 Collin Gagne
Collin Gagne Alexandre Piot
Alexandre Piot Wayne G. Brake
Wayne G. Brake- Department of Psychology, Centre for Studies in Behavioural Neurobiology Concordia University, Montreal, QC, Canada
Ketamine has been shown to acutely and rapidly ameliorate depression symptoms and suicidality. Given that women suffer from major depression at twice the rate of men, it is important to understand how ketamine works in the female brain. This review explores three themes. First, it examines our current understanding of the etiology of depression in women. Second, it examines preclinical research on ketamine's antidepressant effects at a neurobiological level as well as how ovarian hormones present a unique challenge in interpreting these findings. Lastly, the neuroinflammatory hypothesis of depression is highlighted to help better understand how ovarian hormones might interact with ketamine in the female brain.
Ovarian Hormones & Depression
Sex differences in mood disorder prevalence are common and can emerge during adolescence (1, 2). Depression rates are nearly two-fold higher in women than men aged 14–25 (3). Yet, these sex differences decrease with age, such that depression prevalence among men and women tend to be similar around late adulthood (4). A cross-national comparison revealed that women aren't at even more increased risk for depression in countries where sex inequalities are more marked (5) although some caveats were noted in the analysis. This could indicate that potentially confounding factors are not responsible for the differences in prevalence rates. Other findings suggest that sex differences in depression prevalence may be due to inherent factors such as sex steroid hormones in combination external factors such as increased exposure to early life adversity and gender inequities (6).
Estrogens are a family of ovarian hormones, often collectively referred to as estrogens (E), of which there are three subtypes, viz. estrone, estradiol, and estriol. The most potent estrogen, in terms of its affinity for estrogen receptors in both rodents and humans, is 17β- estradiol (7). The ovaries synthesize and release the bulk of E in a cyclical manner, peaking during ovulation. Yet, in both sexes, the adrenal glands as well as neurons and glial cells in the central nervous system also synthesize and release E, albeit in much smaller amounts.
Like E, another ovarian hormone, progesterone (P) is mostly synthesized in the ovaries in a cyclical manner, specifically by the corpus luteum, a temporary gland formed during the ovulatory phase of the human menstrual cycle and after the proestrus phase of the rat estrus cycle. The estrus cycle is the term for the female sex steroid cycle in most non-primate mammals. P is also produced by the placenta once it's formed during gestation, and the adrenal glands, and again by cells in the central nervous system where it's formed by steroidogenesis from cholesterol in both males and females (8, 9).
Douma et al. (10) suggest that a reduction of E levels, a continued deficit of E, or changes in E levels throughout the menstrual cycle are correlated with mood-related distress (11). This idea is also supported by multiple studies demonstrating a link between post-partum depression (PPD) and E levels (12–14). PPD is the emergence of a depressive episode after parturition (i.e., giving birth). This disorder affects between 7 and 20% of women who have given birth and is thought to be caused by fluctuations in E and P levels [see Schiller et al. (15) for review]. While some studies have been unsuccessful in establishing a relationship between hormone levels and PPD in women (16, 17) others have found that treating PPD with estradiol alleviates symptoms (12, 18). Animal models have shown that E and P removal can lead to depressive-like behaviors (19, 20) and hormone replacement can ameliorate these behaviors. It would be wrong, however, to perceive depression as simply linked to a decrease in ovarian hormones in females. Indeed, premenstrual dysphoric disorder (PMDD) illustrates how an increase, rather than decrease, in ovarian hormones is associated with depressive symptoms (21, 22).
PMDD affects 3–8% of premenopausal women and can repeat monthly. Symptoms, such as depressive mood, occur in the luteal phase when P and E are on the rise (23). Importantly, the hormonal profiles of women with PMDD are indistinguishable from those without (24). This suggests that PMDD sufferers are perhaps more reactive to ovarian hormones at a neurobiological level. Furthermore, early menarche, i.e., first menstruation, in girls (prior to 11.5 years of age) has been associated with an increase in E and an increase in depressive disorders in adolescence (25). Other evidence demonstrating an ovarian hormone-depression link comes from menopause and hormone replacement therapy (HRT) in older women.
In the past a substantial proportion of post-menopausal women making use of HRT at one point in their lives, with some estimates being as high as 38% (26). Today, due to efforts demonstrating associations between HRT and various negative health outcomes, post-menopausal women will typically receive HRT between the ages of 50–59 (or <10 y post-menopause) and for shorter time periods (27–29). In such cases HRT typically consists of E, but progestins, such as P, can also be administered either alone or in tandem with E (26, 30). The effects of HRT on depressed mood during menopause are promising. One meta-analysis of 26 studies reveals that E have a considerable effect size (d = 0.69) toward decreasing depressed mood, whereas P solely or in tandem with E was associated with less potent effects (31).
Considering the evidence, it is clear that ovarian hormones may worsen or improve depressive symptoms, but the direction of the relation can change due to various factors. The fluctuating nature of the menstrual cycle, as well as the physiological and cellular changes it entails, renders simplistic generalizations unworkable. To better understand the role of the menstrual cycle in depression one should consider how hormone fluctuations affect the expression of depressive symptoms in tandem with cellular processes, particularly in the brain. To do so, we must firstly consider the neurobiological basis of depression.
Hypotheses of Depression
There are multiple models suggesting what may cause depression, none of which are mutually exclusive, highlighting the complexity of the underlying etiology. One of the more prolific, but dated, theories of depression is the monoamine hypothesis. Put simply, this theory suggest that major depressive disorder (MDD) is caused by depleted levels of the monoamine neurotransmitters dopamine, norepinephrine, or serotonin; or some combination of these (32, 33). While there is evidence in support this theory, for example monoamine depletion diets (34), there is plenty that contradicts it, i.e., the slow onset and moderate efficacy of antidepressants targeting monoamines (35–39).
Another hypothesis, the glutamate hypothesis of depression, posits that MDD is linked to altered glutamate transmission and metabolism in the brain. Studies indicate that there is an increase of glutamate in the plasma of depressed patients compared to healthy controls (40, 41). This can be reversed with the administration of SSRIs (40, 42). This idea suggests that excessive glutamate transmission, associated with a decrease in synaptic clearance by neighboring glial cells, results in cellular toxicity and a reduction in brain volume (43). Brain imaging studies show that individuals suffering from MDD and anxiety-related disorders have smaller hippocampal and prefrontal cortical volumes (44–47). This theory attributes such atrophy to inflammatory glial cells transitioning to an inflammatory state, causing lowered synaptic clearance, neurotoxicity, and phagocytosis of healthy neurons. Moreover, stress, a major predictor of depression (48, 49), has been shown to increase synaptic glutamate in the hippocampus [HPC; (50)] and prefrontal cortex of rats (51). It is proposed that while glial cells carry out the inflammatory response, glutamate metabolism and synaptic clearance may decline, resulting in modified glutamate transmission which could contribute to the emergence of depression. In vivo studies have found lower levels of glutamate metabolites in the cingulate (52) and frontal regions (53) of the prefrontal cortex. These alterations in glutamate metabolism have been intricately associated with depression and treatment resistance (45).
The Neurotrophic Theory of Depression
Another idea is that neurodegeneration and atrophy is of paramount importance to depression's etiology, also known as the neurotrophic hypothesis of depression. Stress can, under some circumstances, cause neuronal apoptosis, dendritic atrophy, and decreases in trophic factors in the HPC, where atrophy and a lower volume is often observed among depressed individuals (54–56). Santarelli et al. (57) showed that induction of hippocampal neurogenesis is necessary for antidepressants to produce behavioral effects in a mouse model of depression. Furthermore, electroconvulsive therapy (ECT), causes increases in trophic factors in the HPC of rats (58).
Brain-derived neurotrophic factor (BDNF) is the most well-studied trophic factor in the context of antidepressants and research supports the idea that BDNF plays an important role in MDD. Post-mortem research shows that the HPC of humans with MDD had lowered levels of BDNF, while those who were taking antidepressants at the time of their death had higher levels (59). Similar effects have been observed in both the HPC and prefrontal cortex (PFC) of suicide victims in comparison to controls (60, 61). It has been proposed that BDNF levels, in both serum (62–65) and plasma (66, 67) are a viable biomarker for MDD and even prescription adherence. This is supported by studies showing that BDNF levels are increased when humans (63, 68–71), or rodents (58, 72) are administered antidepressants. When BDNF itself is administered, either directly to the HPC (73, 74), or peripherally (75), antidepressant-like effects are observed in rodents. Work by Shirayama et al. (73) showed that these effects last for as long as 10 days after the infusion, long after the protein has degraded, suggesting that BDNF triggers mechanisms which sustain its effects on plasticity. Finally, other, non-pharmacological antidepressant interventions, such as exercise (76) and ECT (58) have been shown to increase central BDNF levels in rodents.
The Neuroinflammatory (Cytokine) Theory of Depression
The neuroinflammatory model of depression, also known as the cytokine hypothesis, is comprehensive in the sense that it ties together both the monoamine and neurotrophic theories of depression. To put it simply, this theory posits that depression is caused by inflammatory processes (77–80) that involve microglia.
Microglia, a type of macrophage, can act as part of the brain's immune response and make up approximately 10% of cells in the central nervous system. Microglia are responsible for many processes such as inducing apoptosis in nearby neurons and synaptic pruning, both of which are necessary for healthy brain development and maintaining homeostasis (81–84). A microglial response to threats could entail the release of reactive nitrogen and oxygen species to cause oxidative stress and/or apoptosis in infected neurons (79). Microglia also release small proteins known as cytokines, which can be pro-inflammatory such as; interleukin (IL)−1, 2, 6, and 18 (83, 85, 86), tumor necrosis factor (TNF) α (87) and interferon (IFN) γ (88), or anti-inflammatories, such as IL-4, 10, and 13 (89–91). One signal that microglia use to extend their processes toward a target neuron is adenosine triphosphate [ATP; (92)]. It was found that N-methyl-D-aspartate receptor (NMDA) receptor activation mediates this ATP release. However, it is hypothesized that this mechanism of microglial surveillance has evolved because ATP is also released during apoptosis, thus acting as a “find me” signal for all immune cells (92–94). Importantly, microglia often engage in phagocytosis, when they extend a cup-shaped process to engulf a given target (95). The idea that microglia-mediated neuroinflammation contributes to the etiology of depression is not a new one (96, 97). Sickness behavior, an adaptive behavioral strategy where motivational state is reorganized to optimize coping with illness (98) may be related to depressive behaviors. While sickness behavior clearly demonstrates how depressive behavior is elicited by immune challenges, the neuroinflammatory hypothesis of depression posits that stress is the first step leading to depression (79).
Common psychosocial stressors, such as lack of social support or exam stress have been shown to increase the production and release of pro-inflammatory cytokines (99, 100). Researchers have shown that the Trier social stress test elicits increases in circulating IL-6 (101) and Il-1β (102). Aschbacher et al. (102) showed that peripheral immunoreactivity to stress even predicts future depressive symptoms in a cohort of post-menopausal women and menopause has been associated with an increase in pro-inflammatory cytokines (103). Studies of older adults have also found that levels of peripheral pro-inflammatory IL-1 family cytokines were positively correlated with future depressive symptoms (104, 105).
van den Biggerlaar et al. (105) demonstrated that ex vivo whole blood cytokine production in response to LPS (i.e., lipopolysaccharide, which is used to generate an immune response) administration also predicted future depressive symptoms. Stress-induced increases in pro-inflammatory cytokines, which are increased in people with depression (77–80, 97, 106–108), have been positively correlated with severity of depressive symptoms (109). Pro-inflammatory cytokines affect the CNS through several mechanisms which are hypothesized to then cause depression.
Centrally administered LPS, which causes microglia to release pro-inflammatory cytokines, has been shown to reduce rat hippocampal neurogenesis (110). In fact, the number of activated microglia, indicated by CD68 labeling, was inversely correlated with new neurons. These findings showed that new hippocampal neurons simply do not survive around activated microglia. Conversely, inhibiting microglial activation, via minocycline, gives rise to increases in new neurons. The damaging effects of activated microglia to their neighboring neurons are likely mediated by cytokines such as IL-1β and IL-6 (111, 112). Monje et al. (113) also confirmed that microglial activation is negatively associated with neurogenesis. This work ties together both the neuroinflammatory and neurodegenerative hypotheses of depression.
Common to both the monoamine and neuroinflammatory hypotheses of depression is the kynurenine pathway, which consists of the following reactions: Tryptophan, an essential amino acid and precursor for the monoamine neurotransmitter serotonin [5-HT; (114, 115)], is catabolized into kynurenine by the enzyme, and rate-limiting factor (116), indoleamine 2,3-dioxygenase [IDO; (117)]. Kynurenine is then converted into either quinolinic acid (QUIN), a neurotoxic NMDA-receptor agonist, or kynurenic acid, an NMDA, α-amino-3-hydroxy-5- methyl-4-isoxazolepropionic acid (AMPA), and kainate receptor antagonist, which has been also shown to be protective against excitotoxicity (118, 119). The kynurenine pathway is controlled by the immune system, with the synthesis of certain downstream metabolites, like QUIN, occurring within microglia (120, 121). When this pathway is more actively engaged, for example due to pro-inflammatory cytokines which induce IDO (122–124), less tryptophan is available for 5-HT synthesis (125–129). This directly relates the neuroinflammatory and monoamine hypotheses of depression. Indeed, this process resulting in 5-HT depletion has been implicated in the etiology of depression for decades (130).
