in Tg2576 mice was completely rescued by selegiline, as sub-chronic treatment levels could improve impairments in mesolimbic reward processing by enhancing, in treated Tg2576 mice, the preference for chocolate eating and the amount of consumption (111). Although interpretations of results from animal behavioral studies, particularly in relevance to neuropsychiatric features of human diseases, are inherently complicated, this study could prove a first link between mesocorticolimbic dopamine deficits, neuropsychiatric-like deficits and AD at an early pre-plaque disease stage.
Given the small size of brainstem nuclei like the VTA (approximately 60,000 dopamine neurons in the adult human brain), the resolution power of imaging techniques in humans has been a limiting factor that likely contributed to the neglection of such small brain regions from AD screening in the past. The first study on human subjects showing direct evidence of a functional disconnection of the VTA with brain areas normally affected in AD was the study by Serra, et al. (130). The authors recruited large numbers of amnestic MCI and sporadic AD patients as well as healthy controls and used resting-state functional MRI (rs-fMRI at 3T) to assess the functional connectivity between the VTA and different brain areas (130). The study showed a significant disconnection between the VTA and the thalamus, medial-temporal regions and the parietal lobe in MCI patients. As the disease advanced to the AD stage, patients demonstrated additionally functional disconnection of the posterior parietal cortex with the LC. The disconnection from the VTA was mainly present in the hippocampus and parietal regions in MCI patients, while in AD it involved most regions of the default-mode network (130–132). A parallel study used structural MRI to link the volume of the VTA with typical AD clinical markers, in particular hippocampal size and memory index (133, 134). The main finding of the study was that the VTA volume is strongly associated with the size of the hippocampal formation and memory abilities of both AD patients and healthy controls, thus indicating that a smaller VTA size or shrinkage of the nucleus might correspond to a worse memory performance and smaller hippocampus. This conclusion did not involve the entire midbrain, since no association was observed between the SNpc and the size of the hippocampus in AD patients (133).
Interestingly, the study by Serra, et al. (130) was the first to observe a direct association between functional disconnections of the VTA and early NPS since the MCI stage (Table 1): comparison of patients with and without these symptoms confirmed that patients with apathy, depression or anxiety showed a stronger disconnection between the VTA and the default-mode network. Subsequent studies showed additional deficits that mostly affect the mesocorticolimbic system, while leaving the nigrostriatal system largely intact. For example, by combining structural MRI and 18F-FDG-PET glucose metabolic data, Iaccarino et al., showed significant tissue atrophy, reduced metabolism and widespread loss of gray matter in VTA targets in the medial temporal lobe related to memory performance, but also in targets related to NPS such as the ventral striatum, orbito-frontal cortices and amygdala in both MCI and AD patients (136). On the other hand, regions of the nigrostriatal pathway exhibited fewer alterations (136, 139). Similar results were obtained with a DAT-Scan [(123I)FP-CIT-SPECT], focusing specifically on the dopaminergic innervations (140).
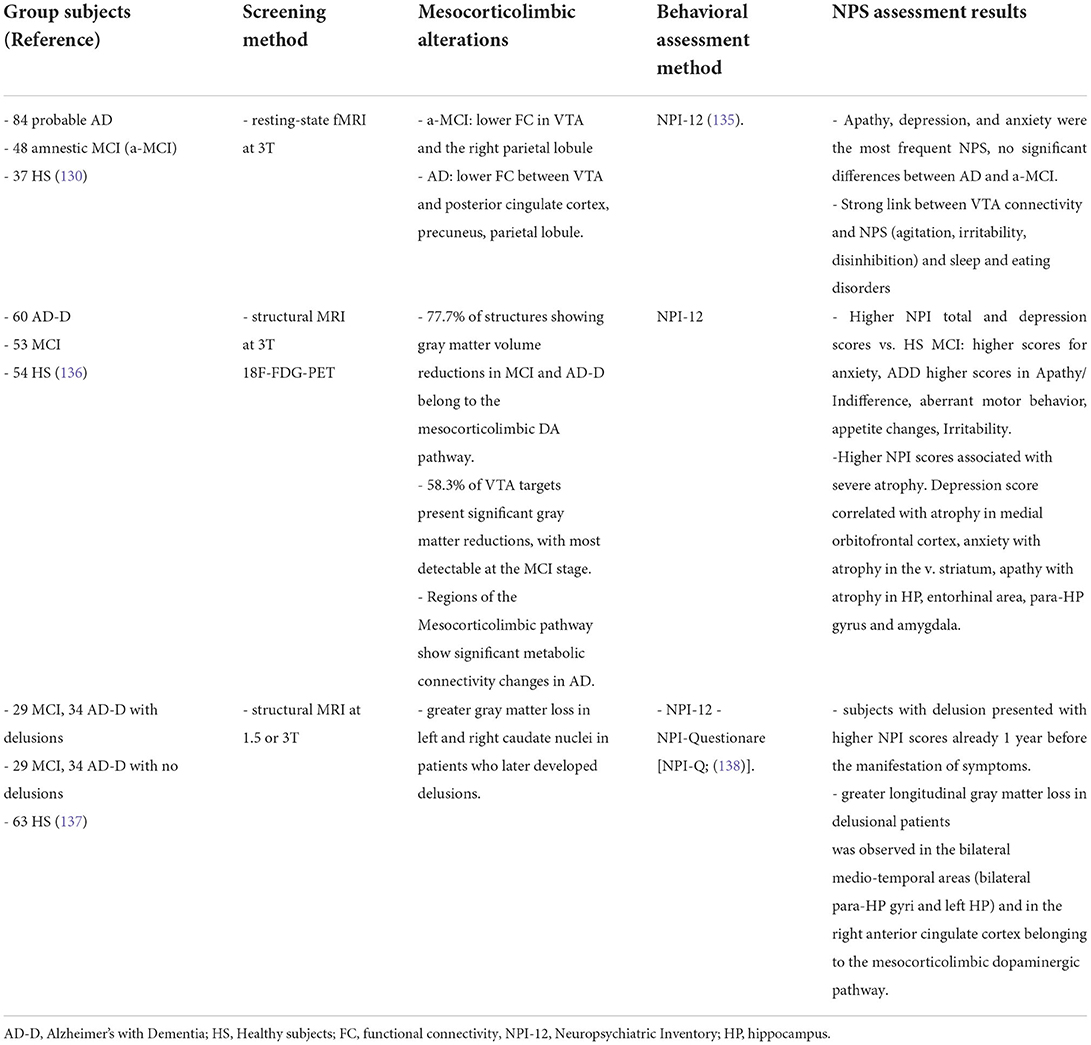
Table 1. Summary of clinical studies investigating direct evidence for an association between deficits in the mesocorticolimbic dopamine system and NPS.