Ketamine: A Novel Antidepressant
The first administration of ketamine to humans occurred at Jackson prison in 1964 soon after its synthesis (131). Decades later, it was discovered to have rapid antidepressant effects among individuals with MDD (132). Historically, ketamine was largely used as an animal anesthetic and sedative and still is today. Additionally, sub-anesthetic doses of ketamine have robust analgesic effects while having little impact on the respiratory system in comparison to opioids, like morphine or fentanyl (133). Ketamine's neuroprotective and anti-inflammatory effects led to its use in emergency medicine for burn patients and its calming and dissociative effects have led it to be administered to patients who are suicidal or in shock (134, 135). When Berman et al. (132) first reported ketamine's antidepressant effects among individuals with MDD, it was largely ignored. It was another 5 years before the study was replicated (136), prompting the original study to become highly cited. Since Zarate et al. (136) replicated Berman's work, ketamine's antidepressant effects have been well-established. A clinical trial was initiated at the National Institute of Mental Health (137), and the medication Spravato (S- ketamine) by Johnson & Johnson was approved by the U.S. Food and Drug Administration as a breakthrough treatment in March 2019, although only indicated for TRD. The neurobiological mechanisms responsible for ketamine's antidepressant effects are not yet fully understood, but several theories have been postulated.
The Glutamate Burst Hypothesis of Ketamine Action
The glutamate burst hypothesis [a.k.a. the disinhibition hypothesis; (138)] posits that ketamine effectively reverses the synaptic and dendritic spine atrophy, notably in the PFC and HPC, known to be associated with stress and depression. Central to this hypothesis is the NMDA receptor (NMDAR).
The NMDA receptor is composed of GluN1 and GluN2 subunits, typically two of each, forming a heterotetramer. The complex can also contain GluN3 subunits (139, 140). The subunits are further divided into isoforms (GluN1A, GluN1B, GluN2A, etc.). NMDARs can be thought of as both ligand and voltage gated. For the channel to open, glycine (or D-serine) must bind to GluN1, glutamate must be bound to GluN2 subunits, and the neuronal membrane must be depolarized to a sufficient degree such that the Mg2+ blockade at the center of the receptor pore is removed. Only when these conditions are met does the channel become permeable to Na+, K+, and importantly Ca2+ ions, producing a plethora of intracellular second messenger cascades (141). Ketamine is a non-competitive NMDAR antagonist, but like other NMDAR ligands, its pharmacological interactions vary based on subunit composition. Ketamine, MK-801, and phencyclidine, block the NMDAR channel at the same site, and are more likely to do so when the receptor is composed of GluN1/GluN2A or GluN1/GluN2B subunit configurations (139).
Gerhard et al. (142) suggest that ketamine leads to a burst of glutamatergic transmission which ultimately reverses synaptic and dendritic spine atrophy, and that this occurs in five steps. The glutamate burst hypothesis: a proposed mechanism of action for ketamine. First, ketamine blocks NMDARs located on inhibitory GABA interneurons. The same research group showed this occurs preferentially through blocking NMDARs on GABA interneurons in mice, specifically in the mPFC (143). This disinhibition occurs preferentially on GABA interneurons likely due to their higher frequency of firing compared to pyramidal neurons. Faster firing allows for NMDARs to be freer of their Mg2+ ion blockade, granting ketamine greater access to the NMDA channel to inhibit its opening (138). This idea is supported by both human research, by evidence showing ketamine increases overall PFC activity in healthy individuals (144), and rodent research, by evidence that MK-801, specifically increases pyramidal neuron-firing in the PFC (145). Second, by blocking the NMDA channels on GABA interneurons, ketamine reduces tonic firing of these interneurons, subsequently resulting in a disinhibition of glutamate neurons, and a burst in glutamate release. Gerhard et al. (143) showed that ketamine's blockade of NMDARs on GABA interneurons in the mPFC caused a disinhibitory net effect in the form of an increase in excitatory postsynaptic currents in layer V primary neurons. Furthermore, the effect appeared to occur through GluN2B-containing NMDARs in male mice only, as was demonstrated via knockdown and genetic deletion models in mice, suggesting sex-differences at a molecular level (143). Third, as a result of a presynaptic glutamate burst, AMPA receptors are activated and upregulated on postsynaptic sites of PFC pyramidal neurons, resulting in greater depolarization of the postsynaptic membrane. Nearby post-synaptic voltage-gated calcium channels open and an influx of calcium occurs. Fourth, BDNF is released by the postsynaptic neuron and binds to tropomyosin receptor kinase B (TrkB) receptors, also on the postsynaptic neuron, initiating several intracellular second messenger cascades. TrkB receptors auto-phosphorylate, allowing them to affect cellular function for extended periods of time. This could explain how centrally administered BDNF produces prolonged antidepressant effects (73). Lastly, targets of rapamycin complex 1 (mTORC1) signaling proteins are rapidly phosphorylated, increasing spine density in the PFC via proteins like postsynaptic density protein (PSD) 95, and synapsin. The importance of the mTORC1 signaling cascade is illustrated by paradigms which block mTORC1 via rapamycin, reducing ketamine's antidepressant-like effects in animal models (138, 142, 146, 147).
Furthermore, convergent pathways which increase synaptogenesis and protein synthesis (such as the mTOR pathway) have been proposed as potential mechanism for the antidepressant properties of psychedelic drugs as such lysergic acid diethylamide, psilocybin, or dimethyltryptamine in addition to ketamine [reviewed in Aleksandrova and Phillips (148)].
Beyond the Glutamate Burst Hypothesis
Another hypothesis of ketamine's mechanism of action–proposed, in part, by several research groups–holds that a glutamate burst is not necessary (149–151). Instead, ketamine's NMDA antagonism blocks spontaneous miniature excitatory postsynaptic currents at rest, which are mediated by NMDA activation. It is suggested that it is the deactivation of eukaryotic elongation factor 2 (eEF2) kinase (a.k.a. CaMKIII), not the activation of mTORC1, at the root of this effect. Deactivation of eEF2 kinase, via ketamine administration, was shown to induce BDNF and dendritic protein translation, including AMPA subunits, in mouse models. This paradigm suggests that NMDA receptor activity at rest allows for eEF2 kinase to chronically phosphorylate eEF2, which then suppresses translation. It is suggested that acutely blocking NMDA, via ketamine, stops eEF2 phosphorylation via eEF2 kinase deactivation, which produces antidepressant-like effects (149–151).
A third ketamine hypothesis (152) posits that NMDA antagonism, and subsequent second-messenger cascades, is not central to ketamine's antidepressant effects. This is because ketamine's R enantiomer produces greater antidepressant-like effects in rodent models than the S enantiomer, despite R-ketamine having an almost four-fold lower affinity (Ki = 2.57) for the NMDA receptor than S-ketamine [Ki = 0.69; (153)]. Research supporting this hypothesis shows that ketamine's antidepressant effects actually occur via its metabolites, specifically 2R and 6R hydroxynorketamine (2R−6R HNK), acting on AMPA receptors. Administering deuterated ketamine, which is not metabolized into other compounds due to a change in its molecular structure, produces no antidepressant-like effects in rodent models. Furthermore, administering 2R-6R HNK with 2,3-dihydroxy-6-nitro-7-sulfamoyl- benzo(f)quinoxaline-2,3-dione (NBQX), an AMPA receptor antagonist, causes no antidepressant-like effects, whereas, without NBQX, the 2R-6R HNK metabolites produces the most potent effects compared to other metabolites (152). Proponents of this theory, therefore suggest ketamine's mechanism of action lies with its ability to activate AMPA receptors (152, 154, 155).
Ketamine in Females
For the above hypotheses of ketamine's antidepressant effects, most of the data has been derived from studies on males. As one might expect, there is a growing body of evidence which suggests that these theories do not hold the same for females. In support of the glutamate burst hypothesis, male rodents with social isolation stress (SIS) have a decrease in sucrose preference, a measure of anhedonia, as well as decreases in medial PFC (mPFC) neuron spine density, PSD- 95, and synapsin. Three hours after a single ketamine infusion all of these effects are reversed. In females, SIS also leads to a decrease in mPFC spine density, PSD-95 and synapsin, yet no changes in sucrose preference and none of these effects were reversed by ketamine (156). Furthermore, ketamine induces hippocampal glutamate in males but not females, and induces aspartate in the mPFC in females but not males (157). All this evidence is contrary to the glutamate burst hypothesis and suggests that the glutamatergic system in females reacts to ketamine differently than in males. Interestingly, female rats require half the minimum dose that males do to produce antidepressant effects, but this effect is only seen when both E2 and P are present (158, 159). This suggests that there are sex differences in the mechanism of action of ketamine and that ovarian hormones may play a role in how it acts in the female brain.
Carrier and Kabbaj (158) have shown that ketamine does not decrease eEF2 in the HPC and PFC of female rats, whereas this decrease has been shown to occur consistently in males (149–151, 158). One theory, supported by evidence in both males and females, places primary importance on ketamine's metabolites. However, while both 2R and 6R-HNK are more abundant in females than males, so too are 2S and 6S-HNK (152, 160). This evidence may explain the sex differences in terms of sensitivity to ketamine's antidepressant-like effects in rodents, but it only muddies the waters in terms of how ketamine may be exerting sex-specific effects. Making the story even less clear, the few meta-analyses conducted among clinically treated individuals have revealed no sex differences in ketamine's antidepressant effects (161), or a slightly higher sensitivity among males, but only at 7 days post-infusion (162). Although more research emphasizing potential sex differences must be carried out to replicate these findings, this suggests that there exists no difference between the sexes among humans in terms of response to ketamine.
When looking to the more clinically relevant S-ketamine, researchers have shown it is eliminated at a faster rate in women than in men, although this investigation was performed in the context of analgesia (163). In fact, there is a dearth of research investigating clinical sex differences when it comes to the efficacy of S-ketamine in comparison to racemic ketamine. One non-inferiority clinical trial (N = 63) looked to compare efficacy between the two molecules and found there were no differences between S-ketamine and racemic ketamine in improving depression. Sex differences were not analyzed but given the size, and results, of the study it is unlikely that sex might have accounted for clinically relevant effects (164). Following this trend, Bahji et al. (165) conducted a meta-analysis to explore whether any differences might exist between the two molecules and examined 24 studies (N = 1,877), but failed to include sex-differences in any analyses or discussion. Importantly, the authors did find that IV racemic ketamine alleviates the symptoms of depression more effectively than intranasal S-ketamine. The literature is lacking a critically important meta-analysis of sex-differences among ketamine clinical trials. Nevertheless, a much more recent and comprehensive theory on the etiology of depression may be the missing link in explaining ketamine's effects in the female brain.
Microglia, Neuroinflammation, and Ketamine
Recently, the kynurenine pathway was suggested as a mechanism through which ketamine might affect neurodegeneration via microglia-mediated neuroinflammation (166). Several studies now have shown that LPS induces QUIN in microglia (167–169). Moreover, higher levels of neurotoxic QUIN was found in the cerebrospinal fluid of suicide victims (170). Furthermore, QUIN was increased specifically in microglia in the post-mortem brains of severely depressed individuals (171).
Establishing ketamine's relevance to this paradigm-shift in our understanding of the neurobiology of depression, Walker's et al. (172) showed that ketamine reverses LPS- induced depressive-like behavior in rodents. What's more, this effect occurred when ketamine was given 10 h after LPS administration, giving the inflammatory and kynurenine pathways enough time to be activated. Pre-treatment with ketamine blocked LPS-induced depressive-like behavior from developing. Interestingly, ketamine had no impact on LPS-induced inflammatory activation in the form of plasma cytokine levels. The researchers hypothesized that ketamine, an NMDA antagonist, was instead blocking QUIN's ability to act as an NMDA-receptor agonist. As mentioned previously, a requirement for ketamine's antidepressant effects to take hold is the upregulation of AMPA-mediated glutamatergic neurotransmission, likely due to ketamine's metabolites (152, 154, 155). Their hypothesis was confirmed via administering NBQX 15 min before ketamine, restoring depressive-like behavior (166, 172).
Verdonk et al. (173) were the first to show that microglia are a direct target of ketamine and, specifically, the production of QUIN within microglia. In a mouse model of LPS-induced depression ketamine induced changes in microglia resulting in a neuroprotective phenotype. They observed that ketamine reversed the LPS-induced increase in QUIN in brain parenchyma, whereas ketamine had no effect on QUIN in control animals. Translating this work to a clinical perspective, the researchers also tested kynurenine pathway metabolite concentration in 15 individuals with TRD in response to ketamine. They found that the KYNA:QUIN ratio before the first ketamine infusion to be a significant predictor of final Montgomery–Åsberg Depression Rating Scale (MADRS) score. This effect was driven primarily by QUIN plasma levels. QUIN concentrations before each ketamine infusion were the best predictor of ketamine efficacy and were the only significant predictor of relative change (i.e., before and after ketamine) in MADRS scores. These findings by Verdonk et al. (173) confirm the hypothesis of Walker et al. (172) that ketamine directly affects microglial QUIN levels, in both mice and humans, resulting in protection from neuroinflammatory processes, and that the degree to which ketamine impacts QUIN levels correlates with relief of depressive symptoms in humans.
Further linking ketamine to inflammatory processes, researchers have shown that ketamine reverses the effects of LPS, in the form of pro-inflammatory cytokine release, in microglia in vitro (174). This makes sense, given that others have shown that NMDA and LPS have very similar effects on microglia. Both produce a release in pro- inflammatory cytokines and the adoption of a more amoeboid-like shape, indicating a more active state (175). NMDA receptors have been implicated in cytokine-induced neurotoxicity in previous research as well. When Chao et al. (176) administered both IL-β and TNF-α to in vitro fetal brain cells, they noticed a marked increase in neuronal injury. When these cytokines were administered in conjunction with MK-801, the increase in neuronal injury was ablated. Recently, researchers showed that ketamine reduced pro-inflammatory cytokines, microglia phagocytic markers in the rat HPC, and depressive-like behavior in a rat model (177), see Figure 1.