An interesting finding was that the degree of loss of dopaminergic inputs in the various brain targets examined was analogous between the MCI and AD-with-dementia stages, suggesting that the dopaminergic dysfunction is an early event that also reaches a plateau early along the disease progression (140). Of note, these studies also confirmed that the extend of gray matter atrophy in mesocorticolimbic dopaminergic targets since the MCI stage was associated with greater severity of NPS, in particular depression linked to the meso-cortical route, anxiety linked to the mesolimbic route, and apathy linked to the mesohippocampal/meso-amygdaloid route (136). Indeed, stronger loss of gray matter in dopaminergic pathways was also paralleled by a worsening not only of episodic memory but also of behavioral alterations, related to the emergence of psychiatric symptoms and delusions in patients with AD (137). Given the link between the mesocorticolimbic dopamine and NPS, and the fact that these symptoms speed up the disease progression, it is not surprising that functional disconnection of the VTA with mesocorticolimbic targets can accelerate the conversion from the MCI stage to clinical AD (137, 141, 142).
Overall, the above mentioned clinical studies show that the various functional, structural or metabolic alterations affecting the dopaminergic VTA and its mesocorticolimbic targets since the MCI stage (Figure 1, panel B) are a very precocious phenomenon in the disease progression, that can be related with the early appearance of NPS. In fact, today the assessment of CSF biomarkers and the application of the Neuropsychiatric Inventory (NPI) for evaluation of NPS are standard clinical practices, with a strong proven link between the two, such as increased levels of total (t-tau) and phosphorylated tau (p-tau) proteins and low levels of Aβ42 correlating with apathy, anxiety, agitation, and irritability (143–148). Combined with these diagnostic tools, early identification of alterations in the mesocorticolimbic dopamine system could provide an additional valid biomarker aimed at the prognosis of NPS and thus prediction of disease acceleration and faster conversion from MCI to AD.
Another aspect to consider is the possibility of early intervention based on a pharmacological treatment against dopamine-related symptoms since the MCI stage, with the aim of slowing down their appearance, improving the quality of life and delaying the conversion to AD. Several dopaminergic drugs have already shown to have positive effects in mild-AD patients (80, 149–153), but their effectiveness as agents that can delay conversion from MCI to AD has yet to be investigated. Also, these earlier clinical trials with dopaminergic drugs have focused mainly on cognitive rather that neuropsychiatric deficits. Yet, today there is increasing demand for the approval of specific drugs for the treatment of NPS in AD, considering also that no such drugs have been approved so far by the FDA. Indeed, drugs usually prescribed to patients with NPS are often anxiolytics, atypical antipsychotics, antidepressants or mood stabilizers, all unapproved for AD, with uncertain efficacy and important adverse effects (154–158). Thus, focused clinical trials that are specific for NPS in MCI and AD are essential. Fortunately, progress in this field is starting to emerge with specific focus on catecholamine reuptake inhibitors like methylphenidate (159–162).
Since disease-modifying treatments have failed, new studies need to focus on a paradigm shift for preventing and treating AD. The emerging experimental and clinical results pinpoint the VTA atrophy as a supportive feature for the diagnosis of probable AD and a target for next pharmacological treatment.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
PK was supported by an under-40 grant from the Italian Association for Alzheimer's Research [AIRALTZH-AGYR2020], by the Italian Ministry of Health [Research Grant: GR-2019-12370446] and by the American Alzheimer's Association [AARG-22-922961]. ES was supported by a PhD Fellowship by Fondazione Melchiorri (IT) and by the Strategic University Projects—Young Researcher Independence Grant [YRG2021] from the University Campus Bio-Medico (Rome, Italy). MD'A was supported by the American Alzheimer's Association [AARG-18-566270; AARG-21-851219], by the Italian Ministry of Health [Research Grant: RF-2018-12365527] and by Fondazione Roma (Rome, Italy).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National institute on aging-Alzheimer's association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc. (2012) 8:1–13. doi: 10.1016/j.jalz.2011.10.007
2. Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet Lond Engl. (2006) 368:387–403. doi: 10.1016/S0140-6736(06)69113-7
3. Canter RG, Penney J, Tsai LH. The road to restoring neural circuits for the treatment of Alzheimer's disease. Nature. (2016) 539:187–96. doi: 10.1038/nature20412
4. Wong DF, Rosenberg PB, Zhou Y, Kumar A, Raymont V, Ravert HT, et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18). J Nucl Med. (2010) 51:913–20. doi: 10.2967/jnumed.109.069088
5. Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. (2004) 55:306–19. doi: 10.1002/ana.20009
6. Jack CR, Wiste HJ, Vemuri P, Weigand SD, Senjem ML, Zeng G, et al. Brain beta-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer's disease. Brain J Neurol. (2010) 133:3336–48. doi: 10.1093/brain/awq277
7. Forsberg A, Engler H, Almkvist O, Blomquist G. Hagm, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. (2008) 29:1456–65. doi: 10.1016/j.neurobiolaging.2007.03.029
8. Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol (Berl). (2011) 121:171–81. doi: 10.1007/s00401-010-0789-4
9. Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. (2002) 58:1791–800. doi: 10.1212/WNL.58.12.1791
10. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (1991) 82:239–59. doi: 10.1007/BF00308809
11. Mortby ME, Burns R, Eramudugolla R, Ismail Z, Anstey KJ. Neuropsychiatric symptoms and cognitive impairment: understanding the importance of co-morbid symptoms. J Alzheimers Dis JAD. (2017) 59:141–53. doi: 10.3233/JAD-170050
12. Mortby ME, Ismail Z, Anstey KJ. Prevalence estimates of mild behavioral impairment in a population-based sample of pre-dementia states and cognitively healthy older adults. Int Psychogeriatr. (2018) 30:221–32. doi: 10.1017/S1041610217001909
13. Peters ME, Schwartz S, Han D, Rabins PV, Steinberg M, Tschanz JT, et al. Neuropsychiatric symptoms as predictors of progression to severe Alzheimer's dementia and death: the cache county dementia progression study. Am J Psychiatry. (2015) 172:460–5. doi: 10.1176/appi.ajp.2014.14040480
14. Lyketsos CG, Carrillo MC, Ryan JM, Khachaturian AS, Trzepacz P, Amatniek J, et al. Neuropsychiatric symptoms in Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc. (2011) 7:532–9. doi: 10.1016/j.jalz.2011.05.2410
15. Miller DS, Robert P, Ereshefsky L, Adler L, Bateman D, Cummings J, et al. Diagnostic criteria for apathy in neurocognitive disorders. Alzheimers Dement J Alzheimers Assoc. (2021) 17:1892–904. doi: 10.1002/alz.12358
16. Ma L. Depression, anxiety, and apathy in mild cognitive impairment: current perspectives. Front Aging Neurosci. (2020) 12:9. doi: 10.3389/fnagi.2020.00009
17. Zhao QF, Tan L, Wang HF, Jiang T, Tan MS, Tan L, et al. The prevalence of neuropsychiatric symptoms in Alzheimer's disease: systematic review and meta-analysis. J Affect Disord. (2016) 190:264–71. doi: 10.1016/j.jad.2015.09.069
18. Sherman C, Liu CS, Herrmann N, Lanctôt KL. Prevalence, neurobiology, and treatments for apathy in prodromal dementia. Int Psychogeriatr. (2018) 30:177–84. doi: 10.1017/S1041610217000527
19. Mega MS, Cummings JL, Fiorello T, Gornbein J. The spectrum of behavioral changes in Alzheimer's disease. Neurology. (1996) 46:130–5. doi: 10.1212/WNL.46.1.130
20. Lanctôt KL, Agüera-Ortiz L, Brodaty H, Francis PT, Geda YE, Ismail Z, et al. Apathy associated with neurocognitive disorders: recent progress and future directions. Alzheimers Dement J Alzheimers Assoc. (2017) 13:84–100. doi: 10.1016/j.jalz.2016.05.008
21. Rog LA, Park LQ, Harvey DJ, Huang CJ, Mackin S, Farias ST. The independent contributions of cognitive impairment and neuropsychiatric symptoms to everyday function in older adults. Clin Neuropsychol. (2014) 28:215–36. doi: 10.1080/13854046.2013.876101
22. Clarke DE, Ko JY, Lyketsos C, Rebok GW, Eaton WW. Apathy and cognitive and functional decline in community-dwelling older adults: results from the Baltimore ECA longitudinal study. Int Psychogeriatr. (2010) 22:819–29. doi: 10.1017/S1041610209991402
23. Barnes DE, Yaffe K, Byers AL, McCormick M, Schaefer C, Whitmer RA. Midlife vs late-life depressive symptoms and risk of dementia: differential effects for Alzheimer disease and vascular dementia. Arch Gen Psychiatry. (2012) 69:493–8. doi: 10.1001/archgenpsychiatry.2011.1481
24. Yang Y, Kwan RYC, Zhai HM, Xu XY, Huang CX, Liang SJ, et al. The association among apathy, leisure activity participation, and severity of dementia in nursing home residents with Alzheimer's disease: a cross-sectional study. Geriatr Nurs N Y N. (2021) 42:1373–8. doi: 10.1016/j.gerinurse.2021.09.009
25. Liew TM. Neuropsychiatric symptoms in early stage of Alzheimer's and non-Alzheimer's dementia, and the risk of progression to severe dementia. Age Ageing. (2021) 50:1709–18. doi: 10.1093/ageing/afab044
26. Geda YE, Roberts RO, Knopman DS, Petersen RC, Christianson TJH, Pankratz VS, et al. Prevalence of neuropsychiatric symptoms in mild cognitive impairment and normal cognitive aging: population-based study. Arch Gen Psychiatry. (2008) 65:1193–8. doi: 10.1001/archpsyc.65.10.1193
27. van der Linde RM, Matthews FE, Dening T, Brayne C. Patterns and persistence of behavioural and psychological symptoms in those with cognitive impairment: the importance of apathy. Int J Geriatr Psychiatry. (2017) 32:306–15. doi: 10.1002/gps.4464
28. Nijsten JMH, Leontjevas R, Pat-El R, Smalbrugge M, Koopmans RTCM, Gerritsen DL. Apathy: risk factor for mortality in nursing home patients. J Am Geriatr Soc. (2017) 65:2182–9. doi: 10.1111/jgs.15007
29. Hölttä EH, Laakkonen ML, Laurila JV, Strandberg TE, Tilvis RS, Pitkälä KH. Apathy: prevalence, associated factors, and prognostic value among frail, older inpatients. J Am Med Dir Assoc. (2012) 13:541–5. doi: 10.1016/j.jamda.2012.04.005
30. Holthoff VA, Beuthien-Baumann B, Kalbe E, Lüdecke S, Lenz O, Zündorf G, et al. Regional cerebral metabolism in early Alzheimer's disease with clinically significant apathy or depression. Biol Psychiatry. (2005) 57:412–21. doi: 10.1016/j.biopsych.2004.11.035
31. Levy R, Dubois B. Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cereb Cortex N Y N 1991. (2006) 16:916–28. doi: 10.1093/cercor/bhj043
32. Chan NK, Gerretsen P, Chakravarty MM, Blumberger DM, Caravaggio F, Brown E, et al. Structural brain differences between cognitively impaired patients with and without apathy. Am J Geriatr Psychiatry. (2021) 29:319–32. doi: 10.1016/j.jagp.2020.12.008