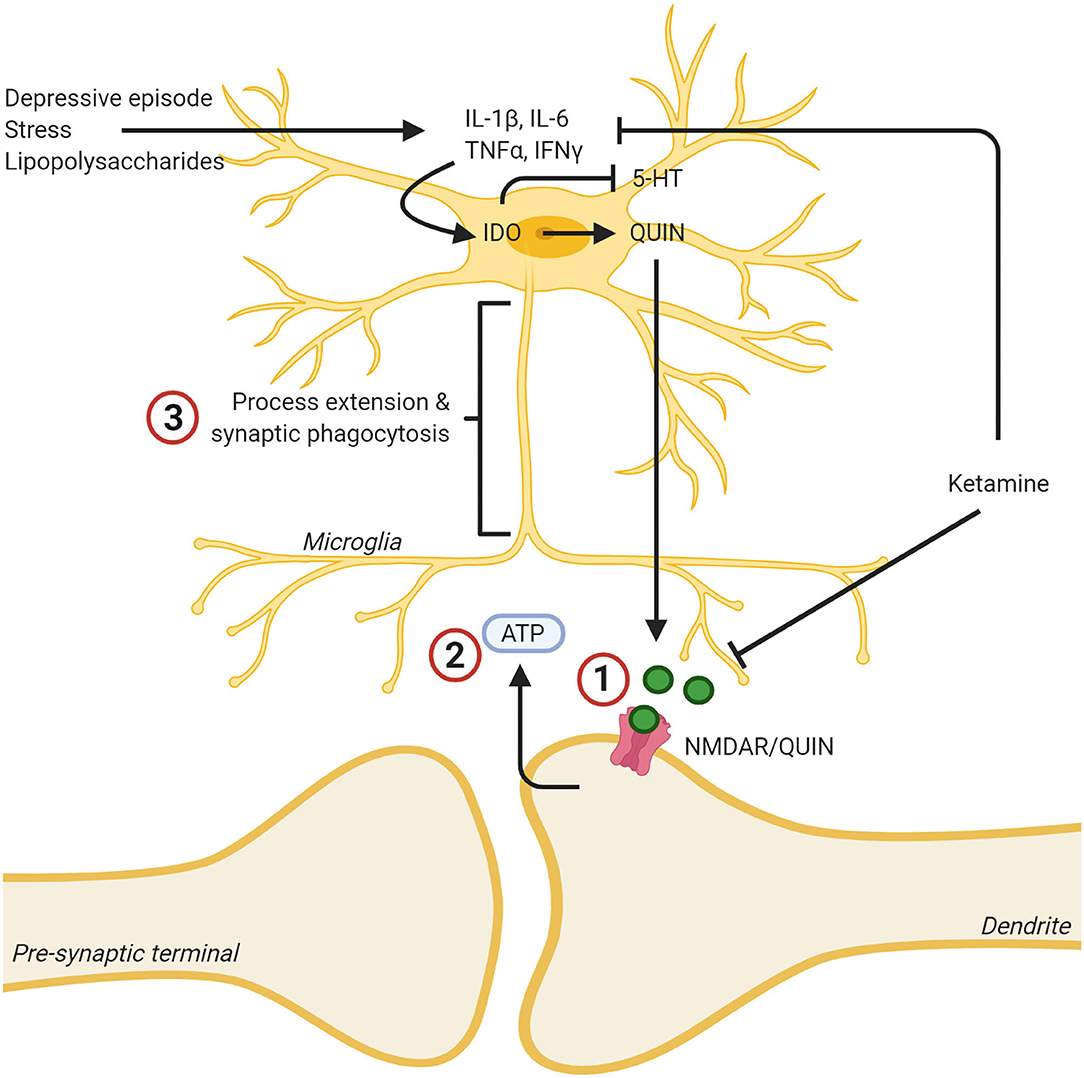
Figure 1. Ketamine's effects on the kynurenine pathway leads to lowered levels of neurotoxic quinolinic acid (QUIN) via inhibition of cytokine production, and lowered effects of QUIN/NMDA binding by blocking the NMDA receptor. (1) Ketamine binding blocks the NMDA receptor, reducing the neurotoxic effects of QUIN released from microglia. (2) This decreased neurotoxicity decreases ATP release from the synapse. (3) As a result, the microglia no longer extend toward this “find me” signal and the inflammatory reaction is dampened, and the microglia assumes a less reactive state. In addition, ketamine was found to mitigate the reaction of microglia to inflammatory stimuli such as stress or LPS and decreased the production of proinflammatory cytokines. Created with BioRender.com.
Much of the evidence presented thus far, points to microglia playing a pivotal role in ketamine's antidepressant effects. Ketamine's NMDA-antagonism make it capable of stopping neuronal ATP release, stopping microglia from potentially damaging nearby neurons, as previously discussed (92). Ketamine reduces the neurotoxic effects of QUIN being produced within microglia (173). Ketamine also reduces the microglial response to otherwise activation-inducing molecules like LPS (174) and stops microglial production of pro-inflammatory cytokines (177). Unfortunately, the vast majority of this research has been conducted using the male sex. Similarly, the theoretical bases which guide such experiments has also, historically, used the male sex. An important consequence of this, beyond the inability of generalizing these findings to 50 percent of the population, is the lack of understanding how ketamine might interact with ovarian hormones. Fortunately, extensive research has been conducted investigating the role of estrogens on microglia.
Ovarian Hormones and Neuroinflammation
The classical mechanism through which lipid-soluble hormones like E2 act on neurons and glia is via nuclear estrogen receptors (ERs). Hormones like E2 diffuse across the cell membrane passively and bind to nuclear receptors in the cytoplasm. These ligand-bound receptors dimerize with other ligand-bound receptors, forming a hetero or homodimer (178–180), and translocate to the nucleus where they act as transcription factors. Nuclear estrogen receptors were thought to only function this way, but more recently membrane-bound estrogen receptors have been discovered, such as G- protein-coupled estrogen receptor 1 (GPER1), and act more rapidly (181). Estrogens, as a whole, have been found to be neuroprotective toward multiple pathologies, most notably those in which microglia are implicated, such as Alzheimer's disease (182–186) and multiple sclerosis (187–190).
One way that E2 impacts microglia is via the nuclear estrogen receptor (ER) α. Researchers have found that systemic administration of E2 reduces LPS-induced microglia activity in a dose-dependent manner by decreasing the expression of proteins associated with phagocytosis, by inhibiting morphological changes, and by inhibiting cell migration. ER knockout mouse models demonstrated that ERα is responsible for these effects. For example, microglia activity was unaffected by the absence or presence E2 administration in ERα-null mice (191). Previous research by Bruce-Keller et al. (192) showed that, in a dose-dependent manner, E2 attenuates microglia phagocytosis, and the release of superoxide, a neurotoxic free radical. These effects were mediated by the phosphorylation of mitogen-activated protein kinase (MAPK). Additionally, Vegeto et al. (193) also found that E2 reduces the buildup of free-radicals, specifically, nitrous oxide, in microglia.
Another way that E2 can impact microglia is via GPER1. E2 binding to GPER1, located predominantly on the endoplasmic reticulum, but also the Golgi apparatus and nuclear membrane, results in the mobilization of intracellular calcium, and the production of nuclear phosphatidylinositol 3,4,5-trisphosphate [PIP3; (194)]. PIP3 is an effector of multiple downstream signaling proteins, particularly the protein kinase AKT which plays a crucial role in several cellular processes, such as cell survival (195–197) and proliferation (198, 199). Zhao et al. (200) were the first to show that GPER1 mediates E2's anti-inflammatory effects on microglia in a rat model of cerebral ischemia. Both E2, or the GPER1 agonist, G1, alone were able to attenuate LPS-induced increases in pro-inflammatory TNF-α and IL-1β. Co-administration of the GPER1 antagonist, G15, with E2 reversed these anti-inflammatory effects, and E2 administration to GPER1- knockdown rats had reduced anti-inflammatory effects. As little is known about the effects of estrogens on microglia, even less is known about the effects of progesterone.
P acts on neurons and glia through classical genomic mechanisms by binding to nuclear progesterone receptor (PR), of which there are two isoforms (PRA, PRB), and via non-classical, a.k.a. non-genomic, membrane-bound progesterone receptors [mPRs; (201)]. Tameh et al. (202) found that a mechanism responsible for P's ability to mediate neuronal survival signaling cascades is through upregulation of certain NMDA subunits, specifically GluN1, GluN2A, and GluN3A, which mediate neuronal survival signaling cascades (203, 204). By upregulating these subunits, P serves to inhibit NMDA-mediated apoptosis.
Work by Bali et al. (205) revealed that P exerts its effects on microglia by binding to progesterone receptor membrane component 1 (PGRMC1, a.k.a. 25-Dx, a.k.a. ventral midline antigen or VEMA). PGRMC1 belongs to neither the classical, nor the membrane-bound progesterone receptor subfamilies, but rather the membrane-associated progesterone receptor (MAPR) family (206). PGRMC1 is the P-binding protein in a single-transmembrane protein complex (207–209) and has been found on the membranes of the Golgi apparatus, endoplasmic reticulum, and mitochondria of CNS cells (210, 211). Both E2 and P upregulate PGRMC1 expression in the ovariectomized rat HPC (212). Activating PGRMC1 with P reinstates neuronal activity (213), enhances spinogenesis (214), and enhances neuronal migration and myelination from Schwann cells in the spinal column, where PGRMC1 has been found in the cell membrane (208, 215). What's more, P has been found to induce BDNF expression, an effect which has been shown to be mediated by both the classical PR in cortical slice explants (216), and by PGRMC1 in cultured glial cells (217). This suggests that ketamine, E2, and P, each on their own, would increase BDNF expression. However, this is contradictory to seminal research which showed that P can antagonize E2's synaptogenesis-inducing effects at certain time points (218).
Prior to the discovery that PGRMC1 is crucial for microglial activation (205), work by the same authors showed that P antagonizes E2-mediated neurite outgrowth in vitro, but only when microglial cells were also present. P had no effect on neurite outgrowth with only neurons and astrocyte cocultures (219). This line of research culminated in several additional important findings. Firstly, activating PGRMC1 induces microglial activation to the same degree as stimulation from LPS, as indicated by CD11b protein expression. Second, when microglia are activated via P-binding to PGRMC1 they inhibit new neurite outgrowth. Third, that P binding to PGRMC1 inhibits BDNF release from astrocytes, further hindering neuritogenesis. Fourth, PGRMC1 knockdown stopped LPS and injury (in vitro scratch-wounding) induced microglia activation (205, 220). Together, these findings suggest that PGRMC1 is critical for microglial activation, and that E2 and P may be acting to antagonize one another, with E2 causing quiescence in microglia, but P activating them. This is of course counter-intuitive when considering P's well-researched neuroprotective effects in the context of ischemia (221–229). Looking to P's broader effects on inflammation could, perhaps, clarify this dichotomy. P also has multiple anti-inflammatory effects in LPS-stimulated microglia in vitro, as indicated by Lei et al. (230). LPS upregulated the pro-inflammatory cytokine TNF-α, inducible nitric oxide synthase (iNOS), an enzyme precursor for the free radical nitric oxide (NO), and cyclooxygenase-2 (COX-2), an enzyme precursor for prostaglandin. All of which are upregulated during inflammation. P attenuated these LPS-induced increases in a dose- dependent manner. Additionally, P decreased the LPS-induced phosphorylation of several other kinases such as p38, c-Jun N-terminal kinase, and extracellular regulated kinase MAPKs.
Importantly, P decreased the LPS-induced activation of the protein complex nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). NF-κB is a transcription factor which controls the expression of various target genes, especially those involved in immune and inflammatory responses (230). NF-κB and pro-inflammatory cytokines have a bidirectional connection: NF-κB is activated by pro-inflammatory cytokines, and directly promotes pro-inflammatory cytokine production by binding to cytokine promoter regions in the genome (231). These pleiotropic effects of P, along with the PGRMC1 activational mechanism, all specific to microglia, demonstrate the relevance of understanding how P, E2, and ketamine might interact with one another. Of course, much more comprehensive research still needs to be done to understand the complexity of these phenomena–see Figure 2.