33. Le Heron C, Apps M a. J, Husain M. The anatomy of apathy: A neurocognitive framework for amotivated behavior. Neuropsychologia. (2018) 118:54–67. doi: 10.1016/j.neuropsychologia.2017.07.003
34. Theleritis C, Politis A, Siarkos K, Lyketsos CG, A. review of neuroimaging findings of apathy in Alzheimer's disease. Int Psychogeriatr. (2014) 26:195–207. doi: 10.1017/S1041610213001725
35. Delrieu J, Desmidt T, Camus V, Sourdet S, Boutoleau-Bretonnière C, Mullin E, et al. Apathy as a feature of prodromal Alzheimer's disease: an FDG-PET ADNI study. Int J Geriatr Psychiatry. (2015) 30:470–7. doi: 10.1002/gps.4161
36. Gatchel JR, Donovan NJ, Locascio JJ, Becker JA, Rentz DM, Sperling RA, et al. Regional 18F-fluorodeoxyglucose hypometabolism is associated with higher apathy scores over time in early Alzheimer disease. Am J Geriatr Psychiatry. (2017) 25:683–93. doi: 10.1016/j.jagp.2016.12.017
37. Bruen PD, McGeown WJ, Shanks MF, Venneri A. Neuroanatomical correlates of neuropsychiatric symptoms in Alzheimer's disease. Brain J Neurol. (2008) 131:2455–63. doi: 10.1093/brain/awn151
38. Mortby ME, Adler L, Agüera-Ortiz L, Bateman DR, Brodaty H, Cantillon M, et al. Apathy as a treatment target in Alzheimer's disease: implications for clinical trials. Am J Geriatr Psychiatry. (2022) 30:119–47. doi: 10.1016/j.jagp.2021.06.016
39. Lavretsky H, Zheng L, Weiner MW, Mungas D, Reed B, Kramer JH, et al. The MRI brain correlates of depressed mood, anhedonia, apathy, and anergia in older adults with and without cognitive impairment or dementia. Int J Geriatr Psychiatry. (2008) 23:1040–50. doi: 10.1002/gps.2030
40. Udo N, Hashimoto N, Toyonaga T, Isoyama T, Oyanagi Y, Narita H, et al. Apathy in Alzheimer's disease correlates with the dopamine transporter level in the caudate nuclei. Dement Geriatr Cogn Disord Extra. (2020) 10:86–93. doi: 10.1159/000509278
41. Morales M, Margolis EB. Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci. (2017) 18:73–85. doi: 10.1038/nrn.2016.165
42. Liss B, Roeper J. Individual dopamine midbrain neurons: functional diversity and flexibility in health and disease. Brain Res Rev. (2008) 58:314–21. doi: 10.1016/j.brainresrev.2007.10.004
43. Skagerberg G, Lindvall O, Björklund A. Origin, course and termination of the mesohabenular dopamine pathway in the rat. Brain Res. (1984) 307:99–108. doi: 10.1016/0006-8993(84)90465-7
44. Björklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. (2007) 30:194–202. doi: 10.1016/j.tins.2007.03.006
45. Björklund A, Dunnett SB. Fifty years of dopamine research. Trends Neurosci. (2007) 30:185–7. doi: 10.1016/j.tins.2007.03.004
46. Sesack SR, Grace AA. Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacol. (2010) 35:27–47. doi: 10.1038/npp.2009.93
47. Yetnikoff L, Lavezzi HN, Reichard RA, Zahm DS. An update on the connections of the ventral mesencephalic dopaminergic complex. Neuroscience. (2014) 282:23–48. doi: 10.1016/j.neuroscience.2014.04.010
48. Gasbarri A, Packard MG, Campana E, Pacitti C. Anterograde and retrograde tracing of projections from the ventral tegmental area to the hippocampal formation in the rat. Brain Res Bull. (1994) 33:445–52. doi: 10.1016/0361-9230(94)90288-7
49. Reynolds LM, Flores C. Mesocorticolimbic dopamine pathways across adolescence: diversity in development. Front Neural Circuits. (2021) 15:735625. doi: 10.3389/fncir.2021.735625
50. Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L, et al. Circuit architecture of VTA dopamine neurons revealed by systematic input-output mapping. Cell. (2015) 162:622–34. doi: 10.1016/j.cell.2015.07.015
51. Berger B, Verney C, Alvarez C, Vigny A, Helle KB. New dopaminergic terminal fields in the motor, visual (area 18b) and retrosplenial cortex in the young and adult rat. Immunocytochemical and catecholamine histochemical analyses. Neuroscience. (1985) 15:983–98. doi: 10.1016/0306-4522(85)90248-9
52. Rosenberg DR, Lewis DA. Changes in the dopaminergic innervation of monkey prefrontal cortex during late postnatal development: a tyrosine hydroxylase immunohistochemical study. Biol Psychiatry. (1994) 36:272–7. doi: 10.1016/0006-3223(94)90610-6
53. Rosenberg DR, Lewis DA. Postnatal maturation of the dopaminergic innervation of monkey prefrontal and motor cortices: a tyrosine hydroxylase immunohistochemical analysis. J Comp Neurol. (1995) 358:383–400. doi: 10.1002/cne.903580306
54. Lewis DA, Sesack SR. Chapter VI dopamine systems in the primate brain. In:Bloom FE, Björklund A, Hökfelt T, , editors Handbook of Chemical Neuroanatomy. Elsevier. (1997). doi: 10.1016/S0924-8196(97)80008-5
55. Barth B, Portella AK, Dubé L, Meaney MJ, Silveira PP. The Interplay Between Dopamine and Environment as the Biological Basis for the Early Origins of Mental Health. In:Vaiserman, A editors. Early Life Origins of Ageing and Longevity Healthy Ageing and Longevity. Springer: Cham (2019). doi: 10.1007/978-3-030-24958-8
56. Reynolds LM, Flores C. Chapter 26 - Adolescent dopamine development: connecting experience with vulnerability or resilience to psychiatric disease. Diagnosis, Management and Modeling of Neurodevelopmental Disorders. Academic Press (2021). p. 295–304. doi: 10.1016/C2018-0-02068-X
57. Hanson JL, Williams AV, Bangasser DA, Peña CJ. Impact of early life stress on reward circuit function and regulation. Front Psychiatry. (2021) 12:744690. doi: 10.3389/fpsyt.2021.744690
58. Hollon NG, Burgeno LM, Phillips PEM. Stress effects on the neural substrates of motivated behavior. Nat Neurosci. (2015) 18:1405–12. doi: 10.1038/nn.4114
59. Roeper J. Dissecting the diversity of midbrain dopamine neurons. Trends Neurosci. (2013) 36:336–42. doi: 10.1016/j.tins.2013.03.003