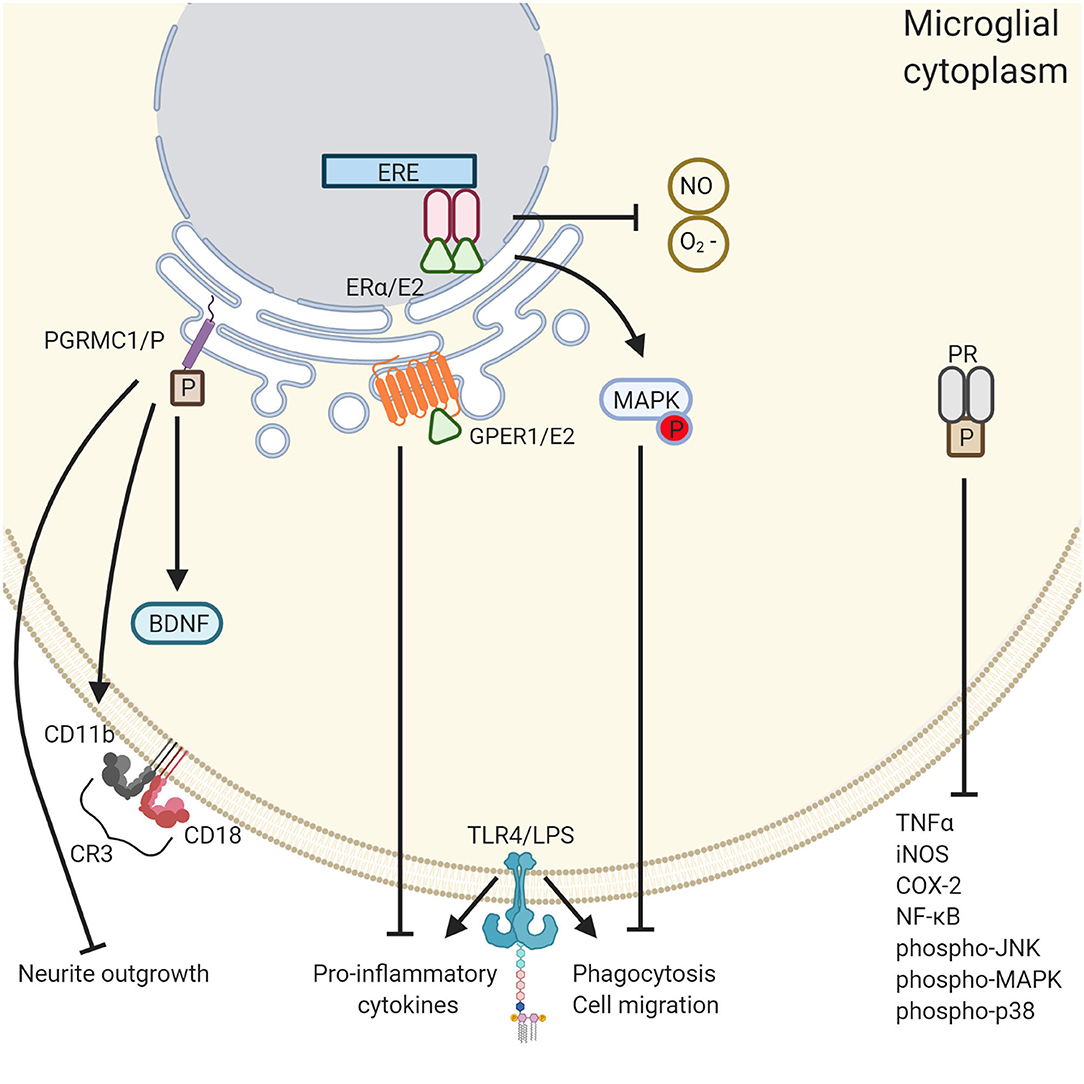
Figure 2. The differential roles of ovarian hormones in the microglial immune response. PGRMC1 activation induces microglial activation whereas PR and E2 quiesces it. PGRMC1 binds to progesterone leading to the inhibition of neurite outgrowth, microglia activation through the expression of CD11b and CD18, and the inhibition of BDNF. Conversely, GPER1 activation by E2 reduced the expression of proinflammatory cytokines TNF-α and IL-1β. In addition, E2 reduces microglial phagocytosis and cell migration through activation of the MAPK pathway and inhibits the release of reactive oxygen species NO and O2−. PR activation also has an anti- inflammatory effect through the inhibition of a various of inflammatory cytokines. Created with BioRender.com.
Ketamine Binds to ERα
It has been shown that estrogens act to mediate enhanced sensitivity to ketamine in depression models using rodents (156–158). Furthermore, both female mice (152) and humans (160) metabolize ketamine to produce higher levels of the crucially important ketamine metabolites 2R-6R/2S-6S HNK than males. It is thought that this occurs due to women having higher levels of the CYP2A6 and CYP2B6 enzymes, responsible for ketamine's metabolism (232). E2 and P are capable of inducing these enzymes (233), with E2 doing so via ERα (234). ERα is therefore critical in understanding how ketamine and ovarian hormones might interact. In most tissues, including the CNS, E2 binding to ERs precipitates an upregulation of PRs (235). Thanks to work by Alves et al. (236) it is known that this occurs via E2 binding to ERα receptors in the HPC of wild- type mice (237). Both E2 and P lead to the induction of PGRMC1 in female ovariectomized rat hippocampal neurons in the CA1, CA3, and DG (212), although it is unclear which ER/PR-binding mechanisms cause this.
Evidence that ketamine binds to ERα, was provided by Ho et al. (232). Radioligand binding assays, coupled with surface plasmon resonance, demonstrated that E2, ketamine, and 2R-6R/2S-6S HNK bind to ERα receptors in cultured astrocytes. They also found that the same compounds act in an additive manner to induce AMPA receptor subunits, again in vitro. This is thought to be crucial for ketamine's antidepressant effects, as previously discussed, and this effect was ablated by ERα knockdown. Given that all three compounds also lead to ERα trafficking to the nucleus, it follows that the authors hypothesized a potential positive feedback loop. Namely, when ERα binds to estrogen-response elements, CYP2A6, CYP2B6, and AMPA subunit transcription is induced, leading to more ketamine metabolism and more AMPA receptors (232). Research has yet to be conducted to determine how PRs might be impacted by these processes but it is likely that, via ERα binding, ketamine and its metabolites cause PR upregulation. Virtually nothing is known of ketamine's ability to impact PGRMC1. How this might be manifested behaviorally is an even greater mystery.
Hippocampus As a Candidate for Ketamine-Hormone Interactions
Depression involves multiple brain regions. The HPC is perhaps the most well-studied brain region in regards to MDD. A meta-analysis of MRI results yielded an average reduction of 8–10% in HPC volume among individuals with unipolar depression (238). The HPC is also a site of continuous neurogenesis as documented in adult macaques (239). That is, while depression may decrease HPC volume, this change may not necessarily be permanent. Outside the context of antidepressant research, it is worth noting that HPC volume declines naturally with age, and that there exists an interactive effect of gender and age on HPC volume decline. As we age, HPC volume declines linearly among men, whereas there is no such correlation among women (240).
Morphological changes in the HPC such as neurogenesis and synaptogenesis are, in part, mediated by BDNF (59, 241, 242). What's more, researchers have shown that peripheral BDNF leads to both hippocampal neurogenesis among adult mice, as well as antidepressant-like effects in the forced swim test [FST; (75)]. Ketamine is well-documented to both increase hippocampal BDNF, and produce antidepressant-like effects in the FST in male rodents (243–246). Furthermore, it is likely that the neuroprotective effects of E2 are partly due to BDNF upregulation (247). Some speculate that the BDNF gene contains an estrogen response element such that E2 could upregulate BDNF directly (248). Others have shown that estrogen receptors are found on BDNF-expressing neurons (249, 250). It is perhaps through this mechanism that E2 mediates dendritic spine growth and synaptogenesis in the HPC, as previously discussed (218, 251).
What makes the HPC particularly interesting in terms of ketamine's effects is, as previously discussed, that the HPC-PFC circuits are likely the most crucial for ketamine's antidepressant effect. Within this circuit ketamine selectively disinhibits GABA interneurons, as per the glutamate burst hypothesis—see Figure 3. With regards to the neuroinflammatory hypothesis, ovarian hormones are known to play an important role in the inflammatory response in the HPC (252, 253) and prefrontal cortex (254). The HPC has been the focus of several research groups who found that ketamine generally suppresses glial inflammation in this area (177, 255, 256).
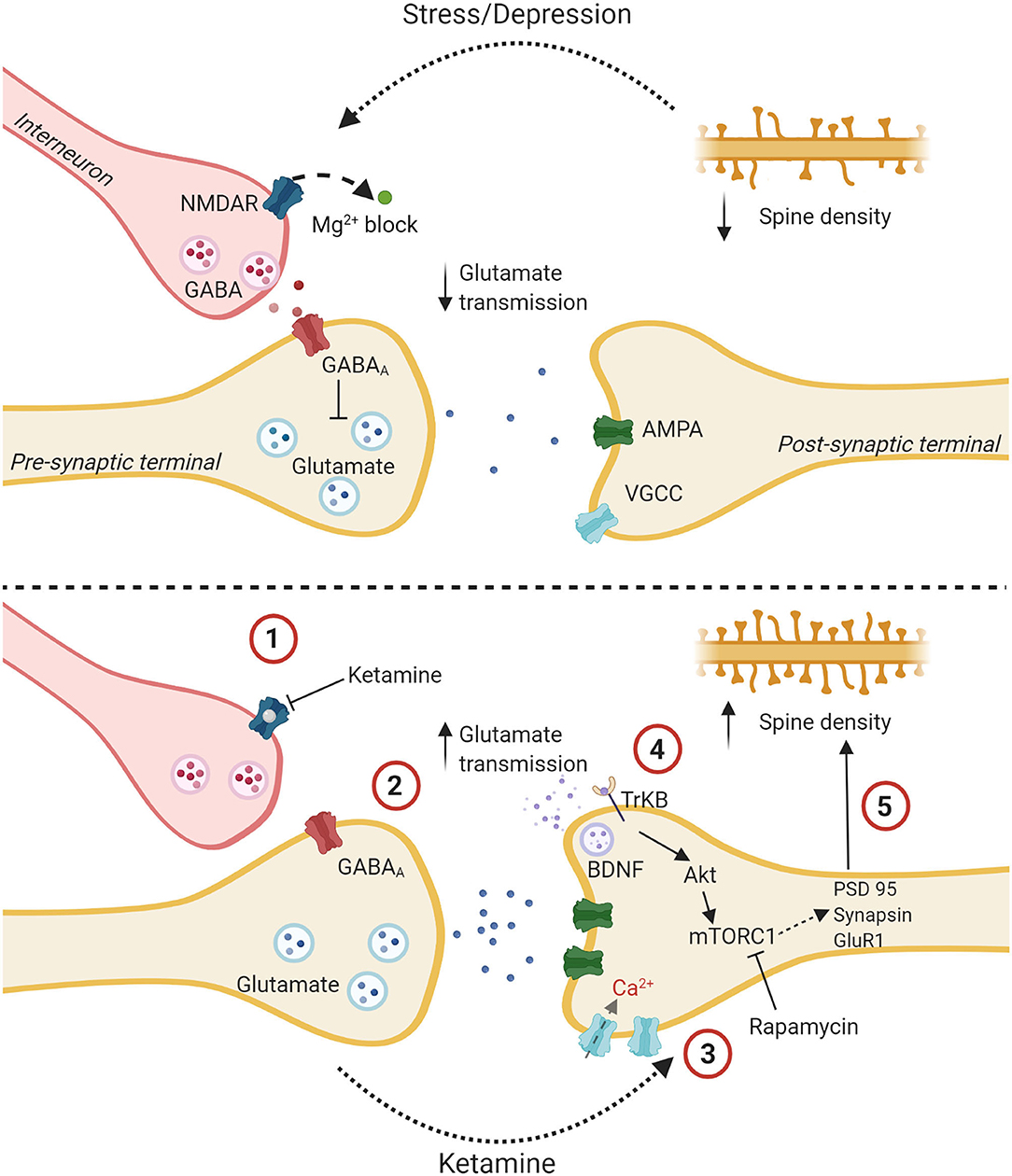
Figure 3. The glutamate burst hypothesis: a proposed mechanism of action for ketamine. The upper panel shows a glutamatergic synapse tonically inhibited by a GABAergic interneuron with decreased spine density and altered glutamatergic transmission due to stress and depression. The lower panel delineates putative steps for ketamine's antidepressant effect. (1) Ketamine binds to an NMDA receptor on a GABAergic interneuron. (2) This reduces the tonic inhibition of a glutamatergic presynaptic terminal which causes the release of bursts of glutamate. (3) Increased signal transmission leads to upregulation of AMPA receptors, increased depolarization, and influx of Ca2+ through Voltage gated calcium channels. (4) Higher intracellular Ca2+ leads to the release of BDNF which binds to the TrKB receptor. The Akt pathway is activated followed by the mTOR pathway. (5) This leads to the synthesis of synaptic proteins and increased spine density. Created with BioRender.com.
Conclusion
The etiology of major depressive disorder is complex, and there is no single explanation for its manifestation. Likewise, it's possible that there are many valid explanations as to why women suffer from MDD more than men, but ovarian hormones are surely implicated. Despite the discovery of ketamine's antidepressant effects being relatively recent, there's plenty of evidence supporting the various hypotheses concerning its mechanisms of action in the CNS. However, evidence specific to the female sex is lacking. That which has been gathered only demonstrates that well-researched theories do not generalize across the sexes. Considering the neuroinflammatory hypothesis of depression may help shed light on ketamine's mechanism of action among females. Indeed, a massive body of literature supports the relevance of this hypothesis, particularly the role of microglia, to novel antidepressants like ketamine. The literature provides more than sufficient evidence to warrant the idea that circulating E2 and P would interact with ketamine's effect on the CNS. In believing some, one might suspect that the three molecules would act additively to reduce depressive symptoms. Believing other lines of research, one might expect E2 to act additively with ketamine, but that P would antagonize this via acting on PGRMC1 on microglia. Evidence which indicates P has neuroinflammatory effects is contradicted by evidence which shows it has anti-inflammatory effects. In vivo experimentation looking specifically at the HPC is the next step in investigating these complex phenomena, as much of the literature uses in vitro models. Exploring this is paramount to the likely eventuality that ketamine, or a similar compound, will be administered to those seeking treatment for MDD on a larger scale.