60. Lammel S, Lim BK, Malenka RC. Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology. (2014) 76:351–9. doi: 10.1016/j.neuropharm.2013.03.019
61. Nguyen C, Mondoloni S, Le Borgne T, Centeno I, Come M, Jehl J, et al. Nicotine inhibits the VTA-to-amygdala dopamine pathway to promote anxiety. Neuron. (2021) 109:2604–15.e9. doi: 10.1016/j.neuron.2021.06.013
62. Poulin JF, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C, et al. Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat Neurosci. (2018) 21:1260–71. doi: 10.1038/s41593-018-0203-4
63. Krashia P, Martini A, Nobili A, Aversa D, D'Amelio M, Berretta N, et al. On the properties of identified dopaminergic neurons in the mouse substantia nigra and ventral tegmental area. Eur J Neurosci. (2017) 45:92–105. doi: 10.1111/ejn.13364
64. Adcock RA, Thangavel A, Whitfield-Gabrieli S, Knutson B, Gabrieli JDE. Reward-motivated learning: mesolimbic activation precedes memory formation. Neuron. (2006) 50:507–17. doi: 10.1016/j.neuron.2006.03.036
65. Brischoux F, Chakraborty S, Brierley DI, Ungless MA. Phasic excitation of dopamine neurons in ventral VTA by noxious stimuli. Proc Natl Acad Sci U S A. (2009) 106:4894–9. doi: 10.1073/pnas.0811507106
66. Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron. (2010) 68:815–34. doi: 10.1016/j.neuron.2010.11.022
67. Schultz W. Getting formal with dopamine and reward. Neuron. (2002) 36:241–63. doi: 10.1016/S0896-6273(02)00967-4
68. Tsai HC, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, et al. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science. (2009) 324:1080–4. doi: 10.1126/science.1168878
69. Puig MV, Rose J, Schmidt R, Freund N. Dopamine modulation of learning and memory in the prefrontal cortex: insights from studies in primates, rodents, and birds. Front Neural Circuits. (2014) 8:93. doi: 10.3389/fncir.2014.00093
70. Narayanan NS, Land BB, Solder JE, Deisseroth K, DiLeone RJ. Prefrontal D1 dopamine signaling is required for temporal control. Proc Natl Acad Sci U S A. (2012) 109:20726–31. doi: 10.1073/pnas.1211258109
71. Land BB, Narayanan NS, Liu RJ, Gianessi CA, Brayton CE, Grimaldi DM, et al. Medial prefrontal D1 dopamine neurons control food intake. Nat Neurosci. (2014) 17:248–53. doi: 10.1038/nn.3625
72. McNamara CG, Tejero-Cantero Á, Trouche S, Campo-Urriza N, Dupret D. Dopaminergic neurons promote hippocampal reactivation and spatial memory persistence. Nat Neurosci. (2014) 17:1658–60. doi: 10.1038/nn.3843
73. Rosen ZB, Cheung S, Siegelbaum SA. Midbrain dopamine neurons bidirectionally regulate CA3-CA1 synaptic drive. Nat Neurosci. (2015) 18:1763–71. doi: 10.1038/nn.4152
74. Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. (2013) 493:532–6. doi: 10.1038/nature11713
75. Lammel S, Tye KM, Warden MR. Progress in understanding mood disorders: optogenetic dissection of neural circuits. Genes Brain Behav. (2014) 13:38–51. doi: 10.1111/gbb.12049
76. Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. (2013) 493:537–41. doi: 10.1038/nature11740
77. Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. (2013) 14:609–25. doi: 10.1038/nrn3381
78. Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. (2011) 70:855–62. doi: 10.1016/j.neuron.2011.03.025
79. Grace AA, Floresco SB, Goto Y, Lodge DJ. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. (2007) 30:220–7. doi: 10.1016/j.tins.2007.03.003
80. Martorana A, Koch G. Is dopamine involved in Alzheimer's disease? Front Aging Neurosci. (2014) 6:252. doi: 10.3389/fnagi.2014.00252
81. Norrara B, Fiuza FP, Arrais AC, Costa IM, Santos JR, Engelberth RCGJ, et al. Pattern of tyrosine hydroxylase expression during aging of mesolimbic pathway of the rat. J Chem Neuroanat. (2018) 92:83–91. doi: 10.1016/j.jchemneu.2018.05.004
82. Bäckman L, Lindenberger U, Li SC, Nyberg L. Linking cognitive aging to alterations in dopamine neurotransmitter functioning: recent data and future avenues. Neurosci Biobehav Rev. (2010) 34:670–7. doi: 10.1016/j.neubiorev.2009.12.008
83. Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley SJ, et al. Dopamine transporters decrease with age. J Nucl Med. (1996) 37:554–9.
84. Attems J, Quass M, Jellinger KA. Tau and alpha-synuclein brainstem pathology in Alzheimer disease: relation with extrapyramidal signs. Acta Neuropathol (Berl). (2007) 113:53–62. doi: 10.1007/s00401-006-0146-9
85. Portet F, Scarmeas N, Cosentino S, Helzner EP, Stern Y. Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch Neurol. (2009) 66:1120–6. doi: 10.1001/archneurol.2009.196
86. Trillo L, Das D, Hsieh W, Medina B, Moghadam S, Lin B, et al. Ascending monoaminergic systems alterations in Alzheimer's disease. Translating basic science into clinical care. Neurosci Biobehav Rev. (2013) 37:1363–79. doi: 10.1016/j.neubiorev.2013.05.008
87. D'Amelio M, Puglisi-Allegra S, Mercuri N. The role of dopaminergic midbrain in Alzheimer's disease: Translating basic science into clinical practice. Pharmacol Res. (2018) 130:414–9. doi: 10.1016/j.phrs.2018.01.016
88. Allard P, Alafuzoff I, Carlsson A, Eriksson K, Ericson E, Gottfries CG, et al. Loss of dopamine uptake sites labeled with [3H]GBR-12935 in Alzheimer's disease. Eur Neurol. (1990) 30:181–5. doi: 10.1159/000117341
89. Joyce JN, Smutzer G, Whitty CJ, Myers A, Bannon MJ. Differential modification of dopamine transporter and tyrosine hydroxylase mRNAs in midbrain of subjects with Parkinson's, Alzheimer's with parkinsonism, and Alzheimer's disease. Mov Disord. (1997) 12:885–97. doi: 10.1002/mds.870120609