Author Contributions
CG wrote the original manuscript as part of his Masters thesis. AP contributed to sections of the manuscript and edited later versions (including figures). WB supervised the process and edited the final document. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Buchanan CM, Eccles JS, Becker JB. Are adolescents the victims of raging hormones: evidence for activational effects of hormones on moods and behavior at adolescence. Psychol Bull. (1992) 111:62–107. doi: 10.1037/0033-2909.111.1.62
2. Seeman MV. Psychopathology in women and men: focus on female hormones. Am J Psychiatry. (1997) 154:1641–7. doi: 10.1176/ajp.154.12.1641
3. Albert PR. Why is depression more prevalent in women?. J Psych Neurosci. (2015) 40:219–21. doi: 10.1503/jpn.150205
4. Bebbington PE, Dunn G, Jenkins R, Lewis G, Brugha T, Farrell M, et al. The influence of age and sex on the prevalence of depressive conditions: report from the national survey of psychiatric morbidity. Int Rev Psychiatry. (2003) 15:74–83. doi: 10.1080/0954026021000045976
5. Salk RH, Hyde JS, Abramson LY. Gender differences in depression in representative national samples: meta-analyses of diagnoses and symptoms. Psychol Bull. (2017) 143:783–822. doi: 10.1037/bul0000102
6. Kuehner C. Why is depression more common among women than among men? Lancet Psychiatry. (2017) 4:146–58. doi: 10.1016/S2215-0366(16)30263-2
7. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. (2007) 87:905–31. doi: 10.1152/physrev.00026.2006
8. González-Orozco JC, Camacho-Arroyo I. progesterone actions during central nervous system development. Front Neurosci. (2019) 13:503. doi: 10.3389/fnins.2019.00503
9. McCarthy MM. Estradiol and the developing brain. Physiol Rev. (2008) 88:91–134. doi: 10.1152/physrev.00010.2007
10. Douma SL, Husband C, O'Donnell ME, Barwin BN, Woodend AK. Estrogen-related mood disorders: reproductive life cycle factors. ANS Adv Nurs Sci. (2005) 28:364–75. doi: 10.1097/00012272-200510000-00008
11. Yim IS, Stapleton LRT, Guardino CM, Hahn-Holbrook J, Schetter CD. Biological and psychosocial predictors of postpartum depression: systematic review and call for integration. Annu Rev Clin Psychol. (2015) 11:99–137. doi: 10.1146/annurev-clinpsy-101414-020426
12. Ahokas A, Kaukoranta J, Wahlbeck K, Aito M. Estrogen deficiency in severe postpartum depression: successful treatment with sublingual physiologic 17beta- estradiol: a preliminary study. J Clin Psychiatry. (2001) 62:332–6. doi: 10.4088/JCP.v62n0504
13. Bloch M, Schmidt PJ, Danaceau M, Murphy J, Nieman L, Rubinow DR. Effects of gonadal steroids in women with a history of postpartum depression. Am J Psychiatry. (2000) 157:924–30. doi: 10.1176/appi.ajp.157.6.924
14. Moses-Kolko EL, Berga SL, Kalro B, Sit DKY, Wisner KL. Transdermal estradiol for postpartum depression: a promising treatment option. Clin Obstet Gynecol. (2009) 52:516–29. doi: 10.1097/GRF.0b013e3181b5a395
15. Schiller CE, Meltzer-Brody S, Rubinow DR. The role of reproductive hormones in postpartum depression. CNS Spectr. (2015) 20:48–59. doi: 10.1017/S1092852914000480
16. O'Hara MW, Schlechte JA, Lewis DA, Varner MW. Controlled prospective study of postpartum mood disorders: psychological, environmental, hormonal variables. J Abnorm Psychol. (1991) 100:63. doi: 10.1037/0021-843X.100.1.63
17. Heidrich A, Schleyer M, Spingler H, Albert P, Knoche M, Fritze J, et al. Postpartum blues: relationship between not-protein bound steroid hormones in plasma and postpartum mood changes. J Affect Disord. (1994) 30:93–8. doi: 10.1016/0165-0327(94)90036-1
18. Gregoire AJP, Kumar R, Everitt B, Henderson AF, Studd JWW. Transdermal oestrogen for treatment of severe postnatal depression. Obstet Gynecol Surv. (1996) 51:510–2. doi: 10.1097/00006254-199609000-00003
19. Stoffel EC, Craft RM. Ovarian hormone withdrawal-induced “depression” in female rats. Physiol Behav. (2004) 83:505–13. doi: 10.1016/j.physbeh.2004.08.033
20. Suda S, Segi-Nishida E, Newton SS, Duman RS. A postpartum model in rat: behavioral and gene expression changes induced by ovarian steroid deprivation. Biol Psychiatry. (2008) 64:311–9. doi: 10.1016/j.biopsych.2008.03.029
21. Pearlstein TB. Hormones and depression: what are the facts about premenstrual syndrome, menopause, and hormone replacement therapy? Am J Obstet Gynecol. (1995) 173:646–53. doi: 10.1016/0002-9378(95)90297-X
22. Steiner M, Pearlstein T. Premenstrual dysphoria and the serotonin system: pathophysiology and treatment. J Clin Psychiatry. (2000) 12:17–21.
23. Rapkin AJ, Lewis EI. Treatment of premenstrual dysphoric disorder. Womens Health. (2013) 9:537–56. doi: 10.2217/WHE.13.62
24. Hantsoo L, Epperson CN. Premenstrual dysphoric disorder: epidemiology and treatment. Curr Psychiatry Rep. (2015) 17:87. doi: 10.1007/s11920-015-0628-3
25. Joinson C, Heron J, Lewis G, Croudace T, Araya R. Timing of menarche and depressive symptoms in adolescent girls from a UK cohort. Br J Psychiatry. (2011) 198:17–23. doi: 10.1192/bjp.bp.110.080861
26. Keating NL, Cleary PD, Rossi AS, Zaslavsky AM, Ayanian JZ. Use of hormone replacement therapy by postmenopausal women in the United States. Ann Intern Med. (1999) 130:545–53. doi: 10.7326/0003-4819-130-7-199904060-00002
27. Chlebowski RT, Anderson GL, Gass M, Lane DS, Aragaki AK, Kuller LH, et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA. (2010) 304:1684–92. doi: 10.1001/jama.2010.1500
28. Lobo RA, Pickar JH, Stevenson JC, Mack WJ, Hodis HN. Back to the future: hormone replacement therapy as part of a prevention strategy for women at the onset of menopause. Atherosclerosis. (2016) 254:282–90. doi: 10.1016/j.atherosclerosis.2016.10.005
29. Santen RJ, Allred DC, Ardoin SP, Archer DF, Boyd N, Braunstein GD, et al. Postmenopausal hormone therapy: an endocrine society scientific statement. J Clin Endocrinol Metab. (2010) 95(7 Suppl. 1):s1–66. doi: 10.1210/jc.2009-2509
30. Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA. (2002) 288:872–81. doi: 10.1001/jama.288.7.872
31. Zweifel JE, O'Brien WH. A meta-analysis of the effect of hormone replacement therapy upon depressed mood. Psychoneuroendocrinology. (1997) 22:189–212. doi: 10.1016/S0306-4530(96)00034-0
32. Hirschfeld RM. History and evolution of the monoamine hypothesis of depression. J Clin Psychiatry. (2000) 61:4–6.
33. Nutt DJ. Relationship of neurotransmitters to the symptoms of major depressive disorder. J Clin Psychiatry. (2008) 69 (Suppl. 1):4–7.
34. Ruhé HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Mol Psychiatry. (2007) 12:331–59. doi: 10.1038/sj.mp.4001949
35. Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR* D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr Serv. (2009) 60:1439–45. doi: 10.1176/ps.2009.60.11.1439
36. Ioannidis JP. Effectiveness of antidepressants: an evidence myth constructed from a thousand randomized trials? Philo Ethics Hum Med. (2008) 3:14. doi: 10.1186/1747-5341-3-14
37. Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. (2002) 34:13–25. doi: 10.1016/S0896-6273(02)00653-0
38. Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. (2006) 163:28–40. doi: 10.1176/appi.ajp.163.1.28
39. Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. (2008) 358:252–60. doi: 10.1056/NEJMsa065779
40. Küçükibrahimoglu E, Saygin MZ, Çalişkan M, Kaplan OK, Ünsal C, Gören MZ. The change in plasma GABA glutamine and glutamate levels in fluoxetine-or S- citalopram-treated female patients with major depression. Eur J Clin Pharmacol. (2009) 65:571–7. doi: 10.1007/s00228-009-0650-7
41. Mitani H, Shirayama Y, Yamada T, Maeda K, Ashby CR Jr, Kawahara R. Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Progr Neuro Psychopharmacol Biol Psychiatry. (2006) 30:1155–8. doi: 10.1016/j.pnpbp.2006.03.036
42. Maes M, Verkerk R, Vandoolaeghe E, Lin A, Scharpe S. Serum levels of excitatory amino acids, serine, glycine, histidine, threonine, taurine, alanine and arginine in treatment-resistant depression: modulation by treatment with antidepressants and prediction of clinical responsivity. Acta Psychiatr Scand. (1998) 97:302–8. doi: 10.1111/j.1600-0447.1998.tb10004.x
43. Cavus I, Pan JW, Hetherington HP, Abi-Saab W, Zaveri HP, Vives KP, et al. Decreased hippocampal volume on MRI is associated with increased extracellular glutamate in epilepsy patients. Epilepsia. (2008) 49:1358–66. doi: 10.1111/j.1528-1167.2008.01603.x
44. Gurvits TV, Shenton ME, Hokama H, Ohta H, Lasko NB, Gilbertson MW, et al. Magnetic resonance imaging study of hippocampal volume in chronic, combat-related posttraumatic stress disorder. Biol Psychiatry. (1996) 40:1091–9. doi: 10.1016/S0006-3223(96)00229-6
45. Sanacora G, Treccani G, Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. (2012) 62:63–77. doi: 10.1016/j.neuropharm.2011.07.036
46. Shah PJ, Ebmeier KP, Glabus MF, Goodwin GM. Cortical grey matter reductions associated with treatment-resistant chronic unipolar depression. Controlled magnetic resonance imaging study. Br J Psychiatry. (1998) 172:527–32. doi: 10.1192/bjp.172.6.527
47. Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA. (1996) 93:3908–13. doi: 10.1073/pnas.93.9.3908
48. Robinson MS, Alloy LB. Negative cognitive styles and stress-reactive rumination interact to predict depression: a prospective study. Cogn Ther Res. (2003) 27:275–91. doi: 10.1023/A:1023914416469
49. Wamsteeker JI, Bains JS. A synaptocentric view of the neuroendocrine response to stress. Euro J Neurosci. (2010) 32:2011–21. doi: 10.1111/j.1460-9568.2010.07513.x
50. Cazakoff BN, Howland JG. Acute stress disrupts paired pulse facilitation and long-term potentiation in rat dorsal hippocampus through activation of glucocorticoid receptors. Hippocampus. (2010) 20:1327–31. doi: 10.1002/hipo.20738
51. Musazzi L, Milanese M, Farisello P, Zappettini S, Tardito D, Barbiero VS, et al. Acute stress increases depolarization-evoked glutamate release in the rat prefrontal/frontal cortex: the dampening action of antidepressants. PLoS ONE. (2010) 5:e8566. doi: 10.1371/journal.pone.0008566
52. Auer DP, Pütz B, Kraft E, Lipinski B, Schill J, Holsboer F. Reduced glutamate in the anterior cingulate cortex in depression: an in vivo proton magnetic resonance spectroscopy study. Biol Psychiatry. (2000) 47:305–13. doi: 10.1016/S0006-3223(99)00159-6
53. Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced prefrontal glutamate/glutamine and γ-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch Gen Psychiatry. (2007) 64:193–200. doi: 10.1001/archpsyc.64.2.193
54. Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. (2006) 59:1116–27. doi: 10.1016/j.biopsych.2006.02.013
55. McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. (1999) 22:105–22. doi: 10.1146/annurev.neuro.22.1.105
56. Sapolsky RM, Krey LC, McEwen BS. Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. J Neurosci. (1985) 5:1222–7. doi: 10.1523/JNEUROSCI.05-05-01222.1985
57. Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. (2003) 301:805–9. doi: 10.1126/science.1083328
58. Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. (1995) 15:7539–47. doi: 10.1523/JNEUROSCI.15-11-07539.1995
59. Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry. (2001) 50:260–5. doi: 10.1016/S0006-3223(01)01083-6
60. Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. (2003) 60:804–15. doi: 10.1001/archpsyc.60.8.804
61. Karege F, Vaudan G, Schwald M, Perroud N, La Harpe R. Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res. (2005) 136:29–37. doi: 10.1016/j.molbrainres.2004.12.020
62. Aydemir C, Yalcin ES, Aksaray S, Kisa C, Yildirim SG, Uzbay T, et al. Brain-derived neurotrophic factor (BDNF) changes in the serum of depressed women. Prog Neuropsychopharmacol Biol Psychiatry. (2006) 30:1256–60. doi: 10.1016/j.pnpbp.2006.03.025
63. Gervasoni N, Aubry J.-M., Bondolfi G, Osiek C, Schwald M, et al. Partial normalization of serum brain-derived neurotrophic factor in remitted patients after a major depressive episode. Neuropsychobiology. (2005) 51:234–8. doi: 10.1159/000085725
64. Karege F, Perret G, Bondolfi G, Schwald M, Bertschy G, Aubry -M., et al. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. (2002) 109:143–8. doi: 10.1016/S0165-1781(02)00005-7
65. Shimizu E, Hashimoto K, Okamura N, Koike K, Komatsu N, Kumakiri C, et al. Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry. (2003) 54:70–5. doi: 10.1016/S0006-3223(03)00181-1
66. Kim YK, Lee HP, Won SD, Park EY, Lee HY, Lee BH, et al. Low plasma BDNF is associated with suicidal behavior in major depression. Prog Neuropsychopharmacol Biol Psychiatry. (2007) 31:78–85. doi: 10.1016/j.pnpbp.2006.06.024
67. Lee BH, Kim H, Park SH, Kim YK. Decreased plasma BDNF level in depressive patients. J Affect Disord. (2007) 101:239–44. doi: 10.1016/j.jad.2006.11.005
68. Aydemir O, Deveci A, Taneli F. The effect of chronic antidepressant treatment on serum brain-derived neurotrophic factor levels in depressed patients: a preliminary study. Prog Neuropsychopharmacol Biol Psychiatry. (2005) 29:261–5. doi: 10.1016/j.pnpbp.2004.11.009
69. Gonul AS, Akdeniz F, Taneli F, Donat O, Eker C, Vahip S. Effect of treatment on serum brain-derived neurotrophic factor levels in depressed patients. Eur Arch Psychiatry Clin Neurosci. (2005) 255:381–6. doi: 10.1007/s00406-005-0578-6
70. Huang TL, Lee CT, Liu YL. Serum brain-derived neurotrophic factor levels in patients with major depression: effects of antidepressants. J Psychiatr Res. (2008) 42:521–5. doi: 10.1016/j.jpsychires.2007.05.007
71. Yoshimura R, Mitoma M, Sugita A, Hori H, Okamoto T, Umene W, et al. Effects of paroxetine or milnacipran on serum brain-derived neurotrophic factor in depressed patients. Prog Neuro Psychopharmacol Biol Psychiatry. (2007) 31:1034–7. doi: 10.1016/j.pnpbp.2007.03.001
72. Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. (1996) 16:2365–72. doi: 10.1523/JNEUROSCI.16-07-02365.1996
73. Shirayama Y, Chen ACH, Nakagawa S, Russell DS, Duman RS. Brain- derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. (2002) 22:3251–61. doi: 10.1523/JNEUROSCI.22-08-03251.2002
74. Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav. (1997) 56:131–7. doi: 10.1016/S0091-3057(96)00169-4
75. Schmidt HD, Duman RS. Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology. (2010) 35:2378–91. doi: 10.1038/npp.2010.114
76. Oliff HS, Berchtold NC, Isackson P, Cotman CW. Exercise-induced regulation of brain-derived neurotrophic factor (BDNF) transcripts in the rat hippocampus. Mol Brain Res. (1998) 61:147–53. doi: 10.1016/S0169-328X(98)00222-8
77. Maes M, Yirmyia R, Noraberg J, Brene S, Hibbeln J, Perini G, et al. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis. (2009) 24:27–53. doi: 10.1007/s11011-008-9118-1
78. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. (2009) 65:732–41. doi: 10.1016/j.biopsych.2008.11.029
79. Miller AH, Raison CL. The role of inflammation in depression: from evolutionaryimperative to modern treatment target. Nat Rev Immunol. (2016) 16:22–34. doi: 10.1038/nri.2015.5
80. Schiepers OJG, Wichers MC, Maes M. Cytokines and major depression. Prog Neuro Psychopharmacol Biol Psychiatry. (2005) 29:201–17. doi: 10.1016/j.pnpbp.2004.11.003
81. Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. (2016) 353:aad8670. doi: 10.1126/science.aad8670
82. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
83. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. (2017) 23:1018–27. doi: 10.1038/nm.4397
84. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
85. Gentleman SM, Leclercq PD, Moyes L, Graham DI, Smith C, Griffin WST, et al. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. (2004) 146:97–104. doi: 10.1016/j.forsciint.2004.06.027
86. Taib T, Leconte C, Van Steenwinckel J, Cho AH, Palmier B, Torsello E, et al. Neuroinflammation, myelin and behavior: temporal patterns following mild traumatic brain injury in mice. PLoS ONE. (2017) 12:e0184811. doi: 10.1371/journal.pone.0184811
87. Ma K, Zhang H, Baloch Z. Pathogenetic and therapeutic applications of tumor necrosis factor-α (TNF-α) in major depressive disorder: a systematic review. Int J Mol Sci. (2016) 17:733. doi: 10.3390/ijms17050733
88. Liu Y, Ho RCM, Mak A. Interleukin (IL)-6, tumour necrosis factor alpha (TNF- α) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: a meta-analysis and meta-regression. J Affect Disord. (2012) 139:230–9. doi: 10.1016/j.jad.2011.08.003
89. Kumar A, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. Microglial/Macrophage polarization dynamics following traumatic brain injury. J Neurotrauma. (2016) 33:1732–50. doi: 10.1089/neu.2015.4268
90. Lively S, Schlichter LC. Microglia responses to pro-inflammatory stimuli (LPS, IFNγ+TNFα) and reprogramming by resolving cytokines (IL-4, IL-10). Front Cell Neurosci. (2018) 12:215. doi: 10.3389/fncel.2018.00215
91. Wojdasiewicz P, Poniatowski ŁA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. (2014) 2014:561459. doi: 10.1155/2014/561459
92. Dissing-Olesen L, LeDue JM, Rungta RL, Hefendehl JK, Choi HB, MacVicar BA. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J Neurosci. (2014) 34:10511–27. doi: 10.1523/JNEUROSCI.0405-14.2014
93. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature. (2010) 467:863–7. doi: 10.1038/nature09413
94. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. (2009) 461:282–6. doi: 10.1038/nature08296
95. Fu R, Shen Q, Xu P, Luo JJ, Tang Y. Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol. (2014) 49:1422–34. doi: 10.1007/s12035-013-8620-6
96. Herbert TB, Cohen S. Depression and immunity: a meta-analytic review. Psychol Bull. (1993) 113:472–86. doi: 10.1037/0033-2909.113.3.472
97. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. (2006) 27:24–31. doi: 10.1016/j.it.2005.11.006
98. Dantzer R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin N Am. (2009) 29:247–64. doi: 10.1016/j.iac.2009.02.002
99. Bouhuys AL, Flentge F, Oldehinkel AJ, van den Berg MD. Potential psychosocial mechanisms linking depression to immune function in elderly subjects. Psychiatry Res. (2004) 127:237–45. doi: 10.1016/j.psychres.2004.05.001
100. Goebel MU, Mills PJ, Irwin MR, Ziegler MG. Interleukin-6 and tumor necrosis factor-α production after acute psychological stress, exercise, and infused isoproterenol:differential effects and pathways. Psychosom Med. (2000) 62:591–8. doi: 10.1097/00006842-200007000-00019
101. Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci USA. (2003) 10:1920–5. doi: 10.1073/pnas.0438019100
102. Aschbacher K, Epel E, Wolkowitz OM, Prather AA, Puterman E, Dhabhar FS. Maintenance of a positive outlook during acute stress protects against pro- inflammatory reactivity and future depressive symptoms. Brain Behav Immun. (2012) 26:346–52. doi: 10.1016/j.bbi.2011.10.010
103. Pfeilschifter J, Köditz R, Pfohl M, Schatz H. Changes in proinflammatory cytokine activity after menopause. Endocr Rev. (2002) 23:90–119. doi: 10.1210/edrv.23.1.0456
104. Milaneschi Y, Corsi AM, Penninx BW, Bandinelli S, Guralnik JM, Ferrucci L. Interleukin-1 receptor antagonist and incident depressive symptoms over 6 years in older persons: the InCHIANTI study. Biol Psychiatry. (2009) 65:973–8. doi: 10.1016/j.biopsych.2008.11.011
105. van den Biggelaar AH, Gussekloo J, de Craen AJ, Frölich M, Stek ML, van der Mast RC, et al. Inflammation and interleukin-1 signaling network contribute to depressive symptoms but not cognitive decline in old age. Exp Gerontol. (2007) 42:693–701. doi: 10.1016/j.exger.2007.01.011
106. Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon- alpha. Biol Psychiatry. (2004) 56:819–24. doi: 10.1016/j.biopsych.2004.02.009
107. Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med. (2009) 71:171–186. doi: 10.1097/PSY.0b013e3181907c1b
108. Mössner R, Mikova O, Koutsilieri E, Saoud M, Ehlis A.-C., et al. Consensus paper of the WFSBP task force on biological markers: biological markers in depression. World J Biol Psychiatry. (2007) 8:141–74. doi: 10.1080/15622970701263303
109. Young JJ, Bruno D, Pomara N. A review of the relationship between proinflammatory cytokines and major depressive disorder. J Affect Disord. (2014) 169:15–20. doi: 10.1016/j.jad.2014.07.032
110. Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci USA. (2003) 100:13632–7. doi: 10.1073/pnas.2234031100
111. Gebicke-Haerter PJ. Microglia in neurodegeneration: molecular aspects. Microsc Res Tech. (2001) 54:47–58. doi: 10.1002/jemt.1120
112. Hanisch UK. Microglia as a source and target of cytokines. Glia. (2002) 40:140–55. doi: 10.1002/glia.10161
113. Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. (2003) 302:1760–5. doi: 10.1126/science.1088417
114. Fernstrom JD. Role of precursor availability in control of monoamine biosynthesis in brain. Physiol Rev. (1983) 63:484–546. doi: 10.1152/physrev.1983.63.2.484
115. Schaechter JD, Wurtman RJ. Serotonin release varies with brain tryptophan levels. Brain Res. (1990) 532:203–10. doi: 10.1016/0006-8993(90)91761-5
116. Soliman H, Mediavilla-Varela M, Antonia S. Indoleamine 2,3-Dioxygenase. Cancer J. (2010) 16:354–9. doi: 10.1097/PPO.0b013e3181eb3343
117. Guillemin GJ, Smith DG, Smythe GA, Armati PJ, Brew BJ. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv Exp Med Biol. (2003) 527:105–12. doi: 10.1007/978-1-4615-0135-0_12
118. Bay-Richter C, Linderholm KR, Lim CK, Samuelsson M, Träskman-Bendz L, Guillemin GJ, et al. A role for inflammatory metabolites as modulators of the glutamate N-methyl-D-aspartate receptor in depression and suicidality. Brain Behav Immun. (2015) 43:110–7. doi: 10.1016/j.bbi.2014.07.012
119. Zunszain PA, Anacker C, Cattaneo A, Choudhury S, Musaelyan K, Myint AM, et al. Interleukin-1β: a new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacology. (2012) 37:939–49. doi: 10.1038/npp.2011.277
120. Schwarcz R, Bruno JP, Muchowski PJ, Wu H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat Rev Neurosci. (2012) 13:465–77. doi: 10.1038/nrn3257
121. Suzuki H, Ohgidani M, Kuwano N, Chrétien F, Lorin de la Grandmaison G, Onaya M, et al. Suicide and microglia: recent findings and future perspectives based on human studies. Front Cell Neurosci. (2019) 13:31. doi: 10.3389/fncel.2019.00031
122. Guillemin GJ. Quinolinic acid, the inescapable neurotoxin. FEBS J. (2012) 279:1356–65. doi: 10.1111/j.1742-4658.2012.08485.x
123. Kindler J, Lim CK, Weickert CS, Boerrigter D, Galletly C, Liu D, et al. Dysregulation of kynurenine metabolism is related to proinflammatory cytokines, attention, and prefrontal cortex volume in schizophrenia. Mol Psychiatry. (2019) 25:2860–72. doi: 10.1038/s41380-019-0401-9
124. O'Connor JC, André C, Wang Y, Lawson MA, Szegedi SS, Lestage J, et al. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci. (2009) 29:4200–9. doi: 10.1523/JNEUROSCI.5032-08.2009
125. Dantzer R, O'Connor JC, Lawson MA, Kelley KW. Inflammation-Associated Depression: From Serotonin to Kynurenine. Psychoneuroendocrinology. (2011) 36:426–436. doi: 10.1016/j.psyneuen.2010.09.012
126. Halaris A, Myint AM, Savant V, Meresh E, Lim E, Guillemin G, et al. Does escitalopram reduce neurotoxicity in major depression? J Psychiatr Res. (2015) 66–67:118–26. doi: 10.1016/j.jpsychires.2015.04.026
127. Maes M, Leonard BE, Myint AM, Kubera M, Verkerk R. The new “5-HT” hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3- dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog Neuropsychopharmacol Biol Psychiatry. (2011) 35:702–21. doi: 10.1016/j.pnpbp.2010.12.017
128. Myint AM. Kynurenines: from the perspective of major psychiatric disorders. FEBS J. (2012) 279:1375–1385. doi: 10.1111/j.1742-4658.2012.08551.x
129. Myint AM, Schwarz MJ, Müller N. The role of the kynurenine metabolism in major depression. J Neural Transm. (2012) 119:245–51. doi: 10.1007/s00702-011-0741-3
130. Lapin IP. Kynurenines as probable participants of depression. Pharmacopsychiatry. (1973) 6:273–9. doi: 10.1055/s-0028-1094391
131. Domino EF. Taming the ketamine tiger. Anesthesiology. (2010) 113:678–84. doi: 10.1097/ALN.0b013e3181ed09a2
132. Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. (2000) 47:351–4. doi: 10.1016/S0006-3223(99)00230-9
133. Vadivelu N, Schermer E, Kodumudi V, Belani K, Urman RD, Kaye AD. Role of ketamine for analgesia in adults and children. J Anaesthesiol Clin Pharmacol. (2016) 32:298–306. doi: 10.4103/0970-9185.168149
134. Larkin GL, Beautrais AL. A preliminary naturalistic study of low-dose ketamine for depression and suicide ideation in the emergency department. Int J Neuropsychopharmacol. (2011) 14:1127–31. doi: 10.1017/S1461145711000629
135. McGown RG. A technique of anaesthesia in haemorrhagic shock. Illustrative case histories and a discussion. Anaesthesia. (1975) 30:616–23. doi: 10.1111/j.1365-2044.1975.tb00921.x
136. Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. (2006) 63:856–64. doi: 10.1001/archpsyc.63.8.856
137. National Institute of Mental Health. Neuropharmacologic Imaging and Biomarker Assessments of Response to Acute and Repeated-Dosed Ketamine Infusions in Major Depressive Disorder. (2017). Available online at: https://www.clinicaltrials.gov/ct2/show/NCT03065335 (accessed November 11, 2019).
138. Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. (2018) 23:801–11. doi: 10.1038/mp.2017.255
139. Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits diversity, development and disease. Curr Opin Neurobiol. (2001) 11:327–35. doi: 10.1016/S0959-4388(00)00215-4
140. Salussolia CL, Prodromou ML, Borker P, Wolmuth LP. Arrangement of subunits in functional NMDA receptors. J Neurosci. (2011) 31:11295–304. doi: 10.1523/JNEUROSCI.5612-10.2011
141. Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. (2005) 438:185–92. doi: 10.1038/nature04089
142. Gerhard DM, Wohleb ES, Duman RS. Emerging treatment mechanisms for depression: focus on glutamate and synaptic plasticity. Drug Discov Today. (2016) 21:454–64. doi: 10.1016/j.drudis.2016.01.016
143. Gerhard DM, Pothula S, Liu RJ, Wu M, Li XY, Girgenti MJ, et al. GABA interneurons are the cellular trigger for ketamine's rapid antidepressant actions. J Clin Invest. (2020) 130:1336–49. doi: 10.1172/JCI130808
144. Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry. (1997) 154:805–11. doi: 10.1176/ajp.154.6.805
145. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. (2007) 27:11496–500. doi: 10.1523/JNEUROSCI.2213-07.2007
146. Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res. (2011) 224:107–11. doi: 10.1016/j.bbr.2011.05.035
147. Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. (2010) 329:959–64. doi: 10.1126/science.1190287
148. Aleksandrova LR, Phillips AG. Neuroplasticity as a convergent mechanism of ketamine and classical psychedelics. Trends Pharmacol Sci. (2021) 42:929–42. doi: 10.1016/j.tips.2021.08.003
149. Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. (2011) 475:91–5. doi: 10.1038/nature10130
150. Kavalali ET, Monteggia LM. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am J Psychiatry. (2012) 169:1150–6. doi: 10.1176/appi.ajp.2012.12040531
151. Nosyreva E, Szabla K, Autry AE, Ryazanov AG, Monteggia LM, Kavalali ET. Acute suppression of spontaneous neurotransmission drives synaptic potentiation. J Neurosci. (2013) 33:6990–7002. doi: 10.1523/JNEUROSCI.4998-12.2013
152. Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. (2016) 533:481–6. doi: 10.1038/nature17998
153. Moaddel R, Abdrakhmanova G, Kozak J, Jozwiak K, Toll L, Jimenez L, et al. Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in α7 nicotinic acetylcholine receptors. Euro J Pharmacol. (2013) 698:228–34. doi: 10.1016/j.ejphar.2012.11.023
154. Maeng S, Zarate CA. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. (2007) 9:467–74. doi: 10.1007/s11920-007-0063-1
155. Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, Ma M, et al. R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry. (2015) 5:e632. doi: 10.1038/tp.2015.136
156. Sarkar A, Kabbaj M. Sex differences in effects of ketamine on behavior, spine density, and synaptic proteins in socially isolated Rats. Biol Psychiatry. (2016) 80:448–56. doi: 10.1016/j.biopsych.2015.12.025
157. Franceschelli A, Sens J, Herchick S, Thelen C, Pitychoutis PM. Sex differences in the rapid and the sustained antidepressant-like effects of ketamine in stress-naïve and “depressed” mice exposed to chronic mild stress. Neuroscience. (2015) 290:49–60. doi: 10.1016/j.neuroscience.2015.01.008
158. Carrier N, Kabbaj M. Sex differences in the antidepressant-like effects of ketamine. Neuropharmacology. (2013) 70:27–34. doi: 10.1016/j.neuropharm.2012.12.009
159. Saland SK, Schoepfer KJ, Kabbaj M. Hedonic sensitivity to low-dose ketamine is modulated by gonadal hormones in a sex-dependent manner. Sci Rep. (2016) 6:21322. doi: 10.1038/srep21322
160. Zarate CA, Brutsche N, Laje G, Luckenbaugh DA, Venkata SLV, Ramamoorthy A, et al. Relationship of ketamine's plasma metabolites with response, diagnosis, and side effects in major depression. Biol Psychiatry. (2012) 72:331–8. doi: 10.1016/j.biopsych.2012.03.004
161. Romeo B, Choucha W, Fossati P, Rotge JY. Meta-analysis of short- and mid- term efficacy of ketamine in unipolar and bipolar depression. Psychiatry Res. (2015) 230:682–8. doi: 10.1016/j.psychres.2015.10.032
162. Coyle CM, Laws KR. The use of ketamine as an antidepressant: a systematic review and meta-analysis. Hum Psychopharmacol Clin Exp. (2015) 30:152–63. doi: 10.1002/hup.2475
163. Sigtermans M, Dahan A, Mooren R, Bauer M, Kest B, Sarton E, et al. S(+)-ketamine effect on experimental pain and cardiac output: a population pharmacokinetic-pharmacodynamic modeling study in healthy volunteers. Anesthesiology. (2009) 111:892–903. doi: 10.1097/ALN.0b013e3181b437b1
164. Correia-Melo FS, Leal GC, Vieira F, Jesus-Nunes AP, Mello RP, Magnavita G, et al. Efficacy and safety of adjunctive therapy using esketamine or racemic ketamine for adult treatment-resistant depression: a randomized, double-blind, non-inferiority study. J Affect Disord. (2020) 264:527–34. doi: 10.1016/j.jad.2019.11.086
165. Bahji A, Vazquez GH, Zarate CA Jr. Comparative efficacy of racemic ketamine and esketamine for depression: a systematic review and meta-analysis. J Affect Disord. (2021) 278:542–55. doi: 10.1016/j.jad.2020.09.071
166. Miller AH. Conceptual confluence: the kynurenine pathway as a common target for ketamine and the convergence of the inflammation and glutamate hypotheses of depression. Neuropsychopharmacology. (2013) 38:1607–8. doi: 10.1038/npp.2013.140
167. Bahrami Z, Firouzi M, Hashemi-Monfared A, Zahednasab H, Harirchian MH. The effect of minocycline on indolamine 2, 3 dioxygenase expression and the levels of kynurenic acid and quinolinic acid in LPS-activated primary rat microglia. Cytokine. (2018) 107:125–9. doi: 10.1016/j.cyto.2017.12.013
168. Garrison AM, Parrott JM, Tuñon A, Delgado J, Redus L, O'Connor JC. Kynurenine pathway metabolic balance influences microglia activity: targeting kynurenine monooxygenase to dampen neuroinflammation. Psychoneuroendocrinology. (2018) 94:1–10. doi: 10.1016/j.psyneuen.2018.04.019
169. Rodrigues FTS, de Souza MRM, Lima CN, de C, da Silva FER, Costa DVS, et al. Major depression model induced by repeated and intermittent lipopolysaccharide administration: long-lasting behavioral, neuroimmune and neuroprogressive alterations. J Psychiatr Res. (2018) 107:57–67. doi: 10.1016/j.jpsychires.2018.10.003
170. Erhardt S, Lim CK, Linderholm KR, Janelidze S, Lindqvist D, Samuelsson M, et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology. (2013) 38:743–52. doi: 10.1038/npp.2012.248
171. Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein H.-G., et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation. (2011) 8:94. doi: 10.1186/1742-2094-8-94
172. Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology. (2013) 38:1609–16. doi: 10.1038/npp.2013.71
173. Verdonk F, Petit A.-C., Abdel-Ahad P, Vinckier F, Jouvion G, et al. Microglial production of quinolinic acid as a target and a biomarker of the antidepressant effect of ketamine. Brain Behav Immun. (2019) 81:361–73. doi: 10.1016/j.bbi.2019.06.033
174. Chang Y, Lee JJ, Hsieh CY, Hsiao G, Chou DS, Sheu JR. Inhibitory effects of ketamine on lipopolysaccharide-induced microglial activation. Mediators Inflamm. (2009) 2009:705379. doi: 10.1155/2009/705379
175. Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Loron G, et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol. (2012) 72:536–49. doi: 10.1002/ana.23626
176. Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N- methyl-D-aspartate receptors. Brain Behav Immun. (1995) 9:355–65. doi: 10.1006/brbi.1995.1033
177. Chen Q, Feng J, Liu L, Wang X, Wan Y, Li M, et al. The effect of ketamine on microglia and proinflammatory cytokines in the hippocampus of depression- like rat. Neuropsychiatry. (2017) 7:77–85. doi: 10.4172/Neuropsychiatry.1000183
178. Cowley SM, Hoare S, Mosselman S, Parker MG. Estrogen receptors alpha and beta form heterodimers on DNA. J Biol Chem. (1997) 272:19858–62. doi: 10.1074/jbc.272.32.19858
179. Pace P, Taylor J, Suntharalingam S, Coombes RC, Ali S. Human estrogen receptor beta binds DNA in a manner similar to and dimerizes with estrogen receptor alpha. J Biol Chem. (1997) 272:25832–8. doi: 10.1074/jbc.272.41.25832
180. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell. (1995) 83:835. doi: 10.1016/0092-8674(95)90199-X
181. Maggiolini M, Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J Endocrinol. (2010) 204:105–114. doi: 10.1677/JOE-09-0242
182. Fillit H, Weinreb H, Cholst I, Luine V, McEwen B, Amador R, et al. Observations in a preliminary open trial of estradiol therapy for senile dementia- Alzheimer's type. Psychoneuroendocrinology. (1986) 11:337–45. doi: 10.1016/0306-4530(86)90019-3
183. Henderson VW, Paganini-Hill A, Emanuel CK, Dunn ME, Buckwalter JG. Estrogen replacement therapy in older women: comparisons between Alzheimer's disease cases and nondemented control subjects. Arch Neurol. (1994) 51:896–900. doi: 10.1001/archneur.1994.00540210068014
184. Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, et al. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore longitudinal study of aging. Neurology. (1997) 48:1517–21. doi: 10.1212/WNL.48.6.1517
185. Paganini-Hill A, Henderson VW. Estrogen deficiency and risk of Alzheimer's disease in women. Am J Epidemiol. (1994) 140:256–61. doi: 10.1093/oxfordjournals.aje.a117244
186. Tang MX, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, et al. Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet. (1996) 348:429–32. doi: 10.1016/S0140-6736(96)03356-9
187. Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. New Engl J Med. (1998) 339:285–91. doi: 10.1056/NEJM199807303390501
188. Gold SM, Voskuhl RR. Estrogen treatment in multiple sclerosis. J Neurol Sci. (2009) 286:99–103. doi: 10.1016/j.jns.2009.05.028
189. Laffont S, Garnier L, Lélu K, Guery JC. Estrogen-mediated protection of experimental autoimmune encephalomyelitis: lessons from the dissection of estrogen receptor-signaling in vivo. Biomed J. (2015) 38:194–205. doi: 10.4103/2319-4170.158509
190. Voskuhl RR, Wang H, Wu TCJ, Sicotte NL, Nakamura K, Kurth F, et al. Estriol combined with glatiramer acetate for women with relapsing- remitting multiple sclerosis: a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. (2016) 15:35–46. doi: 10.1016/S1474-4422(15)00322-1
191. Vegeto E, Belcredito S, Etteri S, Ghisletti S, Brusadelli A, Meda C, et al. Estrogen receptor-α mediates the brain antiinflammatory activity of estradiol. Proc Natl Acad Sci USA. (2003) 100:9614–9. doi: 10.1073/pnas.1531957100
192. Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP. Antiinflammatory effects of estrogen on microglial activation. Endocrinology. (2000) 141:3646–56. doi: 10.1210/endo.141.10.7693
193. Vegeto E, Pollio G, Ciana P, Maggi A. Estrogen blocks inducible nitric oxide synthase accumulation in LPS-activated microglia cells. Exp Gerontol. (2000) 35:1309–16. doi: 10.1016/S0531-5565(00)00161-3
194. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. (2005) 307:1625–30. doi: 10.1126/science.1106943
195. Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. (2004) 15:177–82. doi: 10.1016/j.semcdb.2004.01.002
196. Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. (1998) 273:19929–32. doi: 10.1074/jbc.273.32.19929
197. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. (2005) 9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x
198. Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem. (2008) 283:1882–92. doi: 10.1074/jbc.M709585200
199. Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. (2001) 114:2903–10. doi: 10.1242/jcs.114.16.2903
200. Zhao TZ, Ding Q, Hu J, He SM, Shi F, Ma LT. GPER expressed on microglia mediates the anti-inflammatory effect of estradiol in ischemic stroke. Brain Behav. (2016) 6:e00449. doi: 10.1002/brb3.449
201. Ellmann S, Sticht H, Thiel F, Beckmann MW, Strick R, Strissel PL. Estrogen and progesterone receptors: from molecular structures to clinical targets. Cell Mol Life Sci. (2009) 66:2405–26. doi: 10.1007/s00018-009-0017-3
202. Tameh AA, Karimian M, Zare-Dehghanani Z, Aftabi Y, Beyer C. Role of steroid therapy after ischemic stroke by n-methyl-d-aspartate receptor gene regulation. J Stroke Cerebrovasc Dis. (2018) 27:3066–75. doi: 10.1016/j.jstrokecerebrovasdis.2018.06.041
203. Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. (2007) 27:2846–57. doi: 10.1523/JNEUROSCI.0116-07.2007