90. Kemppainen N, Laine M, Laakso MP, Kaasinen V, Någren K, Vahlberg T, et al. Hippocampal dopamine D2 receptors correlate with memory functions in Alzheimer's disease. Eur J Neurosci. (2003) 18:149–54. doi: 10.1046/j.1460-9568.2003.02716.x
91. Kumar U, Patel SC. Immunohistochemical localization of dopamine receptor subtypes (D1R-D5R) in Alzheimer's disease brain. Brain Res. (2007) 1131:187–96. doi: 10.1016/j.brainres.2006.10.049
92. Murray AM, Weihmueller FB, Marshall JF, Hurtig HI, Gottleib GL, Joyce JN. Damage to dopamine systems differs between Parkinson's disease and Alzheimer's disease with parkinsonism. Ann Neurol. (1995) 37:300–12. doi: 10.1002/ana.410370306
93. Rinne JO, Säkö E, Paljärvi L, Mölsä PK, Rinne UK. Brain dopamine D-1 receptors in senile dementia. J Neurol Sci. (1986) 73:219–30. doi: 10.1016/0022-510X(86)90132-2
94. Rinne JO, Säkö E, Paljärvi L, Mölsä PK, Rinne UK. Brain dopamine D-2 receptors in senile dementia. J Neural Transm. (1986) 65:51–62. doi: 10.1007/BF01249611
95. Burns JM, Galvin JE, Roe CM, Morris JC, McKeel DW. The pathology of the substantia nigra in Alzheimer disease with extrapyramidal signs. Neurology. (2005) 64:1397–403. doi: 10.1212/01.WNL.0000158423.05224.7F
96. Gibb WR, Mountjoy CQ, Mann DM, Lees AJ. The substantia nigra and ventral tegmental area in Alzheimer's disease and Down's syndrome. J Neurol Neurosurg Psychiatry. (1989) 52:193–200. doi: 10.1136/jnnp.52.2.193
97. Storga D, Vrecko K, Birkmayer JG, Reibnegger G. Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neurosci Lett. (1996) 203:29–32. doi: 10.1016/0304-3940(95)12256-7
98. Lopez OL, Wisnieski SR, Becker JT, Boller F, DeKosky ST. Extrapyramidal signs in patients with probable Alzheimer disease. Arch Neurol. (1997) 54:969–75. doi: 10.1001/archneur.1997.00550200033007
99. Braak H, Braak E. Alzheimer's disease: striatal amyloid deposits and neurofibrillary changes. J Neuropathol Exp Neurol. (1990) 49:215–24. doi: 10.1097/00005072-199005000-00003
100. Rudelli RD, Ambler MW, Wisniewski HM. Morphology and distribution of Alzheimer neuritic (senile) and amyloid plaques in striatum and diencephalon. Acta Neuropathol (Berl). (1984) 64:273–81. doi: 10.1007/BF00690393
101. Selden N, Mesulam MM, Geula C. Human striatum: the distribution of neurofibrillary tangles in Alzheimer's disease. Brain Res. (1994) 648:327–31. doi: 10.1016/0006-8993(94)91136-3
102. Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. (1996) 274:99–102. doi: 10.1126/science.274.5284.99
103. Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, et al. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging. (1999) 20:581–9. doi: 10.1016/S0197-4580(99)00065-2
104. Hall AM, Roberson ED. Mouse models of Alzheimer's disease. Brain Res Bull. (2012) 88:3–12. doi: 10.1016/j.brainresbull.2011.11.017
105. Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. (1999) 2:271–6. doi: 10.1038/6374
106. D'Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci. (2011) 14:69–76. doi: 10.1038/nn.2709
107. Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. (2006) 103:5161–6. doi: 10.1073/pnas.0600948103
108. Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, et al. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. (2002) 22:1858–67. doi: 10.1523/JNEUROSCI.22-05-01858.2002
109. Cavallucci V, Berretta N, Nobili A, Nisticò R, Mercuri NB, D'Amelio M. Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer's disease. Neuromolecular Med. (2013) 15:541–8. doi: 10.1007/s12017-013-8241-2
110. Corsetti V, Borreca A, Latina V, Giacovazzo G, Pignataro A, Krashia P, et al. Passive immunotherapy for N-truncated tau ameliorates the cognitive deficits in two mouse Alzheimer's disease models. Brain Commun. (2020) 2:fcaa039. doi: 10.1093/braincomms/fcaa039
111. Nobili A, Latagliata EC, Viscomi MT, Cavallucci V, Cutuli D, Giacovazzo G, et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer's disease. Nat Commun. (2017) 8:14727. doi: 10.1038/ncomms14727
112. Cordella A, Krashia P, Nobili A, Pignataro A, La Barbera L, Viscomi MT, et al. Dopamine loss alters the hippocampus-nucleus accumbens synaptic transmission in the Tg2576 mouse model of Alzheimer's disease. Neurobiol Dis. (2018) 116:142–54. doi: 10.1016/j.nbd.2018.05.006
113. Spoleti E, Krashia P, La Barbera L, Nobili A, Lupascu CA, Giacalone E, et al. Early derailment of firing properties in CA1 pyramidal cells of the ventral hippocampus in an Alzheimer's disease mouse model. Exp Neurol. (2021) 350:113969. doi: 10.1016/j.expneurol.2021.113969
114. Krashia P, Nobili A, D'Amelio M. Unifying hypothesis of dopamine neuron loss in neurodegenerative diseases: focusing on Alzheimer's disease. Front Mol Neurosci. (2019) 12:123. doi: 10.3389/fnmol.2019.00123
115. Ambrée O, Richter H, Sachser N, Lewejohann L, Dere E, de Souza Silva MA, et al. Levodopa ameliorates learning and memory deficits in a murine model of Alzheimer's disease. Neurobiol Aging. (2009) 30:1192–204. doi: 10.1016/j.neurobiolaging.2007.11.010
116. Guzmán-Ramos K, Moreno-Castilla P, Castro-Cruz M, McGaugh JL, Martínez-Coria H, LaFerla FM, et al. Restoration of dopamine release deficits during object recognition memory acquisition attenuates cognitive impairment in a triple transgenic mice model of Alzheimer's disease. Learn Mem Cold Spring Harb N. (2012) 19:453–60. doi: 10.1101/lm.026070.112
117. Hao JR, Sun N, Lei L, Li XY, Yao B, Sun K, et al. L-Stepholidine rescues memory deficit and synaptic plasticity in models of Alzheimer's disease via activating dopamine D1 receptor/PKA signaling pathway. Cell Death Dis. (2015) 6:e1965. doi: 10.1038/cddis.2015.315