204. Nakanishi N, Tu S, Shin Y, Cui J, Kurokawa T, Zhang D, et al. Neuroprotection by the NR3A subunit of the NMDA receptor. J Neurosci. (2009) 29:5260–5. doi: 10.1523/JNEUROSCI.1067-09.2009
205. Bali N, Morgan TE, Finch CE. Pgrmc1: new roles in the microglial mediation of progesterone-antagonism of estradiol-dependent neurite sprouting and in microglial activation. Front Neurosci. (2013) 7:157. doi: 10.3389/fnins.2013.00157
206. Piel RB, Shiferaw MT, Vashisht AA, Marcero J, Praissman J, Phillips JD, et al. A novel role for progesterone receptor membrane component 1 (PGRMC1): a partner and regulator of ferrochelatase. Biochemistry. (2016) 55:5204–17. doi: 10.1021/acs.biochem.6b00756
207. Gellersen B, Fernandes MS, Brosens JJ. Non-genomic progesterone actions in female reproduction. Hum Reprod Update. (2009) 15:119–38. doi: 10.1093/humupd/dmn044
208. Theis V, Theiss C. Progesterone effects in the nervous system. Anat Rec. (2019) 302:1276–86. doi: 10.1002/ar.24121
209. Thomas P. Characteristics of membrane progestin receptor alpha (mPRα) and progesterone membrane receptor component one (PGMRC1) and their roles in mediating rapid progestin actions. Front Neuroendocrinol. (2008) 29:292–312. doi: 10.1016/j.yfrne.2008.01.001
210. Sakamoto H, Ukena K, Takemori H, Okamoto M, Kawata M, Tsutsui K. Expression and localization of 25-Dx, a membrane-associated putative progesterone- binding protein, in the developing purkinje cell. Neuroscience. (2004) 126:325–34. doi: 10.1016/j.neuroscience.2004.04.003
211. Xu J, Zeng C, Chu W, Pan F, Rothfuss JM, Zhang F, et al. Identification of the PGRMC1 protein complex as the putative sigma-2 receptor binding site. Nat Commun. (2011) 2:380. doi: 10.1038/ncomms1386
212. Bali N, Arimoto JM, Iwata N, Lin SW, Zhao L, Brinton RD, et al. Differential responses of progesterone receptor membrane component-1 (Pgrmc1) and the classical progesterone receptor (Pgr) to 17β-estradiol and progesterone in hippocampal subregions that support synaptic remodeling and neurogenesis. Endocrinology. (2012) 153:759–69. doi: 10.1210/en.2011-1699
213. Labombarda F, Gonzalez SL, Gonzalez Deniselle MC, Vinson GP, Schumacher M, De Nicola AF, et al. Effects of injury and progesterone treatment on progesterone receptor and progesterone binding protein 25-Dx expression in the rat spinal cord. J Neurochem. (2003) 87:902–13. doi: 10.1046/j.1471-4159.2003.02055.x
214. Wessel L, Balakrishnan-Renuka A, Henkel C, Meyer HE, Meller K, Brand-Saberi B, et al. Long-term incubation with mifepristone (MLTI) increases the spine density in developing purkinje cells: new insights into progesterone receptor mechanisms. Cell Mol Life Sci. (2014) 71:1723–40. doi: 10.1007/s00018-013-1448-4
215. Castelnovo LF, Magnaghi V, Thomas P. Expression of membrane progesterone receptors (mPRs) in rat peripheral glial cell membranes and their potential role in the modulation of cell migration and protein expression. Steroids. (2019) 142:6–13. doi: 10.1016/j.steroids.2017.09.009
216. Jodhka PK, Kaur P, Underwood W, Lydon JP, Singh M. The differences in neuroprotective efficacy of progesterone and medroxyprogesterone acetate correlate with their effects on brain-derived neurotrophic factor expression. Endocrinology. (2009) 150:3162–8. doi: 10.1210/en.2008-1247
217. Su C, Cunningham RL, Rybalchenko N, Singh M. Progesterone increases the release of brain-derived neurotrophic factor from glia via progesterone receptor membrane component 1 (Pgrmc1)-dependent ERK5 signaling. Endocrinology. (2012) 153:4389–400. doi: 10.1210/en.2011-2177
218. Woolley CS, McEwen BS. Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. J Comp Neurol. (1993) 336:293–306. doi: 10.1002/cne.903360210
219. Wong AM, Rozovsky I, Arimoto JM, Du Y, Wei M, Morgan TE, et al. Progesterone influence on neurite outgrowth involves microglia. Endocrinology. (2009) 150:324–32. doi: 10.1210/en.2008-0988
220. Bali N, Arimoto JM, Morgan TE, Finch CE. Progesterone antagonism of neurite outgrowth depends on microglial activation via Pgrmc1/S2R. Endocrinology. (2013) 154:2468–80. doi: 10.1210/en.2012-2109
221. Cervantes M, González-Vidal MD, Ruelas R, Escobar A, Moralí G. Neuroprotective effects of progesterone on damage elicited by acute global cerebral ischemia in neurons of the caudate nucleus. Arch Med Res. (2002) 33:6–14. doi: 10.1016/S0188-4409(01)00347-2
222. Chen J, Chopp M, Li Y. Neuroprotective effects of progesterone after transient middle cerebral artery occlusion in rat. J Neurol Sci. (1999) 171:24–30. doi: 10.1016/S0022-510X(99)00247-6
223. Dang J, Mitkari B, Kipp M, Beyer C. Gonadal steroids prevent cell damage and stimulate behavioral recovery after transient middle cerebral artery occlusion in male and female rats. Brain Behav Immun. (2011) 25:715–26. doi: 10.1016/j.bbi.2011.01.013
224. Gibson CL, Constantin D, Prior MJW, Bath PMW, Murphy SP. Progesterone suppresses the inflammatory response and nitric oxide synthase-2 expression following cerebral ischemia. Exp Neurol. (2005) 193:522–30. doi: 10.1016/j.expneurol.2005.01.009
225. Gibson CL, Coomber B, Murphy SP. Progesterone is neuroprotective following cerebral ischaemia in reproductively ageing female mice. Brain. (2011) 134 (Pt. 7):2125–33. doi: 10.1093/brain/awr132
226. González-Vidal MD, Cervera-Gaviria M, Ruelas R, Escobar A, Moralí G, Cervantes M. Progesterone: protective effects on the cat hippocampal neuronal damage due to acute global cerebral ischemia. Arch Med Res. (1998) 29:117–24.
227. Ishrat T, Sayeed I, Atif F, Stein DG. Effects of progesterone administration on infarct volume and functional deficits following permanent focal cerebral ischemia in rats. Brain Res. (2009) 1257:94–101. doi: 10.1016/j.brainres.2008.12.048
228. Kumon Y, Kim SC, Tompkins P, Stevens A, Sakaki S, Loftus CM. Neuroprotective effect of postischemic administration of progesterone in spontaneously hypertensive rats with focal cerebral ischemia. J Neurosurg. (2000) 92:848–52. doi: 10.3171/jns.2000.92.5.0848
229. Moralí G, Letechipía-Vallejo G, López-Loeza E, Montes P, Hernández-Morales L, Cervantes M. Post-ischemic administration of progesterone in rats exerts neuroprotective effects on the hippocampus. Neurosci Lett. (2005) 382:286–90. doi: 10.1016/j.neulet.2005.03.066
230. Lei B, Mace B, Dawson HN, Warner DS, Laskowitz DT, James ML. Anti-inflammatory effects of progesterone in lipopolysaccharide-stimulated BV-2 microglia. PLoS ONE. (2014) 9:e103969. doi: 10.1371/journal.pone.0103969
231. Oeckinghaus A, Ghosh S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. (2009) 1:a000034. doi: 10.1101/cshperspect.a000034
232. Ho MF, Correia C, Ingle JN, Kaddurah-Daouk R, Wang L, Kaufmann SH, et al. Ketamine and ketamine metabolites as novel estrogen receptor ligands: induction of cytochrome P450 and AMPA glutamate receptor gene expression. Biochem Pharmacol. (2018) 152:279–92. doi: 10.1016/j.bcp.2018.03.032
233. Choi SY, Koh KH, Jeong H. Isoform-specific regulation of cytochromes P450 expression by estradiol and progesterone. Drug Metab Dispos. (2013) 41:263–9. doi: 10.1124/dmd.112.046276
234. Higashi E, Fukami T, Itoh M, Kyo S, Inoue M, Yokoi T, et al. Human CYP2A6 is induced by estrogen via estrogen receptor. Drug Metab Dispos. (2007) 35:1935–41. doi: 10.1124/dmd.107.016568
235. Graham JD, Clarke CL. Physiological action of progesterone in target tissues. Endocr Rev. (1997) 18:502–19. doi: 10.1210/edrv.18.4.0308
236. Alves SE, McEwen BS, Hayashi S, Korach KS, Pfaff DW, Ogawa S. Estrogen-regulated progestin receptors are found in the midbrain raphe but not hippocampus of estrogen receptor alpha (ER alpha) gene-disrupted mice. J Comp Neurol. (2000) 427:185–95. doi: 10.1002/1096-9861(20001113)427:2<185::AID-CNE2>3.0.CO;2-G
237. Vasudevan N, Pfaff DW. Non-genomic actions of estrogens and their interaction with genomic actions in the brain. Front Neuroendocrinol. (2008) 29:238–57. doi: 10.1016/j.yfrne.2007.08.003
238. Videbech P, Ravnkilde B. Hippocampal Volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. (2004) 161:1957–66. doi: 10.1176/appi.ajp.161.11.1957
239. Kornack DR, Rakic P. Continuation of neurogenesis in the hippocampus of the adult macaque monkey. Proc Natl Acad Sci USA. (1999) 96:5768–73. doi: 10.1073/pnas.96.10.5768
240. Pruessner JC, Collins DL, Pruessner M, Evans AC. Age and gender predict volume decline in the anterior and posterior hippocampus in early adulthood. J Neurosci. (2001) 21:194–200. doi: 10.1523/JNEUROSCI.21-01-00194.2001
241. Liu D, Diorio J, Day JC, Francis DD, Meaney MJ. Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci. (2000) 3:799–806. doi: 10.1038/77702
242. Vaynman S, Ying Z, Gomez-Pinilla F. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur J Neurosci. (2004) 20:2580–90. doi: 10.1111/j.1460-9568.2004.03720.x
243. Garcia LSB, Comim CM, Valvassori SS, Réus GZ, Barbosa LM, Andreazza AC, et al. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. (2008) 32:140–4. doi: 10.1016/j.pnpbp.2007.07.027
244. Réus GZ, Stringari RB, Ribeiro KF, Ferraro AK, Vitto MF, Cesconetto P, et al. Ketamine plus imipramine treatment induces antidepressant- like behavior and increases CREB and BDNF protein levels and PKA and PKC phosphorylation in rat brain. Behav Brain Res. (2011) 221:166–71. doi: 10.1016/j.bbr.2011.02.024
245. Yang C, Hu YM, Zhou ZQ, Zhang GF, Yang JJ. Acute administration of ketamine in rats increases hippocampal BDNF and mTOR levels during forced swimming test. Ups J Med Sci. (2013) 118:3–8. doi: 10.3109/03009734.2012.724118
246. Choi M, Lee SH, Park MH, Kim YS, Son H. Ketamine induces brain-derived neurotrophic factor expression via phosphorylation of histone deacetylase 5 in rats. Biochem Biophys Res Commun. (2017) 489:420–5. doi: 10.1016/j.bbrc.2017.05.157
247. Sato K, Akaishi T, Matsuki N, Ohno Y, Nakazawa K. β-Estradiol induces synaptogenesis in the hippocampus by enhancing brain-derived neurotrophic factor release from dentate gyrus granule cells. Brain Res. (2007) 1150:108–20. doi: 10.1016/j.brainres.2007.02.093
248. Scharfman HE, MacLusky NJ. Estrogen and brain-derived neurotrophic factor (BDNF) in hippocampus: complexity of steroid hormone-growth factor interactions in the adult CNS. Front Neuroendocrinol. (2006) 27:415–35. doi: 10.1016/j.yfrne.2006.09.004
249. Sohrabji F, Lewis DK. Estrogen-BDNF interactions: implications for neurodegenerative diseases. Front Neuroendocrinol. (2006) 27:404–14. doi: 10.1016/j.yfrne.2006.09.003
250. Miranda RC, Sohrabji F, Toran-Allerand CD. Neuronal colocalization of mRNAs for neurotrophins and their receptors in the developing central nervous system suggests a potential for autocrine interactions. Proc Natl Acad Sci USA. (1993) 90:6439–43. doi: 10.1073/pnas.90.14.6439
251. Gould E, Woolley CS, Frankfurt M, McEwen BS. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci. (1990) 10:1286–91. doi: 10.1523/JNEUROSCI.10-04-01286.1990
252. Hudson SP, Jacobson-Pick S, Anisman H. Sex differences in behavior and pro- inflammatory cytokine mRNA expression following stressor exposure and re- exposure. Neuroscience. (2014) 277:239–49. doi: 10.1016/j.neuroscience.2014.07.007
253. Nelson LH, Warden S, Lenz KM. Sex differences in microglial phagocytosis in the neonatal hippocampus. Brain Behav Immun. (2017) 64:11–22. doi: 10.1016/j.bbi.2017.03.010
254. Bollinger JL, Burns CMB, Wellman CL. Differential effects of stress on microglial cell activation in male and female medial prefrontal cortex. Brain Behav Immun. (2016) 52:88–97. doi: 10.1016/j.bbi.2015.10.003
255. Peters AJ, Villasana LE, Schnell E. Ketamine alters hippocampal cell proliferation and improves learning in mice after traumatic brain injury. Anesthesiology. (2018) 129:278–95. doi: 10.1097/ALN.0000000000002197
Keywords: microglia, glutamate, neuroinflamation, sex differences, estradiol (17ß-estradiol)
Citation: Gagne C, Piot A and Brake WG (2022) Depression, Estrogens, and Neuroinflammation: A Preclinical Review of Ketamine Treatment for Mood Disorders in Women. Front. Psychiatry 12:797577. doi: 10.3389/fpsyt.2021.797577
Received: 18 October 2021; Accepted: 24 December 2021;
Published: 18 January 2022.
Edited by:
Ming D. Li, Zhejiang University, ChinaReviewed by:
Yan Li, Apellis Pharmaceuticals, United StatesBernadeta Szewczyk, Maj Institute of Pharmacology, Polish Academy of Sciences (IF PAS), Poland
Copyright © 2022 Gagne, Piot and Brake. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wayne G. Brake, d2F5bmUuYnJha2VAY29uY29yZGlhLmNh