118. Himeno E, Ohyagi Y, Ma L, Nakamura N, Miyoshi K, Sakae N, et al. Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation. Ann Neurol. (2011) 69:248–56. doi: 10.1002/ana.22319
119. Jürgensen S, Antonio LL, Mussi GEA, Brito-Moreira J, Bomfim TR, De Felice FG, et al. Activation of D1/D5 dopamine receptors protects neurons from synapse dysfunction induced by amyloid-beta oligomers. J Biol Chem. (2011) 286:3270–6. doi: 10.1074/jbc.M110.177790
120. Pazini AM, Gomes GM, Villarinho JG, da Cunha C, Pinheiro F, Ferreira APO, et al. Selegiline reverses aβ25?35-induced cognitive deficit in male mice. Neurochem Res. (2013) 38:2287–94. doi: 10.1007/s11064-013-1137-6
121. Tsunekawa H, Noda Y, Mouri A, Yoneda F, Nabeshima T. Synergistic effects of selegiline and donepezil on cognitive impairment induced by amyloid beta (25-35). Behav Brain Res. (2008) 190:224–32. doi: 10.1016/j.bbr.2008.03.002
122. La Barbera L, Vedele F, Nobili A, Krashia P, Spoleti E, Latagliata EC, et al. Nilotinib restores memory function by preventing dopaminergic neuron degeneration in a mouse model of Alzheimer's Disease. Prog Neurobiol. (2021) 202:102031. doi: 10.1016/j.pneurobio.2021.102031
123. Gloria Y, Ceyzériat K, Tsartsalis S, Millet P, Tournier BB. Dopaminergic dysfunction in the 3xTg-AD mice model of Alzheimer's disease. Sci Rep. (2021) 11:19412. doi: 10.1038/s41598-021-99025-1
124. Liu LX, Chen WF, Xie JX, Wong MS. Neuroprotective effects of genistein on dopaminergic neurons in the mice model of Parkinson's disease. Neurosci Res. (2008) 60:156–61. doi: 10.1016/j.neures.2007.10.005
125. Moreno-Castilla P, Rodriguez-Duran LF, Guzman-Ramos K, Barcenas-Femat A, Escobar ML, Bermudez-Rattoni F. Dopaminergic neurotransmission dysfunction induced by amyloid-β transforms cortical long-term potentiation into long-term depression and produces memory impairment. Neurobiol Aging. (2016) 41:187–99. doi: 10.1016/j.neurobiolaging.2016.02.021
126. Vorobyov V, Bakharev B, Medvinskaya N, Nesterova I, Samokhin A, Deev A, et al. Loss of midbrain dopamine neurons and altered apomorphine EEG effects in the 5xFAD mouse model of Alzheimer's disease. J Alzheimers Dis JAD. (2019) 70:241–56. doi: 10.3233/JAD-181246
127. de la Cueva M, Antequera D, Ordoñez-Gutierrez L, Wandosell F, Camins A, Carro E, et al. Amyloid-β impairs mitochondrial dynamics and autophagy in Alzheimer's disease experimental models. Sci Rep. (2022) 12:10092. doi: 10.1038/s41598-022-13683-3
128. Guglielmotto M, Monteleone D, Piras A, Valsecchi V, Tropiano M, Ariano S, et al. Aβ1-42 monomers or oligomers have different effects on autophagy and apoptosis. Autophagy. (2014) 10:1827–43. doi: 10.4161/auto.30001
129. Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. (2012) 76:470–85. doi: 10.1016/j.neuron.2012.10.021
130. Serra L, D'Amelio M, Di Domenico C, Dipasquale O, Marra C, Mercuri NB, et al. In vivo mapping of brainstem nuclei functional connectivity disruption in Alzheimer's disease. Neurobiol Aging. (2018) 72:72–82. doi: 10.1016/j.neurobiolaging.2018.08.012
131. Gili T, Cercignani M, Serra L, Perri R, Giove F, Maraviglia B, et al. Regional brain atrophy and functional disconnection across Alzheimer's disease evolution. J Neurol Neurosurg Psychiatry. (2011) 82:58–66. doi: 10.1136/jnnp.2009.199935
132. Bozzali M, Giulietti G, Basile B, Serra L, Spanò B, Perri R, et al. Damage to the cingulum contributes to Alzheimer's disease pathophysiology by deafferentation mechanism. Hum Brain Mapp. (2012) 33:1295–308. doi: 10.1002/hbm.21287
133. De Marco M, Venneri A. Volume and Connectivity of the ventral tegmental area are linked to neurocognitive signatures of Alzheimer's disease in humans. J Alzheimers Dis JAD. (2018) 63:167–80. doi: 10.3233/JAD-171018
134. D'Amelio M, Serra L, Bozzali M. Ventral tegmental area in prodromal Alzheimer's disease: bridging the gap between mice and humans. J Alzheimers Dis JAD. (2018) 63:181–3. doi: 10.3233/JAD-180094
135. Cummings JL. The neuropsychiatric inventory: assessing psychopathology in dementia patients. Neurology. (1997) 48(5 Suppl. 6):10S−16S. doi: 10.1212/WNL.48.5_Suppl_6.10S
136. Iaccarino L, Sala A, Caminiti SP, Presotto L, Perani D. Alzheimer's Disease Neuroimaging Initiative. In vivo MRI structural and PET metabolic connectivity study of dopamine pathways in Alzheimer's disease. J Alzheimers Dis JAD. (2020) 75:1003–16. doi: 10.3233/JAD-190954
137. Manca R, Valera-Bermejo JM, Venneri A. Alzheimer's disease neuroimaging initiative. Accelerated atrophy in dopaminergic targets and medial temporo-parietal regions precedes the onset of delusions in patients with Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci. (2022). doi: 10.1007/s00406-022-01417-5
138. Kaufer DI, Cummings JL, Ketchel P, Smith V, MacMillan A, Shelley T, et al. Validation of the NPI-Q, a brief clinical form of the neuropsychiatric inventory. J Neuropsychiatry Clin Neurosci. (2000) 12:233–9. doi: 10.1176/jnp.12.2.233
139. Hsu TW, Fuh JL, Wang DW, Chen LF, Chang CJ, Huang WS, et al. Disrupted metabolic connectivity in dopaminergic and cholinergic networks at different stages of dementia from 18F-FDG PET brain persistent homology network. Sci Rep. (2021) 11:5396. doi: 10.1038/s41598-021-84722-8
140. Sala A, Caminiti SP, Presotto L, Pilotto A, Liguori C, Chiaravalloti A, et al. In vivo human molecular neuroimaging of dopaminergic vulnerability along the Alzheimer's disease phases. Alzheimers Res Ther. (2021) 13:187. doi: 10.1186/s13195-021-00925-1
141. Serra L, D'Amelio M, Esposito S, Di Domenico C, Koch G, Marra C, et al. Ventral tegmental area disconnection contributes two years early to correctly classify patients converted to Alzheimer's disease: implications for treatment. J Alzheimers Dis JAD. (2021) 82:985–1000. doi: 10.3233/JAD-210171
142. Venneri A, De Marco M. Reduced monoaminergic nuclei MRI signal detectable in pre-symptomatic older adults with future memory decline. Sci Rep. (2020) 10:18707. doi: 10.1038/s41598-020-71368-1
143. Skogseth R, Mulugeta E, Jones E, Ballard C, Rongve A, Nore S, et al. Neuropsychiatric correlates of cerebrospinal fluid biomarkers in Alzheimer's disease. Dement Geriatr Cogn Disord. (2008) 25:559–63. doi: 10.1159/000137671
144. Scaricamazza E, Colonna I, Sancesario GM, Assogna F, Orfei MD, Franchini F, et al. Neuropsychiatric symptoms differently affect mild cognitive impairment and Alzheimer's disease patients: a retrospective observational study. Neurol Sci. (2019) 40:1377–82. doi: 10.1007/s10072-019-03840-4
145. Ramakers IHGB, Verhey FRJ, Scheltens P, Hampel H, Soininen H, Aalten P, et al. Anxiety is related to Alzheimer cerebrospinal fluid markers in subjects with mild cognitive impairment. Psychol Med. (2013) 43:911–20. doi: 10.1017/S0033291712001870
146. Babulal GM, Ghoshal N, Head D, Vernon EK, Holtzman DM, Benzinger TLS, et al. Mood changes in cognitively normal older adults are linked to alzheimer disease biomarker levels. Am J Geriatr Psychiatry. (2016) 24:1095–104. doi: 10.1016/j.jagp.2016.04.004
147. Johansson M, Stomrud E, Johansson PM, Svenningsson A, Palmqvist S, Janelidze S, et al. Development of apathy, anxiety, and depression in cognitively unimpaired older adults: effects of Alzheimer's disease pathology and cognitive decline. Biol Psychiatry. (2022) 92:34–43. doi: 10.1016/j.biopsych.2022.01.012
148. Sun L, Li W, Li G, Xiao S. Alzheimer's disease neuroimaging initiative. Prefrontal Aβ pathology influencing the pathway from apathy to cognitive decline in non-dementia elderly. Transl Psychiatry. (2021) 11:534. doi: 10.1038/s41398-021-01653-8
149. Koch G, Esposito Z, Codecà C, Mori F, Kusayanagi H, Monteleone F, et al. Altered dopamine modulation of LTD-like plasticity in Alzheimer's disease patients. Clin Neurophysiol. (2011) 122:703–7. doi: 10.1016/j.clinph.2010.10.033
150. Koch G, Di Lorenzo F, Bonnì S, Giacobbe V, Bozzali M, Caltagirone C, et al. Dopaminergic modulation of cortical plasticity in Alzheimer's disease patients. Neuropsychopharmacol. (2014) 39:2654–61. doi: 10.1038/npp.2014.119
151. Monteverde A, Gnemmi P, Rossi F, Monteverde A, Finali GC. Selegiline in the treatment of mild to moderate Alzheimer-type dementia. Clin Ther. (1990) 12:315–22.
152. Koch G, Motta C, Bonnì S, Pellicciari MC, Picazio S, Casula EP, et al. Effect of rotigotine vs placebo on cognitive functions among patients with mild to moderate alzheimer disease: a randomized clinical trial. JAMA Netw Open. (2020) 3:e2010372. doi: 10.1001/jamanetworkopen.2020.10372
153. Matthews DC, Ritter A, Thomas RG, Andrews RD, Lukic AS, Revta C, et al. Rasagiline effects on glucose metabolism, cognition, and tau in Alzheimer's dementia. Alzheimers Dement N Y N. (2021) 7:e12106. doi: 10.1002/trc2.12106
154. Vigen CLP, Mack WJ, Keefe RSE, Sano M, Sultzer DL, Stroup TS, et al. Cognitive effects of atypical antipsychotic medications in patients with Alzheimer's disease: outcomes from CATIE-AD. Am J Psychiatry. (2011) 168:831–9. doi: 10.1176/appi.ajp.2011.08121844
155. Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA. (2005) 293:596–608. doi: 10.1001/jama.293.5.596
156. Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer's disease drug development pipeline: 2021. Alzheimers Dement N Y N. (2021) 7:e12179. doi: 10.1002/trc2.12179
157. Cummings JL, Mackell J, Kaufer D. Behavioral effects of current Alzheimer's disease treatments: a descriptive review. Alzheimers Dement. (2008) 4:49–60. doi: 10.1016/j.jalz.2007.10.011
158. Cummings J. Disease modification is not all - we need symptomatic therapies for Alzheimer disease. Nat Rev Neurol. (2022) 18:3–4. doi: 10.1038/s41582-021-00591-9
159. Mintzer J, Lanctôt KL, Scherer RW, Rosenberg PB, Herrmann N, van Dyck CH, et al. Effect of methylphenidate on apathy in patients with Alzheimer disease: the ADMET 2 randomized clinical trial. JAMA Neurol. (2021) 78:1324–32. doi: 10.1001/jamaneurol.2021.3356
160. Rosenberg PB, Lanctôt KL, Drye LT, Herrmann N, Scherer RW, Bachman DL, et al. Safety and efficacy of methylphenidate for apathy in Alzheimer's disease: a randomized, placebo-controlled trial. J Clin Psychiatry. (2013) 74:810–6. doi: 10.4088/JCP.12m08099
161. Tariot PN, Cummings JL, Soto-Martin ME, Ballard C, Erten-Lyons D, Sultzer DL, et al. Trial of pimavanserin in dementia-related psychosis. N Engl J Med. (2021) 385:309–19. doi: 10.1056/NEJMoa2034634
Keywords: neuropsychiatric symptoms, mesolimbic, mesocortical, ventral tegmental area, midbrain, preclinical AD, DAT Scan, Tg2576
Citation: Krashia P, Spoleti E and D'Amelio M (2022) The VTA dopaminergic system as diagnostic and therapeutical target for Alzheimer's disease. Front. Psychiatry 13:1039725. doi: 10.3389/fpsyt.2022.1039725
Received: 08 September 2022; Accepted: 28 September 2022;
Published: 17 October 2022.
Edited by:
Roberto Coccurello, National Research Council (CNR), ItalyReviewed by:
Ahmad Adam Khundakar, Teesside University, United KingdomCopyright © 2022 Krashia, Spoleti and D'Amelio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paraskevi Krashia, ay5wYXJhc2tldmlAdW5pY2FtcHVzLml0; Marcello D'Amelio, bS5kYW1lbGlvQHVuaWNhbXB1cy5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.